 Shareni Jeyamogan
Shareni Jeyamogan Joseph R. Leventhal
Joseph R. Leventhal James M. Mathew
James M. Mathew Zheng Jenny Zhang
Zheng Jenny Zhang- 1Department of Surgery, Comprehensive Transplant Center Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 2Simpson Querrey Institute for BioNanotechnology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 3Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 4Microsurgery and Pre-Clinical Research Core, Comprehensive Transplant Center, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Despite the advances in therapeutic interventions, solid organ transplantation (SOT) remains the “gold standard” treatment for patients with end-stage organ failure. Recently, vascularized composite allotransplantation (VCA) has reemerged as a feasible treatment option for patients with complex composite tissue defects. In both SOT and VCA, ischemia reperfusion injury (IRI) is inevitable and is a predominant factor that can adversely affect transplant outcome by potentiating early graft dysfunction and/or graft rejection. Restoration of oxygenated blood supply to an organ which was previously hypoxic or ischemic for a period of time triggers cellular oxidative stress, production of both, pro-inflammatory cytokines and chemokines, infiltration of innate immune cells and amplifies adaptive alloimmune responses in the affected allograft. Currently, Food and Drug Administration (FDA) approved drugs for the treatment of IRI are unavailable, therefore an efficacious therapeutic modality to prevent, reduce and/or alleviate allograft damages caused by IRI induced inflammation is warranted to achieve the best-possible transplant outcome among recipients. The tolerogenic capacity of CD4+CD25+FOXP3+ regulatory T cells (Tregs), have been extensively studied in the context of transplant rejection, autoimmunity, and cancer. It was not until recently that Tregs have been recognized as a potential cell therapeutic candidate to be exploited for the prevention and/or treatment of IRI, owing to their immunomodulatory potential. Tregs can mitigate cellular oxidative stress, produce anti-inflammatory cytokines, promote wound healing, and tissue repair and prevent the infiltration of pro-inflammatory immune cells in injured tissues. By using strategic approaches to increase the number of Tregs and to promote targeted delivery, the outcome of SOT and VCA can be improved. This review focuses on two sections: (a) the therapeutic potential of Tregs in preventing and mitigating IRI in the context of SOT and VCA and (b) novel strategies on how Tregs could be utilized for the prevention and/or treatment of IRI.
1 Introduction
The alarming rate at which end-stage organ failure cases are being reported has become a major public health concern, and its burden is expected to double yearly, in part due to the increase in ageing population (1–4). Despite the advances in therapeutic interventions, solid organ transplantation (SOT) is currently the “gold standard” treatment for patients with end-stage organ failure. Moreover, statistics from the Global Observatory on Donation and Transplantation (GODT) from World Health Organization (WHO), demonstrated that kidneys make up the majority of organ transplants followed by liver, heart, lung, pancreas (2, 3, 5) and the percentage of solid organ transplants are projected to increase yearly (2, 3, 5). Unlike SOT, vascularized composite allotransplantation (VCA) involves the transplantation of composite tissues comprising of multiple tissue types such as nerves, muscles, skin, bone, and blood vessels (6, 7). VCA, which is arguably more life-enhancing rather than a life-saving, recently has been deemed a reliable treatment option for patients with complex composite tissue injury (7). In SOT and VCA, besides alloantigen based immunogenicity, the occurrence of ischemia reperfusion injury (IRI) in transplanted allografts resulting in compromised allograft viability, function, and overall transplant outcome, remain a major challenge (8–10). IRI in transplantation, is a complex inflammatory phenomenon triggered by temporary deprivation of blood supply during donor organ procurement and exacerbated following, restoration of oxygenated blood supply after revascularization of the transplanted organ. In both SOT and VCA, IRI is inevitable and is a predominant factor for the onset of several life-threatening complications such as early graft dysfunction and/or graft rejection due to amplified, and prolonged alloantigen immunogenicity induced inflammatory activity (8–11).
To date, Food and Drug Administration (FDA) approved drugs for the treatment of IRI in the context of SOT and VCA are unavailable. Therefore, an efficacious approach or potential therapeutic modality to prevent, reduce and/or to alleviate allograft damages caused by IRI induced inflammation is warranted to achieve the best-possible transplant outcome among transplant recipients. Over the last few years, it has become evident that numerous myeloid lineage regulatory cells (e.g., Dendritic regulatory cells, polymorphonuclear myeloid suppressor cells) and lymphoid lineage regulatory cells (CD4+, CD8+ or B regulatory cells) play crucial roles in the maintenance of immune homeostasis. Among these regulatory cell populations, the discovery and identification of specific CD4+CD25+FOXP3+ regulatory T cells (Tregs) in both human and animal models, had led to unprecedented and extensive investigations on the therapeutic potential of these cells. Studies on autoimmune diseases and organ transplantation had demonstrated the ability of Tregs in inducing immunological tolerance and maintaining immune homeostasis by inhibiting and/or reducing the severity of pro-inflammatory events (12). Furthermore, the natural existence of Tregs in the immune system makes them an excellent candidate to be exploited for the treatment and/or prevention of numerous life-threatening immunological conditions and diseases.
The efficacy of Tregs in suppressing inflammatory responses driven by adaptive immune cells in SOT have been extensively studied and reviewed previously (12, 13). It is important to note that in addition to CD4+ Tregs, CD8+ Tregs are also able to induce immune tolerance and regulatory effects against inflammatory responses. However, due to their low frequency in vivo, the immunomodulatory potential of CD8+ Tregs against IRI have yet to be fully exploited (14). Due to the lack of studies on CD8+ Tregs, we decided to extensively review the therapeutic role of CD4+ Tregs in IRI induced inflammation. In this review, we first provide an overview of the pathophysiology of IRI in the transplant setting. We then review the therapeutic potential and possibilities of Tregs as immunomodulatory and anti-inflammatory mediators, in mitigating IRI induced inflammation. Lastly, we discuss the possible approaches and strategies which could be utilized to optimize the therapeutic efficacy of Tregs for the prevention and/or treatment of IRI in SOT and VCA.
2 Pathogenesis of ischemia and reperfusion injury in transplantation
It has been well established that the inflammatory responses in IRI are initiated during ischemia and are exacerbated during reperfusion (15). For instance, in kidney transplantation, prior to the removal of kidney from the donor, ischemia induced inflammation begins as soon as the donor’s blood supply to the kidney is stopped. During removal, the kidney graft undergoes a period of “warm ischemia” (9, 16) followed by, “cold ischemia” when the removed kidney is stored in a cold preservation solution to protect the viability of kidney cells (15, 17) and to slow down hypoxia mediated cellular stress and inflammatory responses exerted on the graft (18), for a period of time. Once the kidney is implanted into the recipient (before revascularization), the kidney graft experiences a short duration of “warm ischemia” (8–11, 16). Revascularization after implantation, enables the flow of oxygenated blood to the post-ischemic kidney graft (reperfusion), resulting in the activation and initiation of a cascade of pro-inflammatory events, acting interdependently on the affected kidney graft (16, 17). Depending on the severity of IRI, the harmful effects of IRI induced inflammatory responses, may promote graft rejection, graft dysfunction and/or even death (17). The unavoidable occurrence of IRI in both SOT and VCA warrants, a pressing need for alternative approaches and/or strategies to prevent, inhibit or at the very least to reduce IRI induced inflammatory responses against transplanted grafts.
2.1 Initiation of cellular stress during ischemia
Oxygen is a critical component required in cellular metabolism and mitochondrial oxidative phosphorylation to produce energy. Hence, lack of oxygenated blood supply to tissues during hypoxia (ischemic period) impairs both, the mitochondrial energy production metabolic route and cellular metabolic activity (19, 20). The inhibition of mitochondrial oxidative phosphorylation activity caused by the absence of molecular oxygen directly impacts the generation of adenosine triphosphate (ATP) thereby, affecting glycolysis mechanism. As an alternate resort to produce ATP during hypoxia, the aerobic glycolytic pathway is switched to the anaerobic glycolytic pathway to ensure continuity of energy production (19). Consequently, since lactate (also known as lactic acid) is required for the production of ATP in anerobic glycolysis, mass production and accumulation of the extremely acidic lactate in the cytosolic region of mitochondria, compromises the efficacy of the mitochondrial sodium (Na+)-potassium (K+) pump function resulting in, increased mitochondrial membrane permeability followed by decreased mitochondrial viability (8, 20–22). Furthermore, the inhibition of oxidative phosphorylation and inefficacious Na+/K+ pump function also leads to the accumulation of mitochondrial cytosolic calcium (Ca2+). This in turn not only inhibits ATP generation, but also increases the generation of both, reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and superoxide as well as, proteolytic enzymes such as phospholipase A2 (10, 19). At this stage, the damaging effects of ROS and proteolytic enzymes are confined within the internal matrix of the mitochondria due to the mitochondrial permeability transition pores (MPTP) which are initially closed (19, 21) (Figure 1). Subsequently, continuous action of oxidative stress on the mitochondrial membrane results in a completely permeable mitochondrial membrane with widened MPTP. Following this, ROS and other cytotoxic enzymes are released from the mitochondria to the cytosolic region of cells thus, initiating pro-inflammatory responses at the cellular level (8, 20–22).
