 Jiaxin Yong
Jiaxin Yong Songqing Cai1,2
Songqing Cai1,2 Zhaolei Zeng
Zhaolei Zeng- 1State Key Laboratory of Oncology in South China, Guangdong Provincial Clinical Research Center for Cancer, Sun Yat-sen University Cancer Center, Guangzhou, China
- 2Research Unit of Precision Diagnosis and Treatment for Gastrointestinal Cancer, Chinese Academy of Medical Sciences, Guangzhou, China
Nicotinamide adenine dinucleotide (NAD+) is indispensable for various oxidation-reduction reactions in mammalian cells, particularly during energy production. Malignant cells increase the expression levels of NAD+ biosynthesis enzymes for rapid proliferation and biomass production. Furthermore, mounting proof has indicated that NAD-degrading enzymes (NADases) play a role in creating the immunosuppressive tumor microenvironment (TME). Interestingly, both inhibiting NAD+ synthesis and targeting NADase have positive implications for cancer treatment. Here we summarize the detrimental outcomes of increased NAD+ production, the functions of NAD+ metabolic enzymes in creating an immunosuppressive TME, and discuss the progress and clinical translational potential of inhibitors for NAD+ synthesis and therapies targeting NADase.
1 Introduction
NAD+ is necessary for numerous energy production-related oxidation-reduction reactions. To fulfill the need for high rates of growth and biomass generation, cancer cells undergo metabolic reprogramming in unfavorable conditions, such as hypoxia. Warburg effect, a representative type of metabolic reprogramming in tumor cells, refers to a metabolic shift to glycolysis from mitochondrial oxidative phosphorylation (OXPHOS). According to recent research, cells turn to aerobic glycolysis when there is an increasing demand for NAD+ (1). The research offered novel perspectives on aerobic glycolysis in rapid-proliferating cells. Furthermore, NAD+-consuming enzymes (NADases) directly or indirectly impact DNA repair and gene transcription (2, 3).
In mammalian cells, NAD+ is mainly generated through three pathways: the de novo synthesis pathway, the Preiss-Handler (PH) synthesis pathway, and the salvage pathway. Notably, most tissues, except the liver, lack the full array of enzymes to synthesize NAD+ de novo, and thus, peripheral tissues utilize NAM derived from the liver to produce NAD+ through the salvage pathway (4). However, monocyte-derived macrophages (MDMs) can synthesize NAD+ from tryptophan, which delineates the polarization of macrophages and further regulates the innate immune response process of aging and inflammation (5). The most important enzymes involved in the PH pathway and salvage pathway are NAPRT (nicotinic acid phosphoribosyltransferase) and NAMPT (nicotinamide phosphoribosyltransferase), respectively. A recent analysis has shown that the tissue context mainly determines the type of NAD+ anabolic pathway activated in cancer. Because of their dependence on NAD+ production pathways, tumors can be divided into PH-amplified tumors, with a high frequency of tumors showing NAPRT amplification, and salvage-dependent tumors, which undergo NAMPT enhancer remodeling (6). Moreover, there have been multiple categories of NADases discovered, which encompass deacetylases (sirtuins), glycohydrolases (SARM1, CD38, CD157), and poly (ADP-ribose) polymerases (PARPs). The byproduct of all these proteins is NAM, which can be recycled in the salvage pathway to produce NAD+ by NAMPT. The synthesis and degradation of NAD+ in tumor cells is a highly dynamic process (Figure 1). Tumorigenesis and cancer progression have been strongly linked to the upregulation of NAD+ synthesis and dysregulation of NADase, as indicated by a growing number of reviews (7–12).
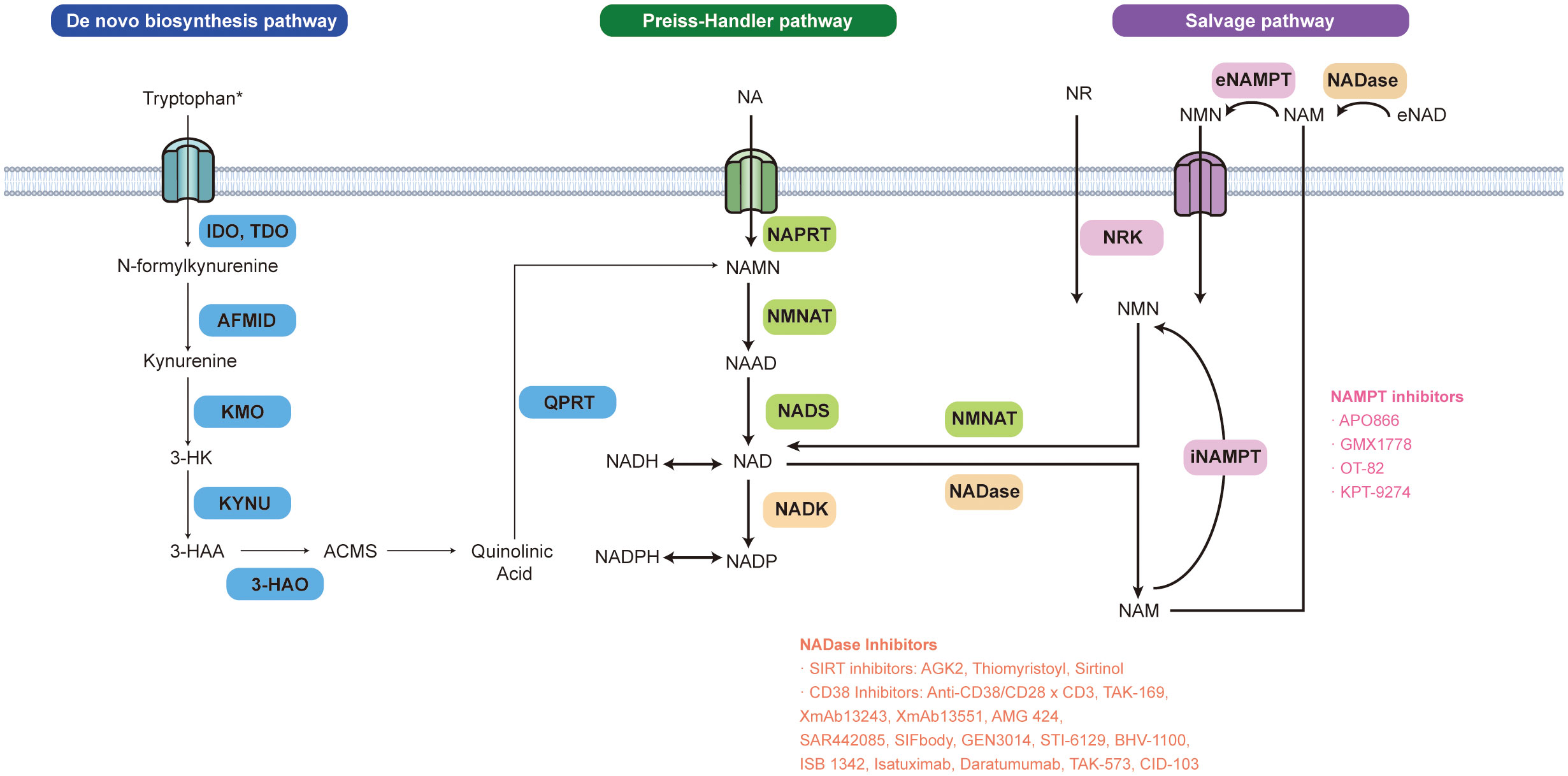
Figure 1 NAD+ biosynthesis pathways and inhibitors related to NAD+ metabolic enzymes. NAD+ can be produced via three pathways in mammalian cells: de novo, Preiss-Handler (PH), and the salvage pathway. The first rate-limiting step of the de novo NAD+ synthesis pathway is catalyzed by indole-2,3-dioxygenase 1,2 (IDO1,2) mainly expressed in the nervous system and tryptophan 2,3-dioxygenase (TDO) predominantly expressed in the liver. In this reaction, tryptophan is oxidized into the N-formyl kynurenine. The arylformamidase (AFMID) next catalyzes the conversion of N-formyl kynurenine to the immunosuppressive tryptophan metabolite kynurenine (Kyn). Kyn can be further metabolized to form quinolinic acid (QA), which is then decarboxylated by QA phosphoribosyl transferase (QPRT), to the nicotinic acid mononucleotide (NAMN) and ultimately to NAD+ via the PH pathway. NAD+ can also be made from metabolite recycling or the dietary uptake of NAD+ precursors (vitamin B3 and its derivatives) via the PH pathway or salvage pathway. Nicotinic acid (NA), the precursor of the PH pathway generates NAMN by nicotinic acid phosphoribosyltransferase (NAPRT). NAMN is transformed into nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylyltransferases (NMNAT) The whole PH pathway ends up with NAD+ synthase (NADS) turning NAAD into NAD+. The NAD+ salvage pathway also interacts with the PH pathway in the human body. A gut microbial nicotinamidase (PncA) can convert ingested nicotinamide (NAM), one of the precursors of NAD+ salvage production, to NA which can enter the PH pathway. Besides dietary uptake, NADases such as CD38 and CD157, cleave NAD+ into NAM which is recycled and transformed into nicotinamide mononucleotide (NMN) by intracellular or extracellular nicotinamide phosphoribosyltransferase (NAMPT). Nicotinamide riboside (NR) generates NMN via nicotinamide riboside kinases 1 and 2 (NRK1,2) as well. Then NMN is transformed into NAD+ through NMNAT. *It is noted that most tissues, except the liver, don’t express the complete array of enzymes to synthesize NAD+ de novo.
Interestingly, inhibiting NAD+ synthesis and targeting NADase have shown positive implications for the treatment of cancer. This may be due to the difference in the main roles played by NAD+ synthetase and degrading enzymes. Enzymes involved in NAD+ synthesis mainly affect tumor internal biological behaviors, while NAD-degrading enzymes participate in the immunosuppressive tumor microenvironment. Here we summarize the detrimental outcomes of increased NAD+ production, the functions of NAD+ metabolic enzymes in creating an immunosuppressive TME, and discuss the progress and clinical translational potential of inhibitors for NAD+ synthesis and therapies targeting NADase.
2 NAD+ anabolism dysregulation and targeted therapy strategies in cancer
Since NAD+ is essential for mediating redox reactions in various metabolic pathways, malignant cells upregulate NAD+ biosynthesis enzyme genes to satisfy the high demands of proliferation and biomass production. A recent analysis has shown that healthy tissues from which NAD+ biosynthetic enzymes were derived determine whether the NAD+ salvage pathway is activated or the PH synthesis pathway is changed (6).
The expression of NAMPT is increased in urothelial carcinoma, breast cancer, stomach cancer, esophageal cancer, colorectal cancer (CRC), ovarian cancer, prostate cancer, glioblastoma, and melanoma. High expression levels of NAMPT are associated with unfavorable overall survival, advanced TNM stage, vascular invasion, and invasion depth (13–16). Furthermore, NAMPT is localized both intracellularly and extracellularly. Extracellular NAMPT (eNAMPT), is also known as visfatin or pre-B-cell colony-enhancing factor. Elevated serum eNAMPT levels indicate poor prognosis (17–21).
According to the upregulated NAD+ synthesis pathway, cancer cells can be divided into PH-amplified tumors, many of which show NAPRT amplification, and salvage-dependent tumors, which are characterized by NAMPT enhancer remodeling (6). PH-amplified cancers include ovarian cancer (22), prostate cancer, and pancreatic cancers (23), which are influenced by the increased expression of NAPRT (13–16, 24). In patients with CRC, a high level of NAPRT is associated with poor prognosis. Furthermore, CRC tissues have shown a correlation with NAPRT and NAMPT (14). On the other hand, gastric, colorectal, and renal carcinoma, along with various leukemia, have shown a low expression level of NAPRT (25). Mechanistically, NAPRT epigenetic expression is inhibited in certain tumors due to the methylation of its promoter (14). The occurrence of gene mutation, amplification, and methylation in NAPRT is more frequent than NAMPT (14, 25). In summary, NAPRT is a critical counterpart to NAMPT in NAD+ synthesis, but the role played by NAPRT is possibly underestimated and is rarely studied. The primary focus of most studies has been on the correlation between NAPRT and susceptibility to NAMPT inhibitors.
