 Rahul Pandey
Rahul Pandey Marina Bakay
Marina Bakay Hakon Hakonarson
Hakon Hakonarson- 1Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA, United States
- 2Department of Pediatrics, The University of Pennsylvania School of Medicine, Philadelphia, PA, United States
Autoimmune diseases arise from atypical immune responses that attack self-tissue epitopes, and their development is intricately connected to the disruption of the JAK-STAT signaling pathway, where SOCS proteins play crucial roles. Conditions such as autoimmune uveitis, psoriasis, lupus, and autoimmune encephalitis exhibit immune system dysfunctions associated with JAK-STAT signaling dysregulation. Emerging therapeutic strategies utilize JAK-STAT inhibitors and SOCS mimetics to modulate immune responses and alleviate autoimmune manifestations. Although more research and clinical studies are required to assess their effectiveness, safety profiles, and potential for personalized therapeutic approaches in autoimmune conditions, JAK-STAT inhibitors and SOCS mimetics show promise as potential treatment options. This review explores the action, effectiveness, safety profiles, and future prospects of JAK inhibitors and SOCS mimetics as therapeutic agents for psoriasis, autoimmune uveitis, systemic lupus erythematosus, and autoimmune encephalitis. The findings underscore the importance of investigating these targeted therapies to advance treatment options for individuals suffering from autoimmune diseases.
Introduction
Genetic predisposition, environmental triggers, and dysregulation of the immune system play significant roles in the origin of autoimmune diseases. Additionally, molecular mimicry, epigenetic modifications, hormonal influences, and gut microbiota composition are also relevant factors in the development of these diseases (1, 2). The incidence of these conditions is increasing, affecting around 3% to 5% of people in Western countries (3). Dysregulated cytokine holds a pivotal position in their pathogenesis, making it an attractive target for treatment (4, 5). Cytokines are diverse proteins that mediate cell signaling within the immune system and other host cells, regulating immune responses and inflammation. Cytokines are grouped into various families (6–15), each serving unique functions by binding to specific receptors on target cells and influencing the behavior and function of the immune system. While many are named as interleukins (IL) with numerical identifiers (e.g., IL-2), some, such as TNF-α, IFN-γ, prolactin, and erythropoietin, do not adhere to this naming convention. As integral components of the innate immune system, interferons provide an early defense against infections and contribute to the overall regulation of the immune response (16, 17).
Precise control of cytokine signaling is essential to maintain immune system homeostasis. Monoclonal antibodies targeting specific pathogenic cytokines have transformed autoimmune disease therapy. Nevertheless, there is a necessity for novel therapeutic approaches to tackle relapses and uncontrollable symptoms in affected individuals. Janus Kinase (JAK) inhibitors offer potential as they effectively target crucial cytokines involved in autoimmune and inflammatory diseases (4). This review presents an in-depth examination of the use of JAK inhibitors and SOCS mimetics in treating autoimmune uveitis, psoriasis, systemic lupus erythematosus, and autoimmune encephalitis.
Janus kinases and signal transducers and activators of transcription
The JAK-STAT pathway is a vital signaling cascade that regulates diverse biological processes, including immune responses, cell growth, and differentiation. It is named after its key components, Janus Kinases (JAKs), which were discovered 30 years ago. The term “Janus” originates from Roman mythology, symbolizing transitions, and duality. JAKs possess two domains: a kinase domain responsible for phosphorylation and a pseudokinase domain acting as a negative regulator, giving them the name “Janus Kinases.” The term “Signal Transducers and Activators of Transcription” describes the primary function of these proteins in the JAK-STAT pathway. Once JAKs phosphorylate STATs, the activated STATs act as signal transducers by relaying the extracellular signal from the cell surface receptors to the cell nucleus. Once in the nucleus, STATs function as transcription factors, activating the transcription (gene expression) of specific target genes (Figure 1).
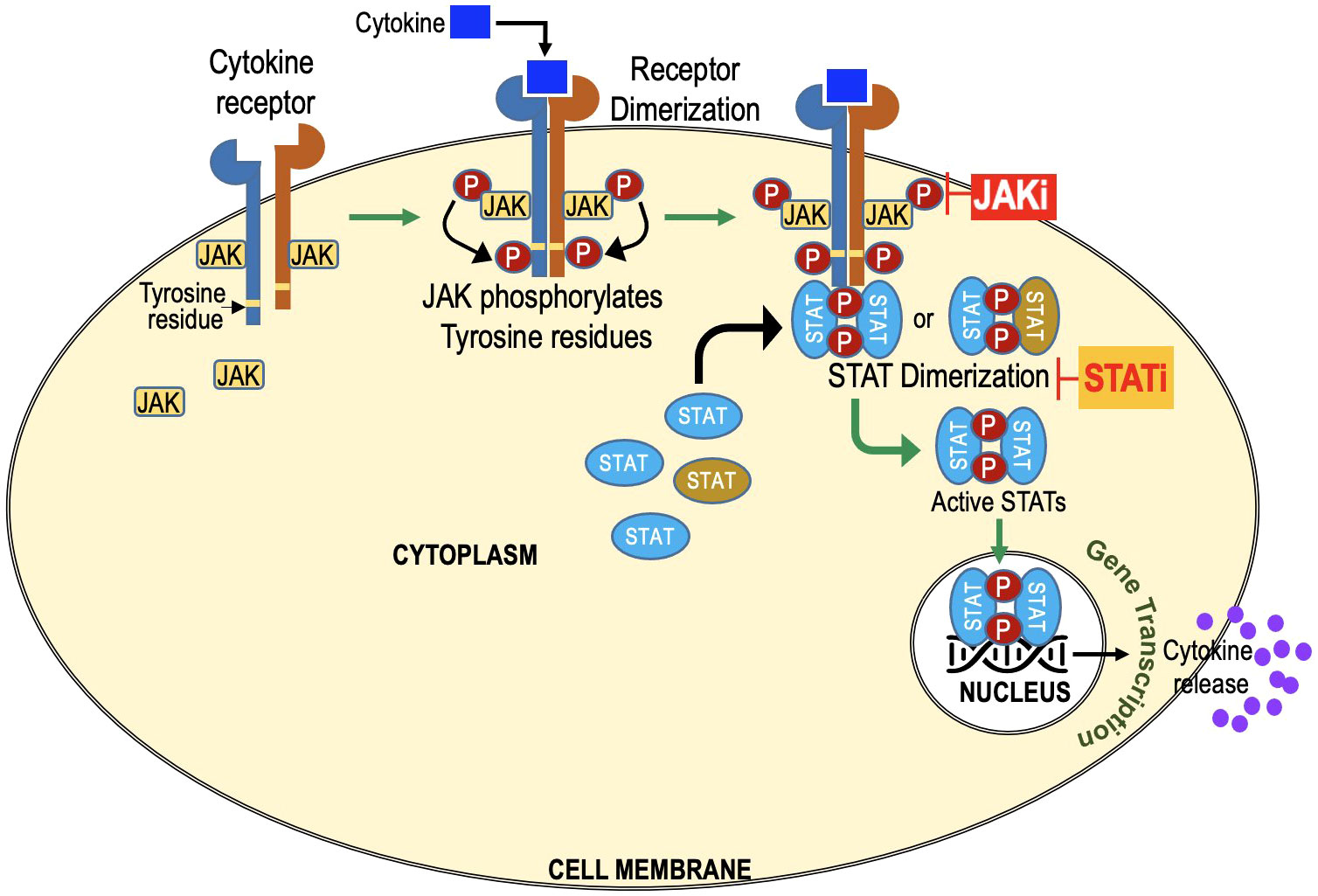
Figure 1 JAK-STAT Signaling Cascade: Key Players in Cellular Regulation and Immune Responses. Extracellular molecules, such as cytokines or growth factors, bind to cell surface receptors, activating Janus Kinases (JAKs). Activated JAKs phosphorylate Signal Transducers and Activators of Transcription (STATs) at specific tyrosine residues, forming homo- or heterodimers in the cytoplasm. STAT dimers then translocate to the cell nucleus and act as transcription factors, regulating gene expression. JAK inhibitors target JAKs. STAT inhibitors target STATs and prevent dimerization and its translocation to nucleus.
The JAK family in humans consists of four members: JAK1, JAK2, JAK3, and TYK2. These four JAK proteins are utilized by over 50 cytokines, leading to substantial overlap in their usage (Figure 2). The STAT family in humans consists of seven members - STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Additionally, some cytokines and chemokines signal through mechanisms independent of JAK-STAT (18). The JAKs and STATs are differentially expressed in various cell types, and their activation can lead to distinct downstream effects (4, 19–24). Selective activation of this pathway enables precise adjustment of cellular reactions to diverse triggers. Nevertheless, in specific cellular scenarios (as illustrated in Figure 2), the JAK-STAT pathway exhibits redundancy. This redundancy guarantees the preservation of vital functions, even when one JAK-STAT axis encounters disruption or inhibition. Such redundancy empowers cells to react to numerous cytokines, ensuring a resilient and flexible immune response.
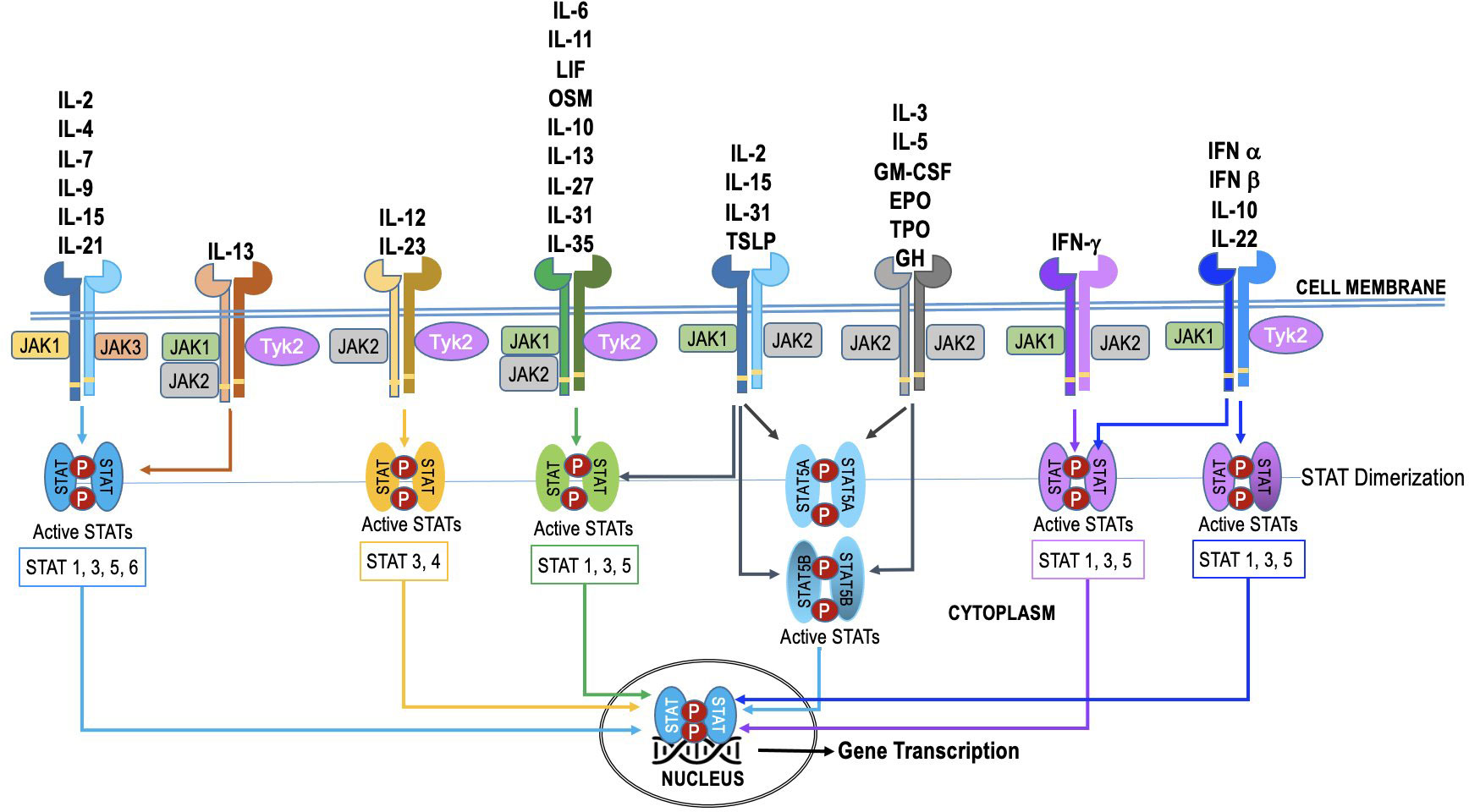
Figure 2 The specificity and redundancy of the JAK-STAT pathway. Different JAK family members are linked to specific cytokine receptors, and they trigger distinct STAT proteins within the pathway. This selective activation allows for fine-tuning of cellular responses to different stimuli. However, the JAK-STAT pathway also demonstrates redundancy, especially in certain cellular contexts. This redundancy ensures that essential functions are maintained even if one JAK-STAT axis is compromised or inhibited. For instance, various cytokines can activate both JAK1 and JAK2, leading to phosphorylation of STAT1 and STAT3, respectively. This redundancy allows cells to respond to multiple cytokines and ensures a robust and adaptable immune response.
The effective functioning of the immune system depends heavily on the JAK-STAT signaling pathway, and disruptions in cytokine-JAK-STAT signaling can result in immunodeficiency (25, 26). If the JAK1 or JAK2 genes are deactivated in the germline, the fetus in non-viable (27, 28). Certain primary immunodeficiencies result from genetic mutations affecting JAK-STAT signaling components, leading to immunological abnormalities and increased susceptibility to infections (29–36). Furthermore, mutations impairing the function of STAT proteins can cause both immunodeficiency and autoimmunity (37–39). The occurrence of autoimmunity is believed to be associated with the disruption of regulatory communication between STAT proteins (40, 41). Job Syndrome (Hyper-IgE Syndrome), a rare genetic disorder caused by impaired functioning of STAT3, leads to compromised JAK-STAT signaling (42, 43). Overall, the loss of cytokine-JAK-STAT signaling interferes with critical immune functions, compromising the development, differentiation, and response of immune cells, as well as the regulation of immune mediators and inflammatory processes. Overactivation of cytokine signaling can have detrimental effects too (25, 26, 44). The overactive JAK-STAT pathway promotes production of pro-inflammatory cytokines and activation of immune cells and contributes to the development of autoimmune diseases. Similarly, in cancer, dysregulated JAK-STAT signaling can play a role in promoting uncontrolled cell growth and survival (4, 45–47).
The SOCS protein family
The Suppressor of Cytokine Signaling (SOCS) protein family is a group of proteins that play a vital role in regulating and inhibiting cytokine signaling pathways. These proteins act as negative feedback regulators, dampening excessive immune responses and maintaining immune system balance. SOCS proteins achieve this by binding to specific signaling proteins, including Janus kinases and cytokine receptors, and interfering with their activity, ultimately controlling the duration and intensity of immune and inflammatory reactions. The SOCS protein family consists of numerous members, namely SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7, and CIS (Cytokine-Inducible SH2-Containing protein), with CIS being the initial SOCS member to be discovered (48). Since their discovery in 1997 simultaneously by three groups (48–50), these have gained widespread recognition due to their prominent role in the negative modulation of signaling pathways subsequent to cytokine engagement with the receptor complex. In the late 1990s, researchers were investigating how cells modulate their responses to cytokines to prevent excessive inflammation and maintain immune system balance. In this regard, retroviral expression screen was developed to investigate how cells modulate their responses to cytokines. During this screen, an identified cDNA sequence encoded a compact protein featuring an SH2 domain, displaying resemblance to the cytokine-inducible SH2-containing (CIS) protein (48). This specific cDNA was designated as SOCS1 and subsequently played a pivotal role in the revelation and cloning of an additional six SOCS family members (SOCS2, 3, 4, 5, 6 and 7). Comparable to CIS, it was observed that SOCS1, SOCS2, and SOCS3 were responsive to cytokine stimulation (50, 51). Studies utilizing a combination of molecular biology techniques, gene expression profiling, and cell culture experiments collectively provided strong evidence that SOCS1 is not solely triggered by cytokine activation but also functions as a standard negative-feedback modulator, effectively limiting JAK signaling (52). The discovery of SOCS1 shed light on a crucial aspect of immune system regulation and paved the way for further research into the broader implications of SOCS proteins in various physiological and pathological contexts. Notably, within autoimmune disorders, multiple SOCS proteins (SOCS1, SOCS3, SOCS5, and CIS) function as potent negative regulators significantly contribute to the underlying mechanisms driving the diseases’ progression. SOCS1 helps to control inflammation, immune responses, and cell differentiation in various autoimmune diseases. SOCS3 targets diverse cytokine pathways by binding to receptors and JAKs and curbs signal transmission, moderating inflammation and immune responses in type 1 diabetes, inflammatory bowel disease (IBD), and psoriasis. CIS competes with STAT proteins for cytokine receptor binding. It fine-tunes cytokine responses and prevents uncontrolled immune reactions. SOCS4-7 extend their functions beyond cytokine signaling, with notable roles in regulating receptor tyrosine kinases that mediate hormonal effects like insulin and growth factors such as epidermal growth factor (EGF) (24, 53). SOCS5 dampens cytokine signaling by interacting with and inhibiting JAKs. While its precise role in autoimmune disorders remains unclear, emerging evidence suggests its potential involvement in autoimmune uveitis (54), type 1 diabetes (55), multiple sclerosis (56), SLE (57) and EAE (58). SOCS-6 was revealed as a suppressor of p56(lck) in yeast two-hybrid screening. By promoting ubiquitin-dependent proteolysis, SOCS-6 acts as a negative regulator of T cell activation (59). In contrast, SOCS7−/− mice exhibited varying immune-related characteristics contingent on their genetic makeup. Precise role of SOCS6 and SOCS7 in the intricate landscape of immune regulation is still emerging (60–62). Although CISH and SOCS1–3 hold evident prominence in the context of the immune system and extensively reviewed in diseases context elsewhere (44, 63), recent investigations suggest that SOCS4–7 might also play a role, underscoring the need for further exploration into these proteins. Although the exact roles they play in distinct autoimmune conditions might necessitate further scrutiny, their significance is progressively acknowledged, offering possibilities for upcoming therapeutic strategies directed at modulating these regulatory pathways.
Structure of SOCS proteins
SOCS (Suppressors of Cytokine Signaling) proteins exhibit a structured composition that encompasses several key elements essential for their regulatory functions (Figure 3). A fundamental feature found in all SOCS family members is the SOCS box, a conserved domain pivotal in protein-protein interactions, especially with components of the ubiquitin ligase complex. SOCS proteins interact with phosphorylated tyrosine residues on target substrates through their SH2 domain (64). The SH2 domains of SOCS proteins possess an additional N-terminal α-helical extension called the extended SH2 domain (ESS) (65). The SOCS controls assembly of E3 ubiquitin ligase complex and contains two motifs: the Elongin B/C (BC) box and the Cullin (Cul) box (66, 67). The N-terminal varies significantly (68). The N-terminus of SOCS4-7 are notably longer (69). Specific motifs within the N-terminal domains have been identified in related SOCS proteins. Notably, SOCS1 and SOCS3 feature a unique kinase inhibitory region (KIR) that binds and inhibits JAKs (70), whereas SOCS3 and CISH contain a PEST motif located between the SH2 domain and SOCS box (48, 65). SOCS4 and SOCS5 have distinct N-terminal conserved region (NTCR) with unknown function (69). SOCS proteins are vital in immune coordination, making them potential targets for therapeutic intervention. The evolutionary relationship between SOCS proteins is reflected in their similarity. This conservation is seen in humans and other mammalian species, which possess equivalent sets of SOCS proteins. Higher vertebrates have homologs for each SOCS protein, and teleost fish have additional duplicates. Recent discoveries have revealed intriguing differences among these proteins, emphasizing their importance in health and disease. SOCS proteins are promptly induced upon cytokine receptor signaling but are rapidly degraded when signaling subsides, remaining inactive in quiescent cells acting as negative feedback regulators. This unique structural arrangement allows SOCS proteins to exert regulation over cytokine signaling pathways, facilitating functions such as competitive binding, targeted protein degradation, and the inhibition of kinase activity. In doing so, SOCS proteins contribute to the maintenance of immune system homeostasis.
