 Meghan A. Berryman
Meghan A. Berryman Jorma Ilonen2
Jorma Ilonen2 Eric W. Triplett
Eric W. Triplett Johnny Ludvigsson
Johnny Ludvigsson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 September 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1270488
This article is part of the Research Topic Novel Understandings of HLA Genes in Pathogenic Mechanisms of Autoimmunity View all 5 articles
Human leukocyte antigen (HLA) genes are associated with more diseases than any other region of the genome. Highly polymorphic HLA genes produce variable haplotypes that are specifically correlated with pathogenically different autoimmunities. Despite differing etiologies, however, many autoimmune disorders share the same risk-associated HLA haplotypes often resulting in comorbidity. This shared risk remains an unanswered question in the field. Yet, several groups have revealed links between gut microbial community composition and autoimmune diseases. Autoimmunity is frequently associated with dysbiosis, resulting in loss of barrier function and permeability of tight junctions, which increases HLA class II expression levels and thus further influences the composition of the gut microbiome. However, autoimmune-risk-associated HLA haplotypes are connected to gut dysbiosis long before autoimmunity even begins. This review evaluates current research on the HLA-microbiome-autoimmunity triplex and proposes that pre-autoimmune bacterial dysbiosis in the gut is an important determinant between autoimmune comorbidities with systemic inflammation as a common denominator.
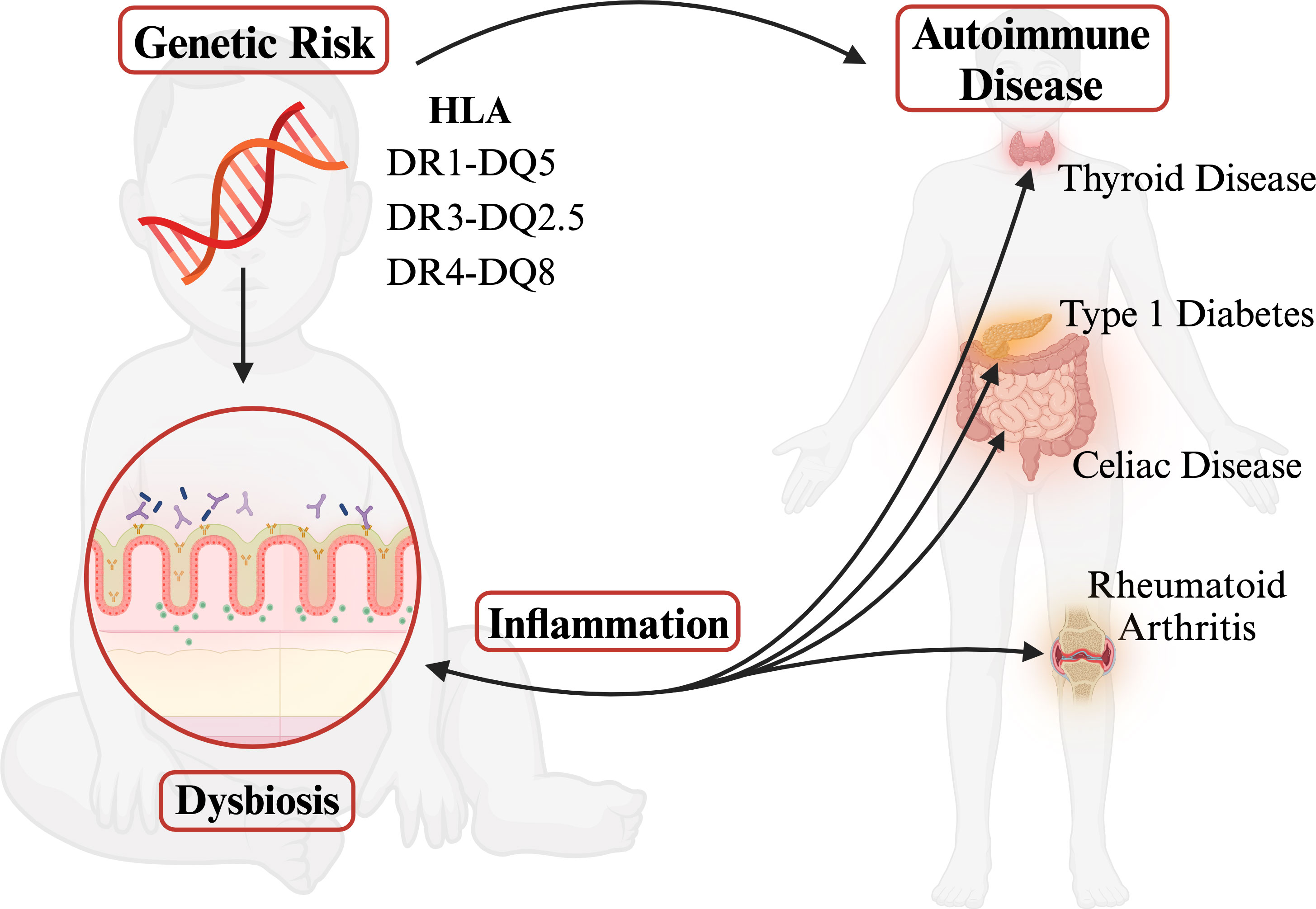
Graphical Abstract Graphical representation of central hypothesis.
The major histocompatibility complex (MHC) has long been established as the human genetic region associated with the greatest number of autoimmune diseases (1, 2). The MHC is broadly categorized into three classes: class I, which encodes for HLA-A, HLA-B, HLA-C, HLA-E, HLA-F, and HLA-G genes; class II, the focus of this review, which encodes for HLA-DR, HLA-DQ, and HLA-DP genes; and class III, which includes components of the complement system, immune regulators, and non-immune associated genes (2–4). Classically, class I HLA are present on all cells, while class II HLA are expressed on the surface of antigen presenting cells (APC) like dendritic cells and macrophages. Class I HLA-peptide combinations bind CD8+ T cell αβ T cell receptors (TCRs) for inspection of internally found antigens, like signals of viral infection and cancer. Class II HLA present externally found peptides to CD4+ T cell TCRs, such as bacteria and other foreign pathogens. However, cross-class presentation has been observed to bypass MHC restriction (4–6). The presentation of externally found antigens to T cells instigates a cascade leading to destruction of the perceived pathogen. HLA-peptide-TCR interaction specificity is fundamental to an effective cell-mediated adaptive immune response (7). The peptide repertoire available for presentation by class II HLA largely depends on the structure of the binding pocket.
HLA DR and DQ loci are highly polymorphic and exhibit an elevated amount of linkage disequilibrium. The combination of these features contribute to creating distinctive and behaviorally differential HLA haplotypes (8). This review will cover autoimmune-risk-associated class II HLA haplotypes DR4-DQ8, DR3-DQ2, and DR1-DQ5. The most polymorphic regions of the DR and DQ molecules are located within extracellular regions making up the peptide-binding cleft, which cause structure-altering changes at the amino acid level (9–12). These structural variations alter peptide-binding and thus antigen-presenting capabilities (7, 13, 14). The structural differences between haplotype molecules result in unique sensitivities and can be the determining factor for many autoimmune diseases, such as type 1 diabetes (T1D), celiac disease (CD), rheumatoid arthritis (RA), and autoimmune thyroid disease (AITD), including Grave’s disease (GD) and Hashimoto’s disease (HD) (9, 15–18).
Epidemiological data show an increase in the frequency of autoimmune diseases over the past few decades that cannot be explained by genetics alone (19–21). Many autoimmune disorders share the same risk-associated HLA haplotypes often resulting in comorbidity despite having differing etiologies (22–24). The combination of high polymorphism and linkage disequilibrium within the gene dense MHC region leads to difficulty in determining the mechanism for the autoimmune associations observed (1, 2). This gap is where the role of the gut microbiome has become increasingly essential in defining the pathogenesis of these autoimmune diseases (25–30). It has been theorized that the dysbiosis seen in autoimmune diseases is associated with systemic inflammation, resulting in loss of barrier function and permeability of tight junctions, allowing for possible increased exposure of HLA proteins to bacterial antigens (31–33). HLA class II proteins are expressed in the upper villi of small intestinal enterocytes at a steady state in the presence of a healthy gut microbiome and are an integral part of maintaining homeostasis; however, dysbiosis and inflammation cause an increase in HLA class II expression in small intestinal crypts and the colonic epithelium, which can in turn influence the composition of the gut microbiome (32, 34–39). Notably, the increase in HLA class II expression levels is active-disease dependent; for example, celiac patients with exposure to gliadin show HLA upregulation whereas celiac patients in remission have HLA class II levels of controls (40). However, certain HLA haplotypes, specifically the known risk HLA discussed here, are associated with gut dysbiosis before autoimmunity occurs (36, 39, 41, 42). Such evidence suggests that certain HLA may be predisposing an individual to systemic inflammation originating from the gut microbiome by clearing beneficial microbes and creating the potential for dysbiosis early in life. The tripartite HLA-microbiome-autoimmunity link is not trivial. This review summarizes current research on the impact class II HLA haplotypes have on the microbiome and its correlation to autoimmune disease onset. Our hypothesis is that bacterial dysbiosis in the gut leads to systemic inflammation which leads to autoimmunity (Graphical Abstract). The sources and types of inflammation can vary, causing different autoimmune disease outcomes.