2.1 Metabolic reprogramming and oxidative stress
Intracellular NAMPT (iNAMPT) plays a pivotal role in the metabolism reprogramming of cancer cells. iNAMPT inhibition leads to impaired glycolytic flux, lactate production, and maximum mitochondrial respiratory capacity, eventually promoting energy abrogation (26). Inhibition of iNAMPT also impacts various metabolic pathways, such as the pathways for serine biosynthesis and one-carbon metabolism (27), amino acid metabolism (28), pentose phosphate pathway (29), lipid metabolism (30), purine and pyrimidine metabolism (28). Inhibiting NAMPT to suppress NAD+ production is a successful approach for modifying cancer cell metabolism and decreasing ATP levels (31). Likewise, NAPRT has a crucial function in controlling cellular levels of NAD+, OXPHOS, energy levels, protein production, and cellular dimensions. It is worth mentioning that the inhibition of NAPRT decreases OXPHOS in cancer cells that overexpress NAPRT and makes them more sensitive to NAMPT inhibitors (NAMPTi).
Oxidative stress results from an imbalance between reactive oxygen species (ROS) and cellular antioxidant capacity. Studies have demonstrated that iNAMPT enhances the antioxidant potential of cancer cells while reducing the harmful consequences of excessive accumulation of ROS (29, 32). In contrast, depletion, or inhibition of NAMPT resulted in antitumor effects by elevating levels of ROS in non-small cell lung cancer (NSCLC) (33), glioblastoma (34), prostate cancer (35), breast cancer (29), and colon cancer (32). In leukemia, NAMPTi induced ROS production, triggering cell death through mitochondria dysfunction and ATP loss (36).
Taken together, these discoveries offer a rationale for focusing on NAMPT/NAPRT as an innovative approach to trigger metabolic impairment resulting in cancer cell death.
2.2 The epithelial-mesenchymal transition (EMT) and cell stemness
Although cancer stem cells (CSCs) only constitute a small subpopulation with self-renewal properties, they are crucial not only to tumorigenesis but also to metastasis and therapy resistance. NAMPT has been described to enhance tumor aggressiveness by modulating the EMT and supporting cancer cell stemness (37, 38). NAMPT has been described to enhance tumor aggressiveness by modulating the EMT and supporting cancer cell stemness (37, 38). Importantly, the role of NAMPT in promoting EMT does not depend on its enzymatic activity (39). Visfatin/eNAMPT also induces EMT to promote cancer cell migration (39, 40). However, pharmacological NAMPT inhibition decreases the expression of SIRT1 and reverses the SIRT1−mediated EMT to inhibit the cell capacity for invasion and metastasis (41). Furthermore, there has been a recent report indicating a correlation between NAMPT activity and the characteristics of stemness in leukemia (42), glioma (43), colon cancer (44), and breast cancers (39).
NAMPTi decreases NAD+ levels and the self-renewal capacity of CSCs and inhibits tumorigenicity in glioblastoma (45). NAMPT suppression reversed the capability of cancer cells to dedifferentiate (46) and potentially decreased the CSC load in metastatic prostate cancer (47).
Mechanistically, NAMPT may modulate SIRT1 and PARP1 activity to regulate stem cell signaling pathways, thus influencing the EMT and tumor cell dedifferentiation (44). Hence, exploring the impact of NAMPT on the EMT and stemness in cancer cells can pave the way for the clinical application of NAMPT inhibitors, PARP inhibitors, and SIRT inhibitors.
2.3 Interactions with oncogenic signaling pathways
Extensive research shows that NAMPT engages in regulating oncogenic signaling pathways. NAMPT upregulation has been reported to be associated with the phosphorylation of AKT (48). NAMPT inhibition suppressed N-MYC expression and reduced the phosphorylation levels of AKT and GSK3 (49). Similarly, NAMPTi decreased the dephosphorylation rate of the EGFR, ERK1/2, MEK1/2, and Akt in NSCLC (50). AKT/PI3K and ERK/MAPK activation by visfatin/eNAMPT promote proliferation and prevent apoptosis in breast cancer, while AKT and ERK1/2 inhibition abrogated these effects (51). In addition, combining NAMPTi with MAPK inhibitors synergistically promoted cell death (52).
In addition, a connection between NAMPT and mTOR activation has been shown in various cancers. NAMPT inhibition suppressed mTOR signaling in multiple myeloma cells (52), hepatocarcinoma cells (53), and pancreatic cancer cells (54). mTOR inactivation induced by NAMPT inhibition may contribute to autophagic death and apoptosis, as reported for adult T-cell leukemia cells (55). Moreover, the effect of decreasing cell viability was more pronounced when NAMPT inhibitors were combined with an AMPK activator and an mTOR inhibitor (54). Further studies are needed to shed light on the mechanisms of action and to verify the potential effectiveness of combinations of NAMPTis and other targeted drugs against cancer.
2.4 Resistance to BRAF inhibitors and chemotherapy
Multiple selective BRAF inhibitors (BRAFis) have been used in combination with MEK inhibitors for the treatment of BRAF-mutated metastatic melanoma (56). However, the clinical benefits of using BRAFis have been limited by drug resistance. Several studies have revealed that increased NAD+ levels and high NAMPT expression were related to the development of drug resistance to BRAFis (57–59). High levels of both iNAMPT and visfatin/eNAMPT were expressed in BRAFi-resistant melanoma cells, while transcriptional downregulation of NAMPT was reported in BRAFi-sensitive cells (57–59). Furthermore, overexpressing NAMPT necessarily and sufficiently recapitulated the BRAFi-resistant phenotype plasticity in MM (59, 60). Nevertheless, pharmacological NAMPT inhibition induced energy abrogation and apoptosis to suppress cell growth in BRAFi-resistant MM cells (57). Thus, NAMPT may become an actionable target in BRAFi-resistant MM patients.
2.5 The regulation of DNA damage repair and sirtuin function
PARPs and sirtuins are NAD+-dependent consuming enzymes that are critical for cellular NAD+ degradation. PARPs participate mainly in DNA repair, while SIRTs constitute a family of deacetylase proteins. Given that NAMPT is involved in the generation of NAD+, NAMPT has an impact on DNA damage repair and sirtuin function.
A functional relationship between NAMPT and PARPs has been proposed based on evidence showing that NAMPTi increased DNA damage because of the loss of PARP activity in Ewing sarcoma and pediatric acute lymphoblastic leukemia (61, 62). The synergistic combination of NAMPT and PARP inhibition increased the inhibitory effect on tumor cells (63, 64). However, other evidence has suggested that PARP1 activation enhanced the antitumor effects of NAMPTi, suggesting a complex and controversial relationship between PARP and NAMPT (36, 65). PARP1 is the best-characterized PARP that is actively involved in chromatin organization and the DNA repair pathway (12). NAMPT is located in both the nucleus and cytoplasm. It has been recently shown that NAMPT and GAPDH form a stable complex that mediates the translocation of NAMPT. NAMPT translocation contributes to the nuclear NAD+ pool, which increases PARP1 activity and response to stressors, such as DNA damage induced by UV light (66, 67). All these studies lay the groundwork for the preclinical development of NAMPTis with modulators of PARP activity.
SIRTs are a group of deacetylase proteins that make use of NAD+ as a coenzyme. Notably, SIRTs are upregulated along with NAMPT in several solid cancers, including breast, prostate, gastric, colorectal, liver, and pancreatic cancers (21). Inhibition of NAMPT led to decreased expression and activity of SIRT1 (41, 68), SIRT2 (69), and SIRT3 (70). Several studies have reported that c-MYC increased the expression and activity of SIRT1 mediated via transcriptional activation of NAMPT to drive tumor cell proliferation and progression (71–73). Moreover, in addition to iNAMPT, visfatin/eNAMPT regulated the expression levels of SIRT1 and P53 to promote cell proliferation (74, 75).
Interestingly, it has been noted that the anticancer effects of NAMPTis in pancreatic cancer are not influenced by either SIRT1 or PARP1, indicating that the impact of NAMPT on PARP1 or sirtuins may vary depending on the type of cancer. Determining the cancer types that are more responsive to NAMPTis is crucial for the future clinical translation of NAMPTis.
3 Enzymes in NAD+ metabolism immunomodulate the tumor microenvironment
3.1 NAMPT and NAPRT
Notably, in contrast to its intracellular form, visfatin/eNAMPT can function as an immunomodulatory cytokine (76, 77). eNAMPT is present in the extracellular space and facilitates strong NF-κB transcriptional stimulation through its interaction with Toll-like receptor 4 (TLR4) (78). eNAMPT induces the release of TNF-α, IL-1α, IL-1β, IL-6, and IL-8, and enhances the expression of the costimulatory molecules CD40, CD54, and CD80 (79–81). Interestingly, extracellular NAPRT (eNAPRT) is structurally and functionally similar to eNAMPT. By binding to TLR4, eNAPRT activates the inflammasome and NF-κB pathway, leading to the release of inflammatory cytokines. Moreover, eNAPRT induces the differentiation of monocytes into macrophages through the enhancement of transcription and secretion of macrophage colony-stimulating factor (M-CSF/CSF1). eNAMPT, when bound to C-C chemokine receptor type 5 as an antagonist, hinders Rantes-dependent calcium signaling in melanoma. The cytokine-like properties appear to be unrelated to its enzymatic function (82).
So far, it has been demonstrated that NAMPT affects myeloid cells as well in the TME. eNAMPT-primed M2 macrophages support the survival of leukemic cells and suppress T-cell responses by increasing the activity of the PD-1/PD-L1 axis. Additionally, NAMPT exerts its effects on myeloid-derived suppressor cells (MDSCs) (83). NAMPT regulates the mobilization of MDSCs and boosts their generation of inhibitory nitric oxide. Inhibiting NAMPT pharmacologically leads to synergistic effects with an anti-PD-1 antibody and releases MDSC restraints on antitumor immunity (84, 85). Moreover, NAMPT, which is downstream of G-CSF (CSF3) receptor signaling, promotes the tumorigenic conversion of tumor-associated neutrophils (TANs), enhancing angiogenesis via SIRT1 activity. TANs from head-and-neck cancer and melanoma patients have been characterized by high NAMPT expression (86).
The mechanisms behind the immunomodulation effects of NAMPT on TME are still being studied. Further studies are needed to shed light on the role of NAMPT in establishing the immunosuppressive tumor microenvironment.
3.2 SIRTs
SIRTs play contradictory and complicated roles in regulating cancer cell growth and proliferation. SIRTs may act as promoters or suppressors in different kinds of cancers (87). Recently, SIRTs have also been reported to play potential roles in antitumor immunity. In an ovarian cancer model, artesunate, a medication used to treat malaria, induced Th1 cell differentiation from CD4+ T cells and showed enhanced proapoptotic effects on ovarian cancer cells by upregulating miR-142 and suppressing SIRT1 levels (88). Furthermore, the overexpression of SIRT2 in TILs of cancer patients has been linked to a negative outcome in terms of their response to immunotherapy. In contrast, SIRT2 inhibition during T-cell activation triggers the hyperacetylation of various metabolic enzymes, resulting in elevated aerobic glycolysis and OXPHOS. In other words, the absence of SIRT2 resulted in hyperreactive T cells with enhanced antitumor activity that overcame the immunological and metabolic barriers within suppressive tumor microenvironments (89). Additionally, there have been reports indicating that SIRT5 has a regulatory function in the differentiation of CD4+ regulatory T (Treg) cells and T helper 1 (Th1) cells during colorectal tumorigenesis (90). SIRT6 increased the levels of Ca2+-mobilizing second messengers and enhanced the expression of proinflammatory cytokines in pancreatic cancer, linking SIRT6 with a cancer cell proinflammatory phenotype and migratory propensity (91). Finally, SIRT7 inhibited the expression of PD-L1 by forming a complex with MEF2D, suggesting that strategies to modulate the activity of SIRT7 may potentially enhance the efficacy of immunotherapies in hepatocellular carcinoma (92). Further investigation is needed to explore the role of SIRT in the TME, despite some previous studies on the regulatory impact of SIRTs on T-cell metabolism and function (93).
