 Wenjun Li
Wenjun Li Jinze Wu
Jinze Wu Yini Zeng
Yini Zeng Wen Zheng
Wen Zheng- Department of Neurology, Third Xiangya Hospital, Central South University, Changsha, China
Epilepsy is a group of enduring neurological disorder characterized by spontaneous and recurrent seizures with heterogeneous etiology, clinical expression, severity, and prognosis. Growing body of research investigates that epileptic seizures are originated from neuronal synchronized and excessive electrical activity. However, the underlying molecular mechanisms of epileptogenesis have not yet been fully elucidated and 30% of epileptic patients still are resistant to the currently available pharmacological treatments with recurrent seizures throughout life. Over the past two decades years accumulated evidences provide strong support to the hypothesis that neuroinflammation, including microglia and astrocytes activation, a cascade of inflammatory mediator releasing, and peripheral immune cells infiltration from blood into brain, is associated with epileptogenesis. Meanwhile, an increasing body of preclinical researches reveal that the anti-inflammatory therapeutics targeting crucial inflammatory components are effective and promising in the treatment of epilepsy. The aim of the present study is to highlight the current understanding of the potential neuroinflammatory mechanisms in epileptogenesis and the potential therapeutic targets against epileptic seizures.
1 Introduction
Epilepsy is a variety of enduring neurological disorder characterized by tendency to spontaneous and recurrent seizures with heterogeneous etiology, pathogenesis, clinical features, and outcomes (1, 2). This disorder is serious global health issue affecting over 50-70 million people in the world (3). Even though a huge number of clinical and basic epileptic researches have been done, approximately 60% of epileptic disorders have unknown pathophysiological basis responsible for the occurrence and recurrence of seizures and 30% of epileptic patients still are resistant to the current pharmacotherapies with recurrent seizures throughout life (4, 5). As a result of these facts, it requires a more thorough understanding of the pathophysiological mechanisms in epileptogenesis, as well as exploring more effective therapeutic strategies for epileptic patients.
Growing body of researches investigate that epileptic seizures are originated from neuronal synchronized and excessive electrical activity, which may be a result of imbalance between glutamatergic and gamma-aminobutyric acid (GABA)ergic signaling induced by pathological factors including aberrant ion channel activation, neurotransmitter release, immune cells activation, etc. (4, 6, 7). Over the past two decades years growing evidences from both clinical and basic studies provide strong support to the conclusion that neuroinflammation is involved in epileptogenesis or a lot of neurological diseases with recurrent epileptic seizures (2, 4, 6).
Neuroinflammation, triggered by a number of exogenous or endogenous conditions including ageing process, gene expression variation, cerebral homeostasis disruption, pathogen infections, autoimmune diseases, brain tumor, stroke, and brain trauma, etc. (1, 4, 8–10), involves in a cascade of innate and adaptive immune responses characterized by the activation of microglia, astrocytes and endothelial cells (9), peripheral recruitment of multiple immune cells including but not limited to leukocytes, monocytes and lymphocytes into brain, and releasing a wide variety of inflammatory molecules, such as cytokines, chemokines, and growth factors, etc. (1, 4, 6, 8–10). Accumulated evidences show neuroinflammation is involved in neuronal hyperexcitability and epileptic seizures, as well prolonged epileptic seizures can trigger a chain of neuroinflammatory reactions (4, 11, 12). Neuroinflammation and recurrent seizures can be the mutual initiation factors and consequent reactions, which collectively contribute to devastating clinical consequences (5, 13). Meanwhile, an increasing body of preclinical researches reveal that the anti-inflammatory therapies specifically targeted a certain inflammatory component are effective and promising in animal models with seizures and epilepsy (14–16).
The aim of the present study is to highlight the update understanding of the underlying inflammatory mechanisms in diverse epileptic seizures, with particular emphasis on the neuroinflammatory pathways in epileptogenesis and potential therapeutic targets in epilepsy.
2 Activated microglia in epilepsy
Microglia, usually known as cerebral macrophages, may activate distinct physiological action in brain following the polarization of pro-inflammatory M1 cell subtype or anti-inflammatory M2 cell subtype, despite the underlying mechanisms and cell interactions between two phenotypes regulating are largely unknown (17–19). The pathophysiological consequences of microglia activation include aggravating inflammation, regulating neuronal activity, endocytosing dead neurons, and inducing epileptic seizures (7, 20–22). Growing evidences show that activated microglia may downregulate the convulsive threshold in experimentally-induced epileptic models (2, 17, 23, 24). The prolonged or excessive microglial activation may secret and release a wide range of inflammatory cytokines and chemokines, which is obviously observed in epileptogenesis (Figure 1) (11, 18, 25). The expression level of these inflammatory molecules is associated with the seizure severity in animal models or epileptic patients (17, 26). In addition, activated microglia may be in cooperation with astrocytes and in turn promote astrocytic glutamate release, which contribute to seizures and cell loss (2, 17).
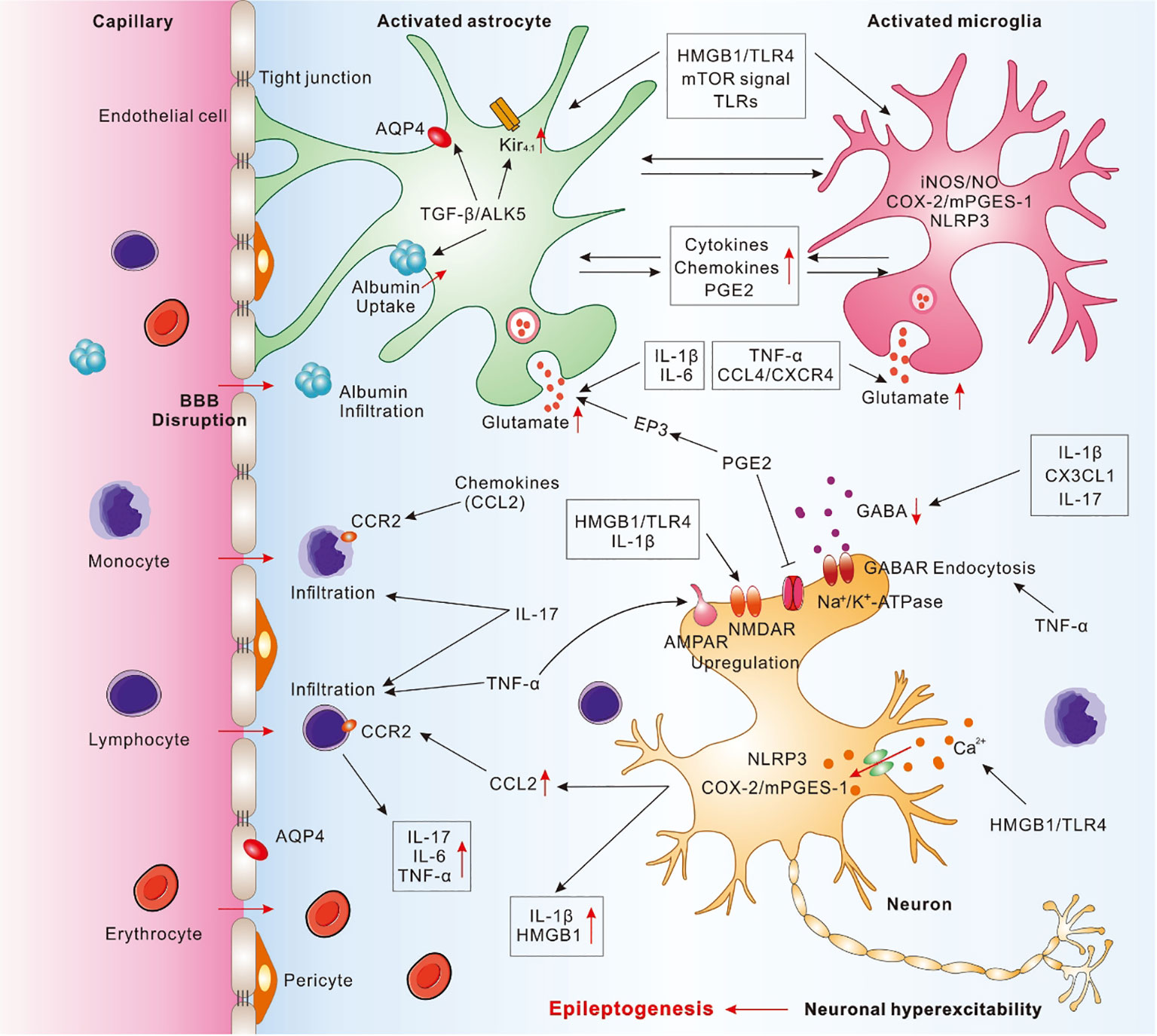
Figure 1 Pathophysiological cascade of inflammatory mechanisms in epilepsy. Activated astrocytes and microglia may cooperate with each other and release a wide range of pro-inflammatory molecules to mediate neuroinflammation, contributed to the neuronal hyperexcitability and epileptogenesis. IL-1β, secreted by activated microglia, astrocytes and neurons, may enhance glutamate release, decrease glutamate reuptake, and decrease GABAA currents. IL-6, secreted from activated astrocytes and microglia, can promote glutamate release. IL-17, secreted from astrocytes, microglia, and T-lymphocytes, may promote the infiltration of peripheral immune cells, inhibit GABA-induced inhibitory synaptic transmission, and result in excitation-inhibition imbalance. TNF-α, secreted by activated microglia and astrocytes, may promote infiltration of peripheral T lymphocytes, trigger microglial glutamate release, and induce GABA receptor endocytosis. HMGB1, released by activated astrocytes, microglia, or neurons, may interact with TLR4 and promote the pro-inflammatory cytokines release, increase Ca2+ influx, upregulate NMDAR, and destruct BBB. Activation of TGF-β/ALK5 signaling in astrocytes may mediate albumin uptake after BBB disruption, downregulate Kir4.1 channel, and impair AQP4. Chemokines, including CCL2, CCL3, CCL4, CX3CL1, and CXCL13, etc., may be secreted from astrocytes, microglia, and endothelial cells and take a crucial role in immune cells migrating from blood to brain through binding to corresponding receptor, such as CCL2-CCR2. PGE2, mainly secreted from activated astrocytes and microglia induced by COX-2/mPGES-1 axis in microglia, neurons, and endothelial cells, may inhibit the neuronal Na+/K+-ATPase. mTOR signal may activate astrocytes and microglia, promote the release of pro-inflammatory cytokines and chemokines, as well as disrupt BBB. TLRs, expressed by microglia and astrocytes, may trigger cerebral innate immune response and promote the release of pro-inflammatory cytokines. NLRP3 inflammasome elevated in neurons and microglia may activate Caspase-1 and increase the release of pro-inflammatory cytokines.