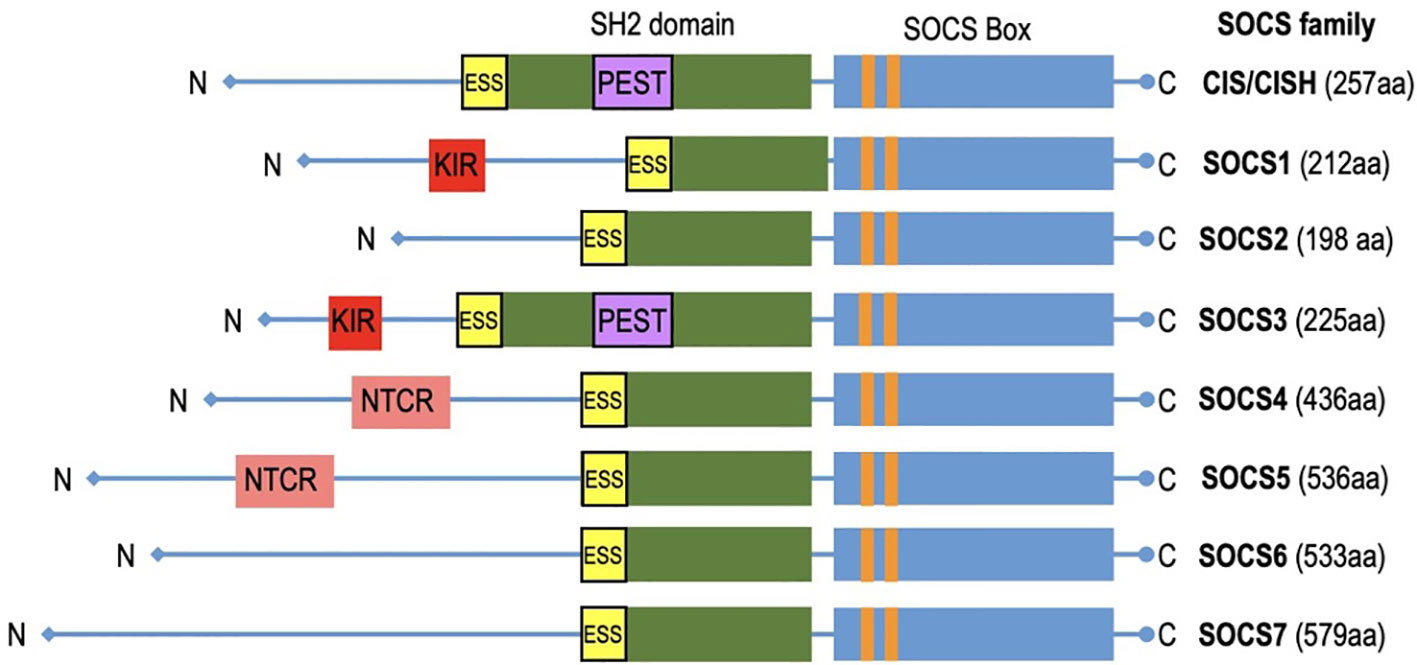
Figure 3 The diagram depicts CIS/SOCS family proteins featuring conserved SOCS box in all of them. Notably, SOCS1 and SOCS3 exhibit unique kinase inhibitory region (KIR) which acts as a pseudo substrate.
Mechanisms of SOCS action
SOCS employ diverse mechanisms, as illustrated in Figure 4, to regulate cytokine signaling, encompassing competitive binding, protein degradation or re-routing, and inhibition of kinase activity. These strategies rely on the presence of specific protein domains and motifs in varying combinations to tightly regulate cytokine signaling and maintain immune system homeostasis. The SH2 domain of SOCS proteins, reviewed elsewhere [24, 92], usually bind to the cytokine receptor signaling complex or downstream signaling proteins by interacting with appropriate motifs containing phosphotyrosine residues (24, 71). Competitive binding hinders the docking of STAT and other proteins, effectively suppressing their subsequent activation through steric interference (24). SOCS proteins degrade target proteins by interacting through their SH2 domain (72, 73) with phosphorylated JAK proteins or receptors, assembling the E3 ubiquitin ligase complex, which transfers ubiquitin to target substrates for degradation (74) and potential re-routing of associated proteins (75). SOCS1 binds to JAKs, while SOCS3 binds to receptors (76, 77), and they both directly inhibit JAK kinase activity by blocking the substrate-binding groove of the JAK kinase domain, acting as a pseudo substrate (78, 79). SOCS1 has a unique nuclear localization signal (NLS) and interacts exclusively with p65 in the nucleus. This interaction effectively curtails prolonged p65 signaling and halts the expression of NF-kB-inducible gene (80–83). SOCS proteins also regulate cytokine-responsive genes by interacting with transcription factors or chromatin modifiers (83).
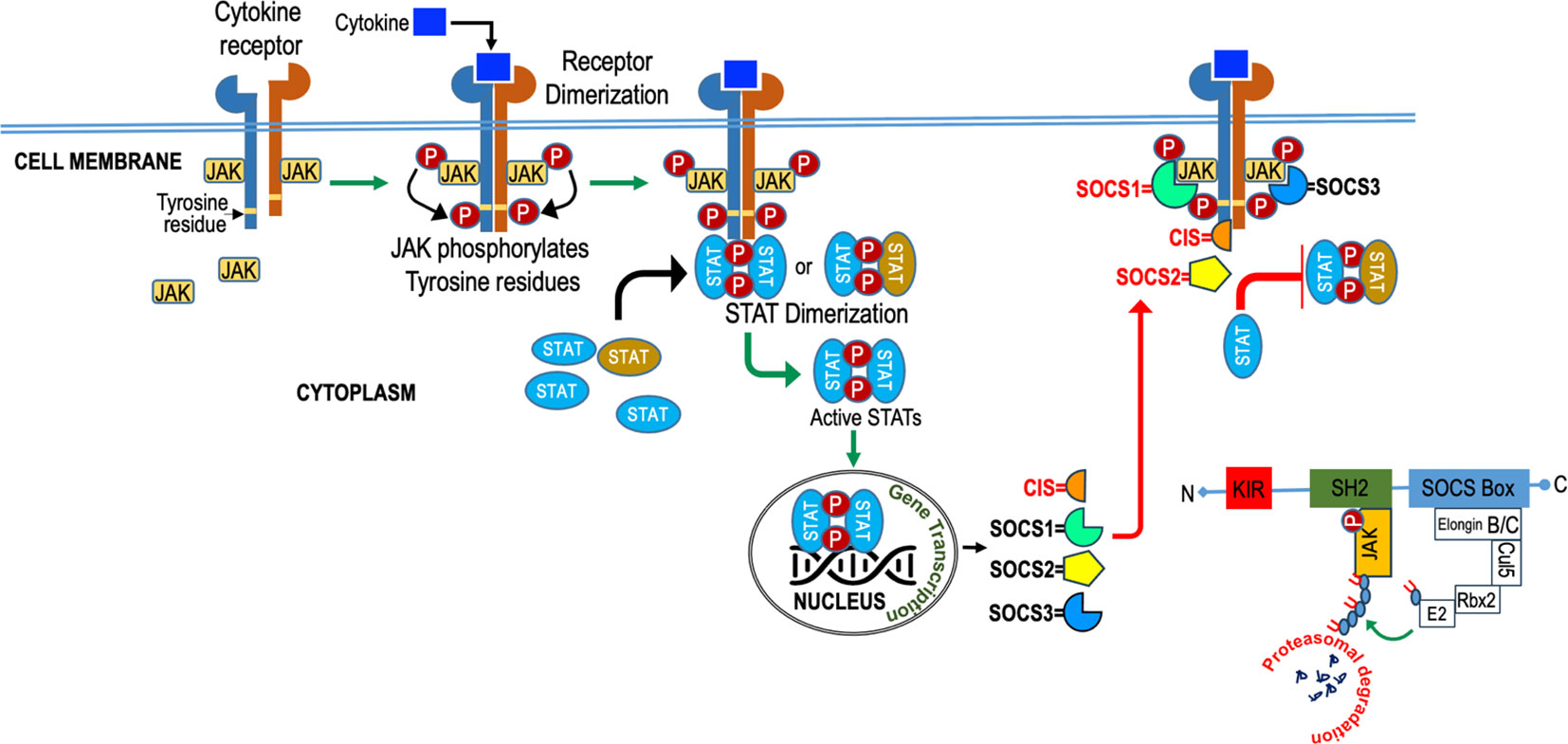
Figure 4 Diagram illustrating the role of SOCS (Suppressor of Cytokine Signaling) in cytokine signaling. SOCS molecules act as crucial regulators, inhibiting excessive cytokine responses to maintain balanced immune function.
At the post-transcriptional level microRNA-155 has been identified as a natural regulator of SOCS1 mRNA (84). miRNA-155 dysregulation has been associated with Inflammation (85–87), heart failure (85), neurodegenerative diseases (88, 89), antiviral immune response in SARS-CoV2 infection (90) and cancers (91–95). Additionally, SOCS1 is regulated post-translationally by several kinases like v-abl, pim1, and pim2. Phosphorylation by these kinases blocks the SOCS Box from binding to Elongin C, a key step in recruiting the E3 complex (96, 97). Numerous cytokines and growth factors stimulate SOCS1, implying its role in moderating and buffering the cellular and inflammatory responses initiated by these molecules suggesting that SOCS1 plays a role in downregulating/buffering the cellular/inflammatory responses triggered by these cytokines (98–102).
SOCS1 in autoimmunity
The SOCS1 gene is located on chromosome 16 alongside CLEC16A (cytogenetic location: 16p13.13; genomic coordinates [GRCh38]: 16:11,254,417-11,256,204). This region, around 530 kb on chromosome 16p13.13, harbors four genes (CIITA-DEXI-CLEC16A-SOCS1) (Figure 5).
Figure 5 Schematic outline representing the genetic region on chromosome 16p13, encompassing CIITA-DEXI-CLEC16A-SOCS1. The genomic coordinates are sourced from Genome Reference Consortium Human Build 38.
In 2007, we initially identified a region on chromosome 16p13 mapping to KIAA0350, now known as C-type lectin-like domain family 16A (CLEC16A), as a novel susceptibility locus for type 1 diabetes (T1D) (103). CLEC16A is situated between two neighboring genes: CIITA, crucial for MHC Class II expression, and SOCS1, a negative regulator of cytokine signaling and DEXI. The susceptibility sites, CIITA-DEXI-CLEC16A-SOCS1, are common across various autoimmune conditions (104–108). SOCS1 and CIITA are recognized contributors to inflammation and autoimmunity (106, 107, 109–111). SOCS1 plays vital role in immune cell homeostasis and modulating inflammation through its intricate modulation of cytokine signaling (63, 112). Variations in SOCS1’s 5’ UTR (rs243324 and rs441349) have been identified as susceptibility factors for multiple sclerosis (MS) in cytokine pathway screenings (105, 113). The CLEC16A locus, encompassing SOCS1, is now associated with 21 autoimmune diseases. These include type 1 diabetes (103, 114–126), multiple sclerosis (MS) (104, 105, 119, 125, 127–140), primary adrenal insufficiency (PAI) (141, 142), systemic lupus erythematosus (SLE) (143–146) Crohn’s disease (CD) (147), selective immunoglobulin A deficiency (IgA) (148), alopecia areata (AA) (149, 150), juvenile idiopathic arthritis (JIA) (151), rheumatoid arthritis (RA) (119, 151), primary biliary cirrhosis (PBC) (152–155), asthma (156–162), Crohn’s disease (CD) (147), allergic rhinitis (AR) (163, 164), autoimmune thyroid diseases (ATD) (115, 165), common variable immunodeficiency (CVID) (166), eosinophilic esophagitis (EE) (167), juvenile idiopathic arthritis (JIA) (151), Selective IgA deficiency (148), Celiac disease (168), systemic sclerosis (169) and even Parkinson’s Disease (PD) (170, 171) as reviewed (1).
The field of autoimmune research is constantly progressing, and over the past three years there has been a major advancement and shift to identify new therapeutic pathways for autoimmunity sufferers. 2020, study highlighted the importance of restoring immune homeostasis and tolerance, with a particular emphasis on therapies aimed at promoting, activating, or delivering regulatory T cells (Tregs). These approaches have shown promise in the pursuit of curing or effectively managing autoimmune diseases (172). 2021, another study discussed the safety and effectiveness and challenges associated with mesenchymal stem cell (MSC) treatment for people with autoimmune liver disease (173). In 2021, a study highlighted the role of patients’ microbiomes in the management of systemic sclerosis and immunoglobulin G4-related disease (IgG4-RD) (174). Recently, we reported an autoimmune and lipodystrophic phenotype using a mouse model, Clec16aΔUBC (175). This study revealed a link between CLEC16A, lipid metabolism, and immune disruptions. Treating Clec16aΔUBC mice with the tofacitinib, partly alleviates the lipodystrophic issue and enhances survival. Tofacitinib affects autophagy and JAK-STAT mediated SOCS signaling (175). The CLEC16A locus role in autophagy (176, 177), mitophagy (178), immune regulation and neurodegeneration (179, 180), makes it a promising target in autoimmune disorders. Genetic interactions and environmental triggers contribute to immune dysregulation, resulting in inflammation, autophagy, and cell death in autoimmune disorders. Exploring the intricate crosstalk and potential synergy between SOCS-mediated cytokine regulation and the contributions of CLEC16A to autoimmune pathogenesis could unveil novel insights into disease mechanisms. Investigating the convergence of SOCS and CLEC16A in the context of autoimmunity might provide a comprehensive understanding of the complex interplay between regulatory and predisposing factors, offering opportunities for innovative treatments.
SOCS-mimetics
The discovery of the SOCS1-KIR binding site on JAK2 has led to the development of SOCS1 mimetics and antagonists with potential immune response enhancement capabilities (181). Specifically, SOCS1-KIR, a unique mimetic peptide, consists solely of the KIR domain, acting as a pseudosubstrate for JAK1, JAK2, and TYK2, but not interacting with JAK3 (78). Several Jakinibs have received FDA approval for specific autoimmune/inflammatory disorders and are currently being evaluated for additional conditions. A significant advantage of SOCS1-KIR as a therapeutic candidate is its structural similarity to natural SOCS1 protein. Nonetheless, mimetic peptide drugs present drawbacks encompassing potential higher costs, restricted permeability, proteolytic vulnerability, short half-life, swift in-vivo clearance, and limited oral bioavailability. To address these limitations, various strategies are currently being employed to improve the properties of peptide drugs (182).
Over the past decade, different types of SOCS mimetics and antagonists have surfaced and been subject to testing. For instance, Tkip (mimetic of SOCS1, WLVFFVIFYFFR) (183, 184) is based on the SOCS-KIR domain. The mimetic peptide showed promising results similar to naturally occurring SOCS1, reduced the inflammatory phenotype in murine encephalomyelitis model (EAE). Tkip inhibits IFN-γ signaling and suppressed the effector functions of T-cells. It compensates for low levels of endogenous SOCS1 and SOCS3 associated with EAE (185). Topical administration of SOCS1-KIR peptide was shown to successfully prevent uveitis and ocular damage (186). Furthermore, a different mimetic peptide, R9-SOCS1-KIR, successfully suppressed autoimmune uveitis EAU in mice by inhibiting the cations of IFN-γ, TNF-α, and IL-17, consequently preventing ocular pathology (187).
Additionally, cell-penetrating forms of SOCS1 (CP-SOCS1) and SOCS3 (CP-SOCS3) have also been developed and tested in various disease models. Controlled, intracellular delivery of recombinant CP-SOCS1 has been shown to suppress the IFN-γ signaling (188). It interacts similar to endogenous SOCS1, and the extent of inhibition depended on the dosage. Another, CP-SOCS3 peptide was developed and tested to treat acute liver injury driven by LPS in mice. It effectively suppressed the inflammation driven by TNF-α and IFN-γ. A previous in-vitro study demonstrated that CP-SOCS3 exhibited similar actions to endogenous SOCS3 (189). Deletion of the SOCS box domain in both CP-SOCS1 and CP-SOCS3 in the mimetic peptide is a strategic modification that yields significant benefits in terms of both activity and longevity. The SOCS box domain is a crucial part of the natural SOCS-1 protein, serving as a recognition site for ubiquitin ligases that lead to the degradation of the protein. By removing this domain, the modified CP-SOCS1 and CP-SOCS3 peptides gain an advantage in their intracellular presence, as they are no longer subject to rapid degradation, ensuring their persistence within the cellular environment to inhibit pro-inflammatory signaling pathways over an extended period. By extending their activity and increasing their potency, the modified peptides provide a more robust and durable anti-inflammatory effect, making them promising candidates for therapeutic interventions aimed at curbing excessive immune responses and managing inflammatory conditions (187–189).
The potential of manipulating SOCS proteins as a therapeutic strategy for immune-related disorders is underscored by these findings. Among the SOCS peptides, SOCS1-KIR stands out as a promising mimetic with unique interactions and potential therapeutic benefits. It holds great promise as an addition to the arsenal of Jakinibs and it is our belief that the development of peptide drugs will persist in the future, offering new avenues for therapeutic interventions with notably fewer adverse events. While peptide drugs face certain limitations, ongoing research and technological advancements offer opportunities to improve their efficacy and overcome challenges related to their absorption, distribution, metabolism, and excretion characteristics. The restoration or enhancement of SOCS1 function is proposed to suppress excessive immune responses in autoimmune and autoinflammatory conditions (71, 108, 190). Table 1 and Figure 6 provide an overview of the application of SOCS mimetics in the therapeutic management of autoimmune uveitis, lupus, and psoriasis. SOCS mimetics have not yet undergone testing for autoimmune encephalitis. However, treatment options for other autoimmune and inflammatory disorders using mimetic peptides can be found in separate literature due to space constrain.
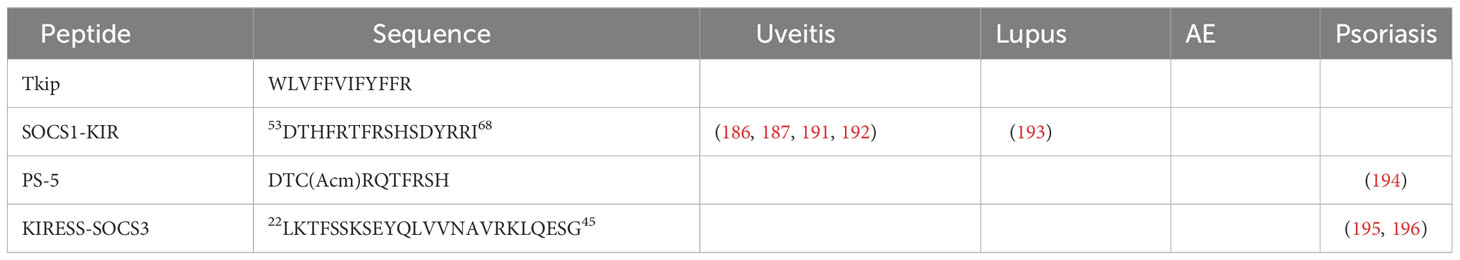
Table 1 SOCS mimetic peptides in autoimmune uveitis, lupus, and psoriasis.
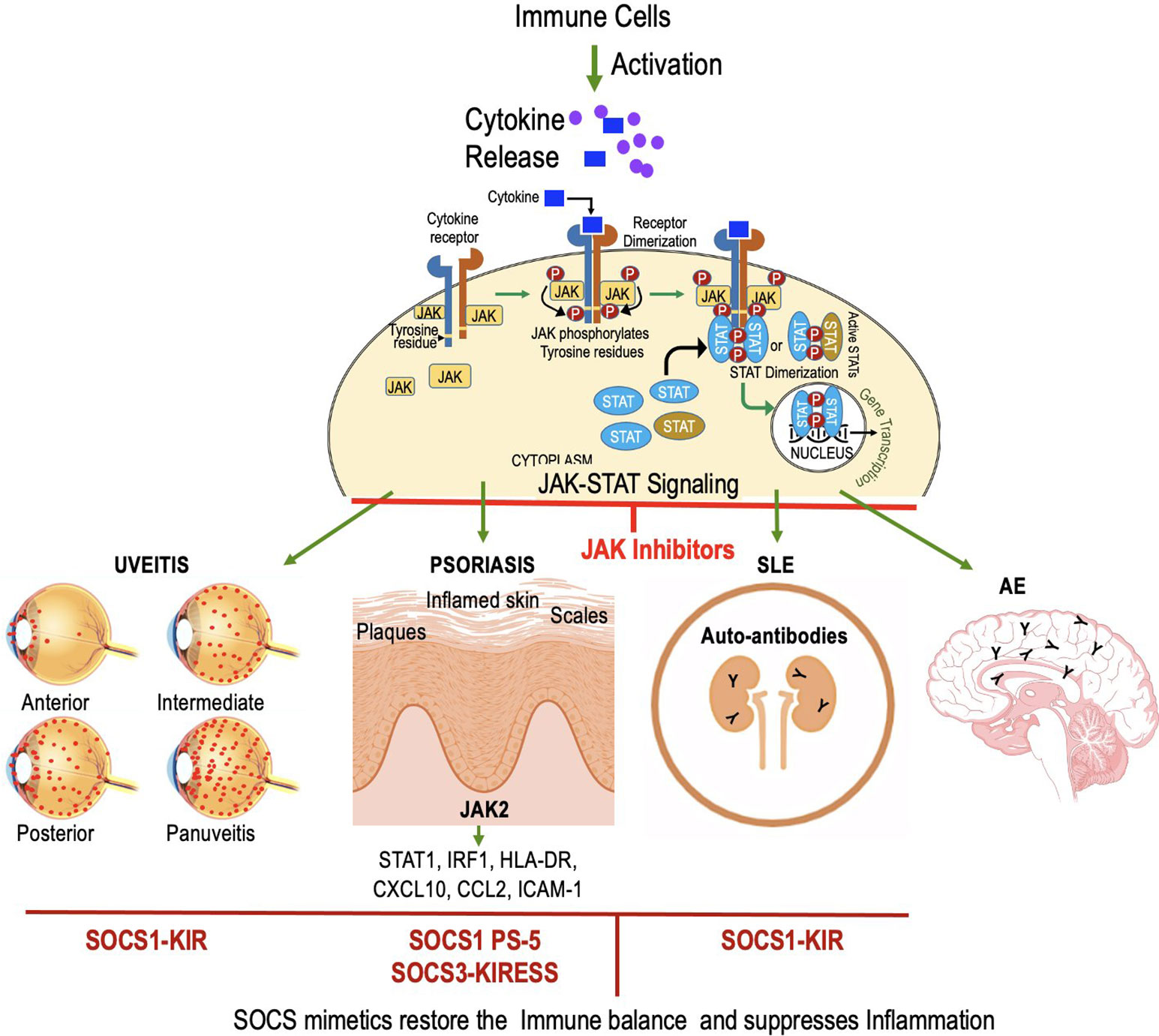
Figure 6 JAK inhibitors and mimetics show promise in treating autoimmune disorders like uveitis, psoriasis, SLE, and AE. SOCS1-KIR peptide mimics SOCS1 to inhibit pro-inflammatory cytokines, reducing uveitis. In psoriasis, characterized by the infiltration of immune cells and excessive keratinocyte proliferation, the action of SOCS1 and the PS-5 peptide involves the inhibition of pro-inflammatory molecules through the targeting of JAK2. In SLE, SOCS1-KIR treatment alters lymphocyte phenotype, potentially mitigating SLE pathology. SOCS1 mimetic has not been tested for AE.