3.3 Glycohydrolases: CD38, CD157 and SARM1
CD38, a transmembrane protein, is widely expressed in endothelial cells (94), fibroblasts (95), smooth muscle cells (96), and various immune cells (97). The main biological role of CD38 is hydrolyzing β-NAD+ into NAM and ADPR. It shows poor ADP-ribosyl cyclase activity, thereby inefficiently producing a minor amount of cyclic ADPR (cADPR). Interestingly, it catalyzes the hydrolysis of cADPR to ADP-ribose (ADPR) (98–100). Both ADPR and cADPR serve as second messengers in the modulation of intracellular calcium signaling (101). CD38/ADPR/Ca2+ signaling immunomodulates neutrophil chemotaxis, lymphocyte proliferation, and T-cell activation (102, 103). On the other hand, in the TME, CD38 participates in the production of noncranial extracellular adenosine (eADO), relying on NAD+ as a substrate to generate ADPR. CD203a processes ADPR into extracellular AMP (eAMP). Then, CD73 catalyzes the hydrolysis of AMP to generate extracellular ADO (eADO). Through its purinergic receptor binding, especially A2A and A2B, adenosine is important in the suppression of both innate and adaptive immune responses. Adenosine restrains the antitumor immune response by inhibiting the anticancer activity of CD8+ T cells and NK cells and recruiting MDSCs and Treg cells. ADO/A2A signaling suppresses T cells producing IFN γ, TNF, granzymes, and perforin (104). There has been speculation that CD38, by participating in the regulation of NAD+ and adenosine homeostasis, could potentially function as an immune checkpoint (104, 105).
CD157 is generated in two forms: in a soluble form (106) and as a glycosylphosphatidylinositol (GPI)-anchored glycoprotein. Similar to its paralog CD38, CD157 shows both glycohydrolase and weak ADP-ribosyl cyclase functions (107–109). It converts β-NAD+ into NAM, substantial ADPR, and a small amount of cADPR. Despite being less efficient than CD38, the ADP-ribosyl cyclase function of CD157 significantly improves under acidic conditions and with the presence of Zn2+ and Mn2+ (108, 109). Considering its role in ADPR generation, CD157 is potentially involved in the CD38/CD157/CD203a/CD73 adenosinergic signaling, increasing ADO levels to favor immunosuppressive TME formation (110). Notably, although it lacks intracellular domains, CD157 in the GPI-anchored form functions as a receptor. The interaction between CD157 and fibronectin plays a crucial role in the formation of a complex with integrins, facilitating the creation of a connected network that transmits signals both inside and outside the cell. This process greatly enhances cell adhesion to the extracellular matrix, promotes cell migration, and ensures cell survival (111, 112). The role played by CD157 as a receptor needs to be further analyzed, particularly its role in regulating the immunosuppressive TME, which has been largely uncharacterized.
SARM1 was initially classified as a negative regulator of TRIF-dependent TLR signaling (113). SARM is mainly expressed in neurons and T lymphocytes, mediating cell death and promoting the neuronal inflammatory response (114–117). SARM1 has recently been designated a new class of NAD+ glycohydrolase in axonal degeneration (118–120). The intrinsic NADase activity of SARM1 depends on the Toll/interleukin receptor domain converting NAD+ to ADPR, cADPR, and NAM, with NAM inhibiting the enzyme in a feedback loop. Notably, NAM also suppresses other NAD+-consuming enzymes (121, 122). Furthermore, its enzymatic activity is essential for bacterial innate immunity (123). However, in contrast to its undisputed pivotal role in axonal degeneration, the immunoregulatory role of NAM remains unclear.
4 The development of drugs targeting NAD+ metabolism
4.1 Targeting NAD+ synthesis
Considering the critical role played by NAMPT in the NAD+ salvage pathway, an increasing number of pharmacological NAMPTis have been developed; these include specific NAMPT inhibitors (APO866, GMX1778, GMX1777, and OT-82) and dual NAMPT inhibitors (KPT-9274). Here, we summarize the most extensively studied NAMPTi and describe its antitumor effects in vitro and in vivo, as well as progress in its clinical applications (124, 125) (Tables 1, 2).
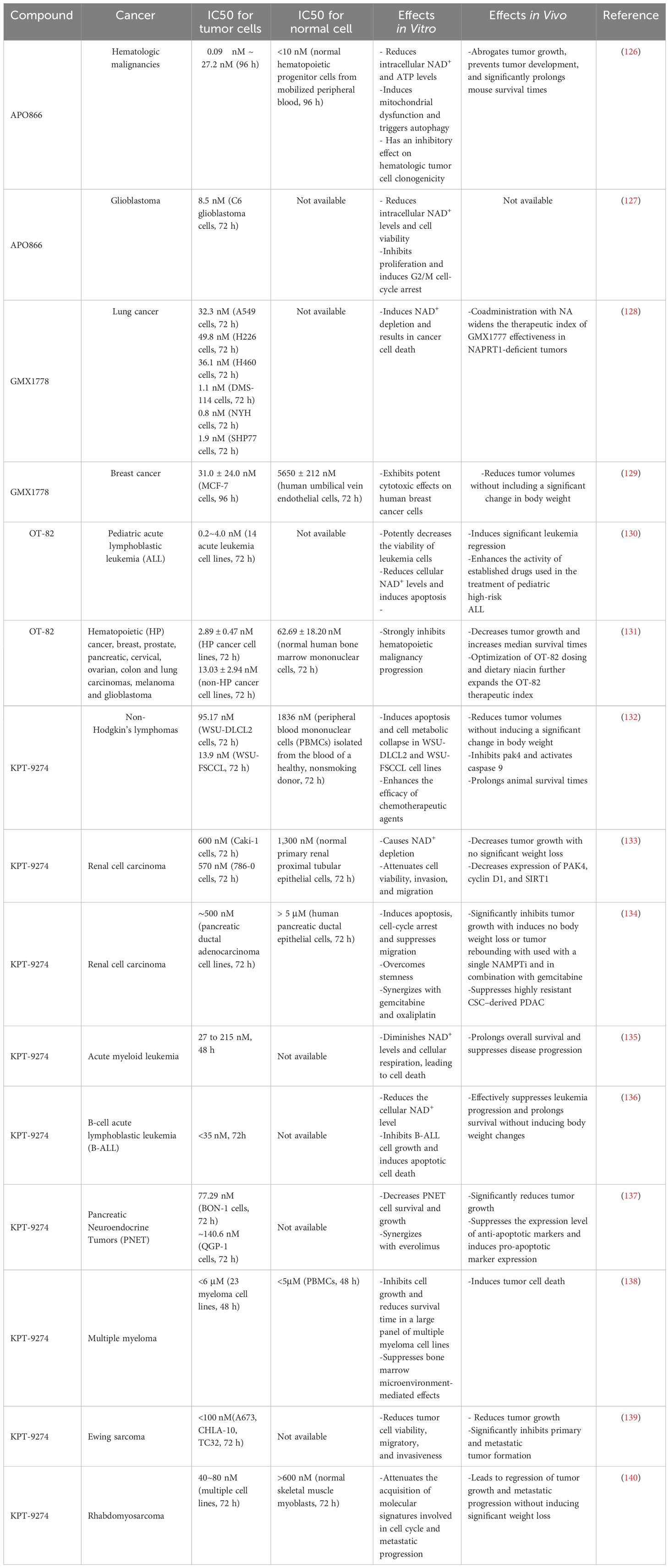
Table 1 Preclinical studies of NAMPT inhibitors in cancer.
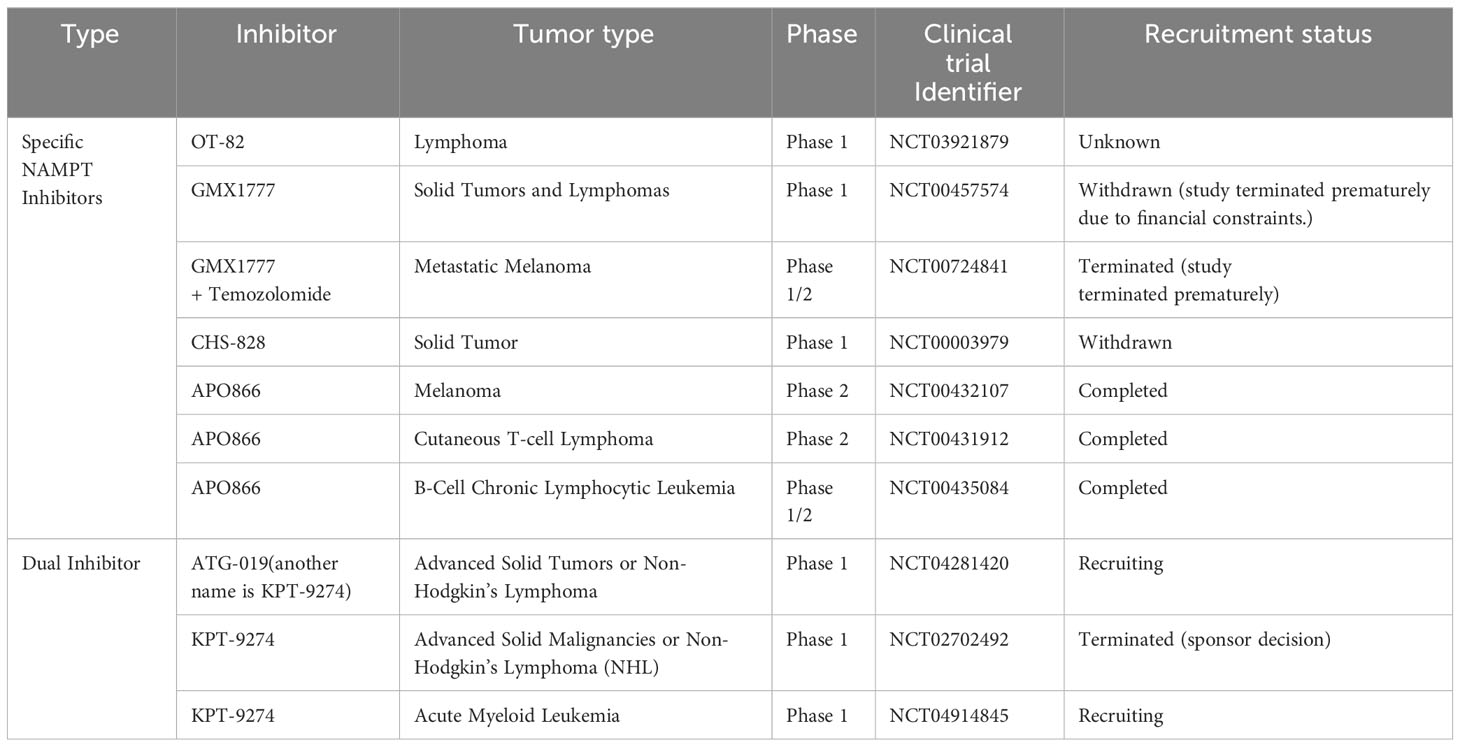
Table 2 Clinical trials with NAMPTis used to treat cancer.
APO866 (FK866), was the initial compound identified as an NAMPTi. It inhibits cancer cell proliferation and induces tumor regression (126, 127). GMX1778 (CHS-828) and OT-82 exert effects similar to each other, highlighting their beneficial effects in the treatment of solid cancers and hematological malignancies (128–131). Interestingly, KPT-9274 (also called ATG-019), which targets both NAMPT and serine/threonine p21-activated kinase 4 (PAK4), also exerts potent anticancer effects (132–140). A Phase I clinical trial is currently enrolling patients diagnosed with lymphoma or solid tumors to assess the efficacy of PAK4 inhibition.
