 Afsar U. Ahmed
Afsar U. Ahmed Saleh Almasabi
Saleh Almasabi Ron Firestein
Ron Firestein Bryan R.G. Williams
Bryan R.G. Williams- Centre for Cancer Research, Hudson Institute of Medical Research, Department of Molecular and Translational Science, Monash University, Clayton, VIC, Australia
Colorectal cancer (CRC) is one of the most common forms of cancer worldwide and treatment options for advanced CRC, which has a low 5-year survival rate, remain limited. Integrin-linked kinase (ILK), a multifunctional, scaffolding, pseudo-kinase regulating many integrin-mediated cellular processes, is highly expressed in many cancers. However, the role of ILK in cancer progression is yet to be fully understood. We have previously uncovered a pro-inflammatory role for myeloid-specific ILK in dextran sodium sulfate (DSS)-induced colitis. To establish a correlation between chronic intestinal inflammation and colorectal cancer (CRC), we investigated the role of myeloid-ILK in mouse models of CRC. When myeloid-ILK deficient mice along with the WT control mice were subjected to colitis-associated and APCmin/+-driven CRC, tumour burden was reduced by myeloid-ILK deficiency in both models. The tumour-promoting phenotype of macrophages, M2 polarization, in vitro was impaired by the ILK deficiency and the number of M2-specific marker CD206-expressing tumour-associated macrophages (TAMs) in vivo were significantly diminished in myeloid-ILK deficient mice. Myeloid-ILK deficient mice showed enhanced tumour infiltration of CD8+ T cells and reduced tumour infiltration of FOXP3+ T cells in colitis-associated and APCmin/+-driven CRC, respectively, with an overall elevated CD8+/FOXP3+ ratio suggesting an anti-tumour immune phenotypes. In patient CRC tissue microarrays we observed elevated ILK+ myeloid (ILK+ CD11b+) cells in tumour sections compared to adjacent normal tissues, suggesting a conserved role for myeloid-ILK in CRC development in both human and animal models. This study identifies myeloid-specific ILK expression as novel driver of CRC, which could be targeted as a potential therapeutic option for advanced disease.
Introduction
Colorectal cancer (CRC) is among the most prevalent forms of cancer worldwide and the second-leading cause of cancer-related deaths in developed countries (1–4). Although colonoscopy screening is an effective way to detect and prevent CRC by surgical excision of precancerous adenomas (5), the 5-year survival rate is extremely low for patients with advanced disease accounting 70% of all detected cases (6). Despite being one of the most common cancers in Western populations, there remain few effective therapies for treating advanced CRC.
CRC originates from intestinal epithelial cells (IECs) due to the loss of tumour suppressor genes such as the adenomatous polyposis coli (Apc) gene (7). While hereditary CRC due to genetic alterations accounts for about 5% of cases (8), most risk factors are related to Western lifestyles causing >75% of CRC in patients with few or no genetic risk factors (8). Several studies implicate chronic inflammatory stimuli as a risk factor for developing CRC, particularly ulcerative colitis (UC), a form of inflammatory bowel disease (IBD), leading to so-called colitis-associated cancer (CAC) (9, 10). There are several animal models that have been developed to study colon cancer (11–14). Among them, CAC provides a ‘proof of concept’ model to better understand how chronic inflammation promote tumour initiation and progression (15) whereas the APCmin/+ mouse carrying a heterozygous germ-line mutation at codon 850 of the Apc gene is one of the most commonly used models to study spontaneous (hereditary) CRC (16).
Tumour-associated immune cells comprise an essential component of the tumour microenvironment (TME) that develops from chronic inflammatory conditions that emerges from germ-line mutations (17). Both innate and adaptive immune cells participate in the immune landscape of the TME. These cells strongly influence tumorigenesis which can be either promoted or suppressed depending on the dominating cell type and polarization profile of the infiltrating immune cells (18). A large number of studies have demonstrated a key role for myeloid cells in CRC as well as other cancer types (18). Depending on their functional status, tumour-infiltrating macrophages can influence both positive and negative outcomes of CRC. Macrophages have been grouped into classically activated M1 and alternatively activated M2 macrophages (19). In the context of TME, M1 macrophages are considered to be tumoricidal whereas M2 macrophages show pro-tumoral phenotype (20). Even though their function seems to vary depending on the tissue, location and TME, tumour-associated macrophages (TAMs) support rather than hinder tumorigenesis. The molecular determinants of the pro-tumoral phenotypes of tumour myeloid cells could lead to novel therapeutic innovations in CRC. However, the detailed molecular mechanisms underlying tumour-promoting macrophage polarization are still poorly understood.
Integrin-linked kinase (ILK) is a multifunctional serine/threonine protein pseudokinase expressed ubiquitously in mammalian tissues and is involved in focal adhesion formation and function (21). ILK regulates many integrin-mediated cellular processes, including growth, proliferation, migration, invasion, and tissue homeostasis (22). Consequently, global genetic deletion of ILK is embryonic lethal due to cytoskeletal and cell attachment defects. Overexpression of ILK is also implicated in pathophysiological conditions, including cancer. ILK overexpression is a feature in the progression of many cancers such as colon, gastric, prostate, breast, melanoma as well as leukaemia (23–26). Moreover, intestinal epithelial cell (IEC) -specific ILK deletion in mice reduced both colitis and CAC, suggesting an integral role for IEC-ILK in intestinal inflammation and CRC (27, 28). Despite considerable evidence linking ILK to cancer, a mechanistic understanding of the role for ILK in cancer progression is yet to be fully realized. Based on our earlier study demonstrating ILK as a pro-inflammatory molecule (29), we investigated the role myeloid-specific ILK in a mouse model of colitis using myeloid-ILK deficient (M-ILK KO) mice., In this model the pathology of experimental colitis was significantly reduced by myeloid ILK deficiency with the pro-inflammatory signalling during intestinal inflammation dependent on myeloid-ILK (30). Given the correlation between chronic intestinal inflammation and CRC, in this study we investigated the effect of myeloid-ILK deficiency in mouse models of CAC and APCmin/+-driven CRC. We show that myeloid-ILK deficiency decreases both AOM/DSS-induced and APCmin/+-driven colon tumour burden. Our results indicate that ILK is required for macrophage M2 polarization in vitro and the expression of M2-specific marker CD206 in tumour-associated macrophages (TAMs) in vivo. Myeloid-ILK deficiency also enhances tumour-infiltration of CD8+ T cells and reduces FOXP3+ T cells in CAC and APCmin/+-driven CRC, respectively, with a significantly elevated CD8+/FOXP3+ ratio indicating a tumoricidal impact in both models. Examination of human CRC tissue microarrays (TMAs), indicate that ILK+ myeloid cells (ILK+ CD11b+) are significantly elevated within the tumour compared to the adjacent normal tissues, suggesting a role of myeloid-ILK in human CRC development consistent with the results in the mouse models. Our study reveals an integrated role for myeloid-specific ILK expression in colon tumorigenesis and highlights the therapeutic potential of targeting ILK in CRC.
Results
ILK expression in myeloid cells promotes colitis-associated and spontaneous colon tumorigenesis
Our earlier studies demonstrated that the myeloid-specific ILK is required for intestinal inflammation during DSS-induced colitis (30). Since chronic colitis is a significant risk factor for development of CRC, we investigated whether myeloid-ILK deficiency ameliorates the colitis-associated cancer (CAC) model, also known as the AOM/DSS model (31). Azoxymethane (AOM) is procarcinogen which is commonly used to induce adenoma growth in the distal colon of rodents and AOM/DSS induces a model of carcinogen-induced intestinal tumorigenesis that reliably induces non-invasive colonic tumours in WT mice.
To investigate the role of myeloid-ILK in colitis-associated tumorigenesis, both wild-type (WT) or M-ILK KO mice were treated with AOM and DSS (Figure 1A). Briefly, mice were given a single i.p. injection of the mutagen AOM, after which they received drinking water containing 1% DSS in three rounds of 5-day periods that were interspersed with periods in which they received normal water. According to the post-mortem examination following the 62-day long AOM/DSS model, the number of visible intestinal polyps and their sizes in M-ILK KO -deficient mice was reduced significantly relative to control mice, suggesting that myeloid-specific ILK loss suppresses colitis-associated colon tumorigenesis (Figures 1B-E).
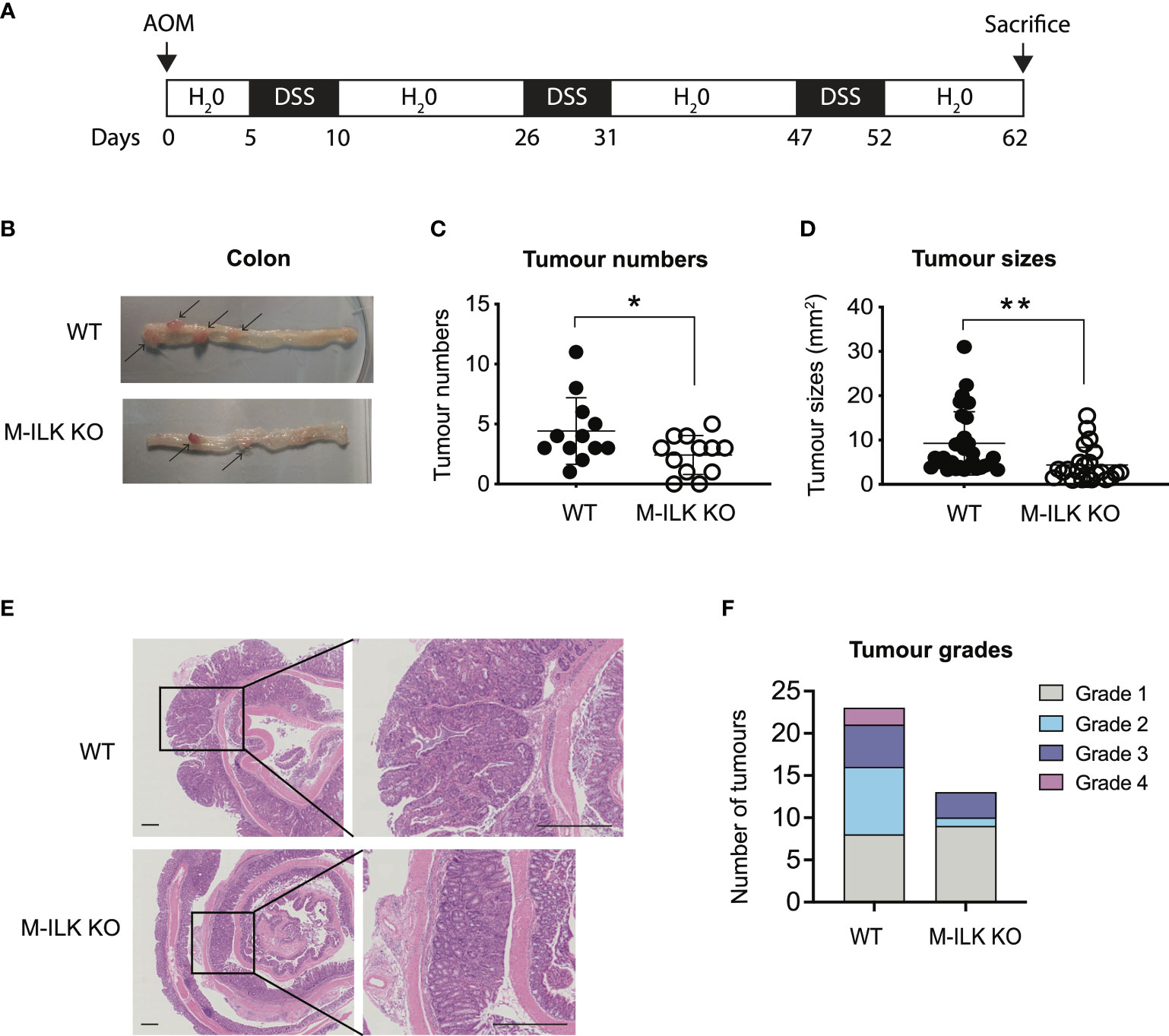
Figure 1 Loss of ILK in myeloid cells reduces colonic tumour formation in AOM/DSS model. (A) A schematic protocol of the AOM/DSS mouse model of CRC used in this study. (B) Representative images of the dissected colon in WT and myeloid-ILK deficient (M-ILK KO) mice following the AOM/DSS model. Arrows indicate polyps. Briefly, 12-week-old mice were subjected to AOM/DSS treatment which is based on a single i.p. injection of a carcinogen (AOM) (12.5 mg/kg) at day 0 followed by 3 cycles of inflammatory agent DSS (2%) treatment in the drinking for 5 days and a subsequent 10-days recovery period without DSS started from day 5 to day 62. All mice were sacrificed at day 62 for post-mortem examination. (C) Numbers of visible polyps in the colon intestine of mice with indicated genotypes are shown. Results are representative of at least three independent experiments and are shown as mean ± SD., n = 12 per group. (D) Colon tumour sizes of mice with the indicated genotypes are shown. Results are representative of at least three independent experiments and are shown as mean ± SD., n = 53 (WT tumours) and n = 29 (M-ILK KO tumours). (E) Representative of H&E-stained sections of colonic tissue with tumours from WT and M-ILK KO mice are shown (scale bars, 200 µm). (F) Blinded histological scoring of colon adenocarcinomas from WT and M-ILK KO mice, n = 23 (WT tumours) and n = 13 (M-ILK KO tumours). *, p < 0.05; **, p < 0.01, Student’s t test.
