 Xu-Dong Zhang
Xu-Dong Zhang Zhong-Yuan Liu
Zhong-Yuan Liu Mao-Sen Wang†
Mao-Sen Wang† Shuai Huang
Shuai Huang Ren-Feng Li
Ren-Feng Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 October 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1269451
Regulation of cell mortality for disease treatment has been the focus of research. Ferroptosis is an iron-dependent regulated cell death whose mechanism has been extensively studied since its discovery. A large number of studies have shown that regulation of ferroptosis brings new strategies for the treatment of various benign and malignant diseases. Iron excess and lipid peroxidation are its primary metabolic features. Therefore, genes involved in iron metabolism and lipid metabolism can regulate iron overload and lipid peroxidation through direct or indirect pathways, thereby regulating ferroptosis. In addition, glutathione (GSH) is the body’s primary non-enzymatic antioxidants and plays a pivotal role in the struggle against lipid peroxidation. GSH functions as an auxiliary substance for glutathione peroxidase 4 (GPX4) to convert toxic lipid peroxides to their corresponding alcohols. Here, we reviewed the researches on the mechanism of ferroptosis in recent years, and comprehensively analyzed the mechanism and regulatory process of ferroptosis from iron metabolism and lipid metabolism, and then described in detail the metabolism of GPX4 and the main non-enzymatic antioxidant GSH in vivo.
Currently there are two types of cell death, according to the most recent recommendations of the 2018 Committee on Nomenclature of Cell Death: accidental cell death (ACD) and regulatory cell death (RCD) (1). ACD is a biologically uncontrolled process triggered by unexpected noxious stimuli that are beyond the regulatory capacity of the cell (2). RCD, also known as programmed cell death (PCD), is a type of cell death process regulated by specific signaling pathways in cells and has biological functions (3). RCD are further subdivided into apoptotic and non-apoptotic types, among which non-apoptotic types are further segmented into ferroptosis, necroptosis, pyroptosis, autophagy, cuproptosis and alkaliptosis, etc. (1, 4), each of which has distinct signal induction, molecular regulation, and disease-causing effects. The discovery of RCD has facilitated progress in the treatment of benign and malignant disorders.
Ferroptosis is caused by the redox imbalance between oxidation and antioxidant, which is driven by the abnormal expression of a variety of redox active enzymes. It is characterized by increased lipid peroxides levels and iron overload (5, 6). Lipid peroxides are highly reactive molecules that can damage cellular components, including lipins, proteins, and DNA, leading to cell death (7). Different from the morphological changes of classic apoptotic cells, such as cell shrinkage, nuclear volume reduction, and highly condensed chromatin to form apoptotic bodies, ferroptosis has its unique morphological characteristics, the density of the mitochondrial membrane increases, the size shrinks, and the structure of the mitochondrial inner cristae disappears (8). Ferroptosis involves complex signaling pathways, since its identification in 2012 (8), it has been a subject of intense study. Numerous studies reveal that ferroptosis is a key regulator in a wide variety of diseases, including cancers, ischemia-reperfusion injury, renal failure, neurological disorders, and hematological conditions (9, 10).
In 2014, Yang et al. (11) used proteomics to find that GPX4 has a regulatory effect on ferroptosis induced by 12 ferroptosis inducers, thus confirming that GPX4 plays a central role in the regulation of ferroptosis. GPX4 is a selenium-dependent enzyme, and it contains a selenium-bound amino acid residue that is essential for its reductive activity (12). GPX4 can reduce lipid peroxides and plays a crucial function in prohibiting lipid peroxidation (13). Actually, GSH is not directly involved in the reduction of lipid peroxides, but it does maintain the reducibility of selenocysteine residues within the active site of GPX4 (13). Cysteine is the rate-limiting precursor of GSH synthesis, and intracellular cysteine content is limited, necessitating the transport of extracellular cystine for intracellular cysteine synthesis (14). Solute carrier family 7 member 11 (SLC7A11) functions as a cystine transporter and thus acting a significant purpose in ferroptosis. Current research has shown that a variety of genes and pharmaceuticals can modulate ferroptosis by targeting the GPX4, GSH or SLC7A11 (15–17).
Due to the fact that ferroptosis entails dysregulation of lipid metabolism, iron metabolism, the oxidation system and the antioxidant system, we reviewed the current research on ferroptosis from these three perspectives. In this publication, the mechanism of ferroptosis was summarized in detail, providing a foundation for future research. However, our understanding of the molecular mechanism of ferroptosis and the usage of ferroptosis in physiological and pathological processes is still limited, constraining the clinical transformation of ferroptosis and highlighting the need for future research in this area.
Iron occurs primarily as two different forms in the human body: trivalent iron ion (Fe3+) and ferrous ion (Fe2+). Iron is absorbed and used in the human body as Fe2+ (18), and it is transported as Fe3+ (19). Iron comes in two main forms in food: heme and nonheme iron. The hemoglobin and myoglobin of meat are the main sources of heme iron, which is Fe2+. Nonheme iron mainly comes from botany, which is Fe3+. Vitamin C and hydrochloric acid can convert nonheme to Fe2+, making it easier to absorb (20). The upper jejunum and duodenum are the primary sites of iron absorption from food. Duodenal cytochrome b (DcytB) is highly expressed in the brush membrane of duodenal epithelial cells, which can convert Fe3+ into Fe2+ (21). As a ferric iron reductase, DcytB plays an irreplaceable role in iron absorption. However, no research has revealed whether DcytB has a regulatory effect on ferroptosis. Patients with hereditary hemochromatosis show progressive iron overload, and most patients have missense mutations in the Hfe gene (C282Y). Herrmann et al. (22) used the Hfe knockout mouse model to prove that increased expression of Dcytb plays a role in the pathogenesis of iron overload in Hfe deficiency. Therefore, we conjecture that DcytB can also regulate ferroptosis, but there hasn’t been any direct study to prove the link between the two.
Then, Fe2+ is taken into the cell via the divalent metal transporter 1 (DMT1, the major transmembrane transporter of divalent metal cations into cells) at the lumen side of tiny intestinal epithelial cells (23). In addition to transporting Fe2+, DMT1 can also transport Zn2+, Mn2+, Co2+, Cd2+, Cu2+, Ni2+, and Pb2+. DMT1 is one of the the principal iron transporters responsible for Fe2+ uptake in the majority of cell types. Therefore, by regulating the expression of DMT1, ferroptosis can be regulated. Iron is essential for normal neurological function, but current studies have found that excess iron in nerve cells is strongly associated with several neurodegenerative diseases. Multiple or prolonged exposure to general anesthesia leads to cognitive deficits in patients, which have been found to be associated with iron overload due to abnormal activation of DMT1 in the brain (24). In addition, isoliquiritigenin, as a classic hepatoprotective drug, induces ferroptosis in hepatic stellate cells by promoting the expression of DMT1, and ultimately alleviates liver fibrosis (25). The above findings demonstrate the essential role of DMT1 in regulating iron metabolism and ferroptosis.
After the absorption of Fe2+ in the small intestine, some Fe2+ can synthesize ferritin in intestinal mucosal epithelial cells, and the other part enters the blood circulation. Ferritin is considered a major iron-storage protein because it can accumulate in large amounts iron ion (26). The ferritin family consists of 4 subfamilies, including the canonical ferritin (FTN), the heme-containing bacterioferritin (BFR), DNA-binding proteins from starved cell (DPS), and, more recently, the discovery of the encapsulated ferritin (27, 28). It exists mainly in the cytoplasm and a small amount in the nucleus, mitochondria and circulating plasma. In humans, ferritin is the canonical ferritin, FTN. BFR and DPS are limited to prokaryotes. FTN is composed of two subunits of 19 kDA ferritin light chain (L) and 21 kDA ferritin heavy chain (H). Poly (RC)-binding proteins (PCBPs) and nuclear receptor coactivator 4 (NCOA4) have been identified as essential regulators of ferritin, which in turn play a role in ferroptosis (29, 30)..
Then, Fe2+ travels via ferroportin (FPN, the only known cellular iron exporting protein) on the basolateral membrane side to the blood circulation (31). FPN is highly express in duodenal enterocytes, liver kupffer cells and splenic red pulp macrophages and plays an extremely important role in the exportation of iron. In Alzheimer’s disease, loss of FPN leads to excess iron in neurons, which eventually promotes disease progression through ferroptosis (32). Hepcidin-FPN rigorously regulates serum iron levels. When iron is deficient in vivo, hepcidin expression decreases, allowing iron to be delivered to plasma via FPN (33). The role of hepcidin in ferroptosis will be described in detail later.
Under the effect of hephaestin (HP) or ceruloplasmin, Fe2+ entering the bloodstream is converted into Fe3+, which combines with transferrin and is transported to the tissues. Each transferrin can carry two Fe3+, making it the primary iron transporter (34). Gao et al. discovered that consumption of transferrin in fetal bovine serum (FBS) can weaken ferroptosis, and that this effect was only seen in iron-carrying transferrin (35). Afterwards, transferrin attaches to transferrin receptor 1 (TFR1) and transferrin receptor 2 (TFR2) on the cell surface of iron-deficient cells and enters the cell in a controlled manner (36, 37). TFR1 is widely expressed on the surface of a variety of cells, and binds to transferrin carrying iron, which then enters the cell through endocytosis, and Fe3+ that enters the cell is reduced to Fe2+ by six-transmembrane epithelial antigen of prostate 3(STEAP3 metal reductase) (38). Unlike TFR1, the expression of TFR2 is more specific. TFR2 is mainly expressed in hepatocytes and interacts with Hfe protein to regulate iron metabolism in the liver (38). TFR1 and TFR2 can also serve as targets for ferroptosis regulation (39, 40). Figure 1 shows roughly how iron is absorbed, transported, stored and transformed in the body.