Activated microglia may generate high levels of nitric oxide (NO) via inducing inducible nitric oxide synthase (iNOS). Subsequently NO activates nicotinamide adenine dinucleotide (NADPH) oxidase and promotes the releasing of pro-inflammatory mediators such as tumor necrosis factor-α (TNF-α) (4). Moreover, the releasing of pro-inflammatory mediators and iNOS may be regulated by intrinsic astrocytic signals, such as the mitogen-activated protein kinase (MAPK) and the nuclear factor-кB (NF-кB) axis (27). Accumulating evidences demonstrate that the expression of NO, iNOS, NF-кB, and MAPK are significantly increased in a variety of epileptic models (4, 28).
Moreover, it has been substantiated that non-inflammatory action mediated by microglia is thus adequate to generate epileptic seizures (22, 29). Several studies report that microglia are involved in the neurogenesis in the epileptic brain. In animal models with KA-induced SE, microglia suppress neurogenesis by Toll-Like Receptor 9 (TLR9)-induced TNF-α secretion and decreasing ectopic granule cells (30). Through suppressing the excessive proliferating of neural stem cells (NSCs) and eliminating adult-born granule cells (GCs), activated microglia restrain the generation of abnormal brain circuits after SE, which may disrupt the balance between excitatory synapses and inhibitory synapses (21, 30). However, a body of other research finds that suppression of microglial activation can inhibit the neurogenesis after SE by decreasing the ectopic granule cells or immature neurons in different animal models (30, 31). These inconsistent results of microglia mediating neurogenesis may be attribute to the different chemically induced animal models and the different pathophysiological conditions. Different gene expression of microglia is observed in several animal models including KA-induced and pilocarpine-induced animal models (20).
3 Activated astrocytes in epilepsy
Reactive astrocytes activation and astrogliosis, including the morphologic alteration, molecular expression, and proliferation, were observed in various cerebral regions of animal models or patients with epileptic seizures (17, 30, 32). By autocrine and paracrine actions, activated astrocytes release pro-inflammatory molecules, including IL-1β, high mobility group box 1 (HMGB1), nuclear factor Kappa-beta (NF-kB), etc., all of which may promote seizure onset and recurrence (17, 23, 25). IL-1β may couple with interleukin 1 receptor type 1 (IL1R1) and activate nuclear factors κB (NF-κB), which strongly contribute to seizure generation and progression (6, 28, 33). HMGB1 may activate IL-1R/Toll-like receptor 4 (TLR4) signals and stimulate NF-κB, which take a crucial role in seizure occurrence and duration (6, 34–37). It is reported that overactivations of astrocytes disturb the synaptic function and lead to neuronal apoptosis (17, 18). On the contrary, astrocytes can be activated by seizures and subsequently mediate neuroinflammation (38).
Apart from the releasing of pro-inflammatory mediators, astrocytes may change neuronal excitability and lead to pathogenesis of epilepsy by regulating the water and K+ flow, which is also involved in the epileptogenesis (17, 39). Aquaporin-4 (AQP4) is a protein expressed in astrocytes and endothelial cells, which take a crucial role in modulating interstitial fluid osmolarity (ISF) and extracellular fluid (ECF) volume. By upregulating the astroglial potassium channel (Kir4.1) expression and AQP4 function, activated astrocytes may result in neuronal hyperexcitability (30). In KA-induced SE model, AQP4 is markedly downregulated, and the homeostatic conditions of water and potassium may be broken in early stage of epileptogenesis (40). AQP4 knockout (KO) mice or AQP4 anchoring the dystrophin-associated protein complex may enhance seizure susceptibility (39).
Several evidences from recent researches suggest that astrocytic enzyme adenosine kinase (ADK) increases in of epileptic animal models, which can lower the seizure threshold by reducing extracellular adenosine (41–43). In addition, growing evidences show that astrocytes regulate the excitatory/inhibitory neurotransmitter homeostasis and control the neuronal activity by releasing, uptake, and storing various mediates (17, 41–43). Astrocytes take part in the removing most of extracellular glutamate via glutamate aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1) (17). GLT-1 KO or selective inhibitor of GLT-1 enhance the concentration of synaptic glutamate and exhibit epileptic hyperexcitability and spontaneous lethal seizures (44, 45).
It is well known that astrocytes regulate neurogenesis, as well glutamate and GABA directly control neurogenesis. The level of nestin in astrocytes is upregulated in animal models with KA-induced severe seizures, which may promote the neuronal migration and location (46). The disruption of homeostasis between glutamate and GABA following astrocyte activation may trigger abnormal neurogenesis (47).
4 Peripheral immune cells in epilepsy
Accumulated evidences from recent researches strongly show that peripheral innate and adaptive immune cells take a major role in the pathogenesis of seizures and epileptogenesis (Figure 1) (2).
Blood monocytes can migrate into the brain through impaired blood-brain barrier (BBB) and subsequently mediate neuroinflammation by differentiating to macrophages or microglia-like cells, which is considered to involve in a wide range of cerebral disorders, like stroke, amyotrophic lateral sclerosis, and epilepsy (18, 22, 48–50). Emerging evidences show that the infiltrating of monocyte amplify the pathological effects of chemically kindled SE (48, 49). The migrate of monocyte depend on the chemokine (C-C motif) ligand 2 (CCL2)/C-C motif chemokine receptor 2 (CCR2) axis (48). CCL2 is extremely upregulated in activated microglia and neurons after SE in animal models or epileptic patients (48, 51). Blocking the CCL2/CCR2 signal pathway may alleviate the seizure occurrence in mice with mesial temporal lobe epilepsy induced by lipopolysaccharide (51). The infiltrating of monocyte may also contribute to the significant microglia activation and microgliosis after SE (50). The activated microglia is the predominant secreting cell of CCL2, and CCL2 subsequently promotes the monocyte recruiting by activating CCR2 (52). Infiltrating monocytes are gradually considered as novel promising therapeutic directions of seizures and epilepsy (18, 22).