While tumours of all genotypes were predominantly low-grade adenocarcinomas, the proportion of tumours that progressed to high-grade adenocarcinomas was relatively higher in WT mice compared to their myeloid-ILK deficient counterparts (Figure 1F), suggesting a role for myeloid-ILK in tumour progression. Based on these observations, we investigated further whether myeloid-ILK plays a role in colitis- independent colon tumour development. Consequently, we used a genetic model of spontaneous adenomatous polyposis driven by APCmin/+. To test the effect of myeloid-ILK deficiency in APCmin/+ model, we generated myeloid-ILK deficient APCmin/+ mice (ILKfl/fl;LysMCre; APCmin/+, referred as APCmin/+ΔM-ILK hereafter). Analysis of 4-month-old APCmin/+ΔM-ILK along with the WT APCmin/+ control mice, showed both tumour incidence and size were significantly decreased in APCmin/+ΔM-ILK compared to the WT control (Figures 2A–D). Similar to the AOM/DSS model, the proportion of APCmin/+ -driven tumours that progressed to high-grade adenocarcinomas was reduced in myeloid-ILK deficient APCmin/+ mice compared to the WT control (Figure 2E). Collectively, these results demonstrate marked tumour suppression by myeloid-ILK deficiency in both carcinogen/inflammation and oncogene-driven models of colon tumorigenesis.
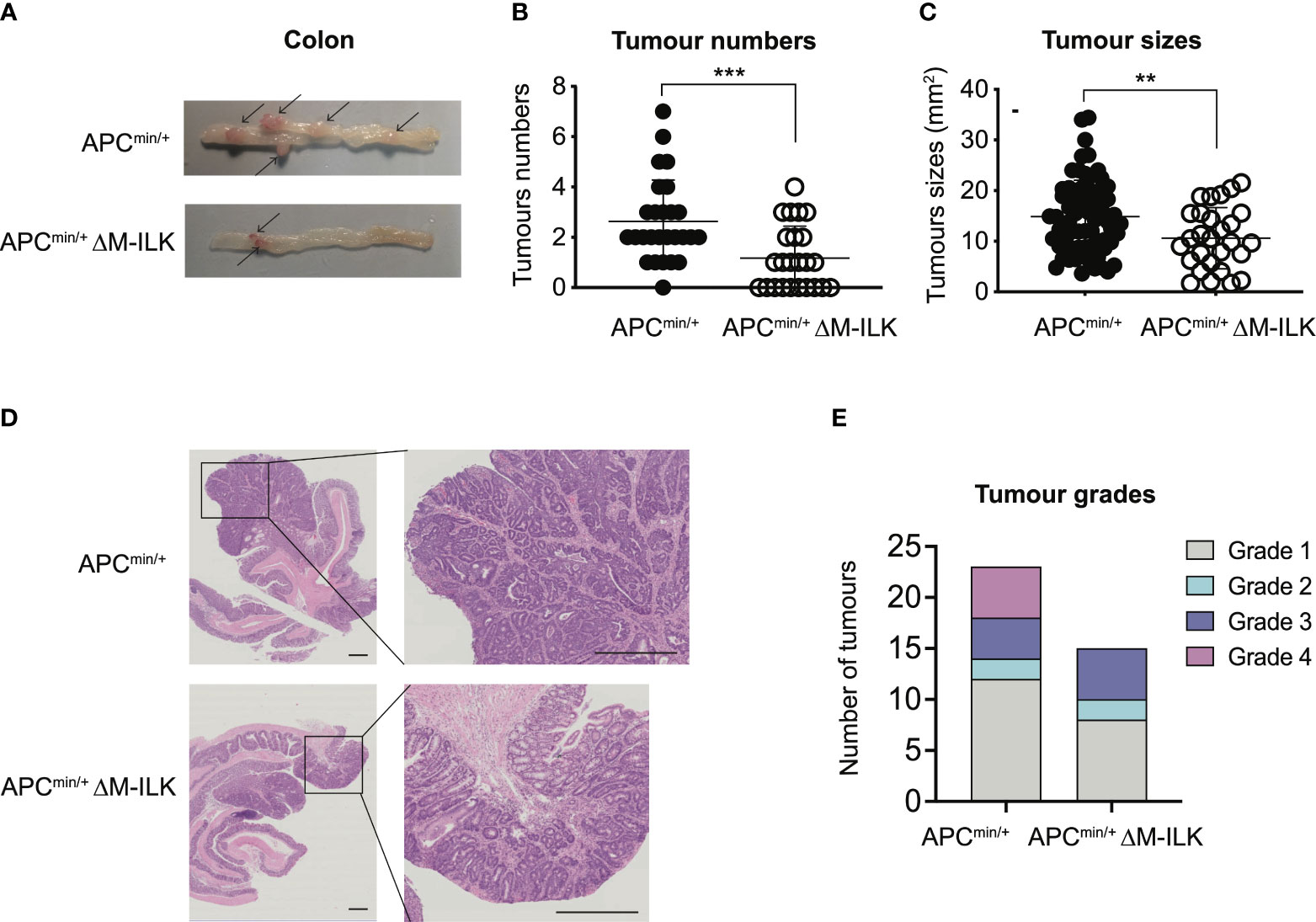
Figure 2 Myeloid-ILK deletion attenuates colonic tumorigenesis in APCmin/+ model. (A) Representative images of the dissected colon in APCmin/+ and APCmin/+ΔM-ILK mice sacrificed at 16-weeks of age. Arrows indicate polyps. (B) Numbers of visible polyps in the colon intestine of mice with indicated genotypes are shown. Results are representative of at least three independent experiments and are shown as mean ± SD, n = 27 (APCmin/+) and n = 23 (APCmin/+ΔM-ILK). (C) Colon tumour sizes of mice with the indicated genotypes are shown. Results are representative of at least three independent experiments and are shown as mean ± SD, n = 71 (APCmin/+) and n = 28 (APCmin/+ΔM-ILK). (D) Representative of H&E-stained sections of colonic tissue with tumours from WT and M-ILK KO mice are shown (scale bars, 200 µm). (E) Blinded histological scoring of colon adenocarcinomas from WT and M-ILK KO mice, n = 23 (APCmin/+) and n = 15 (APCmin/+ΔM-ILK). **, p < 0.01; ***, p < 0.001, Student’s t test.
Myeloid-ILK deficiency impedes tumour-driven body weight loss, poor survival and splenomegaly in APCmin/+ mice
The APCmin/+ mouse is not only an established model of CRC but also presents with cancer cachexia, a wasting syndrome due to body weight loss associated with the skeletal muscle wasting with or without the loss in adipose tissue (32–34). An advantage of this mouse model is the gradual progression of tumour development and muscle wasting that is more physiologically related to human disease. The genetic predisposition in APCmin/+ mice becomes apparent as early as 3 weeks of age and most of the adenomas start to appear in APCmin/+ mice at 5–8 weeks of age (35). In order to monitor the effect of myeloid-ILK deficiency on the development of genetic predisposition in APCmin/+ mice, body weight changes in both APCmin/+ and APCmin/+ΔM-ILK mice were measured from 5 to 16 weeks of age. As shown in Figure 3A, APCmin/+ΔM-ILK mice started to show an elevated body weight compared to APCmin/+ mice from the age of 9 weeks onwards. The body weight of APCmin/+ΔM-ILK mice was increased by approximately 10% from 11 to 16 weeks of age compared to the same aged-APCmin/+ mice.