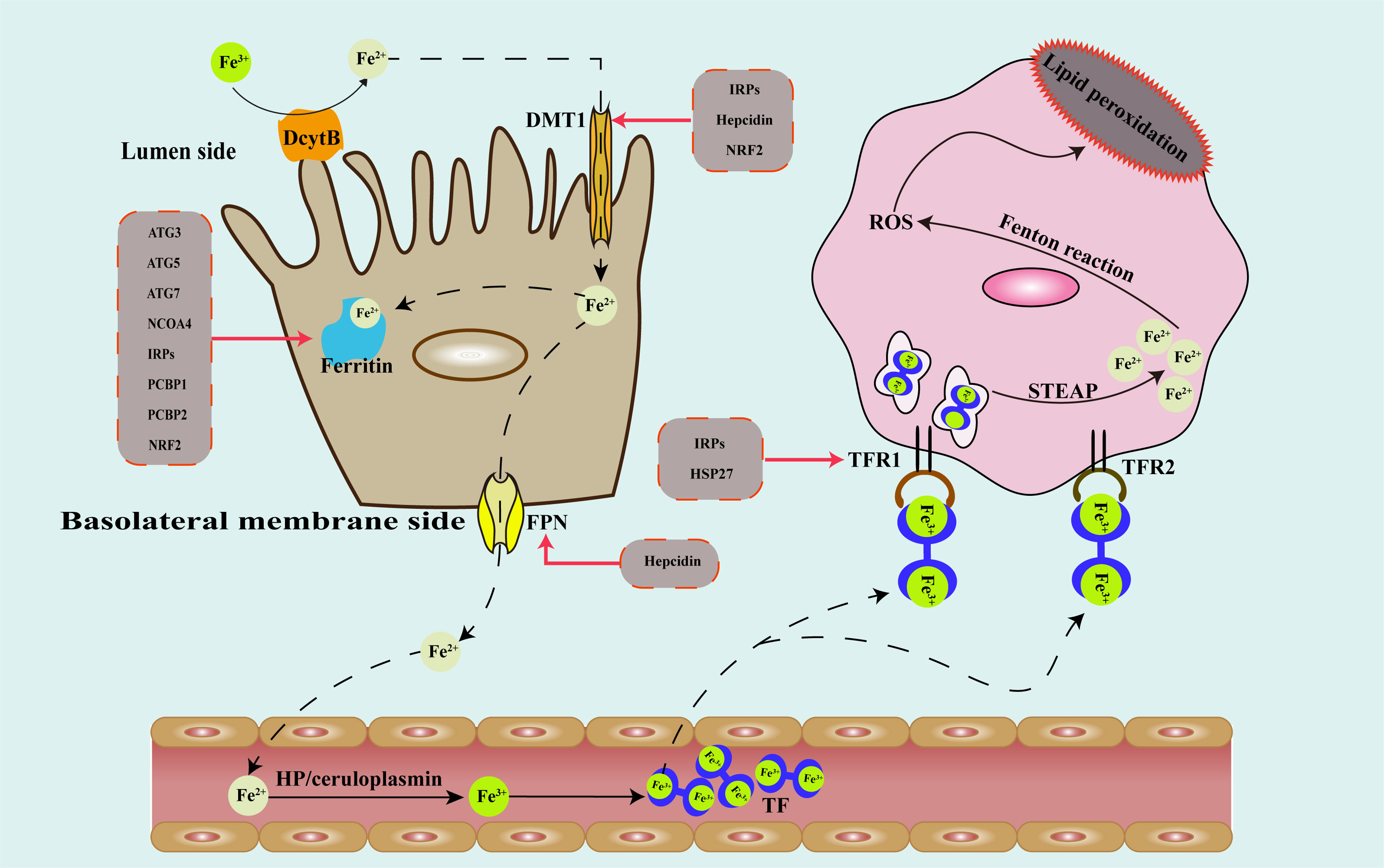
Figure 1 Schematic diagram of the metabolic pathway of iron in the human body and regulatory targets of genes that modulate iron metabolism. DcytB, duodenal cytochrome b; DMT1, divalent metal transporter 1; FPN, ferroportin; HP, hephaestin; TF, transferrin; TFR1, transferrin receptor 1; TFR2, transferrin receptor 2; STEAP3, six-transmembrane epithelial antigen of prostate 3; ROS, reactive oxygen species; ATG, autophagy associated gene; NCOA4, nuclear receptor coactivator 4; IRPs, iron response element binding proteins; PCBP, poly(rC)-binding proteins; HSP, heat shock protein; NRF2, nuclear factor erythroid 2-related factor 2.
As a crucial trace element in the human body, iron plays a crucial part in multiple biological processes. However, iron also has redox-active, it can generate many reactive oxygen species (ROS) through the fenton reaction and other ways (41, 42), further leads to cell damage and death, including ferroptosis. Therefore, iron homeostasis is strictly regulated. In theory, factors that can regulate the absorption, distribution, transport and utilization of iron in the body can regulate ferroptosis.
Lipid peroxidation refers to oxidative deterioration of polyunsaturated fatty acids (PUFAs) and lipids (43). It can disrupt cell membranes, lipoproteins, and other structures containing lipids, hence impairing the proper function of cells. PUFAs refer to straight-chain fatty acids with two or more double bonds and 18 to 22 carbon atoms in the carbon chain, which usually separated into omega-3 and omega-6. Members of the omega-3 fatty acid family, mainly alpha-linolenic acid, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The omega-6 series fatty acids mainly include linoleic acid (LA, octadecadienoic acid) and arachidonic acid (AA, eicosadonic acid). The hydrogen on the methylene group (-CH2-) connected with two double bonds in the PUFAs molecule is more active, so it is more prone to peroxidation (44). Among them, AA and adrenic acid (AdA) are the main substrates of lipid peroxidation in ferroptosis (45). Hydrogen atoms in lipid substances are extracted by active substances to generate lipid free radicals. Then lipid free radicals react with active oxygen to generate lipid peroxy free radicals, which can continue to extract hydrogen atoms from other substances to form lipid peroxides (13). In a study on inflammatory bowel disease, researchers found that dietary supplementation with PUFAs promoted the progression of ferroptosis-associated inflammatory bowel disease in a mouse model (46). Monounsaturated fatty acids (MUFAs) such as oleic acid and palmitoleic acid can effectively inhibit erastin-induced ferroptosis by competing with PUFAs for incorporation into phospholipids (44, 47). This is because MUFAs do not have double Allyl group position, therefore are not simple to peroxidized. Mitochondrial 2,4-dienoyl-CoA reductase (DECR1) is one of the key enzymes for PUFAs β-oxidation. Studies have found that in prostate cancer, inhibiting the expression of DECR1 can inhibit PUFAs β-oxidation and increase the content of PUFAs in cells, thereby enhancing mitochondrial stress and lipid peroxidation (48, 49).
This indicates that fatty acid β-oxidation plays a crucial function in balancing PUFAs and MUFAs. Lipid peroxidation to form lipid peroxidation products such as malondialdehyde (MDA) and 4-hydroxynonenoic (4-HNE). Therefore, people often judge the level of ferroptosis in cells by detecting the levels of MDA and 4-HNE in cells (50).
Enzymatic and non-enzymatic lipid peroxidation are the two forms of lipid peroxidation (51). Non-enzymatic lipid peroxidation is driven mainly by hydroxyl radicals generated by the interaction of Fe2+, Cu2+, Co2+, and other transition metal ions with ROS (52). Despite being a potent oxidizing agent, molecular oxygen does not directly participate in nonenzymatic lipid peroxidation reactions. Lipoxygenases (LOXs) is the key enzymes involved in enzymatic lipid peroxidation. There are mainly 6 kinds of LOXs in the human body, namely LOX5, LOX12, LOX12B, LOX15, LOX15B and LOXE3. In addition, oxidases such as cytochrome p-450 oxidoreductase (POR) can also promote lipid peroxidation (53). POR is a NADPH-dependent, FMN-containing oxidoreductase. Zou et al. (53) found that POR has a pro-ferroptosis role in different cancer lineages and used systematic lipidomic analysis to demonstrate that POR promotes ferroptosis by upregulating peroxidation of membrane polyunsaturated phospholipids. A recent study found that COX-2 can regulate ferroptosis through prostaglandins (54). However, Wan Seok et al. (11) used indomethacin (a COX-1/COX-2 inhibitor) to handle BJeLR cells treated with erastin or RSL3 and found that ferroptosis caused by erastin or RSL3 was not affected by indomethacin treatment. This result indicates that COXs play no function in ferroptosis. Therefore, whether COXs have a regulatory effect on ferroptosis needs further study.
Degradation and synthesis are tightly regulated in eukaryotic cells due to the continual turnover of substances. There are two main degradation systems in mammalian cells, lysosome (also called autophagy) and the proteasome (55). Autophagy is further divided into three categories, namely macroautophagy, microautophagy and chaperone-mediated autophagy. Since macroautophagy is the most prevalent kind of autophagy, it has been the subject of the most research. Studies have revealed that the regulation of ferroptosis involves autophagy in a complicated way. On the one hand, autophagy can protect cells from ferroptosis by eliminating damaged cellular components and decreasing iron content in cells (56). Additionally, excessive autophagy can promote ferroptosis (57, 58). Here, we introduce the relevant autophagy regulatory molecules and their function in ferroptosis.
Our understanding of the process and function of autophagy has considerably improved as a result of the discovery of ATGs (59). So far, more than 40 kinds of ATGs have been found in yeast cells. ATGs participate in multiple autophagy-activating stages, including initiation, nucleation, elongation, maturation, and fusion (60). Numerous investigations have discovered that autophagy is crucial to ferroptosis (61). In 2016, Daolin Tang et al. (62) found that knockout of ATG5 or ATG7 could inhibit erastin-induced ferroptosis, and further studies demonstrated that activation of the autophagy pathway promoted ferroptosis by degrading ferritin. Artesunate can induce ferroptosis in activated hepatic stellate cells, and up-regulation of ATG3, ATG5, ATG6/beclin1, ATG12 was observed (63). By triggering ferroptosis in clear cell ovarian cancer tissues, superparamagnetic iron oxide nanoparticles (SPIONs) suppress the activity of human ovarian cancer stem cells. Mechanism studies have found that ferroptosis is activated by reducing the autophagy activity of ovarian cancer stem cells. PCR analysis showed that SPIONs treated human ovarian cancer stem cells autophagy-related factors (ATG3, ATG5, MAP1ALC3a, MAP1ALC3b, and MAP1ALC3c) expression decreased (64). Unfortunately, the authors did not further explore the mechanism by which SPIONs promote ferroptosis, and whether they interact directly with ATG3 or ATG5. The above findings suggest that ferroptosis is the result of the joint action of multiple regulatory factors, rather than being regulated by a single factor.
NCOA4 is a protein that plays significant roles in cellular processes related to iron homeostasis and autophagy. Alec C Kimmelman et al. (65) discovered that NCOA4 was substantially enriched in autophagosomes using quantitative proteomics in 2014. Further research found that NCOA4 is essential for the transfer of ferritin to lysosomes, and cells missing NCOA4 are incapable of degrading ferritin. Thus identifying NCOA4 is a selective cargo receptor for ferritinophagy, which is vital for iron homeostasis (65, 66). Murphy et al. demonstrated that the interaction between NCOA4 and ferritin was exclusive to ferritin heavy chain (FTH1) specifically (66). By disrupting the NCOA4-FTH1 protein-protein interaction, it does reduce the amount of iron available to organisms in the cell, thus inhibiting ferroptosis (67). Blocking intestinal NCOA4 protects against iron overloading in a mouse model of hemochromatosis. And NCOA4-mediated intestinal ferritin phagosomes are regulated by HIF2α (68). Because NCOA4 plays a crucial part in ferroptosis, treating illnesses by targeting NCOA4 has become an area of intense research (69, 70).
IRPs regulate iron metabolism at the post-transcriptional level by binding to iron response elements (IREs) of target transcripts, such as TFR1and ferritin H and L subunits (71). Back in the 1990s, researchers discovered that IRPs regulate the amount of iron in cells (72). Initially IRP1 can increase the expression of TFR1and increase cells’ ability to absorb iron (73). Additionally, Galy et al. (74) used Cre/Lox technology to simultaneously ablate IRP1 and IRP2 in a tissue-specific manner, and found that IRP deficiency significantly reduced DMT1 mRNA expression. IRPs can also regulate ferritin expression. Under iron-deficient conditions, the expression of IRPs is increased and the translation of ferritin mRNA is inhibited; while in iron-rich cells, the binding of IRPs to the IRE of ferritin is reduced (75). Due to its important function in cellular iron homeostasis, its relationship with ferroptosis has attracted much attention in recent years. α-Enolase 1 (ENO1) is an essential glycolytic enzyme. Zhang et al. (76) discovered that ENO1 inhibits mitochondrial ferroptosis by inhibiting IRP1, and demonstrated through in vitro and in vivo experiments and clinical sample analysis that IRP1 exerts an anti-tumor effect in liver cancer cells by inducing ferroptosis. miRNAs are an essential post-transcriptional regulator involved in numerous physiological and pathological processes. By inhibiting IRP2 expression, MiR-19a inhibits ferroptosis in colorectal cancer cells, thereby promoting the progression of colorectal cancer (77). Studies have shown that elevated iron content in the liver is one of the main causes of liver steatosis. It was found that the levels of IRP2 mRNA and protein were increased in steatosis liver. Further research has found that mir-29a-3p can reduce the expression of IRP2, thereby lowering Fe2+ levels, thus inhibiting ferroptosis and improving cell damage, according to additional research (78). These studies indicate that modulating ferroptosis at the post-transcriptional level by regulating IRPs is conducive to the treatment of diseases.