As well as monocyte, lymphocytes are recruited into brain in epileptic neuroinflammation and mediate the adaptive immune response (26, 53). It was observed that pro-inflammatory blood-borne CD4+ and CD8+ T cells are recruited into neocortex and hippocampus in seizure animals or patients with epilepsy (54). Pro-inflammatory γδ T cells are increased in epileptic lesions, and their numbers are correspondingly associated with clinical outcomes in pediatric epileptic patients (55). It is noteworthy that regulatory T cells (Tregs), a kind of lymphocyte involved in immune regulation by inhibiting inflammation, may increase significantly following cerebral inflammation, which may alleviate seizure severity (56). The mice lacked T and B cells by silencing recombination activating gene 1 (RAG1) show significant neurodegenerating and recurrent seizures after KA administration (57). It is also reported that the deficiency of both γδ T cell- and IL-17RA aggravates KA-kindled SE in mice, as same as Tregs depletion (55).
5 Pro-inflammatory molecules in epilepsy
Pro-inflammatory molecules, such as cytokines, chemokines, and growth factors, etc., modulate neuroinflammatory processes and mediate the occurrence and development of epileptic seizures (Figure 1) (16, 18, 33, 58). A growing body of study has demonstrated cytokines, including IL-1β, IL-6, IL-17, TNF-α, and transforming growth factor-beta1(TGF-β1), etc., are significantly upregulated in brain following epileptic seizures, primarily secreted by glial cells and neurons (18, 59–61). Such crucial cytokines and homologous receptors involve in various pro-inflammatory pathways and result in neuronal hyperexcitability (59).
5.1 IL-1β
As a typical extracellular inflammatory cytokine, IL-1β is secreted by innate immune cells, including monocyte and macrophage. But in central nervous system (CNS), IL-1β is predominantly secreted by activated microglia, astrocytes, neurons, and oligodendrocytes (6, 62). The level of IL-1β can be instantly elevated in response to serious pathological courses via cleaving pro-IL-1β, which subsequently combines with IL1R1 and activates NF-κB to mediate the inflammatory process (6, 14, 34, 62). Compelling evidence has shown that IL-1β upregulated in serum, cerebrospinal fluid (CSF) and multiple brain regions of patients with epilepsy or in epileptic animal models (63). During the acute course of SE or the chronic course of epileptic seizures, IL-1β is predominantly released by activated microglia or astrocytes, which may exacerbate the epileptic seizure and cause more serious consequences (33). It is also demonstrated that exogenous IL-1β significantly enhances seizure severity and duration in rodents with KA-induced seizures (64). On the contrary, downregulation of IL-1β in rat hippocampus delays seizure occurrence and reduces seizure severity (65).
A wider breadth of research documented that IL-1β may disturb the balance between excitatory transmission and inhibitory transmission by enhancing glutamate releasing and decreasing glutamate re-uptake (66), which may lead to neuronal hyperexcitability and excitotoxicity (6). IL-1β can enhance the glutamate transmission of excitatory neuron by activating of the N-methyl-D-aspartate (NMDA) receptor subunit 2B (GluN2B) (67). In addition, overexpression of IL-1β may decrease the GABAA current (up to 30%) in temporal lobe epilepsy and rats with chronic epilepsy (68).
5.2 IL-6
As a pro-inflammatory cytokine, IL-6 takes a crucial modulating role in neuroinflammatory responses (69). IL-6 expresses in lower level in normal physiological conditions, but is significantly secreted from activated astrocytes and microglia, which may be regulated by different cytokines including TNF-α, IL-Iβ, and IL-17, etc. (70, 71). In epileptic patients and multitudinous seizure models, the expression of IL-6 obviously increases in various cerebral region like hippocampus, cortex, thalamus, and hypothalamus, as well as in blood (6). Different from other pro-inflammatory cytokines, the plasma level of IL-6 may increase significantly up to 24 h after seizures (72).
IL-6 can upregulate glutamate release, inhibit long-term potentiation (LTP), decrease hippocampal neurogenesis, and promote glosis, which significantly enhance neuronal excitability and mediate epileptogenesis (6, 70). A growing body of researches have demonstrated that IL-6 facilitate the seizure occurrence and exacerbation of epilepsy (73, 74). The mice overexpressed IL-6 in astrocytes is more subject to seizures after injection of KA (70, 73). Exogenous IL-6 may distinctly increase the severity of pentylenetetrazol (PTZ)-induced seizures (70, 74). The increasing of IL-6 and IL-1β in hippocampus during pregnancy may lead to neuronal hyperexcitability and development of seizures in offspring (75). However, the molecular mechanism underlying IL-6 contributing to seizures is not complete understood. Some other studies showed that genetic ablation or knockout of IL-6 activated oxidative stress-related signaling, increased neuronal damages, enhanced the susceptibility to seizures, and leaded to higher mortality rates in multiple chemoconvulsant models (76). Therefore, a large amount of research is required to identify the crucial roles and mechanisms of IL-6 in epileptogenesis and disease-modifying therapy.
5.3 IL-17
IL-17 is a large family of cytokines consisting of IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F (77). The IL-17A and IL-17F cooperate to compose a dimer and subsequently bind to receptor complex IL-17RA/C, including IL-17 receptor A and IL-17 receptor C. The activating of IL-17 signal pathways may recruit adaptor molecule actin-related gene 1 (Act1) to the SEFIR domain of IL-17RA/C complex, subsequently recruit TNF receptor associated factor 6 (TRAF6) and activate a wide spectrum of pro-inflammatory signal pathways in brain (78).
Several previous studies found that IL-17 is primarily secreted from CD4 + T cells, however, it is demonstrated that astrocytes are the predominant secreting cells of IL-17 in brain (79). Recent researches reveal that IL-17 is obviously increased in multiple innate and adaptive immune cells or nonimmune cells, such as dysmorphic neuron, balloon cell, giant cell, microglia, and T-lymphocyte, following the serious epileptic seizures (80). The elevated IL-17 in brain can activate microglia, promote the infiltrating of peripheral immune cells, and increase the secretion of pro-inflammatory mediators, including IL-6, IL-1β, and TNF-α, etc. (58, 81). At the same time, the upregulated pro-inflammatory mediators may inversely induce the releasing of IL-17 (82).
Accumulated evidences from recent studies support that IL-17 is closely related to epileptogenicity, and the IL-17 signal pathways may be an underlying antiepileptic treatment target (5). Increased IL-17 results in neuronal hyperexcitability in brain slice cultures (55). In variable types of epileptic patients, increased IL-17 in serum or CSF positively associated with seizure severity (53). A recent research observes that the numbers of IL-17-secreting γδ T cell locating in epileptogenic lesions are closely related to seizure severity (5). The deletion of IL-17R in KA-induce SE animal model decreases neuronal excitability and alleviates the seizure activity (55). IL-17 may result in an imbalance between excitatory neural circuit and inhibitory neural circuit and induce neuronal excessive discharge by regulating neurotransmitters releasing (83). Another study demonstrates that IL-17 inhibits GABA-mediated inhibitory postsynaptic electrical activity and leads to neuronal hyperexcitability (79).
5.4 TNF-α
TNF-α is a pro-inflammatory cytokine predominantly secreted by activated microglia and activated astrocytes reacting to inflammatory conditions (84). Overexpression of TNF-α is associated with seizures, ataxia, and paresis in mice, accompanied by inflammatory pathological alterations in brain, including T lymphocyte infiltrating, reactive astrogliosis and microgliosis, and focal demyelination (85). In rats with amygdala kindling or KA-inducing seizures, the expression TNF-α is elevated and persists for up to several days in hippocampus, amygdala and cortex, followed by extensive neuronal damage (61).