Autoimmune uveitis
Uveitis refers to inflammation in any part of the uveal tract. Uveitis can stem from localized eye issues or systemic disorders affecting the body like autoimmune conditions (e.g., Bechet syndrome, ankylosing spondylitis) or infections (e.g., tuberculosis, herpes) (197–199). Uveitis can affect a single or multiple uveal structures of the eye and may also involve adjacent ocular tissues. Injuries to the eye, certain medications, and environmental factors play roles. Uveitis is a significant cause of ocular morbidity, leading to legal blindness in developed societies, with prevalence influenced by geography, ethnicity, and unknown factors in 30-60% of patients. Autoimmune or autoinflammatory uveitis is prevalent in developed countries, with different forms based on the location of inflammation within the uveal tract (anterior, intermediate, posterior, or panuveitis). The pathogenesis involves complex interactions between genetic, environmental, and immunological factors, and prompt diagnosis and treatment are crucial to prevent severe vision impairment or blindness (200).
EAU pathogenesis
Experimental autoimmune uveitis (EAU) is a valuable model for studying the pathogenesis of human autoimmune uveitis (192, 197, 199, 201–203). EAU is primarily triggered by a dysregulated immune response directed against ocular antigens, specifically retinal antigens. Antigen-presenting cells, such as dendritic cells, capture retinal antigens and present them to autoreactive T cells in the draining lymph nodes. This interaction leads to the activation of pathogenic T cell subsets, particularly Th1 and Th17 cells, which migrate back to the eye, triggering an inflammatory cascade and attract secondary effectors (204–206). In EAU models, genetic factors influence the immune response (207). Human leucocyte antigens (HLA) association with different forms of uveitis have been identified since the early 1970s (208). Recent progress in genetics of Uveitis has shown novel associations of AAU, BD and BSCR with HLA-B27, HLA-B51, and HLA-A29, respectively (207). Inflammation within the eye involves the recruitment of various immune cells, including neutrophils, macrophages, and T cells, leading to tissue damage, breakdown of the blood-retinal barrier, and retinal destruction. The release of pro-inflammatory cytokines, chemokines, and growth factors further amplifies the immune response, leading to the characteristic clinical features of uveitis (209–213). The intricate interplay between immune cells, cytokines, and ocular tissues in EAU provides valuable insights into the complex pathogenesis of autoimmune uveitis in humans, guiding the development of targeted therapies to mitigate the detrimental effects of the disease. The integration of human studies, equine models, and mouse uveitis models highlights Th17 cells and the JAK-STAT pathway’s significance in uveitis pathogenesis, with SOCS molecules playing crucial roles (192, 213).
Treatment of autoimmune uveitis
The JAK-STAT pathway regulates uveitis inflammation. Uveitis treatment can be categorized into local therapy, systemic therapy, or a combination of both. STAT3 inhibitors like ORLL-NIH001, blocking lymphocyte entry into the retina, shows promise for uveitis treatment (214).
Novel approaches to uveitis treatment aim to improve outcomes and mitigate its effects. These therapies include targeting cytokines and cytokine receptors, Janus kinases, the STAT3 pathway (via synthetic inhibitors), SOCS mimetic peptide therapy, and therapeutic cytokines like IL-35. Employing immunosuppressive cytokines such as IL-27 and IL-35, as well as using small molecule inhibitors to counteract proinflammatory cytokines, are noteworthy therapeutic strategies. For instance, ustekinumab specifically targets IL-12 and IL-23, while anakinra focuses on IL-1. IL-35 treatment was protective in mice with EAU. Conversely, mice lacking IL-35 (p35 knockout, or KO, mice) or with impaired IL-35 signaling (IL-12Rβ2 KO mice) experienced severe uveitis (215). Additionally, IL-35 exosomes effectively combat instability and short lifespan concerns, providing protection against severe uveitis in mice (216). These findings suggest role of IL-35 in modulating Uveitis through cytokine therapy. In mice, the absence of STAT3 within the CD19 B cell compartment (known as CD19-STAT3KO mice) worsens uveitis EAU (217). Another aspect influenced by STAT3 is miR-155-5p (miR-155), which is linked to ocular inflammation. STAT3 activates miR-155, and this interplay between STAT3 and miR-155 contributes to severe uveitis by fostering the expansion of pathogenic Th17 cells (218, 219). This suggests STAT3 pathway employed by Th17 cells is a potential strategy for uveitis.
Currently, TNF-α inhibitors are the main biologic agents used (220). Uveitis clinical trials of JAK inhibitors to date are listed in Table 2. The search for a potentially innovative uveitis therapeutic option continues. There are three clinical uveitis trials with JAK-STAT inhibitors now underway: Brepocitinib (NCT05523765) and Baricitinib (NCT04088409 and NCT05651880).
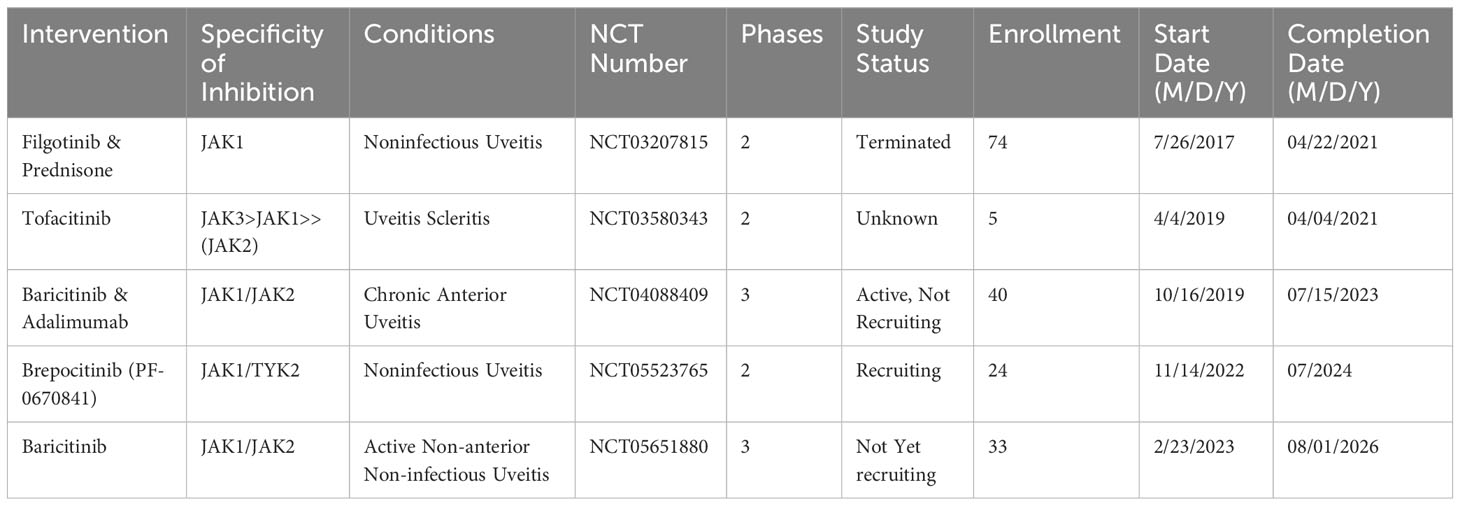
Table 2 Uveitis clinical trials of JAK inhibitors, chronological by the start date.
SOCS1 mimetics in autoimmune uveitis
The JAK-STAT pathway is a crucial regulator in NIU, and SOCS1 mimetics are promising to address SOCS1 deficiency. The SOCS1-KIR peptide (53DTHFRTFRSHSDYRRI68) is a noteworthy candidate due to its similarity to natural SOCS1 and its ability to inhibit JAK1/2 and TYK2, extending survival in SOCS1-deficient mice (183, 184, 221, 222). SOCS1 expression in the retina reduces inflammation and lymphocyte recruitment (223). Locally administered SOCS1-KIR peptide shows positive effects in mitigating disease symptoms in rodents (186, 187, 191).
A pilot study recently examined the safety and effectiveness of the SOCS1 mimetic peptide for treating equine recurrent uveitis (ERU), which serves as a model for human recurrent uveitis (RU) (192). RU is a debilitating autoimmune disease that can cause visual impairment in both horses and humans, despite existing treatments. Cases unresponsive to current treatments remain a significant concern. In ERU, inflammatory process driven by cytokines utilizes JAK2 signaling and contributes to blindness. SOCS1, which limits JAK2 activation, is often deficient in autoimmune disease patients. Bioinformatics and binding assays confirmed the SOCS1-KIR peptide’s potential to bind to equine JAK2. The safety of SOCS1-KIR peptide in equine eyes was initially assessed in a pilot study using healthy horses without equine recurrent uveitis. Two experimental horses received topical administration of SOCS1-KIR or a carrier for two weeks, while two other horses received intravitreal injection of SOCS1-KIR. Ophthalmic, physical exams, and electroretinography were conducted, showing that both topical and intravitreal SOCS1-KIR were safe for equine eyes (192). The results suggest that topical SOCS1-KIR treatment may restore immune tolerance in the eye and inhibit antigen presentation, a factor in triggering inflammation. Further testing in human and equine samples is essential for a comprehensive understanding of the mechanisms of action. These findings strongly encourage additional exploration of the mechanistic aspects of SOCS mimetics in uveitis.
Psoriasis
Psoriasis is a chronic, inflammatory skin condition with genetic and environmental roots, impacting about 2-3% of the global population (224) (225, 226). Often underdiagnosed, it has serious effects on quality of life and is associated with medical and psychiatric issues. Although treatments alleviate symptoms, accurate diagnosis and classification of its specific type are crucial for effective management. The most prevalent form is plaque psoriasis, ranging from mild to severe (227–229). Psoriasis’s cause involves complex interactions within the immune system, particularly T helper cells like Th1 and Th17, which lead to inflammation and thickened skin (230–234). Key pathways include JAK/STAT, NF-κB, and MAPK. IL-12 and IL-23 signaling sustains chronic inflammation by promoting cytokines (233, 235). This imbalance results in scaly plaques due to excessive keratinocyte growth (231–234).
Genetic factors significantly contribute to psoriasis. Certain genes, like HLA-C, IL23R, IL12B, and LCE3B/3C, increase susceptibility to psoriasis. Environmental triggers like infections, skin trauma, and medications exacerbate this condition in genetically predisposed individuals by activating immune responses. Targeting the JAK/STAT pathway with inhibitors, such as SOCS1-KIR, shows promise for alleviating psoriatic inflammation.
Genetic factors significantly contribute to psoriasis. Certain genes, like HLA-C, IL23R, IL12B, and LCE3B/3C, increase susceptibility to psoriasis (236, 237). Environmental factors, such as infections (streptococcal infections), trauma to the skin, and certain medications, can trigger or exacerbate psoriasis in individuals with a genetic predisposition (238, 239). These triggers can activate immune responses and further perpetuate the inflammatory processes in psoriasis. Targeting the JAK/STAT pathway with inhibitors including SOCS1-KIR, shows promise for alleviating psoriatic inflammation.
Preclinical studies - animal models
Multiple animal models have been developed to study psoriasis and its mechanisms (spontaneous, xenograft, genetically engineered, intradermal injection of cytokines (L-23 or IL-17), and topical application of irritant imiquimod (IMQ)) resulting in skin inflammation, hyperproliferation of keratinocytes, and immune cell infiltration, mimicking key aspects of human psoriasis (240). Cytokines like IL-17, IL-23, and TNF-α are crucial in psoriasis, driving inflammation and abnormal cell proliferation. In light of this, targeting these cytokines could revolutionize the psoriasis treatment.
Tofacitinib have been investigated in several psoriasis mouse models. The topical application of Tofacitinib effectively reduces IL-22 expression in imiquimod-treated mouse ear skin (241) and IL-31 in mice with allergic dermatitis from toluene-2,4-diisocyanate (242). In-vitro tofacitinib has been shown to inhibit IL-23 expression in dendritic cells, suppress IL-22 expression in Th17 cells, and impede the differentiation of CD4+ T cells into Th2 cells, which are responsible for releasing IL-31 (243). Systemic delivery of tofacitinib has been shown to reduce the itch-related behaviors in the mice by controlling cytokines (IL-22, IL-23, and IL-31) and modulating epidermal nerves. Tofacitinib also influenced the expression of TSLP and IL-23, affecting both downstream and upstream signals of JAK (elevating Tslp mRNA expression and decreasing IL-23 mRNA expression). Although IL-17A expression, regulated by IL-23, remained unaffected, tofacitinib increased the density of peptidergic epidermal nerves (244). Mouse models provide valuable initial insights, but clinical trials are essential to assess JAK inhibitors’ safety and efficacy in treating human psoriasis.
Treatment - clinical trials
Psoriasis treatment has evolved from ancient remedies to modern therapies like UV light, immunosuppressants, biologics, and personalized approaches. Ongoing research aims for better, safer options, offering hope for improved management and quality of life for patients with a specific emphasis on targeting cytokines. Tofacitinib has undergone extensive clinical trials for psoriasis treatment. In a 12-week Phase 2b trial, tofacitinib displayed notable clinical improvement and good tolerance for moderate-to-severe chronic plaque psoriasis (245, 246). Another small phase 2 clinical trials (NCT01710046) further supported its effectiveness by attenuating the pathological immune pathways in psoriasis (247). Phase 3 trials confirmed tofacitinib’s efficacy, showing it to be comparable to etanercept and superior to placebo in treating moderate-to-severe plaque psoriasis, offering a viable treatment option (248). Tofacitinib effectively treats nail psoriasis, showing continued efficacy in a two-year extension study (249). Additionally, in Asian patients with plaque psoriasis, tofacitinib was found to be more effective than placebo, with its efficacy maintained throughout a 52-week study period (250, 251). These findings support the potential of tofacitinib as a promising treatment for psoriasis, offering hope for improved management of this chronic skin disorder.
Solcitinib (GSK2586184), an oral JAK1 inhibitor, showed promise in a phase 2a trial (NCT01782664) for moderate-to-severe plaque psoriasis. In this trial, patients across different doses (100, 200, or 400 mg) experienced significant PASI 75 response rates at week 12, along with improved itch and quality of life. Adverse events were consistent across treatment groups, without a dose-related trend. Overall, Solcitinib demonstrated clinical improvement and good tolerability in treating moderate-to-severe plaque psoriasis over 12 weeks (252). Another oral JAK1 inhibitor, Abrocitinib (PF-04965842), showed efficacy and good tolerability in treating moderate-to-severe plaque psoriasis in a phase 2 trial with 59 patients (NCT02201524). Patients receiving different doses (200 or 400 mg) demonstrated improved symptoms compared to placebo. Although some abnormal laboratory test results were observed, no serious infections or bleeding events were reported. The study was terminated despite positive results (253).
Brepocitinib (PF-0670841), a dual TYK2/JAK1 inhibitor (254, 255), has undergone three clinical trials for psoriasis treatment. In a phase 1 trial (NCT02310750), it was well-tolerated up to 200 mg in healthy subjects and 100 mg in psoriasis patients, showing significant improvements in PASI scores (256). In a phase 2a trial (NCT02969018), it reduced PASI scores compared to placebo in multiple treatment groups, with continuous treatment at 30 mg once daily showing the greatest improvement (257). However, in a recent phase 2b trial (NCT03850483) using topical brepocitinib cream, it was well-tolerated but did not show significant changes compared to the vehicle in treating mild-to-moderate plaque psoriasis [288].
Ropsacitinib (PF-06826647), a dual oral TYK2/JAK2 inhibitor targeting IL-12 and IL-23 signaling (258) shows promising results in treating moderate-to-severe plaque psoriasis (259) with significant improvements seen in a phase 2b, 2020 (260). A network meta-analysis of JAK inhibitors (included eight randomized clinical trials involving tofacitinib, peficitinib, solcitinib, baricitinib, abrocitinib, and deucravacitinib) for psoriasis found tofacitinib to be the most effective, despite not being FDA-approved for psoriasis due to side effects (261). Tofacitinib and deucravacitinib showed the best responses in both efficacy and safety, supporting JAK inhibitors as a promising treatment option for moderate-to-severe plaque psoriasis. Table 3 lists all JAK inhibitors that have been used in clinical trials for psoriasis.
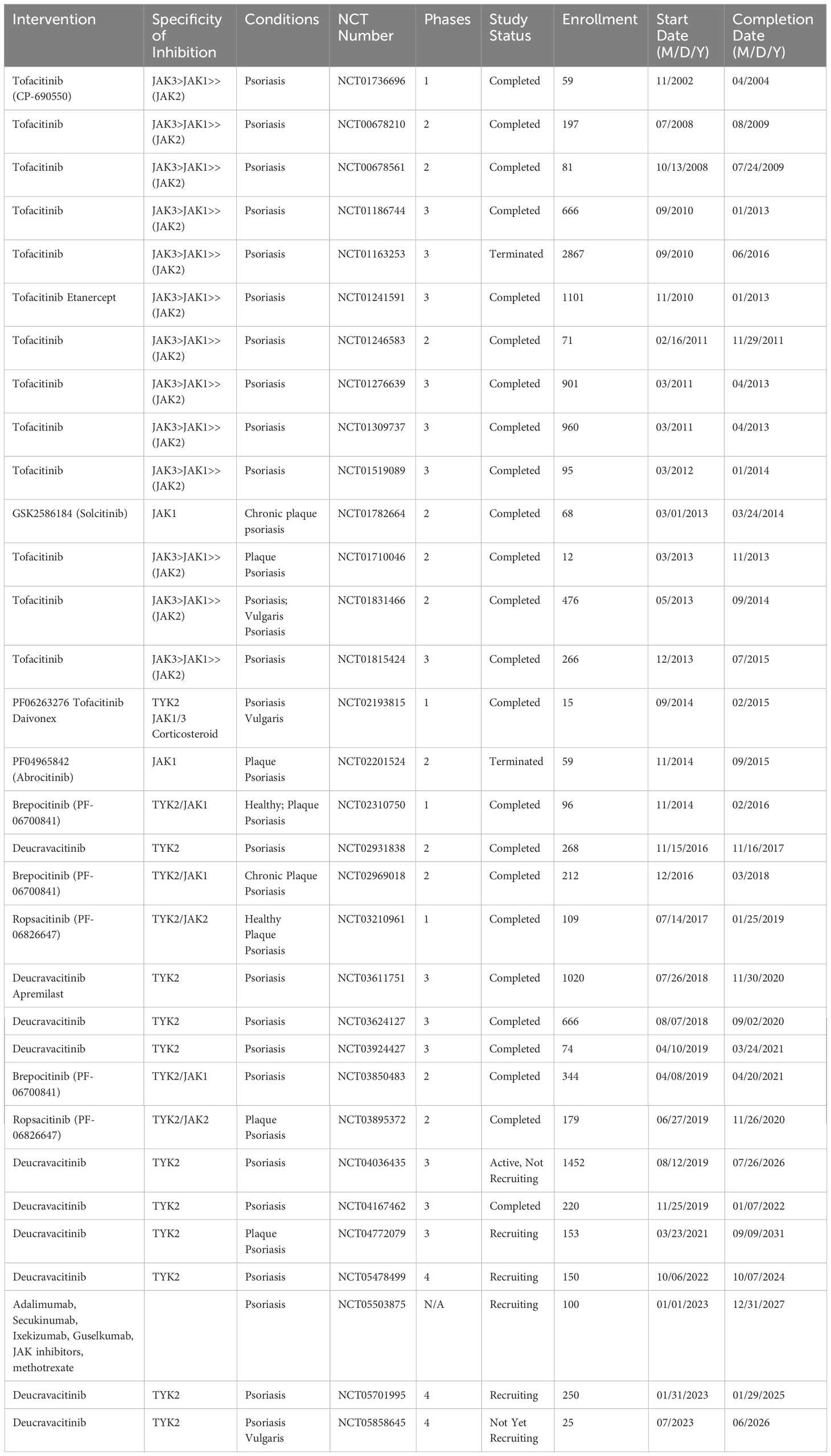
Table 3 Psoriasis clinical trials of JAK inhibitors, chronological by the start date.