HSPs are a class of important molecular chaperone proteins, including HSP110 (HSPH), HSP90 (HSPC), HSP70 (HSPA), HSP60(HSPD/E), HSP40 (DNAJ), and small heat shock proteins (HSPB), which are involved in a variety of physiological and pathological processes (79, 80).
HSP27, a key component of HSPB, protects cells from oxidative stress by inhibiting TFR1-mediated iron uptake and decreasing intracellular active iron (81). After ferroptosis was discovered, researchers quickly focused on the function of HSP27 in ferroptosis due to its critical role in controlling oxidative stress. Inhibiting HSP27 expression in vitro and in vivo can increase the anticancer activity of erastin mediated ferroptosis (82). Dihydroartemisinin (DHA) has cytotoxicity to various malignant tumors, but DHA attenuates ferroptosis in glioma cells through the PERK/ATF4/HSPA5 pathway (83). In pancreatic cancer, HSPA5 negatively regulated ferroptosis in human pancreatic ductal adenocarcinoma cells. In mechanism, HSPA5 reduces GPX4 protein degradation and subsequent lipid peroxidation, thus limiting the anticancer activity of gemcitabine (84). In addition, Zhou et al. (85) demonstrated that overexpression of DNAJB6 in esophageal cancer can promote the degradation of GSH, downregulate GPX4, enhance lipid peroxidation, and promote ferroptosis. Additionally, researchers have discovered that HSP90 and HSPA8 can promote GPX4 degradation via autophagy, thus promoting ferroptosis (86, 87). This indicates that the HSP family regulates ferroptosis through multiple pathways. More avenues may be discovered in the future.
The activation of heat shock factor 1 (HSF1) is triggered when cells are driven by external causes such as nutritional shortage, hypoxia, oxidative stress, and other similar conditions (88). Upon activation, HSF1 will translocate to the nucleus, thereby facilitating the transcriptional upregulation of HSPs (89). Since HSPs are directly regulated by HSF1, HSF1 plays a regulatory role in ferroptosis by regulating HSPs (90). Sun et al. (82) were the first to describe the regulatory role of the HSF1-HSPs signaling axis in ferroptosis in 2015. The researchers discovered that the suppression of the HSF1-HSPB1 pathway resulted in an augmentation of erastin-induced ferroptosis (82). A recent study discovered that HSF1 enhances prostate cancer cell resistance to ferroptosis by modulating HSPE1, and that HSF1 knockdown can promote prostate cancer cell sensitivity to RSL3 therapy (91). Furthermore, inhibiting HSF1 specifically can reverse the ferroptosis resistance generated by GPX4 suppression and greatly improve the in vitro sensitivity of resistant cancer cells and tumors to ferroptosis (92). Thus, it may be concluded that HSF1 is a promising target for ferroptosis.
Current researches have only revealed the function of some HSPs in iron metabolism and ferroptosis, and further researches on the effects of other HSPs are required in the future.
The PCBPs family includes five protein members, but current research mainly focuses on PCBP1 and PCBP2. As cytoplasmic iron chaperone protein, PCBP1 and PCBP2 bind to ferritin in the body, promoting iron loading into ferritin, thereby increasing the amount of iron loaded into ferritin (29, 93). The study discovered that mice lacking PCBP1 in hepatocytes exhibited defects in hepatic iron homeostasis, and ferroptosis occurred even in the absence of iron overload (94). In addition, the latest research found that PCBP1 can also inhibit the activation of ferritin autophagy by binding to BECN1 mRNA and inhibit the PUFAs peroxidation by restraining the expression of ALOX15 mRNA (95). As an RNA-binding protein, PCBP2 is able to bind and stabilize the expression of SLC7A11 mRNA, inhibiting malignancy ferroptosis and promoting tumor progression (96). In the future, enhancing the expression of PCBP1 and PCBP2 in malignant diseases may become a novel strategy for treating cancers.
Hepcidin is an iron-regulating hormone secreted by the liver. FPN, which is the only known cellular iron exporter, is found in duodenal epithelial cells, macrophages, and liver cells and can transport intracellular iron into plasma. The discovery of the hepcidin-FPN axis is a significant development in the field of systemic iron homeostasis. Hepcidin regulates FPN expression in two ways: inducing ubiquitination and endocytosis of FPN (97) and blocking FPN from opening the central cavity (98). A recent study revealed that hepcidin binding to FPN is coupled to iron binding, and the affinity of hepcidin increases 80 times in the presence of iron (99). Binding of hepcidin to FPN restricts iron export even if the transporter is in an outwardly open state (99). Subsequently, people began to pay attention to the role of hepcidin in diseases. It has been discovered that cigarette tar can accelerate the progression of atherosclerosis (AS) by inducing macrophage ferroptosis. Further research has confirmed that tar can upregulate the expression of macrophage hepcidin in AS plaques and downregulate the expression of FPN and SLC7A11. And this change can be reversed by ferroptosis inhibitors Fer-1 and DFO (100). Chlorogenic acid (CGA) is an intestinal protective agent. Studies have found that CGA reduces hepcidin production and increases the expression of FPN in the duodenum by inhibiting the liver IL-6/JAK2/STAT3 pathway, thereby restoring iron homeostasis and inhibiting ferroptosis (101). Current research on hepcidin and ferroptosis focuses primarily on benign diseases; in the future, it will be necessary to strengthen hepcidin research in malignant diseases.
NRF2 is a transcription factor that is crucial for cellular defense against oxidative stress and iron homeostasis. NRF2 regulates iron metabolism primarily via three pathways: Inhibiting the release of free iron from ferritin (102) and the expression of hepcidin (103); Promoting the expression of iron-related enzymes such as heme oxygenase-1 (HO-1) (104); Downregulating the expression of DMT1 to reduce iron uptake (105). Because it plays an important regulatory role in iron metabolism, NRF2 can also regulate ferroptosis. The expression of NRF2 is regulated by the p62-Keap1-NRF2 pathway. In 2016, Sun et al. (106) confirmed that activating the p62-Keap1-NRF2 pathway can inhibit ferroptosis in liver cancer cells. P62 is an autophagic protein, also known as SQSTM1 protein. The latest study found that autophagy inhibition can protect HepG2 cells from alcohol induced ferroptosis by activating the p62-Keap1-Nrf2 pathway (107). Theoretically, because NRF2 plays multiple functions in iron metabolism, it can theoretically regulate ferroptosis via various mechanisms. Finally, we summarize the mechanisms by which different genes modulate iron metabolism and their influence on ferroptosis (Table 1).
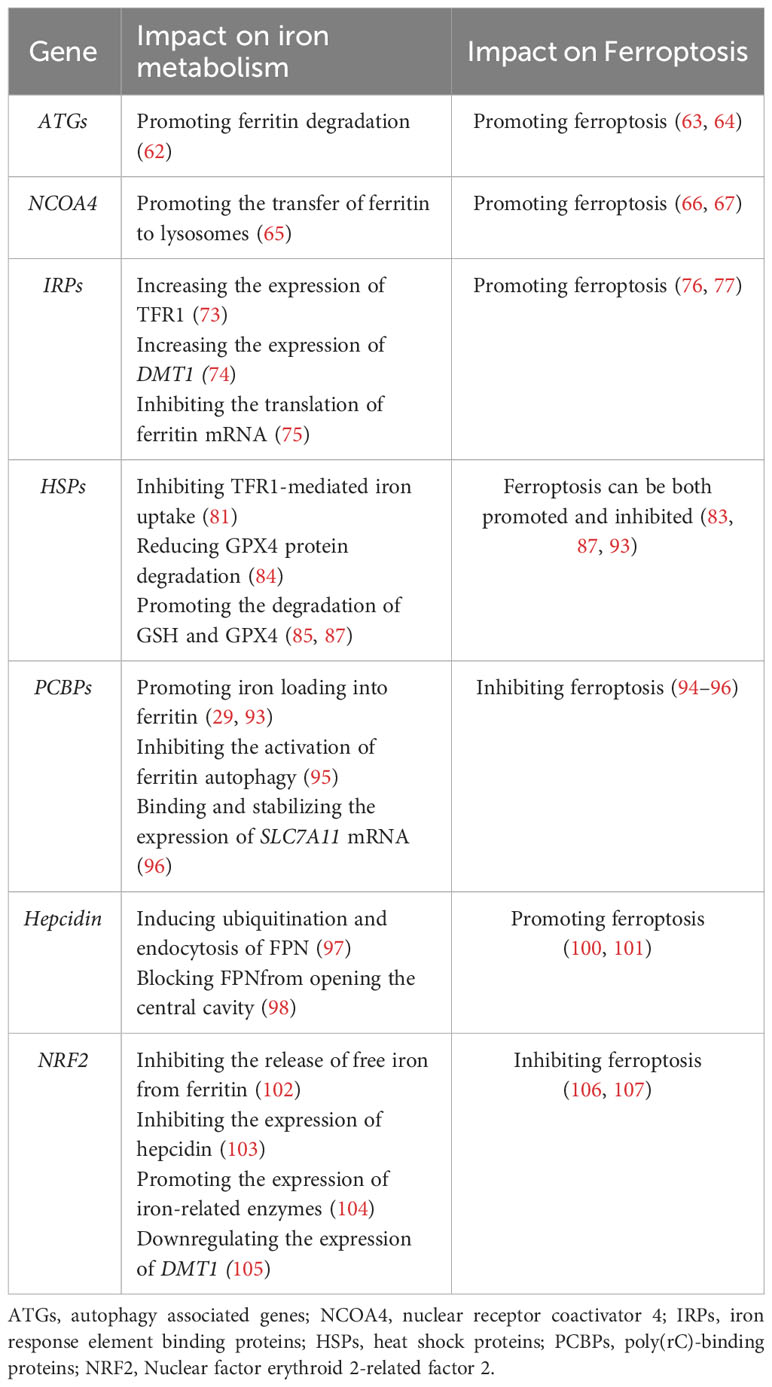
Table 1 Genes regulating iron metabolism and their regulation of ferroptosis.
LOXs are widely distributed in eukaryotes and prokaryotes, and it has received a lot of attention in eukaryotes. LOXs are enzymes that non-heme iron (or in some cases manganese) dependent, which can trigger the peroxidation of PUFAs to fatty acid hydroperoxides (108). Although most mammalian LOXs catalyze arachidonic acid and linoleic acid, some can also convert α-linolenic acid, eicosapentaenoic acid, and docosahexaenoic acid. There are six functional LOX genes in the human genome, designated LOX5, LOX12, LOX12B, LOX15, LOX15B and LOXE3. Multiple subtypes and subunits comprise LOXs, which are generalised lipoxygenases. Current research demonstrates that ALOXs is crucial for regulating ferroptosis. Microsomal glutathione S-transferase 1 (MGST1) is a membrane-bound transferase that not only inhibits apoptosis (109), but further studies have found that MGST1 reduces lipid peroxidation by combining with ALOX5, thereby inhibiting ferroptosis (110). ALOX15 can catalyze arachidonic acid to generate 15-Hydroperoxyeicosatetraenoic acid (15-HpETE), and 15-HpETE can promote the triggering of cardiomyocyte ferroptosis (111). In addition, the researchers discovered that the downregulation of ALOX15 expression in gastric cancer resulted in a decrease in ROS production, which ultimately led to a significant inhibition of ferroptosis, fostering tumor growth, and decreasing sensitivity to cisplatin and paclitaxel (112). In summary, ALOXs can play a significant role in the occurrence and regulation of ferroptosis by promoting the generation of phospholipid peroxidation products, boosting the production of ROS, and partaking in lipid peroxidation. Furthermore, ALOXs system-mediated ferroptosis is also regulated by P53 (113, 114).