TNF-α signaling can provoke glutamate release from microglia by upregulating glutaminase and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (86). Upregulated AMPA receptors contribute to calcium uptake and neurotoxicity (24). In addition, TNF-α may decrease inhibitory receptors of postsynaptic membrane via inducing GABA receptor endocytosis (24, 87). Furthermore, it is reported TNF-α regulated adhesion molecule N-cadherin and mediated the generation and development of both excitatory synaptic networks and inhibitory synaptic networks (88). These multiple roles of TNF-α on the glutamate receptors and GABA receptors, as well as excitatory and inhibitory synaptic networks, synergistically lead to neuronal hyperexcitability and epileptogenesis (59).
Although the mounting evidences show that TNF-α takes an important role in epileptogenesis, it is also reported that recombinant TNF-α inhibits seizure occurrence induced by PTZ (89). The available researches demonstrate that two different tumor necrosis factor receptor (TNFR) subtypes, TNFR1 and TNFR2, mediate these dichotomous pro-convulsive and anti-convulsive function (87). Compared with TNFR2, TNFR1 has higher binding ability to TNF-α (90). The low expression TNF-α predominantly binds to TNFR1 and takes a pro-convulsive action, and the continually accumulation of TNF-α further activates TNFR2 and triggers an anti-convulsive mechanism in epileptic brain (6). In preclinical researches, certain ligands of TNF receptor have come to exploring as underlying treatment targets in epilepsy, while it is still under controversy whether the therapeutics against epilepsy targeting TNF-α signaling may generate the serious risks of infection and tumorigenesis (59).
5.5 Transforming growth factor β
TGF-β is a multifunctional cytokine regulating a wide range of downstream signaling through activating TGF-beta type II receptor (TβRII), TGF-beta type I receptor (TβR1), activin-like kinase 5 (ALK5), SMAD protein complexes, and multiple mitogen-activated protein kinase (MAPK), etc. (91). TGF-β mediates a series of biological activities such as inflammatory responses, cell proliferation, differentiation, apoptosis, and embryogenesis (91).
Recent mounting evidences show that TGF-β signaling pathways are intricated in epileptogenesis (92). In resected brain regions from epileptic patients and animal models, the increased expression of TβR1 is observed in cortex, amygdala, and hippocampus, particularly in the piriform cortex (61, 93). Activation of TGF-β signaling pathways in astrocytes can mediate albumin uptake, potassium (K+) buffering, glutamate metabolism, pro-inflammatory mediators releasing, and synaptogenesis, which collectively lead to neuronal hyperexcitability and epileptogenesis (94, 95). TGF-β/ALK5 signaling pathways activated by intracerebroventricular injection of albumin may induce excitatory synaptogenesis and spontaneous seizures in mice (96). As well, activation of TGF-β/ALK5 signaling pathways in transgenic ALK5CA mice regulates the late stages of adult hippocampal neurogenesis (96, 97). Both excitatory synaptogenesis and hippocampal neurogenesis may be involved in epileptogenesis. It is also demonstrated that TGF-β signaling pathways mediates astrocytes to uptake serum albumin through disrupted BBB, downregulates of potassium channel Kir4.1 in astrocytes, and reduces extracellular K+ buffering, which may induce NMDA receptor-induced neuronal hyperexcitability and seizure-like discharges (94). Another research found that activation of the TGF-β signaling pathways in hippocampus upregulates glutamatergic associated genes and downregulated GABAergic associated genes in reactive astrocytes, followed by glia-neuron communication and hippocampal-kindling epileptiform activity in rats (92). The recurrent seizures mediated by TGF-β signaling pathways can be abolished by homologous pharmacological inhibitor or antagonist, such as SJN2511, SB431542, and losartan (97), which suggests targeting TGF-β signaling pathways is a prospective therapeutic strategy for acquired epilepsy.
5.6 HMGB1
HMGB1, a ubiquitous chromatin-binding protein, contains two DNA-binding domains (box A and box B) in N-terminus and take part in regulating transcription, translation, and repair of DNA (37). As a pro-inflammatory domain, box B can bind to Toll-like receptors (TLRs) to initiate a neuroinflammatory response (98, 99). Oppositely, box A is a competitive anti-inflammatory response domain to alleviate the inflammatory activity (98).
HMGB1 may be a potential clinical biomarker for prediction, diagnosing, and prognosing of seizures or drug-resistant epilepsy (100–102). Numerous studies demonstrate that serum HMGB1 increases after seizures according to the severity of seizures and epileptic discharges (34, 35, 102). It is also reported that the levels of HMGB1 in CSF are upregulated in patients with autoimmune epilepsy, accompanied by other upregulated pro-inflammatory cytokines and chemokines (103). In mice model with KA-induced seizures, the high levels of HMGB1 are observed in hippocampal, and the antagonists of HMGB1 or anti-HMGB1 monoclonal antibodies are able to alleviate occurrence and severity of seizures (25, 101, 104).
A growing body of research demonstrates that HMGB1 is generated from activated astrocytes and microglia, or neuron, subsequently interacts with the primary receptor TLR4 to involve in the pathophysiology of epilepsy (2, 34). The levels of HMGB1 and TLR4 increase in cerebral regions of patients with mesial temporal lobe epilepsy, focal cortical dysplasia type II (FCD-II) with epilepsy, or drug-refractory epilepsy, correlating with the severity of epilepsy (35, 100). Binding to TLR4, extracellular HMGB1 can activate IL-1β/IL-1R and TLR4 signals, subsequently promote NF-κB transporting into nucleus and result in release of numerous pro-inflammatory cytokines, which may be involved in epileptogenesis (6, 35–37). It is also reported that HMGB1/TLR4 signaling pathway significantly increases the Ca2+ influx and activates the NMDA receptor by phosphorylating NR2B subunit (105), which usually contribute to neuronal hyperexcitability and epileptogenesis (106). In KA-induced animal models, HMGB1 downregulates the level of glutamate decarboxylase 67 (GAD67), glutamate dehydrogenase 1 (GLUD1), and glutamate dehydrogenase 2 (GLUD2), upregulates the levels of intracellular glutamate and major histocompatibility complex II (MHC II), which collectively contribute to increasing the neuronal excitability and inducing epileptic seizures (107). Another mechanism of extracellular HMGB1 contributing to epilepsy is based on the destruction of the BBB (108).
5.7 Chemokines
Chemokines are a large family of small proteins released by astrocytes, microglia, and endothelial cells (38), which is primarily stimulated by pro-inflammatory cytokines (109). Through acting on specific G protein‐coupled receptors (GPCRs), chemokines take a crucial role in infiltration of immune cells from blood to brain, as well as modulating electrophysiology via regulating neurotransmitter releasing and voltage-dependent channels or G-protein-gated channels (6, 110, 111). Several researches observed that chemokines, including CCL2, CCL3, CCL4, fractalkine/CX3C chemokine ligand 1 (CX3CL1), chemokine ligand 13 (CXCL13), and the corresponding receptor CCR2, CCR5, C-X-C chemokine receptor 4 (CXCR4), CXCR5, highly upregulated in hippocampus or other cerebral regions of patients and animal models with epilepsy (6, 59). In patients with intractable epilepsy, chemokine CCL2 significantly upregulates in neuron, astrocyte, microglia, neural progenitor cell, and microvascular endothelial cell (111). In KA-kindled SE, high-level CCL2 in hippocampus activates CCR2 and leads to microglia activation, monocyte infiltration, and neuron death by triggering transcription 3 (STAT3) and IL-1β signals (48). In TLE patients, the upregulation of CXCR4 on microglia and astrocytes induces microglia to release TNF-α and glutamate (110). In the pilocarpine-induced SE, knockout of CCR2 prevents the monocyte migration and decreases the secreting of pro-inflammatory cytokines (49). In addition, inhibition of CCL2/CCR2 signals significantly alleviate the severity of seizures in the KA-induce SE model (51). In pilocarpine SE models, exogenous CX3CL1 enhances SE-induced neuronal loss, which can be reversed by the antibodies of CX3CL1 or CX3CR1 (112). In KA-induce rat models, downregulation of CCR5 expression delays seizure onset and decreases seizure severity, neuron damage, and neuroinflammation (113). All of these accumulating evidences suggest that the chemokines and downstream signal pathways mediate the interaction between neuroinflammation and epileptogenesis (51, 113).