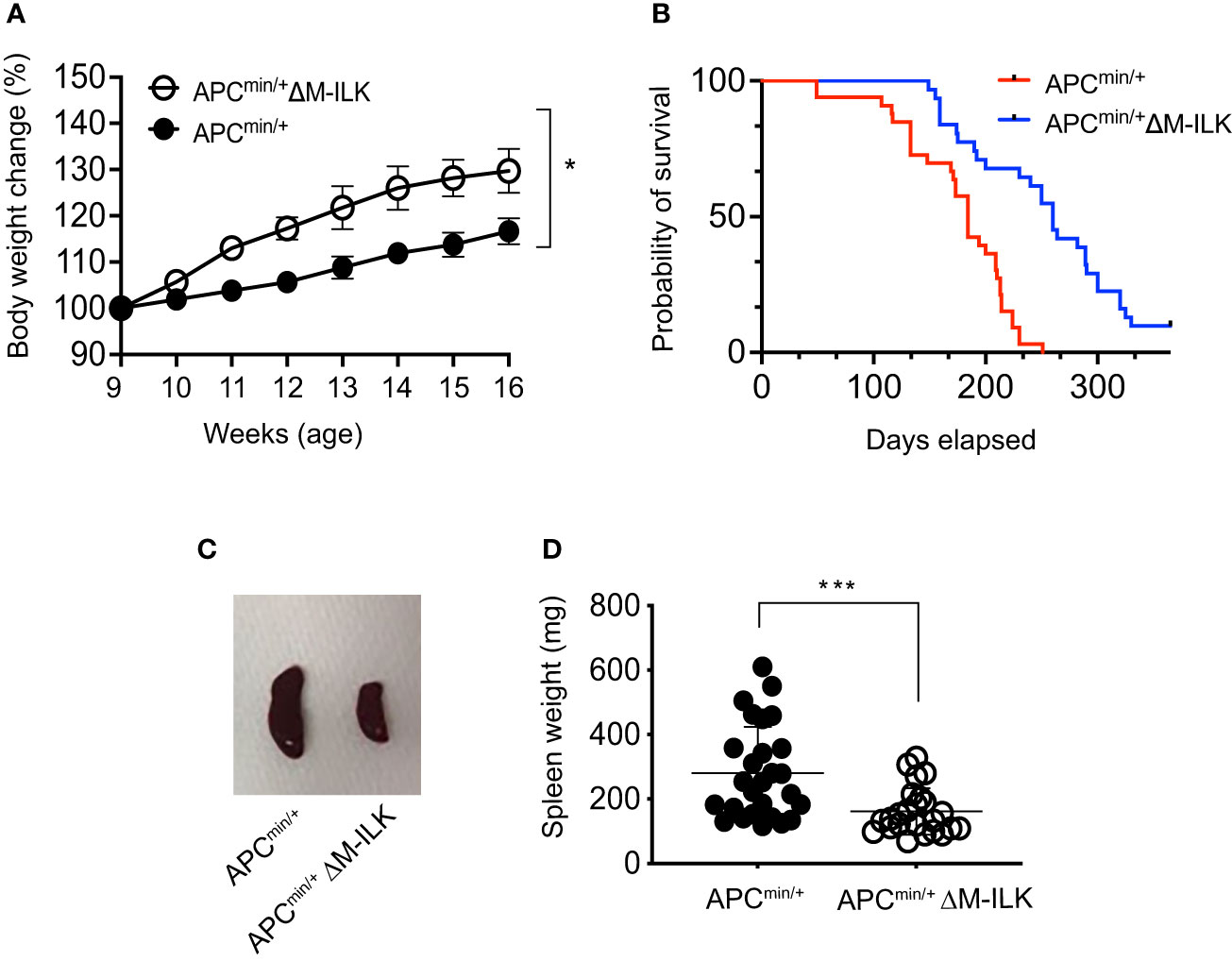
Figure 3 Myeloid-ILK deficiency reverses body weight loss, improves survival and reduces splenomegaly in Apcmin/+ mice. (A) Changes in body weight in both APCmin/+ and APCmin/+ΔM-ILK mice are expressed as percentage of body weight at 9 weeks of age. Body weights of randomly selected mice from each genotype were measured weekly from week 9 to week 17, n = 10 per group. Results are representative of at least three independent experiments and are shown as mean ± SD. (B) Both APCmin/+ and APCmin/+ΔM-ILK mice were followed for long-term survival. Survival probability was analyzed using the Kaplan-Meier estimate, and differences were evaluated using the log-rank test, n = 33 (APCmin/+) and n = 31 (APCmin/+ΔM-ILK). (C) Representative images of spleen from APCmin/+ and APCmin/+ΔM-ILK mice sacrificed at 16-weeks of age. (D) Spleen weights (mg) are shown for both APCmin/+ and APCmin/+ΔM-ILK mice sacrificed at 16-weeks of age, n = 27 (APCmin/+) and n = 24 (APCmin/+ΔM-ILK). Results are representative of at least three independent experiments and are shown as mean ± SD. *, p < 0.05; ***, p < 0.001, Student’s t test.
Since myeloid-ILK deficiency improves body weight loss in APCmin/+ mice (Figure 3A) and cancer-associated cachexia is associated with a poor outcomes with a reduced survival rate (33, 36), it was of interest to determine whether myeloid-ILK deficiency confers a protective effect and prolonged survival in APCmin/+ mice. As shown in Figure 3B, the mortality rate of APCmin/+ΔM-ILK mice was significantly reduced relative to APCmin/+ control mice. APCmin/+ΔM-ILK mice had a median survival of 253.7 days compared to 174.5 days for APCmin/+ mice. It was previously reported that the spleens were enlarged (known as splenomegaly) in tumour-laden APCmin/+ mice due to vigorous splenic haematopoiesis (37, 38). In fact, the splenomegaly process was positive correlated with the tumour development in the APCmin/+ mice (39). Similarly, we observed splenomegaly in APCmin/+ mice following post mortem examination at 16 weeks of age. The spleen size and weight between APCmin/+ and APCmin/+ΔM-ILK mice, (sizes shown as representative images in Figure 3C) were significantly reduced in APCmin/+ΔM-ILK mice compared to the ILK-sufficient APCmin/+ control mice (Figure 3D). Overall, these results demonstrate that myeloid-ILK pays critical roles in APCmin/+-driven physiological abnormalities including body weight loss, splenomegaly and poor survival.
ILK promotes M2 macrophage polarization
The impact of myeloid-ILK deficiency on colon tumour development suggests a macrophage-intrinsic role of ILK affecting tumour growth via the regulation of macrophage phenotypes. Macrophages exhibit a wide phenotypic diversity and exert multiple pathobiological effects on tumour growth. The ability of macrophages to adapt to their environment has led to the identification of two main polarized phenotypes – classically activated macrophages (M1) and alternatively activated macrophages (M2) (19). M1 macrophages are characterized by the expression of many pro-inflammatory cytokines and a high tumoricidal capacity, while alternatively activated M2 macrophages perform immunosuppressive functions and tumour promotion. The distinction between M1 and M2 cells has been verified using gene profiles, which support this phenotypic and functional change (40). The classical M1 activation is driven by the stimulation of TLR ligands (e.g. LPS) and IFN-γ (41, 42). Classically activated macrophages upregulate the expression of proinflammatory cytokines (e.g. TNF-α) and mediators such as iNOS and CCL2. Alternative/M2 polarization is induced by exposure to IL-4 and IL-13 cytokines that bind to a common receptor subunit, IL-4R (43). M2 macrophages up-regulate expression of the mannose receptor (CD206) as well as macrophage-specific genes including Arg1, Ym1 and Fizz1 (44). To examine whether ILK plays any role in macrophage polarization, both peritoneal and bone-marrow derived macrophages (BMDMs) from WT and M-ILK KO mice were isolated and polarized in vitro to the M1 phenotype by exposure to LPS and IFN-γ as well as the M2 phenotype by exposure to IL-4 and IL-13 cytokines. As per our observation, during M1 polarization, only TNF-α expression was affected by ILK deletion in both BMDMs and peritoneal macrophages which correlates with our earlier observation (29) but the expression levels of both iNOS and CCL2 remained unaffected, indicating that macrophage M1 polarization is only partially impacted by ILK deletion (Supplementary Figure 1). Western blot analysis of iNOS protein levels in both WT and ILK-KO BMDMs during M1 polarization further confirms that ILK is dispensable for iNOS expression (Supplementary Figure 2). Next, we examined whether the tumour-promoting M2 phenotypes are affected by deletion of ILK in macrophages. Accordingly, Analyses of M2-specific gene expression markers (Arg1, Ym1 and Fizz1) showed a consistent impairment in ILK KO cells compared to the WT cells (Figure 4), suggesting impeded M2 polarization in ILK KO cells. Reduced expression levels of Arg1 observed in ILK-KO cells also correlated with reduced levels of Arginase-1 protein in ILK-KO BMDMs during M2 polarization (Supplementary Figure 3). Our results also correspond with our earlier report indicating a correlation between ILK expression and markers of M2 macrophages (45). These observations establish that ILK is required for macrophage M2 polarization.
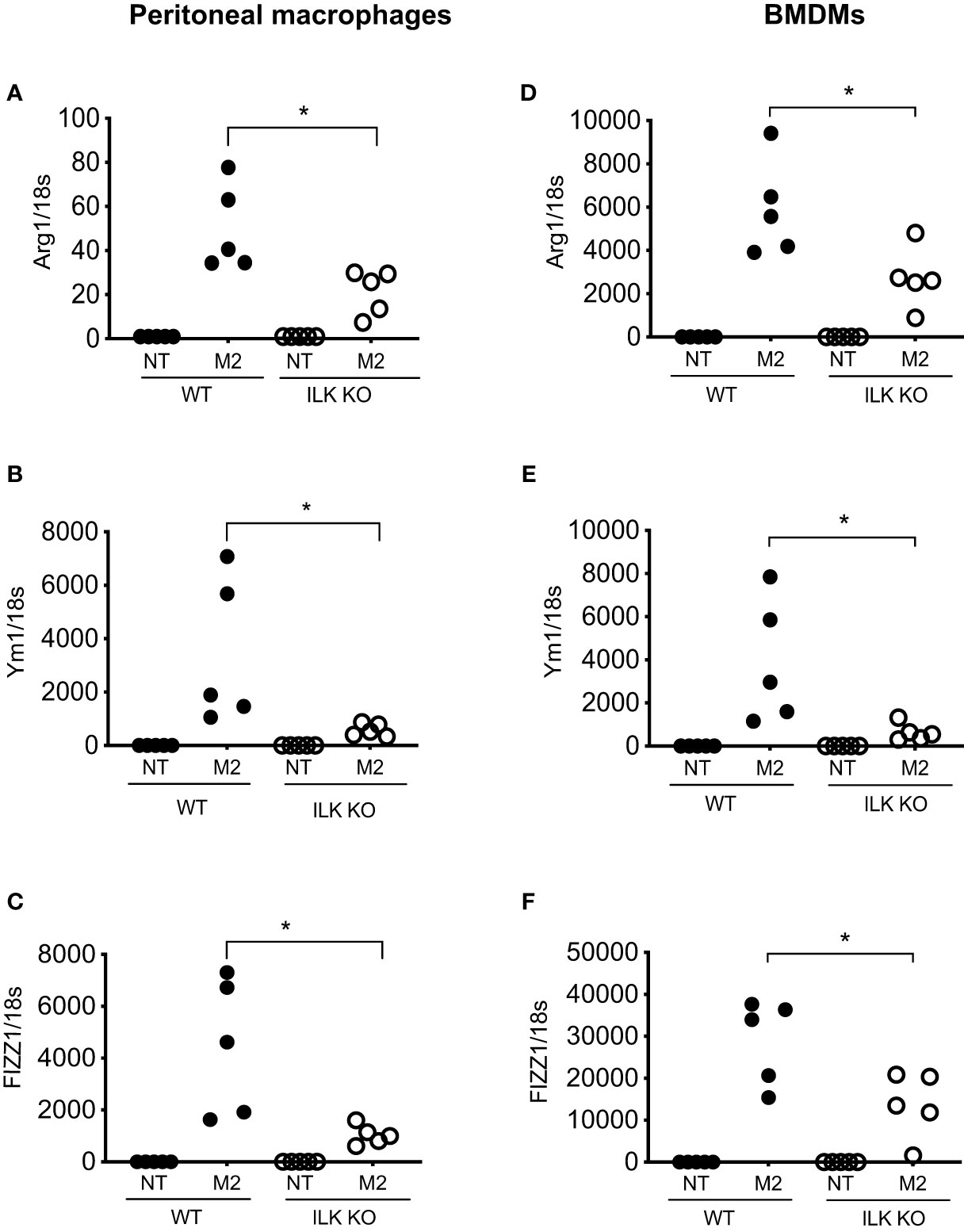
Figure 4 ILK promotes M2 macrophage polarization. q-RT-PCR analyses of M2-specific genes Arg1, Ym1 and FIZZ1 in both peritoneal (A–C) and bone-marrow derived macrophages (BMDM) (D–F) isolated from WT and myeloid-ILK deficient (M-ILK KO) mice are shown. Analyses were carried out under both resting and M2-polarised conditions (refer to Materials and Method section). Results are representative of three independent experiments and are shown as mean ± SD. n = 5 per genotype. *, p < 0.05; Student’s t test.