ACSL can catalyze the combination of long-chain fatty acids and coenzyme A (CoA) to form long-chain acyl-CoA (acyl-CoA). Acyl-CoA participates in fatty acid metabolism, membrane modification, and numerous other physiological processes. There are five members of the animal ACSL family: ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6. According to the current research, ACSL3 and ACSL4 are capable of regulating ferroptosis (115). Oleic acid protects melanoma cells from ferroptosis in an ACSL3-dependent manner, thereby facilitating melanoma metastasis (116). Similarly, in triple-negative breast cancer cells, the secretion of oleic acid from adipocytes increased in the presence of ACSL3, which can inhibit lipid peroxidation and ferroptosis in breast cancer cells (117). Furthermore, in a mouse model of myocardial infarction, the release of miR-223-3p, a platelet-enriched miRNA, was increased. Whereas miR-223-3p leads to decreased secretion of stearic acid-phosphatidylcholine in cardiomyocytes by targeting ACSL3, and stearic acid-phosphatidylcholine can protect cardiomyocytes from ferroptosis (118). Consequently, ACSL3 can induce cells to develop ferroptosis resistance. In contrast, ACSL4 is a positive regulator of ferroptosis. In a study, Sebastian et al. found that in ACSL4 KO cells, lipid peroxidation was not detected even when cells were treated with RSL3, indicating that ACSL4 can promote ferroptosis (119). Phosphatidylethanolamine (PE), containing arachidonic acid (AA) and adrenic acid (AdA), was found to be the preferred substrate for oxidation by lipidomics approach, catalyzting the formation of Acyl-CoA, and then activates the corresponding fatty acids for lipid peroxidation (119, 120). Furthermore, ACSL4 levels are elevated in the livers of NAFLD patients, thereby promoting the fatty acid β-oxidation capacity of hepatocytes with a goal to reduce fat buildup (121). And studies have found that inhibition of fatty acid β-oxidation can induce ferroptosis (122). These findings suggest that ACSL4 can regulate ferroptosis through fatty acid β-oxidation. Due to the different catalysis of ACSL3 and ACSL4 within cells, they play opposite roles in ferroptosis.
MBOAT family is a group of genes that encode proteins playing crucial roles in lipid metabolism (123). These enzymes are involved in various aspects of lipid processing and modification within cellular membranes. According to the latest studies, MBOAT1 and MBOAT2 can inhibit ferroptosis (124). Jiang et al. (124) used genome-wide CRISPR activation screening technology to demonstrate that MBOAT1 and MBOAT2 can selectively convert MUFAs into lyso-phosphatidylethanolamine (lyso-PE), thereby reducing the content of intracellular PUFAs. Moreover, MOBAT1 and MOBAT2 are directly regulated by sex hormones. This discovery not only provides a new target for regulating ferroptosis, but also suggests that sex hormones play an essential role in ferroptosis.
LPCAT is one of the most important enzymes for maintaining phosphatidylcholine (PC) homeostasis. In fact, LPCAT is a subtype of enzyme in the MBOAT family (123). It controls phospholipid fatty acyl composition by catalyzing lysophospholipid sn-2 reacylation (125). Currently, four varieties of LPCAT have been described in the literature: LPCAT1, LPCAT2, LPCAT3, LPCAT4. Lpcat3’s role in lipid metabolism is by far the most evident. LPCAT3 is widely distributed in the human liver, intestines, and adipocytes (126, 127). LPCAT3 can regulate intestinal lipid absorption, lipoprotein secretion, and liver fat synthesis to maintain systemic lipid homeostasis. In a 2018 study, researchers found that knockdown of LPCAT3 in 3T3-L1 preadipocytes can reduce levels of polyunsaturated phospholipids and reduce the expression of genes related to fat production (128). In subsequent years, people began to pay attention to whether LPCAT3 has a regulatory effect on ferroptosis due to the antioxidant properties of polyunsaturated phospholipids. In 2022, Benjamin F Cravatt et al. (129) found that inhibiting the expression of LPCAT3 can inhibit the occurrence of ferroptosis. Researchers discovered in a recent study that LPCAT3 can promote the esterification of PUFAsinto phospholipids, provide raw materials for lipid peroxidation, and then promote ferroptosis (130). Furthermore, it has been established that ZEB promotes LPCAT3 transcription in a YAP-dependent manner (130). And YAP can also modulate the expression of the ACSL4 (131). The above research results suggest that regulating the expression of LPCAT3 and ACSL4 by targeting YAP expression, thus regulating ferroptosis, may be of great value in disease treatment.
Since its discovery in 1979, P53, a tumor suppressor gene, has been extensively studied for its role in tumors. P53 is essential for maintaining genome integrity, modulating cell cycle, and inhibiting angiogenesis (132, 133). In the past period, academics have discovered that P53 can regulate not only cell necrosis (134), apoptosis (135), and autophagy (136), but also ferroptosis (137). Complex signaling pathways are involved in the regulation of P53 on ferroptosis, and the results of distinct signaling pathways are inconsistent.
Already in 2015, researchers found that P533KR, a P53 acetylation deficient mutant, lost the function of regulating cell cycle, aging and apoptosis, but retained its tumor inhibitory function and the ability to regulate the expression of metabolic targets. It can inhibit cystine uptake and make cells sensitive to ferroptosis by inhibiting the transcription of SLC7A11 (a component of cystine/glutamate reverse transporter) (15). In recent years, it has also been discovered that P53 can block the expression of SLC7A11 in conditions such ovarian cancer (138) and acute lung injury (139). In the metabolism of polyamines, SAT1 (spermidine/spermine N1-acetyltransferase 1) plays a key role. According to Ou et al. (113), SAT1 deletion can prevent P53-mediated ferroptosis. Mechanistic analysis reveals that SAT1 induces ALOX15 expression to enhance ferroptosis (113). Additionally, P53 can control ferroptosis via ALOX12 (114). However, the specific regulatory mechanism has not been further explored. In a recent study, researchers found that vitamin K epoxide reductase complex subunit 1 like 1 (VKORC1L1) is a direct transcriptional target for P53 (140). The VKORC1L1 enzyme facilitates the synthesis of vitamin K hydroquinone, which is a completely reduced variant of vitamin K that effectively removes lipid peroxides (140, 141). And P53 promotes ferroptosis by inhibiting the expression of VKORC1L1 (140). Glutaminase 2 (GLS2) is a key enzyme in the catabolism of glutamine. Under the action of GLS2, glutamine is hydrolyzed into glutamic acid. In 2010, researchers discovered that GLS2, as the target gene of P53, can increase the content of GSH in cells and reduce the level of ROS, thereby protecting cells from oxidative stress damage (142, 143). The role of GLS2 in ferroptosis was not considered at the time because the phenomenon of ferroptosis had not yet been discovered. Interestingly, however, in 2015, researchers found that downregulation of GLS2 could inhibit ferroptosis induced by total amino acid or cystine deprivation (35). Moreover, Suzuki et al. (144) found that both GLS2-knockout mouse models and GLS2-deficient hepatocellular carcinoma cells confer significant resistance to ferroptosis. Further study of the mechanism found that GLS2 increases the generation of lipid ROSby promoting the conversion of glutamate to α-ketoglutarate, thereby promoting ferroptosis (144). Obviously, the conclusions of previous studies on the role of GLS2 in ferroptosis are contrary to the conclusions of recent studies. This phenomenon is worth pondering, and further research is needed to explain this paradox in the future.
The above described P53 positive regulation of ferroptosis related pathways, and then P53 expression can also inhibit the sensitivity of ferroptosis. In a 2019 study, Laura D et al. (145) used CRISPR/Cas9 genome editing, small molecule probes, and high-resolution time-lapse imaging techniques to find that P53 delays the response of cystine deprivation to ferroptosis through transcriptional regulation of P21. Therefore, the role of P53-P21 signal axis in ferroptosis was clarified. Subsequently, Venkatesh et al. (146) demonstrated again in tumor cells that unstable p53 inhibits the activity and expression of GPX4 by inhibiting p21 protein. In addition, P21 can also regulate ferroptosis independently of P53 (147). In colorectal cancer, P53 regulates ferroptosis by regulating the subcellular localization of dipeptidyl-peptidase-4 (DPP4) protein. P53 is able to prevent the nuclear accumulation of DPP4. In the absence of P53, DPP4 localizes to the plasma membrane and forms a complex with NADPH oxidase 1 on the plasma membrane, thereby increasing lipid peroxidation and iron deposition (148).
Regulation of ferroptosis by p53 is highly context-specific. Although current studies have shown that P53 plays an important role in regulating ferroptosis, the specific molecular mechanisms and interrelationships still need further exploration.
GPX4, which utilizes reduced GSH as a cofactor to detoxify lipid peroxides as lipid alcohols, thereby inhibiting ferroptosis, is one of the most notable defence mechanisms that cells have evolved to metabolise hazardous lipid peroxides (13). In addition, system x- c, which plays a core role in providing raw materials for the biosynthesis of GSH in cells, is also of concern (149). Therefore, GPX4 and SLC7A11 play a central role in resistance to ferroptosis.
GSH is a tripeptide found in all mammalian tissues, notably the liver, and is composed of glutamic acid, cysteine, and glycine held together by peptide bonds. Glutamate, cysteine and glycine are synthesized in the cytoplasm of liver cells under the catalysis of glutamate cysteine ligase (GCL) and glutathione synthase (GS) (150). GSH is the most abundant antioxidant in organisms, most of which are distributed in the cytoplasm, and a small part is distributed in the mitochondria and endoplasmic reticulum (151). There are two forms of glutathione in the human body: thiol-reduced (GSH) and disulfide-oxidized (GSSG) (152). Under normal circumstances, the content of GSSH in the human body is very low, and GSH mainly plays an antioxidant role.
GSH is essential for GPX4-catalyzed reactions. There are 8 kinds of GPX4 proteins in mammals, but only GPX4 has the effect of resisting lipid peroxidation (153). GPX4 is a selenocysteine-containing and glutathione-dependent enzyme. By means of GSH, it can catalyze the conversion of particular lipid hydroperoxides into lipid alcohols.
Selenocysteine is the active site of GPX4, and selenocysteine shuttles back and forth between the reduced state (Se-H) and the oxidized state (Se-OH) to complete the biological function. This process is mainly divided into three steps: First, the reduced GPX4(GPX4-Se-H) is oxidized to the oxidized GPX4(GPX4-Se-OH) by lipid peroxides, which are reduced to the corresponding alcohols; Then, GPX4-Se-OH is reduced by the reducing substrate GSH to generate selenium- glutathione adduct (GPX4-Se-SG); Finally, GPX4-Se-SG reacts with GSH again, transforms into GPX4-Se-H, and produces GSSH (13, 153). After the cycle described above, GSH as an electronics body, toxic lipid peroxides are converted into benign alcohols, so GPX4 is known as the gatekeeper of ferroptosis and plays an imperative part in suppressing lipid peroxidation. Finally, electrons are provided by NADPH, and GSSH is reduced to GSH under the catalysis of glutathione reductase (GR). The process of ubiquitination and dephosphorylation of GPX4 has also been found to facilitate the initiation of ferroptosis (154, 155).
The synthesis of GSH requires cysteine, and cysteine itself also has reducibility (156). However, extracellular cysteine is highly unstable and rapidly oxidised to cystine (the oxidised dimer form of cysteine) owing to the high level of oxidation in the extracellular environment. Therefore, most cells need cystine transporter system x- c to transport extracellular cystine to the intracellular synthesis of GSH.