Class II HLA DR and DQ loci represent the greatest genetic determinants of multiple autoimmune diseases. HLA-DR is a heterodimer consisting of an α (DRA) and β (DRB) chain, each of which have two extracellular domains, an intramembranous domain, and a cytoplasmic tail (Figure 1). DRA has two potential α polypeptide chains for the HLA-DR heterodimer, but the allelic differences do not result in function-altering polymorphisms (43, 44). The HLA-DR β chain can be encoded by DRB1, DRB2 (pseudogene), DRB3, DRB4, and DRB5 genes (43). Many DRB1 allelic variations are associated with multiple autoimmune diseases and are the basis for the HLA-DR naming system. For example, HLA-DR4 is the name for the DRB1*04 allele group. HLA-DQ is also a highly variable αβ heterodimer forming a type 1 membrane protein. DQA and DQB can both be encoded by two paralogs: DQA1, DQA2, DQB1, DQB2, respectively. Both DQA1 and DQB1 are highly polymorphic resulting in hundreds of possible combinations (43).
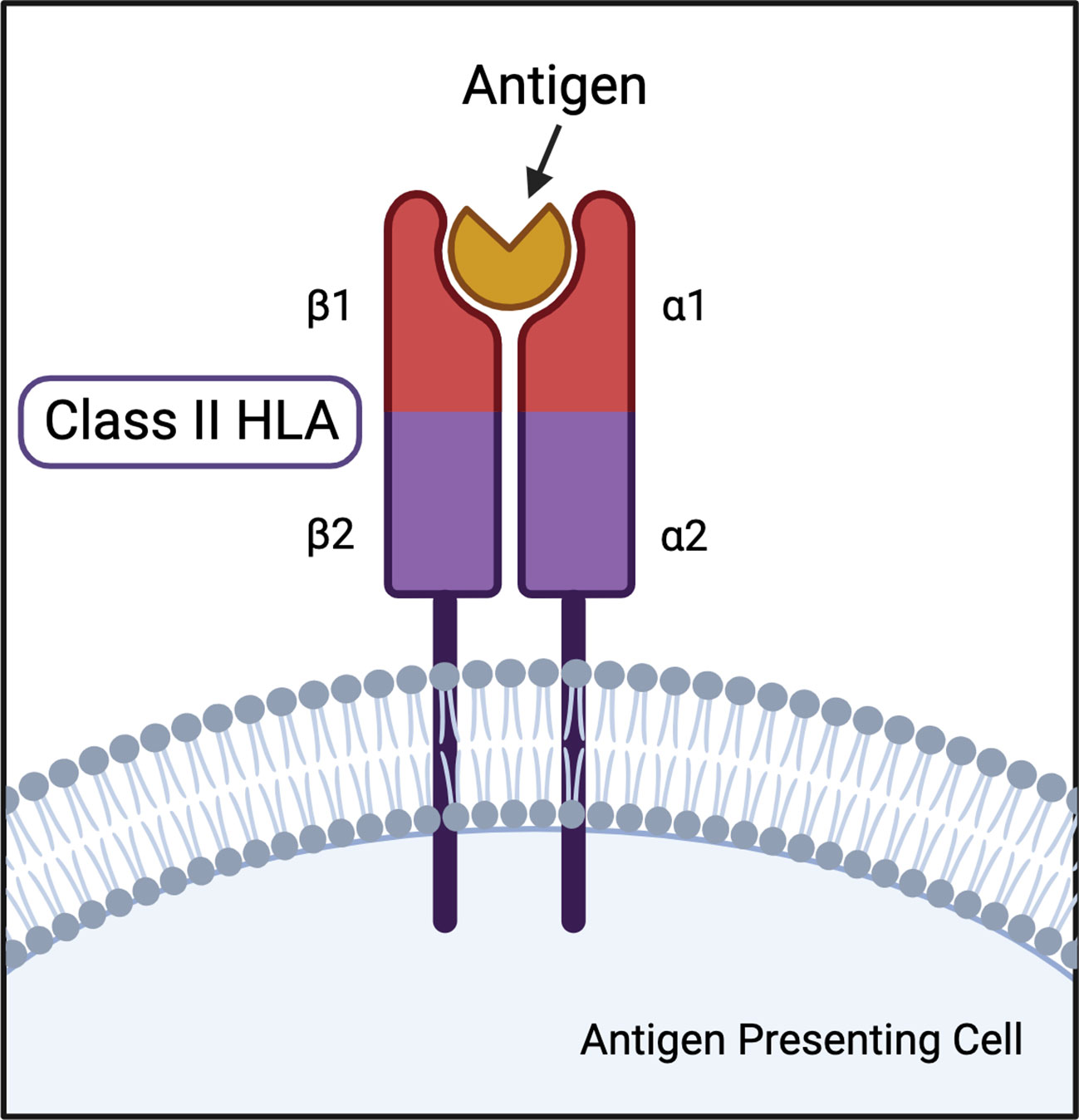
Figure 1 Illustration of class II HLA protein structure with antigen in peptide binding pocket.
HLA DR4-DQ8 is the nomenclature used to represent that an individual has the gene products of HLA DRA1-DRB1*04:01/02/04/05/08 and DQA1*03:01-DQB1*03:02/04 (11). DR3-DQ2.5 represents gene products of DRB1*03:01/02/03/04-DQA1*05:01-DQB1*02:01. DR1-DQ5 represents DRB1*01:01/02-DQA1*01:01-DQB1*05:01. DR5-DQ7.5 represents DRB1*05:01-DQA1*05:05-DQB1*03:01. For clarification, the HLA naming system (e.g., DRB1*04:01) is the gene locus name (e.g., DRB1), followed by an asterisk, the serologic designation of the allelic group (e.g., 04), a colon, and then the numeric designation of the specific HLA protein (e.g., 01). The naming system can be further expanded to a six-digit identifier that includes another colon followed by a two-digit number that represents a synonymous DNA substitution in the coding region (e.g., DRB1*04:01:01). For this review, the four-digit naming system will be sufficient.
Because DQA1 and DQB1 can both have polymorphisms, unique DQ heterodimers can be formed by pairing α and β chains from the same chromosome (cis) or opposite chromosomes (trans). While the cis form of DQ has been studied predominantly, trans variants are functional and surface expressed (45). This trans molecular formation means that a person heterozygous for DR4-DQ8 and DR3-DQ2.5 can produce a DQ2.3 (DQA1*03:01-DQB1*02:01) molecules from the α chain of DQ8 and the β chain of DQ2.5 (46). For this review, all DQ can be assumed to be cis unless specifically reported as trans.
Type 1 diabetes (T1D) is characterized by autoimmune destruction of pancreatic β-cells, resulting in a lifelong exogenous insulin dependency that affects millions of people worldwide (47). While there are over 50 known regions of the genome to show association with genetic risk for T1D, the greatest genetic determinants are MHC class II DRB1, DQA1, and DQB1 (48). The haplotypes most strongly associated with T1D susceptibility in those with European ancestry are DRB1*04:01-DQA1*03:01-DQB*03:02, DRB1*04:05-DQA1*03:01-DQB1*03:02, and DRB1*04:02-DQA1*03:01-DQB1*03:02, and then DRB1*04:04-DQA1*03:01-DQB1*03:02 conferring weaker disease susceptibility (9, 49). These well-known associations have predominantly been studied in European-centric populations and they may not be translatable to other ethnic backgrounds. For example in the Japanese population DR4-DQ8 is not prevalent; DRB1*04:05-DQA1*03:03-DQB1*04:01 (DR4-DQ4) and DRB1*09:01-DQA1*03:02-DQB1*03:03 (DR9-DQ9) are the most susceptible haplotypes for T1D (50). Three haplotypes, DR4-DQ8, DR3-DQ2.5, and DR4-DQ4, which combine the risk haplotypes for Caucasian and Japanese populations are the most highly associated with T1D in the Taiwanese population (51). For the Caucasian population, the development of diabetes-associated autoantibodies and T1D is significantly more common in children with heterozygous HLA DR3-DQ2/DR4-DQ8 compared to homozygous DR4-DQ8/DR4-DQ8 and DR3-DQ2.5/DR3-DQ2.5 (11, 12, 52). In Finland, children with high-risk HLA DR3-DQ2/DR4-DQ8 genotypes have a 45-fold increased risk for T1D compared to those with neutral or protective genotypes (17). The age of T1D diagnosis and type of autoantibody first observed are also strongly associated with HLA genotypes (53, 54). Insulin autoantibodies (IAA) and insulinoma-associated-2 autoantibodies (IA2A) are strongly correlated with DR4-DQ8 (12, 55). Approximately 1 in 20 heterozygous for DR3-DQ2/DR4-DQ8 will be diagnosed with T1D by the age of 15 (11). The proportion of HLA DR3-DQ2/DR4-DQ8 heterozygous T1D subjects decreases with increasing age at diagnosis (12). Seroconversion and development of T1D is associated with specific residues at both the DR and the DQ loci, the motif lysine, alanine, glycine at DRB1 β71, β74, β86 residues, which corresponds with DRB1*04:01, and glutamine, alanine, and aspartic acid at DQ α44, β57, and β135 residues, respectively, which correspond to DQA1*03:01-DQB1*03:02 (56, 57). An alanine at DQ β57 is most strongly associated with T1D (58).