5.8 Cyclooxygenase-2, microsomal prostaglandin E synthase-1, prostaglandin E2 and PGE2 receptors
Cyclooxygenase (COX) is the crucial enzyme in biosynthesis of prostanoid, including prostaglandin D2 (PGD2), PGE2, prostaglandin F2 alpha (PGF2α), prostaglandin I2 (PGI2), and thromboxane A2 (TXA2) (14). COX-1 isoform, expressed widely in body, primarily take a key role in homeostatic prostaglandins (14). COX-2 isoform, a major pro-inflammatory factor, is usually expressed at very low levels in normal cerebral tissue, but which significantly increases following inflammatory conditions, fever, and seizures (114, 115). Prostaglandin E synthase (PGES), existing in three isoforms including mPGES-1, mPGES-2, and cytosolic PGES (cPGES), is a downstream enzyme of COX cascade and contribute to synthesis of PGE2 from prostaglandin H2 (PGH2) directly (116). mPGES-1 combines with COX-2 to co-induce the synthesizing of PGE2 and promote inflammatory responses (116, 117).
Both COX-2 and mPGES-1 are observed to be synchronously upregulated in activated microglia, neurons, and endothelial cells, in response to a wide range of neurological disorders, including seizures and strokes (118, 119). In mice with pilocarpine-induced SE, the expressions of COX-2 and mPGES-1 increase in the hippocampus in very synchronous time-courses, along with the elevating of PGE2 (115, 120). The selective inhibitors of both COX-2 and mPGES-1 may largely reduce brain inflammation, shorten seizure duration, and reduce the seizure incidence in epileptic animal models (12, 117). In mice models with chemoconvulsant seizures, the deficiency of mPGES-1 may decrease the levels of PGE2, which blocks the releasing of pro-inflammatory cytokines, attenuates occurrence of seizures and neuronal damage in brain (12). In addition, emerging evidences show that the upregulation of mPGES-1 in endothelial cell promotes astrocytic glutamate releasing and results in excitotoxicity (121). It is also reported by several studies that COX-2 inhibitors including NS-398, indomethacin, celecoxib and mPGES-1 inhibitor BI1029539 prevent P-glycoprotein upregulation after seizures (122, 123), which may be associated to the development of drug-resistance seizures (124).
PGE2 is induced by COX-2/mPGES-1 axis and mainly secreted from activated astrocytes and microglia, which couple with homologous receptors, including EP1, EP2, EP3, and EP4, and mediate neuroinflammation, neuronal hyperexcitability and excitotoxicity by infiltrating of immune cells and upregulating of many pro-inflammatory factors (125, 126). A growing body of researches have investigated the roles of PGE2 and its receptors in the development of seizures. By mediating of EP1, EP3 or EP4 receptor, PGE2 takes an inhibitory action on Na+/K+-ATPase to upregulate neuronal excitability (127). In addition, PGE2 binding to receptor EP3 promotes astrocytic glutamate release, which disrupts the balance between both glutamatergic signaling and GABAergic signaling (125). In multiple chemically kindled epileptic models, the antagonists of EP1, EP3 or EP4 decrease epileptic susceptibility and delays seizure induction by decreasing extracellular glutamate concentrations (125, 128–130).
Differing from the EP1, EP3 or EP4 receptors, EP2 receptor usually affords beneficial and positive roles in brain after cerebral injury (6). The activation of EP2 may block brain-derived neurotrophic factor (BDNF)/tyrosine kinase receptor B (TrkB) signal pathway and prevent from acquired epileptogenesis after pilocarpine administration in mice (120, 131). However, recently accumulated evidences strongly demonstrate that EP2 activating may induce the secondary toxicity and damage in animal models with neuroinflammation or neurodegeneration (126, 132–134). Meanwhile, EP2 antagonist decreases neuronal death, reduces mortality, attenuates BBB breakdown, alleviates gliosis in the hippocampus of SE animal models (6, 135–137). In addition, several other studies show some inconsistent findings in COX-2 inhibitors, including nimesulide, rofecoxib, parecoxib, and SC-58236, which have no effect on seizure, even exacerbate seizure severity and increase mortality in animal models (14, 138–141). These contradictory findings in different studies suggest the complexity of neuroinflammatory signal pathways in epileptogenesis.
5.9 Mammalian/mechanistic target of rapamycin signaling
The mTOR signaling plays acritical roles in neurodevelopment and neural circuit formation by regulation the protein synthesis and autophagy (18). Recently, several lines of evidences support that mTOR signaling is implicated in neuroinflammation via regulating the function of immune cells and mediating the releasing of pro-inflammatory cytokines and chemokines (142, 143). mTOR signaling may promote the generating of monocytes and macrophages in marrow cavity and promote the transformation of monocytes into macrophages by downregulating the expression of macrophage colony-stimulating factor receptor CD115 (144, 145). mTOR signaling also regulates differentiating and activating of multiple lymphocytes, including T cells and Th cells (18, 146–148). In brain, blocking of mTOR signaling may decrease the formation of autophagosome, increase lipopolysaccharides (LPS)-induced pro-inflammatory cytokines in microglia, attenuate the microglial activation, mitigate astrocyte migration and proliferation, and alleviate seizure severity (18). On the contrary, the activation of mTOR signaling may result in microglial activation and BBB disruption, which facilitate the infiltrating of peripheral immune cells into CNS (149, 150). It is also reported that mTOR signaling regulates the differentiation of Th17 cells and mediates the expression of IL-1β, IL-17 and TNF-α, all of which have been identified to be involved in the epileptogenesis (151–155). Recently, evidences from both lab investigations and clinical trials show treatments targeting mTOR signaling present promising anti-seizure effects, but usually associate with serious adverse events (156).
5.10 TLRs
TLRs (TLR 1, 2, 3), a kind of transmembrane proteins expressed by immune cells like microglia and astrocytes, take in important role in recognizing conserved motifs of pathogens or endogenous dangerous molecules secreted by pathological cells. The activating of TLRs subsequently provokes a cascade of innate and adaptive immune response, followed by neuronal hyperexcitability and epileptogenesis (157, 158). The activating of TLRs can be largely enhanced by HMGB1 (159).
Due to the key role on neuroinflammation and regulation of neuronal excitability, TLRs may be potential therapeutic targets in epileptic treatment, which is be reported by several preclinical studies. In a pilocarpine-induced SE model, deletion of TLR3 reduces spontaneous recurrent seizures, inhibits the microglial activation, decreases the levels of pro-inflammatory factors including TNF-α and interferon-β (IFN-β) (160). It is also reported that FOX3P, expressed by microglia and taking part in T cell differentiation, attenuates seizure activity by inhibiting TLR4 signaling pathway (161).
5.11 Nucleotide-binding domain-like receptor family pyrin domain containing 3 inflammasome
The NLRP3 inflammasome is an inflammatory complex composed of three proteins, such as NLRP3, apoptosis associated speck like protein containing a CARD (ASC), and cysteinyl aspartate specific proteinase-1 (Caspase-1) (162). By interacting with ASC, NLRP3 inflammasome may induce caspase-1 proteolysis and promote the secretion of a wide spectrum of pro-inflammatory molecules following brain injury in various cerebral diseases (15, 163). In the epileptic brain, NLRP3 is observed to primarily increased in neurons and microglia (164). A growing body of researches show the activating of NLRP3 inflammasome may be involved in epileptic neuron loss, seizures progression, and the therapeutic effects of antiseizure drugs (ASDs) by regulating innate immunity and neuroinflammation (165–169). In rats with amygdala kindling SE, inhibition of the NLRP3 inflammasome downregulates the levels of IL-1β and IL-18, alleviates the severity of epileptic seizures, and decreases hippocampal neuronal apoptosis during the chronic epileptic phase (170). It is also reported that GPR120 downregulates neuroinflammation and alleviates epileptic seizure activity via NLRP3/Caspase-1/IL-1β signals in KA-induced temporal lobe epilepsy (171).
5.12 Conclusion
The neuroinflammation, evoked by pathogens, self-antigens, or tissue injury in various cerebral diseases, involves the activation of microglia and astrocytes, a cascade of inflammatory mediator releasing, and infiltration of peripheral immune cells and serum albumin from blood into brain, which subsequently induce the imbalance between glutamatergic signaling and GABAergic signaling, lower seizure threshold and contribute to epileptogenesis (6, 172). Moreover, frequent and continual epileptic seizures also have undesirable consequences on neuroinflammation, thus deteriorating the frequency and severity of epilepsy. During the past two decades, plenty of evidences depending on the animal model or epileptic patients have been demonstrated that neuroinflammation is intricate with the occurrence and development of epileptic seizures.