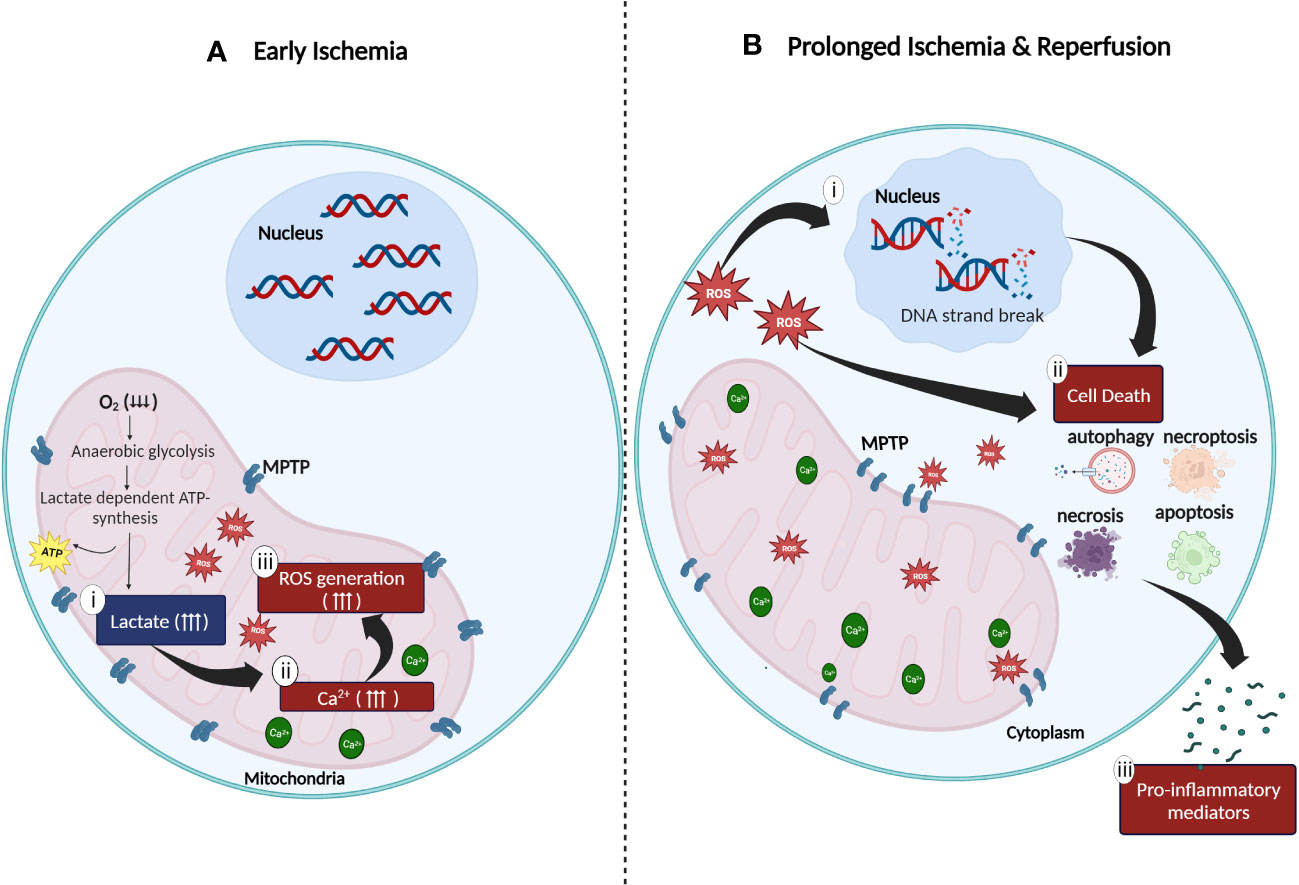
Figure 1 Cellular oxidative stress during (A) ischemia and (B) reperfusion injury. (A) Anaerobic metabolic activity during ischemia generates energy via the lactate dependent ATP pathway resulting in (i) lactate acidosis followed by (ii) accumulation of cytosolic calcium in the mitochondrial matrix due to compromised function of the sodium (Na+)-potassium (K+) pump. Increased concentration of mitochondrial Ca2+ generates (iii) high amount of ROS which is trapped in the mitochondria due to the closed mitochondrial permeability transition pores (MPTP). (B) Continuous stress on MPTP leads to the widening of MPTP which results in the release of ROS to the cytoplasm of cells. The release of ROS to the surrounding matrix leads to the (i) destruction of deoxyribonucleic acid (DNA) single strands and (ii) activation of cell death enzymes such as nuclear enzyme poly (ADP-ribose) synthetase which promotes apoptosis, necrosis, autophagy, and necroptosis. (iii) Excessive cell death mechanisms contribute to the release of pro-inflammatory mediators which further aggravates the inflammatory response (created with BioRender.com).
2.2 Reperfusion triggers pro-inflammatory events
Restoration of oxygen to hypoxic tissues during reperfusion, further exacerbates the existing pro-inflammatory activities initiated during ischemic stress in the mitochondria. This is driven by four major factors: (i) enhanced ROS production, (ii) overexpression of adhesion molecules, (iii) excessive infiltration of pro-inflammatory immune cells and (iv) damaged microvascular endothelial cells. The influx of molecular oxygen during reperfusion, accelerates the production of ROS (23, 24). Enzymes such as xanthine oxidase (23, 25), Nicotinamide Adenine Dinucleotide Phosphate oxidase (NADPH) (23, 25), monoamine oxidase (23, 26) and cytochrome P450 (23, 26) also mediates the production of ROS in an accelerated manner. As discussed previously, during ischemia, excessive accumulation of Ca2+, ROS and proteolytic enzymes in the mitochondria causes widening of MPTP resulting in the release of large amounts of ROS, proteolytic enzymes, and pro-apoptotic enzymes such as cytochrome C into the cytoplasm of cells (24). The release of these molecules and enzymes leads to a cascade of events including deoxyribonucleic acid (DNA) single strand breakage, increased production of intracellular pro-inflammatory chemokines and cytokines and overexpression of cell surface adhesion molecules (e.g., Intracellular Adhesion Molecule-1, ICAM-1; Vascular Cell Adhesion Molecule-1, VCAM-1; E- selection and P-selectin) on the injured endothelial cells (27–29). Spontaneously, the release of danger-associated molecular pattern signals (DAMPs), pro-inflammatory cytokines (e.g., Tumor Necrosis Factor-α, TNF-α; Interleukin-6, IL-6) and chemokines (e.g., C-C Motif Chemokine Ligand 2, CCL2) responsible for the activation of Toll-like receptors (TLR) in ischemic tissues, accelerates the inflammatory responses in IRI (19, 30) (Figure 1). Endoplasmic reticulum (ER) stress, characterized by the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum lumen during IRI is another major problem caused by IRI (31, 32). ER stress affects the function of unfolded protein responses (UPRs), resulting in compromised protein synthesis and cell function (29, 31, 32). Aphoristically, persistent IRI, if not treated, initiates a milieu of cell death pathways such as necrosis, apoptosis, autophagy, and necroptosis (33, 34) (Figure 1). The long-term persistence of these damaging mechanisms eventually leads to graft dysfunction and rejection (8, 22, 27, 28, 35).
2.3 Recruitment of innate immune cells during IRI
Detection of the above-mentioned pro-inflammatory signals and cell surface adhesion molecules by circulating leukocytes leads to increased leukocyte receptor binding affinity, resulting in the migration and infiltration of leukocytes across the endothelium of the injured tissues (29, 30, 35). The mechanism of leukocyte infiltration into injured tissues during IRI comprises of 4 major steps: (i) tethering/rolling, (ii) adhesion, (iii) transmigration/extravasation and (iv) infiltration (36–39). During reperfusion, the overexpression of selectins such as L-selectin, P-selectin and E- selectin, on the surface of blood vessel endothelial cells of transplanted grafts, mediates the tethering and rolling of leukocytes towards the endothelial cell ligands (36). Following that, cells expressing integrins such as lymphocyte function-associated antigen-1 (LFA-1), α4 integrins and β1 integrins, adheres to the adhesion receptor molecules which are abundantly expressed on the surface of injured tissues (37, 38). In tissues with IRI, prominently expressed adhesion molecules (e.g., ICAM-1, VCAM-1) promote migration and adhesion of LFA-1 expressing leukocytes to the injured tissue within the first 30 to 60 minutes after reperfusion (36, 37). Pro-inflammatory chemoattractants and/or chemokines released during ischemia and reperfusion, also attract circulating leukocytes towards injured tissues, thus promoting increased chances of adhesion to the surface of injured tissues. Upon firm and successful adhesion to endothelial cells, leukocytes undergo polarization and diapedesis to successfully infiltrate the injured tissue (36, 38). Following successful leukocyte infiltration/extravasation into the injured organ/tissue, cytotoxic signals in the form of cytokines and chemokines are released into the circulation. The release of these signals drives the maturation of lymph node penetrating tissue resident dendritic cells (DC), resulting in increased activation and migration of leukocytes to the site of inflammation (37, 38).
Neutrophils, being the most abundant leukocyte in the circulatory system, are not only the first LFA-1 expressing cells to migrate, adhere and infiltrate the injured tissue but also are key players in augmenting IRI inflammation. Infiltration of neutrophils accelerates the production of pro-inflammatory cytotoxic mediators such as ROS, myeloperoxidases, and Interleukin-17 (IL-17) cytokines (40, 41). Besides neutrophils, activation of C-C chemokine receptor type 2 (CCR2) and CXC3 chemokine receptor 1 (CX3CR1) in IRI promotes the migration, adhesion, and infiltration of pro-inflammatory type M1 macrophages at the injured site (41, 42). Infiltration of M1 macrophages, results in the release of inflammatory-promoting molecules such as IL-6, TNF-α, nitric oxide (NO) leading to continuous inflammatory responses (41, 42). Conversely, the ability of pro-inflammatory M1 macrophages in differentiating into anti-inflammatory M2 macrophages, resulting in the reduction of IRI induced inflammation and recovery of renal function in IRI experimental models were observed (43). However, the exact mechanism responsible for the “switch” remains unknown and must be investigated before macrophages can be considered as potential therapeutic agents along with Tregs for the attenuation of IRI induced inflammation. Furthermore, maturation of resident monocytes (macrophage and dendritic cell precursors) has also been observed in kidneys with IRI. Under inflammatory conditions, “resident” CCR2−CX3CR1highGR-1−Ly6C− monocytes are differentiated into CCR2+CX3CR1lowGR-1+Ly6Chigh pro-inflammatory macrophages and DCs (44, 45). Therefore, the reduction and/or inhibition of pro-inflammatory mediators might offer a potential strategy for the treatment of IRI by reducing the infiltration of pro-inflammatory immune cells.
2.4 IRI augments alloimmunity via adaptive cell infiltration
Inflammatory responses leading to the onset of graft dysfunction and rejection in the presence of alloantigens are major challenges in organ/composite tissue transplantation. Upon implantation of the donor graft into recipient, allograft immune recognition is initiated by both, preformed alloantibodies, if any, and recipient T-cell receptor engagements. Generally, T-cell recognition occurs via 3 pathways: direct pathway, indirect pathway, and semi-indirect pathway (12, 13). The direct allorecognition pathway involves the activation of recipient T cells by peptides presented on donor Major Histocompatibility Complex (MHC) of donor antigen presenting cells (APCs) whereas, the indirect allorecognition pathway involves recipient T cell recognition of donor peptides presented by recipient MHCs on recipient APCs (12, 13). The semi-direct allorecognition pathway involves a hybrid of the two with the recipient T cell recognizing donor MHC peptides presented by donor MHC but on recipient APCs (12, 13). Apart from donor MHC and MHC peptides, other antigenic moieties generated during IRI, such as damaged DNA strands, danger associated molecular pattern (DAMPs) like Heat-Shock Proteins (HSP) and released cytotoxic molecules from dying cells, are presented to the recipient T cells by donor APCs thus activating the direct allorecognition pathway (11, 39). Concurrently, the signals and products released during cell death are also presented to recipient T cells via the indirect and semi-direct pathway. As a consequence, upon successful implantation of allografts, the presence of both allorecognition induced inflammation and IRI induced inflammation magnify the allograft infiltration rate of CD4+ and CD8+ T cells as well as inflammatory macrophages, resulting in the release of abundant pro-inflammatory interleukin cytokines (e.g., IL-6, IL-1, IL-17a) and chemokines from the CXC and CC groups (e.g., CXCL1, CXCL2, CCL2) (11). Additionally, increase of thrombin expression in IRI allografts leads to the increase of CCL2 and CCL3 expression, resulting in increased infiltration of CD4+ T cells and decreased infiltration of existing endogenous Tregs (46). The heavy influx of inflammatory events due to increased production of inflammatory mediators, presence of alloantigens, and inflammatory cell infiltration further exacerbates the severity of inflammatory responses whilst compromising the viability of the transplanted allograft.