The system x- c consists of two subunits connected by a disulfide bond, the light chain subunit SLC7A11 (also commonly known as xCT) and the heavy chain subunit solute carrier family 3 member 2 (SLC3A2) respectively. SLC7A11 is a 12-transmembrane protein with both N- and C-termini inside the cell (157). SLC7A11 is highly specific for cystine and glutamate, and is responsible for the main transport work, importing cystine into the cell at a ratio of 1:1 and exporting glutamate outside the cell (149). SLC3A2 is a single transmembrane protein (157), as a chaperone, it is critical to maintain the stability and membrane localization of SLC7A11 protein (158, 159). The cystine transported into the cell is reduced to cysteine by NADPH providing electrons, and then participates in the synthesis of GSH (160). Therefore, the overexpression of SLC7A11 in tumour cells can stimulate GSH synthesis and result in ferroptosis resistance (15). Figure 2 shows the role of SLC7A11 in GSH synthesis, and the metabolic process of GSH and GPX4 in cells.
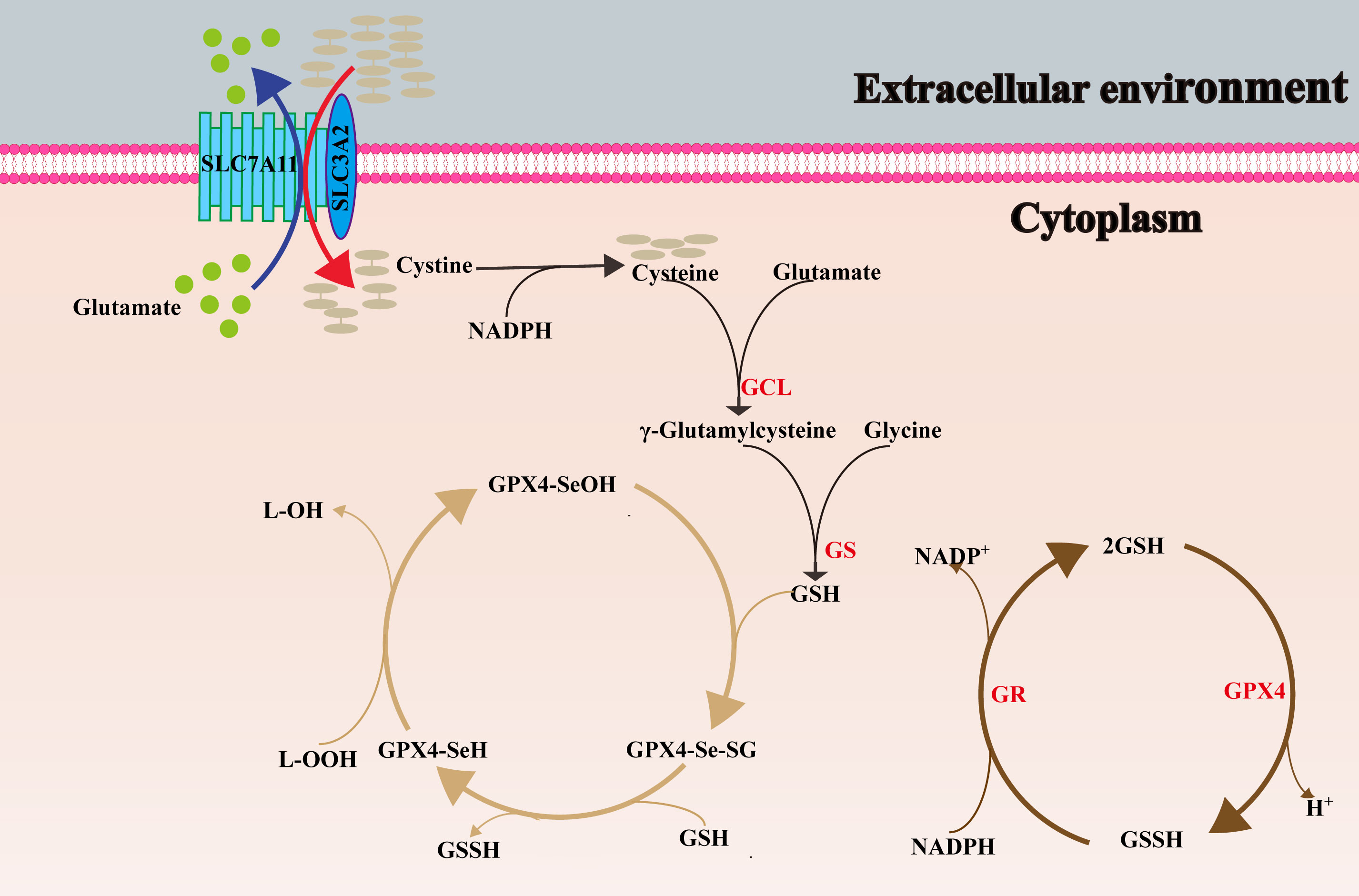
Figure 2 The synthesis process of GSH, and the conversion between oxidized and reduced GPX4. SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GCL, glutamate cysteine ligase; GS, glutathione synthase; L–OOH, lipid peroxides; L-OH, alcohol; SeOH, selenenic acid; Se–H, selenol; Se-SG, selenium- glutathione adduct; GR, glutathione reductase.
The prevailing perspective in the early stages of research was that GPX4/GSH played a significant role as the primary mechanism for monitoring and regulating ferroptosis. Nevertheless, as research progresses, it has become evident that the responsiveness of various cancer cell lines to GPX4 inhibitors exhibits significant variability (161). This observation implies the existence of other variables that govern the susceptibility of cells to ferroptosis. To verify this result, Sebastian Doll et al. (162) extracted a cDNA expression library from the ferroptosis-resistant cell line MCF7 and screened complementary genes for GPX4 deletion, thereby identifying FSP1 as an unidentified anti-ferroptosis gene. Subsequent investigation has revealed that myristoylation of FSP1 is seemingly required for its anti-ferroptotic function (162). Myristoylation facilitates the recruitment of FSP1 to the plasma membrane (163). Once localized, FSP1 employs NAD(P)H to facilitate the catalytic regeneration of non-mitochondrial coenzyme Q10 (CoQ10). Ultimately, FSP1 mediates the prevention of ferroptosis by means of CoQ10 (162). In 2019, a groundbreaking discovery was made by researchers, revealing the coordination between the NAD(P)H-FSP1-CoQ10 axis and NAD(P)H-GPX4-GSH in the regulation of ferroptosis (162, 163). Furthermore, in recent years, scholars have made significant progress in the development of diverse small molecule inhibitors targeting FSP1 (162, 164). This achievement has laid a solid foundation for the potential utilization of FSP1 as a promising therapeutic approach for various diseases.
ROS are residual products of aerobic metabolism in the organism, including superoxide anion(O2–), hydroxyl radicals (•OH), peroxyl free radical (ROO•), hydrogen peroxide (H2O2) and singlet oxygen (1O2). ROS are produced primarily by mitochondrial metabolism (165) and NADPH oxidase on the cell membrane (166). Appropriate levels of ROS are crucial signaling molecules that play essential roles in signal transduction (167), cellular immune function (168), cell proliferation and cell repair (169). Nonetheless, excessive levels of ROS are toxic, triggering DNA damage (170), inflammatory responses (171), and cell death (172). Lipids dominated by PUFAs are oxidized many times by ROS to produce lipid peroxide, which leads to ferroptosis (13). Therefore, reducing intracellular ROS sources has become a strategy for disease treatment. Finally, as shown in Figure 3, we summarized the mechanism and process of ferroptosis.
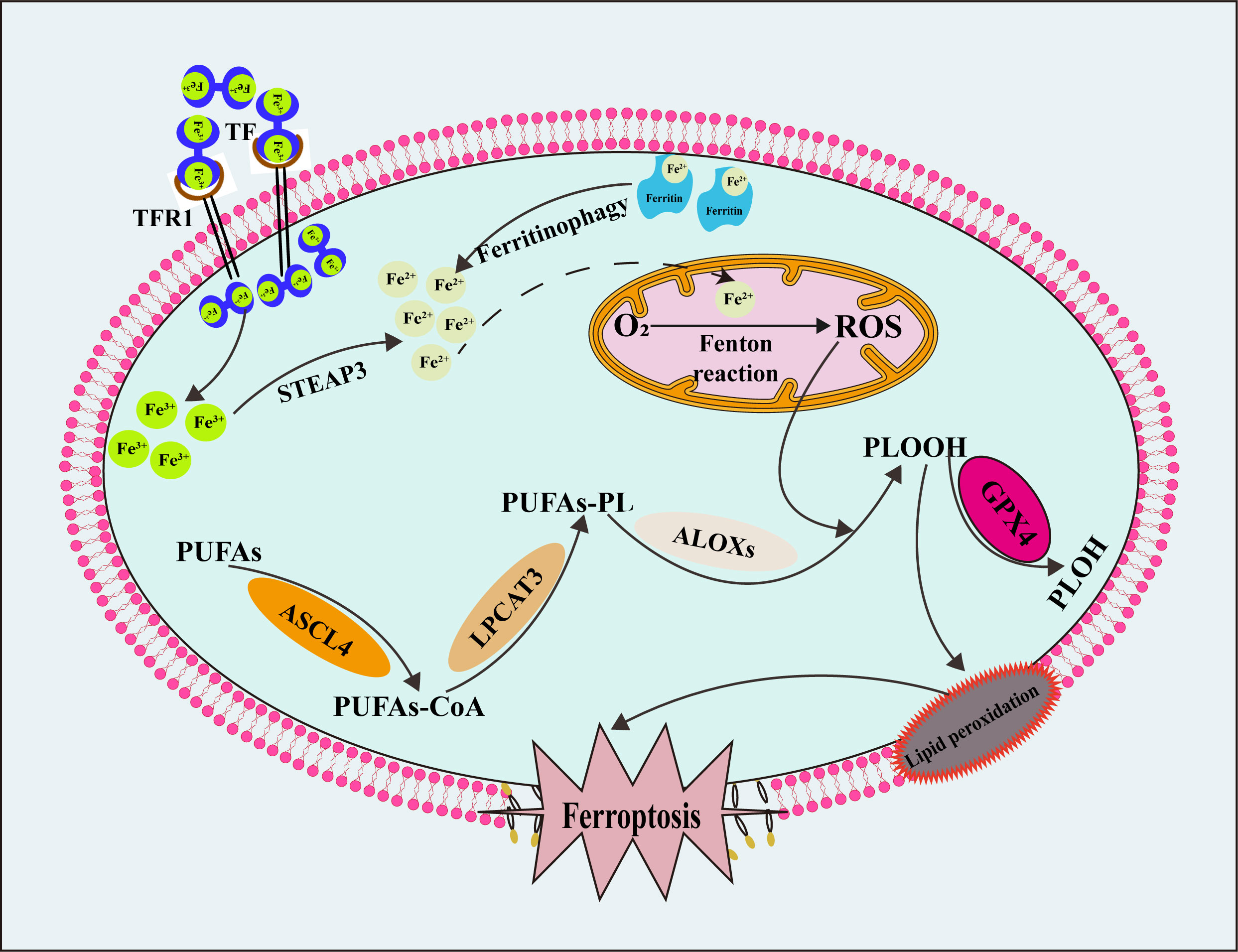
Figure 3 The mechanism and process of ferroptosis. TF, transferrin; TFR1, transferrin receptor 1; STEAP3, six transmembrane epithelial antigen of prostate 3; ROS, reactive oxygen species; PUFAs, polyunsaturated fatty acids; PUFAs-CoA, polyunsaturated fatty acids- coenzyme A; PUFAs-PL, polyunsaturated fatty acids- phospholipid; PLOOH, phospholipid peroxides; PLOH, phospholipid alcohol; ASCL4, long-chain acyl-CoA synthetase 4; LPCAT3, phosphatidylcholine acyltransferase 3; ALOXs, arachidonate lipoxygenases.