SOCS mimetics in psoriasis
In 2012, Doti et al. introduced PS-5, a new peptide inhibitor of JAK2, which is a mimetic of the kinase-inhibitory region of SOCS1 (262). This peptide (DTC(Acm)RQTFRSH) has a distinct amino acid composition and length compared to SOCS1-KIR. Specific amino acid substitutions in PS-5, involving phenylalanine and arginine residues, led to improved JAK2 binding by establishing enhanced electrostatic interactions with the negative phosphate moiety on Y1007. PS-5, containing a nonnatural residue (Cys(Acm)), exhibited greater protease stability and effectively inhibited STAT1 phosphorylation, leading to reduced interferon regulatory factor-1 (IRF-1) expression through binding-assay screening (262). In a 2013 study, Madonna et al. explored the therapeutic potential of PS-5 in managing IFN-γ-mediated skin pathogenesis. The research compared PS-5 with the full kinase inhibitory region of SOCS1 protein for its ability to suppress inflammatory gene expression in IFN-γ-treated human keratinocytes. PS-5 mimetic showed comparable effectiveness to the kinase inhibitory region peptide, inhibiting Jak, IFN-γRα, and STAT1 phosphorylation, along with the expression of ICAM-1, HLA-DR, CXCL10, and CCL2 (194). These findings highlight PS-5 as a promising novel therapeutic strategy for IFN-γ-induced skin pathogenesis, as depicted in Figure 6.
A distinct SOCS3 mimetic peptide, referred to as the KIRESS peptide (sequence: 22LKTFSSKSEYQLVVNAVRKLQESG45), was synthesized and assessed in vitro. This peptide spans both the KIR (Kinase Inhibitory Region) and ESS (Extended SH2-Substrate Binding) regions (195). KIRESS peptide was shown to inhibit the IL-22 signaling pathway by regulating the STAT3 and ERK 1/2 signaling, along with suppression of STAT3 expression in keratinocytes. In-vivo KIRESS peptide effectively suppressed tumor growth and increased STAT3 activation in athymic nude mice harboring squamous cell carcinoma (SCC) xenografts (196). While, these results are encouraging, the specificity and efficacy of these mimetic peptides in an in-vivo setting requires further investigation, necessitating additional studies.
Autoimmune encephalitis
Autoimmune encephalitis (AE) is a rare, severe neurological disorder involving brain inflammation from an autoimmune response against synaptic antigens (263). The immune system mistakenly targets and attacks healthy brain tissue, leading to a range of neurological symptoms. A list of commonly reported AE autoantibodies includes: N-methyl-D-aspartate receptor (NMDAR) (264), anti-leucine-rich glioma-inactivated protein 1 (LGI1) (265), contactin associated protein-like 2 receptors (CASPR2) (266), gamma aminobutyric acid (GABA) (267, 268); alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) (269), dipeptidyl-peptidase-like protein-6 (DPPX) (270), and myelin oligodendrocyte glycoprotein (MOG) (271). Currently, NMDAR encephalitis is the most common and well-studied subtype of AE (272).
AE presents cognitive impairments, seizures, memory problems, behavioral changes, movement disorders, and psychiatric symptoms (273). It require careful diagnosis and appropriate treatment, often involving immunosuppressive therapies (274). Antibodies in AE are intrinsically pathogenic. They target synaptic proteins and induce conformational changes leading to widespread inflammation (275). A prior infection that caused inflammation and neurological symptoms is a common precursor to AE. Herpes simplex virus encephalitis (HSVE) has been shown to trigger immune response causing NMDAR encephalitis (276). Moreover, the human leukocyte antigen (HLA) is linked to the production of antibodies that trigger autoimmune responses (277). Up to the present time, a considerable number of comparable syndromes, often termed AE, have been discovered. In neuron cultures, pathogenic antibody effects have been shown for various AE types. These effects include receptor blocking (GABABR), receptor cross-linking and internalization (NMDAR) (278, 279), and disruption of protein-protein interactions (LGI1) (280). Yet, the lack of suitable animal models continues to restrict our comprehension and development of novel therapies. Anti‐NMDAR encephalitis is a prevalent (281) and best studied subtype of AE, for which a few mouse models were developed (75, 282, 283). Studies have explored the pathogenic effects of patient-derived or a human recombinant antibody in passive-transfer animal models. Planaguma et al. demonstrated that infusing CSF from anti-NMDAR encephalitis patients altered memory and behavior in mice. The antibodies disrupted NMDAR interaction with the ephrin-B2 receptor, leading to receptor internalization and impaired synaptic plasticity, memory, anhedonia, and depressive behavior. After the antibody infusion was terminated, these changes gradually resolved (75). Other studies indicated that passive NMDAR antibody transfer from patients to mice could induce seizures (283) and psychotic behavior in mice (282). These findings demonstrate the potential of AE patient autoantibodies for creating precise mouse models, providing insights across multiple levels, including cellular, synaptic, and neural networks, and facilitating novel therapy testing. A postinfectious autoimmune encephalitis mouse model developed by performing multiple intranasal infections with live group A Streptococcus (GAS) shows Th17 cells migration from the nose into the brain, resulting in the disruption of the blood-brain barrier (BBB) and the inflow of autoantibodies into the CNS (284). Later, the same group highlighted the essential role of Th17 lymphocytes in enabling selective CNS autoantibody entry, microglial activation, and neural circuit impairment in postinfectious AE. Mice lacking Th17 cells exhibited reduced BBB leakage, microglial activation, CNS antibody infiltration, and partial olfactory function restoration (285). In AE, the JAK-STAT pathway can become dysregulated, leading to abnormal immune responses and inflammation within the central nervous system (CNS). Tofacitinib’s ability to cross the blood-brain barrier and modulate cytokine receptors positions it as a potential therapy for AE and refractory AE, offering hope for more targeted treatments (286, 287).
Preclinical studies - animal models
Autoimmune Encephalitis (AE) is predominantly a human condition and is less frequently observed in mice. Animal models that mimic aspects of AE have been instrumental in providing direct evidence of the pathogenicity of autoantibodies. For instance, researchers have created these models by transferring cerebrospinal fluid (CSF) or immunoglobulins from patients with anti-NMDA receptor encephalitis to mice (283). These studies have shed light on a critical process: the continuous production of autoantibodies by self-reactive B cells. Two major pathogenic pathways have been documented in autoimmune encephalitis: one involving the selective and reversible reduction of NMDA receptor surface density and synaptic localization upon exposure to autoantibodies from anti-NMDAR encephalitis patients (279, 288), and the other pathway involves complement activation, demonstrated in CASPR2 antibody-associated encephalitis cases (289). Despite extensive research and increasing clinical insights, a notable proportion of patients still do not benefit from existing treatments. A recent study reported a translational rodent model of NMDARE (n-methyl-D-aspartic acid receptor (NMDR) encephalitis), using active immunization and offers a valuable tool for delving into the pathophysiology of AE (290). This development holds promise for advancing the diagnosis and treatment of this debilitating neuropsychiatric condition with a relatively rapid onset of the phenotype, enabling in-depth investigations into its pathophysiology. Additionally, the NMDARE mouse model has the potential to serve as an effective translational platform for pre-clinical testing of both existing and future therapeutic interventions.
Clinical trials
Autoimmune encephalitis (AE) is a rare and serious medical condition that involves inflammation of the brain (51, 291), and treatment options are based on autoimmune disease management principles (274, 277, 292). Immunotherapy, such as corticosteroids, intravenous immunoglobulins, and plasmapheresis, is the first-line therapy, rituximab and cyclophosphamide (second line therapy) and other immunosuppressive drugs used when needed (293). Controlled clinical trials for AE are lacking due to the rarity of the disease (294–296). However, ongoing trials, such as one evaluating bortezomib (NCT03993262), a proteasome inhibitor, in severe AE, hold promise for establishing guidelines and advancing to larger phase III trials (295). In refractory AE, blood-brain barrier penetrating novel immunotherapies are crucial (297). Tofacitinib, a JAK3/1 inhibitor, has shown potential in treating refractory immune-mediated diseases, and recent studies suggest it could be a promising option for some AE patients (298). However, further research with a larger patient group is needed to fully understand its effectiveness in AE. As tofacitinib penetrates the BBB (242, 299) it has the potential to be effective in the CNS autoimmune disorders, however further analysis of much larger group of patients is required to make a conclusion for the use of tofacitinib in AE. Despite progress in understanding AE (300), the exact pathological mechanisms remain unclear, and therapeutic options are currently limited.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a complex autoimmune disorder with a wide-ranging impact on the body. Common manifestations include joint pain, skin rashes (especially the characteristic “butterfly” rash on the face), fatigue, and fever. SLE is more frequent in women. The precise mechanism of SLE pathogenesis is still unknown. SLE highlights the intricate interplay between genetics, environmental factors, and immune dysregulation. Cytokines are crucial in the development of SLE’s pathophysiology (301, 302). Cytokines, including interleukin-10 (IL-10) and B-cell activating factor (BAFF), have a pivotal role in the activation, survival, and differentiation of B cells. Additionally, cytokines such as interleukin-2 (IL-2) and interleukin-21 (IL-21) impact T-cell function and differentiation. Elevated levels of specific cytokines such as interleukin-6 (IL-6), interleukin-17 (IL-17), and tumor necrosis factor-alpha (TNF-alpha) are a common occurrence in individuals with SLE (303, 304). Elevated type I interferons (IFNs) correlate strongly with active SLE, driving autoantibody production, immune complex formation, and tissue damage. JAK1 and TYK2, downstream signals of IFN, show significant associations with SLE, especially TYK2 polymorphism (305). [357]. In patients with SLE, there is a substantial increase in the expression of CXCR4, a crucial receptor involved in immune regulation (306, 307). This heightened expression is intricately associated with the activation of the JAK/STAT pathway, fostering the infiltration of immune cells into the kidneys, thereby aggravating the progression of the disease. These findings suggest the potential use of JAK inhibitors for treating lupus (308).
Preclinical studies - animal models
Over time, a diverse range of murine models for lupus has been created and utilized, each with its own limitations, contributing to our understanding of systemic lupus erythematosus (SLE) (309). Among these models, the MRL/lpr mice distinguish themselves by their exceptional capacity to generate an extensive spectrum of lupus-associated autoantibodies (ANA, anti-dsDNA, anti-Sm, anti-Ro, and anti-La) alongside exhibiting features such as arthritis, cerebritis, skin rash, and vasculitis (310).
Promising outcomes in mouse models have been demonstrated by various JAK inhibitors. Ruxolitinib reduced severe skin lesions in MRL/lpr mice, but its effect on other lupus manifestations remains unclear, requiring further research (311). Tofacitinib demonstrated reduced disease activity, nephritis, and autoantibody titer in MRL/lpr and NZB/NZWF1 mice, suggesting potential as a therapy for SLE (312–315). Baricitinib, a selective JAK1/2 inhibitor, significantly suppressed lupus-like symptoms and restored disrupted podocyte structures (316). Another potential therapy, Deucravacitinib (BMS-986165), showed reduced IFN expression in SLE patient cells and decreased type I IFN-regulated gene expression in NZB/W mice (317). These results underscore the promise of JAK inhibitors as innovative solutions for SLE, yet additional research and clinical trials are required in human patients.
Treatment of SLE patient – clinical trials
Immunosuppressive medications and glucocorticoids are the two most common current SLE treatments, marked by notable side effects and limited efficacy. Under these circumstances, SLE still has a high rate of morbidity and mortality. Identification of bioactive agents has positioned the JAK/STAT pathway as a more suitable contender for SLE’s pathogenesis.
The first JAK1 inhibitor, GSK2586184 (Solcitinib), was assessed for its effectiveness, safety, and tolerability in adults with SLE (ClinicalTrials.gov NCT01777256). However, the study revealed various safety issues in participants (318, 319). Consequently, the trial was deemed futile, and recruitment was stopped after 50 patients. Based on the safety and efficacy statistics from the study, further research on GSK2586184 in SLE patients is not recommended. Tofacitinib (CP-690550) was then evaluated and showed promise in treating SLE [380]. Another recent randomized, double-blind, placebo-controlled study (NCT02535689) demonstrated that tofacitinib is safe and well-tolerated in patients with mild-to-moderate SLE. Tofacitinib reduced IFN type I signature, improved lipid profile, restored endothelial function, and enhanced cardiometabolic and immunologic parameters associated with premature atherosclerosis. Additionally, the drug’s protective benefits were more pronounced in individuals with the STAT4 risk allele, which is linked to more severe SLE and an increased risk of cardiovascular (CV) events (320). A case report evaluating the efficacy of tofacitinib in three patients with recalcitrant cutaneous lupus reported favorable outcomes, showing significant improvement (321). Ongoing and approved trials of tofacitinib in SLE aim to gather more data on targeting the JAK pathway’s effectiveness, with potential implications for cardiovascular function across autoimmune disorders and the broader population.
Baricitinib selectively inhibits JAK1 and JAK3 subtypes. A double-blind placebo-controlled study in 314 lupus patients with skin and joint manifestations (NCT02708095) showed positive results. After 24 weeks of baricitinib (4 mg) treatment, 67% of patients achieved resolution of arthritis or rash (322), along with reduced expression of key lupus-related cytokines, improved SLE disease activity, anti-dsDNA antibody levels, and diminished swollen and sore joints (323). It showed potential in treating cutaneous and articular involvements (322, 324, 325), but its efficacy in lupus nephritis is still being investigated in a phase 2/3 trial (NCT05686746). Phase III trials for moderate to severe SLE adult patients (NCT03616964 & NCT03616912) had inconclusive outcomes. A follow-up study for long-term safety (NCT03843125) was terminated due to insufficient evidence of positive benefit. Further pre-clinical data are needed to better understand baricitinib potential efficacy and mechanism in treating lupus-related phenotypes.
Filgotinib (GLPG0634) treatment, a selective JAK1 inhibitor, showed disappointing outcomes in a cutaneous lupus erythematosus trial, failing to significantly improve CLASI scores (326). Its use in lupus membranous nephropathy yielded unsatisfactory results with only limited conclusions due to a small number of participants (327). Presently, no planned trials involve filgotinib in SLE patients. In contrast, Upadacitinib (ABT-494), a second-generation selective JAK1 inhibitor, has been approved for conditions like rheumatoid arthritis (328), psoriatic arthritis (329), and atopic dermatitis. Limited data on its efficacy in SLE exists, with only one case report showing resolution of accelerated nodulosis (330). Phase II trials (NCT04451772) have been completed to evaluate the safety and efficacy of upadacitinib in SLE, and phase III (NCT05843643) trials anticipated to commence soon [www.clinicaltrials.gov last accessed June 6, 2023].
In healthy volunteers, Deucravacitinib demonstrates favorable pharmacokinetics and safety traits (331) and is now FDA approved to manage moderate-to-severe plaque psoriasis (332). A recent phase II trial in adult SLE patients (NCT03252587) reported positive outcomes, with a higher SLE Responder Index 4 (SRI-4) response compared to placebo (333). The trial met all secondary endpoints, indicating deucravacitinib efficacy and acceptable safety. The ongoing phase 3 trials, (NCT05617677 & NCT05620407) are assessing its efficacy and safety. A study (NCT03920267) is also assessing its long-term safety and efficacy in SLE.
JAK inhibitors of the first generation address multiple JAK isoforms, yielding diverse impacts yet also triggering a spectrum of side effects (330). Although SLE clinical trials have demonstrated potential benefits, none of the JAK-STAT inhibitors listed in Table 4 have yet garnered approval for use in SLE clinical practice. Tofacitinib and baricitinib have shown the most promising outcomes so far. Nonetheless, while their safety and effectiveness profiles present encouraging data, further clinical trials are imperative for future validation.
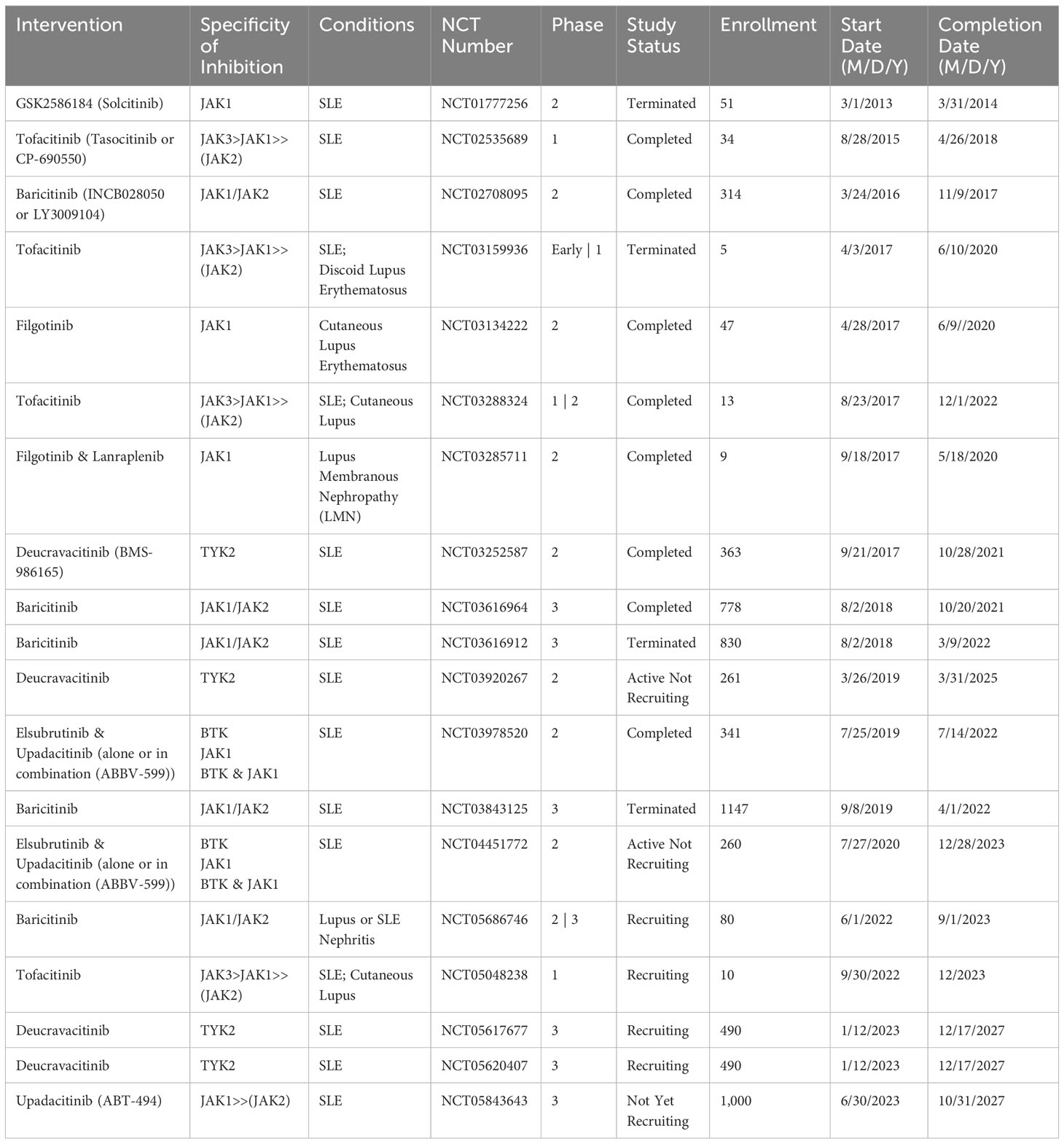
Table 4 SLE clinical trials of JAK inhibitors, chronological by the start date.
SOCS mimetics in SLE
Cytokine imbalance and the diminished SOCS1 expression both hold significant roles in the advancement of SLE. Restoring or enhancing SOCS1 expression or function could potentially modulate cytokine signaling, reduce inflammation, and attenuate the progression of SLE. SOCS1 mimetics could be a safe alternative.
Reduced SOCS1 expression in individuals with systemic lupus erythematosus (SLE) and in lupus mouse models is established (334–336). This decline in SOCS1 expression exhibits a negative correlation with the extent of inflammation. The decreased expression of SOCS1 contributes to the dysregulated cytokine signaling and excessive immune activation seen in SLE. Hematologic abnormalities, including abnormal blood cell counts, and the generation of autoantibodies are influenced by SOCS1 insufficiency. Aberrations in SOCS1 also contribute to lupus nephritis, a grave complication of SLE, influencing organ performance in regions like the skin, central nervous system, heart, and kidneys (335, 337).
Research by Sukka-Ganesh and Larkin was aimed to identify a therapeutic potential to target SOCS1 in the treatment of SLE by using mice and studying SLE patients and clinical samples. The study findings revealed that after 48 hrs. of stimulation with anti-CD3, LPS, or IFN-γ, SOCS1 and SOCS3 expression peaked in murine splenic samples. Furthermore, the peripheral blood mononuclear cells (PBMCs) from SLE patients exhibited notably lower levels of both SOCS1 and SOCS3 mRNA and protein compared to control subjects. The study also demonstrated that reduced SOCS1 levels in SLE patients were associated with increased inflammatory markers and upregulated expression of major histocompatibility complex (MHC) class II molecules. Moreover, patients receiving steroid exhibited elevated levels of SOCS1 compared to untreated, and the human PBMCs treated with steroid showed dose- and time-dependent upregulation of SOCS1 mRNA (336).