3 Therapeutic potential of Tregs against ischemia and reperfusion injury
Numerous studies have shown the involvement of Tregs in driving a “self-regulatory defense” mechanism following the onset of inflammation (47) however, the exact anti-inflammatory mechanism exerted by Tregs in mitigating ischemic reperfusion injury remain to be elucidated. Tregs are phenotypically heterogenous and are classified into various subsets such as natural Tregs (thymus derived Tregs), inducible Tregs (iTregs), CD8+ Tregs, IL-17 producing Tregs, co-stimulatory Tregs (ICOS+ Tregs), and Type 1 regulatory T (Tr1) cells. Despite the phenotypic difference, these Tregs subsets possess similar characteristics such as FOXP3+ expression, IL-2 cytokine dependency for survival and production of anti-inflammatory cytokine such as IL-10 (48), except for Tr1 cells, which secret high IL-10 but do not express Foxp3 (49, 50). CD4+ CD25+ Foxp3 Tregs (both nTregs and iTregs) have been mostly investigated regarding their role in modulating and mitigating the harmful effects of inflammatory immune responses (47, 51); findings from these studies will be highlighted in the following sections.
The efficacy of Tregs in promoting graft (e.g., kidney, heart, liver and vascularized composite tissue) protection and tolerance against the detrimental effects of IRI induced inflammation, besides maintaining immune homeostasis has been well evidenced in numerous in vitro and in vivo studies (51–54). Jun et al. (2014) showed that, at 72 hours post-reperfusion, the population of circulating endogenous Tregs significantly increased and the majority of Tregs accumulated at injured kidneys. Moreover, the accumulation of Tregs at injured kidneys led to the declination of blood urea nitrogen (BUN) and serum creatinine (sCr) values (55), suggesting the presence of both kidney functional recovery and a pro-reparative mechanism. On the contrary, deterioration of kidney function was observed in experimental groups which were treated with PC61 (anti-CD25 monoclonal antibody) to deplete the presence of endogenous Tregs (55, 56). Treg-mediated immunoregulatory activity was also observed when ex vivo autologous expanded Tregs were infused in hepatic IRI experimental models. The levels of serum alanine aminotransferase, aspartate aminotransferase, Interferon-γ (IFN-γ) and IL-17 were significantly reduced in IRI-induced livers receiving Tregs, as compared to non-treated control groups (53). Similarly, Kinsey et al. (55), demonstrated the presence of IL-10 overexpression on Tregs which co-localized at IRI kidneys when, ex vivo expanded autologous Tregs were administered prior to the onset of IRI (56).
Findings from the above studies, displayed the involvement of Tregs in attenuating and/or reducing the severity of IRI induced inflammation possibly, by four ways: (i) mitigating cellular oxidative stress, (ii) initiating healing, tissue repair and recovery mechanisms, (iii) preventing and/or reducing the adhesion and infiltration of both, innate immune cells, and adaptive T cells at the injured tissue and (iv) producing anti-inflammatory cytokines (Figure 2). Research progress in transplant immunology and autoimmune diseases, has confirmed the immunosuppressive and anti-inflammatory potential of Tregs though, the exact role and mechanisms exerted by Tregs in inhibiting and/or diminishing the progression of IRI induced inflammatory activity remains to be elucidated (55, 56).
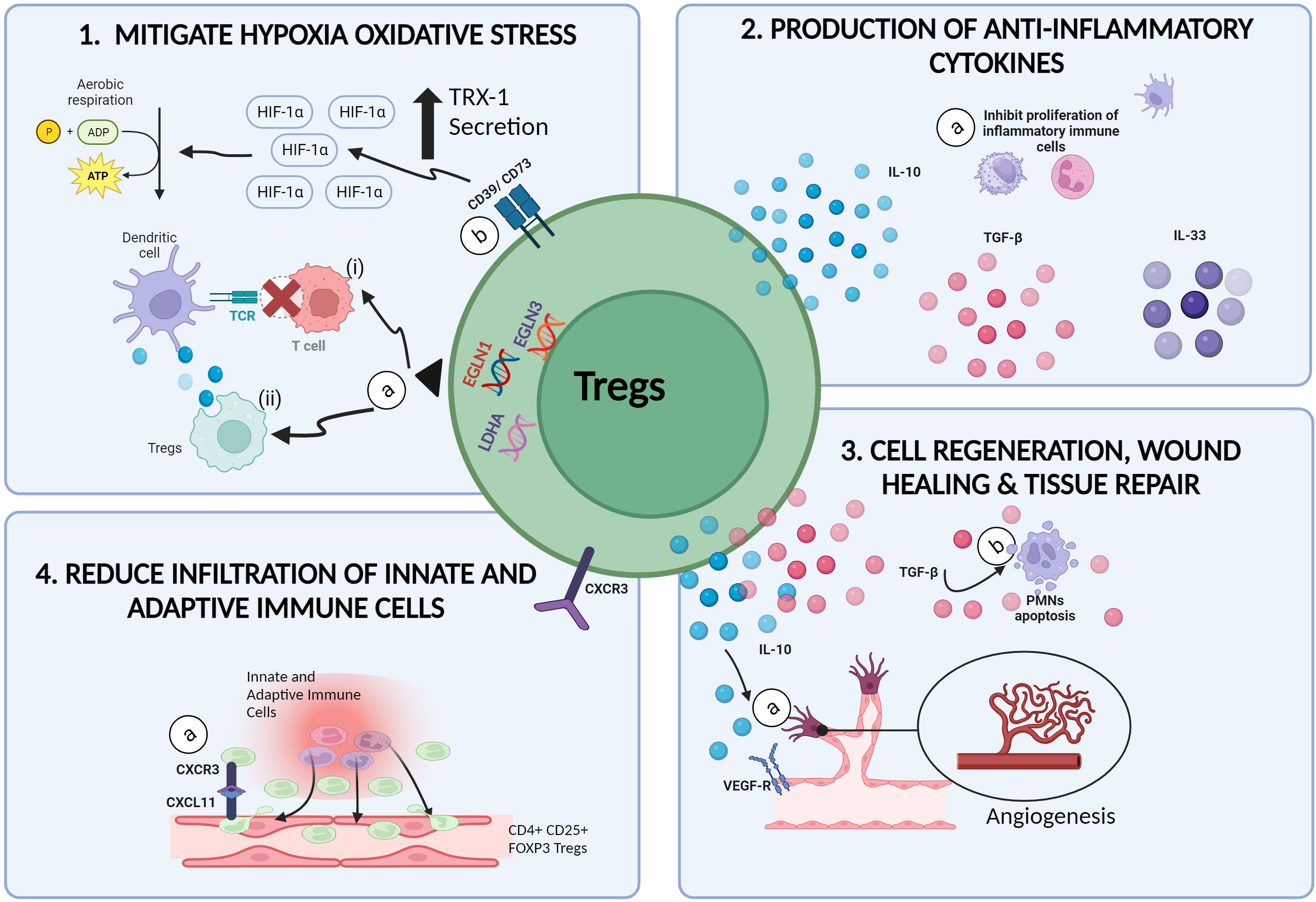
Figure 2 Therapeutic potential of Tregs in mitigating IRI. (1a) In hypoxic microenvironments, the upregulation of EGLN1, EGLN3 and LDHA genes in Tregs, (i) inhibits the communication between alloantigen loaded DCs with naïve T cells, hence preventing the onset of alloimmune immunogenicity induced inflammation. (ii) Tregs are also able to extract and ingest cellular materials from DCs in a process called trogocytosis, resulting in reduced MHCII expression of DCs. (1b) CD39/CD73 expressing Tregs promotes the upregulation of H1F1A expression, resulting in the switch from anaerobic glycolysis to aerobic glycolysis and reduced ROS production. (2a) The secretion of anti-inflammatory cytokines such as IL-10, TGF-β and IL-35 by Tregs promotes the inactivation and growth of inflammatory immune cells such as NKs, DCs and PMNs. (3a) Tregs promote angiogenesis by activating VEGFR-2 receptors via the secretion of IL-10 cytokines. (3b) Tregs are also capable of promoting wound healing and tissue repair in injured tissues by inducing apoptosis and cell death among PMNs via the production of TGF-β. (4a) Increased expression of CXCR3 on Tregs allows the binding of Tregs to the CXCL11 ligands on injured tissue, thus reducing the infiltration of other inflammatory cells (created with BioRender.com).
3.1 Tregs mitigate cellular oxidative stress
The initiation and progression of hypoxia induced damage in tissues deprived of nutrients and oxygen during ischemia, is driven by increased toxic metabolite production (57), oxidative stress (8, 57), mitochondrial dysfunction (8, 22) and overexpression of pro-inflammatory signals released during cell death mechanisms. The continuous cycle of pro-inflammatory events eventually results in allograft rejection and/or organ dysfunction (8, 22, 57). Findings from research on cancer immunotherapies had provided insights on the superiority of Tregs as opposed to other immune cells, in being tolerant and resistant to hypoxia induced stress (58, 59). In hypoxic microenvironments, which is a common scenario in cancer cases, the upregulation of genes such as Egl-9 Family Hypoxia Inducible Factor 1 (EGLN1), Egl-9 Family Hypoxia Inducible Factor 3 (EGLN3) and Lactate Dehydrogenase A (LDHA) in Tregs, represses the antigen presenting efficacy of type -2 conventional dendritic cells (cDC2) thus, inducing immune tolerance and homeostasis (59). The antigen presenting efficiency of DCs can be compromised by Tregs via several pathways. Firstly, Tregs can inhibit the communication between alloantigen loaded DCs with naïve T cells, hence preventing the onset of alloimmune immunogenicity induced inflammation (60). Secondly, Tregs are able to extract and ingest cellular materials from DCs in a process called trogocytosis, resulting in reduced MHCII expression of DCs (61, 62) (Figure 2). Furthermore, the generation of ROS were shown to stimulate the expression of Programmed Death-Ligand 1 (PD-L1) proteins however the mechanism responsible for this correlation remains vague (63). Hypoxia induced upregulation of PD-L1 expressing DCs, were previously shown to induce differentiation of naïve T cells to Tregs (63), increase the production of chemokines responsible for the infiltration of Tregs (e.g., CXCL5, CCL20) (64) and increase the proliferation of CD169+ macrophages responsible for the recruitment of Tregs at the site of inflammation (59). In a nutshell, the hypoxic resistant characteristics of Tregs, allows Tregs to hit two birds with a stone: (i) thriving throughout ischemia whilst (ii) promoting anti-inflammatory activities during ischemia.
As opposed to most T cells, Tregs are dependent on fatty acid oxidation for persistence and functionality. Depletion of glucose during ischemia promotes proliferation of Tregs at high speed due to the presence of lipid in the circulation. As a result, the increased number of Tregs in the circulation would enable the therapeutic and protective effect of Tregs on the ischemic organ. Interestingly, the inhibition of glycolysis was shown to induce the differentiation of T cells to Tregs due to the increased presence of HIF-1α (65). Therefore, taking into account, both the hypoxic resistant characteristics of Tregs and the presence of Treg-favoring mechanisms during hypoxic oxidative stress, a strategy to manipulate and use these mechanisms to induce Treg mediated immune tolerance may hold the key for the treatment of IRI.
As discussed previously, the overproduction of lactic acid during anaerobic metabolism, impairs the Na+-K+ pump function, leading to the accumulation of Ca2+ and increased ROS production in the mitochondrial matrix (8, 20, 22). The continuous production of ROS during the ischemic episode, reperfusion episode and inflammatory responses in IRI is a major concern as the overexpression of ROS, irreversibly damages the mitochondria, cells, and can even lead to the onset of cancer (58, 64). As compared to other immune cells, the ability of Tregs to withstand the damaging effects of oxidative stress makes them a potential therapeutic option for the treatment and/or management of allografts with IRI. In the presence of oxidative stress, Tregs are protected from undergoing apoptosis due to the ability in producing and secreting high concentrations of thioredoxin-1 (Trx-1) (Figure 2). Trx-1 is a cysteine-containing redox-active protein which can scavenge H2O2, resulting in reduced production and release of oxidative-stress harmful by-products (58, 66, 67).