Despite promising results in preclinical studies, NAMPTi monotherapy led to few objective tumor responses in Phase I/II clinical trials (141–145). These outcomes may indicate that before NAMPTi application, biomarkers may be needed to select patients who may benefit from NAMPT inhibitors. Indeed, due to their dependency on the NAD+ salvage pathway, salvage-dependent cancers with NAPRT deficiency are sensitive to treatment with NAMPT inhibitors (128, 146). However, PH-dependent cancers with upregulated NAPRT expression are resistant to NAMPT inhibitors (22). An increasing number of studies show that tumors that rely on NAPRT for NAD+ synthesis are initially resistant to NAMPT inhibitors but become sensitive to NAMPT inhibitors after NAPRT downregulation, illustrating the need for NAPRT inhibitor development (22, 147, 148). Unfortunately, very few NAPRT inhibitors have been reported to date. Among the few NAPRT inhibitors, 2-Hydroxynicotinic acid (2-HNA) can sensitize PH-amplified pancreatic and ovarian cancer cells to NAMPT inhibitors. Other compounds with NAPRT-inhibitory activity include several nonsteroidal anti-inflammatory compounds (149–151). Recently, several new small-molecule NAPRT inhibitors (NAPRTis) have been identified in silico. These NAPRTis administered with the NAMPTi FK866 exerted a synergistic effect, suggesting the therapeutic potential of these compounds (152, 153). However, more studies are required to verify the efficacy of these NAPRT inhibitors.
In addition, to overcome the limitations of available NAMPTis, targets for drug development such as antibody-drug conjugates (ADCs), dual-target inhibitors, and proteolysis-targeting chimera (PROTAC) technology may increase the efficacy and sensitivity of NAMPT inhibitors. For instance, based on PROTAC technology, the NAMPT PROTAC A7 inhibited tumor-infiltrating MDSCs and boosted antitumor efficacy by degrading both iNAMPT and eNAMPT (85). Similarly pharmacological NAMPT inhibitors administered with anti-PD-1 antibodies exhibited synergy and released MDSC suppression to induce antitumor immune responses (84). All of these studies indicate that NAMPTis are promising anticancer drug candidates when used in combination with immunotherapy to enhance their effects.
4.2 Targeting NADase
4.2.1 SIRTs
SIRTs play contradictory and complicated roles in the regulation of cancer cell growth and proliferation. SIRTs may function as promoters or suppressors in different kinds of cancers (87). SIRTs, both activators and inhibitors, have been reported to exert anticancer effects. As mentioned above, SIRTs participate in the modulation of the TME. In this review, we focus mainly on the roles of SIRT actions in antitumor immunity. SIRT inhibitors may improve anticancer immunotherapy (Table 3). In particular, SIRT7 disruption enhanced the efficacy of PD-1 blockade therapy in hepatocellular carcinoma cells. Unfortunately, since no specific SIRT7 inhibitors have been developed, researchers use CRISPR/Cas9 to knock out SIRT7 to mimic the effects of SIRT7 inhibitors. We firmly believe that other SIRT modulators may play roles in immunotherapy and can increase the efficacy of immunotherapy. However, more research is still needed to further understand the roles played by SIRTs in antitumor immunity and develop more specific SIRT inhibitors.
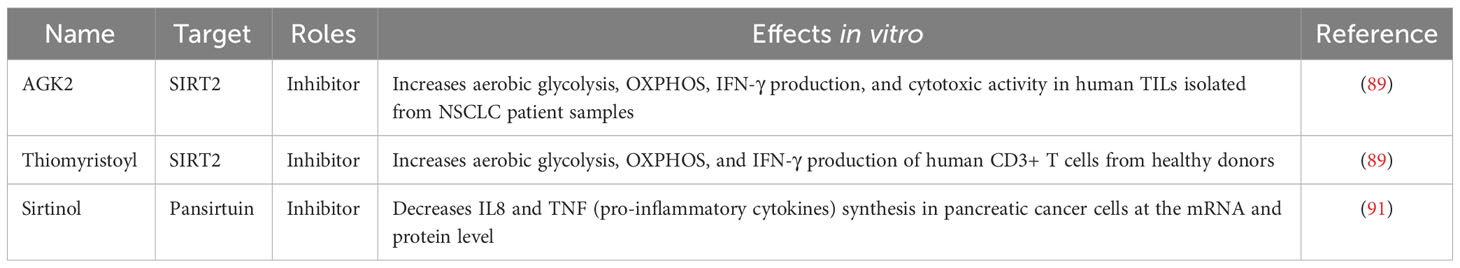
Table 3 Studies on the inhibition mediated by SIRTs in anticancer immunity.
4.2.2 CD38 and CD157
Multiple myeloma(MM) cells express CD38 in high levels, whereas the expression levels of CD38 are lower in normal myeloid and lymphoid cells as well as some non-hematopoietic tissues, driving the development of anti-CD38 monoclonal antibodies (mAbs) in the treatment of MM. Also, anti-CD38 mAbs have been shown to enhance T cell function and suppress Treg cell proliferation in the tumor microenvironment in vivo. In November 2015, Daratumumab, the first human CD38 IgG1κ monoclonal antibody, was approved by the Food and Drug Administration (FDA) as a monotherapy for patients with MM who have received ≥3 prior therapies. Daratumumab has shown significant clinical activity in patients with relapsed or refractory MM when used as either a monotherapy (154) or in combination with lenalidomide and dexamethasone (155). Targeting CD38 by daratumumab induced robust increases in helper and cytotoxic T cell numbers and depleted CD38+ Tregs (156).
Another well-studied CD38 mAb is isatuximab (SAR650894). It induces potent proapoptotic activity in MM cells and elicits complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC) mediated by macrophages (157). Isatuximab also preferentially blocks the induction and function of immunosuppressive CD38+ Tregs and restores the immune effector function of NK and CD8+ T effector cells in MM (158). CD38 mAbs, including daratumumab, isatuximab, felzartamab (MOR202), and Ab79 (Millenium/Takeda), show similar mechanisms by binding cells and inducing ADCC. However, they differ in their abilities to inhibit CD38 as well as to mediate apoptosis, ADCP, and especially CDC (159). Other strategies based on the inhibition of CD38 are being developed and evaluated, such as antibody-drug conjugates (ADCs), antibody-recruiting molecules (ARMs), engineered toxin bodies (ETBs), bispecific T-cell engagers (BiTEs) and XmAb Fc domain technology. Many preclinical studies and clinical trials evaluating CD38 inhibition are ongoing or have been completed with patients harboring either hematological malignancies or solid tumors (Tables 4, 5).
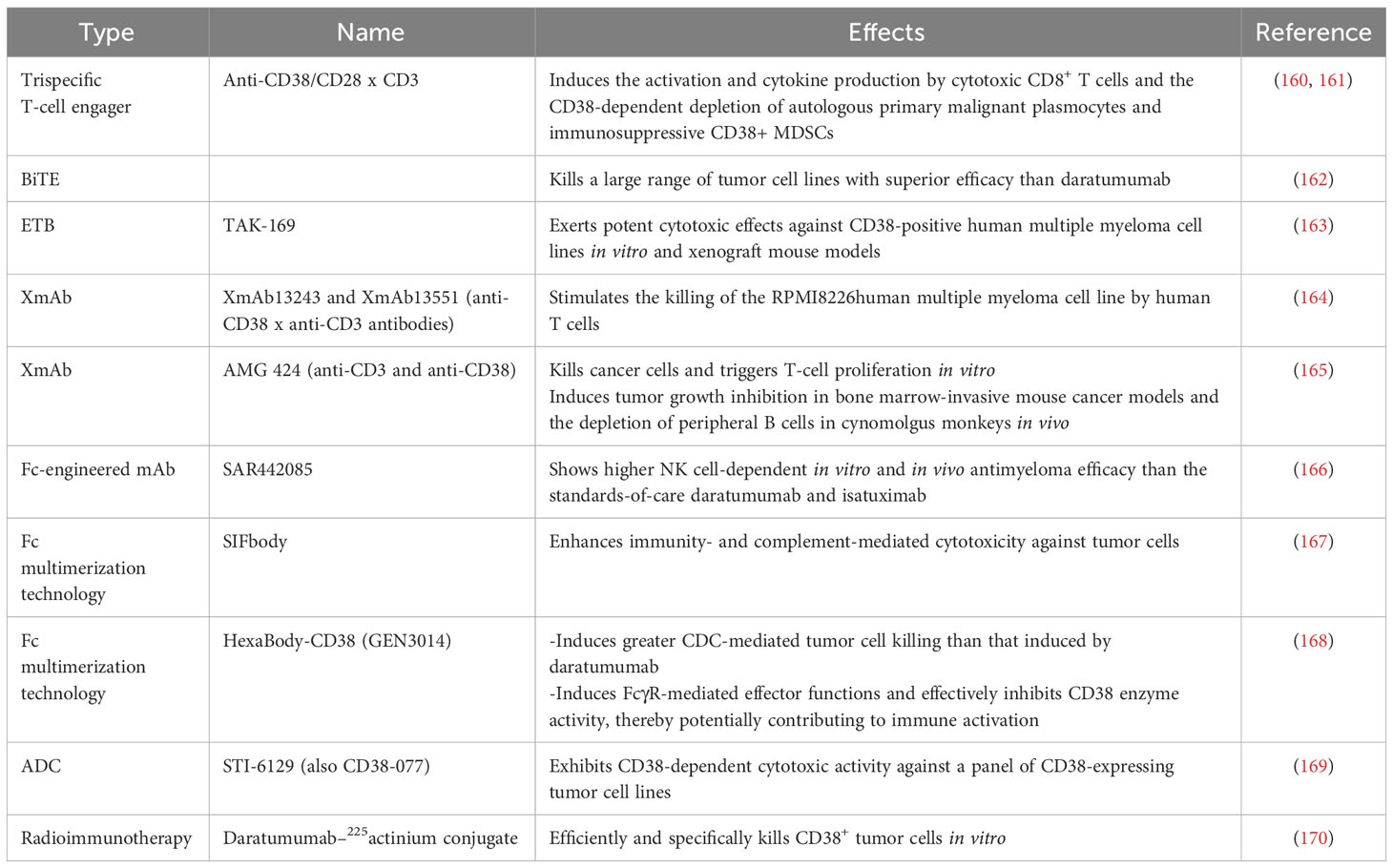
Table 4 Preclinical studies evaluating the inhibition of CD38 in anticancer immunity.
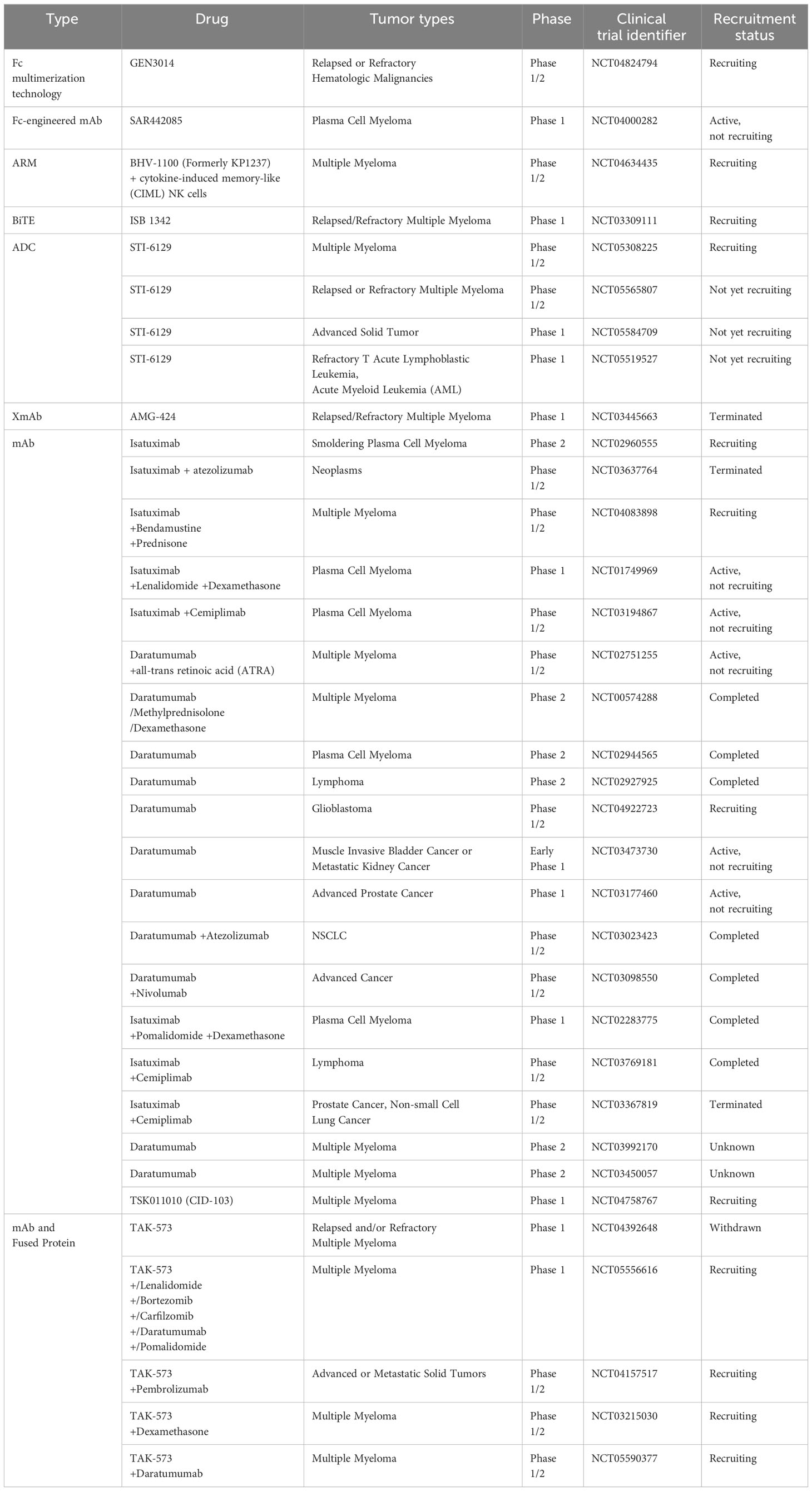
Table 5 Clinical trials to evaluate drug effects on CD38 inhibition in cancer treatments.