The therapeutic potential of SOCS1-KIR in modulating lupus-associated pathologies has been recently evaluated in Fas-deficient MRL/lpr mice. The application of SOCS1-KIR led to diminished skin lesion severity, lowered production of autoantibodies, and moderate enhancements in kidney pathology. At the cellular level, the introduction of SOCS1-KIR through peritoneal administration augmented the expression of Foxp3 in overall splenic and follicular regulatory T cells, lowered the ratio of effector memory to naive T lymphocytes in both CD4+ and CD8+ cell populations, and diminished the occurrence of germinal center B cells. These observations suggest that SOCS1-KIR treatment triggers changes in lymphocyte dynamics, hinting at the therapeutic potential of peptide administration for alleviating SLE-related pathology (193) (Figure 6).
FDA and EMA approved JAK inhibitors
JAK inhibitors, also known as Jakinibs, belong to a class of small-molecule medications. These inhibitors hold promise in treating autoimmune conditions and have proven effectiveness against inflammatory diseases such as rheumatoid arthritis and psoriasis, among others. Administered orally, they initially targeted multiple JAK enzymes, but newer iterations are more discerning. Despite their swift action, drawbacks exist, encompassing side effects (gastrointestinal problems, liver irregularities, anemia, changes in blood lipids), high expense, and heightened susceptibility to infections and malignancies. Long-term safety data is still being collected. Figure 7 roadmap showcases the expanding use of JAK inhibitors in treating distinct autoimmune and inflammatory disorders, underlining their growing significance in therapeutic interventions that have obtained approval from both the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) in chronological order based on first approval: Ruxolitinib (338–340), Tofacitinib (341–344), Baricitinib (343, 345, 346), Fedratinib (347), Peficitinib (348), Upadacitinib (328, 329, 349, 350), Delgocitinib (351), Filgotinib (352), Abrocitinib (353), Deucravacitinib (332), Pacritinib (354).
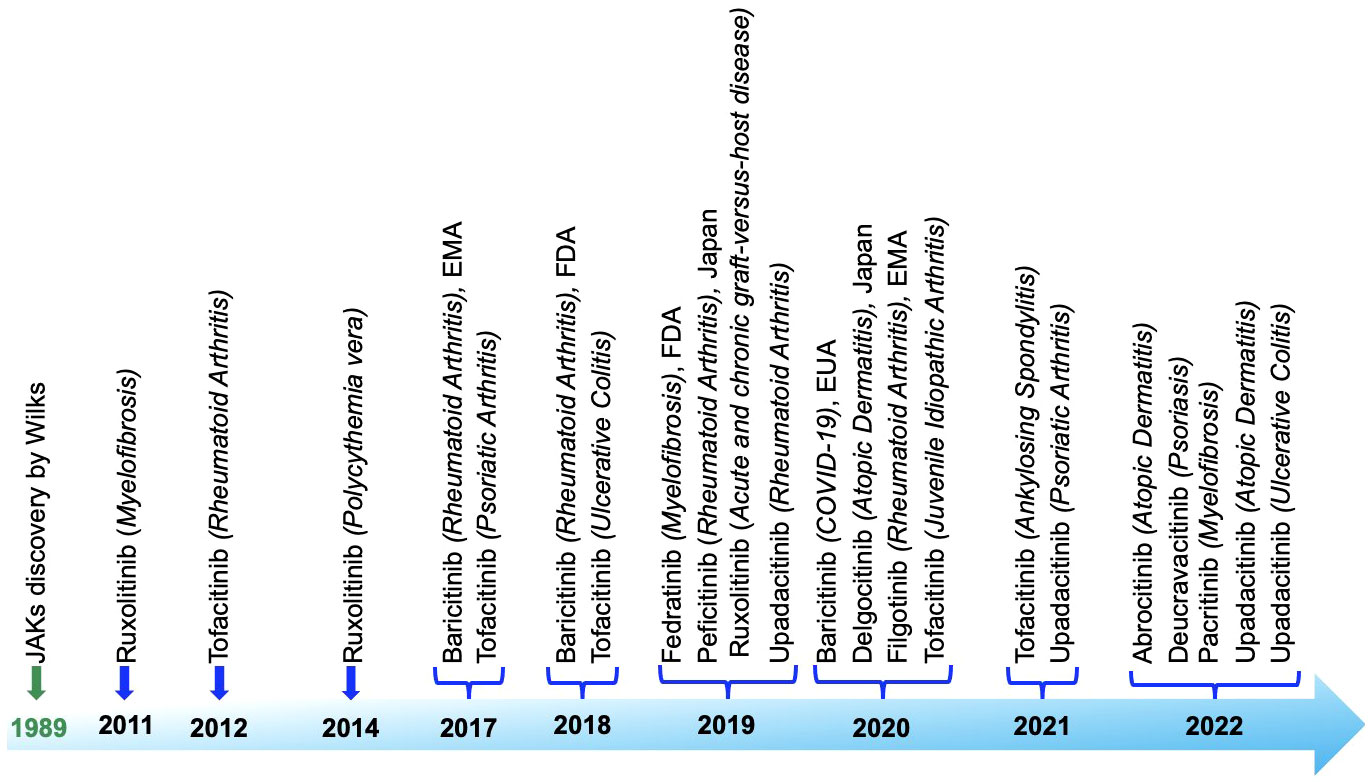
Figure 7 Roadmap detailing the discovery and approval of Janus kinase inhibitors, highlighting the key milestones and stages in the process.
Conclusions
The field of autoimmune research is constantly progressing. Currently, there are 2,254 planned studies focused on autoimmune diseases (https://clinicaltrials.gov). Examining and refining Jakinibs through development and testing has illuminated the specific functions that different JAKs play in various human diseases. SOCS-KIR peptides show promise as cutting-edge therapeutic options for autoimmune disorders, thanks to their small size, stability, and low immunogenicity, which make them favorable candidates for safe therapeutic development. SOCS1-KIR, when internalized by cells, has shown strong therapeutic potential in EAE, autoimmune uveitis, psoriasis, and diabetes models. In contrast, SOCS1 antagonist (pJAK2 (1001–1013)) has been shown to enhance immune responses against various viruses. SOCS mimetics and antagonists hold promise as potential therapeutics for regulating the immune system in both negative and positive ways. However, it remains imperative to conduct additional research and clinical trials to gain a comprehensive understanding of their mechanisms of action, safety, and effectiveness across various autoimmune conditions before considering their widespread utilization in clinical practice. Scientists and the medical community are diligently striving to devise and apply inventive methodologies aimed at targeting JAK-STAT pathways and creating mimetics that target SOCS. SOCS1 mimetics hold promise for treating disorders associated with excess inflammation or SOCS1 deficiency upon fully established safety studies.
Author contributions
RP: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. MB: Conceptualization, Methodology, Writing – review & editing. HH: Conceptualization, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
HH, MB, and RP currently have three patents under consideration: ‘Innovative weight reduction therapies targeting CLEC16A,’ ‘Utilization of CLEC16A and SOCS1 as therapeutic strategies in autoimmunity using a UBC-Cre-Clec16aloxP Phenotype Mouse model,’ and ‘Probucol’s beneficial effects on the autoimmune, lipodystrophic, and neurodegenerative phenotypes in Clec16a knockout mice.’
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pandey R, Bakay M, Hakonarson H. CLEC16A-an emerging master regulator of autoimmunity and neurodegeneration. Int J Mol Sci (2023) 24(9):8224. doi: 10.3390/ijms24098224
2. Kapoor B, Gulati M, Gupta R, Singla RK. Microbiota dysbiosis and myasthenia gravis: Do all roads lead to Rome? Autoimmun Rev (2023) 22(5):103313. doi: 10.1016/j.autrev.2023.103313
3. Mazzucca CB, Raineri D, Cappellano G, Chiocchetti A. How to tackle the relationship between autoimmune diseases and diet: well begun is half-done. Nutrients (2021) 13(11):3956. doi: 10.3390/nu13113956
4. Xue C, Yao Q, Gu X, Shi Q, Yuan X, Chu Q, et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signal Transduct Target Ther (2023) 8(1):204. doi: 10.1038/s41392-023-01468-7
5. Telliez JB, Gadina M, Ghoreschi K, Silvennoinen O, Spinelli FR. Editorial: JAK inhibition in autoimmune and inflammatory diseases. Front Immunol (2022) 13:1120281. doi: 10.3389/fimmu.2022.1120281
6. Srivastava S, Rasool M. Underpinning IL-6 biology and emphasizing selective JAK blockade as the potential alternate therapeutic intervention for rheumatoid arthritis. Life Sci (2022) 298:120516. doi: 10.1016/j.lfs.2022.120516
7. Kallen KJ, Galle PR, Rose-John S. New developments in IL-6 dependent biology and therapy: where do we stand and what are the options? Expert Opin Investig Drugs (1999) 8(9):1327–49. doi: 10.1517/13543784.8.9.1327
8. Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta (2002) 1592(3):251–63. doi: 10.1016/S0167-4889(02)00319-1
9. Guo Z, Ma Y, Wang Y, Xiang H, Yang SY, Guo Z, et al. The role of IL-6 and TMEM100 in lumbar discogenic pain and the mechanism of the glycine-serine-threonine metabolic axis: A metabolomic and molecular biology study. J Pain Res (2023) 16:437–61. doi: 10.2147/JPR.S400871
10. Dittrich A, Hessenkemper W, Schaper F. Systems biology of IL-6, IL-12 family cytokines. Cytokine Growth Factor Rev (2015) 26(5):595–602. doi: 10.1016/j.cytogfr.2015.07.002
11. Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol (2020) 16(6):335–45. doi: 10.1038/s41584-020-0419-z
12. Akira S, Hirano T, Taga T, Kishimoto T. Biology of multifunctional cytokines: IL 6 and related molecules (IL 1 and TNF). FASEB J (1990) 4(11):2860–7. doi: 10.1096/fasebj.4.11.2199284
13. Abbas AK, Trotta E D, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol (2018) 3(25):1482. doi: 10.1126/sciimmunol.aat1482
14. Martinez-Moczygemba M, Huston DP. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol (2003) 112(4):653–65; quiz 66. doi: 10.1016/S0091
15. Deconinck E, Herve P. [Biology and clinical applications of the principal hematopoietic cytokines (GM-CSF, G-CSF, IL-3, IL-6, IL-1)]. Rev Fr Transfus Hemobiol. (1990) 33(4):259–90. doi: 10.1016/s1140-4639(05)80052-8
16. Mertowska P, Smolak K, Mertowski S, Grywalska E. Immunomodulatory role of interferons in viral and bacterial infections. Int J Mol Sci (2023) 24(12):10115. doi: 10.3390/ijms241210115
17. Yang Q, Shu HB. Deciphering the pathways to antiviral innate immunity and inflammation. Adv Immunol (2020) 145:1–36. doi: 10.1016/bs.ai.2019.11.001
18. Damsky W, Peterson D, Ramseier J, Al-Bawardy B, Chun H, Proctor D, et al. The emerging role of Janus kinase inhibitors in the treatment of autoimmune and inflammatory diseases. J Allergy Clin Immunol (2021) 147(3):814–26. doi: 10.1016/j.jaci.2020.10.022
19. Yang M, Wu S, Cai W, Ming X, Zhou Y, Chen X. Hypoxia-induced MIF induces dysregulation of lipid metabolism in Hep2 laryngocarcinoma through the IL-6/JAK-STAT pathway. Lipids Health Dis (2022) 21(1):82. doi: 10.1186/s12944-022-01693-z
20. Westerberg J, Tideholm E, Piersiala K, Drakskog C, Kumlien Georen S, Maki-Torkko E, et al. JAK/STAT dysregulation with SOCS1 overexpression in acquired cholesteatoma-adjacent mucosa. Otol Neurotol (2021) 42(1):e94–e100. doi: 10.1097/MAO.0000000000002850
21. Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological Malignancies and JAK inhibitors for clinical application. biomark Res (2013) 1(1):5. doi: 10.1186/2050-7771-1-5
22. Chim CS, Wong AS, Kwong YL. Epigenetic dysregulation of the Jak/STAT pathway by frequent aberrant methylation of SHP1 but not SOCS1 in acute leukaemias. Ann Hematol (2004) 83(8):527–32. doi: 10.1007/s00277-004-0843-1
23. Amoyel M, Anderson AM, Bach EA. JAK/STAT pathway dysregulation in tumors: a Drosophila perspective. Semin Cell Dev Biol (2014) 28:96–103. doi: 10.1016/j.semcdb.2014.03.023
24. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci (2018) 27(12):1984–2009. doi: 10.1002/pro.3519
25. Luo Y, Alexander M, Gadina M, O’Shea JJ, Meylan F, Schwartz DM. JAK-STAT signaling in human disease: From genetic syndromes to clinical inhibition. J Allergy Clin Immunol (2021) 148(4):911–25. doi: 10.1016/j.jaci.2021.08.004
26. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
27. Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu SN, Knoops L, et al. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica (2011) 96(6):845–53. doi: 10.3324/haematol.2010.036350
28. Gunawan AS, McLornan DP, Wilkins B, Waghorn K, Hoade Y, Cross NCP, et al. Ruxolitinib, a potent JAK1/JAK2 inhibitor, induces temporary reductions in the allelic burden of concurrent CSF3R mutations in chronic neutrophilic leukemia. Haematologica (2017) 102(6):e238–e40. doi: 10.3324/haematol.2017.163790
29. Pan Y, Pan H, Lian C, Wu B, Lin J, Huang G, et al. Case Report: Mutations in JAK3 causing severe combined immunodeficiency complicated by disseminated Bacille Calmette-Guerin disease and Pneumocystis pneumonia. Front Immunol (2022) 13:1055607. doi: 10.3389/fimmu.2022.1055607
30. O’Shea JJ, Husa M, Li D, Hofmann SR, Watford W, Roberts JL, et al. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol Immunol (2004) 41(6-7):727–37. doi: 10.1016/j.molimm.2004.04.014
31. Candotti F, Oakes SA, Johnston JA, Notarangelo LD, O’Shea JJ, Blaese RM. In vitro correction of JAK3-deficient severe combined immunodeficiency by retroviral-mediated gene transduction. J Exp Med (1996) 183(6):2687–92. doi: 10.1084/jem.183.6.2687
32. Bozzi F, Lefranc G, Villa A, Badolato R, Schumacher RF, Khalil G, et al. Molecular and biochemical characterization of JAK3 deficiency in a patient with severe combined immunodeficiency over 20 years after bone marrow transplantation: implications for treatment. Br J Haematol (1998) 102(5):1363–6. doi: 10.1111/j.1365-2141.1998.tb08990.x
33. Abolhassani H, Cheraghi T, Rezaei N, Aghamohammadi A, Hammarstrom L. Common variable immunodeficiency or late-onset combined immunodeficiency: A new hypomorphic JAK3 patient and review of the literature. J Investig Allergol Clin Immunol (2015) 25(3):218–20.
34. Tassara C, Pepper AE, Puck JM. Intronic point mutation in the IL2RG gene causing X-linked severe combined immunodeficiency. Hum Mol Genet (1995) 4(9):1693–5. doi: 10.1093/hmg/4.9.1693
35. Mou W, He J, Chen X, Zhang H, Ren X, Wu X, et al. A novel deletion mutation in IL2RG gene results in X-linked severe combined immunodeficiency with an atypical phenotype. Immunogenetics (2017) 69(1):29–38. doi: 10.1007/s00251-016-0949-3
36. Hou Y, Gratz HP, Urena-Bailen G, Gratz PG, Schilbach-Stuckle K, Renno T, et al. Somatic reversion of a novel IL2RG mutation resulting in atypical X-linked combined immunodeficiency. Genes (Basel) (2021) 13(1):35. doi: 10.3390/genes13010035
37. Kryworuchko M, Pasquier V, Theze J. Human immunodeficiency virus-1 envelope glycoproteins and anti-CD4 antibodies inhibit interleukin-2-induced Jak/STAT signalling in human CD4 T lymphocytes. Clin Exp Immunol (2003) 131(3):422–7. doi: 10.1046/j.1365-2249.2003.02065.x
38. Holland SM. Interferon gamma, IL-12, IL-12R and STAT-1 immunodeficiency diseases: disorders of the interface of innate and adaptive immunity. Immunol Res (2007) 38(1-3):342–6. doi: 10.1007/s12026-007-0045-8
39. Cornez I, Yajnanarayana SP, Wolf AM, Wolf D. JAK/STAT disruption induces immuno-deficiency: Rationale for the development of JAK inhibitors as immunosuppressive drugs. Mol Cell Endocrinol (2017) 451:88–96. doi: 10.1016/j.mce.2017.01.035
40. Land KJ, Moll JS, Kaplan MH, Seetharamaiah GS. Signal transducer and activator of transcription (Stat)-6-dependent, but not Stat4-dependent, immunity is required for the development of autoimmunity in Graves’ hyperthyroidism. Endocrinology (2004) 145(8):3724–30. doi: 10.1210/en.2004-0352
41. Cosio MG. Autoimmunity, T-cells and STAT-4 in the pathogenesis of chronic obstructive pulmonary disease. Eur Respir J (2004) 24(1):3–5. doi: 10.1183/09031936.04.00043104
42. Zhang Y, Lin T, Leung HM, Zhang C, Wilson-Mifsud B, Feldman MB, et al. STAT3 mutation-associated airway epithelial defects in Job syndrome. J Allergy Clin Immunol (2023) 152(2):538–50. doi: 10.1016/j.jaci.2022.12.821
43. Renner ED, Torgerson TR, Rylaarsdam S, Anover-Sombke S, Golob K, LaFlam T, et al. STAT3 mutation in the original patient with Job’s syndrome. N Engl J Med (2007) 357(16):1667–8. doi: 10.1056/NEJMc076367
44. Agashe RP, Lippman SM, Kurzrock R. JAK: not just another kinase. Mol Cancer Ther (2022) 21(12):1757–64. doi: 10.1158/1535-7163.MCT-22-0323
45. Pencik J, Pham HT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, et al. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine (2016) 87:26–36. doi: 10.1016/j.cyto.2016.06.017
46. Constantinescu SN, Girardot M, Pecquet C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci (2008) 33(3):122–31. doi: 10.1016/j.tibs.2007.12.002
47. Brooks AJ, Putoczki T. JAK-STAT signalling pathway in cancer. Cancers (Basel). (2020) 12(7):1971. doi: 10.3390/cancers12071971
48. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J (1995) 14(12):2816–26. doi: 10.1002/j.1460-2075.1995.tb07281.x
49. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature (1997) 387(6636):921–4. doi: 10.1038/43213
50. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature (1997) 387(6636):917–21. doi: 10.1038/43206
51. Hoftberger R, Titulaer MJ, Sabater L, Dome B, Rozsas A, Hegedus B, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology (2013) 81(17):1500–6. doi: 10.1212/WNL.0b013e3182a9585f
52. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature (1997) 387(6636):924–9. doi: 10.1038/43219
53. Wang B, Wangkahart E, Secombes CJ, Wang T. Insights into the evolution of the suppressors of cytokine signaling (SOCS) gene family in vertebrates. Mol Biol Evol (2019) 36(2):393–411. doi: 10.1093/molbev/msy230
54. Wang H, Zhou ZC, Peng XY, Zhao M. [Expression of suppressor of cytokine signaling in retina of experimental autoimmune uveoretinitis]. Zhonghua Yan Ke Za Zhi. (2008) 44(10):876–82.
55. Ni R, Ihara K, Miyako K, Takemoto M, Ishimura M, Kohno H, et al. Association study of polymorphisms in SOCS family genes with type 1 diabetes mellitus. Int J Immunogenet. (2006) 33(1):7–10. doi: 10.1111/j.1744-313X.2006.00563.x
56. Rojas-Morales E, Santos-Lopez G, Hernandez-Cabanas S, Arcega-Revilla R, Rosas-Murrieta N, Jasso-Miranda C, et al. Differential transcription of SOCS5 and SOCS7 in multiple sclerosis patients treated with interferon beta or glatiramer acetate. Int J Mol Sci (2019) 21(1):218. doi: 10.3390/ijms21010218
57. Zhang J, Guo Y, Sun Y, Chang L, Wang X. Inhibition of microRNA-448 suppresses CD4(+) T cell inflammatory activation via up-regulating suppressor of cytokine signaling 5 in systemic lupus erythematosus. Biochem Biophys Res Commun (2022) 596:88–96. doi: 10.1016/j.bbrc.2022.01.097
58. Wu L, Xia J, Li D, Kang Y, Fang W, Huang P. Mechanisms of M2 macrophage-derived exosomal long non-coding RNA PVT1 in regulating th17 cell response in experimental autoimmune encephalomyelitisa. Front Immunol (2020) 11:1934. doi: 10.3389/fimmu.2020.01934
59. Choi YB, Son M, Park M, Shin J, Yun Y. SOCS-6 negatively regulates T cell activation through targeting p56lck to proteasomal degradation. J Biol Chem (2010) 285(10):7271–80. doi: 10.1074/jbc.M109.073726
60. Abbas MN, Kausar S, Sun YX, Tian JW, Zhu BJ, Liu CL. Suppressor of cytokine signaling 6 can enhance epidermal growth factor receptor signaling pathway in Bombyx mori (Dazao). Dev Comp Immunol (2018) 81:187–92. doi: 10.1016/j.dci.2017.12.003
61. Fu B, Yin S, Lin X, Shi L, Wang Y, Zhang S, et al. PTPN14 aggravates inflammation through promoting proteasomal degradation of SOCS7 in acute liver failure. Cell Death Dis (2020) 11(9):803. doi: 10.1038/s41419-020-03014-7
62. Liu HS, Li LC, Wang M, Liu DS, Su Q, Zhang QH. Differentiated expressed miRNAs in splenic monocyte induced by burn injury in mice. Int Wound J (2023). doi: 10.1111/iwj.14288
63. Sobah ML, Liongue C, Ward AC. SOCS proteins in immunity, inflammatory diseases, and immune-related cancer. Front Med (Lausanne). (2021) 8:727987. doi: 10.3389/fmed.2021.727987
64. Kukenshoner T, Schmit NE, Bouda E, Sha F, Pojer F, Koide A, et al. Selective targeting of SH2 domain-phosphotyrosine interactions of Src family tyrosine kinases with monobodies. J Mol Biol (2017) 429(9):1364–80. doi: 10.1016/j.jmb.2017.03.023
65. Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell (2006) 22(2):205–16. doi: 10.1016/j.molcel.2006.03.024
66. Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev (2004) 18(24):3055–65. doi: 10.1101/gad.1252404
67. Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol (2009) 387(1):162–74. doi: 10.1016/j.jmb.2009.01.024
68. Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol (2013) 2(1):1–29.