In addition to DR4-DQ8, among the highest risk HLA haplotypes for T1D is DRB1*0301-DQA1*05:01-DQB1*02:01 (DR3-DQ2.5) (9). For the Caucasian population, the development of diabetes-associated autoantibodies is significantly associated with both the homozygous DR3-DQ2.5 and heterozygous DR3-DQ2.5/DR4-DQ8 (11, 12, 52). As previously discussed, the age of T1D diagnosis and type of autoantibody first observed are also strongly associated HLA haplotype; autoantibodies, IAA and IA2A, are associated with DR4-DQ8 and early seroconversion, but glutamic acid decarboxylase autoantibodies (GADA) are found in individuals have later seroconversion and are strongly associated with DR3-DQ2.5 (12, 52–55). Recent evidence suggests that GAD peptides bind to DR3-DQ2.5 molecules and in turn induce CD4+ T cell cytokine expression (59). In addition to the cis DR3-DQ2.5/DR4-DQ8 heterodimer, the heterozygote DR3-DQ2.5/DR4-DQ8 in trans heterodimer form encoded by DQA1*05:01-DQB1*03:02 is also very high risk for T1D (9). At a young age the risk for T1D is highest with heterozygous HLA-DR3-DQ2.5/DR4-DQ8, approximately 1 in 20 with this HLA haplotype pairing will be diagnosed with T1D by the age of 15 (11). The strongest amino acid association with T1D onset is an alanine at residue DQ β57, which is seen in both DQ2 and DQ8 (58, 60).
Celiac disease (CD) is autoimmune enteropathy characterized by the immunogenicity of gliadin peptides derived from dietary gluten. The chief HLA determinant of CD development is HLA-DQ (61). While HLA-DQ2.5 discussed in the next section has the highest risk association, DQ8 specifically, DQA1*03:01-DQB1*03:02, represents about 2–10% of the Caucasian CD population (62). The determining factor for HLA-associated CD susceptibility is the preferential binding of negatively charged gliadin-derived glutamate residues to certain binding pockets of DQ molecules, specifically P1 and P9 for DQ8 and P4, P6 and P7 for DQ2.5 and DQ2.2 (61, 63).
Globally, CD seroprevalence is 1.4% and 1.8% in North America. However, a study recently found that the highest prevalence is 3.1% within those from northwest India (64, 65). Within the Caucasian population, DQ2.5 is the predominant HLA predictor of CD susceptibility, approximately 95% of those with CD are positive for DQA1*05:01-DQB1*02:01 (DQ2.5); the remaining CD population has either DQA1*03:01-DQB1*03:02 (DQ8) or DQA1*02:01-DQB1*02:02 (DQ2.2) (66). DQ2.5 also has the strongest risk association with CD within the Indian population (67). DQ2.5 was the greatest CD-associated HLA in Moroccan, Libyan, Greek, and Italian CD populations; however, approximately 73.9%, 80.7%, 81.3%, and 78.2% of the populations, respectively, were DQ2.5 positive (68). The determining factor for HLA-associated CD susceptibility is the preferential binding of negatively charged gliadin-derived glutamate residues to certain binding pockets of DQ molecules, specifically P4, P6 and P7 for DQ2.5 and DQ2.2 (63). While DQ2.5 is the predominant isoform seen in association with CD susceptibility and the majority of the remaining CD population carry DQ8, DQA1*02:01-DQB1*02:02 (DQ2.2), linked to DRB1*07:01 (DR7), represents about 3.4% of the CD population (69). Generally, DQ2.2 is associated with very low CD risk; however, when heterozygous with DQ2.5 or trans configuration creates DQ2.5, a high risk association is observed (69, 70). Risk for CD is associated with DQ2.2 predominantly when individuals also carry DR3-DQ2.5 or DR5-DQ7.5 because the DQA1*05:05 of DQ7.5 is nearly identical to the DQA1*05:01 of DQ2.5 which means the DQB1*02:02 of DQ2.2 and DQA1*05:05 of DQ7.5 can make a DQA1*05:05-DQB1*02:02 (DQ2.5) heterodimer (70).
Autoimmune thyroid diseases (AITD) include both Graves’ disease (GD), which is an autoimmune hyperthyroidism, and Hashimoto’s disease (HD), which is an autoimmune hypothyroidism. HD is characterized by positive autoantibodies to thyroglobulin and thyroid peroxidase, whereas GD is characterized by autoantibodies against the thyroid stimulating hormone receptor, thyroglobulin, and thyroid peroxidase (71). The chief genetic determinant of GD susceptibility is DRB1*03:04-DQA1*05:01-DQB1*02:01 (DR3-DQ2.5); however, the DQB1*02:01 locus appears to be associated through linkage disequilibrium as opposed to actual influence on susceptibility (18, 72, 73). The strongest amino acid association with GD is an arginine at residue DR β74, which is integral to the binding and presentation of thyroglobulin (18, 72, 74). The positive charge provided by arginine in this positive likely facilitates auto-antigen presentation (75). Though less convincingly, DR3 is also associated risk of HD susceptibility and, in those with HD and T1D, DR3 is responsible for joint susceptibility (18, 75).
The association of DRB1*04 and genetic predisposition for rheumatoid arthritis (RA) has been observed since the late 1980s (76, 77). RA is the autoimmune destruction of the synovium in the small joints characterized by the presence of autoantibodies: rheumatoid factor, anti-cyclic citrullinated peptide-2, and anti-carbamylated protein (78, 79). Within the Caucasian population of RA patients, significant associations are seen with DRB1*04:01, DRB1*04:04, DRB1*04:05 and 95% of those with severe arthritis expressed DRB1*04:01 (80, 81). Those homozygous for DR4 have the highest risk association for RA (82). While allotypes of DR4 are high risk for RA, DRB1*04:02 is not associated with the disease (80). The determining factor for HLA-associated RA susceptibility is a positive charge at the DRβ71 amino acid residue—DRB1*04:01 and DRB1*04:04 have a positively charged lysine or arginine, respectively, at this position whereas DRB1*04:02 has a negatively charged glutamic acid (80). It is important to note that while certain DR4 alleles have long been observed in Caucasian RA studies, there is no statistical significance in the prevalence of DR4 in RA patients versus controls within the Iranian population and no association between RA-associated autoantibodies and risk HLA in the Japanese population (79, 83).
For RA patients who lack DR4, DRB1*01:01/02 (DR1) is notably associated with RA susceptibility (77). Like DRB1*04:01/04, DRB1*0101 carries a positively charged arginine at the DRβ71 amino acid residue, which is a determining factor for HLA-associated RA susceptibility (80). A small study shows that the majority multi-drug resistant RA patients have DRB1*01:01/02 (84). HLA-DRB1*04:01, DRB1*04:05, and DRB1*01:01 share a common motif at residues β11, β13, β71, and β74, specifically an alanine at position 74 and a positively charged lysine or arginine at position 71, influencing the DRB1 P4 binding pocket (85). Also, homozygote DR1-DQ5 is also strongly associated with juvenile idiopathic arthritis (JIA) with an odds ratio of 3.6, which increases to 6.4 when individual was breastfed for fewer than 8 months (86). JIA was also associated with DR5-DQ7 in individuals who breastfed under 8 months (86).
Despite differing etiologies, as discussed, many autoimmune disorders share the same risk-associated HLA haplotypes often resulting in comorbidity. Individuals with T1D are 4.9 times more likely to have RA as adults than the general population (87). A 2011 study suggests that 12.3% of the T1D population assessed have AITD and 24.6% have CD (88). However, that number was an overestimation. A 2023 study found that while 18.6% of the T1D population tests positive for CD, 12.6% were serologically false positive and only 6% are actually confirmed CD patients (89), which is in agreement with prevalence found in many other studies (90). Globally, biopsy-confirmed CD prevalence is 0.7%; however, biopsy-confirmed CD prevalence is 1.6% in the general AITD population and 2.6% in the hyperthyroid community (64, 91). For those with CD, 26% of the population have AITD compared to 2–5% of the general population; individuals with CD are 2.4 times more likely to develop an AITD and 5.9 times more likely if they are female (92, 93). The odds of having RA is also higher in CD, occurring nearly 2 times as often compared to the general population (94).
While T1D is caused by autoreactive T cells, a link between T1D and notable microbial patterns and intestinal inflammation is evident (36, 95, 96). The microbiome of T1D children lack diversity and have higher levels of butyrate-producing and mucin-degrading bacteria than healthy children (38, 97). Microbiome differences of those with future T1D diagnosis can be seen as early as one year of age (42). Research from the Finnish Type 1 Diabetes Prediction and Prevention Study (DIPP), a prospective, general-population cohort, shows high abundance of Bacteroides dorei and Bacteroides vulgatus between 12 and 15 months before seroconversion (37). Active T1D cases are associated with higher relative abundance of Ruminococcus and Prevotella copri and lower relative abundance of Bifidobacterium, Lactobacillus, Roseburia, and Faecalibacterium (Table 1) (98–100). The microbial composition observed in T1D patients likely leads to intestinal permeability, causing intestinal inflammation (101). Intestinal permeability results in increased exposure of intestinal immune cells to bacteria antigens. Intestinal biopsies from T1D children showed an increase in class II HLA molecule expression, and high levels of CD25+ cells (102). Increased exposure to commensal bacteria and excessive immune response over time could result in aberrant self-tolerance mechanisms. T1D patients exhibit immune dysregulation with higher percentages of Th1, Th17, and TNFa+ T cells (103).