Furthermore, CD38, as a NADase, regulates a wide range of NAD-dependent cellular processes as mentioned above. Although most anti-CD38 mAbs have been reported to mainly function via their cytolytic effects, isatuximab and daratumumab also inhibit enzymatic activities of CD38 to some extent (154, 171, 172). Their therapeutic effects are unknown related to inhibition of CD38 enzymatic activity as well. In fact, given the role of CD38 metabolism in solid tumor microenvironment, metabolism reprogramming of NAD+ regulation via CD38 inhibition has attracted attention as a strategy for immunotherapy, either with anti-CD38 antibodies alone or in combination with immunomodulatory drugs. For instance, in a preclinical melanoma model, Chatterjee et al. demonstrated that blocking CD38 expression on T cells boosts NAD+ levels and improves the efficacy of adoptive transferred T cells (173). Moreover, targeting the eADO pathway can increase the antitumor effects of drugs through various mechanisms, such as by enhancing T-cell and natural killer cell functions, promoting antigen presentation, and inhibiting the immunosuppressive effects of MDSC (104). In particular, we identified CD73, a nucleotidase in the eADO pathway, as an immunotherapy response predictor and prognostic biomarker in head and neck squamous cell carcinoma (174). Similarly, CD38, which also participates in the adenosinergic pathway, has been reported to be an effective predictor of anti-PD-1 antibody-based checkpoint immunotherapy responses in hepatocellular carcinoma (175).
In addition, apart from monoclonal antibodies, there are currently more than 200 small-molecule inhibitors that suppress the enzymatic activity of CD38 (176). Although these small molecule inhibitors of CD38 have not been reported in the literature for their use in tumor treatment, we strongly believe that these inhibitors will at least become powerful tools for unveiling the role of the enzymatic function of CD38 in tumors and solid TME.
Finally, although the regulatory mechanism underlying CD157 effects in the immunosuppressive TME remains unclear, an Fc-optimized antibody against BST1/CD157, MEN1112/OBT357NF, exhibited effective ADCC in vitro (177). A Phase 1 clinical trial to evaluate MEN1112 in patients with relapsed or refractory acute myeloid leukemia (NCT02353143) is ongoing.
5 Conclusions
In summary, the enzymes involved in NAD+ metabolism are extensively involved in various intracellular signaling pathways and play a nonnegligible role in the development of immunosuppressive TME.
On the one hand, upregulated NAD+ anabolism contributes to metabolic reprogramming, the EMT, cell stemness, etc. Pharmacological inhibition of key enzymes involved in NAD+ production, mainly NAMPTis and a few NAPRTis, that impair NAD+ generation has exhibited promising results in cancer cell lines and preclinical studies. Unfortunately, NAMPTi monotherapy has been proven to be limited in clinical trials. Considering that different tumors vary in their dependence on the NAD+ synthesis pathway, the development of only NAMPT inhibitors has been an insufficient approach to date. Potent-specific NAPRT inhibitors need to also be developed. Selecting NAMPTis or NAPRTis according to the dependence of cancers on NAD+ synthesis may further increase the effectiveness of targeted NAD+ synthesis therapy. In addition, extracellular NAMPT and NAPRT are involved in regulating the immunosuppressive TME; therefore, specifically targeting extracellular NAMPTis and NAPRTis may increase the effectiveness of NAD+ inhibition therapy and potentially enhance the efficacy of immunotherapy.
In addition, NAD-degrading enzymes, especially CD38, play critical roles in tumor cell immune evasion via the adenosinergic pathway. CD38 inhibition has attracted attention as a strategy for immunotherapy, either with anti-CD38 antibodies alone or in combination with other immunomodulatory drugs. Indeed, many preclinical studies and clinical trials to evaluate CD38 inhibition in both solid tumors and hematological malignancies are ongoing or have been completed. Targeting CD38 is considered to have been a breakthrough in multiple myeloma immunotherapies. But from another perspective, there are still many unresolved issues in the future application of anti-CD38 monoclonal antibodies. The first is whether, in addition to isatuximab and daratumumab, other anti-CD38 monoclonal antibodies inhibit CD38 enzymatic activity. Also, the impact of CD38 enzymatic activity on the immune system and tumor microenvironment still needs to be elucidated, besides participating in adenosine pathway. Secondly, the CD38 monoclonal antibody inhibits the extracellular NADase activity of CD38 and increases the level of extracellular NAD+. On the other hand, the increase in intracellular NAD+ is beneficial to tumor growth. Therefore, it still needs to be clarified whether the CD38 monoclonal antibody leads to an increase in the intracellular NAD+ level and whether it plays a double-edged sword role in tumor treatment. Finally, small molecule inhibitors of CD38 can inhibit the enzymatic activity of CD38. Using these tools, we can better understand the role of CD38 enzymatic activity in tumors. Also, apart from CD38, other NADases may also promote tumor cell immune escape, but to understand the specific role they play, further research is still needed.
Author contributions
JY: Conceptualization, Resources, Writing – original draft, Writing – review & editing. SC: Resources, Visualization, Writing – review & editing. ZZ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The National Natural Science Foundation of China (82072612, 81930065, 82173128) and the CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-036) provide funding for this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell (2021) 81(4):691–707.e6. doi: 10.1016/j.molcel.2020.12.012
2. Giblin W, Bringman-Rodenbarger L, Guo AH, Kumar S, Monovich AC, Mostafa AM, et al. The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics. J Clin Invest (2021) 131(12):e138926. doi: 10.1172/JCI138926
3. Bilokapic S, Suskiewicz MJ, Ahel I, Halic M. Bridging of DNA breaks activates PARP2-HPF1 to modify chromatin. Nature (2020) 585(7826):609–13. doi: 10.1038/s41586-020-2725-7
4. Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab (2018) 27(5):1067–80.e5. doi: 10.1016/j.cmet.2018.03.018
5. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol (2019) 20(1):50–63. doi: 10.1038/s41590-018-0255-3
6. Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature (2019) 569(7757):570–5. doi: 10.1038/s41586-019-1150-2
7. Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer (2012) 12(11):741–52. doi: 10.1038/nrc3340
8. Poniewierska-Baran A, Warias P, Zgutka K. Sirtuins (SIRTs) as a novel target in gastric cancer. Int J Mol Sci (2022) 23(23):15119. doi: 10.3390/ijms232315119
9. Gao L, Du X, Li J, Qin FX. Evolving roles of CD38 metabolism in solid tumour microenvironment. Br J Cancer (2022) 128(4):492–504. doi: 10.1038/s41416-022-02052-6
10. Wo YJ, Gan ASP, Lim X, Tay ISY, Lim S, Lim JCT, et al. The roles of CD38 and CD157 in the solid tumor microenvironment and cancer immunotherapy. Cells (2019) 9(1):26. doi: 10.3390/cells9010026
11. Morale MG, Tamura RE, Cintra R, Araújo NM, Villa LL. TLR4 and SARM1 modulate survival and chemoresistance in an HPV-positive cervical cancer cell line. Sci Rep (2022) 12(1):6714. doi: 10.1038/s41598-022-09980-6
12. Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discovery (2020) 19(10):711–36. doi: 10.1038/s41573-020-0076-6
13. Ji C, Cong R, Wang Y, Wang Y, Zhang Q, Zhou X, et al. Relationship between NAMPT/PBEF/visfatin and prognosis of patients with Malignant tumors: a systematic review and meta-analysis. Ann Transl Med (2019) 7(23):785. doi: 10.21037/atm.2019.11.32
14. Li XQ, Lei J, Mao LH, Wang QL, Xu F, Ran T, et al. NAMPT and NAPRT, key enzymes in NAD salvage synthesis pathway, are of negative prognostic value in colorectal cancer. Front Oncol (2019) 9:736. doi: 10.3389/fonc.2019.00736
15. Guo Q, Han N, Shi L, Yang L, Zhang X, Zhou Y, et al. NAMPT: A potential prognostic and therapeutic biomarker in patients with glioblastoma. Oncol Rep (2019) 42(3):963–72. doi: 10.3892/or.2019.7227
16. Lin T-C. Updated functional roles of NAMPT in carcinogenesis and therapeutic niches. Cancers (2022) 14(9):2059. doi: 10.3390/cancers14092059
17. Sun Y, Zhu S, Wu Z, Huang Y, Liu C, Tang S, et al. Elevated serum visfatin levels are associated with poor prognosis of hepatocellular carcinoma. Oncotarget (2017) 8(14):23427–35. doi: 10.18632/oncotarget.15080
18. Yang J, Zhang K, Song H, Wu M, Li J, Yong Z, et al. Visfatin is involved in promotion of colorectal carcinoma Malignancy through an inducing EMT mechanism. Oncotarget (2016) 7(22):32306–17. doi: 10.18632/oncotarget.8615
19. Hung AC, Lo S, Hou M-F, Lee Y-C, Tsai C-H, Chen Y-Y, et al. Extracellular visfatin-promoted Malignant behavior in breast cancer is mediated through c-Abl and STAT3 activation. Clin Cancer Res (2016) 22(17):4478–90. doi: 10.1158/1078-0432.CCR-15-2704
20. Zhou SJ, Bi TQ, Qin CX, Yang XQ, Pang K. Expression of NAMPT is associated with breast invasive ductal carcinoma development and prognosis. Oncol Lett (2018) 15(5):6648–54. doi: 10.3892/ol.2018.8164
21. Dalamaga M, Christodoulatos GS, Mantzoros CS. The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: Diagnostic and therapeutic perspectives and challenges. Metabolism (2018) 82:72–87. doi: 10.1016/j.metabol.2018.01.001
22. Piacente F, Caffa I, Ravera S, Sociali G, Passalacqua M, Vellone VG, et al. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors, and DNA repair. Cancer Res (2017) 77(14):3857–69. doi: 10.1158/0008-5472.CAN-16-3079
23. Audrito V, Messana VG, Deaglio S. NAMPT and NAPRT: two metabolic enzymes with key roles in inflammation. Front Oncol (2020) 10:358. doi: 10.3389/fonc.2020.00358
24. Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in Malignancy: a review. Genes Cancer (2013) 4(11-12):447–56. doi: 10.1177/1947601913507576
25. Duarte-Pereira S, Pereira-Castro I, Silva SS, Correia MG, Neto C, da Costa LT, et al. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget (2016) 7(2):1973–83. doi: 10.18632/oncotarget.6538
26. Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res (2014) 20(1):120–30. doi: 10.1158/1078-0432.CCR-13-0150
27. Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP, et al. The NAD(+) salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep (2018) 24(9):2381–91.e5. doi: 10.1016/j.celrep.2018.07.086
28. Tolstikov V, Nikolayev A, Dong S, Zhao G, Kuo MS. Metabolomics analysis of metabolic effects of nicotinamide phosphoribosyltransferase (NAMPT) inhibition on human cancer cells. PLoS One (2014) 9(12):e114019. doi: 10.1371/journal.pone.0114019
29. Hong SM, Park CW, Kim SW, Nam YJ, Yu JH, Shin JH, et al. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene (2016) 35(27):3544–54. doi: 10.1038/onc.2015.415
30. Bowlby SC, Thomas MJ, D'Agostino RB Jr., Kridel SJ. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One (2012) 7(6):e40195. doi: 10.1371/journal.pone.0040195
31. Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PWK, et al. NAD+ salvage pathway in cancer metabolism and therapy. Pharmacol Res (2016) 114:274–83. doi: 10.1016/j.phrs.2016.10.027
32. Hong SM, Hwang SW, Wang T, Park CW, Ryu YM, Jung JH, et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci (2019) 110(2):629–38. doi: 10.1111/cas.13886
33. Xiao Y, Kwong M, Daemen A, Belvin M, Liang X, Hatzivassiliou G, et al. Metabolic response to NAD depletion across cell lines is highly variable. PLoS One (2016) 11(10):e0164166. doi: 10.1371/journal.pone.0164166
34. Feng J, Yan PF, Zhao HY, Zhang FC, Zhao WH, Feng M. Inhibitor of nicotinamide phosphoribosyltransferase sensitizes glioblastoma cells to temozolomide via activating ROS/JNK signaling pathway. BioMed Res Int (2016) 2016:1450843. doi: 10.1155/2016/1450843
35. Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene (2011) 30(8):907–21. doi: 10.1038/onc.2010.468
36. Cloux AJ, Aubry D, Heulot M, Widmann C, ElMokh O, Piacente F, et al. Reactive oxygen/nitrogen species contribute substantially to the antileukemia effect of APO866, a NAD lowering agent. Oncotarget (2019) 10(62):6723–38. doi: 10.18632/oncotarget.27336
37. Navas LE, Carnero A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct Target Ther (2021) 6(1):2. doi: 10.1038/s41392-020-00354-w
38. Heske CM. Beyond energy metabolism: exploiting the additional roles of NAMPT for cancer therapy. Front Oncol (2019) 9:1514. doi: 10.3389/fonc.2019.01514
39. Soncini D, Caffa I, Zoppoli G, Cea M, Cagnetta A, Passalacqua M, et al. Nicotinamide phosphoribosyltransferase promotes epithelial-to-mesenchymal transition as a soluble factor independent of its enzymatic activity *. J Biol Chem (2014) 289(49):34189–204. doi: 10.1074/jbc.M114.594721
40. Cheng G, Liu C, Sun X, Zhang L, Liu L, Ouyang J, et al. Visfatin promotes osteosarcoma cell migration and invasion via induction of epithelial-mesenchymal transition. Oncol Rep (2015) 34(2):987–94. doi: 10.3892/or.2015.4053
41. Zhang B, Shi D, Zhang X, Liang G, Liu W, Qiao S. FK866 inhibits the epithelial−mesenchymal transition of hepatocarcinoma MHCC97−H cells. Oncol Lett (2018) 16(6):7231–8. doi: 10.3892/ol.2018.9541
42. Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, et al. Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell (2020) 27(5):748–64.e4. doi: 10.1016/j.stem.2020.07.021
43. Lucena-Cacace A, Umeda M, Navas LE, Carnero A. NAMPT as a dedifferentiation-inducer gene: NAD(+) as core axis for glioma cancer stem-like cells maintenance. Front Oncol (2019) 9:292. doi: 10.3389/fonc.2019.00292
44. Lucena-Cacace A, Otero-Albiol D, Jiménez-García MP, Muñoz-Galvan S, Carnero A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through sirt1 and PARP. Clin Cancer Res (2018) 24(5):1202–15. doi: 10.1158/1078-0432.CCR-17-2575
45. Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y, et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci U S A (2016) 113(51):E8247–e56. doi: 10.1073/pnas.1610921114
46. Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, Tabibzadeh S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S–Nampt. Exp Cell Res (2015) 330(1):135–50. doi: 10.1016/j.yexcr.2014.09.027
47. Mazumder S, Mitra Ghosh T, Mukherjee UK, Chakravarti S, Amiri F, Waliagha RS, et al. Integrating pharmacogenomics data-driven computational drug prediction with single-cell RNAseq to demonstrate the efficacy of a NAMPT inhibitor against aggressive, taxane-resistant, and stem-like cells in lethal prostate cancer. Cancers (2022) 14(23):6009. doi: 10.3390/cancers14236009
48. Ge X, Zhao Y, Dong L, Seng J, Zhang X, Dou D. NAMPT regulates PKM2 nuclear location through 14-3-3ζ: Conferring resistance to tamoxifen in breast cancer. J Cell Physiol (2019) 234(12):23409–20. doi: 10.1002/jcp.28910
49. Vallejo FA, Sanchez A, Cuglievan B, Walters WM, De Angulo G, Vanni S, et al. NAMPT inhibition induces neuroblastoma cell death and blocks tumor growth. Front Oncol (2022) 12:883318. doi: 10.3389/fonc.2022.883318
50. Okumura S, Sasaki T, Minami Y, Ohsaki Y. Nicotinamide phosphoribosyltransferase: A potent therapeutic target in non-small cell lung cancer with epidermal growth factor receptor-gene mutation. J Thorac Oncol (2012) 7(1):49–56. doi: 10.1097/JTO.0b013e318233d686
51. Gholinejad Z, Kheiripour N, Nourbakhsh M, Ilbeigi D, Behroozfar K, Hesari Z, et al. Extracellular NAMPT/Visfatin induces proliferation through ERK1/2 and AKT and inhibits apoptosis in breast cancer cells. Peptides (2017) 92:9–15. doi: 10.1016/j.peptides.2017.04.007
52. Cea M, Cagnetta A, Fulciniti M, Tai Y-T, Hideshima T, Chauhan D, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood (2012) 120(17):3519–29. doi: 10.1182/blood-2012-03-416776
53. Schuster S, Penke M, Gorski T, Gebhardt R, Weiss TS, Kiess W, et al. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem Biophys Res Commun (2015) 458(2):334–40. doi: 10.1016/j.bbrc.2015.01.111
54. Espindola-Netto JM, Chini CCS, Tarragó M, Wang E, Dutta S, Pal K, et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget (2017) 8(49):85054–67. doi: 10.18632/oncotarget.18841
55. Kozako T, Aikawa A, Ohsugi T, Uchida Y-i, Kato N, Sato K, et al. High expression of NAMPT in adult T-cell leukemia/lymphoma and anti-tumor activity of a NAMPT inhibitor. Eur J Pharmacol (2019) 865:172738. doi: 10.1016/j.ejphar.2019.172738
56. Cohen JV, Sullivan RJ. Developments in the space of new MAPK pathway inhibitors for BRAF-mutant melanoma. Clin Cancer Res (2019) 25(19):5735–42. doi: 10.1158/1078-0432.CCR-18-0836
57. Audrito V, Managò A, La Vecchia S, Zamporlini F, Vitale N, Baroni G, et al. Nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic target in BRAF-mutated metastatic melanoma. JNCI: J Natl Cancer Institute (2017) 110(3):290–303. doi: 10.1093/jnci/djx198
58. Audrito V, Managò A, Zamporlini F, Rulli E, Gaudino F, Madonna G, et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is a novel marker for patients with BRAF-mutated metastatic melanoma. Oncotarget (2018) 9(27):18997–9005. doi: 10.18632/oncotarget.24871
59. Ohanna M, Cerezo M, Nottet N, Bille K, Didier R, Beranger G, et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev (2018) 32(5-6):448–61. doi: 10.1101/gad.305854.117
60. Audrito V, Messana VG, Moiso E, Vitale N, Arruga F, Brandimarte L, et al. NAMPT over-expression recapitulates the BRAF inhibitor resistant phenotype plasticity in melanoma. Cancers (2020) 12(12):3855. doi: 10.3390/cancers12123855
61. Gibson AE, Yeung C, Issaq SH, Collins VJ, Gouzoulis M, Zhang Y, et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) with OT-82 induces DNA damage, cell death, and suppression of tumor growth in preclinical models of Ewing sarcoma. Oncogenesis (2020) 9(9):80. doi: 10.1038/s41389-020-00264-0
62. Somers K, Evans K, Cheung L, Korotchkina L, Chernova O, Gudkov A, et al. PO-028 Effective targeting of NAD+biosynthesis in patient-derived xenograft models of high-risk paediatric acute lymphoblastic leukaemia. ESMO Open (2018) 3:A238. doi: 10.1136/esmoopen-2018-EACR25.563
63. Heske CM, Davis MI, Baumgart JT, Wilson K, Gormally MV, Chen L, et al. Matrix screen identifies synergistic combination of PARP inhibitors and nicotinamide phosphoribosyltransferase (NAMPT) inhibitors in Ewing sarcoma. Clin Cancer Res (2017) 23(23):7301–11. doi: 10.1158/1078-0432.CCR-17-1121
64. Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med (2012) 4(10):1087–96. doi: 10.1002/emmm.201201250
65. Chan M, Gravel M, Bramoullé A, Bridon G, Avizonis D, Shore GC, et al. Synergy between the NAMPT inhibitor GMX1777(8) and pemetrexed in non–small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Res (2014) 74(21):5948–54. doi: 10.1158/0008-5472.CAN-14-0809
66. Grolla AA, Miggiano R, Di Marino D, Bianchi M, Gori A, Orsomando G, et al. A nicotinamide phosphoribosyltransferase-GAPDH interaction sustains the stress-induced NMN/NAD+ salvage pathway in the nucleus. J Biol Chem (2020) 295(11):3635–51. doi: 10.1074/jbc.RA119.010571
67. Svoboda P, Krizova E, Sestakova S, Vapenkova K, Knejzlik Z, Rimpelova S, et al. Nuclear transport of nicotinamide phosphoribosyltransferase is cell cycle–dependent in mammalian cells, and its inhibition slows cell growth. J Biol Chem (2019) 294(22):8676–89. doi: 10.1074/jbc.RA118.003505
68. Liu H, Liu N, Zhao Y, Zhu X, Wang C, Liu Q, et al. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging (Albany NY) (2019) 11(21):9643–60. doi: 10.18632/aging.102410
69. Grohmann T, Penke M, Petzold-Quinque S, Schuster S, Richter S, Kiess W, et al. Inhibition of NAMPT sensitizes MOLT4 leukemia cells for etoposide treatment through the SIRT2-p53 pathway. Leukemia Res (2018) 69:39–46. doi: 10.1016/j.leukres.2018.04.004
70. Bergaggio E, Riganti C, Garaffo G, Vitale N, Mereu E, Bandini C, et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological Malignancies. Blood (2019) 133(2):156–67. doi: 10.1182/blood-2018-05-850826
71. Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Lüscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci (2012) 109(4):E187–E96. doi: 10.1073/pnas.1105304109
72. Brandl L, Zhang Y, Kirstein N, Sendelhofert A, Boos SL, Jung P, et al. Targeting c-MYC through Interference with NAMPT and SIRT1 and Their Association to Oncogenic Drivers in Murine Serrated Intestinal Tumorigenesis. Neoplasia (2019) 21(10):974–88. doi: 10.1016/j.neo.2019.07.009
73. Brandl L, Kirstein N, Neumann J, Sendelhofert A, Vieth M, Kirchner T, et al. The c-MYC/NAMPT/SIRT1 feedback loop is activated in early classical and serrated route colorectal cancer and represents a therapeutic target. Med Oncol (2018) 36(1):5. doi: 10.1007/s12032-018-1225-1