69. Feng ZP, Chandrashekaran IR, Low A, Speed TP, Nicholson SE, Norton RS. The N-terminal domains of SOCS proteins: a conserved region in the disordered N-termini of SOCS4 and 5. Proteins (2012) 80(3):946–57. doi: 10.1002/prot.23252
70. Ushiki T, Huntington ND, Glaser SP, Kiu H, Georgiou A, Zhang JG, et al. Rapid inflammation in mice lacking both SOCS1 and SOCS3 in hematopoietic cells. PloS One (2016) 11(9):e0162111. doi: 10.1371/journal.pone.0162111
71. Yoshimura A, Ito M, Mise-Omata S, Ando M. SOCS: negative regulators of cytokine signaling for immune tolerance. Int Immunol (2021) 33(12):711–6. doi: 10.1093/intimm/dxab055
72. Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life (2012) 64(4):316–23. doi: 10.1002/iub.1011
73. Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HM, Hilton DJ. The SOCS box: a tale of destruction and degradation. Trends Biochem Sci (2002) 27(5):235–41. doi: 10.1016/S0968-0004(02)02085-6
74. Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science (1999) 284(5414):657–61. doi: 10.1126/science.284.5414.657
75. Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain (2015) 138(Pt 1):94–109. doi: 10.1093/brain/awu310
76. Kazi JU, Kabir NN, Flores-Morales A, Ronnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci (2014) 71(17):3297–310. doi: 10.1007/s00018-014-1619-y
77. Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev (2008) 19(5-6):371–81. doi: 10.1016/j.cytogfr.2008.08.006
78. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun (2018) 9(1):1558. doi: 10.1038/s41467-018-04013-1
79. Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol (2014) 26(1):13–9. doi: 10.1016/j.smim.2013.12.004
80. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J (2011) 25(3):863–74. doi: 10.1096/fj.10-170597
81. Prele CM, Woodward EA, Bisley J, Keith-Magee A, Nicholson SE, Hart PH. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages. J Immunol (2008) 181(11):8018–26. doi: 10.4049/jimmunol.181.11.8018
82. Chen Y, Zhong W, Xie Z, Li B, Li H, Gao K, et al. Suppressor of cytokine signaling 1 (SOCS1) inhibits antiviral responses to facilitate Senecavirus A infection by regulating the NF-kappaB signaling pathway. Virus Res (2022) 313:198748. doi: 10.1016/j.virusres.2022.198748
83. Coelho DR, Palma FR, Paviani V, LaFond KM, Huang Y, Wang D, et al. SOCS1 regulates a subset of NFkappaB-target genes through direct chromatin binding and defines macrophage functional phenotypes. iScience (2023) 26(4):106442. doi: 10.1016/j.isci.2023.106442
84. Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PloS One (2012) 7(10):e46082. doi: 10.1371/journal.pone.0046082
85. Ye J, Guo R, Shi Y, Qi F, Guo C, Yang L. miR-155 regulated inflammation response by the SOCS1-STAT3-PDCD4 axis in atherogenesis. Mediators Inflamm (2016) 2016:8060182. doi: 10.1155/2016/8060182
86. Gao Y, Wang R, Li L, He Y, Yuan D, Zhang Y, et al. Total saponins from Panax japonicus reduce inflammation in adipocytes through the miR155/SOCS1/NFkappaB signaling pathway. Phytomedicine (2023) 115:154827. doi: 10.1016/j.phymed.2023.154827
87. Qayum AA, Paranjape A, Abebayehu D, Kolawole EM, Haque TT, McLeod JJ, et al. IL-10-induced miR-155 targets SOCS1 to enhance IgE-mediated mast cell function. J Immunol (2016) 196(11):4457–67. doi: 10.4049/jimmunol.1502240
88. Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology (2012) 135(1):73–88. doi: 10.1111/j.1365-2567.2011.03514.x
89. Kim JH, Jou I, Joe EH. Suppression of miR-155 expression in IFN-gamma-treated astrocytes and microglia by DJ-1: A possible mechanism for maintaining SOCS1 expression. Exp Neurobiol (2014) 23(2):148–54. doi: 10.5607/en.2014.23.2.148
90. Soltani-Zangbar MS, Hajivalili M, Daneshdoust D, Ghadir S, Savari G, Zolfaghari M, et al. SARS-CoV2 infection induce miR-155 expression and skewed Th17/Treg balance by changing SOCS1 level: A clinical study. Cytokine (2023) 169:156248. doi: 10.1016/j.cyto.2023.156248
91. Iranparast S, Tahmasebi-Birgani M, Motamedfar A, Amari A, Ghafourian M. Altered expression levels of microRNA-155 and SOCS-1 in peripheral blood mononuclear cells of newly diagnosed breast cancer patients. Iran J Allergy Asthma Immunol (2022) 21(1):12–9. doi: 10.18502/ijaai.v21i1.8608
92. di Iasio MG, Norcio A, Melloni E, Zauli G. SOCS1 is significantly up-regulated in Nutlin-3-treated p53wild-type B chronic lymphocytic leukemia (B-CLL) samples and shows an inverse correlation with miR-155. Invest New Drugs (2012) 30(6):2403–6. doi: 10.1007/s10637-011-9786-2
93. Huang C, Li H, Wu W, Jiang T, Qiu Z. Regulation of miR-155 affects pancreatic cancer cell invasiveness and migration by modulating the STAT3 signaling pathway through SOCS1. Oncol Rep (2013) 30(3):1223–30. doi: 10.3892/or.2013.2576
94. Mignacca L, Saint-Germain E, Benoit A, Bourdeau V, Moro A, Ferbeyre G. Sponges against miR-19 and miR-155 reactivate the p53-Socs1 axis in hematopoietic cancers. Cytokine (2016) 82:80–6. doi: 10.1016/j.cyto.2016.01.015
95. Zhang W, Chen CJ, Guo GL. MiR-155 promotes the proliferation and migration of breast cancer cells via targeting SOCS1 and MMP16. Eur Rev Med Pharmacol Sci (2018) 22(21):7323–32. doi: 10.26355/eurrev_201811_16269
96. Limnander A, Danial NN, Rothman PB. v-Abl signaling disrupts SOCS-1 function in transformed pre-B cells. Mol Cell (2004) 15(3):329–41. doi: 10.1016/j.molcel.2004.06.041
97. Chen XP, Losman JA, Cowan S, Donahue E, Fay S, Vuong BQ, et al. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc Natl Acad Sci U S A. (2002) 99(4):2175–80. doi: 10.1073/pnas.042035699
98. Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood (2001) 97(1):221–6. doi: 10.1182/blood.V97.1.221
99. Federici M, Giustizieri ML, Scarponi C, Girolomoni G, Albanesi C. Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J Immunol (2002) 169(1):434–42. doi: 10.4049/jimmunol.169.1.434
100. Cornish AL, Chong MM, Davey GM, Darwiche R, Nicola NA, Hilton DJ, et al. Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other gamma c-dependent cytokines in peripheral T cells. J Biol Chem (2003) 278(25):22755–61. doi: 10.1074/jbc.M303021200
101. Ding Y, Chen D, Tarcsafalvi A, Su R, Qin L, Bromberg JS. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J Immunol (2003) 170(3):1383–91. doi: 10.4049/jimmunol.170.3.1383
102. van de Geijn GJ, Gits J, Touw IP. Distinct activities of suppressor of cytokine signaling (SOCS) proteins and involvement of the SOCS box in controlling G-CSF signaling. J Leukoc Biol (2004) 76(1):237–44. doi: 10.1189/jlb.0104041
103. Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature (2007) 448(7153):591–4. doi: 10.1038/nature06010
104. Mero IL, Ban M, Lorentzen AR, Smestad C, Celius EG, Saether H, et al. Exploring the CLEC16A gene reveals a MS-associated variant with correlation to the relative expression of CLEC16A isoforms in thymus. Genes Immun (2011) 12(3):191–8. doi: 10.1038/gene.2010.59
105. Zuvich RL, Bush WS, McCauley JL, Beecham AH, De Jager PL, International Multiple Sclerosis Genetics C, et al. Interrogating the complex role of chromosome 16p13.13 in multiple sclerosis susceptibility: independent genetic signals in the CIITA-CLEC16A-SOCS1 gene complex. Hum Mol Genet (2011) 20(17):3517–24. doi: 10.1093/hmg/ddr250
106. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun (2020) 11(1):5341. doi: 10.1038/s41467-020-18925-4
107. Wang H, Wang J, Xia Y. Defective suppressor of cytokine signaling 1 signaling contributes to the pathogenesis of systemic lupus erythematosus. Front Immunol (2017) 8:1292. doi: 10.3389/fimmu.2017.01292
108. Sharma J, Larkin J3. Therapeutic implication of SOCS1 modulation in the treatment of autoimmunity and cancer. Front Pharmacol (2019) 10:324. doi: 10.3389/fphar.2019.00324
109. Cooney RN. Suppressors of cytokine signaling (SOCS): inhibitors of the JAK/STAT pathway. Shock (2002) 17(2):83–90. doi: 10.1097/00024382-200202000-00001
110. Leikfoss IS, Mero IL, Dahle MK, Lie BA, Harbo HF, Spurkland A, et al. Multiple sclerosis-associated single-nucleotide polymorphisms in CLEC16A correlate with reduced SOCS1 and DEXI expression in the thymus. Genes Immun (2013) 14(1):62–6. doi: 10.1038/gene.2012.52
111. Kim H, Suh JM, Hwang ES, Kim DW, Chung HK, Song JH, et al. Thyrotropin-mediated repression of class II trans-activator expression in thyroid cells: involvement of STAT3 and suppressor of cytokine signaling. J Immunol (2003) 171(2):616–27. doi: 10.4049/jimmunol.171.2.616
112. Yoshimura A, Aki D, Ito M. SOCS. SPRED, and NR4a: Negative regulators of cytokine signaling and transcription in immune tolerance. Proc Jpn Acad Ser B Phys Biol Sci (2021) 97(6):277–91. doi: 10.2183/pjab.97.016
113. Pahlevan Kakhki M, Rakhshi N, Heidary M, Behmanesh M, Nikravesh A. Expression of suppressor of cytokine signaling 1 (SOCS1) gene dramatically increases in relapsing-remitting multiple sclerosis. J Neurol Sci (2015) 350(1-2):40–5. doi: 10.1016/j.jns.2015.02.005
114. The Wellcome Trust Case Control Consortium, Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447(7145):661–78. doi: 10.1038/nature05911
115. Awata T, Kawasaki E, Tanaka S, Ikegami H, Maruyama T, Shimada A, et al. Association of type 1 diabetes with two Loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J Clin Endocrinol Metab (2009) 94(1):231–5. doi: 10.1210/jc.2008-0718
116. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
117. Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet (2008) 40(12):1399–401. doi: 10.1038/ng.249
118. Howson JM, Rosinger S, Smyth DJ, Boehm BO, Todd JA. Genetic analysis of adult-onset autoimmune diabetes. Diabetes (2011) 60(10):2645–53. doi: 10.2337/db11-0364
119. Martinez A, Perdigones N, Cenit MC, Espino L, Varade J, Lamas JR, et al. Chromosomal region 16p13: further evidence of increased predisposition to immune diseases. Ann rheumatic diseases. (2010) 69(1):309–11. doi: 10.1136/ard.2008.098376
120. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47(4):381–6. doi: 10.1038/ng.3245
121. Sang Y, Zong W, Yan J, Liu M. The correlation between the CLEC16A gene and genetic susceptibility to type 1 diabetes in Chinese children. Int J Endocrinol (2012) 2012:245384. doi: 10.1155/2012/245384
122. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39(7):857–64. doi: 10.1038/ng2068
123. Wu X, Zhu X, Wang X, Ma J, Zhu S, Li J, et al. Intron polymorphism in the KIAA0350 gene is reproducibly associated with susceptibility to type 1 diabetes (T1D) in the Han Chinese population. Clin endocrinology. (2009) 71(1):46–9. doi: 10.1111/j.1365-2265.2008.03437.x
124. Yamashita H, Awata T, Kawasaki E, Ikegami H, Tanaka S, Maruyama T, et al. Analysis of the HLA and non-HLA susceptibility loci in Japanese type 1 diabetes. Diabetes/metabolism Res Rev (2011) 27(8):844–8. doi: 10.1002/dmrr.1234
125. Zoledziewska M, Costa G, Pitzalis M, Cocco E, Melis C, Moi L, et al. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun (2009) 10(1):15–7. doi: 10.1038/gene.2008.84
126. Plagnol V, Howson JM, Smyth DJ, Walker N, Hafler JP, Wallace C, et al. Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PloS Genet (2011) 7(8):e1002216. doi: 10.1371/journal.pgen.1002216
127. Steri M, Orru V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med (2017) 376(17):1615–26. doi: 10.1056/NEJMoa1610528
128. Johnson BA, Wang J, Taylor EM, Caillier SJ, Herbert J, Khan OA, et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun (2010) 11(4):343–50. doi: 10.1038/gene.2009.81
129. International Multiple Sclerosis Genetics Consortium (IMSGC), Booth DR, Heard RN, Stewart GJ, Goris A, Dobosi R, et al. International Multiple Sclerosis Genetics C. The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun (2009) 10(1):11–4. doi: 10.1038/gene.2008.83
130. De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet (2009) 41(7):776–82. doi: 10.1038/ng.401
131. D’Netto MJ, Ward H, Morrison KM, Ramagopalan SV, Dyment DA, DeLuca GC, et al. Risk alleles for multiple sclerosis in multiplex families. Neurology (2009) 72(23):1984–8. doi: 10.1212/WNL.0b013e3181a92c25
132. International Multiple Sclerosis Genetics C, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet (2013) 45(11):1353–60. doi: 10.1038/ng.2770
133. Andlauer TF, Buck D, Antony G, Bayas A, Bechmann L, Berthele A, et al. Novel multiple sclerosis susceptibility loci implicated in epigenetic regulation. Sci Adv (2016) 2(6):e1501678. doi: 10.1126/sciadv.1501678
134. Pandit L, Ban M, Sawcer S, Singhal B, Nair S, Radhakrishnan K, et al. Evaluation of the established non-MHC multiple sclerosis loci in an Indian population. Mult Scler. (2011) 17(2):139–43. doi: 10.1177/1352458510384011
135. Perera D, Stankovich J, Butzkueven H, Taylor BV, Foote SJ, Kilpatrick TJ, et al. Fine mapping of multiple sclerosis susceptibility genes provides evidence of allelic heterogeneity at the IL2RA locus. J Neuroimmunol. (2009) 211(1-2):105–9. doi: 10.1016/j.jneuroim.2009.03.010
136. Rubio JP, Stankovich J, Field J, Tubridy N, Marriott M, Chapman C, et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun (2008) 9(7):624–30. doi: 10.1038/gene.2008.59
137. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genomewide study. New Engl J Med (2007) 357(9):851–62. doi: 10.1056/NEJMoa073493
138. Hoppenbrouwers IA, Aulchenko YS, Janssens AC, Ramagopalan SV, Broer L, Kayser M, et al. Replication of CD58 and CLEC16A as genome-wide significant risk genes for multiple sclerosis. J Hum Genet (2009) 54(11):676–80. doi: 10.1038/jhg.2009.96
139. Nischwitz S, Cepok S, Kroner A, Wolf C, Knop M, Muller-Sarnowski F, et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol Scand (2011) 123(6):400–6. doi: 10.1111/j.1600-0404.2010.01421.x
140. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476(7359):214–9. doi: 10.1038/nature10251
141. Eriksson D, Bianchi M, Landegren N, Dalin F, Skov J, Hultin-Rosenberg L, et al. Common genetic variation in the autoimmune regulator (AIRE) locus is associated with autoimmune Addison’s disease in Sweden. Sci Rep (2018) 8(1):8395. doi: 10.1038/s41598-018-26842-2
142. Skinningsrud B, Husebye ES, Pearce SH, McDonald DO, Brandal K, Wolff AB, et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab (2008) 93(9):3310–7. doi: 10.1210/jc.2008-0821
143. Langefeld CD, Ainsworth HC, Cunninghame Graham DS, Kelly JA, Comeau ME, Marion MC, et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun (2017) 8:16021. doi: 10.1038/ncomms16021
144. Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet (2016) 48(8):940–6. doi: 10.1038/ng.3603
145. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet (2015) 47(12):1457–64. doi: 10.1038/ng.3434
146. Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet (2013) 92(1):41–51. doi: 10.1016/j.ajhg.2012.11.018
147. Marquez A, Varade J, Robledo G, Martinez A, Mendoza JL, Taxonera C, et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(-) Crohn’s disease patients. Eur J Hum Genet (2009) 17(10):1304–8. doi: 10.1038/ejhg.2009.50
148. Bronson PG, Chang D, Bhangale T, Seldin MF, Ortmann W, Ferreira RC, et al. Common variants at PVT1, ATG13-AMBRA1, AHI1 and CLEC16A are associated with selective IgA deficiency. Nat Genet (2016) 48(11):1425–9. doi: 10.1038/ng.3675
149. Betz RC, Petukhova L, Ripke S, Huang H, Menelaou A, Redler S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun (2015) 6:5966. doi: 10.1038/ncomms6966
150. Jagielska D, Redler S, Brockschmidt FF, Herold C, Pasternack SM, Garcia Bartels N, et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J Invest Dermatol (2012) 132(9):2192–7. doi: 10.1038/jid.2012.129
151. Skinningsrud B, Lie BA, Husebye ES, Kvien TK, Forre O, Flato B, et al. A CLEC16A variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann Rheum Dis (2010) 69(8):1471–4. doi: 10.1136/ard.2009.114934
152. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun (2015) 6:8019. doi: 10.1038/ncomms9019
153. Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet (2012) 44(10):1137–41. doi: 10.1038/ng.2395
154. Hirschfield GM, Xie G, Lu E, Sun Y, Juran BD, Chellappa V, et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun (2012) 13(4):328–35. doi: 10.1038/gene.2011.89
155. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet (2011) 43(4):329–32. doi: 10.1038/ng.789
156. Valette K, Li Z, Bon-Baret V, Chignon A, Berube JC, Eslami A, et al. Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK Biobank. Commun Biol (2021) 4(1):700. doi: 10.1038/s42003-021-02227-6
157. Shrine N, Portelli MA, John C, Soler Artigas M, Bennett N, Hall R, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med (2019) 7(1):20–34. doi: 10.1016/S2213-2600(18)30389-8
158. Pividori M, Schoettler N, Nicolae DL, Ober C, Im HK. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med (2019) 7(6):509–22. doi: 10.1016/S2213-2600(19)30055-4
159. Zhu Z, Lee PH, Chaffin MD, Chung W, Loh PR, Lu Q, et al. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat Genet (2018) 50(6):857–64. doi: 10.1038/s41588-018-0121-0
160. Demenais F, Margaritte-Jeannin P, Barnes KC, Cookson WOC, Altmuller J, Ang W, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet (2018) 50(1):42–53. doi: 10.1038/s41588-017-0014-7
161. Almoguera B, Vazquez L, Mentch F, Connolly J, Pacheco JA, Sundaresan AS, et al. Identification of four novel loci in asthma in European American and African American populations. Am J Respir Crit Care Med (2017) 195(4):456–63. doi: 10.1164/rccm.201604-0861OC
162. Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet (2016) 48(7):709–17. doi: 10.1038/ng.3570
163. Niu Y, Wang H, Li Z, Shamsi BH, Liu M, Liu J, et al. CLEC16A variants conferred a decreased risk to allergic rhinitis in the Chinese population. Front Genet (2022) 13:1053761. doi: 10.3389/fgene.2022.1053761
164. Gao Y, Li J, Zhang Y, Zhang L. Replication study of susceptibility variants associated with allergic rhinitis and allergy in Han Chinese. Allergy Asthma Clin Immunol (2020) 16:13. doi: 10.1186/s13223-020-0411-9
165. Muhali FS, Cai TT, Zhu JL, Qin Q, Xu J, He ST, et al. Polymorphisms of CLEC16A region and autoimmune thyroid diseases. G3 (Bethesda) (2014) 4(6):973–7. doi: 10.1534/g3.114.010926
166. Li J, Jorgensen SF, Maggadottir SM, Bakay M, Warnatz K, Glessner J, et al. Association of CLEC16A with human common variable immunodeficiency disorder and role in murine B cells. Nat Commun (2015) 6:6804. doi: 10.1038/ncomms7804
167. Kottyan LC, Maddox A, Braxton JR, Stucke EM, Mukkada V, Putnam PE, et al. Genetic variants at the 16p13 locus confer risk for eosinophilic esophagitis. Genes Immun (2019) 20(4):281–92. doi: 10.1038/s41435-018-0034-z
168. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet (2010) 42(4):295–302. doi: 10.1038/ng.543
169. Gorlova OY, Li Y, Gorlov I, Ying J, Chen WV, Assassi S, et al. Gene-level association analysis of systemic sclerosis: A comparison of African-Americans and White populations. PloS One (2018) 13(1):e0189498. doi: 10.1371/journal.pone.0189498
170. Strafella C, Caputo V, Termine A, Assogna F, Pellicano C, Pontieri FE, et al. Immune system and neuroinflammation in idiopathic parkinson’s disease: association analysis of genetic variants and miRNAs interactions. Front Genet (2021) 12:651971. doi: 10.3389/fgene.2021.651971
171. Fan HH, Cui L, Jiang XX, Song YD, Liu SS, Wu KY, et al. Autoimmune disease associated CLEC16A variants convey risk of parkinson’s disease in Han Chinese. Front Genet (2022) 13:856493. doi: 10.3389/fgene.2022.856493
172. Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci (2020) 21(19):7015. doi: 10.3390/ijms21197015
173. He C, Yang Y, Zheng K, Chen Y, Liu S, Li Y, et al. Mesenchymal stem cell-based treatment in autoimmune liver diseases: underlying roles, advantages and challenges. Ther Adv Chronic Dis (2021) 12:2040622321993442. doi: 10.1177/2040622321993442
174. Plichta DR, Somani J, Pichaud M, Wallace ZS, Fernandes AD, Perugino CA, et al. Congruent microbiome signatures in fibrosis-prone autoimmune diseases: IgG4-related disease and systemic sclerosis. Genome Med (2021) 13(1):35. doi: 10.1186/s13073-021-00853-7
175. Pandey R, Bakay M, Strenkowski BP, Hain HS, Hakonarson H. JAK/STAT inhibitor therapy partially rescues the lipodystrophic autoimmune phenotype in Clec16a KO mice. Sci Rep (2021) 11(1):7372. doi: 10.1038/s41598-021-86493-8
176. Tam RC, Li MW, Gao YP, Pang YT, Yan S, Ge W, et al. Human CLEC16A regulates autophagy through modulating mTOR activity. Exp Cell Res (2017) 352(2):304–12. doi: 10.1016/j.yexcr.2017.02.017
177. Kim S, Naylor SA, DiAntonio A. Drosophila Golgi membrane protein Ema promotes autophagosomal growth and function. Proc Natl Acad Sci U S A. (2012) 109(18):E1072–81. doi: 10.1073/pnas.1120320109
178. Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell (2014) 157(7):1577–90. doi: 10.1016/j.cell.2014.05.016
179. Hain HS, Pandey R, Bakay M, Strenkowski BP, Harrington D, Romer M, et al. Inducible knockout of Clec16a in mice results in sensory neurodegeneration. Sci Rep (2021) 11(1):9319. doi: 10.1038/s41598-021-88895-0
180. Smits DJ, Dekker J, Schot R, Tabarki B, Alhashem A, Demmers JAA, et al. CLEC16A interacts with retromer and TRIM27, and its loss impairs endosomal trafficking and neurodevelopment. Hum Genet (2023) 142(3):379–97. doi: 10.1007/s00439-022-02511-3
181. Ahmed CM, Larkin J 3rd, Johnson HM. SOCS1 mimetics and antagonists: A complementary approach to positive and negative regulation of immune function. Front Immunol (2015) 6:183. doi: 10.3389/fimmu.2015.00183
182. Lee MF, Poh CL. Strategies to improve the physicochemical properties of peptide-based drugs. Pharm Res (2023) 40(3):617–32. doi: 10.1007/s11095-023-03486-0
183. Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SM, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol (2004) 172(12):7510–8. doi: 10.4049/jimmunol.172.12.7510
184. Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol (2007) 178(8):5058–68. doi: 10.4049/jimmunol.178.8.5058
185. Jager LD, Dabelic R, Waiboci LW, Lau K, Haider MS, Ahmed CM, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol. (2011) 232(1-2):108–18. doi: 10.1016/j.jneuroim.2010.10.018
186. He C, Yu CR, Mattapallil MJ, Sun L, Larkin Iii J, Egwuagu CE. SOCS1 mimetic peptide suppresses chronic intraocular inflammatory disease (Uveitis). Mediators Inflamm (2016) 2016:2939370. doi: 10.1155/2016/2939370
187. Ahmed CM, Massengill MT, Brown EE, Ildefonso CJ, Johnson HM, Lewin AS. A cell penetrating peptide from SOCS-1 prevents ocular damage in experimental autoimmune uveitis. Exp Eye Res (2018) 177:12–22. doi: 10.1016/j.exer.2018.07.020
188. DiGiandomenico A, Wylezinski LS, Hawiger J. Intracellular delivery of a cell-penetrating SOCS1 that targets IFN-gamma signaling. Sci Signal (2009) 2(80):ra37. doi: 10.1126/scisignal.1162191
189. Fletcher TC, DiGiandomenico A, Hawiger J. Extended anti-inflammatory action of a degradation-resistant mutant of cell-penetrating suppressor of cytokine signaling 3. J Biol Chem (2010) 285(24):18727–36. doi: 10.1074/jbc.M109.095216
190. Egwuagu CE, Larkin Iii J. Therapeutic targeting of STAT pathways in CNS autoimmune diseases. JAKSTAT (2013) 2(1):e24134. doi: 10.4161/jkst.24134
191. He C, Yu CR, Sun L, Mahdi RM, Larkin J 3rd, Egwuagu CE. Topical administration of a suppressor of cytokine signaling-1 (SOCS1) mimetic peptide inhibits ocular inflammation and mitigates ocular pathology during mouse uveitis. J Autoimmun (2015) 62:31–8. doi: 10.1016/j.jaut.2015.05.011
192. Plummer CE, Polk T, Sharma J, Bae SS, Barr O, Jones A, et al. Open label safety and efficacy pilot to study mitigation of equine recurrent uveitis through topical suppressor of cytokine signaling-1 mimetic peptide. Sci Rep (2022) 12(1):7177. doi: 10.1038/s41598-022-11338-x
193. Sharma J, Collins TD, Roach T, Mishra S, Lam BK, Mohamed ZS, et al. Suppressor of cytokine signaling-1 mimetic peptides attenuate lymphocyte activation in the MRL/lpr mouse autoimmune model. Sci Rep (2021) 11(1):6354. doi: 10.1038/s41598-021-86017-4
194. Madonna S, Scarponi C, Doti N, Carbone T, Cavani A, Scognamiglio PL, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol (2013) 43(7):1883–95. doi: 10.1002/eji.201343370
195. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol (2013) 20(4):469–76. doi: 10.1038/nsmb.2519
196. Madonna S, Scarponi C, Morelli M, Sestito R, Scognamiglio PL, Marasco D, et al. SOCS3 inhibits the pathological effects of IL-22 in non-melanoma skin tumor-derived keratinocytes. Oncotarget (2017) 8(15):24652–67. doi: 10.18632/oncotarget.15629
197. Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. (2010) 120(9):3073–83. doi: 10.1172/JCI42440
198. Caspi RR. Immunogenetic aspects of clinical and experimental uveitis. Reg Immunol (1992) 4(5):321–30.
199. Diedrichs-Mohring M, Kaufmann U, Wildner G. The immunopathogenesis of chronic and relapsing autoimmune uveitis - Lessons from experimental rat models. Prog Retin Eye Res (2018) 65:107–26. doi: 10.1016/j.preteyeres.2018.02.003
200. Rosenbaum JT, Bodaghi B, Couto C, Zierhut M, Acharya N, Pavesio C, et al. New observations and emerging ideas in diagnosis and management of non-infectious uveitis: A review. Semin Arthritis Rheumatol (2019) 49(3):438–45. doi: 10.1016/j.semarthrit.2019.06.004
201. Witkowski L, Cywinska A, Paschalis-Trela K, Crisman M, Kita J. Multiple etiologies of equine recurrent uveitis–A natural model for human autoimmune uveitis: A brief review. Comp Immunol Microbiol Infect Dis (2016) 44:14–20. doi: 10.1016/j.cimid.2015.11.004
202. Bansal S, Barathi VA, Iwata D, Agrawal R. Experimental autoimmune uveitis and other animal models of uveitis: An update. Indian J Ophthalmol (2015) 63(3):211–8. doi: 10.4103/0301-4738.156914
203. Egwuagu CE, Alhakeem SA, Mbanefo EC. Uveitis: molecular pathogenesis and emerging therapies. Front Immunol (2021) 12:623725. doi: 10.3389/fimmu.2021.623725
204. Rodriguez-Fernandez CA, Iglesias MB, de Domingo B, Conde-Perez K, Vallejo JA, Rodriguez-Martinez L, et al. Microbiome in immune-mediated uveitis. Int J Mol Sci (2022) 23(13):7020. doi: 10.3390/ijms23137020
205. Chen J, Qian H, Horai R, Chan CC, Caspi RR. Mouse models of experimental autoimmune uveitis: comparative analysis of adjuvant-induced vs spontaneous models of uveitis. Curr Mol Med (2015) 15(6):550–7. doi: 10.2174/1566524015666150731100318
206. Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med (2007) 13(6):711–8. doi: 10.1038/nm1585
207. Huang XF, Brown MA. Progress in the genetics of uveitis. Genes Immun (2022) 23(2):57–65. doi: 10.1038/s41435-022-00168-6
208. Brewerton DA, Caffrey M, Nicholls A, Walters D, James DC. Acute anterior uveitis and HL-A 27. Lancet (1973) 302(7836):994–6. doi: 10.1016/S0140-6736(73)91090-8
209. Horai R, Chong WP, Zhou R, Chen J, Silver PB, Agarwal RK, et al. Spontaneous ocular autoimmunity in mice expressing a transgenic T cell receptor specific to retina: A tool to dissect mechanisms of uveitis. Curr Mol Med (2015) 15(6):511–6. doi: 10.2174/1566524015666150731095201
210. Mesquida M, Molins B, Llorenc V, de la Maza MS, Adan A. Targeting interleukin-6 in autoimmune uveitis. Autoimmun Rev (2017) 16(10):1079–89. doi: 10.1016/j.autrev.2017.08.002
211. Sota J, Rigante D, Lopalco G, Frediani B, Franceschini R, Galeazzi M, et al. Biological therapies for the treatment of Behcet’s disease-related uveitis beyond TNF-alpha blockade: a narrative review. Rheumatol Int (2018) 38(1):25–35. doi: 10.1007/s00296-017-3775-5
212. Chen YH, Lightman S, Calder VL. CD4(+) T-cell plasticity in non-infectious retinal inflammatory disease. Int J Mol Sci (2021) 22(17):9584. doi: 10.3390/ijms22179584
213. Su Y, Tao T, Liu X, Su W. JAK-STAT signaling pathway in non-infectious uveitis. Biochem Pharmacol (2022) 204:115236. doi: 10.1016/j.bcp.2022.115236
214. Yu CR, Lee YS, Mahdi RM, Surendran N, Egwuagu CE. Therapeutic targeting of STAT3 (signal transducers and activators of transcription 3) pathway inhibits experimental autoimmune uveitis. PloS One (2012) 7(1):e29742. doi: 10.1371/journal.pone.0029742
215. Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med (2014) 20(6):633–41. doi: 10.1038/nm.3554
216. Kang M, Choi JK, Jittayasothorn Y, Egwuagu CE. Interleukin 35-producing exosomes suppress neuroinflammation and autoimmune uveitis. Front Immunol (2020) 11:1051. doi: 10.3389/fimmu.2020.01051
217. Oladipupo FO, Yu CR, Olumuyide E, Jittaysothorn Y, Choi JK, Egwuagu CE. STAT3 deficiency in B cells exacerbates uveitis by promoting expansion of pathogenic lymphocytes and suppressing regulatory B cells (Bregs) and Tregs. Sci Rep (2020) 10(1):16188. doi: 10.1038/s41598-020-73093-1
218. Muhammad F, Trivett A, Wang D, Lee DJ. Tissue-specific production of MicroRNA-155 inhibits melanocortin 5 receptor-dependent suppressor macrophages to promote experimental autoimmune uveitis. Eur J Immunol (2019) 49(11):2074–82. doi: 10.1002/eji.201848073
219. Escobar T, Yu CR, Muljo SA, Egwuagu CE. STAT3 activates miR-155 in Th17 cells and acts in concert to promote experimental autoimmune uveitis. Invest Ophthalmol Vis Sci (2013) 54(6):4017–25. doi: 10.1167/iovs.13-11937
220. Burkholder BM, Jabs DA. Uveitis for the non-ophthalmologist. BMJ (2021) 372:m4979. doi: 10.1136/bmj.m4979
221. Collins EL, Jager LD, Dabelic R, Benitez P, Holdstein K, Lau K, et al. Inhibition of SOCS1-/- lethal autoinflammatory disease correlated to enhanced peripheral Foxp3+ regulatory T cell homeostasis. J Immunol (2011) 187(5):2666–76. doi: 10.4049/jimmunol.1003819
222. Recio C, Oguiza A, Lazaro I, Mallavia B, Egido J, Gomez-Guerrero C. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler Thromb Vasc Biol (2014) 34(9):1953–60. doi: 10.1161/ATVBAHA.114.304144
223. Yu CR, Mahdi RR, Oh HM, Amadi-Obi A, Levy-Clarke G, Burton J, et al. Suppressor of cytokine signaling-1 (SOCS1) inhibits lymphocyte recruitment into the retina and protects SOCS1 transgenic rats and mice from ocular inflammation. Invest Ophthalmol Vis Sci (2011) 52(9):6978–86. doi: 10.1167/iovs.11-7688
224. Nabieva K, Vender R. Quality of life and body region affected by psoriasis: A systematic review. Actas Dermosifiliogr. (2023) 114(1):T33–T8. doi: 10.1016/j.ad.2022.07.021
225. Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ (2020) 369:m1590. doi: 10.1136/bmj.m1590
226. Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. (2017) 31(2):205–12. doi: 10.1111/jdv.13854
227. Tinggaard AB, Hjuler KF, Andersen IT, Winther S, Iversen L, Bottcher M. Prevalence and severity of coronary artery disease linked to prognosis in psoriasis and psoriatic arthritis patients: a multi-centre cohort study. J Intern Med (2021) 290(3):693–703. doi: 10.1111/joim.13311
228. Merola JF, Tian H, Patil D, Richardson C, Scott A, Chen YH, et al. Incidence and prevalence of psoriatic arthritis in patients with psoriasis stratified by psoriasis disease severity: Retrospective analysis of an electronic health records database in the United States. J Am Acad Dermatol (2022) 86(4):748–57. doi: 10.1016/j.jaad.2021.09.019
229. Lebwohl MG, Kavanaugh A, Armstrong AW, Van Voorhees AS. US perspectives in the management of psoriasis and psoriatic arthritis: patient and physician results from the population-based multinational assessment of psoriasis and psoriatic arthritis (MAPP) survey. Am J Clin Dermatol (2016) 17(1):87–97. doi: 10.1007/s40257-015-0169-x
230. Miot HA, Criado PR, de Castro CCS, Ianhez M, Talhari C, Ramos PM. JAK-STAT pathway inhibitors in dermatology. Bras Dermatol (2023) 98(5):656–77. doi: 10.1016/j.abd.2023.03.001
231. Tokarski JS, Zupa-Fernandez A, Tredup JA, Pike K, Chang C, Xie D, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem (2015) 290(17):11061–74. doi: 10.1074/jbc.M114.619502
232. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev (2009) 228(1):273–87. doi: 10.1111/j.1600-065X.2008.00754.x
233. Xie J, LeBaron MJ, Nevalainen MT, Rui H. Role of tyrosine kinase Jak2 in prolactin-induced differentiation and growth of mammary epithelial cells. J Biol Chem (2002) 277(16):14020–30. doi: 10.1074/jbc.M112399200
234. Papadopoullos M, Yesudian PD. Understanding psoriasis: the development of the immune pathogenesis. Clin Exp Dermatol (2022) 47(11):2072–3. doi: 10.1111/ced.15360
235. Rusinol L, Puig L. Tyk2 targeting in immune-mediated inflammatory diseases. Int J Mol Sci (2023) 24(4):3391. doi: 10.3390/ijms24043391
236. Dand N, Mahil SK, Capon F, Smith CH, Simpson MA, Barker JN. Psoriasis and genetics. Acta Derm Venereol. (2020) 100(3):adv00030. doi: 10.2340/00015555-3384
237. Ran D, Cai M, Zhang X. Genetics of psoriasis: a basis for precision medicine. Precis Clin Med (2019) 2(2):120–30. doi: 10.1093/pcmedi/pbz011
238. Zeng J, Luo S, Huang Y, Lu Q. Critical role of environmental factors in the pathogenesis of psoriasis. J Dermatol (2017) 44(8):863–72. doi: 10.1111/1346-8138.13806
239. Roszkiewicz M, Dopytalska K, Szymanska E, Jakimiuk A, Walecka I. Environmental risk factors and epigenetic alternations in psoriasis. Ann Agric Environ Med (2020) 27(3):335–42. doi: 10.26444/aaem/112107
240. Gangwar RS, Gudjonsson JE, Ward NL. Mouse models of psoriasis: A comprehensive review. J Invest Dermatol (2022) 142(3 Pt B):884–97. doi: 10.1016/j.jid.2021.06.019
241. Ma HL, Masek-Hammerman K, Fish S, Napierata L, Nagiec E. Attenuating janus kinases (JAK) by tofacitinib effectively prevented psoriasis pathology in various mouse skin inflammation models. J Clin Cell Immunol (2013) 4(6):176–83. doi: 10.4172/2155-9899.1000176
242. Fukuyama T, Tschernig T, Qi Y, Volmer DA, Baumer W. Aggression behaviour induced by oral administration of the Janus-kinase inhibitor tofacitinib, but not oclacitinib, under stressful conditions. Eur J Pharmacol (2015) 764:278–82. doi: 10.1016/j.ejphar.2015.06.060
243. Zhou Y, Leng X, Luo S, Su Z, Luo X, Guo H, et al. Tolerogenic dendritic cells generated with tofacitinib ameliorate experimental autoimmune encephalomyelitis through modulation of Th17/Treg balance. J Immunol Res (2016) 2016:5021537. doi: 10.1155/2016/5021537
244. Hashimoto T, Sakai K, Sanders KM, Yosipovitch G, Akiyama T. Antipruritic effects of Janus kinase inhibitor tofacitinib in a mouse model of psoriasis. Acta Derm Venereol. (2019) 99(3):298–303. doi: 10.2340/00015555-3086
245. Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol (2012) 167(3):668–77. doi: 10.1111/j.1365-2133.2012.11168.x
246. Mamolo C, Harness J, Tan H, Menter A. Tofacitinib (CP-690,550), an oral Janus kinase inhibitor, improves patient-reported outcomes in a phase 2b, randomized, double-blind, placebo-controlled study in patients with moderate-to-severe psoriasis. J Eur Acad Dermatol Venereol. (2014) 28(2):192–203. doi: 10.1111/jdv.12081
247. Krueger J, Clark JD, Suarez-Farinas M, Fuentes-Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: A randomized phase 2 study. J Allergy Clin Immunol (2016) 137(4):1079–90. doi: 10.1016/j.jaci.2015.12.1318
248. Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet (2015) 386(9993):552–61. doi: 10.1016/S0140-6736(14)62113-9
249. Merola JF, Elewski B, Tatulych S, Lan S, Tallman A, Kaur M. Efficacy of tofacitinib for the treatment of nail psoriasis: Two 52-week, randomized, controlled phase 3 studies in patients with moderate-to-severe plaque psoriasis. J Am Acad Dermatol (2017) 77(1):79–87 e1. doi: 10.1016/j.jaad.2017.01.053
250. Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol (2016) 74(5):841–50. doi: 10.1016/j.jaad.2016.01.013
251. Zhang J, Tsai TF, Lee MG, Zheng M, Wang G, Jin H, et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: A Phase 3, randomized, double-blind, placebo-controlled study. J Dermatol Sci (2017) 88(1):36–45. doi: 10.1016/j.jdermsci.2017.05.004
252. Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br J Dermatol (2016) 174(5):985–95. doi: 10.1111/bjd.14399
253. Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br J Dermatol (2018) 179(1):54–62. doi: 10.1111/bjd.16004
254. Fensome A, Ambler CM, Arnold E, Banker ME, Brown MF, Chrencik J, et al. Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: discovery of ((S)-2,2-difluorocyclopropyl)((1 R,5 S)-3-(2-((1-methyl-1 H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J Med Chem (2018) 61(19):8597–612. doi: 10.1021/acs.jmedchem.8b00917
255. Jo CE, Gooderham M, Beecker J. TYK 2 inhibitors for the treatment of dermatologic conditions: the evolution of JAK inhibitors. Int J Dermatol (2022) 61(2):139–47. doi: 10.1111/ijd.15605
256. Banfield C, Scaramozza M, Zhang W, Kieras E, Page KM, Fensome A, et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of a TYK2/JAK1 inhibitor (PF-06700841) in healthy subjects and patients with plaque psoriasis. J Clin Pharmacol (2018) 58(4):434–47. doi: 10.1002/jcph.1046
257. Forman SB, Pariser DM, Poulin Y, Vincent MS, Gilbert SA, Kieras EM, et al. TYK2/JAK1 inhibitor PF-06700841 in patients with plaque psoriasis: phase IIa, randomized, double-blind, placebo-controlled trial. J Invest Dermatol (2020) 140(12):2359–70 e5. doi: 10.1016/j.jid.2020.03.962
258. Danese S, Peyrin-Biroulet L. Selective tyrosine kinase 2 inhibition for treatment of inflammatory bowel disease: new hope on the rise. Inflammation Bowel Dis (2021) 27(12):2023–30. doi: 10.1093/ibd/izab135