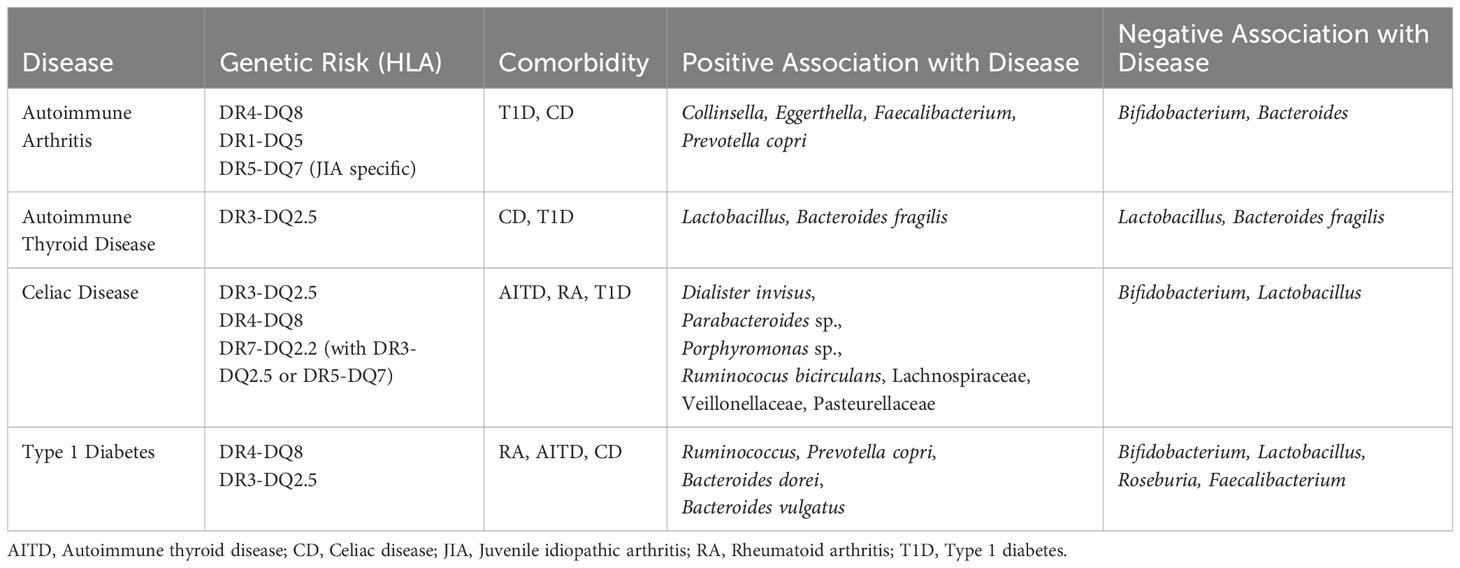
Table 1 Autoimmune disease association with HLA genetics, bacteria, and each other.
CD has obvious connections to gastrointestinal distress, gliadin peptides induce upregulation of zonulin and the shielding of gliadin peptides from destruction by lysosomes increases peptide secretion into the intestinal lamina propria, perpetuating inflammation and intestinal permeability (29, 63). Studies focusing on fecal microbiota show lower abundance of Bifidobacterium and Lactobacillus species in CD patients than healthy controls, both of which are considered to have anti-inflammatory effects (29, 104–106). Research from the Celiac Disease Genomic Environmental Microbiome and Metabolomic (CDGEMM) study, a prospective cohort of healthy infants with a first-degree relative who has CD, shows microbiome composition patterns up to 15 months before disease onset—increased abundance of Dialister invisus, Parabacteroides sp., Porphyromonas sp., Ruminococus bicirculans, Lachnospiraceae and decreased abundance of Streptococcus thermophilus, Faecalibacterium prausnitzii, and Clostridium clostridioforme (107). Microbiome differences between those with a future diagnosis of CD and healthy matched controls can be seen as early as one year of age (41). Children progressed to CD diagnosis not only have a distinct microbiome composition compared to healthy controls but also have an increased IgA response, resulting in more IgA-coated bacteria, suggesting altered bacterial clearance (108). A recent study shows that changes in the gut microbiome, specifically abundance of Veillonellaceae, may have causal effects on CD development, while Pasteurellaceae abundance differences may be caused by the disease itself (109). Since CD occurs in the small intestine, intestinal location specific studies reveal the importance of location in microbial composition—higher abundance of Escherichia coli, Prevotella salivae, and Neisseria are associated with CD when sampling the duodenum (110).
The gut-thyroid axis is a relatively new discussion point in study of autoimmune thyroid disease (AITD). Evidence that suggests dysbiosis is seen across AITD patients and the disruption of the gut microbial composition affects thyroid hormone metabolism (111, 112). Serum lipopolysaccharide and zonulin are significantly higher in GD patients than healthy controls and fecal transplant from GD patients into a mouse model significantly increases the incidence of GD (113, 114). However, specific microbiome community dynamics and potential for microbial biomarkers remains conflicted. The ratio of Firmicutes to Bacteroidetes is seen elevated in one study but significantly decreased in the AITD patients of a different study (115). Specifically, Bacteroides fragilis is observed as both higher and lower in abundance in AITD patients compared to healthy controls depending on the study (Table 1) (113, 115). Lactobacillus is proposed as both a potential probiotic for AITD amelioration and a potential major player in AITD pathogenesis (115, 116). Larger and more extensive microbiome studies may be required if a potential microbial biomarker for the gut-thyroid axis is determined.
Despite emphasis on joint inflammation, the majority of RA patients also exhibit gastrointestinal disorders and significant gut microbiome differences are observed in RA patients versus controls (Table 1) (117–119). Patients with RA have a decreased microbial diversity and distinct microbial patterns (120). Collinsella, Eggerthella, and Faecalibacterium segregate with RA in random forest modeling (121) Collinsella and Eggerthella correlate with increased intestinal permeability, mucosal inflammation, and immune response, and Collinsella is confirmed to correlate with increased proinflammatory cytokine IL-17A, gut permeability, and RA disease severity (119, 121). Specifically, Prevotella copri is more abundant in new-onset RA patients, while Bacteroides and Bifidobacterium are decreased in the same population (120). Prevotella is also substantially more prevalent in children with a future diagnosis of JIA, while Bifidobacterium and Bacteroides species are reduced in the same cohort (86). A recent study in mice and colonic tissue shows increased intestinal permeability and zonulin-1 expression upon exposure to fecal bacteria from pre-RA human individuals (122). A recent small human RA study following bowel cleansing and fasting found a link between intestinal microbes and inflammation specific to RA, suggesting dysbiosis as a primary player in disease activity (123).
The fundamental role of class II HLA is to bind to foreign peptides and present them on the plasma membrane for recognition by CD4+ T helper cells. Within the gut, this antigen presentation leads to B cell production of secretory IgA. IgA mediates microbial composition by inhibiting bacterial adhesion to epithelial cells, regulating bacterial epitope expression, and facilitating the elimination of bacteria from the gut via peristaltic and mucociliary actions (124, 125). Structural variety associated with HLA allelic polymorphisms alters microbiome composition by linking MHC-peptide binding affinity differences to which bacteria get eliminated by IgA (36, 126, 127). With regard to gut microbiome composition, HLA polymorphisms significantly alter biases in antibody-mediated selection against microbiota and in turn correlate to unique microbial communities (14). Evidence shows that populations with functionally similar HLA also feature similar microbial patterns (128). Many studies group HLA haplotypes by autoimmune risk group. Therefore, most analysis available look at both DR3-DQ2 and DR4-DQ8 as either pooled homozygotes and/or heterozygotes. Future research could benefit from analyzing risk haplotypes against each other to verify their similarities and differences in influence over the microbiome. Compared to low-risk or neutral haplotypes, high-risk HLA DR3-DQ2 and DR4-DQ8 are associated with higher abundance of Prevotella copri at the species level, Agathobacter, Bacteroides, Blautia, Dorea, Enterococcus, Intestinimonas, Klebsiella, Veillonella at the genus level, and Enterobacteriaceae, which includes Klebsiella, Lachnospiraceae, which includes Agathobacter, Blautia, and Dorea, and Ruminococcaceae, which includes Intestinimonas, at the family level (36, 129–132). Bifidobacterium and Lactobacillus stand out as either negatively associated or in lower abundance in DR3-DQ2 and DR4-DQ8 compared to protective or neutral alleles (36, 39, 133, 134). Of note, the large general population cohort, All Babies in Southeat Sweden (ABIS), found that when controlling for breastfeeding, DR5-DQ7 is a significant factor in an infant’s likelihood to be colonized by Lactobacillus at all (135). This correlation may be associated with DQ7.5 trans configuration with DQ2.2 to create DQ2.5, the primary risk allele for CD. Aside from the association of increased relative abundance of Bifidobacterium in homozygous the DR1-DQ5 population (39), there has been limited examination into the role of DR1-DQ5 in microbiome community constructs to date.
Many autoimmune disorders share the same risk-associated HLA haplotypes often resulting in comorbidity despite differing etiologies (22–24). The role of the gut microbiome has become increasingly essential in defining the pathogenesis of these autoimmune diseases (25–30). It has been theorized that dysbiosis seen in autoimmune diseases is associated with systemic inflammation, resulting in loss of barrier function and permeability of tight junctions, allowing for possible increased exposure of HLA proteins to bacterial antigens (31–33). HLA class II proteins are expressed in the upper villi of small intestinal enterocytes at a steady state in the presence of a healthy gut microbiome and are an integral part of maintaining homeostasis. However, dysbiosis and inflammation cause an increase in HLA class II expression in small intestinal crypts and the colonic epithelium, which can in turn influence the composition of the gut microbiome (32, 34–39). The fundamental role of class II HLA is to bind to foreign peptides and present them on the plasma membrane for recognition by CD4+ T helper cells. Within the gut, this antigen presentation leads to B cell production of secretory IgA. IgA mediates microbial composition by inhibiting bacterial adhesion to epithelial cells, regulating bacterial epitope expression, and facilitating the elimination of bacteria from the gut via peristaltic and mucociliary actions (124, 125). Structural differences associated with HLA allelic polymorphisms alter microbiome composition by linking HLA-peptide binding affinity differences to which bacteria get eliminated by IgA (36, 126, 127). The precedence for HLA molecular “preference” for specific peptides can be seen in celiac disease, where HLA DQ2 and DQ8 affinity for negatively charged residues results in class II MHC molecules binding and presenting gliadin peptides, leading to autoimmunity. However, certain HLA haplotypes, specifically the known risk HLA alleles discussed here, are associated with microbiome community dynamics that implicate dysbiosis before autoimmunity occurs (36, 39, 41, 42). Such evidence suggests that certain HLA may be predisposing an individual to systemic inflammation originating from the gut microbiome by clearing beneficial microbes and creating the potential for dysbiosis early in life. Increased exposure to commensal bacteria and excessive immune response over time could result in aberrant self-tolerance mechanisms.