CD39/CD73 expressing Tregs have been shown to display enhanced tolerogenicity against oxidative stress whilst inhibiting inflammation, in the presence of hypoxia-inducible factor 1-alpha (HIF1A) (68) (Table 1). Previous studies have demonstrated both, the upregulation of HIFIA under hypoxia and the protective role of HIF1A on kidneys (68, 80). Since one of the major causes of IRI is related to the presence of oxygen deprivation, utilizing the benefits which HIF1A can exert on kidneys during this episode would help in the effort to reduce the harmful effects of IRI in kidney allografts post transplantation. Recently, HIFIA were shown to protect cardiac cells against IRI induced inflammation by modulating mitochondrial metabolism and promoting angiogenesis (20). Jarvis et al. (68) had developed an optimized protocol for the expansion of Tregs with significant expression of CD39 and CD73 markers. The administration of CD73 expressing Tregs in recipients with kidney IRI, led to the upregulation of H1F1A expression in kidney cells, resulting in the switch of anaerobic glycolysis to aerobic glycolysis (68). In the context of IRI, this metabolic switch would result in the reduction of lactic acid production whilst preventing damages to the mitochondria and cells. Although this approach seems promising, it is crucial to determine the “right amount/optimal amount” of HIF1A-CD73 expressing Tregs interaction for the induction of immune tolerance as this hypoxic factor can also contribute to the onset of cancer.
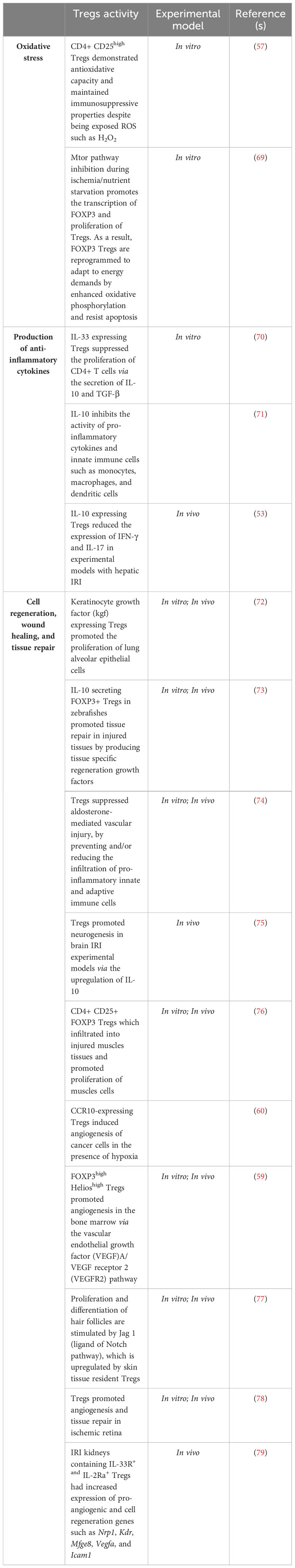
Table 1 Therapeutic role of Tregs in mitigating IRI induced inflammation.
Overall, Tregs can counter oxidative stress via several routes. As discussed earlier, besides producing high levels of Trx-1, Tregs are also able to limit the expression of γ-glutamylcysteine synthetase and glutathione synthesis which is crucial for the proliferation and activation of pro-inflammatory T cells by DCs (67, 81). Developments in autoimmune disease findings, had also revealed the potency of Tregs in reducing and/or inhibiting the production of harmful oxidative stress by-products. A decrease in serum oxidized low-density lipoprotein (ox-LDL), an oxidative stress product, were observed in Amyotrophic lateral sclerosis patients administered with ex vivo expanded autologous Tregs, as compared to the untreated control group (82). It is important to note that, anti-oxidative mechanisms are commonly modulated by antioxidant enzymes rather than endogenous cells itself. Therefore, identifying the exact anti-oxidative enzymes manipulated by Tregs for the suppression of IRI induced oxidative stress would hold the key to treat and/or to prevent oxidative stress in many other diseases, besides graft IRI. To date, in vivo data demonstrating the reduction and/or inhibition of IRI induced oxidative stress by Tregs remain insufficient therefore, understanding the route taken by Tregs in reducing oxidative stress may offer a potential strategy to prevent the onset of IRI in allografts instead of treating IRI grafts.
3.2 Tregs facilitates cell regeneration, wound healing and tissue repair
Even though the occurrence of inflammatory activity is part of the defense mechanism in tissue injury, excessive and persistent pro-inflammatory activity in IRI grafts can lead to the formation of fibrosis and scar tissues (e.g., heart), acute kidney injury (AKI) (e.g., kidney), graft dysfunction and/or graft failure. The indispensable roles of Tregs in promoting angiogenesis, wound healing, and tissue repair in addition to maintaining immune homeostasis, makes them an excellent candidate to be exploited for the treatment of grafts with IRI. To date, the cellular and molecular pro-reparative mechanisms mediated by Tregs in allografts with IRI has not been fully elucidated albeit, evidenced in numerous studies (Table 1). Tregs were reported to facilitate skin healing and wound repair by decreasing both, the expression of IFN-γ and pro-inflammatory macrophage accumulation at injured tissues via the Epidermal Growth Factor Receptor (EGFR) pathway (83). Wang et al. (75), demonstrated the ability of Tregs in promoting neurogenesis in brain IRI models via the upregulation of IL-10 (75).
The formation of new blood vessels, a process known as angiogenesis, plays an important role in tissue repair and wound healing (Figure 2). Generally, Tregs mediated pro-angiogenesis activity are driven by (i) increased production of anti-inflammatory cytokines, (ii) upregulation of angiogenesis promoting genes and proteins and (iii) activation of growth factor receptors to produce growth factors (75, 79, 84). Secretion of IL-10 anti-inflammatory cytokines and activation of Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) for the production of VEGFs (85) by Tregs in renal cell carcinoma, were shown to promote aggressive angiogenesis resulting in cancer cell expansion and metastasis (84, 86, 87). Considering this, the secretion of pro-angiogenic stimulators such as IL-10 and VEGF in IRI grafts, may promote angiogenesis which would eventually result in cell regeneration and tissue repair. The upregulation of pro-angiogenic and cell regeneration genes (e.g., Nrp1, Kdr, Mfge8, Vegfa, Icam1) by IL33R+ and IL-2Ra+ expressing Tregs in IRI kidneys, demonstrated by do Valle Duraes et al. (79) supported the above-mentioned hypothesis (79).
Nonetheless, Tregs were also shown to negatively regulate angiogenesis in certain tissues and conditions (84, 88, 89). Tregs inhibited endothelial cell angiogenesis by inducing the downregulation of VEGF and upregulation of the pro-apoptotic Notch signaling pathway (88). The administration of exogenous Tregs in CD28 Knock out (KO) mice with hindlimb IRI resulted in reduced neovascularization rate following IRI (89). Despite the numerous evidence of angiogenesis inhibition by Tregs, it is noteworthy that dysfunctional Tregs, rather than functional Tregs were reported to be involved in ischemia induced inflammation (90). In ischemic cardiomyopathy, excessive proliferation of immunomodulatory lacking- dysfunctional Tregs led to the overexpression of pro-inflammatory cytokines, anti-angiogenic activity, and formation of fibrosis (90). Adoptive transfer of functional Tregs to replace the dysfunctional Tregs in the context of ischemic cardiomyopathy may be a fruitful approach to overcome the complications in ischemic cardiomyopathy cases.
Tregs are also able to inhibit and/or manipulate the behavior of certain pro-inflammatory innate immune cells to promote tissue repair (91). In the presence of activated Tregs, polymorphonuclear neutrophils (PMNs) decreased the production of IL-6 pro-inflammatory cytokines and increased the production of anti-inflammatory molecules like IL-10, TGF-β1, heme oxygenase-1 (HO-1) and the suppressor of cytokine signaling 3 molecule (SOCS3) (91). Moreover, activated Tregs were also able to promote accelerated wound healing and tissue repair in injured lungs by inducing apoptosis and cell death among PMNs via the production of TGF-β (92). Aside from neutrophils, Tregs are also able to manipulate and reduce the pro-inflammatory activity exerted by other innate immune cells such as macrophages. Tregs promoted anti-inflammatory activity in cardiac tissues and inhibited the formation of scar tissues by preventing extracellular collagen degradation, as well as increasing the secretion of cytokines responsible for the proliferation of anti-inflammatory M2 macrophages (59, 93).
Owing to this finding, additional and detailed studies to confirm the exact mechanisms and/or trigger factors responsible for Treg mediated angiogenesis and anti-angiogenesis is warranted before these cells can be used as a therapeutic option in IRI.
3.3 Tregs secretes anti-inflammatory cytokines
The secretion of anti-inflammatory cytokines such as Interleukin-10 (IL-10) (56, 94–96), Interleukin-35 (IL-35) (96), Interleukin-4 (IL-4) (97, 98) and Transforming growth factor-beta (TGF-β) (96) by Tregs are the few major drivers responsible for the induction of Treg-mediated IRI inflammatory suppression (Figure 2) (Table 1). The attenuation of IRI inflammation observed in Recombination Activation Gene-1 (RAG-1) KO mice administered with Tregs as opposed to IL-10 KO Tregs (56), emphasized the importance of IL-10 in Tregs for the dampening of aberrant hypoxia induced inflammatory response. IL-10 exerts immunosuppressive effects by inhibiting the pro-inflammatory activity of macrophages and monocytes via the JAK/STAT signaling pathway, stimulating the production of anti-inflammatory molecules such as Interleukin-1 receptor antagonist (IL-1RA), soluble Tumor necrosis factor-alpha (TNF-α) and Interleukin- 27 (IL-27) as well as enhancing the function of Tregs (99). TGF-β on the other hand, is crucial for the differentiation of naïve T cells into Tregs, regulation of FOXP3+ expression and maintenance of functional Tregs via the IL-2/STAT5 signaling pathway (100, 101). IL-10 and TGF-β expressing Tregs also induces immune tolerance by decreasing the expression of pro-inflammatory cytokines, CC and CXC chemokines as well as adhesion molecules (56, 94–96, 99, 102). Consequently, the proliferation and activation of innate immune cells namely natural killer (NK) cells, granulocytes, DCs, monocytes, effector T cells and B cells are hampered. IL-4 was previously shown to exert both, negative and positive effect against Tregs. On a positive note, IL-4 are able to enhance the immunosuppressive activity and prolong the survival of Tregs (97, 98). Yang et al. (2017), showed the ability of IL-4 stimulated Tregs in inducing immune suppression via the secretion of granzymes. On the contrary, IL-4 and granzyme production also prolongs inflammatory events by inducing apoptosis among cytotoxic lymphocytes (103) which would eventually compromise the function of the transplanted graft.