74. Pan JH, Zhou H, Zhu SB, Huang JL, Zhao XX, Ding H, et al. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis marker in colorectal cancer. J Cell Physiol (2019) 234(4):4385–95. doi: 10.1002/jcp.27228
75. Behrouzfar K, Alaee M, Nourbakhsh M, Gholinejad Z, Golestani A. Extracellular NAMPT/visfatin causes p53 deacetylation via NAD production and SIRT1 activation in breast cancer cells. Cell Biochem Funct (2017) 35(6):327–33. doi: 10.1002/cbf.3279
76. Samal BB, Sun Y, Stearns G, Xie C, Suggs SV, McNiece IK. Cloning and characterization of the cDNA encoding a novel human pre-B- cell colony-enhancing factor. Mol Cell Biol (1994) 14(2):1431–37. doi: 10.1128/MCB.14.2.1431
77. Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, et al. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol (2002) 32(11):3225–34. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L
78. Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, et al. Unique toll-like receptor 4 activation by NAMPT/PBEF induces NFκB signaling and inflammatory lung injury. Sci Rep (2015) 5:13135. doi: 10.1038/srep13135
79. Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PloS One (2009) 4(11):e7897. doi: 10.1371/journal.pone.0007897
80. Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PloS One (2008) 3(5):e2267. doi: 10.1371/journal.pone.0002267
81. Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol (2007) 178(3):1748–58. doi: 10.4049/jimmunol.178.3.1748
82. Torretta S, Colombo G, Travelli C, Boumya S, Lim D, Genazzani AA, et al. The cytokine nicotinamide phosphoribosyltransferase (eNAMPT; PBEF; Visfatin) acts as a natural antagonist of C-C chemokine receptor type 5 (CCR5). Cells (2020) 9(2):496. doi: 10.3390/cells9020496
83. Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood (2015) 125(1):111–23. doi: 10.1182/blood-2014-07-589069
84. Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide phosphoribosyltransferase acts as a metabolic gate for mobilization of myeloid-derived suppressor cells. Cancer Res (2019) 79(8):1938–51. doi: 10.1158/0008-5472.CAN-18-1544
85. Wu Y, Pu C, Fu Y, Dong G, Huang M, Sheng C. NAMPT-targeting PROTAC promotes antitumor immunity via suppressing myeloid-derived suppressor cell expansion. Acta Pharm Sin B (2022) 12(6):2859–68. doi: 10.1016/j.apsb.2021.12.017
86. Pylaeva E, Harati MD, Spyra I, Bordbari S, Strachan S, Thakur BK, et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. Int J Cancer (2019) 144(1):136–49. doi: 10.1002/ijc.31808
87. Zhao E, Hou J, Ke X, Abbas MN, Kausar S, Zhang L, et al. The roles of Sirtuin family proteins in cancer progression. Cancers (2019) 11(12):1949. doi: 10.3390/cancers11121949
88. Chen X, Zhang XL, Zhang GH, Gao YF. Artesunate promotes Th1 differentiation from CD4+ T cells to enhance cell apoptosis in ovarian cancer via miR-142. Braz J Med Biol Res (2019) 52(5):e7992. doi: 10.1590/1414-431x20197992
89. Hamaidi I, Zhang L, Kim N, Wang M-H, Iclozan C, Fang B, et al. Sirt2 inhibition enhances metabolic fitness and effector functions of tumor-reactive T cells. Cell Metab (2020) 32(3):420–36.e12. doi: 10.1016/j.cmet.2020.07.008
90. Wang K, Hu Z, Zhang C, Yang L, Feng L, Yang P, et al. SIRT5 contributes to colorectal cancer growth by regulating T cell activity. J Immunol Res (2020) 2020:3792409. doi: 10.1155/2020/3792409
91. Bauer I, Grozio A, Lasigliè D, Basile G, Sturla L, Magnone M, et al. The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J Biol Chem (2012) 287(49):40924–37. doi: 10.1074/jbc.M112.405837
92. Xiang J, Zhang N, Sun H, Su L, Zhang C, Xu H, et al. Disruption of SIRT7 increases the efficacy of checkpoint inhibitor via MEF2D regulation of programmed cell death 1 ligand 1 in hepatocellular carcinoma cells. Gastroenterology (2020) 158(3):664–78.e24. doi: 10.1053/j.gastro.2019.10.025
93. Hamaidi I, Kim S. Sirtuins are crucial regulators of T cell metabolism and functions. Exp Mol Med (2022) 54(3):207–15. doi: 10.1038/s12276-022-00739-7
94. Boslett J, Hemann C, Christofi FL, Zweier JL. Characterization of CD38 in the major cell types of the heart: endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am J Physiol Cell Physiol (2018) 314(3):C297–c309. doi: 10.1152/ajpcell.00139.2017
95. Ben Baruch B, Mantsur E, Franco-Barraza J, Blacher E, Cukierman E, Stein R. CD38 in cancer-associated fibroblasts promotes pro-tumoral activity. Lab Invest (2020) 100(12):1517–31. doi: 10.1038/s41374-020-0458-8
96. Barata H, Thompson M, Zielinska W, Han YS, Mantilla CB, Prakash YS, et al. The role of cyclic-ADP-ribose-signaling pathway in oxytocin-induced Ca2+ transients in human myometrium cells. Endocrinology (2004) 145(2):881–9. doi: 10.1210/en.2003-0774
97. Chini CCS, Peclat TR, Warner GM, Kashyap S, Espindola-Netto JM, de Oliveira GC, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab (2020) 2(11):1284–304. doi: 10.1038/s42255-020-00298-z
98. Takasawa S, Tohgo A, Noguchi N, Koguma T, Nata K, Sugimoto T, et al. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J Biol Chem (1993) 268(35):26052–4. doi: 10.1016/S0021-9258(19)74275-6
99. Kim H, Jacobson EL, Jacobson MK. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science (1993) 261(5126):1330–3. doi: 10.1126/science.8395705
100. Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RME, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science (1993) 262(5136):1056–9. doi: 10.1126/science.8235624
101. Fliegert R, Gasser A, Guse AH. Regulation of calcium signalling by adenine-based second messengers. Biochem Soc Trans (2007) 35(Pt 1):109–14. doi: 10.1042/BST0350109
102. Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med (2001) 7(11):1209–16. doi: 10.1038/nm1101-1209
103. Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol (2019) 10:1187. doi: 10.3389/fimmu.2019.01187
104. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol (2020) 17(10):611–29. doi: 10.1038/s41571-020-0382-2
105. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-Arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol (2020) 11:597959. doi: 10.3389/fimmu.2020.597959
106. Lee BO, Ishihara K, Denno K, Kobune Y, Itoh M, Muraoka O, et al. Elevated levels of the soluble form of bone marrow stromal cell antigen 1 in the sera of patients with severe rheumatoid arthritis. Arthritis Rheumatism (1996) 39(4):629–37. doi: 10.1002/art.1780390414
107. Ferrero E, Malavasi F. Human CD38, a leukocyte receptor and ectoenzyme, is a member of a novel eukaryotic gene family of nicotinamide adenine dinucleotide+-converting enzymes: extensive structural homology with the genes for murine bone marrow stromal cell antigen 1 and aplysian ADP-ribosyl cyclase. J Immunol (1997) 159(8):3858–65. doi: 10.4049/jimmunol.159.8.3858
108. Yamamoto-Katayama S, Ariyoshi M, Ishihara K, Hirano T, Jingami H, Morikawa K. Crystallographic studies on human BST-1/CD157 with ADP-ribosyl cyclase and NAD glycohydrolase activities1 1Edited by R. Huber J Mol Biol (2002) 316(3):711–23. doi: 10.1006/jmbi.2001.5386
109. Hirata Y, Kimura N, Sato K, Ohsugi Y, Takasawa S, Okamoto H, et al. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Letters (1994) 356(2):244–8. doi: 10.1016/0014-5793(94)01279-2
110. Morandi F, Morandi B, Horenstein AL, Chillemi A, Quarona V, Zaccarello G, et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget (2015) 6(28):25602–18. doi: 10.18632/oncotarget.4693
111. Ortolan E, Augeri S, Fissolo G, Musso I, Funaro A. CD157: From immunoregulatory protein to potential therapeutic target. Immunol Lett (2019) 205:59–64. doi: 10.1016/j.imlet.2018.06.007
112. Aomatsu E, Takahashi N, Sawada S, Okubo N, Hasegawa T, Taira M, et al. Novel SCRG1/BST1 axis regulates self-renewal, migration, and osteogenic differentiation potential in mesenchymal stem cells. Sci Rep (2014) 4:3652. doi: 10.1038/srep03652
113. Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF–dependent Toll-like receptor signaling. Nat Immunol (2006) 7(10):1074–81. doi: 10.1038/ni1382
114. Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, et al. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ (2013) 20(3):478–89. doi: 10.1038/cdd.2012.144
115. Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, et al. A cell-permeant mimetic of NMN activates SARM1 to produce cyclic ADP-ribose and induce non-apoptotic cell death. iScience (2019) 15:452–66. doi: 10.1016/j.isci.2019.05.001
116. Kim Y, Zhou P, Qian L, Chuang J-Z, Lee J, Li C, et al. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med (2007) 204(9):2063–74. doi: 10.1084/jem.20070868
117. Wang Q, Zhang S, Liu T, Wang H, Liu K, Wang Q, et al. Sarm1/myd88-5 regulates neuronal intrinsic immune response to traumatic axonal injuries. Cell Rep (2018) 23(3):716–24. doi: 10.1016/j.celrep.2018.03.071
118. Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc Natl Acad Sci U S A (2016) 113(41):E6271–e80. doi: 10.1073/pnas.1601506113
119. Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science (2015) 348(6233):453–7. doi: 10.1126/science.1258366
120. Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron (2017) 93(6):1334–43.e5. doi: 10.1016/j.neuron.2017.02.022
121. Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell (2005) 17(6):855–68. doi: 10.1016/j.molcel.2005.02.022
122. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol (2012) 13(7):411–24. doi: 10.1038/nrm3376
123. DiAntonio A, Milbrandt J, Figley MD. The SARM1 TIR NADase: mechanistic similarities to bacterial phage defense and toxin-antitoxin systems. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.752898
124. Wei Y, Xiang H, Zhang W. Review of various NAMPT inhibitors for the treatment of cancer. Front Pharmacol (2022) 13. doi: 10.3389/fphar.2022.970553
125. Ghanem MS, Monacelli F, Nencioni A. Advances in NAD-lowering agents for cancer treatment. Nutrients (2021) 13(5):1665. doi: 10.3390/nu13051665
126. Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic Malignancies. Blood (2009) 113(14):3276–86. doi: 10.1182/blood-2008-08-173369
127. Zhang L-Y, Liu L-Y, Qie L-L, Ling K-N, Xu L-H, Wang F, et al. Anti-proliferation effect of APO866 on C6 glioblastoma cells by inhibiting nicotinamide phosphoribosyltransferase. Eur J Pharmacol (2012) 674(2):163–70. doi: 10.1016/j.ejphar.2011.11.017
128. Watson M, Roulston A, Bélec L, Billot X, Marcellus R, Bédard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol (2009) 29(21):5872–88. doi: 10.1128/MCB.00112-09
129. Vig Hjarnaa P-J, Jonsson E, Latini S, Dhar S, Larsson R, Bramm E, et al. CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res (1999) 59(22):5751–7.