Myeloid-ILK deficiency is associated with reduced infiltration of CD206+ cells in tumours
Cancer progression is a complex multistep process which involves interactions of neoplastic cells with various immune and non-immune cells (46). TAMs that infiltrate into TME play a complex role in tumour progression through their interactions with neoplastic cells (47, 48). TAMs are therefore characterized as M2 polarized macrophages in response to cues from tumour cells and activated lymphocytes. The mannose receptor (CD206), a proteolytic and widely used marker for alternatively activated type 2 (M2) macrophages (44), is highly expressed on TAMs. Since ILK deficiency impairs macrophage M2 polarization in vitro (Figure 4), we wanted to investigate further whether myeloid-ILK deficiency impairs TAMs M2 polarization in vivo. To do this, we quantified the tumour-infiltrated CD206+ cells via IHC using tumour sections. As shown in Figure 5, the tumour infiltration of CD206+ cells was significantly reduced in both AOM/DSS and APCmin/+-driven models of CRC, suggesting that ILK is required for the tumour promoting role of TAMs.
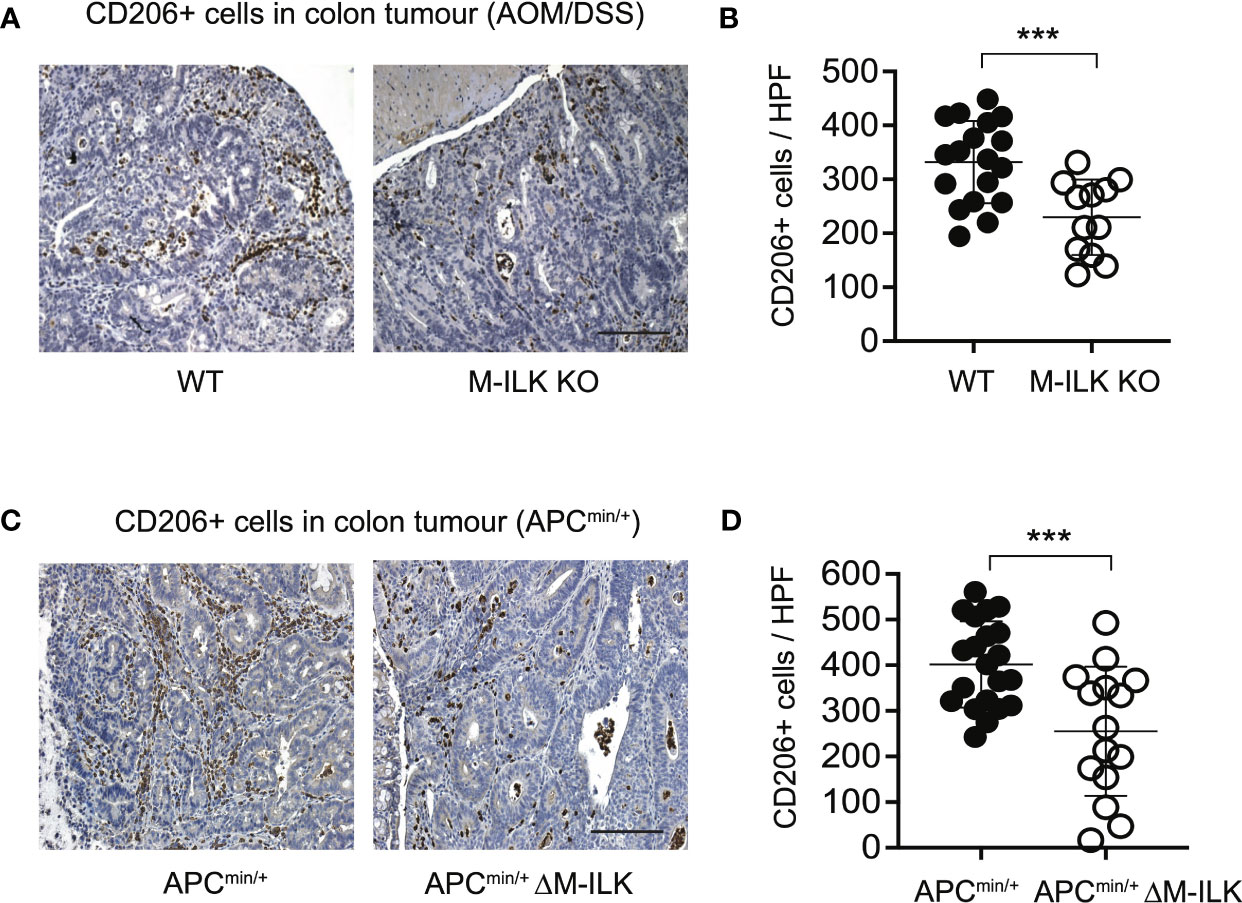
Figure 5 Myeloid-ILK deficiency is associated with reduced infiltration of CD206+ cells in tumours. (A) Representative immunohistochemistry images of colon tumours showing CD206+ cells of indicated mice subjected to the AOM/DSS model and (B) quantification of CD206+ cells in high-powered fields (HPFs, original magnification x400) across 5 mice per genotype, n = 18 (WT) and n = 18 (M-ILK KO) HPFs. (C) Representative immunohistochemistry images of colon tumours showing CD206+ cells of APCmin/+ and APCmin/+ΔM-ILK mice, and (D) quantification of CD206+ cells in high-powered fields (HPFs) across 5 mice per genotype, n = 21 (APCmin/+) and n = 15 (APCmin/+ΔM-ILK) HPFs. Results are shown as mean ± SD. Scale bars, 100 µm; ***, p < 0.001, Student’s t test.
Myeloid-ILK deficient mice demonstrate enhanced tumour-infiltration of CD8+ T cells in AOM/DSS and reduced tumour-infiltration of FOXP3+ T cells in APCmin/+ models
Several studies have suggested integrally connected roles of myeloid cells with other immune cells including adaptive immune cells. For examples, based on 3D visualization of the CRC immune landscape, macrophages were found to be associated with the T cell network within tumours (49). Myeloid-derived suppressor cells (MDSCs) were shown to promote tumour growth by suppressing CD8+ T cell cytotoxic activity in the AOM/DSS model of CRC (15). Antibody-mediated depletion of macrophages decreased tumour-infiltrating both T cells and NK cells, thereby affecting anti-tumour immunity in a mouse model of cancer immunotherapy (50). Accordingly, to examine the effect of myeloid-ILK deficiency on the tumour-infiltration of T cells, the tumour-infiltrated regulatory FOXP3+ and cytotoxic CD8+ T cells in both models of CRC were quantified. As shown in Figure 6 (A–C), myeloid-ILK deficiency resulted in enhanced filtration of CD8+ T cells without any impact of the infiltration of FOXP3+ T cells in the AOM/DSS model. In contrast, for the APCmin/+ model, the tumour infiltration of FOXP3+ T cells were reduced by myeloid-ILK deficiency whereas the infiltration of CD8+ T cells remained unaffected (Figures 6E–G). In both models, the ratio of CD8+/FOXP3+ T cells in tumours were significantly elevated in myeloid-ILK deficient mice compared the ILK-sufficient controls (Figures 6D, H). Overall, these results indicate a role of myeloid-ILK in tumour promotion through regulation of the tumour-infiltration of T cells either by restraining tumour-suppressing CD8+ T cells or by enhancing tumour-promoting FOXP3+ T cells.
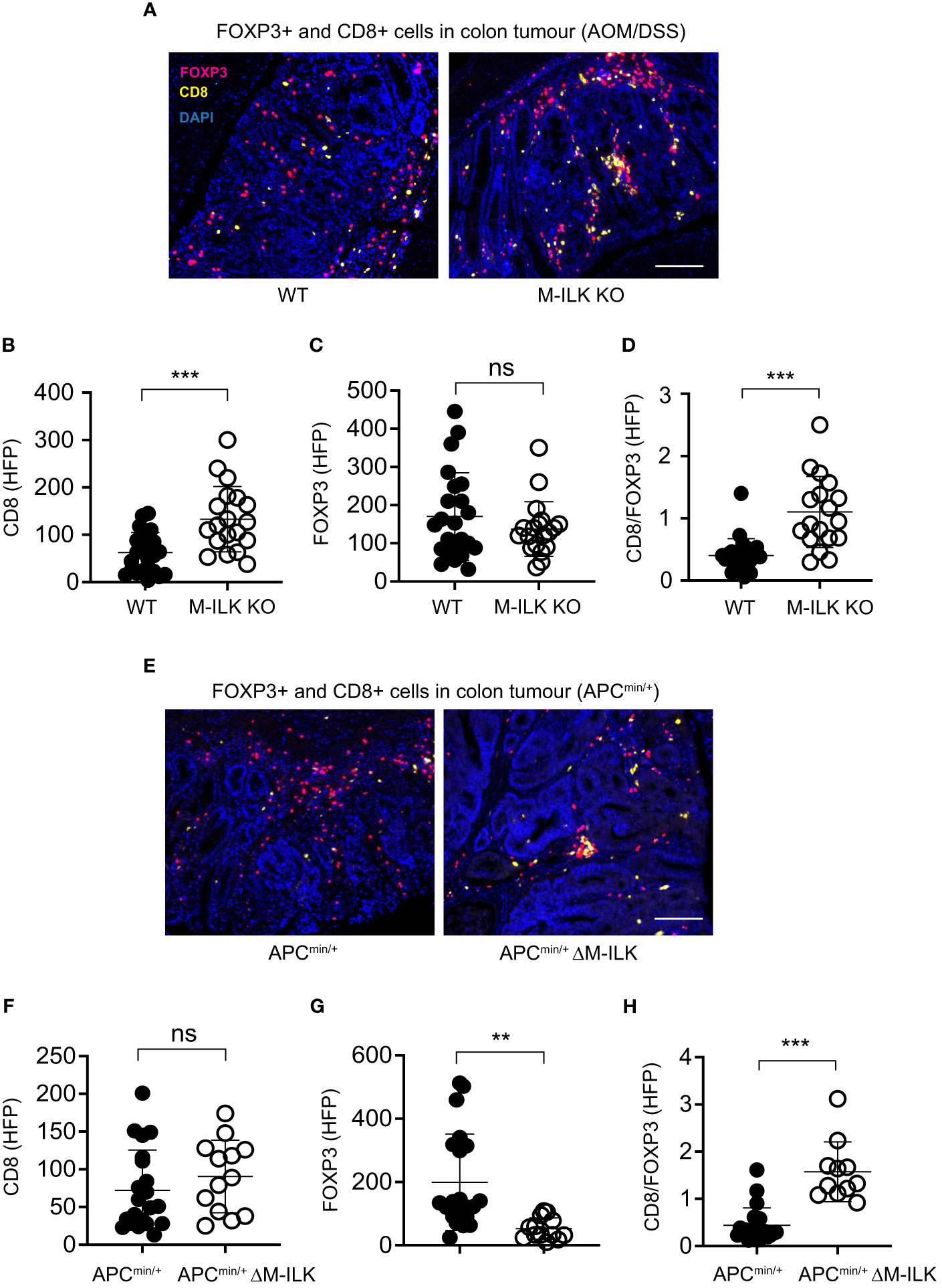
Figure 6 Myeloid-ILK deficiency enhances tumour-infiltration of CD8+ T in AOM/DSS model and reduces tumour-infiltration of FOXP3+ T cells in APCmin/+ model. (A) Representative immunofluorescence images of colon tumours showing infiltration of FOXP3+ and CD8+ T cells in tumours of indicated mice subjected to the AOM/DSS model. Pink, FOXP3+ cells; yellow, CD8+ cells; and blue, DAPI. Quantification of CD8+ T cells (B), FOXP3+ T cells (C), and the ratio of CD8+/FOXP3+ cells (D) in the AOM/DSS-driven colon tumours using the high-powered fields (HPFs, original magnification x400) across 4 mice per genotype, n = 23 (WT) and n = 19 (M-ILK KO) HPFs. (E) Representative immunofluorescence images of colon tumours showing infiltration of FOXP3+ and CD8+ T cells in tumours of APCmin/+ and APCmin/+ΔM-ILK mice. Pink, FOXP3+ cells; yellow, CD8+ cells; and blue, DAPI. (B) Quantification of CD8+ T cells (F), FOXP3+ T cells (G), and the ratio of CD8+/FOXP3+ cells (H) in APCmin/+-driven colon tumours using high-powered fields (HPFs) across 4 mice per genotype, n = 21 (APCmin/+) and n = 13 (APCmin/+ΔM-ILK) HPFs. Results are shown as mean ± SD. Scale bars, 100 µm; **, p < 0.01; ***, p < 0.001, ns, not significant, Student’s t test.