3.4 Tregs reduces the infiltration of pro-inflammatory immune cells
Tregs are also able to decrease the progression of IRI induced inflammatory responses and induce immune tolerance by adhering to the adhesion molecules expressed on injured endothelial cells henceforth, reducing the infiltration of pro-inflammatory leukocytes (Figure 2). Overexpression of G-protein-coupled receptor 9 (CD183), commonly known as C-X-C motif chemokine receptor 3 (CXCR3) on Tregs were shown to be responsible in promoting Tregs mediated anti-inflammatory responses in IRI kidneys (52, 104, 105). Overexpression of CXCR3 on Tregs, were shown to increase both, rate of migration and binding and/or adhesion probabilities of Tregs to the CXCL11 ligand at injured tissues as compared to other pro-inflammatory leukocytes henceforward, reducing the severity of IRI inflammation (104, 105). Increased expression of CXCR3 on Tregs also led to the overexpression of anti-inflammatory cytokines (e.g., Interleukin-10), antioxidants (e.g., Superoxide dismutase, SOD and glutathione peroxidase, GSH-Px) and downregulated the expression of pro-inflammatory cytokines (e.g., IL-6 and TNF-α) in experimental models with renal IRI (105). However, although IRI induced inflammation can be attenuated or reduced by the binding of CXCR3 expressing Tregs to CXCL11 ligand, it is important to note that the binding of CXCR3 to other ligands such as CXCL9 and CXCL10 would increase the recruitment and infiltration rate of neutrophils and natural killer T cells, resulting in the worsening of pro-inflammatory events (52, 105, 106). Therefore, inhibition of these pro-inflammatory ligands and administration of CXCR3 rich Tregs in IRI renal grafts may promote tolerance against IRI induced inflammation. Another possible mechanism utilized by Tregs in reducing IRI induced inflammation is by binding to a class IV semaphorin known as, Semaphorin 4A (Sema4A) (107). The binding of neuropilin-1 (Nrp-1) on Tregs to Sema4A before the onset of IRI, resulted in accelerated expansion of endogenous Tregs which in turn, decreased the infiltration rate of innate immune cells such as macrophages and neutrophils into injured tissues leading to the alleviation of renal IRI inflammation (56, 107).
4 Treg based cellular therapy: challenges and stratagems to combat ischemia and reperfusion injury
The fundamental role played by Tregs in promoting immune tolerance, controlling pro-inflammatory activities, and maintaining immune homeostasis in both, alloantigen immunogenicity and inflammation has been well evidenced (52, 55, 56). The significant increase of endogenous Tregs proliferation and enhanced trafficking of Tregs at the site of inflammation, resulting in improved allograft function post IRI, demonstrated an obvious anti-inflammatory “antagonistic” relationship between Tregs against IRI progression. Regardless of the promising existence of Treg-mediated anti-inflammatory activity in allografts against IRI induced inflammation and alloantigen immunogenicity, depending completely on “endogenous” Tregs alone, seems unreasonable due to a number of reasons. Firstly, the number of endogenous Tregs in the circulation to drive anti-inflammatory responses is insufficient, as they only make up 5-10% of the total CD4+ T cell population (12, 108). Secondly, since Tregs in the circulation exists in small numbers, the time taken for the proliferation of Tregs to reach optimal levels before, initiating an anti-inflammatory response, might be too long. Grafts with IRI, will be severely damaged due to the co-existence of several pro-inflammatory events taking place simultaneously against the graft (108). Thirdly, the number of circulating endogenous Tregs are too little to compete, reduce and/or to prevent the adhesion and infiltration of other inflammatory driving leukocytes such as DCs, macrophages, granulocytes, and T cells at organs with IRI. Taking into consideration of the above-mentioned limitations of endogenous Tregs- feasible, reasonable, and strategic approaches to (i) increase the number of Tregs in the circulation, (ii) to enhance the survival of Tregs whilst, (iii) ensuring targeted delivery of Tregs are necessary to prevent, reduce and/or to alleviate IRI induced inflammation in allografts.
4.1 Adoptive transfer of ex-vivo expanded Tregs
Adoptive transfer of Tregs is a direct, convenient, and promising therapeutic strategy to increase the number of Tregs in the circulation for the prevention, inhibition and/or alleviation of IRI induced inflammatory responses in transplanted grafts. Several in vitro, in vivo, pre-clinical and clinical studies had demonstrated the efficacy of adoptive Treg cellular therapy for the treatment of autoimmune disorders (12, 109) and induction of immune tolerance in organ transplantation (12, 110). AKI was prevented in kidney grafts of recipients who received an adoptive transfer of polyclonally expanded, autologous Tregs before the onset of IRI (55). The kidneys of lymphocyte deficient RAG-1 KO mice were protected from IRI when polyclonally expanded, autologous Tregs were infused 2 weeks prior to the onset of IRI (56). Tregs reduced and/or prevented the progression of IRI-induced pro-inflammatory responses in RAG-1 KO mice kidneys by producing IL-10 cytokines. Despite understanding the therapeutic effect of IL-10 producing Tregs in IRI, it is noteworthy that the absence of both, endogenous T cells and B cells in RAG-1 KO mice, does not display the precise effect of IL-10 producing Tregs in treating IRI. Besides IL-10, TGF-β producing Tregs were reported to suppress and/or to prevent IRI-induced inflammation of the liver, when administered into C57BL/6 mice 24 hours before the onset of IRI (111). Gandolfo et al. (112) on the other hand, demonstrated increased trafficking of Tregs at the interstitium of IRI kidney outer medulla, when polyclonally expanded autologous Tregs were infused 24 hours after IRI. The heavy trafficking of Tregs at the site of injury, prevented and/or reduced the infiltration percentage of pro-inflammatory innate leukocytes such as CD11c+ F4/80 DCs as well as lymphoid T cells (112). As a result, abundant anti-inflammatory cytokines released by activated Tregs suppresses the rate of IRI-induced pro-inflammatory responses hence, resulting in a lesser extend of kidney damage as well as promoting kidney recovery (112). Overall, it can be presumed that adoptive cellular transfer of Tregs before and/or after the onset of IRI, may promote protection against transplanted grafts by ameliorating IRI-induced inflammatory effects. Although polyclonally expanded Tregs significantly demonstrated protection of renal grafts against the detrimental effects of IRI, the functional stability and increased probability of polyclonal Tregs in exhibiting pan-immunosuppression against broad spectrum of unspecific targets remain a concern (47, 113).
The existing limitation of using polyclonally expanded Tregs, although convenient, led to the discovery and development of donor antigen specific Tregs (ds-Tregs). In transplanted grafts, pro-inflammatory events are triggered by both, alloantigen recognition and IRI. In addition to that, IRI-induced inflammatory responses have also been shown to augment the severity of alloimmune response, resulting in graft rejection, and/or graft failure. As opposed to polyclonal Tregs, ds-Tregs offer a more potent and targeted approach in controlling inflammation and inducing immune tolerance since these cells can migrate and act specifically on the donor tissue containing the recognized alloantigens (12, 113). We hypothesize that, by utilizing ds-Tregs, the pro-inflammatory responses induced by allo-antigen recognition could be controlled and/or alleviated hence, indirectly suppressing oxidative stress, ROS generation and cell damaging mechanisms exerted on the transplanted graft, in addition to IRI. The anti-inflammatory and immunomodulatory potential of ds-Tregs in SOT has been extensively studied however, to the best of our knowledge, the potency of ds-Tregs for the prevention and/or treatment of IRI are yet to be discovered. Despite the efficacy of ds-Tregs in promoting immune tolerance, the purity and yield of expanded ds-Tregs remains a concern (12, 47, 114). Administration of low purity ds-tregs into transplant patients, may further aggravate the existing inflammation due to the presence of non-Tregs pro-inflammatory cells. Recently, Mathew et al. (114), established a protocol for the expansion of FOXP3 rich, immunosuppressive ds-tregs by using recipient Tregs stimulated with donor B cells. The expanded ds-tregs significantly, inhibited the proliferation of recipient T cells at a Tregs to responder cell ratio of (1:10) to (1:250) (114). Furthermore, as opposed to non-specific polyclonal Tregs, only a small number of ds-tregs are required to induce immunosuppression (113).
4.2 Endogenous Tregs proliferative stimulants
Alternatively, the immunomodulatory benefits of Tregs can also be exploited indirectly for the prevention and/or treatment of IRI by using non-cell-based therapeutic approaches. Cytokines such as TGF-β and Interleukin -2 (IL-2), play a crucial role in the survival, development, and immunosuppressive potential of Tregs (12, 115). The transcriptional identity of Tregs, characterized by the presence of CD25high FOXP3high is upregulated by Signal Transducer and Activator of Transcription 5 (STAT5) activation under the influence of IL-2 expression (115). As compared to other pro-inflammatory leukocytes such as T cells and NKs, CD25 proteins are significantly overexpressed on Tregs. Therefore, administration of IL-2, even in low doses, promotes selective targeting of IL-2 on CD25 receptors which are abundantly expressed on Tregs (115–117), resulting in the survival and proliferation of highly immunosuppressive CD25high FOXP3high Tregs. Owing to this theory, administration of low dose IL-2 along with ex vivo expanded autologous Tregs, may enhance the proliferation and prolong the survival of both, endogenous and infused Tregs in allografts with IRI. Additionally, inhibition of thrombin expression on injured endothelium, were shown to enhance the accumulation of both, endogenous and ex-vivo infused Tregs as well as M2 anti-inflammatory macrophages at the site of inflammation (46). Furthermore, recent studies in the application of normothermic and/or hypothermic machine perfusion of donor organs prior to transplant have shown promising results in reducing the extent of IRI induced damage on allografts (118). It is reasonable to speculate that lesser extent of IRI damage on the allograft would enhance the accumulation of Tregs at the injured site in a shorter period resulting in fast recovery. Enhanced migration, accumulation, and infiltration of Tregs at the site of inflammation would reduce and/or prevent both, the infiltration of other non-Tregs pro-inflammatory leukocytes and production of pro-inflammatory cytokines (117). The secretion of anti-inflammatory cytokines by Tregs at the site of inflammation would eventually aid in the alleviation of IRI induced inflammatory responses and accelerated healing of the injured graft.
Immunosuppressive (IS) agents, from the family of steroids, calcineurin inhibitors and mTOR inhibitors are routinely administered in transplantation to prevent the onset of aggressive inflammatory responses (116, 119). Interestingly, mTOR inhibitor IS drugs were found to selectively promote the proliferation, survival, and enhancement of suppressive phenotypes on Tregs whilst preventing the proliferation of effector T cells (116, 120). Therefore, the administration of mTOR inhibitor IS drugs such as Rapamycin, Sirolimus (121) and Everolimus (120) in SOT/VCA alone or along with ex vivo expanded autologous Tregs, preferably ds-Tregs, may prevent and/or alleviate the detrimental effects of IRI against grafts. While the effects of IS drugs from the mTOR inhibitor family on Tregs are extremely promising, the line which differentiates the “yin effect” from the “yan effect” of mTOR inhibitors against Tregs must be investigated in detail since precise mechanisms underlying mTOR inhibitor tolerogenic activity remains vague. The involvement of mTOR inhibitors in the progression of cancer, is one of the major reasons these IS drugs, are not the first choice of option during transplantation (122). In addition to this, IS drugs have also been reported to affect the expression of FOXP3 in Tregs therefore, the optimal dosage for optimal Tregs immunosuppression induction must be understood before they could be applied in the clinical setting. Alternatively, other compounds such as N,N-dimethylsphingosine (DMS) (123, 124), Histone deacetylase inhibitors (125, 126), HMG-CoA reductase inhibitors (127) and Sphingosine-1-phosphate receptor agonists (128) were also reported to protect organs against the detrimental effects of IRI by increasing the proliferation rate of immunosuppressive Tregs however, more detailed investigations on the exact underlying mechanisms of the above-mentioned molecules should be understood.