Patterns emerge when investigating the overlap between HLA-associated and autoimmune-associated microbiomes. These relationships are unsurprising when considering the common risk-associated HLA haplotypes by autoimmunities. For example, Prevotella copri is more abundant in RA, JIA, and T1D patients compared to controls and is also associated HLA DR3-DQ2 and DR4-DQ8 (98, 99, 120, 131). This pattern makes sense when considering that HLA DR4-DQ8 is a risk-associated genotype for both RA and T1D. Higher abundances of inflammatory microbes, like Klebsiella and Veillonella, are associated with autoimmunity and risk; while conversely, lower abundances of known anti-inflammatory microbes like Bifidobacterium and Lactobacillus are associated with both autoimmune disorders and risk HLA (29, 104–106, 135–137). Causal relationships between microbiome composition and autoimmune onset are starting to be investigated. A recent study shows that abundance of Veillonellaceae may have causal effects on CD development (109). Prior to seroconversion, significantly higher abundance of Bacteroides species are observed in children with future T1D and JIA autoantibody seroconversion (37, 86). Both of these bacteria are associated with risk HLA (36, 138) Microbiome community differences can be seen as early as one year of age between those who go on to acquire an autoimmune disease versus those who do not (37, 41, 42). It is possible that the common denominator here is the introduction of early-life inflammation caused by HLA-specific dysbiosis.
It is important to note that all these studies focus on fecal microbiota, meaning the microbial composition is likely exclusively colonic and does not represent the small intestines. The field would benefit from microbial sampling from within a variety of locations in the gut. Also, there is limited current research into the gut-thyroid axis. Larger and more extensive microbiome studies may be required if a potential microbial biomarker for the gut-thyroid axis is to be determined. Many of the large cohort studies in this review focus on high-genetic-risk communities. To truly determine the impact of genetics on the gut, the field would benefit from general population studies that can compare risk vs. non-risk groups.
To validate the hypothesis that gut dysbiosis leads to early-life inflammation and elevate the link between gut microbiome composition and autoimmune disease onset, we propose an organ-on-a-chip model of human intestines. Within this model system, HLA-specific intestinal cultures could be generated to establish phenotypic differences between risk, neutral, and protection-associated tissue. To investigate innate immune response, we suggest quantifying cytokine secretion and examining zonulin, mucin, and permeability levels at baseline and following co-culture with either specific microbes of interest or a bacterial community culture (139, 140). It would be of interest to explore an adaptive immune response, as well. It is possible to characterize HLA-specific T cell response to commensal gut bacterial peptides through the presentation of secreted bacterial peptides to T cell stimulation assays measured with flow cytometry and ELISpot (141). An HLA-specific intestinal organ-on-a-chip model could also be used to measure T cell response by co-culturing peripheral blood mononuclear cells within the bottom chamber of the microfluidic chip and assessing T cell stimulation from the peptides that make it through the epithelial barrier in the microfluidic system.
The tripartite HLA-microbiome-autoimmunity link is not trivial. Risk HLA may be predisposing an individual early in life to dysbiosis originating in the clearance of beneficial microbes and/or promotion of inflammatory microbes, creating the potential systemic inflammation later in life. While it may be enticing to put emphasis on the dysbiosis and inflammation seen after autoimmune onset because of the clear evidence that autoimmune-induced permeability of tight junctions allows for increased exposure of HLA proteins to bacterial antigens, it is important to consider genetics and the initial role haplotype-specific peptide binding affinities may play in defining an individual’s microbiome.
MB: Conceptualization, Data curation, Writing – original draft. ET: Conceptualization, Supervision, Writing – review & editing. JI: Supervision, Validation, Writing – review & editing. JL: Conceptualization, Project administration, Supervision, Writing – review & editing.
ABIS is supported by Barndiabetesfonden (Swedish Child Diabetes Foundation); Swedish Council for Working Life and Social Research, Grant/Award Numbers: FAS2004-1775, FAS2004–1775; Swedish Research Council, Grant/Award Numbers: K2005-72X-11242-11A and K2008-69X-20826-01-4, K2008-69X-20826-01-4; Östgöta Brandstodsbolag; Medical Research Council of Southeast Sweden (FORSS); JDRF Wallenberg Foundation, Grant/Award Number: K 98-99D-12813-01A; ALF and LfoU grants from Region Östergötland and Linköping University, Sweden; Joanna Cocozza Foundation.
All figures created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet (2013) 14:301–23. doi: 10.1146/annurev-genom-091212-153455
2. Dendrou CA, Petersen J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol (2018) 18:325–39. doi: 10.1038/nri.2017.143
3. Schott G, Garcia-Blanco MA. MHC class III RNA binding proteins and immunity. RNA Biol (2021) 18(5):640–6. doi: 10.1080/15476286.2020.1860388
4. Miles JJ, McCluskey J, Rossjohn J, Gras S. Understanding the complexity and malleability of T-cell recognition. Immunol Cell Biol (2015) 93:433–41. doi: 10.1038/icb.2014.112
5. Singh NK, Alonso JA, Devlin JR, Keller GLJ, Gray GI, Chiranjivi AK, et al. A class-mismatched TCR bypasses MHC restriction via an unorthodox but fully functional binding geometry. Nat Commun (2022) 13:7189. doi: 10.1038/s41467-022-34896-0
6. Rist M, Smith C, Bell MJ, Burrows SR, Khanna R. Cross-recognition of HLA DR4 alloantigen by virus-specific CD8+ T cells: a new paradigm for self-/nonself-recognition. Blood (2009) 114:2244–53. doi: 10.1182/blood-2009-05-222596
7. Abualrous ET, Sticht J, Freund C. Major histocompatibility complex (MHC) class I and class II proteins: impact of polymorphism on antigen presentation. Curr Opin Immunol (2021) 70:95–104. doi: 10.1016/j.coi.2021.04.009
8. Djilali-Saiah I, Benini V, Daniel S, Assan R, Bach JF, Caillat-Zucman S. Linkage disequilibrium between HLA class II (DR, DQ, DP) and antigen processing (LMP, TAP, DM) genes of the major histocompatibility complex. Tissue Antigens (1996) 48:87–92. doi: 10.1111/j.1399-0039.1996.tb02612.x
9. Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk. Diabetes (2008) 57:1084–92. doi: 10.2337/db07-1331
10. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2:a007732. doi: 10.1101/cshperspect.a007732
11. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diabetes Rep (2011) 11:533–42. doi: 10.1007/s11892-011-0223-x
12. Jerram ST, Leslie RD. The genetic architecture of type 1 diabetes. Genes (Basel) (2017) 8(8):209. doi: 10.3390/genes8080209
13. Rappazzo CG, Huisman BD, Birnbaum ME. Repertoire-scale determination of class II MHC peptide binding via yeast display improves antigen prediction. Nat Commun (2020) 11. doi: 10.1038/s41467-020-18204-2
14. Kubinak JL, Stephens WZ, Soto R, Petersen C, Chiaro T, Gogokhia L, et al. MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat Commun (2015) 6. doi: 10.1038/ncomms9642
15. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
16. Bodis G, Toth V, Schwarting A. Role of human leukocyte antigens (HLA) in autoimmune diseases. Rheumatol Ther (2018) 5:5–20. doi: 10.1007/s40744-018-0100-z