259. Singh RSP, Pradhan V, Roberts ES, Scaramozza M, Kieras E, Gale JD, et al. Safety and pharmacokinetics of the oral TYK2 inhibitor PF-06826647: A phase I, randomized, double-blind, placebo-controlled, dose-escalation study. Clin Transl Sci (2021) 14(2):671–82. doi: 10.1111/cts.12929
260. Tehlirian C, Singh RSP, Pradhan V, Roberts ES, Tarabar S, Peeva E, et al. Oral tyrosine kinase 2 inhibitor PF-06826647 demonstrates efficacy and an acceptable safety profile in participants with moderate-to-severe plaque psoriasis in a phase 2b, randomized, double-blind, placebo-controlled study. J Am Acad Dermatol (2022) 87(2):333–42. doi: 10.1016/j.jaad.2022.03.059
261. Zhang L, Guo L, Wang L, Jiang X. The efficacy and safety of tofacitinib, peficitinib, solcitinib, baricitinib, abrocitinib and deucravacitinib in plaque psoriasis - A network meta-analysis. J Eur Acad Dermatol Venereol. (2022) 36(11):1937–46. doi: 10.1111/jdv.18263
262. Doti N, Scognamiglio PL, Madonna S, Scarponi C, Ruvo M, Perretta G, et al. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem J (2012) 443(1):231–40. doi: 10.1042/BJ20111647
263. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med (2018) 378(9):840–51. doi: 10.1056/NEJMra1708712
264. Al-Diwani A, Handel A, Townsend L, Pollak T, Leite MI, Harrison PJ, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry (2019) 6(3):235–46. doi: 10.1016/S2215-0366(19)30001-X
265. van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology (2016) 87(14):1449–56. doi: 10.1212/WNL.0000000000003173
266. van Sonderen A, Arino H, Petit-Pedrol M, Leypoldt F, Kortvelyessy P, Wandinger KP, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology (2016) 87(5):521–8. doi: 10.1212/WNL.0000000000002917
267. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol (2010) 9(1):67–76. doi: 10.1016/S1474-4422(09)70324-2
268. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol (2014) 13(3):276–86. doi: 10.1016/S1474-4422(13)70299-0
269. Laurido-Soto O, Brier MR, Simon LE, McCullough A, Bucelli RC, Day GS. Patient characteristics and outcome associations in AMPA receptor encephalitis. J Neurol (2019) 266(2):450–60. doi: 10.1007/s00415-018-9153-8
270. Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology (2014) 83(20):1797–803. doi: 10.1212/WNL.0000000000000991
271. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein igG disease vs aquaporin 4 igG disease. JAMA Neurol (2018) 75(1):65–71. doi: 10.1001/jamaneurol.2017.3196
272. Kanniah G, Kumar R, Subramaniam G. Anti-NMDA receptor encephalitis: A challenge in psychiatric settings. J Psychiatr Pract (2022) 28(1):78–83. doi: 10.1097/PRA.0000000000000602
273. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol (2016) 15(4):391–404. doi: 10.1016/S1474-4422(15)00401-9
274. Esposito S, Principi N, Calabresi P, Rigante D. An evolving redefinition of autoimmune encephalitis. Autoimmun Rev (2019) 18(2):155–63. doi: 10.1016/j.autrev.2018.08.009
275. Hebert J, Muccilli A, Wennberg RA, Tang-Wai DF. Autoimmune encephalitis and autoantibodies: A review of clinical implications. J Appl Lab Med (2022) 7(1):81–98. doi: 10.1093/jalm/jfab102
276. Armangue T, Spatola M, Vlagea A, Mattozzi S, Carceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol (2018) 17(9):760–72. doi: 10.1016/S1474-4422(18)30244-8
277. Cabezudo-Garcia P, Mena-Vazquez N, Estivill Torrus G, Serrano-Castro P. Response to immunotherapy in anti-IgLON5 disease: A systematic review. Acta Neurol Scand (2020) 141(4):263–70. doi: 10.1111/ane.13207
278. Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain (2016) 139(Pt 10):2641–52. doi: 10.1093/brain/aww208
279. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30(17):5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
280. Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci (2013) 33(46):18161–74. doi: 10.1523/JNEUROSCI.3506-13.2013
281. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis (2012) 54(7):899–904. doi: 10.1093/cid/cir1038
282. Carceles-Cordon M, Mannara F, Aguilar E, Castellanos A, Planaguma J, Dalmau J. NMDAR antibodies alter dopamine receptors and cause psychotic behavior in mice. Ann Neurol (2020) 88(3):603–13. doi: 10.1002/ana.25829
283. Taraschenko O, Fox HS, Pittock SJ, Zekeridou A, Gafurova M, Eldridge E, et al. A mouse model of seizures in anti-N-methyl-d-aspartate receptor encephalitis. Epilepsia (2019) 60(3):452–63. doi: 10.1111/epi.14662
284. Dileepan T, Smith ED, Knowland D, Hsu M, Platt M, Bittner-Eddy P, et al. Group A Streptococcus intranasal infection promotes CNS infiltration by streptococcal-specific Th17 cells. J Clin Invest. (2016) 126(1):303–17. doi: 10.1172/JCI80792
285. Platt MP, Bolding KA, Wayne CR, Chaudhry S, Cutforth T, Franks KM, et al. Th17 lymphocytes drive vascular and neuronal deficits in a mouse model of postinfectious autoimmune encephalitis. Proc Natl Acad Sci U S A. (2020) 117(12):6708–16. doi: 10.1073/pnas.1911097117
286. Jang Y, Lee WJ, Lee HS, Chu K, Lee SK, Lee ST. Tofacitinib treatment for refractory autoimmune encephalitis. Epilepsia (2021) 62(4):e53–e9. doi: 10.1111/epi.16848
287. Dinoto A, Ferrari S, Mariotto S. Treatment options in refractory autoimmune encephalitis. CNS Drugs (2022) 36(9):919–31. doi: 10.1007/s40263-022-00943-z
288. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol (2014) 76(1):108–19. doi: 10.1002/ana.24195
289. Kortvelyessy P, Bauer J, Stoppel CM, Bruck W, Gerth I, Vielhaber S, et al. Complement-associated neuronal loss in a patient with CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm. (2015) 2(2):e75. doi: 10.1212/NXI.0000000000000075
290. Linnoila J, Jalali Motlagh N, Jachimiec G, Lin CJ, Kullenberg E, Wojtkiewicz G, et al. Optimizing animal models of autoimmune encephalitis using active immunization. Front Immunol (2023) 14:1177672. doi: 10.3389/fimmu.2023.1177672
291. Dai Y, Zhang J, Ren H, Zhou X, Chen J, Cui L, et al. Surgical outcomes in patients with anti-N-methyl D-aspartate receptor encephalitis with ovarian teratoma. Am J Obstet Gynecol. (2019) 221(5):485 e1– e10. doi: 10.1016/j.ajog.2019.05.026
292. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol (2013) 12(2):157–65. doi: 10.1016/S1474-4422(12)70310-1
293. Li Q, Fu N, Han Y, Qin J. Pediatric autoimmune encephalitis and its relationship with infection. Pediatr Neurol (2021) 120:27–32. doi: 10.1016/j.pediatrneurol.2021.04.001
294. Dubey D, Britton J, McKeon A, Gadoth A, Zekeridou A, Lopez Chiriboga SA, et al. Randomized placebo-controlled trial of intravenous immunoglobulin in autoimmune LGI1/CASPR2 epilepsy. Ann Neurol (2020) 87(2):313–23. doi: 10.1002/ana.25655
295. Wickel J, Chung HY, Platzer S, Lehmann T, Pruss H, Leypoldt F, et al. Generate-Boost: study protocol for a prospective, multicenter, randomized controlled, double-blinded phase II trial to evaluate efficacy and safety of bortezomib in patients with severe autoimmune encephalitis. Trials (2020) 21(1):625. doi: 10.1186/s13063-020-04516-7
296. Blackburn KM, Denney DA, Hopkins SC, Vernino SA. Low recruitment in a double-blind, placebo-controlled trial of ocrelizumab for autoimmune encephalitis: A case series and review of lessons learned. Neurol Ther (2022) 11(2):893–903. doi: 10.1007/s40120-022-00327-x
297. Dalmau J, Armangue T, Planaguma J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol (2019) 18(11):1045–57. doi: 10.1016/S1474-4422(19)30244-3
298. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Seve P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev (2019) 18(11):102390. doi: 10.1016/j.autrev.2019.102390
299. Dowty ME, Lin J, Ryder TF, Wang W, Walker GS, Vaz A, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab Dispos (2014) 42(4):759–73. doi: 10.1124/dmd.113.054940
300. Yang J, Liu X. Immunotherapy for refractory autoimmune encephalitis. Front Immunol (2021) 12:790962. doi: 10.3389/fimmu.2021.790962
301. Pathak S, Mohan C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Ther (2011) 13(5):241. doi: 10.1186/ar3465
302. Uhm WS, Na K, Song GW, Jung SS, Lee T, Park MH, et al. Cytokine balance in kidney tissue from lupus nephritis patients. Rheumatol (Oxford). (2003) 42(8):935–8. doi: 10.1093/rheumatology/keg255
303. Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol (2014) 47(2):148–62. doi: 10.1007/s12016-013-8403-x
304. Lettre G, Rioux JD. Autoimmune diseases: insights from genome-wide association studies. Hum Mol Genet (2008) 17(R2):R116–21. doi: 10.1093/hmg/ddn246
305. Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet (2005) 76(3):528–37. doi: 10.1086/428480
306. Chong BF, Mohan C. Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin Ther Targets. (2009) 13(10):1147–53. doi: 10.1517/14728220903196761
307. Wang A, Fairhurst AM, Tus K, Subramanian S, Liu Y, Lin F, et al. CXCR4/CXCL12 hyperexpression plays a pivotal role in the pathogenesis of lupus. J Immunol (2009) 182(7):4448–58. doi: 10.4049/jimmunol.0801920
308. Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, Moreno-Ortiz MC, Martinez AC, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J (1999) 13(13):1699–710. doi: 10.1096/fasebj.13.13.1699
309. Richard ML, Gilkeson G. Mouse models of lupus: what they tell us and what they don’t. Lupus Sci Med (2018) 5(1):e000199. doi: 10.1136/lupus-2016-000199
310. Reilly CM, Gilkeson GS. Use of genetic knockouts to modulate disease expression in a murine model of lupus, MRL/lpr mice. Immunol Res (2002) 25(2):143–53. doi: 10.1385/IR:25:2:143
311. Chan ES, Herlitz LC, Jabbari A. Ruxolitinib attenuates cutaneous lupus development in a mouse lupus model. J Invest Dermatol (2015) 135(7):1912–5. doi: 10.1038/jid.2015.107
312. Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol (2012) 189(2):988–1001. doi: 10.4049/jimmunol.1103031
313. Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol (2017) 69(1):148–60. doi: 10.1002/art.39818
314. Ikeda K, Hayakawa K, Fujishiro M, Kawasaki M, Hirai T, Tsushima H, et al. JAK inhibitor has the amelioration effect in lupus-prone mice: the involvement of IFN signature gene downregulation. BMC Immunol (2017) 18(1):41. doi: 10.1186/s12865-017-0225-9
315. Yan Q, Chen W, Song H, Long X, Zhang Z, Tang X, et al. Tofacitinib ameliorates lupus through suppression of T cell activation mediated by TGF-beta type I receptor. Front Immunol (2021) 12:675542. doi: 10.3389/fimmu.2021.675542
316. Lee J, Park Y, Jang SG, Hong SM, Song YS, Kim MJ, et al. Baricitinib attenuates autoimmune phenotype and podocyte injury in a murine model of systemic lupus erythematosus. Front Immunol (2021) 12:704526. doi: 10.3389/fimmu.2021.704526
317. Burke JR, Cheng L, Gillooly KM, Strnad J, Zupa-Fernandez A, Catlett IM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med (2019) 11(502):1736. doi: 10.1126/scitranslmed.aaw1736
318. van Vollenhoven RF, Layton M, Kahl L, Schifano L, Hachulla E, MaChado D, et al. DRESS syndrome and reversible liver function abnormalities in patients with systemic lupus erythematosus treated with the highly selective JAK-1 inhibitor GSK2586184. Lupus (2015) 24(6):648–9. doi: 10.1177/0961203315573347
319. Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus (2016) 25(13):1420–30. doi: 10.1177/0961203316640910
320. Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun (2021) 12(1):3391. doi: 10.1038/s41467-021-23361-z
321. Bonnardeaux E, Dutz JP. Oral tofacitinib citrate for recalcitrant cutaneous lupus. JAAD Case Rep (2022) 20:61–4. doi: 10.1016/j.jdcr.2021.09.030
322. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet (2018) 392(10143):222–31. doi: 10.1016/S0140-6736(18)31363-1
323. Dorner T, Tanaka Y, Dow ER, Koch AE, Silk M, Ross Terres JA, et al. Mechanism of action of baricitinib and identification of biomarkers and key immune pathways in patients with active systemic lupus erythematosus. Ann Rheum Dis (2022) 81(9):1267–72. doi: 10.1136/annrheumdis-2022-222335
324. Maeshima K, Shibata H. Efficacy of JAK 1/2 inhibition in the treatment of diffuse non-scarring alopecia due to systemic lupus erythematosus. Ann Rheum Dis (2020) 79(5):674–5. doi: 10.1136/annrheumdis-2019-216571
325. Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective janus kinase 1 inhibition is a promising therapeutic approach for lupus erythematosus skin lesions. Front Immunol (2020) 11:344. doi: 10.3389/fimmu.2020.00344
326. Werth VP, Fleischmann R, Robern M, Touma Z, Tiamiyu I, Gurtovaya O, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Rheumatol (Oxford). (2022) 61(6):2413–23. doi: 10.1093/rheumatology/keab685
327. Baker M, Chaichian Y, Genovese M, Derebail V, Rao P, Chatham W, et al. Phase II, randomised, double-blind, multicentre study evaluating the safety and efficacy of filgotinib and lanraplenib in patients with lupus membranous nephropathy. RMD Open (2020) 6(3):e001490. doi: 10.1136/rmdopen-2020-001490
328. Duggan S, Keam SJ. Upadacitinib: first approval. Drugs (2019) 79(16):1819–28. doi: 10.1007/s40265-019-01211-z
329. Muensterman E, Engelhardt B, Gopalakrishnan S, Anderson JK, Mohamed MF. Upadacitinib pharmacokinetics and exposure-response analyses of efficacy and safety in psoriatic arthritis patients - Analyses of phase III clinical trials. Clin Transl Sci (2022) 15(1):267–78. doi: 10.1111/cts.13146
330. Michailidou D, Long TH, Argenyi ZB, Noss EH. Resolution of accelerated nodulosis with upadacitinib in a patient with systemic lupus erythematosus and Jaccoud’s arthropathy. Clin Exp Rheumatol (2022) 135(9):11–2. doi: 10.55563/clinexprheumatol/hkokxs
331. Catlett IM, Aras U, Hansen L, Liu Y, Bei D, Girgis IG, et al. First-in-human study of deucravacitinib: A selective, potent, allosteric small-molecule inhibitor of tyrosine kinase 2. Clin Transl Sci (2023) 16(1):151–64. doi: 10.1111/cts.13435
332. Hoy SM. Deucravacitinib: first approval. Drugs (2022) 82(17):1671–9. doi: 10.1007/s40265-022-01796-y
333. Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: A phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol (2023) 75(2):242–52. doi: 10.1002/art.42391
334. Qiu LJ, Xu K, Liang Y, Cen H, Zhang M, Wen PF, et al. Decreased SOCS1 mRNA expression levels in peripheral blood mononuclear cells from patients with systemic lupus erythematosus in a Chinese population. Clin Exp Med (2015) 15(3):261–7. doi: 10.1007/s10238-014-0309-2
335. Li J, Zhao S, Yi M, Hu X, Li J, Xie H, et al. Activation of JAK-STAT1 signal transduction pathway in lesional skin and monocytes from patients with systemic lupus erythematosus. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2011) 36(2):109–15. doi: 10.3969/j.issn.1672-7347.2011.02.003
336. Sukka-Ganesh B, Larkin J 3rd. Therapeutic potential for targeting the suppressor of cytokine signalling-1 pathway for the treatment of SLE. Scand J Immunol (2016) 84(5):299–309. doi: 10.1111/sji.12475
337. Chen J, Jia F, Ren K, Luo M, Min X, Wang P, et al. Inhibition of suppressor of cytokine signaling 1 mediates the profibrotic effect of TWEAK/Fn14 signaling on kidney cells. Cell Signal (2020) 71:109615. doi: 10.1016/j.cellsig.2020.109615
338. Mascarenhas J, Hoffman R. Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin Cancer Res (2012) 18(11):3008–14. doi: 10.1158/1078-0432.CCR-11-3145
339. Raedler LA. Jakafi (Ruxolitinib): first FDA-approved medication for the treatment of patients with polycythemia vera. Am Health Drug Benefits. (2015) 8(Spec Feature):75–9.
340. Yang W, Zhu G, Qin M, Li Z, Wang B, Yang J, et al. The effectiveness of ruxolitinib for acute/chronic graft-versus-host disease in children: A retrospective study. Drug Des Devel Ther (2021) 15:743–52. doi: 10.2147/DDDT.S287218
341. Kostik MM, Raupov RK, Suspitsin EN, Isupova EA, Gaidar EV, Gabrusskaya TV, et al. The safety and efficacy of tofacitinib in 24 cases of pediatric rheumatic diseases: single centre experience. Front Pediatr (2022) 10:820586. doi: 10.3389/fped.2022.820586
342. Ayala-Aguilera CC, Valero T, Lorente-Macias A, Baillache DJ, Croke S, Unciti-Broceta A. Small molecule kinase inhibitor drugs (1995-2021): medical indication, pharmacology, and synthesis. J Med Chem (2022) 65(2):1047–131. doi: 10.1021/acs.jmedchem.1c00963
343. Coricello A, Mesiti F, Lupia A, Maruca A, Alcaro S. Inside perspective of the synthetic and computational toolbox of JAK inhibitors: recent updates. Molecules (2020) 25(15):3321. doi: 10.3390/molecules25153321
344. Mohanakrishnan R, Beier S, Deodhar A. Tofacitinib for the treatment of active ankylosing spondylitis in adults. Expert Rev Clin Immunol (2022) 18(3):273–80. doi: 10.1080/1744666X.2022.2038134
345. Markham A. Baricitinib: first global approval. Drugs (2017) 77(6):697–704. doi: 10.1007/s40265-017-0723-3
346. FDA Fact Sheet for Healthcare Providers Emergency use Authorization (EUA) of Baricitinib [press release] (2022).
347. Blair HA. Fedratinib: first approval. Drugs (2019) 79(15):1719–25. doi: 10.1007/s40265-019-01205-x
348. Markham A, Keam SJ. Peficitinib: first global approval. Drugs (2019) 79(8):887–91. doi: 10.1007/s40265-019-01131-y
349. U.S. FDA approves RINVOQ® (upadacitinib) to treat adults and children 12 years and older with refractory, moderate to severe atopic dermatitis [press release] (2022).
350. RINVOQ® (upadacitinib) receives FDA approval for the treatment of adults with moderately to severely active ulcerative colitis [press release] (2022).
351. Dhillon S. Delgocitinib: first approval. Drugs (2020) 80(6):609–15. doi: 10.1007/s40265-020-01291-2
352. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs (2020) 80(18):1987–97. doi: 10.1007/s40265-020-01439-0
353. Deeks ED, Duggan S. Abrocitinib: first approval. Drugs (2021) 81(18):2149–57. doi: 10.1007/s40265-021-01638-3
Keywords: JAK-STAT, inflammation, autoimmunity, SOCS (suppressor of cytokine signaling), psoriasis, uveitis, autoimmune encephalitis (AE), lupus
Citation: Pandey R, Bakay M and Hakonarson H (2023) SOCS-JAK-STAT inhibitors and SOCS mimetics as treatment options for autoimmune uveitis, psoriasis, lupus, and autoimmune encephalitis. Front. Immunol. 14:1271102. doi: 10.3389/fimmu.2023.1271102
Received: 01 August 2023; Accepted: 02 October 2023;
Published: 26 October 2023.
Edited by:
Kamran Ghoreschi, Charité University Medicine Berlin, GermanyReviewed by:
Thierry Mp Gauthier, National Institutes of Health (NIH), United StatesHoward M. Johnson, University of Florida, United States
Copyright © 2023 Pandey, Bakay and Hakonarson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hakon Hakonarson, aGFrb25hcnNvbkBjaG9wLmVkdQ==