4.3 Localized administration route
To the best of our knowledge, the route used for the adoptive transfer of ex vivo expanded Tregs in the majority of current experimental models and clinical trials are via the convenient and minimally invasive intravenous (IV) route (129, 130). The route used for the delivery of therapeutic agents in general, plays a huge role in determining the effectiveness of a therapy. Previously, it was shown that the majority of IV administered therapeutic Mesenchymal Stem Cells (MSCs) were trapped within the pulmonary capillaries thus preventing optimal distribution to the targeted organs (131). On the contrary, IV-infused MSCs in recipients treated with sodium nitroprusside vasodilators, were able to reach the targeted organs without being trapped in the lungs, suggesting that the entrapment of MSCs could be due to their large size (129, 130, 132, 133). IV infusion of actively proliferating ex vivo expanded Tregs, may undergo the same problem resulting in lesser accumulation of Tregs at the IRI organ. In addition to size, other factors such as cellular adhesion to the vascular endothelium may contribute to the incidence of pulmonary cell trapping (133). In support of this, Lyu et al. (134), demonstrated the presence of CD49d lung homing marker on umbilical cord blood derived Tregs which resulted in the accumulation of Tregs at the lungs, post infusion (134). IV infused cells were also found to be distributed to other organs such as liver and spleen, after by-passing the lungs resulting, in increased cell loss and lesser accumulation at the targeted organ (129, 131). This limitation would result in the inability of Tregs in suppressing and/or alleviating IRI induced inflammation due to lesser number of Tregs accumulation at the site of inflammation (130). In order to overcome this problem, an alternative route is required to enhance the delivery of Tregs at the targeted organ. Direct injection of Tregs on grafts with IRI may offer a more promising outcome although, this approach may introduce more injury to the stressed organ/tissue (130).
4.4 Targeted delivery of Tregs using nanotechnology
Developing efficient cell delivery methods using nanomaterials could also significantly improve the effective implementation and outcomes of Treg-based cell therapy in IRI. The attachment of nanomaterials on the extracellular surface of Tregs is an approach which could be investigated for the treatment of IRI. As discussed earlier, the overexpression of adhesion molecules such as ICAM-1, VCAM-1 and MAdCAM-1 on the surface of IRI grafts following reperfusion, initiates the migration and infiltration of pro-inflammatory leukocytes at the site of injury (36–38), triggering a cascade of pro-inflammatory responses. In order to prevent this, the coating and/or binding of Tregs to nanoparticles which could selectively bind to adhesion molecules commonly upregulated in IRI, would reduce the infiltration of pro-inflammatory leukocytes at the site of inflammation. The usage of nanoparticles to aid and/or to enhance the efficacy of Treg based cellular therapy however warrants in-depth investigation to assess the costs, accuracy, and toxicity of nanoparticles against Tregs as well as other organs in the system.
5 Conclusion
Cellular adoptive therapy using Tregs for the induction of immune tolerance are a preferred choice as compared to synthetic drugs, since they are naturally existent and are non-toxic to our system. Although the advancements in immunology-based research have enabled us to isolate, identify and understand some of the pathways utilized by Tregs in alleviating and/or reducing the detrimental effects of IRI induced inflammatory events, all that glitters are not gold. One of the major challenges faced in the utilization of Tregs as a therapeutic option, is the ability of culturing and expanding ex vivo Tregs-expressing markers such CD73 which aids in the induction of immune tolerance, specifically in an IRI setting. Furthermore, it has been brought to attention that, a subset of FOXP3+ Tregs were able to produce and secrete pro-inflammatory cytokines under certain conditions (135). Therefore, the second challenge in using Tregs as a therapeutic option for the treatment of IRI is the lack of data on the role of certain stimulatory factors responsible for the differentiation and plasticity of Tregs. The ability to overcome at least a few of the challenges reviewed in this review, can open alternative and effective pathways in using Tregs for the treatment of IRI in clinical trials. This review is timely and further investigation is warranted to explore the various methods Tregs could be manipulated for the treatment of IRI.
Author contributions
SJ: Writing – original draft, Writing – review & editing. JM: Funding acquisition, Supervision, Writing – review & editing. ZZ: Funding acquisition, Supervision, Writing – review & editing, Conceptualization. JL: Writing – review & editing, Supervision, Conceptualization, Validation.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. The authors were supported by the funding from the Department of Defense, W81XWH2110862 ZJZ and W81XWH2110911 (JM).
Acknowledgments
All figures in this manuscript were created using biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APCs, Antigen Presenting Cells; ATP, Adenosine Triphosphate; BUN, Blood Urea Nitrogen; Ca2+, Calcium; cDC2, type-2 conventional dendritic cells; DAMPs, Danger-Associated Molecular Patterns; DNA, Deoxyribonucleic acid; EGFR, Epidermal Growth Factor Receptor; EGLN1, Egl-9 Family Hypoxia Inducible Factor 1; EGLN3, Egl-9 Family Hypoxia Inducible Factor 3; ER, Endoplasmic reticulum; FDA, Food and Drug Administration; Foxp3, forkhead box p3; GODT, Global Observatory on Donation and Transplantation; HSP, Heat Shock Proteins; H1FIA, Hypoxia-inducible factor 1-alpha; H2O2, Hydrogen peroxide; ICAM-1, Intracellular Adhesion Molecule-1; IFN-γ, Interferon γ; IL, Interleukin; IRI, ischemic reperfusion injury; K+, Potassium; KO, knockout; LDHA, Lactate Dehydrogenase A; LFA-1, Lymphocyte function-associated antigen-1; MHC, major histocompatibility complex; MPTP, Mitochondrial Permeability Transition Pores; Na+, Sodium; NADPH, Nicotinamide adenine dinucleotide phosphate; NO, Nitric oxide; PD-L1, Programmed Death-Ligand 1; RAG-1, recombinant activation gene-1; ROS, Reactive Oxygen Species; sCr, serum creatinine; SOT, Solid Organ Transplantation; STAT-5, Signal Transducer and Activator of Transcription 5; TGF-β, Transforming Growth Factor-βeta; TLR, Toll Like Receptors; TNF-α, Tumor Necrosis Factor-alpha; Tregs, regulatory T cells; UPR, Unfolded Protein Response; VCA, Vascularized Composite Allotransplantation; VCAM-1, Vascular Cell Adhesion Molecule-1; VEGF, Vascular Endothelial Growth Factor; WHO, World Health Organization.
References
1. Fung E, Kurella Tamura M. Epidemiology and public health concerns of CKD in older adults. Adv Chronic Kidney Dis (2016) 23(1):8–11. doi: 10.1053/j.ackd.2015.10.001
2. Data of the WHO-ONT Global Observatory on Donation and Transplantation. Organ donation and transplantation activities report. (GODT) GOoDaT. Global Observatory on Donation and Transplantation (2021).
3. Mudiayi D, Shojai S, Okpechi I, Christie EA, Wen K, Kamaleldin M, et al. Global estimates of capacity for kidney transplantation in world countries and regions. Transplantation (2022) 106(6):1113–22. doi: 10.1097/TP.0000000000003943
4. Triolo F, Gridelli B. End-stage organ failure: will regenerative medicine keep its promise? Cell Transplant (2006) 15 Suppl 1:S3–10. doi: 10.3727/000000006783982340
5. Matesanz R, Mahillo B, Alvarez M, Carmona M. Global observatory and database on donation and transplantation: world overview on transplantation activities. Transplant Proc (2009) 41(6):2297–301. doi: 10.1016/j.transproceed.2009.05.004
6. Anggelia MR, Cheng HY, Lai PC, Hsieh YH, Lin CH, Lin CH. Cell therapy in vascularized composite allotransplantation. BioMed J (2022) 45(3):454–64. doi: 10.1016/j.bj.2022.01.005
7. Kadono K, Gruszynski M, Azari K, Kupiec-Weglinski JW. Vascularized composite allotransplantation versus solid organ transplantation: innate-adaptive immune interphase. Curr Opin Organ Transplant (2019) 24(6):714–20. doi: 10.1097/MOT.0000000000000705
8. Salvadori M, Rosso G, Bertoni E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J Transplant (2015) 5(2):52–67. doi: 10.5500/wjt.v5.i2.52
9. Halazun KJ, Al-Mukhtar A, Aldouri A, Willis S, Ahmad N. Warm ischemia in transplantation: search for a consensus definition. Transplant Proc (2007) 39(5):1329–31. doi: 10.1016/j.transproceed.2007.02.061
10. Malek M, Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Inj Prev (2015) 4(2):20–7. doi: 10.12861/jrip.2015.06
11. Uehara M, Solhjou Z, Banouni N, Kasinath V, Xiaqun Y, Dai L, et al. Ischemia augments alloimmune injury through IL-6-driven CD4(+) alloreactivity. Sci Rep (2018) 8(1):2461. doi: 10.1038/s41598-018-20858-4
12. Sanders JM, Jeyamogan S, Mathew JM, Leventhal JR. Foxp3+ regulatory T cell therapy for tolerance in autoimmunity and solid organ transplantation. Front Immunol (2022) 13:1055466. doi: 10.3389/fimmu.2022.1055466
13. Goswami TK, Singh M, Dhawan M, Mitra S, Emran TB, Rabaan AA, et al. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders - Advances and challenges. Hum Vaccin Immunother (2022) 18(1):2035117. doi: 10.1080/21645515.2022.2035117
14. Bolivar-Wagers S, Larson JH, Jin S, Blazar BR. Cytolytic CD4(+) and CD8(+) regulatory T-cells and implications for developing immunotherapies to combat graft-versus-host disease. Front Immunol (2022) 13:864748. doi: 10.3389/fimmu.2022.864748
15. Nieto-Rios JF, Ochoa-Garcia CL, Serna-Campuzano A, Benavides-Hermosa B, Calderon-Puentes LL, Aristizabal-Alzate A, et al. Time of cold ischemia and delayed graft function in a cohort of renal transplant patients in a reference center. Indian J Nephrol (2019) 29(1):8–14. doi: 10.4103/ijn.IJN_162_18
16. Kaminska D, Koscielska-Kasprzak K, Chudoba P, Halon A, Mazanowska O, Gomolkiewicz A, et al. The influence of warm ischemia elimination on kidney injury during transplantation - clinical and molecular study. Sci Rep (2016) 6:36118. doi: 10.1038/srep36118
17. Ponticelli CE. The impact of cold ischemia time on renal transplant outcome. Kidney Int (2015) 87(2):272–5. doi: 10.1038/ki.2014.359
18. Guibert EE, Petrenko AY, Balaban CL, Somov AY, Rodriguez JV, Fuller BJ. Organ preservation: current concepts and new strategies for the next decade. Transfus Med Hemother (2011) 38(2):125–42. doi: 10.1159/000327033