17. Taka A-M, But A, Lempainen J, Vatanen T, Härkönen T, Ilonen J, et al. Finnish children carrying the high-risk HLA genotype have a 45-fold increased risk of type 1 diabetes compared to peers with neutral or protective genotypes. Diabetes Res Clin Pract (2023) 197:110256. doi: 10.1016/j.diabres.2023.110256
18. Jacobson EM, Huber A, Tomer Y. THE HLA GENE COMPLEX IN THYROID AUTOIMMUNITY: FROM EPIDEMIOLOGY TO ETIOLOGY. J Autoimmun (2008) 30:58–62. doi: 10.1016/j.jaut.2007.11.010
19. Tuomilehto J. The emerging global epidemic of type 1 diabetes. Curr Diabetes Rep (2013) 13:795–804. doi: 10.1007/s11892-013-0433-5
20. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis (2015) 3:151–5. doi: 10.12691/ijcd-3-4-8
21. Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet (2016) 387:2340–8. doi: 10.1016/S0140-6736(16)30507-4
22. Alshiekh S, Maziarz M, Geraghty DE, Larsson HE, Agardh D. High-resolution genotyping indicates that children with type 1 diabetes and celiac disease share three HLA class II loci in DRB3, DRB4 and DRB5 genes. HLA (2021) 97:44–51. doi: 10.1111/tan.14105
23. Koning F, Thomas R, Rossjohn J, Toes RE. Coeliac disease and rheumatoid arthritis: similar mechanisms, different antigens. Nat Rev Rheumatol (2015) 11:450–61. doi: 10.1038/nrrheum.2015.59
24. Miyadera H, Tokunaga K. Associations of human leukocyte antigens with autoimmune diseases: challenges in identifying the mechanism. J Hum Genet (2015) 60:697–702. doi: 10.1038/jhg.2015.100
25. Heidt C, Kämmerer U, Fobker M, Rüffer A, Marquardt T, Reuss-Borst M. Assessment of intestinal permeability and inflammation bio-markers in patients with rheumatoid arthritis. Nutrients (2023) 15:2386. doi: 10.3390/nu15102386
26. Piccioni A, Rosa F, Mannucci S, Manca F, Merra G, Chiloiro S, et al. Gut microbiota, LADA, and type 1 diabetes mellitus: an evolving relationship. Biomedicines (2023) 11:707. doi: 10.3390/biomedicines11030707
27. Monticolo M, Mucha K, Foroncewicz B. Lupus nephritis and dysbiosis. Biomedicines (2023) 11:1165. doi: 10.3390/biomedicines11041165
28. Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol (2016) 12:154–67. doi: 10.1038/nrendo.2015.218
29. Rossi RE, Dispinzieri G, Elvevi A, Massironi S. Interaction between gut microbiota and celiac disease: from pathogenesis to treatment. Cells (2023) 12:823. doi: 10.3390/cells12060823
30. Xu Q, Ni J-J, Han B-X, Yan S-S, Wei X-T, Feng G-J, et al. Causal relationship between gut microbiota and autoimmune diseases: A two-sample mendelian randomization study. Front Immunol (2022) 12:746998. doi: 10.3389/fimmu.2021.746998
31. de Oliveira GLV, Cardoso CRDB, Taneja V, Fasano A. Editorial: intestinal dysbiosis in inflammatory diseases. Front Immunol (2021) 12:727485. doi: 10.3389/fimmu.2021.727485
32. Davis-Richardson AG, Triplett EW. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia (2015) 58:1386–93. doi: 10.1007/s00125-015-3614-8
33. Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol (2008) 24:701–6. doi: 10.1097/MOG.0b013e32830e6d98
34. Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hülsdünker J, Varelias A, et al. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity (2019) 51:885–898.e7. doi: 10.1016/j.immuni.2019.08.011
35. Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol (2018) 9:2144. doi: 10.3389/fimmu.2018.02144
36. Russell JT, Roesch LFW, Ördberg M, Ilonen J, Atkinson MA, Schatz DA, et al. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat Commun (2019) 10:3621. doi: 10.1038/s41467-019-11460-x
37. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol (2014) 5:678. doi: 10.3389/fmicb.2014.00678
38. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med (2013) 11:46. doi: 10.1186/1741-7015-11-46
39. Berryman MA, Milletich PL, Petrone JR, Roesch LFW, Ilonen J, Triplett EW, et al. Autoimmune-associated genetics impact probiotic colonization of the infant gut. J Autoimmun (2022) 133:102943. doi: 10.1016/j.jaut.2022.102943
40. Arnaud-Battandier F, Cerf-Bensussan N, Amsellem R, Schmitz J. Increased HLA-DR expression by enterocytes in children with celiac disease. Gastroenterology (1986) 91:1206–12. doi: 10.1016/s0016-5085(86)80018-x
41. Milletich PL, Ahrens AP, Petrone JR, Russell JT, Berryman MA, Agardh D, et al. Gut microbiome markers in subgroups of HLA class II genotyped infants signal future celiac disease in the general population: ABIS study. Front Microbiol (2022) 12. doi: 10.3389/fcimb.2022.920735
42. Bélteky M, Milletich PL, Ahrens AP, Triplett EW, Ludvigsson J. Infant gut microbiome composition correlated with type 1 diabetes acquisition in the general population: the ABIS study. Diabetologia (2023) 66:1116–28. doi: 10.1007/s00125-023-05895-7
43. Bondinas GP, Moustakas AK, Papadopoulos GK. The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics (2007) 59:539–53. doi: 10.1007/s00251-007-0224-8
44. Pinet V, Eliaou JF, Clot J. Description of a polymorphism in the regulatory region of the HLA-DRA gene. Hum Immunol (1991) 32:162–9. doi: 10.1016/0198-8859(91)90052-b
45. Nilsson JB, Kaabinejadian S, Yari H, Peters B, Barra C, Gragert L, et al. Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome. Commun Biol (2023) 6:442. doi: 10.1038/s42003-023-04749-7
46. Tollefsen S, Hotta K, Chen X, Simonsen B, Swaminathan K, Mathews II, et al. Structural and functional studies of trans-encoded HLA-DQ2.3 (DQA1*03:01/DQB1*02:01) protein molecule. J Biol Chem (2012) 287:13611–9. doi: 10.1074/jbc.M111.320374
47. Gregory GA, Robinson TIG, Linklater SE, Wang F, Colagiuri S, de Beaufort C, et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol (2022) 10:741–60. doi: 10.1016/S2213-8587(22)00218-2
48. Robertson CC, Rich SS. Genetics of type 1 diabetes. Curr Opin Genet Dev (2018) 50:7–16. doi: 10.1016/j.gde.2018.01.006
49. Ilonen J, Kiviniemi M, Lempainen J, Simell O, Toppari J, Veijola R, et al. Genetic susceptibility to type 1 diabetes in childhood – estimation of HLA class II associated disease risk and class II effect in various phases of islet autoimmunity. Pediatr Diabetes (2016) 17:8–16. doi: 10.1111/pedi.12327
50. Kawabata Y, Ikegami H, Kawaguchi Y, Fujisawa T, Shintani M, Ono M, et al. Asian-specific HLA haplotypes reveal heterogeneity of the contribution of HLA-DR and -DQ haplotypes to susceptibility to type 1 diabetes. Diabetes (2002) 51:545–51. doi: 10.2337/diabetes.51.2.545
51. Hu CY, Allen M, Chuang LM, Lin BJ, Gyllensten U. Association of insulin-dependent diabetes mellitus in Taiwan with HLA class II DQB1 and DRB1 alleles. Hum Immunol (1993) 38:105–14. doi: 10.1016/0198-8859(93)90526-7
52. Krischer JP, Lynch KF, Lernmark Å, Hagopian WA, Rewers MJ, She J-X, et al. Genetic and environmental interactions modify the risk of diabetes-related autoimmunity by 6 years of age: the TEDDY study. Diabetes Care (2017) 40:1194–202. doi: 10.2337/dc17-0238
53. Fava D, Gardner S, Pyke D, Leslie RD. Evidence that the age at diagnosis of IDDM is genetically determined. Diabetes Care (1998) 21:925–9. doi: 10.2337/diacare.21.6.925
54. Mikk M-L, Pfeiffer S, Kiviniemi M, Laine A-P, Lempainen J, Härkönen T, et al. HLA-DR-DQ haplotypes and specificity of the initial autoantibody in islet specific autoimmunity. Pediatr Diabetes (2020) 21:1218–26. doi: 10.1111/pedi.13073
55. Regnell SE, Lernmark Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia (2017) 60:1370–81. doi: 10.1007/s00125-017-4308-1
56. Zhao LP, Papadopoulos GK, Lybrand TP, Moustakas AK, Bondinas GP, Carlsson A, et al. The KAG motif of HLA-DRB1 (β71, β74, β86) predicts seroconversion and development of type 1 diabetes. EBioMedicine (2021) 69:103431. doi: 10.1016/j.ebiom.2021.103431
57. Zhao LP, Papadopoulos GK, Kwok WW, Moustakas AK, Bondinas GP, Larsson HE, et al. Motifs of three HLA-DQ amino acid residues (α44, β57, β135) capture full association with the risk of type 1 diabetes in DQ2 and DQ8 children. Diabetes (2020) 69:1573–87. doi: 10.2337/db20-0075
58. Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen W-M, et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet (2015) 47:898–905. doi: 10.1038/ng.3353
59. Chuzho N, Mishra N, Tandon N, Kanga U, Mishra G, Sharma A, et al. HLA-DR3 mediated CD4 T cell response against GAD65 in type 1 diabetes patients. J Diabetes (2023) 15:607–21. doi: 10.1111/1753-0407.13406
60. Rønningen K S, Iwe T, Halstensen TS, Spurkland A, Thorsby E. The amino acid at position 57 of the HLA-DQB chain and susceptibility to develop insulin-dependent diabetes mellitus. Hum Immunol (1989) 26:215–25. doi: 10.1016/0198-8859(89)90040-2
61. Sollid LM. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics (2017) 69:605–16. doi: 10.1007/s00251-017-0985-7
62. Lundin KE, Scott H, Fausa O, Thorsby E, Sollid LM. T cells from the small intestinal mucosa of a DR4, DQ7/DR4, DQ8 celiac disease patient preferentially recognize gliadin when presented by DQ8. Hum Immunol (1994) 41:285–91. doi: 10.1016/0198-8859(94)90047-7
63. Qiao S-W, Sollid LM, Blumberg RS. Antigen presentation in celiac disease. Curr Opin Immunol (2009) 21:111. doi: 10.1016/j.coi.2009.03.004
64. Silvester JA, Therrien A, Kelly CP. Celiac disease: fallacies and facts. Am J Gastroenterol (2021) 116:1148–55. doi: 10.14309/ajg.0000000000001218
65. Krigel A, Turner KO, Makharia GK, Green PH, Genta RM, Lebwohl B. Ethnic variations in duodenal villous atrophy consistent with celiac disease in the United States. Clin Gastroenterol Hepatol (2016) 14:1105–11. doi: 10.1016/j.cgh.2016.04.032
66. Banerjee P, Chaudhary R, Singh AK, Parulekar P, Kumar S, Senapati S. Specific genetic polymorphisms contributing in differential binding of gliadin peptides to HLA-DQ and TCR to elicit immunogenicity in celiac disease. Biochem Genet (2023). doi: 10.1007/s10528-023-10377-x
67. Ramakrishna BS, Venugopal G, Singh A, Pugazhendhi S, Dutta S, Ahuja V, et al. Human Leukocyte Antigen DQ (HLA-DQ) genotypes and haplotypes and their association with phenotype in patients with celiac disease in India. JGH Open (2021) 5:1190–6. doi: 10.1002/jgh3.12651
68. Piancatelli D, Ben El Barhdadi I, Oumhani K, Sebastiani P, Colanardi A, Essaid A. HLA typing and celiac disease in Moroccans. Med Sci (Basel) (2017) 5:2. doi: 10.3390/medsci5010002
69. Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, et al. A new genomic blueprint of the human gut microbiota. Nature (2019) 568:499–504. doi: 10.1038/s41586-019-0965-1
70. Pisapia L, Picascia S, Farina F, Barba P, Gianfrani C, Del Pozzo G. Differential expression of predisposing HLA-DQ2.5 alleles in DR5/DR7 celiac disease patients affects the pathological immune response to gluten. Sci Rep (2020) 10:17227. doi: 10.1038/s41598-020-73907-2
71. Zeitlin AA, Heward JM, Newby PR, Carr-Smith JD, Franklyn JA, Gough SCL, et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun (2008) 9:358–63. doi: 10.1038/gene.2008.26
72. Simmonds MJ, Howson JMM, Heward JM, Cordell HJ, Foxall H, Carr-Smith J, et al. Regression mapping of association between the human leukocyte antigen region and graves disease. Am J Hum Genet (2005) 76:157–63. doi: 10.1086/426947
73. Heward JM, Allahabadia A, Daykin J, Carr-Smith J, Daly A, Armitage M, et al. Linkage disequilibrium between the human leukocyte antigen class II region of the major histocompatibility complex and graves’ Disease: replication using a population case control and family-based study1. J Clin Endocrinol Metab (1998) 83:3394–7. doi: 10.1210/jcem.83.10.5137
74. Li CW, Osman R, Menconi F, Hou H, Schechter C, Kozhakhmetova A, et al. Effective inhibition of thyroid antigen presentation using retro-inverso peptides in experimental autoimmune thyroiditis: A pathway toward immune therapies of thyroid autoimmunity. Thyroid (2023) 33:492–500. doi: 10.1089/thy.2022.0511
75. Lee HJ, Stefan-Lifshitz M, Li CW, Tomer Y. GENETICS AND EPIGENETICS OF AUTOIMMUNE THYROID DISEASES: TRANSLATIONAL IMPLICATIONS. Best Pract Res Clin Endocrinol Metab (2023) 37:101661. doi: 10.1016/j.beem.2022.101661
76. Nepom GT, Byers P, Seyfried C, Healey LA, Wilske KR, Stage D, et al. HLA genes associated with rheumatoid arthritis. Identification of susceptibility alleles using specific oligonucleotide probes. Arthritis Rheum (1989) 32:15–21. doi: 10.1002/anr.1780320104
77. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheumatism (1987) 30:1205–13. doi: 10.1002/art.1780301102
78. Wang M, Wu J, Lei S, Mo X. Genome-wide identification of RNA modification-related single nucleotide polymorphisms associated with rheumatoid arthritis. BMC Genomics (2023) 24:153. doi: 10.1186/s12864-023-09227-2
79. de Moel EC, Derksen VFAM, Trouw LA, Bang H, Collée G, Lard LR, et al. In rheumatoid arthritis, changes in autoantibody levels reflect intensity of immunosuppression, not subsequent treatment response. Arthritis Res Ther (2019) 21:28. doi: 10.1186/s13075-019-1815-0
80. Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity (1997) 7:473–81. doi: 10.1016/S1074-7613(00)80369-6
81. Roudier J. HLA-DRB1 genes and extraarticular rheumatoid arthritis. Arthritis Res Ther (2006) 8:103. doi: 10.1186/ar1886
82. Jawaheer D, Thomson W, Macgregor AJ, Carthy D, Davidson J, Dyer PA, et al. “Homozygosity” for the HLA–DR shared epitope contributes the highest risk for rheumatoid arthritis concordance in identical twins. Arthritis Rheumatism (1994) 37:681–6. doi: 10.1002/art.1780370511
83. Salesi M, Boroujeni GT, Salehi M, Farzamnia S. Evaluation of differences in HLA-DR4 gene and its subtypes prevalence among healthy people and RA patients in Isfahan province population. Adv BioMed Res (2016) 5:11. doi: 10.4103/2277-9175.175244
84. Tesolin P, Bertinetto FE, Sonaglia A, Cappellani S, Concas MP, Morgan A, et al. High throughput genetic characterisation of caucasian patients affected by multi-drug resistant rheumatoid or psoriatic arthritis. J Pers Med (2022) 12:1618. doi: 10.3390/jpm12101618
85. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee H-S, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet (2012) 44:291–6. doi: 10.1038/ng.1076
86. Kindgren E, Ahrens AP, Triplett EW, Ludvigsson J. Infant gut microbiota and environment associate with juvenile idiopathic arthritis many years prior to disease onset, especially in genetically vulnerable children. eBioMedicine (2023) 93:104654. doi: 10.1016/j.ebiom.2023.104654
87. Liao KP, Gunnarsson M, Källberg H, Ding B, Plenge RM, Padyukov L, et al. A specific association exists between type 1 diabetes and anti-CCP positive rheumatoid arthritis. Arthritis rheumatism (2009) 60:653. doi: 10.1002/art.24362
88. Triolo TM, Armstrong TK, McFann K, Yu L, Rewers MJ, Klingensmith GJ, et al. Additional autoimmune disease found in 33% of patients at type 1 diabetes onset. Diabetes Care (2011) 34:1211–3. doi: 10.2337/dc10-1756
89. Department of Pediatric Gastroenterology, Adana City Training and Research Hospital, Adana, Turkey, Gulcu Taskin D, Ata A, Department of Pediatric Endocrinology, Adana City Training and Research Hospital, Adana, Turkey. The screening of celiac serology in pediatric patients diagnosed with type 1 diabetes mellitus. Turk J Gastroenterol (2023) 34:293–7. doi: 10.5152/tjg.2023.22775
90. Kurppa K, Laitinen A, Agardh D. Coeliac disease in children with type 1 diabetes. Lancet Child Adolesc Health (2018) 2:133–43. doi: 10.1016/S2352-4642(17)30172-4
91. Roy A, Laszkowska M, Sundström J, Lebwohl B, Green PHR, Kämpe O, et al. Prevalence of celiac disease in patients with autoimmune thyroid disease: A meta-analysis. Thyroid® (2016) 26:880–90. doi: 10.1089/thy.2016.0108
92. Franco J-S, Amaya-Amaya J, Anaya J-M. Thyroid disease and autoimmune diseases, in: Autoimmunity: from bench to bedside (2013). El Rosario University Press. Available at: https://www.ncbi.nlm.nih.gov/books/NBK459466/ (Accessed July 19, 2023).