CRC tumours from patients exhibit high ILK expression in both epithelial cells and infiltrating CD11b+ myeloid cells
In order to determine whether the observations in the above mouse studies correlate with human CRC, a CRC tissue microarray consisting of tumours and corresponding adjacent normal tissues of individual CRC patients (n=13) was examined. Based on immunofluorescence analyses of ILK+ and CD11b+ (myeloid) cells in both tumours and adjacent normal tissues, elevated expression of ILK was apparent not only in tumour epithelium but also in the tumour-infiltrating myeloid (CD11b+) cells (Figure 7). While the elevated ILK expression in tumour epithelium compared to the adjacent normal tissue is supported by published reports (27), we observed higher tumour-infiltration of ILK-positive myeloid (CD11b+ ILK+) cells as indicated by merged images from both CD11b+ and ILK+ cells (Figure 7A). In accordance with published literature highlighting a critical involvement of myeloid cells (51), we observed an elevated infiltration of myeloid cells in tumours (data not shown). Among the tumour-infiltrating myeloid (CD11b+) cells, our results indicate that the major proportion of these infiltrated CD11b+ cells are ILK+ compared to the ILK-deficient myeloid cells which are comparable to those in adjacent normal tissues (Figures 7C, D), suggesting myeloid-ILK as a prognostic marker for CRC in humans. Overall, these observations show that ILK is an integral player in the tumour-promoting role of myeloid cells.
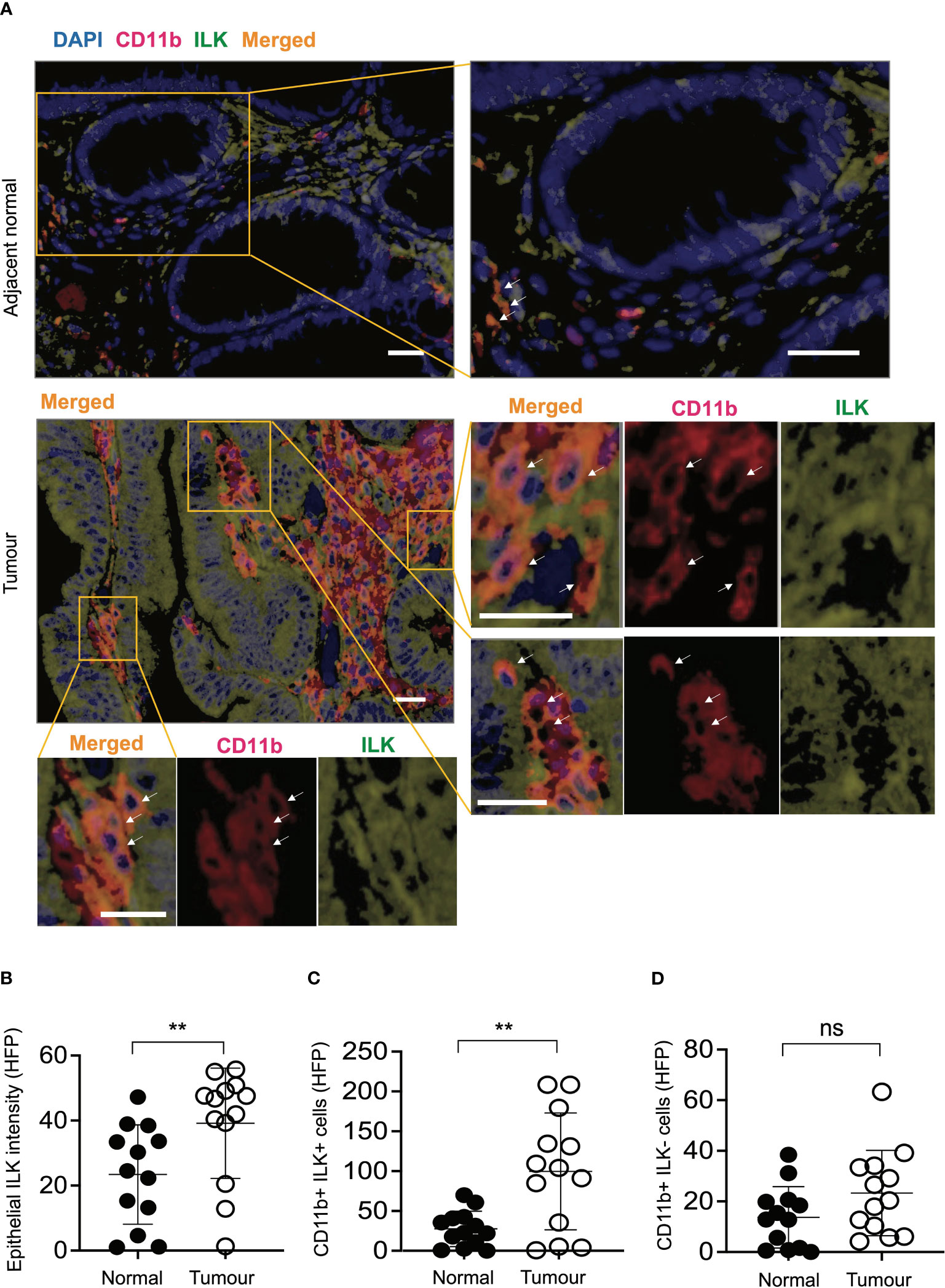
Figure 7 ILK expression is elevated in human CRC epithelial cells and infiltrating CD11b+ cells compared to the adjacent normal tissues. (A) CRC tissue microarray from patients (n=13) consisting of both tumours and their adjacent normal tissues were stained with immunofluorescence to investigate ILK expression (green) and CD11b marker for myeloid cell infiltration (red) in addition to the DAPI nuclear stain (blue). Orange colour in the merged image is an expression of both red CD11b and green ILK. The top panel is a representative microscopic image of adjacent normal tissue and the bottom panel is the tumour tissue showing ILK expression and infiltrating CD11b+ myeloid cells. Single-coloured images within the tumour section corresponding to CD11b+ cells (red) and ILK+ cells (green) show that merged signals (orange) are originated from the combined signals of both CD11b and ILK. Arrows indicate myeloid cells in both CD11b+ and merged images. The scale bar is 20μm. (B-D) Quantifications of epithelial ILK intensity (B), CD11b+ ILK+ cells (C) and CD11b+ ILK- cells (D) in the tumours and their adjacent normal tissues. Results are shown as mean ± SD. Scale bars, 20 µm; **, p < 0.01, ns, not significant, Student’s t test.
Discussion
Over the past several decades our understanding has been rapidly expanded in terms of epidemiology, aetiology, molecular biology and clinical aspects of CRC. Nevertheless, the incidence of CRC remains elevated with an annual 1.8 million new cases being diagnosed worldwide (52). Furthermore, CRC is no longer considered a disease primarily of Western populations only since rapid rises in incidence are also occurring in developing countries undergoing changes in diet and lifestyle (52). Colonoscopy screening is a gold standard method for detecting early stage CRC and nutrient-based chemoprevention such as calcium supplementation, vitamin D and folate could also curb CRC risk safely (5, 53, 54). While low dose aspirin has also been associated with a reduced risk of colorectal cancer in chemopreventative studies (55), there is a need for novel and efficacious drugs for advanced CRC. However, the existing medical treatments and preventive options are both currently insufficient to control the escalating rise in CRC (52). Therefore, it is critically important to fully understand the molecular drivers of CRC for the development of treatment and prevention strategies to counter the rise. In this study, we have identified myeloid-specific ILK as a critical driver of CRC which could potentially be developed as a as a novel therapeutic. However, the development of novel therapeutic options for CRC requires a deeper understanding of the mechanisms underlying tumour progression. Accumulating evidence suggests that tumour progression is largely governed by the complex interactions between tumours and their microenvironment. The involvement of immune cells in CRC progression and recurrence has been extensively demonstrated and the immune infiltrate composition has been characterised as the most heterogeneous and dynamic feature of CRC as its composition changes across tumour stages. Both adaptive and innate immune cells showed differential infiltration patterns along with tumour progression, implying the prognostic features of infiltrated immune cells (49). Further studies have defined a scoring system, termed Immunoscore (IS), based on the quantification of infiltrated cytotoxic and memory T cells. Interestingly, IS strongly predicts both tumour recurrence and patient survival (56). Moreover, higher IS was also associated with fewer metastases and, for the least-infiltrated metastasis, IS was also predictive of patient survival (57). Our study demonstrates the ratios of CD8/FOXP3 correlate with the tumour burdens in both AOM/DSS and APCmin/+ models, indicating an impact of myeloid-ILK deficiency on the differential tumour-infiltration of T cells. The increased numbers of CD8 T cells showed a strong correlation with the reduced tumour burdens in both AOM/DSS- and APCmin/+ -driven mouse models, indicating an active status of CD8 T cells with an anti-cancer property. Further verification of active CD8 T cells status comprises the assessment of more than just one marker such as CD69, HLA-DR, IFN-γ, IL-2 and IL-12 (58, 59) and is unlikely to add anything new to our conclusions. Since the multiplex immunostaining will be challenging with additional markers along with existing CD8 and DAPI, it will require a completely different approach of extracting and flow cytometry analysis of tumour-infiltrated T cells and will be a significantly time-consuming exercise considering these mouse models. Overall, our observation also suggests a potential of role for myeloid-ILK in modulating the IS.