130. Somers K, Evans K, Cheung L, Karsa M, Pritchard T, Kosciolek A, et al. Effective targeting of NAMPT in patient-derived xenograft models of high-risk pediatric acute lymphoblastic leukemia. Leukemia (2020) 34(6):1524–39. doi: 10.1038/s41375-019-0683-6
131. Korotchkina L, Kazyulkin D, Komarov PG, Polinsky A, Andrianova EL, Joshi S, et al. OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological Malignancies on NAD biosynthesis. Leukemia (2020) 34(7):1828–39. doi: 10.1038/s41375-019-0692-5
132. Khan HY, Uddin MH, Balasubramanian SK, Sulaiman N, Iqbal M, Chaker M, et al. PAK4 and NAMPT as novel therapeutic targets in diffuse large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma. Cancers (Basel) (2021) 14(1):160. doi: 10.3390/cancers14010160
133. Abu Aboud O, Chen C-H, Senapedis W, Baloglu E, Argueta C, Weiss RH. Dual and specific inhibition of NAMPT and PAK4 by KPT-9274 decreases kidney cancer growth. Mol Cancer Ther (2016) 15(9):2119–29. doi: 10.1158/1535-7163.MCT-16-0197
134. Aboukameel A, Muqbil I, Senapedis W, Baloglu E, Landesman Y, Shacham S, et al. Novel p21-activated kinase 4 (PAK4) allosteric modulators overcome drug resistance and stemness in pancreatic ductal adenocarcinoma. Mol Cancer Ther (2017) 16(1):76–87. doi: 10.1158/1535-7163.MCT-16-0205
135. Mitchell SR, Larkin K, Grieselhuber NR, Lai T-H, Cannon M, Orwick S, et al. Selective targeting of NAMPT by KPT-9274 in acute myeloid leukemia. Blood Advances (2019) 3(3):242–55. doi: 10.1182/bloodadvances.2018024182
136. Takao S, Chien W, Madan V, Lin DC, Ding LW, Sun QY, et al. Targeting the vulnerability to NAD+ depletion in B-cell acute lymphoblastic leukemia. Leukemia (2018) 32(3):616–25. doi: 10.1038/leu.2017.281
137. Mpilla G, Aboukameel A, Muqbil I, Kim S, Beydoun R, Philip PA, et al. PAK4-NAMPT dual inhibition as a novel strategy for therapy resistant pancreatic neuroendocrine tumors. Cancers (2019) 11(12):1902. doi: 10.3390/cancers11121902
138. Fulciniti M, Martinez-Lopez J, Senapedis W, Oliva S, Lakshmi Bandi R, Amodio N, et al. Functional role and therapeutic targeting of p21-activated kinase 4 in multiple myeloma. Blood (2017) 129(16):2233–45. doi: 10.1182/blood-2016-06-724831
139. Qasim SL, Sierra L, Shuck R, Kurenbekova L, Patel TD, Rajapakshe K, et al. p21-activated kinases as viable therapeutic targets for the treatment of high-risk Ewing sarcoma. Oncogene (2021) 40(6):1176–90. doi: 10.1038/s41388-020-01600-9
140. Dasgupta A, Sierra L, Tsang SV, Kurenbekova L, Patel T, Rajapakse K, et al. Targeting PAK4 inhibits Ras-mediated signaling and multiple oncogenic pathways in high-risk rhabdomyosarcoma. Cancer Res (2021) 81(1):199–212. doi: 10.1158/0008-5472.CAN-20-0854
141. Ravaud A, Cerny T, Terret C, Wanders J, Bui BN, Hess D, et al. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: an ECSG/EORTC study. Eur J Cancer (2005) 41(5):702–7. doi: 10.1016/j.ejca.2004.12.023
142. Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs (2008) 26(1):45–51. doi: 10.1007/s10637-007-9083-2
143. Hovstadius P, Larsson R, Jonsson E, Skov T, Kissmeyer AM, Krasilnikoff K, et al. A Phase I study of CHS 828 in patients with solid tumor Malignancy. Clin Cancer Res (2002) 8(9):2843–50.
144. von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol (2010) 65(6):1165–72. doi: 10.1007/s00280-009-1125-3
145. Goldinger SM, Gobbi Bischof S, Fink-Puches R, Klemke C-D, Dréno B, Bagot M, et al. Efficacy and safety of APO866 in patients with refractory or relapsed cutaneous T-cell lymphoma: A phase 2 clinical trial. JAMA Dermatol (2016) 152(7):837–9. doi: 10.1001/jamadermatol.2016.0401
146. Franceschini N, Oosting J, Tamsma M, Niessen B, Bruijn I-d, van den Akker B, et al. Targeting the NAD salvage synthesis pathway as a novel therapeutic strategy for osteosarcomas with low NAPRT expression. Int J Mol Sci (2021) 22(12):6273. doi: 10.3390/ijms22126273
147. Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res (2013) 19(24):6912–23. doi: 10.1158/1078-0432.CCR-13-1186
148. Fons NR, Sundaram RK, Breuer GA, Peng S, McLean RL, Kalathil AN, et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat Commun (2019) 10(1):3790. doi: 10.1038/s41467-019-11732-6
149. Gaut ZN, Solomon HM. Inhibition of nicotinate phosphoribosyl transferase by nonsteroidal anti-inflammatory drugs: A possible mechanism of action. J Pharm Sci (1971) 60(12):1887–8. doi: 10.1002/jps.2600601230
150. Gaut ZN, Solomon HM. Uptake and metabolism of nicotinic acid by human blood platelets effects of structure analogs and metabolic inhibitors. Biochim Biophys Acta (BBA) - Gen Subjects (1970) 201(2):316–22. doi: 10.1016/0304-4165(70)90306-5
151. Gaut ZN, Solomon HM. Inhibition of nicotinate phosphoribosyltransferase in human platelet lysate by nicotinic acid analogs. Biochem Pharmacol (1971) 20(10):2903–6. doi: 10.1016/0006-2952(71)90202-4
152. Franco J, Piacente F, Walter M, Fratta S, Ghanem M, Benzi A, et al. Structure-based identification and biological characterization of new NAPRT inhibitors. Pharmaceuticals (2022) 15(7):855. doi: 10.3390/ph15070855
153. Ghanem MS, Caffa I, Del Rio A, Franco J, Parenti MD, Monacelli F, et al. Identification of NAPRT inhibitors with anti-cancer properties by in silico drug discovery. Pharm (Basel) (2022) 15(7):848. doi: 10.3390/ph15070848
154. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. New Engl J Med (2015) 373(13):1207–19. doi: 10.1056/NEJMoa1506348
155. Dimopoulos MA, San-Miguel J, Belch A, White D, Benboubker L, Cook G, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica (2018) 103(12):2088–96. doi: 10.3324/haematol.2018.194282
156. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood (2016) 128(3):384–94. doi: 10.1182/blood-2015-12-687749
157. Deckert J, Wetzel M-C, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F, et al. SAR650984, A novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ Hematologic Malignancies. Clin Cancer Res (2014) 20(17):4574–83. doi: 10.1158/1078-0432.CCR-14-0695
158. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res (2017) 23(15):4290–300. doi: 10.1158/1078-0432.CCR-16-3192
159. Kinder M, Bahlis NJ, Malavasi F, De Goeij B, Babich A, Sendecki J, et al. Comparison of CD38 antibodies in vitro and ex vivo mechanisms of action in multiple myeloma. Haematologica (2021) 106(7):2004–8. doi: 10.3324/haematol.2020.268656
160. El-Murr N, Kassem S, Carrié N, Henry C, Virone-Oddos A, Yang Z-y, et al. Abstract 1887: Anti-CD38/CD28xCD3 trispecific T cell engager induces proliferation of primary T cells and mediates potent killing of primary Malignant plasma cells isolated from Multiple Myeloma bone marrow aspirates. Cancer Res (2021) 81(13_Supplement):1887–. doi: 10.1158/1538-7445.AM2021-1887
161. Wu L, Seung E, Xu L, Rao E, Lord DM, Wei RR, et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat Cancer (2020) 1(1):86–98. doi: 10.1038/s43018-019-0004-z
162. Doucey M-A, Pouleau B, Estoppey C, Stutz C, Croset A, Laurendon A, et al. ISB 1342: A first-in-class CD38 T cell engager for the treatment of relapsed refractory multiple myeloma. J Clin Oncol (2021) 39(15_suppl):8044–. doi: 10.1200/JCO.2021.39.15_suppl.8044
163. Bruins WSC, Zheng W, Higgins JP, Willert EK, Newcomb J, Dash AB, et al. TAK-169, a novel recombinant immunotoxin specific for CD38, induces powerful preclinical activity against patient-derived multiple myeloma cells. Blood (2020) 136(Supplement 1):11–2. doi: 10.1182/blood-2020-136928
164. Chu SY, Miranda Y, Phung S, Chen H, Rashid R, Endo NA, et al. Immunotherapy with long-lived anti-CD38 × Anti-CD3 bispecific antibodies stimulates potent T cell-mediated killing of human myeloma cell lines and CD38+ Cells in monkeys: A potential therapy for multiple myeloma. Blood (2014) 124(21):4727. doi: 10.1182/blood.V124.21.4727.4727
165. Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res (2019) 25(13):3921–33. doi: 10.1158/1078-0432.CCR-18-2752
166. Kassem S, Diallo BK, El-Murr N, Carrié N, Tang A, Fournier A, et al. SAR442085, a novel anti-CD38 antibody with enhanced antitumor activity against multiple myeloma. Blood (2022) 139(8):1160–76. doi: 10.1182/blood.2021012448
167. Choudhury A, Ortiz DF, Argueta S, Garofalo K, Lansing JC, Jetley U, et al. Abstract 561: Discovery of a potential best-in-class anti-CD38 therapeutic utilizing Fc multimerization. Cancer Res (2019) 79(13_Supplement):561–. doi: 10.1158/1538-7445.AM2019-561
168. De Goeij BECG, Janmaat ML, Andringa G, Kil L, Van Kessel B, Frerichs KA, et al. Hexabody-CD38, a novel CD38 antibody with a hexamerization enhancing mutation, demonstrates enhanced complement-dependent cytotoxicity and shows potent anti-tumor activity in preclinical models of hematological Malignancies. Blood (2019) 134:3106. doi: 10.1182/blood-2019-125788
169. Li L, Hau A, Tong W, Lau M, Zhu T, Fells K, et al. Abstract LB-227: Preclinical development and characterization of STI-6129, an anti-CD38 antibody-drug conjugate, as a new therapeutic agent for multiple myeloma. Cancer Res (2020) 80(16_Supplement):LB–227-LB-. doi: 10.1158/1538-7445.AM2020-LB-227
170. Dawicki W, Allen KJH, Jiao R, Malo ME, Helal M, Berger MS, et al. Daratumumab-(225)Actinium conjugate demonstrates greatly enhanced antitumor activity against experimental multiple myeloma tumors. Oncoimmunology (2019) 8(8):1607673. doi: 10.1080/2162402X.2019.1607673
171. Lammerts van Bueren J, Jakobs D, Kaldenhoven N, Roza M, Hiddingh S, Meesters J, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood (2014) 124(21):3474. doi: 10.1182/blood.V124.21.3474.3474
172. Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood (2017) 129(25):3294–303. doi: 10.1182/blood-2016-09-740787
173. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD(+)Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab (2018) 27(1):85–100.e8. doi: 10.1016/j.cmet.2017.10.006
174. Shen A, Ye Y, Chen F, Xu Y, Zhang Z, Zhao Q, et al. Integrated multi-omics analysis identifies CD73 as a prognostic biomarker and immunotherapy response predictor in head and neck squamous cell carcinoma. Front Immunol (2022) 13:969034. doi: 10.3389/fimmu.2022.969034
175. Goh S, Ng HHM, Chew V, Sim XN, Li H, Lim S, et al. CD38 is a good predictor of anti-PD-1 immunotherapy responsiveness in hepatocellular carcinoma. bioRxiv (2019) 638981. doi: 10.1101/638981
176. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci (2018) 39(4):424–36. doi: 10.1016/j.tips.2018.02.001
Keywords: NAD+ metabolism, tumor microenvironment, cancer treatment, NAMPT inhibitor, CD38, cancer immunotherapy
Citation: Yong J, Cai S and Zeng Z (2023) Targeting NAD+ metabolism: dual roles in cancer treatment. Front. Immunol. 14:1269896. doi: 10.3389/fimmu.2023.1269896
Received: 31 July 2023; Accepted: 20 November 2023;
Published: 05 December 2023.
Edited by:
Di Zhao, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Eduardo Nunes Chini, Mayo Clinic, United StatesHao Fan, The University of Chicago, United States
Copyright © 2023 Yong, Cai and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaolei Zeng, emVuZ3pobEBzeXN1Y2Mub3JnLmNu