19. Zhang H, Yan Q, Wang X, Chen X, Chen Y, Du J, et al. The role of mitochondria in liver ischemia-reperfusion injury: from aspects of mitochondrial oxidative stress, mitochondrial fission, mitochondrial membrane permeable transport pore formation, mitophagy, and mitochondria-related protective measures. Oxid Med Cell Longev (2021) 2021:6670579. doi: 10.1155/2021/6670579
20. Walkon LL, Strubbe-Rivera JO, Bazil JN. Calcium overload and mitochondrial metabolism. Biomolecules (2022) 12(12):1891. doi: 10.3390/biom12121891
21. Naito H, Nojima T, Fujisaki N, Tsukahara K, Yamamoto H, Yamada T, et al. Therapeutic strategies for ischemia reperfusion injury in emergency medicine. Acute Med Surg (2020) 7(1):e501. doi: 10.1002/ams2.501
22. Kezic A, Stajic N, Thaiss F. Innate immune response in kidney ischemia/reperfusion injury: potential target for therapy. J Immunol Res (2017) 2017:6305439. doi: 10.1155/2017/6305439
23. Adzigbli L, Sokolov EP, Wimmers K, Sokolova IM, Ponsuksili S. Effects of hypoxia and reoxygenation on mitochondrial functions and transcriptional profiles of isolated brain and muscle porcine cells. Sci Rep (2022) 12(1):19881. doi: 10.1038/s41598-022-24386-0
24. Sun K, Kiss E, Bedke J, Stojanovic T, Li Y, Gwinner W, et al. Role of xanthine oxidoreductase in experimental acute renal-allograft rejection. Transplantation (2004) 77(11):1683–92. doi: 10.1097/01.TP.0000131169.29553.B1
25. Kunduzova OR, Bianchi P, Pizzinat N, Escourrou G, Seguelas MH, Parini A, et al. Regulation of JNK/ERK activation, cell apoptosis, and tissue regeneration by monoamine oxidases after renal ischemia-reperfusion. FASEB J (2002) 16(9):1129–31. doi: 10.1096/fj.01-1008fje
26. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol (2019) 25:101084. doi: 10.1016/j.redox.2018.101084
27. Chimenti MS, Sunzini F, Fiorucci L, Botti E, Fonti GL, Conigliaro P, et al. Potential role of cytochrome c and tryptase in psoriasis and psoriatic arthritis pathogenesis: focus on resistance to apoptosis and oxidative stress. Front Immunol (2018) 9:2363. doi: 10.3389/fimmu.2018.02363
28. Thurman JM. Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol (2007) 123(1):7–13. doi: 10.1016/j.clim.2006.09.008
29. Qiu L, Zhang ZJ. Therapeutic strategies of kidney transplant ischemia reperfusion injury: insight from mouse models. BioMed J Sci Tech Res (2019) 14(5). doi: 10.26717/BJSTR.2019.14.002617
30. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
31. Qiu L, Zheng X, Jaishankar D, Green R, Fang D, Nadig S, et al. Beyond UPR: cell-specific roles of ER stress sensor IRE1alpha in kidney ischemic injury and transplant rejection. Kidney Int (2023) 104(3):463–469. doi: 10.1016/j.kint.2023.06.016
32. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi: 10.1038/nrm3270
33. Lee SR, Lo EH. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. J Cereb Blood Flow Metab (2004) 24(7):720–7. doi: 10.1097/01.WCB.0000122747.72175.47
34. Chaturvedi S, Robinson LA. Slit2-Robo signaling in inflammation and kidney injury. Pediatr Nephrol (2015) 30(4):561–6. doi: 10.1007/s00467-014-2825-4
35. Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A (1999) 96(24):13978–82. doi: 10.1073/pnas.96.24.13978
36. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol (2005) 6(12):1182–90. doi: 10.1038/ni1275
37. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest (2011) 121(11):4210–21. doi: 10.1172/JCI45161
38. Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA. Neutrophils–a key component of ischemia-reperfusion injury. Shock (2013) 40(6):463–70. doi: 10.1097/SHK.0000000000000044
39. Rabb H, Daniels F, O'Donnell M, Haq M, Saba SR, Keane W, et al. Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol (2000) 279(3):F525–31. doi: 10.1152/ajprenal.2000.279.3.F525
40. Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE Jr., et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int (2008) 74(12):1526–37. doi: 10.1038/ki.2008.500
41. Cisbani G, Le Behot A, Plante MM, Prefontaine P, Lecordier M, Rivest S. Role of the chemokine receptors CCR2 and CX3CR1 in an experimental model of thrombotic stroke. Brain Behav Immun (2018) 70:280–92. doi: 10.1016/j.bbi.2018.03.008
42. Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiol (Bethesda) (2015) 30(3):183–94. doi: 10.1152/physiol.00046.2014
43. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ Cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res (2019) 124(2):263–78. doi: 10.1161/CIRCRESAHA.118.314028
44. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol (2017) 37(1):35–42. doi: 10.1161/ATVBAHA.116.308198
45. Carnel N, Lancia HH, Guinier C, Benichou G. Pathways of antigen recognition by T cells in allograft rejection. Transplantation (2023) 107(4):827–37. doi: 10.1097/TP.0000000000004420
46. Peng Q, Nowocin A, Ratnasothy K, Smith RA, Smyth LA, Lechler RI, et al. Inhibition of thrombin on endothelium enhances recruitment of regulatory T cells during IRI and when combined with adoptive Treg transfer, significantly protects against acute tissue injury and prolongs allograft survival. Front Immunol (2022) 13:980462. doi: 10.3389/fimmu.2022.980462
47. Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJ, Joosten I. Clinical grade Treg: GMP isolation, improvement of purity by CD127 Depletion, Treg expansion, and Treg cryopreservation. PloS One (2008) 3(9):e3161. doi: 10.1371/journal.pone.0003161
48. Boonpiyathad T, Sozener ZC, Akdis M, Akdis CA. The role of Treg cell subsets in allergic disease. Asian Pac J Allergy Immunol (2020) 38(3):139–49. doi: 10.12932/AP-030220-0754
49. Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med (1996) 184(1):19–29. doi: 10.1084/jem.184.1.19
50. Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389(6652):737–42. doi: 10.1038/39614
51. Jun C, Ke W, Qingshu L, Ping L, Jun D, Jie L, et al. Protective effect of CD4(+)CD25(high)CD127(low) regulatory T cells in renal ischemia-reperfusion injury. Cell Immunol (2014) 289(1-2):106–11. doi: 10.1016/j.cellimm.2014.04.002
52. Jun C, Qingshu L, Ke W, Ping L, Jun D, Jie L, et al. Protective effect of CXCR3(+)CD4(+)CD25(+)Foxp3(+) regulatory T cells in renal ischemia-reperfusion injury. Mediators Inflamm (2015) 2015:360973. doi: 10.1155/2015/360973
53. Lu L, Li G, Rao J, Pu L, Yu Y, Wang X, et al. In vitro induced CD4(+)CD25(+)Foxp3(+) Tregs attenuate hepatic ischemia-reperfusion injury. Int Immunopharmacol (2009) 9(5):549–52. doi: 10.1016/j.intimp.2009.01.020
54. Lin JY, Tsai FC, Wallace CG, Huang WC, Wei FC, Liao SK. Combined treatment with regulatory T cells and vascularized bone marrow transplantation creates mixed chimerism and induces donor-specific tolerance to vascularized composite allografts without cytoreductive conditioning. Chimerism (2013) 4(1):20–2. doi: 10.4161/chim.23349
55. Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol (2009) 20(8):1744–53. doi: 10.1681/ASN.2008111160
56. Barry WH. Mechanisms of myocardial cell injury during ischemia and reperfusion. J Card Surg (1987) 2(3):375–83. doi: 10.1111/j.1540-8191.1987.tb00196.x
57. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113(15):3542–5. doi: 10.1182/blood-2008-09-181040
58. Suthen S, Lim CJ, Nguyen PHD, Dutertre CA, Lai HLH, Wasser M, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology (2022) 76(5):1329–44. doi: 10.1002/hep.32419
59. Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med (2006) 203(3):505–11. doi: 10.1084/jem.20050783
60. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol (2019) 20(2):218–31. doi: 10.1038/s41590-018-0280-2
61. Miyake K, Karasuyama H. The role of trogocytosis in the modulation of immune cell functions. Cells (2021) 10(5):1255. doi: 10.3390/cells10051255
62. Bailly C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci (2020) 246:117403. doi: 10.1016/j.lfs.2020.117403
63. Niu M, Liu Y, Yi M, Jiao D, Wu K. Biological characteristics and clinical significance of soluble PD-1/PD-L1 and exosomal PD-L1 in cancer. Front Immunol (2022) 13:827921. doi: 10.3389/fimmu.2022.827921
64. Bouhamida E, Morciano G, Perrone M, Kahsay AE, Della Sala M, Wieckowski MR, et al. The interplay of hypoxia signaling on mitochondrial dysfunction and inflammation in cardiovascular diseases and cancer: from molecular mechanisms to therapeutic approaches. Biol (Basel) (2022) 11(2):300. doi: 10.3390/biology11020300
65. Kempkes RWM, Joosten I, Koenen H, He X. Metabolic pathways involved in regulatory T cell functionality. Front Immunol (2019) 10:2839. doi: 10.3389/fimmu.2019.02839
66. Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood (2011) 117(3):857–61. doi: 10.1182/blood-2010-09-307041
67. Kesarwani P, Murali AK, Al-Khami AA, Mehrotra S. Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal (2013) 18(12):1497–534. doi: 10.1089/ars.2011.4073
68. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically expanded human regulatory T-cells are super-suppressive due to HIF1A induced expression of CD73. Commun Biol (2021) 4(1):1186. doi: 10.1038/s42003-021-02721-x
69. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
70. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta release. PloS One (2016) 11(8):e0161507. doi: 10.1371/journal.pone.0161507
71. O'Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest (2004) 114(10):1372–8. doi: 10.1172/JCI23215
72. Dial CF, Tune MK, Doerschuk CM, Mock JR. Foxp3(+) regulatory T cell expression of keratinocyte growth factor enhances lung epithelial proliferation. Am J Respir Cell Mol Biol (2017) 57(2):162–73. doi: 10.1165/rcmb.2017-0019OC
73. Hui SP, Sheng DZ, Sugimoto K, Gonzalez-Rajal A, Nakagawa S, Hesselson D, et al. Zebrafish regulatory T cells mediate organ-specific regenerative programs. Dev Cell (2017) 43(6):659–72 e5. doi: 10.1016/j.devcel.2017.11.010
74. Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension (2012) 59(2):324–30. doi: 10.1161/HYPERTENSIONAHA.111.181123
75. Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, et al. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front Cell Neurosci (2015) 9:361. doi: 10.3389/fncel.2015.00361
76. Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PloS One (2015) 10(6):e0128094. doi: 10.1371/journal.pone.0128094
77. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell (2017) 169(6):1119–29 e11. doi: 10.1016/j.cell.2017.05.002
78. Deliyanti D, Talia DM, Zhu T, Maxwell MJ, Agrotis A, Jerome JR, et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat Commun (2017) 8(1):748. doi: 10.1038/s41467-017-00751-w
79. do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight (2020) 5(3). doi: 10.1172/jci.insight.130651
80. Hung TW, Liou JH, Yeh KT, Tsai JP, Wu SW, Tai HC, et al. Renal expression of hypoxia inducible factor-1alpha in patients with chronic kidney disease: a clinicopathologic study from nephrectomized kidneys. Indian J Med Res (2013) 137(1):102–10. doi: 10.1111/tri.12475