ILK is a focal adhesion protein which is ubiquitously expressed in both immune and non-immune cells. As an adaptor protein, it links the extracellular matrix with downstream signalling pathways and thereby it is implicated in many cellular processes such as survival, differentiation, proliferation, and gene expression. While under homeostasis ILK activation is transient as a biological switch for many signal transduction pathways, in pathological conditions (e.g. cancers) ILK is constitutively activated. Overexpression or constitutive activation of ILK is required for cell survival through the phosphorylation of Akt on Ser473, which also involve the activation of nuclear factor B (NF-κB) pathway (23, 24). Despite its central role in cell-extracellular matrix interactions, the mechanistic details of ILK upregulation is not fully understood yet but is considered to be cell type- and context-dependent (60, 61). For examples, our earlier report demonstrates a correlation between ILK overexpression and markers of M2 macrophages (45), indicating a cancer-promoting cue driving ILK upregulation. Even the mechanistic details could also vary within the same cell type depending on the stimulation. For example, we demonstrated earlier that ILK-driven phosphorylation of p65 on Ser536 and TNF-α synthesis are independent of PI3K/Akt pathway under LPS stimulation but they are dependent on the PI3K/Akt pathway during H. pylori infection (29). While the mechanistic details of ILK expression in myeloid cells are yet to be revealed, our study demonstrates a tumour-promoting role of myeloid-specific ILK expression.
CRC development is a complex, multistep process involving an intricate array of gene alterations. While it is not possible to have an universal mouse model to address this heterogenous disease, a number CRC mouse models were established which differ in their underlying mechanisms of tumorigenesis (62, 63). For example, AOM/DSS model induces several pathogenic pathways such as Kras, c-Myc and Apc/β-catenin. On the other hand, Apc mutation results in the activation of a complex together with Apc, Axin and GS3K, which leads to WNT/β-catenin signalling pathway. Regardless of the differences in the mechanisms of tumorigenesis, the hallmarks of immune cell infiltration within tumours seem to be mostly conserved given their impact on the TME and correlate with the tumorigenesis process (49). Accordingly, the overall CD8/FOXP3 ratio that we measured in the study correlates well with the tumour burden in both models. However, the differences in the number of infiltrated CD8+ and FOXP3+ cells that we observed between these models is most likely related to the underlying mechanism of tumorigenesis associated with these models. With respect to the details of tumour-associated immune signalling, there is not enough studies dedicated to comparison between these models. Nevertheless, the critical involvement of immune signalling in CRC development and progression is highlighted by recent literature showing a pivotal role of gut microbiota in both AOM/DSS and APCmin/+ models (63, 64). Even though the microbiota could vary from human to mice as well as within mice under various housing conditions, the outcomes of these models hold some relevance to the pathology of human CRC due to the randomness of carcinogen-induced mutations as well as the Apc mutation observed in over 70% of human CRC.
Among tumour-infiltrating immune cells, myeloid cells have been reported to be the most significant and abundant cell type. Several recent studies suggest a significant involvement of myeloid cells in tumour progression. For examples, the massive infiltration of granulocytic MDSCs (G-MDSCs) from the circulatory system to colonic mucosa is essential for the development of CAC tumorigenesis since the inhibition of G-MDSCs infiltration via CXCR2 deletion attenuated colitis-associated tumorigenesis (15). The levels of granulocytic myeloid cells were found to be elevated in both circulation and tumours of CRC patients (65). Myeloid-specific deletion of EGFR in mice resulted in fewer and smaller tumours in both APCmin/+ and AOM/DSS models, and EGFR expression in myeloid cells but not in intestinal epithelial cells was associated with tumour metastasis and shorter patient survival time (66). The loss of IKKα kinase activity protected mice from intestinal tumour development via the suppression of M1-like myeloid cell infiltration rather than a direct impact on tumour cell proliferation (67). Increased oxidative stress in myeloid cells also promotes AOM-initiated colon tumorigenesis (68). Myeloid-specific p38α controls the tumour-promoting inflammatory microenvironment and promotes CAC (69). In a colorectal cancer liver metastasis model, the premetastatic niche in the liver was formed by MDSCs driven by the S1PR1–STAT3 signalling pathway in cancer cells (70). ScRNA-seq analyses on immune and stromal populations from CRC patients and mouse tumour models highlight conserved myeloid subsets as potential therapeutic targets (71). Transcriptomic analysis of tumour-infiltrating MDSCs also identified gene signatures in MDSC of poor prognosis (72). Tumor-resident Aspergillus sydowii promotes lung adenocarcinoma by inducing MDSCs expansion and activation (73), indicating a role of myeloid cells in intratumour microbiome-mediated lung cancer progression. Based on these diverse roles of myeloid cells integrated with cancer progression, the future prospect of myeloid cells as a potential therapeutic option has been widely recognised (51, 74). Our study further emphasises the critical involvement of myeloid cell biology in cancer progression and we demonstrate that ILK is required for macrophage M2 polarization. Macrophage polarization is a result of temporal changes in diverse signalling (NF-κB, IRF3, IRF5, STAT1/3/6, MEK and PPARχ/δ) and metabolic pathways (glucose, glycosaminoglycan and retinoic acid signalling) (41, 42, 75). Particularly, a shift in cellular metabolism including mitochondrial energy metabolism is more closely linked to macrophage functional changes. Future investigations on a diverse signalling pathways along with quantitative proteomics will be required to fully understand the mechanism of ILK-driven M2 polarization.
Over the last decade, immune cell therapeutic approaches to cancer have been dominated by both adoptive transfer of T cells and CAR T cell therapy, which have shown clinical efficacy only in the treatment of blood malignancies while the majority of solid tumours remains largely unresponsive. The lack of trafficking to the tumour site and the immunosuppressive nature of the TME have been key limitations to T cell-based therapies. Unlike T cells, the adoptive transfer of myeloid cells can overcome some of these limitations by ensuing more efficient trafficking to the TME. In relation to the immunosuppressive nature of TME, certain myeloid cell subsets (such as TAMs and MDSCs) are critical determinants of resistant to cancer therapies (76). As a result, targeting myeloid cell subsets as a potential therapeutic option for cancer has recently been the subject of intense research. However, developmental heterogeneity and functional redundancy of myeloid cells along with the lack of well-defined target specificity have made this approach challenging (51). In this study, we have identified the myeloid-specific ILK expression as a potential target for solid tumours such as CRC. Since integrins are the main cellular adhesion receptors that are implicated in nearly every step of cancer progression such as cancer cell migration and invasion, anchorage-independent survival and colonization of metastatic sites, the targeting of the integrin signalling has emerged as a therapeutic strategy in cancer (77). While the therapeutic potential of blocking integrin signalling has primarily been focused on cancer cells, its implication in the tumour-promoting roles of immune cells is yet to be fully explored. By identifying the myeloid-specific ILK as a potential driver of CRC, our study reveals a mechanism through which the tumour-promoting roles of myeloid cells could be exploited for clinical applications.
Materials and methods
Mice
ILKfl/fl;LysMCre (referred as myeloid-ILK deficient or M-ILK KO) mice were generated as described previously (30). ILKfl/fl;LysMCre;APCmin/+ (referred as myeloid-ILK deficient APCmin/+ or APCmin/+ΔM-ILK) mice were generated by inter-breeding mice carrying ILKfl/fl;LysMCre and APCmin/+. Aged-matched ILK-sufficient LysMCre and LysMCre;APCmin/+ mice were used as wild-type (WT) controls for M-ILK KO and APCmin/+ΔM-ILK mice, respectively. Genotypes of mouse litters were confirmed via PCR analysis using the tail DNA as described previously (30). The PCR analyses for ILK and LysMCre were carried out as described previously (30). Genotyping for APCmin/+ was carried out using the following primers: APC-mutant (APCmin) primer: 5′-TTCTGAGAAAGACAGAAGTTA-3′ and APC-common primer: 5′-TTCCACTTTGGCATAAGGC-3′, detecting the mutant APC allele (313 bp) which is only present in heterozygous APC mice. The PCR protocol for APC mice consists of an initial denaturing step of 5 min at 94 °C, followed by 30 cycles of 94 °C for 35 sec, 60 °C for 30 sec and 72 °C for 40 sec, and then a final extension at 72 °C for 5 min. All mice were generated and maintained in the same facility under pathogen-free conditions with a 12 hr light/dark cycle at the Monash Medical Centre Animal Facility, Melbourne, Australia (30). All animal breeding and experiment protocols were approved by the Monash University Animal Ethics Committee and adhered to the national guidelines provided by the Bureau of Animal Welfare, Victoria, Australia (30).
Macrophage isolation and culture in conditioned media
BMDMs were isolated and cultured as described previously (30). Briefly, primary bone marrow cells were flushed from femurs of 8-weeks old mice and were differentiated to macrophages in RPMI 1640 supplemented with 20% L929–conditioned media, 10% FBS Glutamax and Pen/Strep over 7 d. Half of the culture media refreshed on day 5 and adherent macrophages were harvested by scraping on day 7. Peritoneal macrophages were harvested in PBS from unchallenged 8-weeks old mice, cultured in RPMI 1640 supplemented with 10% FBS Glutamax and Pen/Strep and adherent cells were used for experiments. The purity of the isolated macrophages was confirmed via the estimation of FITC-positive cells using FACS analysis. For M2 polarization, macrophages were stimulated with recombinant mouse IL-4 (50 ng/mL, Peprotech) and IL-13 (50 ng/mL, Peprotech) in RPMI 1640 supplemented with 10% FBS Glutamax and Pen/Strep. For M1 polarization, macrophages were stimulated with lipopolysaccharide (LPS) (100 ng/ml, Sigma) and recombinant mouse IFN-γ (50 ng/mL, Peprotech) in RPMI 1640 supplemented with 10% FBS Glutamax and Pen/Strep. After 24 hours, wells were washed with ice-cold PBS and adherent macrophages were detached from plates using a cell-scraper.
Mouse models of colon cancer
The AOM/DSS model was performed as described by Tanaka et al. (31), with slight modifications. Briefly, 8-weeks old mice were given a single i.p. injection of the mutagen AOM (Sigma-Aldrich) at 12.5 mg/kg at day 0, after which at day 5 they received drinking water containing 1% DSS (30- 50 kDa, colitis grade, MP Biomedicals) over three cycles of 5-day periods that were interspersed with 16-day periods in which they received normal water. The entire model takes 62 days and all mice were sacrificed at day 62 for post-mortem examination. After the administration of DSS, all experimental mice were monitored daily for both general well-being and symptoms including weight loss, abnormal stool consistency, blood in stool and rectal prolapse. To minimize the degree of pain for experimental mice, the mouse intervention criteria were followed as per the guidelines of the ethics committee. For the APCmin/+-driven colon cancer model, age- and gender-matched experimental APCmin/+ΔM-ILK mice along with their WT counterparts were monitored once a week for both general well-being and symptoms related to tumour burden such as chronic weight loss, pale toes/feet and rectal prolapse. In case of any of these symptoms, mice were monitored daily and subjected to same intervention criteria as per the guidelines of the ethics committee. all experimental APCmin/+ mice were sacrificed at 18 weeks of age for the post- mortem analysis of tumours. For the survival study, experimental APCmin/+ mice were kept alive for up to 12 months as long as they remained healthy. At the end of both models, the colons were extracted, flushed with ice-cold phosphate-buffered saline (PBS) and processed for histological analysis.