In the past decade, ferroptosis research has exploded, substantially enhancing scientists’ understanding of the phenomenon. Ferroptosis is the result of the joint action of many biological pathways in the body. The present investigations have unveiled the existence of three primary biological axes that play a crucial role in the regulation of ferroptosis. The first is the biological axis of iron metabolism. The biological axis of iron metabolism encompasses three key components: the regulation of iron absorption, involving proteins such as IRPs and HSPs (73, 81); the regulation of iron transport, involving the protein hepcidin (97); and the regulation of iron storage, involving genes associated with autophagy (ATGs and NCOA4) (62, 65), as well as PCBPs and NRF2 (93, 102). The second biological axis involved in lipid metabolism encompasses significant genes such as ACSL4, LPCAT3, ALOXs, P53, and the recently identified MBOAT1 and MBOAT2 (111, 113, 122, 124, 128). Lastly, the antioxidant biological axis encompasses components NAD(P)H-FSP1-CoQ10 axis and NAD(P)H-GPX4-GSH (153, 162, 163). Therefore, we summarized the mechanisms and regulations of ferroptosis from these three aspects. It should be pointed out that even a specific gene can affect multiple links in ferroptosis metabolism. IRPs, for instance, can not only promote the expression of DMT1 and TFR1 (73, 74), but also inhibit the synthesis of ferritin (75), and finally increase the amount of iron in the labile iron pool in cells by acting on three aspects of iron metabolism simultaneously. Among them, the most complex one is P53, which can both promote ferroptosis and inhibit ferroptosis through various ways. But the conclusions of the study on GLS2 are puzzling. In early studies, researchers found that GLS2 can play an antioxidant role (142). In recent years, studies have found that GLS2 can promote lipid peroxidation in a variety of cells (144). Different signaling pathways, intracellular microenvironment, and post-transcriptional modification may account for this difference. All of the aforementioned phenomena demonstrate that ferroptosis is regulated by a complex network in cells, and it is insufficient to explain this phenomenon from a single perspective. Furthermore, many studies are limited to the cellular level, but not the in vivo level, ignoring the influence of the complex physiological environment and the natural disease development process.
The correlation between ferroptosis and diseases has also been extensively studied. As a potential therapeutic strategy, promoting ferroptosis in malignant diseases and inhibiting ferroptosis in benign diseases has been investigated. Sorafenib is a first-line drug for targeted therapy of hepatocellular carcinoma. Later studies have found that sorafenib can promote ferroptosis in hepatocellular carcinoma (173). The anticancer actions of sorafenib are manifested through the inhibition of the x- c system (174). Additionally, the research investigation revealed a correlation between resistance to sorafenib and cellular resistance to ferroptosis (175). Therefore, in recent studies, researchers have studied the synergistic effect of various chemicals and sorafenib on Ferroptosis, hoping to achieve therapeutic effect by promoting ferroptosis of hepatocellular carcinoma (176, 177). Similarly, in osteoarthritis, researchers found that the occurrence of ferroptosis in chondrocytes promoted the progress of osteoarthritis, so inhibiting ferroptosis in animal models significantly reduced the sensitivity of chondrocytes to oxidative stress, and improved the progress of the disease (178). D-mannose is a monosaccharide naturally occurring in plants. Zhou et al. (179) found that D-mannose reduced the sensitivity of chondrocytes to ferroptosis by inhibiting HIF-2α, thereby alleviating osteoarthritis progression and cartilage degeneration. Therefore, the future development of D-mannose-based drugs to treat osteoarthritis will be beneficial for patients. Furthermore, current studies have established that GPX4 and SLC7A11 play an important role in limiting lipid peroxidation; therefore, inhibiting the expression of GPX4 and SLC7A11 in tumor patients can promote tumor ferroptosis in theory. Unfortunately, however, early embryonic lethality was found in GPX4 knockout mice (180, 181), suggesting that in addition to playing a central role in limiting ferroptosis, GPX4 also plays other irreplaceable functions in vivo. Although some experiments have achieved ideal results in cell and animal experiments, the translation of theoretical results to clinical practice is still far away.
So far, no specific and rapid monitoring indicators of ferroptosis in living cells or tissues have been found, so one of the urgent problems is to find a gold standard for detecting ferroptosis. BODIPY-C11 and LiperFluo are currently relatively high-accuracy detection methods for detecting intracellular lipid peroxidation (182, 183). Other methods include detection of ferroptosis-related metabolic indicators, such as intracellular Fe2+, GPX4, GSH, ROS, MDA, and 4-HNE levels. However, changes in the above indicators may also occur in other types of cell death, such as apoptosis and autophagy (43). Therefore, in actual scientific research work, researchers use a variety of small molecule regulators of cell death to exclude the influence of other forms of cell death. Moreover, different types of cell death may have similar initiators and regulators, for example, GPX4 can also regulate apoptosis and necrosis (184, 185). Moreover, inhibition of ferroptosis may activate other cell death signaling pathways, which ultimately lead to cell death as well (186).
Current research on ferroptosis still faces many challenges and knowledge gaps, but scientists are continually working to improve our understanding of this mode of cell death. With further research and exploration, we can expect a deeper understanding of ferroptosis and the development of relevant therapeutic strategies and applications.
XZ: Writing – original draft, Writing – review & editing. ZL: Methodology, Writing – review & editing. MW: Methodology, Writing – review & editing. YG: Methodology, Writing – review & editing. XW: Visualization, Writing – review & editing. KL: Methodology, Writing – review & editing. SH: Visualization, Writing – review & editing. RL: Writing – original draft, Writing – review & editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This manuscript was supported by 2022 Henan Province Medical Science and Technology Joint Construction Project (LHGJ20220317), 2023 Provincial Natural Science Foundation Project of Henan Province (232300420038), 2023 Provincial Natural Science Foundation Youth Fund Project of Henan Province (232300420249) and The present study was supported by The Key Scientific Research Project Plan of Henan University (Grant No.20A320037) and 2022 Henan Province Key R&D and Promotion Special Support Project (The role of OSBP2 in the regulation of malignant phenotype of pancreatic cancer).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
2. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
3. Koren E, Fuchs Y. Modes of regulated cell death in cancer. Cancer Discovery (2021) 11:245–65. doi: 10.1158/2159-8290.CD-20-0789
4. Cobine PA, Brady DC. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell (2022) 82:1786–7. doi: 10.1016/j.molcel.2022.05.001
5. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
6. Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest (2013) 123:4423–34. doi: 10.1172/JCI67867
7. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal (2018) 29:61–74. doi: 10.1089/ars.2017.7115
8. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
9. Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med (2021) 13:e14351. doi: 10.15252/emmm.202114351
10. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer (2022) 21:47. doi: 10.1186/s12943-022-01530-y
11. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
12. Rohr-Udilova N, Sieghart W, Eferl R, Stoiber D, Björkhem-Bergman L, Eriksson LC, et al. Antagonistic effects of selenium and lipid peroxides on growth control in early hepatocellular carcinoma. Hepatology (2012) 55:1112–21. doi: 10.1002/hep.24808
13. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics (2019) 19:e1800311. doi: 10.1002/pmic.201800311
14. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12:599–620. doi: 10.1007/s13238-020-00789-5
15. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520:57–62. doi: 10.1038/nature14344
16. Yuan Y, Zhai Y, Chen J, Xu X, Wang H. Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating nrf2/SLC7A11/GPX4 axis. Biomolecules (2021) 11:923. doi: 10.3390/biom11070923
17. Xu Y, Li Y, Li J, Chen W. Ethyl carbamate triggers ferroptosis in liver through inhibiting GSH synthesis and suppressing Nrf2 activation. Redox Biol (2022) 53:102349. doi: 10.1016/j.redox.2022.102349
18. Mackenzie B, Garrick MD, Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol (2005) 289:G981–986. doi: 10.1152/ajpgi.00363.2005
19. de Jong G, van Dijk JP, van Eijk HG. The biology of transferrin. Clin Chim Acta (1990) 190:1–46. doi: 10.1016/0009-8981(90)90278-Z
20. Lynch SR, Cook JD. Interaction of vitamin C and iron. Ann N Y Acad Sci (1980) 355:32–44. doi: 10.1111/j.1749-6632.1980.tb21325.x
21. McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science (2001) 291:1755–9. doi: 10.1126/science.1057206
22. Herrmann T, Muckenthaler M, van der Hoeven F, Brennan K, Gehrke SG, Hubert N, et al. Iron overload in adult Hfe-deficient mice independent of changes in the steady-state expression of the duodenal iron transporters DMT1 and Ireg1/ferroportin. J Mol Med (Berl) (2004) 82:39–48. doi: 10.1007/s00109-003-0508-x
23. Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med (2019) 133:55–63. doi: 10.1016/j.freeradbiomed.2018.07.020
24. Wu J, Yang JJ, Cao Y, Li H, Zhao H, Yang S, et al. Iron overload contributes to general anaesthesia-induced neurotoxicity and cognitive deficits. J Neuroinflamm (2020) 17:110. doi: 10.1186/s12974-020-01777-6
25. Huang S, Wang Y, Xie S, Lai Y, Mo C, Zeng T, et al. Isoliquiritigenin alleviates liver fibrosis through caveolin-1-mediated hepatic stellate cells ferroptosis in zebrafish and mice. Phytomedicine (2022) 101:154117. doi: 10.1016/j.phymed.2022.154117
26. Ford GC, Harrison PM, Rice DW, Smith JM, Treffry A, White JL, et al. Ferritin: design and formation of an iron-storage molecule. Philos Trans R Soc Lond B Biol Sci (1984) 304:551–65. doi: 10.1098/rstb.1984.0046
27. Andrews SC. The Ferritin-like superfamily: Evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim Biophys Acta (2010) 1800:691–705. doi: 10.1016/j.bbagen.2010.05.010
28. He D, Hughes S, Vanden-Hehir S, Georgiev A, Altenbach K, Tarrant E, et al. Structural characterization of encapsulated ferritin provides insight into iron storage in bacterial nanocompartments. Elife (2016) 5:e18972. doi: 10.7554/eLife.18972
29. Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem (2013) 288:17791–802. doi: 10.1074/jbc.M113.460253
30. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
31. Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab (2015) 22:777–87. doi: 10.1016/j.cmet.2015.09.006
32. Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ (2021) 28:1548–62. doi: 10.1038/s41418-020-00685-9
33. Nemeth E, Ganz T. Hepcidin-ferroportin interaction controls systemic iron homeostasis. Int J Mol Sci (2021) 22:6493. doi: 10.3390/ijms22126493
34. Dev S, Babitt JL. Overview of iron metabolism in health and disease. Hemodial Int (2017) 21 Suppl 1:S6–s20. doi: 10.1111/hdi.12542
35. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
36. Lambert LA, Mitchell SL. Molecular evolution of the transferrin receptor/glutamate carboxypeptidase II family. J Mol Evol (2007) 64:113–28. doi: 10.1007/s00239-006-0137-4
37. Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem (1999) 274:20826–32. doi: 10.1074/jbc.274.30.20826
38. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol (2007) 69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337
39. Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci (2020) 260:118305. doi: 10.1016/j.lfs.2020.118305
40. Tong S, Hong Y, Xu Y, Sun Q, Ye L, Cai J, et al. TFR2 regulates ferroptosis and enhances temozolomide chemo-sensitization in gliomas. Exp Cell Res (2023) 424:113474. doi: 10.1016/j.yexcr.2023.113474
41. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
42. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev (2014) 2014:360438. doi: 10.1155/2014/360438
43. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev (2019) 2019:5080843. doi: 10.1155/2019/5080843
44. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U.S.A. (2016) 113:E4966–4975. doi: 10.1073/pnas.1603244113
45. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
46. Mayr L, Grabherr F, Schwärzler J, Reitmeier I, Sommer F, Gehmacher T, et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun (2020) 11:1775. doi: 10.1038/s41467-020-15646-6
47. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol (2019) 26:420–432.e429. doi: 10.1016/j.chembiol.2018.11.016
48. Blomme A, Ford CA, Mui E, Patel R, Ntala C, Jamieson LE, et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat Commun (2020) 11:2508. doi: 10.1038/s41467-020-16126-7
49. Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM, et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife (2020) 9:e54166. doi: 10.7554/eLife.54166
50. Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol (2021) 9:637162. doi: 10.3389/fcell.2021.637162
51. Berger S, Weichert H, Porzel A, Wasternack C, Kühn H, Feussner I. Enzymatic and non-enzymatic lipid peroxidation in leaf development. Biochim Biophys Acta (2001) 1533:266–76. doi: 10.1016/S1388-1981(01)00161-5
52. Anthonymuthu TS, Kenny EM, Bayır H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res (2016) 1640:57–76. doi: 10.1016/j.brainres.2016.02.006
53. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
54. Xu Y, Liu Y, Li K, Yuan D, Yang S, Zhou L, et al. COX-2/PGE2 pathway inhibits the ferroptosis induced by cerebral ischemia reperfusion. Mol Neurobiol (2022) 59:1619–31. doi: 10.1007/s12035-021-02706-1
55. Hanna J, Guerra-Moreno A, Ang J, Micoogullari Y. Protein degradation and the pathologic basis of disease. Am J Pathol (2019) 189:94–103. doi: 10.1016/j.ajpath.2018.09.004
56. Cui S, Liu X, Liu Y, Hu W, Ma K, Huang Q, et al. Autophagosomes defeat ferroptosis by decreasing generation and increasing discharge of free fe(2+) in skin repair cells to accelerate diabetic wound healing. Adv Sci (Weinh) (2023) 10:e2300414. doi: 10.1002/advs.202300414
57. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis (2019) 10:822. doi: 10.1038/s41419-019-2064-5
58. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
59. Klionsky DJ, Cregg JM, Dunn WA Jr., Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell (2003) 5:539–45. doi: 10.1016/S1534-5807(03)00296-X
60. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer (2020) 19:12. doi: 10.1186/s12943-020-1138-4
61. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
62. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
63. Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. BioMed Pharmacother (2019) 109:2043–53. doi: 10.1016/j.biopha.2018.11.030
64. Huang Y, Lin J, Xiong Y, Chen J, Du X, Liu Q, et al. Superparamagnetic iron oxide nanoparticles induce ferroptosis of human ovarian cancer stem cells by weakening cellular autophagy. J BioMed Nanotechnol (2020) 16:1612–22. doi: 10.1166/jbn.2020.2991
65. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509:105–9. doi: 10.1038/nature13148
66. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol (2014) 16:1069–79. doi: 10.1038/ncb3053
67. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: A new mechanism of action. ACS Cent Sci (2021) 7:980–9. doi: 10.1021/acscentsci.0c01592
68. Das NK, Jain C, Sankar A, Schwartz AJ, Santana-Codina N, Solanki S, et al. Modulation of the HIF2α-NCOA4 axis in enterocytes attenuates iron loading in a mouse model of hemochromatosis. Blood (2022) 139:2547–52. doi: 10.1182/blood.2021013452
69. Li W, Li W, Wang Y, Leng Y, Xia Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Discovery (2021) 7:267. doi: 10.1038/s41420-021-00656-0
70. Zhang Y, Kong Y, Ma Y, Ni S, Wikerholmen T, Xi K, et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene (2021) 40:1425–39. doi: 10.1038/s41388-020-01622-3
71. Ghosh MC, Tong WH, Zhang D, Ollivierre-Wilson H, Singh A, Krishna MC, et al. Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice. Proc Natl Acad Sci U.S.A. (2008) 105:12028–33. doi: 10.1073/pnas.0805361105
72. Philpott CC, Klausner RD, Rouault TA. The bifunctional iron-responsive element binding protein/cytosolic aconitase: the role of active-site residues in ligand binding and regulation. Proc Natl Acad Sci U.S.A. (1994) 91:7321–5. doi: 10.1073/pnas.91.15.7321
73. Jeong SM, Lee J, Finley LW, Schmidt PJ, Fleming MD, Haigis MC. SIRT3 regulates cellular iron metabolism and cancer growth by repressing iron regulatory protein 1. Oncogene (2015) 34:2115–24. doi: 10.1038/onc.2014.124
74. Galy B, Ferring-Appel D, Kaden S, Gröne HJ, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab (2008) 7:79–85. doi: 10.1016/j.cmet.2007.10.006
75. Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci (2004) 1012:1–13. doi: 10.1196/annals.1306.001
76. Zhang T, Sun L, Hao Y, Suo C, Shen S, Wei H, et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat Cancer (2022) 3:75–89. doi: 10.1038/s43018-021-00299-1
77. Fan H, Ai R, Mu S, Niu X, Guo Z, Liu L. MiR-19a suppresses ferroptosis of colorectal cancer cells by targeting IREB2. Bioengineered (2022) 13:12021–9. doi: 10.1080/21655979.2022.2054194
78. Li X, Wu L, Tian X, Zheng W, Yuan M, Tian X, et al. miR-29a-3p in exosomes from heme oxygenase-1 modified bone marrow mesenchymal stem cells alleviates steatotic liver ischemia-reperfusion injury in rats by suppressing ferroptosis via iron responsive element binding protein 2. Oxid Med Cell Longev (2022) 2022:6520789. doi: 10.1155/2022/6520789
79. Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci (2017) 38:226–56. doi: 10.1016/j.tips.2016.11.009
80. Zuehlke AD, Moses MA, Neckers L. Heat shock protein 90: its inhibition and function. Philos Trans R Soc Lond B Biol Sci (2018) 373:20160527. doi: 10.1098/rstb.2016.0527
81. Chen H, Zheng C, Zhang Y, Chang YZ, Qian ZM, Shen X. Heat shock protein 27 downregulates the transferrin receptor 1-mediated iron uptake. Int J Biochem Cell Biol (2006) 38:1402–16. doi: 10.1016/j.biocel.2006.02.006
82. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene (2015) 34:5617–25. doi: 10.1038/onc.2015.32
83. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res (2019) 38:402. doi: 10.1186/s13046-019-1413-7
84. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
85. Jiang B, Zhao Y, Shi M, Song L, Wang Q, Qin Q, et al. DNAJB6 promotes ferroptosis in esophageal squamous cell carcinoma. Dig Dis Sci (2020) 65:1999–2008. doi: 10.1007/s10620-019-05929-4
86. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U.S.A. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
87. Zhou J, Tan Y, Hu L, Fu J, Li D, Chen J, et al. Inhibition of HSPA8 by rifampicin contributes to ferroptosis via enhancing autophagy. Liver Int (2022) 42:2889–99. doi: 10.1111/liv.15459
88. Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol (2010) 11:545–55. doi: 10.1038/nrm2938
89. Neef DW, Jaeger AM, Gomez-Pastor R, Willmund F, Frydman J, Thiele DJ. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep (2014) 9:955–66. doi: 10.1016/j.celrep.2014.09.056
90. Aolymat I, Hatmal MM, Olaimat AN. The emerging role of heat shock factor 1 (HSF1) and heat shock proteins (HSPs) in ferroptosis. Pathophysiology (2023) 30:63–82. doi: 10.3390/pathophysiology30010007
91. Jia G, Wu W, Chen L, Yu Y, Tang Q, Liu H, et al. HSF1 is a novel prognostic biomarker in high-risk prostate cancer that correlates with ferroptosis. Discovery Oncol (2023) 14:107. doi: 10.1007/s12672-023-00715-1
92. Brown CW, Chhoy P, Mukhopadhyay D, Karner ER, Mercurio AM. Targeting prominin2 transcription to overcome ferroptosis resistance in cancer. EMBO Mol Med (2021) 13:e13792. doi: 10.15252/emmm.202013792
93. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science (2008) 320:1207–10. doi: 10.1126/science.1157643
94. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology (2021) 73:1176–93. doi: 10.1002/hep.31328
95. Lee J, You JH, Roh JL. Poly(rC)-binding protein 1 represses ferritinophagy-mediated ferroptosis in head and neck cancer. Redox Biol (2022) 51:102276. doi: 10.1016/j.redox.2022.102276
96. Shen L, Zhang J, Zheng Z, Yang F, Liu S, Wu Y, et al. PHGDH inhibits ferroptosis and promotes Malignant progression by upregulating SLC7A11 in bladder cancer. Int J Biol Sci (2022) 18:5459–74. doi: 10.7150/ijbs.74546
97. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (2004) 306:2090–3. doi: 10.1126/science.1104742
98. Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood (2018) 131:899–910. doi: 10.1182/blood-2017-05-786590
99. Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature (2020) 586:807–11. doi: 10.1038/s41586-020-2668-z
100. Bao X, Luo X, Bai X, Lv Y, Weng X, Zhang S, et al. Cigarette tar mediates macrophage ferroptosis in atherosclerosis through the hepcidin/FPN/SLC7A11 signaling pathway. Free Radic Biol Med (2023) 201:76–88. doi: 10.1016/j.freeradbiomed.2023.03.006
101. Zhao Y, Wang C, Yang T, Wang H, Zhao S, Sun N, et al. Chlorogenic acid alleviates chronic stress-induced duodenal ferroptosis via the inhibition of the IL-6/JAK2/STAT3 signaling pathway in rats. J Agric Food Chem (2022) 70:4353–61. doi: 10.1021/acs.jafc.2c01196
102. Anandhan A, Dodson M, Shakya A, Chen J, Liu P, Wei Y, et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv (2023) 9:eade9585. doi: 10.1126/sciadv.ade9585
103. Lim PJ, Duarte TL, Arezes J, Garcia-Santos D, Hamdi A, Pasricha SR, et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat Metab (2019) 1:519–31. doi: 10.1038/s42255-019-0063-6
104. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci (2016) 73:3221–47. doi: 10.1007/s00018-016-2223-0
105. Huang S, Wang Y, Xie S, Lai Y, Mo C, Zeng T, et al. Hepatic TGFβr1 deficiency attenuates lipopolysaccharide/D-galactosamine-induced acute liver failure through inhibiting GSK3β-nrf2-mediated hepatocyte apoptosis and ferroptosis. Cell Mol Gastroenterol Hepatol (2022) 13:1649–72. doi: 10.1016/j.jcmgh.2022.02.009
106. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology (2016) 63:173–84. doi: 10.1002/hep.28251
107. Zhao Y, Lu J, Mao A, Zhang R, Guan S. Autophagy Inhibition Plays a Protective Role in Ferroptosis Induced by Alcohol via the p62-Keap1-Nrf2 Pathway. J Agric Food Chem (2021) 69:9671–83. doi: 10.1021/acs.jafc.1c03751
108. Chrisnasari R, Hennebelle M, Vincken JP, van Berkel WJH, Ewing TA. Bacterial lipoxygenases: Biochemical characteristics, molecular structure and potential applications. Biotechnol Adv (2022) 61:108046. doi: 10.1016/j.biotechadv.2022.108046
109. Zeng B, Ge C, Li R, Zhang Z, Fu Q, Li Z, et al. Knockdown of microsomal glutathione S-transferase 1 inhibits lung adenocarcinoma cell proliferation and induces apoptosis. BioMed Pharmacother (2020) 121:109562. doi: 10.1016/j.biopha.2019.109562