81. Yan Z, Garg SK, Banerjee R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J Biol Chem (2010) 285(53):41525–32. doi: 10.1074/jbc.M110.189944
82. Beers DR, Thonhoff JR, Faridar A, Thome AD, Zhao W, Wen S, et al. Tregs attenuate peripheral oxidative stress and acute phase proteins in ALS. Ann Neurol (2022) 92(2):195–200. doi: 10.1002/ana.26375
83. Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, et al. Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol (2016) 196(5):2010–4. doi: 10.4049/jimmunol.1502139
84. Luznik Z, Anchouche S, Dana R, Yin J. Regulatory T cells in angiogenesis. J Immunol (2020) 205(10):2557–65. doi: 10.4049/jimmunol.2000574
85. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol (2016) 17(10):611–25. doi: 10.1038/nrm.2016.87
86. Li X, Li D, Shi Q, Huang X, Ju X. Umbilical cord blood−derived Helios−positive regulatory T cells promote angiogenesis in acute lymphoblastic leukemia in mice via CCL22 and the VEGFA−VEGFR2 pathway. Mol Med Rep (2019) 19(5):4195–204. doi: 10.3892/mmr.2019.10074
87. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
88. Huang MT, Dai YS, Chou YB, Juan YH, Wang CC, Chiang BL. Regulatory T cells negatively regulate neovasculature of airway remodeling via DLL4-Notch signaling. J Immunol (2009) 183(7):4745–54. doi: 10.4049/jimmunol.0804371
89. Zouggari Y, Ait-Oufella H, Waeckel L, Vilar J, Loinard C, Cochain C, et al. Regulatory T cells modulate postischemic neovascularization. Circulation (2009) 120(14):1415–25. doi: 10.1161/CIRCULATIONAHA.109.875583
90. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation (2019) 139(2):206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
91. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil–CD4+CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology (2013) 218(4):455–64. doi: 10.1016/j.imbio.2012.05.029
92. D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest (2009) 119(10):2898–913. doi: 10.1172/JCI36498
93. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol (2012) 107(1):232. doi: 10.1007/s00395-011-0232-6
94. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
95. Ng TH, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol (2013) 4:129. doi: 10.3389/fimmu.2013.00129
96. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4(1):29–37. doi: 10.1093/jmcb/mjr033
97. Yang WC, Hwang YS, Chen YY, Liu CL, Shen CN, Hong WH, et al. Interleukin-4 supports the suppressive immune responses elicited by regulatory T cells. Front Immunol (2017) 8:1508. doi: 10.3389/fimmu.2017.01508
98. Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol (2004) 34(2):366–76. doi: 10.1002/eji.200324455
99. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol (2012) 32(1):23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
100. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178(1):280–90. doi: 10.4049/jimmunol.178.1.280
101. Yoshimura A, Muto G. TGF-beta function in immune suppression. Curr Top Microbiol Immunol (2011) 350:127–47. doi: 10.1007/82_2010_87
102. Beebe AM, Cua DJ, de Waal Malefyt R. The role of interleukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev (2002) 13(4-5):403–12. doi: 10.1016/S1359-6101(02)00025-4
103. Hiebert PR, Granville DJ. Granzyme B in injury, inflammation, and repair. Trends Mol Med (2012) 18(12):732–41. doi: 10.1016/j.molmed.2012.09.009
104. Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, et al. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest (2018) 128(3):1200–1. doi: 10.1172/JCI120358
105. Xian W, Wu J, Li Q, Du X, Wang N, Chen D, et al. CXCR3 alleviates renal ischemia−reperfusion injury via increase of Tregs. Mol Med Rep (2021) 24(1):1–9. doi: 10.3892/mmr.2021.12180
106. Hakimi H, Zainodini N, Khorramdelazad H, Kazemi Arababadi M, Hassanshahi G. Seminal levels of pro-inflammatory (CXCL1, CXCL9, CXCL10) and homeostatic (CXCL12) chemokines in men with asymptomatic chlamydia trachomatis infection. Jundishapur J Microbiol (2014) 7(12):e11152. doi: 10.5812/jjm.11152
107. Xu J, Li X, Yuan Q, Wang C, Xu L, Wei X, et al. The semaphorin 4A-neuropilin 1 axis alleviates kidney ischemia reperfusion injury by promoting the stability and function of regulatory T cells. Kidney Int (2021) 100(6):1268–81. doi: 10.1016/j.kint.2021.08.023
108. Hu X, Li P, Chen J. Pro: Regulatory T cells are protective in ischemic stroke. Stroke (2013) 44(8):e85–6. doi: 10.1161/STROKEAHA.113.001267
109. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
110. Sanchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant (2020) 20(4):1125–36. doi: 10.1111/ajt.15700
111. Feng M, Wang Q, Zhang F, Lu L. Ex vivo induced regulatory T cells regulate inflammatory response of Kupffer cells by TGF-beta and attenuate liver ischemia reperfusion injury. Int Immunopharmacol (2012) 12(1):189–96. doi: 10.1016/j.intimp.2011.11.010
112. Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int (2009) 76(7):717–29. doi: 10.1038/ki.2009.259
113. Jin X, Hu M, Gong L, Li H, Wang Y, Ji M, et al. Adoptive transfer of xenoantigen−stimulated T cell receptor Vbeta−restricted human regulatory T cells prevents porcine islet xenograft rejection in humanized mice. Mol Med Rep (2018) 18(5):4457–67. doi: 10.3892/mmr.2018.9471
114. Mathew JM, Voss JH, McEwen ST, Konieczna I, Chakraborty A, Huang X, et al. Generation and characterization of alloantigen-specific regulatory T cells for clinical transplant tolerance. Sci Rep (2018) 8(1):1136. doi: 10.1038/s41598-018-19621-6
115. Toomer KH, Lui JB, Altman NH, Ban Y, Chen X, Malek TR. Essential and non-overlapping IL-2Ralpha-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat Commun (2019) 10(1):1037. doi: 10.1038/s41467-019-08960-1
116. Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci (2020) 21(19):7015. doi: 10.3390/ijms21197015
117. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis (2020) 79(1):141–9. doi: 10.1136/annrheumdis-2019-215396
118. Bhogal RH, Mirza DF, Afford SC, Mergental H. Biomarkers of liver injury during transplantation in an era of machine perfusion. Int J Mol Sci (2020) 21(5):1578. doi: 10.3390/ijms21051578
119. Parlakpinar H, Gunata M. Transplantation and immunosuppression: a review of novel transplant-related immunosuppressant drugs. Immunopharmacol Immunotoxicol (2021) 43(6):651–65. doi: 10.1080/08923973.2021.1966033
120. Barjon C, Dahlqvist G, Ghazal K, Saliba F, Durand F, Duvoux C, et al. Influence of everolimus-based treatment on circulating regulatory T cells after liver transplantation: Comparative study with tacrolimus-based therapy. Clin Res Hepatol Gastroenterol (2021) 45(5):101559. doi: 10.1016/j.clinre.2020.10.004
121. Ma A, Qi S, Wang Z, Massicotte E, Dupuis M, Daloze P, et al. Combined therapy of CD4(+)CD25(+) regulatory T cells with low-dose sirolimus, but not calcineurin inhibitors, preserves suppressive function of regulatory T cells and prolongs allograft survival in mice. Int Immunopharmacol (2009) 9(5):553–63. doi: 10.1016/j.intimp.2009.01.033
122. Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discovery Today (2007) 12(3-4):112–24. doi: 10.1016/j.drudis.2006.12.008
123. Lai LW, Yong KC, Lien YH. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int (2012) 81(10):983–92. doi: 10.1038/ki.2011.412
124. Fang J, Hu F, Ke D, Yan Y, Liao Z, Yuan X, et al. N,N-dimethylsphingosine attenuates myocardial ischemia-reperfusion injury by recruiting regulatory T cells through PI3K/Akt pathway in mice. Basic Res Cardiol (2016) 111(3):32. doi: 10.1007/s00395-016-0548-3
125. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13(11):1299–307. doi: 10.1038/nm1652
126. Lee JH, Kim HS, Jang SW, Lee GR. Histone deacetylase 6 plays an important role in TGF-beta-induced murine Treg cell differentiation by regulating cell proliferation. Sci Rep (2022) 12(1):22550. doi: 10.1038/s41598-022-27230-7
127. Mausner-Fainberg K, Luboshits G, Mor A, Maysel-Auslender S, Rubinstein A, Keren G, et al. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis (2008) 197(2):829–39. doi: 10.1016/j.atherosclerosis.2007.07.031
128. Kim MG, Lee SY, Ko YS, Lee HY, Jo SK, Cho WY, et al. CD4+ CD25+ regulatory T cells partially mediate the beneficial effects of FTY720, a sphingosine-1-phosphate analogue, during ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant (2011) 26(1):111–24. doi: 10.1093/ndt/gfq480
129. Schmuck EG, Koch JM, Centanni JM, Hacker TA, Braun RK, Eldridge M, et al. Biodistribution and clearance of human mesenchymal stem cells by quantitative three-dimensional cryo-imaging after intravenous infusion in a rat lung injury model. Stem Cells Transl Med (2016) 5(12):1668–75. doi: 10.5966/sctm.2015-0379
130. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol (2012) 3:297. doi: 10.3389/fimmu.2012.00297
131. Sanchez-Diaz M, Quinones-Vico MI, Sanabria de la Torre R, Montero-Vilchez T, Sierra-Sanchez A, Molina-Leyva A, et al. Biodistribution of mesenchymal stromal cells after administration in animal models and humans: A systematic review. J Clin Med (2021) 10(13):2925. doi: 10.3390/jcm10132925
132. Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. Stem cell transplantation: the lung barrier. Transplant Proc (2007) 39(2):573–6. doi: 10.1016/j.transproceed.2006.12.019
133. Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev (2009) 18(5):683–92. doi: 10.1089/scd.2008.0253
134. Lyu MA, Huang M, Zeng K, Li L, Khoury JD, Nishimoto M, et al. Allogeneic cord blood regulatory T cells can resolve lung inflammation. Cytotherapy (2023) 25(3):245–53. doi: 10.1016/j.jcyt.2022.10.009
Keywords: ischemia, reperfusion, regulatory T cells, anti-inflammation, oxidative stress, allografts, immune tolerance
Citation: Jeyamogan S, Leventhal JR, Mathew JM and Zhang ZJ (2023) CD4+CD25+FOXP3+ regulatory T cells: a potential “armor” to shield “transplanted allografts” in the war against ischemia reperfusion injury. Front. Immunol. 14:1270300. doi: 10.3389/fimmu.2023.1270300
Received: 31 July 2023; Accepted: 25 September 2023;
Published: 06 October 2023.
Edited by:
Marco Romano, King’s College London, United KingdomReviewed by:
Austin Schenk, The Ohio State University, United StatesMo Atif, Sorbonne Universités, France
Copyright © 2023 Jeyamogan, Leventhal, Mathew and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng Jenny Zhang, emp6aGFuZ0Bub3J0aHdlc3Rlcm4uZWR1; James M. Mathew, amFtZXMtbWF0aGV3QG5vcnRod2VzdGVybi5lZHU=
†ORCID: Shareni Jeyamogan, orcid.org/0000-0003-1308-7689
Joseph Leventhal, orcid.org/0000-0003-1604-9207
James M. Mathew, orcid.org/0000-0002-5667-750X
Zheng Jenny Zhang, orcid.org/0000-0001-6469-5778