Histology
Mouse colon tissues were fixed in 10% buffered formalin for 4 h as ““swiss rolls”, which were further dehydrated, embedded in paraffin, and sectioned at 5 μm thickness. Histological scoring of tumour grades was performed on Hematoxylin and eosin (H&E) stained tissue sections in a blinded fashion according to a previously described protocol (30).
Immunohistochemistry and immunofluorescence
Immunohistochemistry and immunofluorescence of paraffin-embedded sections were performed as previously described (30). For immunohistochemistry, anti-CD206 (Cat# ab64693, Abcam) was used as a primary antibody (Ab) followed by biotin secondary and HRP-conjugated Abs. Immunohistochemical staining was performed with DAB followed by counterstaining with hematoxylin. Quantification of positive cells (CD206+) was performed with ImageJ, a public domain Java image-processing program (National Institutes of Health) as per the procedure described previously (30). For multiplex immunofluorescence of mouse colon tissues, anti-CD8 (Cat# 38553, Cell Signaling Technology) and anti-FOXP3 (Cat# 62072, Cell Signaling Technology) were used as primary Abs. For multiplex immunofluorescence of CRC TMA, anti-ILK (Cat# ab52480, Abcam) and anti-CD11b (Cat# 49420, Cell Signaling Technology) were used as primary Abs. The SignalStain Boost IHC Detection Reagent (HRP, rabbit, 8114; Cell Signaling Technology) was used as a secondary Ab, and the staining was performed with Opal 670 (PerkinElmer, Boston, MA) for Anti-CD8 and Opal 570 (PerkinElmer) for Anti-FOXP3. All slides were mounted with SlowFade Gold Antifade Mountant with DAPI (ThermoFisher Scientific). Stained serial sections of the whole colon was scanned on a Aperio CS2 (Leica). Quantification of CD8+, FOXP3+, ILK+ and CD11b+ cells was performed manually on the high-powered fields (HPFs) of the stained serial sections of the whole colon or TMA using ImageJ, as described previously (30). All images were quantified in a blinded fashion.
Real-time quantitative PCR
Total RNA was extracted by using the RNA mini columns (GE Healthcare) according to the manufacturer’s protocol. The concentration and purity of RNA was determined based on the absorbance ratio at 260/280 (nm). 1 μg RNA was used for cDNA synthesis in a total volume of 20 ul using Superscript First Strand Synthesis System (Invitrogen) according to the manufacturer’s protocol. Quantitative real time PCR (qRT-PCR) was performed as previously described (ref). Briefly, qRT-PCR was performed in 10 ul containing 10 ul Power SYBR Green PCR master mix (Applied Biosystems), 0.5 uM of each primer (forward and reverse), and 1–2 ul of cDNA template. qRT-PCT performed in the 7900HT Fast Real-Time PCR System (Applied Biosystems) and reactions were performed at 90°C for 8 min 30 s, followed by 40 cycles of 94°C for 15 s, and 60°C for 1 min. A melting curve was generated at the end to ensure uniformity of the product and the fold changes were obtained by converting the logarithmic scale to an exponential scale (2-ΔΔCT). Expression levels were normalized based on the levels of the housekeeping gene 18s. The following primers were used:
ARG1forward primer, 5´-CAGAAGAATGGAAGAGTCAG-3´; ARG1reverse primer, 5´-CAGATATGCAGGGAGTCACC-3´; YM1 forward primer, 5´- GAAGCTCTCCAGAAGCAATCCT-3´; YM1 reverse primer, 5´- GCCTGTCCTTAGCCCAACTG-3´; FIZZ1 forward primer, 5´- CCTGCTGGGATGACTGCTAC-3´; FIZZ1reverse primer, 5´- CCACTCTGGATCTCCCAAGA-3´; 18s forward primer, 5´- GTAACCCGTTGAACCCCATT-3´, and 18s reverse primer, 5´- CCATCCAATCGGTAGTAGCG -3´.
Western blot analysis
Western blot analysis was carried out as described previously (29, 30). The membrane was immunoblotted with the following primary antibodies: rabbit polyclonal antibody against Arginase-1 (Cat. No. 9819, Cell Signaling Technology, Danvers, MA), iNOS (Cat. No. 2982, Cell Signaling Technology, Danvers, MA) and pan-actin mouse monoclonal antibody (NeoMarker, Fremont, CA). Secondary antibodies used were anti-rabbit Alexa-680- and anti-mouse Alexa-760-conjugated antibodies (Invitrogen, Carlsbad, CA). An Odyssey® Infrared Imaging System (LI-COR® Biosciences, Lincoln, NE) was used to capture the fluorescence signals which were converted into grayscale images with a white background.
Human primary tumour samples and tissue microarray
Tissue microarray slides containing primary human colon tumours and adjacent normal tissues were obtained from the Monash Biobank facility of Monash Health, Monash Medical Centre, Melbourne, Australia. All samples derived from the Monash Biobank facility were obtained with informed consent under institutional review board–approved protocols. For IHC analysis, 13 cases of tumours along with adjacent normal tissues were examined. Quantification of ILK+ and CD11b+ cells was performed manually on the high-powered fields (HPFs) of the stained sections using ImageJ, as described previously (30).
Statistical analysis
All data are presented as mean ± SD. Student’s t test or paired t test was performed for inter-group comparisons. Kaplan-Meier method was used to estimate median overall survival time, and P values were calculated using log-rank test. “n” represents number of mice or cells used in each experiments, as indicated in the figure legends. Quantifications on blinded histological samples were performed by counting/measuring microscopic fields (high-powered fields, HPFs, where indicated) as indicated in the legends. Statistical analysis was performed using Prism 9.0 (GraphPad Software La Jolla, California USA).
Data availability statement
The data presented in the study are deposited in the figshare depository (https://figshare.com), accession code is 10.6084/m9.figshare.24534073.
Ethics statement
The studies involving humans were approved by Monash Biobank facility of Monash Health, Monash Medical Centre, Melbourne, Australia. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from gifted from another research group. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by Monash University Animal Ethics Committee, Monash University, Australia. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft. SA: Data curation, Formal Analysis, Writing – review & editing. RF: Resources, Writing – review & editing. BW: Validation, Writing – review & editing.
Acknowledgments
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AA was supported by a Ron Evans Cancer Research Fellowship. SA was supported by a post-graduate scholarship from Najran University, Najran, Saudi Arabia. Research at the Hudson Institute of Medical Research is supported by the Victorian Government’s Operational Infrastructure Support Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1270194/full#supplementary-material
Supplementary Figure 1 | ILK is required for partial M1 macrophage polarization. q-RT-PCR analyses of M1-specific genes encoding TNF-α, iNOS and CCL2 in both peritoneal (A, B, C) and bone-marrow derived macrophages (BMDM) (D, E, F) isolated from WT and myeloid-ILK deficient (M-ILK KO) mice are shown. Analyses were carried out under both resting and M1-polarised conditions (refer to Materials and Method section). Results are representative of three independent experiments and are shown as mean ± SD. n = 5 per genotype. **, p < 0.01; ***, p < 0.001; Student’s t test.
Supplementary Figure 2 | ILK is not required for iNOS expression during M1 macrophage polarization. Western blot analysis showing iNOS protein levels in WT and ILK-KO bone-marrow derived macrophages (BMDMs) undergone M1 polarization as described in Materials and Methods. n = 3 per genotype.
Supplementary Figure 3 | ILK is required for Arginase-1 expression during M2 macrophage polarization. Western blot analysis showing Arginase-1 protein levels in WT and ILK-KO bone-marrow derived macrophages (BMDMs) undergone M2 polarization as described in Materials and Methods. n = 3 per genotype.
References
1. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin (2014) 64:252–71. doi: 10.3322/caac.21235
2. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev (2010) 19:1893–907. doi: 10.1158/1055-9965.EPI-10-0437
3. Labianca R, Merelli B. Screening and diagnosis for colorectal cancer: present and future. Tumori (2010) 96:889–901. doi: 10.1177/548.6506
4. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin (2014) 64:104–17. doi: 10.3322/caac.21220
5. Zauber AG, Winawer SJ, O'Brien MJ, Lansdorp-Vogelaar I, van Ballegooijen M, Hankey BF, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med (2012) 366:687–96. doi: 10.1056/NEJMoa1100370
6. Yamashita K, Watanabe M. Clinical significance of tumor markers and an emerging perspective on colorectal cancer. Cancer Sci (2009) 100:195–9. doi: 10.1111/j.1349-7006.2008.01022.x
7. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell (1996) 87:159–70. doi: 10.1016/S0092-8674(00)81333-1
8. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet (2014) 383:1490–502. doi: 10.1016/S0140-6736(13)61649-9
9. Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med (1990) 323:1228–33. doi: 10.1056/NEJM199011013231802
10. Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology (2010) 138:2101–2114.e2105. doi: 10.1053/j.gastro.2010.01.058
11. McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract (2008) 204:479–90. doi: 10.1016/j.prp.2008.03.004
12. Roper J, Hung KE. Priceless GEMMs: genetically engineered mouse models for colorectal cancer drug development. Trends Pharmacol Sci (2012) 33:449–55. doi: 10.1016/j.tips.2012.05.001
13. Suzui M, Morioka T, Yoshimi N. Colon preneoplastic lesions in animal models. J Toxicol Pathol (2013) 26:335–41. doi: 10.1293/tox.2013-0028
14. Terracina KP, Aoyagi T, Huang WC, Nagahashi M, Yamada A, Aoki K, et al. Development of a metastatic murine colon cancer model. J Surg Res (2015) 199:106–14. doi: 10.1016/j.jss.2015.04.030
15. Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell (2013) 24:631–44. doi: 10.1016/j.ccr.2013.10.009
16. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science (1992) 256:668–70. doi: 10.1126/science.1350108
17. Quante M, Varga J, Wang TC, Greten FR. The gastrointestinal tumor microenvironment. Gastroenterology (2013) 145:63–78. doi: 10.1053/j.gastro.2013.03.052
18. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
19. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5:953–64. doi: 10.1038/nri1733
20. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
21. Hannigan GE, McDonald PC, Walsh MP, Dedhar S. Integrin-linked kinase: not so 'pseudo' after all. Oncogene (2011) 30:4375–85. doi: 10.1038/onc.2011.177
22. Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol (2006) 7:20–31. doi: 10.1038/nrm1789
23. Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer (2005) 5:51–63. doi: 10.1038/nrc1524
24. McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase–essential roles in physiology and cancer biology. J Cell Sci (2008) 121:3121–32. doi: 10.1242/jcs.017996
25. Rothe K, Babaian A, Nakamichi N, Chen M, Chafe SC, Watanabe A, et al. Integrin-Linked kinase mediates therapeutic resistance of quiescent CML stem cells to tyrosine kinase inhibitors. Cell Stem Cell (2020) 27:110–124.e119. doi: 10.1016/j.stem.2020.04.005
26. Zheng CC, Hu HF, Hong P, Zhang QH, Xu WW, He QY, et al. Significance of integrin-linked kinase (ILK) in tumorigenesis and its potential implication as a biomarker and therapeutic target for human cancer. Am J Cancer Res (2019) 9:186–97.