93. Dore MP, Fanciulli G, Rouatbi M, Mereu S, Pes GM. Autoimmune thyroid disorders are more prevalent in patients with celiac disease: A retrospective case-control study. J Clin Med (2022) 11:6027. doi: 10.3390/jcm11206027
94. Doyle JB, Lebwohl B, Askling J, Forss A, Green PHR, Roelstraete B, et al. Risk of juvenile idiopathic arthritis and rheumatoid arthritis in patients with celiac disease: A population-based cohort study. Off J Am Coll Gastroenterol | ACG (2022) 117:1971. doi: 10.14309/ajg.0000000000002014
95. Higuchi BS, Rodrigues N, Gonzaga MI, Paiolo JCC, Stefanutto N, Omori WP, et al. Intestinal dysbiosis in autoimmune diabetes is correlated with poor glycemic control and increased interleukin-6: A pilot study. Front Immunol (2018) 9:1689. doi: 10.3389/fimmu.2018.01689
96. Kemppainen KM, Ardissone AN, Davis-Richardson AG, Fagen JR, Gano KA, León-Novelo LG, et al. Early childhood gut microbiomes show strong geographic differences among subjects at high risk for type 1 diabetes. Diabetes Care (2015) 38:329–32. doi: 10.2337/dc14-0850
97. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J (2011) 5:82–91. doi: 10.1038/ismej.2010.92
98. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature (2018) 562:583–8. doi: 10.1038/s41586-018-0617-x
99. Traversi D, Scaioli G, Rabbone I, Carletto G, Ferro A, Franchitti E, et al. Gut microbiota, behavior, and nutrition after type 1 diabetes diagnosis: A longitudinal study for supporting data in the metabolic control. Front Nutr (2022) 9:968068. doi: 10.3389/fnut.2022.968068
100. Matos J, Matos I, Calha M, Santos P, Duarte I, Cardoso Y, et al. Insights from bacteroides species in children with type 1 diabetes. Microorganisms (2021) 9:1436. doi: 10.3390/microorganisms9071436
101. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia (2006) 49:2824–7. doi: 10.1007/s00125-006-0465-3
102. Vaarala O, Atkinson MA, Neu J. The “Perfect storm” for type 1 diabetes. Diabetes (2008) 57:2555–62. doi: 10.2337/db08-0331
103. Lo Conte M, Cosorich I, Ferrarese R, Antonini Cencicchio M, Nobili A, Palmieri V, et al. Alterations of the intestinal mucus layer correlate with dysbiosis and immune dysregulation in human Type 1 Diabetes. EBioMedicine (2023) 91:104567. doi: 10.1016/j.ebiom.2023.104567
104. Sanz Y, Sánchez E, Marzotto M, Calabuig M, Torriani S, Dellaglio F. Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol Med Microbiol (2007) 51:562–8. doi: 10.1111/j.1574-695X.2007.00337.x
105. Lorenzo Pisarello MJ, Vintiñi EO, González SN, Pagani F, Medina MS. Decrease in lactobacilli in the intestinal microbiota of celiac children with a gluten-free diet, and selection of potentially probiotic strains. Can J Microbiol (2015) 61:32–7. doi: 10.1139/cjm-2014-0472
106. Olivares M, Benítez-Páez A, de Palma G, Capilla A, Nova E, Castillejo G, et al. Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: The PROFICEL study. Gut Microbes (2018) 9:551–8. doi: 10.1080/19490976.2018.1451276
107. Leonard MM, Valitutti F, Karathia H, Pujolassos M, Kenyon V, Fanelli B, et al. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc Natl Acad Sci U.S.A. (2021) 118:e2020322118. doi: 10.1073/pnas.2020322118
108. Girdhar K, Dogru YD, Huang Q, Yang Y, Tolstikov V, Raisingani A, et al. Dynamics of the gut microbiome, IgA response, and plasma metabolome in the development of pediatric celiac disease. Microbiome (2023) 11:9. doi: 10.1186/s40168-022-01429-2
109. González-García BP, Marí S, Cilleros-Portet A, Hernangomez-Laderas A, Fernandez-Jimenez N, García-Santisteban I, et al. Two-Sample Mendelian Randomization detects bidirectional causality between gut microbiota and celiac disease in individuals with high genetic risk. Front Immunol (2023) 14:1082862. doi: 10.3389/fimmu.2023.1082862
110. Constante M, Libertucci J, Galipeau HJ, Szamosi JC, Rueda G, Miranda PM, et al. Biogeographic variation and functional pathways of the gut microbiota in celiac disease. Gastroenterology (2022) 163:1351–1363.e15. doi: 10.1053/j.gastro.2022.06.088
111. Knezevic J, Starchl C, Tmava Berisha A, Amrein K. Thyroid-gut-axis: how does the microbiota influence thyroid function? Nutrients (2020) 12:1769. doi: 10.3390/nu12061769
112. Fenneman AC, Bruinstroop E, Nieuwdorp M, van der Spek AH, Boelen A. A comprehensive review of thyroid hormone metabolism in the gut and its clinical implications. Thyroid® (2023) 33:32–44. doi: 10.1089/thy.2022.0491
113. Su X, Yin X, Liu Y, Yan X, Zhang S, Wang X, et al. Gut dysbiosis contributes to the imbalance of treg and th17 cells in graves’ Disease patients by propionic acid. J Clin Endocrinol Metab (2020) 105:3526–47. doi: 10.1210/clinem/dgaa511
114. Zheng D, Liao H, Chen S, Liu X, Mao C, Zhang C, et al. Elevated levels of circulating biomarkers related to leaky gut syndrome and bacterial translocation are associated with graves’ Disease. Front Endocrinol (Lausanne) (2021) 12:796212. doi: 10.3389/fendo.2021.796212
115. Gong B, Wang C, Meng F, Wang H, Song B, Yang Y, et al. Association between gut microbiota and autoimmune thyroid disease: A systematic review and meta-analysis. Front Endocrinol (Lausanne) (2021) 12:774362. doi: 10.3389/fendo.2021.774362
116. Jiang W, Yu X, Kosik RO, Song Y, Qiao T, Tong J, et al. Gut microbiota may play a significant role in the pathogenesis of graves’ Disease. Thyroid (2021) 31:810–20. doi: 10.1089/thy.2020.0193
117. Terato K, Do CT, Shionoya H. Slipping through the cracks: linking low immune function and intestinal bacterial imbalance to the etiology of rheumatoid arthritis. Autoimmune Dis (2015) 2015:e636207. doi: 10.1155/2015/636207
118. Chen J, Li S, Zhu J, Su W, Jian C, Zhang J, et al. Multi-omics profiling reveals potential alterations in rheumatoid arthritis with different disease activity levels. Arthritis Res Ther (2023) 25:74. doi: 10.1186/s13075-023-03049-z
119. Blenkinsopp HC, Seidler K, Barrow M. Microbial imbalance and intestinal permeability in the pathogenesis of rheumatoid arthritis: A mechanism review with a focus on bacterial translocation, citrullination, and probiotic intervention. J Am Nutr Assoc (2023), 1–18. doi: 10.1080/27697061.2023.2211129
120. Meng X, Zhou H-Y, Shen H-H, Lufumpa E, Li X-M, Guo B, et al. Microbe-metabolite-host axis, two-way action in the pathogenesis and treatment of human autoimmunity. Autoimmun Rev (2019) 18:455–75. doi: 10.1016/j.autrev.2019.03.006
121. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med (2016) 8:43. doi: 10.1186/s13073-016-0299-7
122. Luo Y, Tong Y, Wu L, Niu H, Li Y, Su LC, et al. Alteration of gut microbiota in high-risk individuals for rheumatoid arthritis is associated with disturbed metabolome and initiates arthritis by triggering mucosal immunity imbalance. Arthritis Rheumatol (2023). doi: 10.1002/art.42616
123. Häupl T, Sörensen T, Smiljanovic B, Darcy M, Scheder-Bieschin J, Steckhan N, et al. Intestinal microbiota reduction followed by fasting discloses microbial triggering of inflammation in rheumatoid arthritis. J Clin Med (2023) 12:4359. doi: 10.3390/jcm12134359
124. Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol (2011) 4:603–11. doi: 10.1038/mi.2011.41
125. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14:141–53. doi: 10.1038/nri3608
126. Roland MM, Mohammed AD, Kubinak JL. How MHCII signaling promotes benign host-microbiota interactions. PloS Pathog (2020) 16. doi: 10.1371/journal.ppat.1008558
127. Silverman M, Kua L, Tanca A, Pala M, Palomba A, Tanes C, et al. Protective major histocompatibility complex allele prevents type 1 diabetes by shaping the intestinal microbiota early in ontogeny. Proc Natl Acad Sci U.S.A. (2017) 114:9671–6. doi: 10.1073/pnas.1712280114
128. Andeweg SP, Keşmir C, Dutilh BE. Quantifying the impact of human leukocyte antigen on the human gut microbiota. mSphere (2021) 6:e00476–21. doi: 10.1128/mSphere.00476-21
129. Leonard MM, Karathia H, Pujolassos M, Troisi J, Valitutti F, Subramanian P, et al. Multi-omics analysis reveals the influence of genetic and environmental risk factors on developing gut microbiota in infants at risk of celiac disease. Microbiome (2020) 8:130. doi: 10.1186/s40168-020-00906-w
130. Aguayo-Patrón SV, Trujillo-Rivera OA, Cornejo-Granados F, Ochoa-Leyva A, Calderón de la Barca AM. HLA-haplotypes influence microbiota structure in northwestern mexican schoolchildren predisposed for celiac disease or type 1 diabetes. Microorganisms (2023) 11:1412. doi: 10.3390/microorganisms11061412
131. Yue T, Tan H, Wang C, Liu Z, Yang D, Ding Y, et al. High-risk genotypes for type 1 diabetes are associated with the imbalance of gut microbiome and serum metabolites. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1033393
132. Clancy RM, Marion MC, Ainsworth HC, Chang M, Howard TD, Izmirly PM, et al. Gut dysbiosis and the clinical spectrum in anti-Ro positive mothers of children with neonatal lupus. Gut Microbes (2022) 14:2081474. doi: 10.1080/19490976.2022.2081474
133. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut (2015) 64:406–17. doi: 10.1136/gutjnl-2014-306931
134. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
135. Berryman MA, Triplett EW, Ludvigsson J. Human leucocyte antigen-dependent colonization of lactobacillus in the early-life gut. Front Microbiomes (2023) 2:1192773. doi: 10.3389/frmbi.2023.1192773
136. Rashid T, Ebringer A, Wilson C. The role of Klebsiella in Crohn’s disease with a potential for the use of antimicrobial measures. Int J Rheumatol (2013) 2013:e610393. doi: 10.1155/2013/610393
137. Zhan Z, Liu W, Pan L, Bao Y, Yan Z, Hong L. Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway. Cell Death Discovery (2022) 8:251. doi: 10.1038/s41420-022-01015-3
138. Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome (2016) 4:17. doi: 10.1186/s40168-016-0163-4
139. Šuligoj T, Vigsnæs LK, den Abbeele PV, Apostolou A, Karalis K, Savva GM, et al. Effects of human milk oligosaccharides on the adult gut microbiota and barrier function. Nutrients (2020) 12:2808. doi: 10.3390/nu12092808
140. Pediaditakis I, Kodella KR, Manatakis DV, Le CY, Barthakur S, Sorets A, et al. A microengineered Brain-Chip to model neuroinflammation in humans. iScience (2022) 25. doi: 10.1016/j.isci.2022.104813
Keywords: ABIS, type 1 diabetes, celiac disease, rheumatoid arthritis, autoimmune thyroid disease, HLA-DR, HLA-DQ
Citation: Berryman MA, Ilonen J, Triplett EW and Ludvigsson J (2023) Important denominator between autoimmune comorbidities: a review of class II HLA, autoimmune disease, and the gut. Front. Immunol. 14:1270488. doi: 10.3389/fimmu.2023.1270488
Received: 31 July 2023; Accepted: 11 September 2023;
Published: 26 September 2023.
Edited by:
Fernando Gabriel Chirdo, CONICET Instituto de Estudios Inmunológicos y Fisiopatalógicos (IIFP), ArgentinaReviewed by:
Ana Maria Calderon De La Barca, National Council of Science and Technology (CONACYT), MexicoCopyright © 2023 Berryman, Ilonen, Triplett and Ludvigsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric W. Triplett, ZXd0QHVmbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.