110. Kuang F, Liu J, Xie Y, Tang D, Kang R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol (2021) 28:765–775.e765. doi: 10.1016/j.chembiol.2021.01.006
111. Cai W, Liu L, Shi X, Liu Y, Wang J, Fang X, et al. Alox15/15-hpETE aggravates myocardial ischemia-reperfusion injury by promoting cardiomyocyte ferroptosis. Circulation (2023) 147:1444–60. doi: 10.1161/CIRCULATIONAHA.122.060257
112. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer (2020) 19:43. doi: 10.1186/s12943-020-01168-8
113. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U.S.A. (2016) 113:E6806–e6812. doi: 10.1073/pnas.1607152113
114. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol (2019) 21:579–91. doi: 10.1038/s41556-019-0305-6
115. Yang Y, Zhu T, Wang X, Xiong F, Hu Z, Qiao X, et al. ACSL3 and ACSL4, distinct roles in ferroptosis and cancers. Cancers (Basel) (2022) 14:5896. doi: 10.3390/cancers14235896
116. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature (2020) 585:113–8. doi: 10.1038/s41586-020-2623-z
117. Xie Y, Wang B, Zhao Y, Tao Z, Wang Y, Chen G, et al. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J Hematol Oncol (2022) 15:72. doi: 10.1186/s13045-022-01297-1
118. Miao S, Zhang Q, Ding W, Hou B, Su Z, Li M, et al. Platelet internalization mediates ferroptosis in myocardial infarction. Arterioscler Thromb Vasc Biol (2023) 43:218–30. doi: 10.1161/ATVBAHA.122.318161
119. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
120. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol (2022) 24:88–98. doi: 10.1038/s41556-021-00818-3
121. Duan J, Wang Z, Duan R, Yang C, Zhao R, Feng Q, et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology (2022) 75:140–53. doi: 10.1002/hep.32148
122. Chen J, Wu K, Lei Y, Huang M, Cheng L, Guan H, et al. Inhibition of fatty acid β-oxidation by fatty acid binding protein 4 induces ferroptosis in HK2 cells under high glucose conditions. Endocrinol Metab (Seoul) (2023) 38:226–44. doi: 10.3803/EnM.2022.1604
123. Pierce MR, Hougland JL. A rising tide lifts all MBOATs: recent progress in structural and functional understanding of membrane bound O-acyltransferases. Front Physiol (2023) 14:1167873. doi: 10.3389/fphys.2023.1167873
124. Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell (2023) 186:2748–2764.e2722. doi: 10.1016/j.cell.2023.05.003
125. Soupene E, Fyrst H, Kuypers FA. Mammalian acyl-CoA:lysophosphatidylcholine acyltransferase enzymes. Proc Natl Acad Sci U.S.A. (2008) 105:88–93. doi: 10.1073/pnas.0709737104
126. Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U.S.A. (2008) 105:2830–5. doi: 10.1073/pnas.0712245105
127. Zhao Y, Chen YQ, Bonacci TM, Bredt DS, Li S, Bensch WR, et al. Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J Biol Chem (2008) 283:8258–65. doi: 10.1074/jbc.M710422200
128. Feng C, Lou B, Dong J, Li Z, Chen Y, Li Y, et al. Lysophosphatidylcholine acyltransferase 3 deficiency impairs 3T3L1 cell adipogenesis through activating Wnt/β-catenin pathway. Biochim Biophys Acta Mol Cell Biol Lipids (2018) 1863:834–43. doi: 10.1016/j.bbalip.2018.04.008
129. Reed A, Ichu TA, Milosevich N, Melillo B, Schafroth MA, Otsuka Y, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol (2022) 17:1607–18. doi: 10.1021/acschembio.2c00317
130. Cui J, Wang Y, Tian X, Miao Y, Ma L, Zhang C, et al. LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid Redox Signal (2023) 39:491–511. doi: 10.1089/ars.2023.0237
131. He S, Li R, Peng Y, Wang Z, Huang J, Meng H, et al. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J Cachexia Sarcopenia Muscle (2022) 13:1717–30. doi: 10.1002/jcsm.12953
132. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
133. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell (1997) 88:323–31. doi: 10.1016/S0092-8674(00)81871-1
134. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell (2012) 149:1536–48. doi: 10.1016/j.cell.2012.05.014
135. Gottlieb TM, Oren M. p53 and apoptosis. Semin Cancer Biol (1998) 8:359–68. doi: 10.1006/scbi.1998.0098
136. Balaburski GM, Hontz RD, Murphy ME. p53 and ARF: unexpected players in autophagy. Trends Cell Biol (2010) 20:363–9. doi: 10.1016/j.tcb.2010.02.007
137. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
138. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol (2021) 42:101928. doi: 10.1016/j.redox.2021.101928
139. Yang Y, Ma Y, Li Q, Ling Y, Zhou Y, Chu K, et al. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway. Cell Death Dis (2022) 13:530. doi: 10.1038/s41419-022-04971-x
140. Yang X, Wang Z, Zandkarimi F, Liu Y, Duan S, Li Z, et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab (2023) 35:1474–1490.e1478. doi: 10.1016/j.cmet.2023.06.014
141. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
142. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U.S.A. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
143. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U.S.A. (2010) 107:7461–6. doi: 10.1073/pnas.1002459107
144. Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res (2022) 82:3209–22. doi: 10.1158/0008-5472.CAN-21-3914
145. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep (2018) 22:569–75. doi: 10.1016/j.celrep.2017.12.077
146. Gao Y, Chen B, Wang R, Xu A, Wu L, Lu H, et al. Knockdown of RRM1 in tumor cells promotes radio-/chemotherapy induced ferroptosis by regulating p53 ubiquitination and p21-GPX4 signaling axis. Cell Death Discovery (2022) 8:343. doi: 10.1038/s41420-022-01140-z
147. Venkatesh D, Stockwell BR, Prives C. p21 can be a barrier to ferroptosis independent of p53. Aging (Albany NY) (2020) 12:17800–14. doi: 10.18632/aging.103961
148. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
149. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) (2018) 38:12. doi: 10.1186/s40880-018-0288-x
150. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med (2009) 30:42–59. doi: 10.1016/j.mam.2008.05.005
151. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science (1992) 257:1496–502. doi: 10.1126/science.1523409
152. Akerboom TP, Bilzer M, Sies H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J Biol Chem (1982) 257:4248–52. doi: 10.1016/S0021-9258(18)34713-6
153. Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta (2013) 1830:3289–303. doi: 10.1016/j.bbagen.2012.11.020
154. Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol (2021) 14:19. doi: 10.1186/s13045-020-01016-8
155. Qian B, Che L, Du ZB, Guo NJ, Wu XM, Yang L, et al. Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling. Theranostics (2023) 13:4288–302. doi: 10.7150/thno.82132
156. Combs JA, DeNicola GM. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers (Basel) (2019) 11:678. doi: 10.3390/cancers11050678
157. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274:11455–8. doi: 10.1074/jbc.274.17.11455
158. Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, et al. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem (1999) 274:3009–16. doi: 10.1074/jbc.274.5.3009
159. Shin CS, Mishra P, Watrous JD, Carelli V, D’Aurelio M, Jain M, et al. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun (2017) 8:15074. doi: 10.1038/ncomms15074
160. Stipanuk MH, Dominy JE Jr., Lee JI, Coloso RM. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr (2006) 136:1652s–9s. doi: 10.1093/jn/136.6.1652S
161. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
162. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
163. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
164. Nakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of FSP1 promotes ferroptosis. Nature (2023) 619:371–7. doi: 10.1038/s41586-023-06255-6
165. Perry JJ, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta (2010) 1804:245–62. doi: 10.1016/j.bbapap.2009.11.004
166. Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G, et al. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal (2014) 20:2873–89. doi: 10.1089/ars.2013.5603
167. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev (2016) 2016:4350965. doi: 10.1155/2016/4350965
168. Koch RE, Josefson CC, Hill GE. Mitochondrial function, ornamentation, and immunocompetence. Biol Rev Camb Philos Soc (2017) 92:1459–74. doi: 10.1111/brv.12291
169. Medda N, De SK, Maiti S. Different mechanisms of arsenic related signaling in cellular proliferation, apoptosis and neo-plastic transformation. Ecotoxicol Environ Saf (2021) 208:111752. doi: 10.1016/j.ecoenv.2020.111752
170. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol (2019) 25:101084. doi: 10.1016/j.redox.2018.101084
171. Li F, Li J, Wang PH, Yang N, Huang J, Ou J, et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim Biophys Acta Mol Basis Dis (2021) 1867:166260. doi: 10.1016/j.bbadis.2021.166260
172. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol (2022) 13:1039241. doi: 10.3389/fimmu.2022.1039241
173. Huang CY, Chen LJ, Chen G, Chao TI, Wang CY. SHP-1/STAT3-signaling-axis-regulated coupling between BECN1 and SLC7A11 contributes to sorafenib-induced ferroptosis in hepatocellular carcinoma. Int J Mol Sci (2022) 23:11092. doi: 10.3390/ijms231911092
174. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
175. Garten A, Grohmann T, Kluckova K, Lavery GG, Kiess W, Penke M. Sorafenib-induced apoptosis in hepatocellular carcinoma is reversed by SIRT1. Int J Mol Sci (2019) 20:4048. doi: 10.3390/ijms20164048
176. Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH, Wang ZX, et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol Sin (2021) 42:301–10. doi: 10.1038/s41401-020-0478-3
177. Cui Z, Wang H, Li S, Qin T, Shi H, Ma J, et al. Dihydroartemisinin enhances the inhibitory effect of sorafenib on HepG2 cells by inducing ferroptosis and inhibiting energy metabolism. J Pharmacol Sci (2022) 148:73–85. doi: 10.1016/j.jphs.2021.09.008
178. Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine (2022) 76:103847. doi: 10.1016/j.ebiom.2022.103847
179. Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, et al. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif (2021) 54:e13134. doi: 10.1111/cpr.13134
180. Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun (2003) 305:278–86. doi: 10.1016/S0006-291X(03)00734-4
181. Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem (2005) 280:37423–9. doi: 10.1074/jbc.M506439200
182. Drummen GP, van Liebergen LC, Op den Kamp JA, Post JA. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic Biol Med (2002) 33:473–90. doi: 10.1016/S0891-5849(02)00848-1
183. Westberg M, Bregnhøj M, Banerjee C, Blázquez-Castro A, Breitenbach T, Ogilby PR. Exerting better control and specificity with singlet oxygen experiments in live mammalian cells. Methods (2016) 109:81–91. doi: 10.1016/j.ymeth.2016.07.001
184. Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ 2nd, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem (2004) 279:55137–46. doi: 10.1074/jbc.M410387200
185. Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood (2016) 127:139–48. doi: 10.1182/blood-2015-06-654194
Keywords: ferroptosis, iron metabolism, lipid metabolism, lipid peroxidation, autophagy
Citation: Zhang X-D, Liu Z-Y, Wang M-S, Guo Y-X, Wang X-K, Luo K, Huang S and Li R-F (2023) Mechanisms and regulations of ferroptosis. Front. Immunol. 14:1269451. doi: 10.3389/fimmu.2023.1269451
Received: 30 July 2023; Accepted: 25 September 2023;
Published: 06 October 2023.
Edited by:
Daolin Tang, University of Texas Southwestern Medical Center, United StatesReviewed by:
Jiao Liu, Third Affiliated Hospital of Guangzhou Medical University, ChinaCopyright © 2023 Zhang, Liu, Wang, Guo, Wang, Luo, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ren-Feng Li, bGlyZW5mZW5nZG9nQDEyNi5jb20=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.