27. Assi K, Mills J, Owen D, Ong C, St Arnaud R, Dedhar S, et al. Integrin-linked kinase regulates cell proliferation and tumour growth in murine colitis-associated carcinogenesis. Gut (2008) 57:931–40. doi: 10.1136/gut.2007.142778
28. Assi K, Patterson S, Dedhar S, Owen D, Levings M, Salh B. Role of epithelial integrin-linked kinase in promoting intestinal inflammation: effects on CCL2, fibronectin and the T cell repertoire. BMC Immunol (2011) 12:42. doi: 10.1186/1471-2172-12-42
29. Ahmed AU, Sarvestani ST, Gantier MP, Williams BR, Hannigan GE. Integrin-linked kinase modulates lipopolysaccharide- and Helicobacter pylori-induced nuclear factor kappaB-activated tumor necrosis factor-alpha production via regulation of p65 serine 536 phosphorylation. J Biol Chem (2014) 289:27776–93. doi: 10.1074/jbc.M114.574541
30. Ahmed AU, Yim HCH, Alorro M, Ernst M, Williams BRG. Integrin-linked kinase expression in myeloid cells promotes inflammatory signaling during experimental colitis. J Immunol (2017) 199:2128–39. doi: 10.4049/jimmunol.1700125
31. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci (2003) 94:965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x
32. Baltgalvis KA, Berger FG, Peña MM, Davis JM, White JP, Carson JA. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc ( Min/+ ) mouse. Pflugers Arch (2009) 457:989–1001. doi: 10.1007/s00424-008-0574-6
33. Tisdale MJ. Cancer cachexia. Curr Opin Gastroenterol (2010) 26:146–51. doi: 10.1097/MOG.0b013e3283347e77
34. White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PloS One (2011) 6:e24650. doi: 10.1371/journal.pone.0024650
35. Kettunen HL, Kettunen AS, Rautonen NE. Intestinal immune responses in wild-type and Apcmin/+ mouse, a model for colon cancer. Cancer Res (2003) 63:5136–42.
36. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach-Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet Muscle (2016) 6:26. doi: 10.1186/s13395-016-0098-2
37. Lane SW, Sykes SM, Al-Shahrour F, Shterental S, Paktinat M, Lo Celso C, et al. The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood (2010) 115:3489–97. doi: 10.1182/blood-2009-11-251728
38. You S, Ohmori M, Peña MM, Nassri B, Quiton J, Al-Assad ZA, et al. Developmental abnormalities in multiple proliferative tissues of Apc(Min/+) mice. Int J Exp Pathol (2006) 87:227–36. doi: 10.1111/j.1365-2613.2006.00477.x
39. Hodgson A, Wier EM, Fu K, Sun X, Wan F. Ultrasound imaging of splenomegaly as a proxy to monitor colon tumor development in Apc(min716/+) mice. Cancer Med (2016) 5:2469–76. doi: 10.1002/cam4.842
40. Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol (2009) 174:1048–64. doi: 10.2353/ajpath.2009.080676
41. Peng Y, Zhou M, Yang H, Qu R, Qiu Y, Hao J, et al. Regulatory mechanism of M1/M2 macrophage polarization in the development of autoimmune diseases. Mediators Inflammation (2023) 2023:8821610. doi: 10.1155/2023/8821610
42. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
43. Kotowicz K, Callard RE, Friedrich K, Matthews DJ, Klein N. Biological activity of IL-4 and IL-13 on human endothelial cells: functional evidence that both cytokines act through the same receptor. Int Immunol (1996) 8:1915–25. doi: 10.1093/intimm/8.12.1915
44. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi: 10.1038/nri978
45. Almasabi S, Boyd R, Ahmed AU, Williams BRG. Integrin-Linked kinase expression characterizes the immunosuppressive tumor microenvironment in colorectal cancer and regulates PD-L1 expression and immune cell cytotoxicity. Front Oncol (2022) 12:836005. doi: 10.3389/fonc.2022.836005
46. Landskron G, de la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi: 10.1155/2014/149185
47. Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest (2006) 116:2132–41. doi: 10.1172/JCI27648
48. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today (1992) 13:265–70. doi: 10.1016/0167-5699(92)90008-U
49. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
50. Sagnella SM, Yang L, Stubbs GE, Boslem E, Martino-Echarri E, Smolarczyk K, et al. Cyto-immuno-therapy for cancer: A pathway elicited by tumor-targeted, cytotoxic drug-packaged bacterially derived nanocells. Cancer Cell (2020) 37:354–370.e357. doi: 10.1016/j.ccell.2020.02.001
51. Goswami S, Anandhan S, Raychaudhuri D, Sharma P. Myeloid cell-targeted therapies for solid tumours. Nat Rev Immunol (2022) 23(2):106–120. doi: 10.1038/s41577-022-00737-w
52. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol (2019) 16:713–32. doi: 10.1038/s41575-019-0189-8
53. Dulai PS, Singh S, Marquez E, Khera R, Prokop LJ, Limburg PJ, et al. Chemoprevention of colorectal cancer in individuals with previous colorectal neoplasia: systematic review and network meta-analysis. Bmj (2016) 355:i6188. doi: 10.1136/bmj.i6188
54. Keum N, Lee DH, Greenwood DC, Zhang X, Giovannucci EL. Calcium intake and colorectal adenoma risk: dose-response meta-analysis of prospective observational studies. Int J Cancer (2015) 136:1680–7. doi: 10.1002/ijc.29164
55. Patrono C. Cyclooxygenase inhibitors and cancer: the missing pieces. J Pharmacol Exp Ther (2023) 386:181–9. doi: 10.1124/jpet.122.001631
56. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity (2016) 44:698–711. doi: 10.1016/j.immuni.2016.02.025
57. Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell (2018) 34:1012–1026.e1013. doi: 10.1016/j.ccell.2018.11.003
58. Arneth BM. Activation of CD4 and CD8 T cell receptors and regulatory T cells in response to human proteins. PeerJ (2018) 6:e4462. doi: 10.7717/peerj.4462
59. Keppler SJ, Rosenits K, Koegl T, Vucikuja S, Aichele P. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PloS One (2012) 7:e40865. doi: 10.1371/journal.pone.0040865
60. Alasseiri M, Ahmed AU, Williams BRG. Mechanisms and consequences of constitutive activation of integrin-linked kinase in acute myeloid leukemia. Cytokine Growth Factor Rev (2018) 43:1–7. doi: 10.1016/j.cytogfr.2018.06.001
61. Almasabi S, Ahmed AU, Boyd R, Williams BRG. A potential role for integrin-linked kinase in colorectal cancer growth and progression via regulating senescence and immunity. Front Genet (2021) 12:638558. doi: 10.3389/fgene.2021.638558
62. Kucherlapati MH. Mouse models in colon cancer, inferences, and implications. iScience (2023) 26:106958. doi: 10.1016/j.isci.2023.106958
63. Li C, Lau HC, Zhang X, Yu J. Mouse models for application in colorectal cancer: understanding the pathogenesis and relevance to the human condition. Biomedicines (2022) 10. doi: 10.3390/biomedicines10071710
64. Yuan C, Zhao X, Wangmo D, Alshareef D, Gates TJ, Subramanian S. Tumor models to assess immune response and tumor-microbiome interactions in colorectal cancer. Pharmacol Ther (2022) 231:107981. doi: 10.1016/j.pharmthera.2021.107981
65. Toor SM, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, et al. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol (2016) 7:560. doi: 10.3389/fimmu.2016.00560
66. Srivatsa S, Paul MC, Cardone C, Holcmann M, Amberg N, Pathria P, et al. EGFR in tumor-associated myeloid cells promotes development of colorectal cancer in mice and associates with outcomes of patients. Gastroenterology (2017) 153:178–190.e110. doi: 10.1053/j.gastro.2017.03.053
67. Göktuna SI, Canli O, Bollrath J, Fingerle AA, Horst D, Diamanti MA, et al. IKKα promotes intestinal tumorigenesis by limiting recruitment of M1-like polarized myeloid cells. Cell Rep (2014) 7:1914–25. doi: 10.1016/j.celrep.2014.05.006
68. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell (2017) 32:869–883.e865. doi: 10.1016/j.ccell.2017.11.004
69. Youssif C, Cubillos-Rojas M, Comalada M, Llonch E, Perna C, Djouder N, et al. Myeloid p38α signaling promotes intestinal IGF-1 production and inflammation-associated tumorigenesis. EMBO Mol Med (2018) 10. doi: 10.15252/emmm.201708403
70. Lin Q, Ren L, Jian M, Xu P, Li J, Zheng P, et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid-derived suppressor cells induced by the S1PR1-STAT3 signaling pathway. Cell Death Dis (2019) 10:693. doi: 10.1038/s41419-019-1922-5
71. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181:442–459.e429. doi: 10.1016/j.cell.2020.03.048
72. Saleh R, Sasidharan Nair V, Al-Dhaheri M, Khawar M, Abu Nada M, Alajez NM, et al. RNA-seq analysis of colorectal tumor-infiltrating myeloid-derived suppressor cell subsets revealed gene signatures of poor prognosis. Front Oncol (2020) 10:604906. doi: 10.3389/fonc.2020.604906
73. Liu NN, Yi CX, Wei LQ, Zhou JA, Jiang T, Hu CC, et al. The intratumor mycobiome promotes lung cancer progression via myeloid-derived suppressor cells. Cancer Cell (2023) 41:1–18. doi: 10.1016/j.ccell.2023.08.012
74. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer (2016) 16:447–62. doi: 10.1038/nrc.2016.54
75. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep (2021) 37:109955. doi: 10.1016/j.celrep.2021.109955
76. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
Keywords: ILK, myeloid, CRC, CAC, Apc min/+, M2 polarization, CD8, Foxp3
Citation: Ahmed AU, Almasabi S, Firestein R and Williams BRG (2023) Integrin-linked kinase expression in myeloid cells promotes colon tumorigenesis. Front. Immunol. 14:1270194. doi: 10.3389/fimmu.2023.1270194
Received: 31 July 2023; Accepted: 30 October 2023;
Published: 21 November 2023.
Edited by:
Lvyun Zhu, National University of Defense Technology, ChinaReviewed by:
Xing Chen, Case Western Reserve University, United StatesGuangchuan Wang, Jinzhou Medical University, China
Copyright © 2023 Ahmed, Almasabi, Firestein and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afsar U. Ahmed, YWZzYXIuYWhtZWRAbGF0cm9iZS5lZHUuYXU=; Bryan R.G. Williams, YnJ5YW4ud2lsbGlhbXNAaHVkc29uLm9yZy5hdQ==
†Present addresses: Afsar U. Ahmed, Department of Microbiology, Anatomy, Physiology and Pharmacology, School of Agriculture, Biomedicine and Environment (SABE), La Trobe University, Bundoora, VIC, Australia
Saleh Almasabi, Clinical Laboratory Sciences, Applied Medical Sciences, Najran University, Najran, Saudi Arabia