Although above several established and novel mechanisms are reported to explore the effects of inflammation on neuronal hyperexcitability and the occurrence of epileptic seizure, the current researches predominantly focused on the co-occurrence of both neuroinflammation and epileptic seizures, but could not give a complete insight into the cause-effect relationship between inflammatory molecules and epileptic seizures. The exact role and mechanism of neuroinflammation in epilepsy are far from illustrating. Thus, more in-depth studies are required and indispensable to elucidate the inflammatory mechanisms in epileptogenesis. In future, extensive efforts should be made to further explore the potential crosstalk action between different cells such as activated microglia, astrocytes, peripheral immune cells, and neurons in neuroinflammation, as well as the key crosstalk role of pro-inflammatory factors such as cytokines, chemokines, bioactive lipids, growth factors, etc. In addition, the elevated inflammatory mediators in serum and CSF associated with the seizure severity and recurrence may potentially be explored as biomarkers of refractory epilepsy or epileptogenesis.
Base on the crucial role of inflammation in epileptogenesis, immune modulation and anti-inflammation are excepted to be effective therapeutics for epilepsy. To date, substantial number of preclinical studies have been done and showed that combating neuroinflammation to blocking the development and progression of epilepsy may be an attractive therapeutic strategy for the management of epilepsy. Recently, a wide range of ASDs are available in hospital, but it can’t be ignored the fact that the current ASDs are mainly block the occurrence of seizures but do not act on the underlying pathophysiologic mechanism and epilepsy development (modifying the epileptic process). Different from ASDs, treatment with anti-inflammatory agents may obtain more beneficial effects from neuroprotection to functional recovery in patients with epilepsy, as well as improving the pharmaco-sensitivity to ASDs, but which need further clinical investigations to be done. Therefore, future translational researches should be more focus on evaluating the combined treatments including anti-neuroinflammatory agents and ASDs in patients with epilepsy.
Author contributions
WL: Writing – original draft. JW: Writing – original draft. YZ: Writing – original draft. WZ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the General project of Provincial Natural Science Foundation of Hunan, China (No. 2020JJ4865); the research funding of CAAE, China (No. CX-A-2021-09); the Project of Health Commission of Hunan Province, China (No. 202203073989); and the Project from Natural Science Foundation of Changsha, China (No. kq2202420).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Uczay M, Pfluger P, Picada JN, de Oliveira JDM, da SilvaTorres IL, Medeiros HR, et al. Geniposide and asperuloside alter the COX-2 and GluN2B receptor expression after pilocarpine-induced seizures in mice. Naunyn Schmiedebergs Arch Pharmacol (2023) 396:951–62.doi: 10.1007/s00210-022-02367-4
2. Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol (2011) 7:31–40. doi: 10.1038/nrneurol.2010.178
3. Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet (2019) 393:689–701. doi: 10.1016/S0140-6736(18)32596-0
4. Rabidas SS, Prakash C, Tyagi J, Suryavanshi J, Kumar P, Bhattacharya J, et al. A comprehensive review on anti-inflammatory response of flavonoids in experimentally-induced epileptic seizures. Brain Sci (2023) 13, 102. doi: 10.3390/brainsci13010102
5. An J, Li H, Xia D, Xu B, Wang J, Qiu H, et al. The role of interleukin-17 in epilepsy. Epilepsy Res (2022) 186:107001. doi: 10.1016/j.eplepsyres.2022.107001
6. Chen Y, Nagib MM, Yasmen N, Sluter MN, Littlejohn TL, Yu Y, et al. Neuroinflammatory mediators in acquired epilepsy: an update. Inflammation Res (2023) 72:683–701. doi: 10.1007/s00011-023-01700-8
7. Hiragi T, Ikegaya Y, Koyama R. Microglia after seizures and in epilepsy. Cells (2018) 7, 26. doi: 10.3390/cells7040026
8. Gilhus NE, Deuschl G. Neuroinflammation - a common thread in neurological disorders. Nat Rev Neurol (2019) 15:429–30. doi: 10.1038/s41582-019-0227-8
9. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener (2020) 9:42. doi: 10.1186/s40035-020-00221-2
10. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol (2019) 10:1008. doi: 10.3389/fphar.2019.01008
11. Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, Gorter JA, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia (2017) 58 Suppl 3:27–38. doi: 10.1111/epi.13783
12. Yasmen N, Sluter MN, Li L, Yu Y, Jiang J. Transient inhibition of microsomal prostaglandin E synthase-1 after status epilepticus blunts brain inflammation and is neuroprotective. Mol Brain (2023) 16:14. doi: 10.1186/s13041-023-01008-y
13. Marchi N, Granata T, Janigro D. Inflammatory pathways of seizure disorders. Trends Neurosci (2014) 37:55–65. doi: 10.1016/j.tins.2013.11.002
14. Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti-inflammatory small molecules to treat seizures and epilepsy: from bench to bedside. Trends Pharmacol Sci (2016) 37:463–84. doi: 10.1016/j.tips.2016.03.001
15. Gong L, Han Y, Chen R, Yang P, Zhang C. LncRNA ZNF883-mediated NLRP3 inflammasome activation and epilepsy development involve USP47 upregulation. Mol Neurobiol (2022) 59:5207–21. doi: 10.1007/s12035-022-02902-7
16. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol (2019) 15:459–72. doi: 10.1038/s41582-019-0217-x
17. Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci (2013) 36:174–84. doi: 10.1016/j.tins.2012.11.008
18. Zeng C, Hu J, Chen F, Huang T, Zhang L. The coordination of mTOR signaling and non-coding RNA in regulating epileptic neuroinflammation. Front Immunol (2022) 13:924642. doi: 10.3389/fimmu.2022.924642
19. Valmori D, Tosello V, Souleimanian NE, Godefroy E, Scotto L, Wang Y, et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J Immunol (2006) 177:944–9. doi: 10.4049/jimmunol.177.2.944
20. Benson MJ, Manzanero S, Borges K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia (2015) 56:895–905. doi: 10.1111/epi.12960
21. Luo C, Koyama R, Ikegaya Y. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia (2016) 64:1508–17. doi: 10.1002/glia.23018
22. Bosco DB, Tian DS, Wu LJ. Neuroimmune interaction in seizures and epilepsy: focusing on monocyte infiltration. FEBS J (2020) 287:4822–37. doi: 10.1111/febs.15428
23. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol (2013) 244:11–21. doi: 10.1016/j.expneurol.2011.09.033
24. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol (2012) 33:116–25. doi: 10.1016/j.yfrne.2011.12.002
25. Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med (2010) 16:413–9. doi: 10.1038/nm.2127
26. Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis (2008) 29:142–60. doi: 10.1016/j.nbd.2007.08.012
27. Spencer JP, Vafeiadou K, Williams RJ, Vauzour D. Neuroinflammation: modulation by flavonoids and mechanisms of action. Mol Aspects Med (2012) 33:83–97. doi: 10.1016/j.mam.2011.10.016
28. Zhang Y, Zhang M, Zhu W, Pan X, Wang Q, Gao X, et al. Role of elevated thrombospondin-1 in kainic acid-induced status epilepticus. Neurosci Bull (2020) 36:263–76. doi: 10.1007/s12264-019-00437-x
29. Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep (2018) 22:2080–93. doi: 10.1016/j.celrep.2018.02.004
30. Araki T, Ikegaya Y, Koyama R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur J Neurosci (2021) 54:5880–901. doi: 10.1111/ejn.14969
31. Yang F, Liu ZR, Chen J, Zhang SJ, Quan QY, Huang YG, et al. Roles of astrocytes and microglia in seizure-induced aberrant neurogenesis in the hippocampus of adult rats. J Neurosci Res (2010) 88:519–29. doi: 10.1002/jnr.22224
32. Thom M. Review: Hippocampal sclerosis in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol (2014) 40:520–43. doi: 10.1111/nan.12150
33. van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol (2018) 44:91–111. doi: 10.1111/nan.12444
34. Zhang S, Chen F, Zhai F, Liang S. Role of HMGB1/TLR4 and IL-1beta/IL-1R1 signaling pathways in epilepsy. Front Neurol (2022) 13:904225. doi: 10.3389/fneur.2022.904225
35. Kan M, Song L, Zhang X, Zhang J, Fang P. Circulating high mobility group box-1 and toll-like receptor 4 expressions increase the risk and severity of epilepsy. Braz J Med Biol Res (2019) 52:e7374. doi: 10.1590/1414-431x20197374
36. Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano A, Aleksovska K, et al. HMGB1: A common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Neurosci (2018) 12:628. doi: 10.3389/fnins.2018.00628
37. Shi Y, Zhang L, Teng J, Miao W. HMGB1 mediates microglia activation via the TLR4/NF-kappaB pathway in coriaria lactone induced epilepsy. Mol Med Rep (2018) 17:5125–31. doi: 10.3892/mmr.2018.8485
38. Mukherjee S, Arisi GM, Mims K, Hollingsworth G, O'Neil K, Shapiro LA. Neuroinflammatory mechanisms of post-traumatic epilepsy. J Neuroinflamm (2020) 17:193. doi: 10.1186/s12974-020-01854-w
39. Binder DK, Nagelhus EA, Ottersen OP. Aquaporin-4 and epilepsy. Glia (2012) 60:1203–14. doi: 10.1002/glia.22317
40. Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol (2012) 235:246–55. doi: 10.1016/j.expneurol.2012.02.002
42. Li T, Quan Lan J, Fredholm BB, Simon RP, Boison D. Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biol (2007) 3:353–66. doi: 10.1017/S1740925X0800015X
43. de Groot M, Iyer A, Zurolo E, Anink J, Heimans JJ, Boison D, et al. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia (2012) 53:58–66. doi: 10.1111/j.1528-1167.2011.03306.x
44. Campbell SL, Hablitz JJ. Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol Dis (2008) 32:254–61. doi: 10.1016/j.nbd.2008.07.003
45. David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci (2009) 29:10588–99. doi: 10.1523/JNEUROSCI.2323-09.2009
46. Yang F, Wang JC, Han JL, Zhao G, Jiang W. Different effects of mild and severe seizures on hippocampal neurogenesis in adult rats. Hippocampus (2008) 18:460–8. doi: 10.1002/hipo.20409
47. Nakamichi N, Takarada T, Yoneda Y. Neurogenesis mediated by gamma-aminobutyric acid and glutamate signaling. J Pharmacol Sci (2009) 110:133–49. doi: 10.1254/jphs.08R03CR
48. Tian DS, Peng J, Murugan M, Feng LJ, Liu JL, Eyo UB, et al. Chemokine CCL2-CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1beta Production after Status Epilepticus. J Neurosci (2017) 37:7878–92. doi: 10.1523/JNEUROSCI.0315-17.2017
49. Varvel NH, Neher JJ, Bosch A, Wang W, Ransohoff RM, Miller RJ, et al. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U.S.A. (2016) 113:E5665–74. doi: 10.1073/pnas.1604263113
50. Feng L, Murugan M, Bosco DB, Liu Y, Peng J, Worrell GA, et al. Microglial proliferation and monocyte infiltration contribute to microgliosis following status epilepticus. Glia (2019) 67:1434–48. doi: 10.1002/glia.23616
51. Cerri C, Genovesi S, Allegra M, Pistillo F, Puntener U, Guglielmotti A, et al. The chemokine CCL2 mediates the seizure-enhancing effects of systemic inflammation. J Neurosci (2016) 36:3777–88. doi: 10.1523/JNEUROSCI.0451-15.2016
52. Melik-Parsadaniantz S, Rostene W. Chemokines and neuromodulation. J Neuroimmunol (2008) 198:62–8. doi: 10.1016/j.jneuroim.2008.04.022
53. Kumar P, Shih DCW, Lim A, Paleja B, Ling S, Li Yun L, et al. Pro-inflammatory, IL-17 pathways dominate the architecture of the immunome in pediatric refractory epilepsy. JCI Insight (2019) 5(8):e126337. doi: 10.1172/jci.insight.126337
54. Silverberg J, Ginsburg D, Orman R, Amassian V, Durkin HG, Stewart M. Lymphocyte infiltration of neocortex and hippocampus after a single brief seizure in mice. Brain Behav Immun (2010) 24:263–72. doi: 10.1016/j.bbi.2009.10.006
55. Xu D, Robinson AP, Ishii T, Duncan DS, Alden TD, Goings GE, et al. Peripherally derived T regulatory and gammadelta T cells have opposing roles in the pathogenesis of intractable pediatric epilepsy. J Exp Med (2018) 215:1169–86. doi: 10.1084/jem.20171285
56. Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence entry, maintenance, and exit in adult stem cells. Int J Mol Sci (2019) 20(9):2158. doi: 10.3390/ijms20092158
57. Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci (2011) 31:4037–50. doi: 10.1523/JNEUROSCI.6210-10.2011
58. de Vries EE, van den Munckhof B, Braun KP, van Royen-Kerkhof A, de Jager W, Jansen FE. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci Biobehav Rev (2016) 63:177–90. doi: 10.1016/j.neubiorev.2016.02.007
59. Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflamm (2018) 15:144. doi: 10.1186/s12974-018-1192-7
60. Han T, Qin Y, Mou C, Wang M, Jiang M, Liu B. Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1beta receptor through PI3K/Akt pathway. Am J Transl Res (2016) 8:4499–509.
61. Plata-Salaman CR, Ilyin SE, Turrin NP, Gayle D, Flynn MC, Romanovitch AE, et al. Kindling modulates the IL-1beta system, TNF-alpha, TGF-beta1, and neuropeptide mRNAs in specific brain regions. Brain Res Mol Brain Res (2000) 75:248–58. doi: 10.1016/S0169-328X(99)00306-X
62. Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun (2008) 22:797–803. doi: 10.1016/j.bbi.2008.03.009
63. Shi LM, Chen RJ, Zhang H, Jiang CM, Gong J. Cerebrospinal fluid neuron specific enolase, interleukin-1beta and erythropoietin concentrations in children after seizures. Childs Nerv Syst (2017) 33:805–11. doi: 10.1007/s00381-017-3359-4
64. Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T, et al. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia (2002) 43 Suppl 5:30–5. doi: 10.1046/j.1528-1157.43.s.5.14.x
65. Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, et al. Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics (2011) 8:304–15. doi: 10.1007/s13311-011-0039-z
66. Alyu F, Dikmen M. Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr (2017) 29:1–16. doi: 10.1017/neu.2016.47
67. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci (2003) 23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
68. Roseti C, van Vliet EA, Cifelli P, Ruffolo G, Baayen JC, Di Castro MA, et al. GABAA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis (2015) 82:311–20. doi: 10.1016/j.nbd.2015.07.003
69. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther (2006) 8 Suppl 2:S3. doi: 10.1186/ar1917
70. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci (2012) 8:1254–66. doi: 10.7150/ijbs.4679
71. Gruol DL. IL-6 regulation of synaptic function in the CNS. Neuropharmacology (2015) 96:42–54. doi: 10.1016/j.neuropharm.2014.10.023
72. Alapirtti T, Lehtimaki K, Nieminen R, Makinen R, Raitanen J, Moilanen E, et al. The production of IL-6 in acute epileptic seizure: A video-EEG study. J Neuroimmunol (2018) 316:50–5. doi: 10.1016/j.jneuroim.2017.12.008
73. Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res (2003) 73:176–87. doi: 10.1002/jnr.10635
74. Kalueff AV, Lehtimaki KA, Ylinen A, Honkaniemi J, Peltola J. Intranasal administration of human IL-6 increases the severity of chemically induced seizures in rats. Neurosci Lett (2004) 365:106–10. doi: 10.1016/j.neulet.2004.04.061
75. Pineda E, Shin D, You SJ, Auvin S, Sankar R, Mazarati A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann Neurol (2013) 74:11–9. doi: 10.1002/ana.23898
76. De Sarro G, Russo E, Ferreri G, Giuseppe B, Flocco MA, Di Paola ED, et al. Seizure susceptibility to various convulsant stimuli of knockout interleukin-6 mice. Pharmacol Biochem Behav (2004) 77:761–6. doi: 10.1016/j.pbb.2004.01.012
77. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
78. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol (2007) 8:247–56. doi: 10.1038/ni1439
79. Luo H, Liu HZ, Zhang WW, Matsuda M, Lv N, Chen G, et al. Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep (2019) 29:2384–97 e5. doi: 10.1016/j.celrep.2019.10.085
80. He JJ, Sun FJ, Wang Y, Luo XQ, Lei P, Zhou J, et al. Increased expression of interleukin 17 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J Neuroimmunol (2016) 298:153–9. doi: 10.1016/j.jneuroim.2016.07.017
81. Knebel A, Kampe A, Carlson R, Rohn K, Tipold A. Th17 cell-mediated immune response in a subpopulation of dogs with idiopathic epilepsy. PloS One (2022) 17:e0262285. doi: 10.1371/journal.pone.0262285
82. Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol (2010) 184:4898–906. doi: 10.4049/jimmunol.1000142
83. Kostic M, Zivkovic N, Cvetanovic A, Stojanovic I, Colic M. IL-17 signalling in astrocytes promotes glutamate excitotoxicity: Indications for the link between inflammatory and neurodegenerative events in multiple sclerosis. Mult Scler Relat Disord (2017) 11:12–7. doi: 10.1016/j.msard.2016.11.006
84. Olmos G, Llado J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflammation (2014) 2014:861231. doi: 10.1155/2014/861231
85. Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci U.S.A. (1995) 92:11294–8.
86. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem (2006) 281:21362–8. doi: 10.1074/jbc.M600504200
87. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci (2005) 25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
88. Kubota K, Inoue K, Hashimoto R, Kumamoto N, Kosuga A, Tatsumi M, et al. Tumor necrosis factor receptor-associated protein 1 regulates cell adhesion and synaptic morphology via modulation of N-cadherin expression. J Neurochem (2009) 110:496–508. doi: 10.1111/j.1471-4159.2009.06099.x
89. Yuhas Y, Weizman A, Ashkenazi S. Bidirectional concentration-dependent effects of tumor necrosis factor alpha in Shigella dysenteriae-related seizures. Infect Immun (2003) 71:2288–91. doi: 10.1128/IAI.71.4.2288-2291.2003
90. Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U.S.A. (1998) 95:570–5.
91. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discovery (2004) 3:1011–22. doi: 10.1038/nrd1580
92. Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, et al. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci (2009) 29:8927–35. doi: 10.1523/JNEUROSCI.0430-09.2009
93. Yu W, Zou Y, Du Y, Luo J, Zhang M, Yang W, et al. Altered cerebrospinal fluid concentrations of TGFbeta1 in patients with drug-resistant epilepsy. Neurochem Res (2014) 39:2211–7. doi: 10.1007/s11064-014-1422-z
94. Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain (2007) 130:535–47. doi: 10.1093/brain/awl317
95. Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia (2012) 60:1251–7. doi: 10.1002/glia.22311
96. He Y, Zhang H, Yung A, Villeda SA, Jaeger PA, Olayiwola O, et al. ALK5-dependent TGF-beta signaling is a major determinant of late-stage adult neurogenesis. Nat Neurosci (2014) 17:943–52. doi: 10.1038/nn.3732
97. Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis (2015) 78:115–25. doi: 10.1016/j.nbd.2015.02.029
98. Andersson U, Tracey KJ, Yang H. Post-translational modification of HMGB1 disulfide bonds in stimulating and inhibiting inflammation. Cells (2021) 10(12):3323. doi: 10.3390/cells10123323
99. Zhao L, Liu P, Kepp O, Kroemer G. Methods for measuring HMGB1 release during immunogenic cell death. Methods Enzymol (2019) 629:177–93. doi: 10.1016/bs.mie.2019.05.001
100. Pauletti A, Terrone G, Shekh-Ahmad T, Salamone A, Ravizza T, Rizzi M, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain (2019) 142:e39. doi: 10.1093/brain/awz130
101. Walker LE, Frigerio F, Ravizza T, Ricci E, Tse K, Jenkins RE, et al. Molecular isoforms of high-mobility group box 1 are mechanistic biomarkers for epilepsy. J Clin Invest (2019) 129:2166. doi: 10.1172/JCI129285
102. Zhu M, Chen J, Guo H, Ding L, Zhang Y, Xu Y. High mobility group protein B1 (HMGB1) and interleukin-1beta as prognostic biomarkers of epilepsy in children. J Child Neurol (2018) 33:909–17. doi: 10.1177/0883073818801654
103. Han Y, Yang L, Liu X, Feng Y, Pang Z, Lin Y. HMGB1/CXCL12-mediated immunity and th17 cells might underlie highly suspected autoimmune epilepsy in elderly individuals. Neuropsychiatr Dis Treat (2020) 16:1285–93. doi: 10.2147/NDT.S242766
104. Zhao J, Zheng Y, Liu K, Chen J, Lai N, Fei F, et al. HMGB1 is a therapeutic target and biomarker in diazepam-refractory status epilepticus with wide time window. Neurotherapeutics (2020) 17:710–21. doi: 10.1007/s13311-019-00815-3
105. Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal (2014) 21:1726–40. doi: 10.1089/ars.2013.5349
106. Terrone G, Balosso S, Pauletti A, Ravizza T, Vezzani A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology (2020) 167:107742. doi: 10.1016/j.neuropharm.2019.107742
107. Kaneko Y, Pappas C, Malapira T, Vale FL, Tajiri N, Borlongan CV. Extracellular HMGB1 modulates glutamate metabolism associated with kainic acid-induced epilepsy-like hyperactivity in primary rat neural cells. Cell Physiol Biochem (2017) 41:947–59. doi: 10.1159/000460513
108. Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol (2018) 135:311–36. doi: 10.1007/s00401-018-1815-1
109. Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med (2008) 14:1377–83. doi: 10.1038/nm.1878
110. Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. J Neuroimmunol (2010) 224:22–7. doi: 10.1016/j.jneuroim.2010.05.016
111. Bozzi Y, Caleo M. Epilepsy, seizures, and inflammation: role of the C-C motif ligand 2 chemokine. DNA Cell Biol (2016) 35:257–60. doi: 10.1089/dna.2016.3345
112. Yeo SI, Kim JE, Ryu HJ, Seo CH, Lee BC, Choi IG, et al. The roles of fractalkine/CX3CR1 system in neuronal death following pilocarpine-induced status epilepticus. J Neuroimmunol (2011) 234:93–102. doi: 10.1016/j.jneuroim.2011.03.005
113. Louboutin JP, Chekmasova A, Marusich E, Agrawal L, Strayer DS. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J (2011) 25:737–53. doi: 10.1096/fj.10-161851
114. Serrano GE, Lelutiu N, Rojas A, Cochi S, Shaw R, Makinson CD, et al. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci (2011) 31:14850–60. doi: 10.1523/JNEUROSCI.3922-11.2011
115. Jiang J, Yang MS, Quan Y, Gueorguieva P, Ganesh T, Dingledine R. Therapeutic window for cyclooxygenase-2 related anti-inflammatory therapy after status epilepticus. Neurobiol Dis (2015) 76:126–36. doi: 10.1016/j.nbd.2014.12.032
116. Ikeda-Matsuo Y. The role of mPGES-1 in inflammatory brain diseases. Biol Pharm Bull (2017) 40:557–63. doi: 10.1248/bpb.b16-01026
117. Takemiya T, Maehara M, Matsumura K, Yasuda S, Sugiura H, Yamagata K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci Res (2006) 56:103–10. doi: 10.1016/j.neures.2006.06.003
118. O'Banion MK. Prostaglandin E2 synthases in neurologic homeostasis and disease. Prostaglandins Other Lipid Mediat (2010) 91:113–7. doi: 10.1016/j.prostaglandins.2009.04.008
119. Li L, Yasmen N, Hou R, Yang S, Lee JY, Hao J, et al. Inducible prostaglandin E synthase as a pharmacological target for ischemic stroke. Neurotherapeutics (2022) 19:366–85. doi: 10.1007/s13311-022-01191-1
120. Yu Y, Jiang J. COX-2/PGE(2) axis regulates hippocampal BDNF/TrkB signaling via EP2 receptor after prolonged seizures. Epilepsia Open (2020) 5:418–31. doi: 10.1002/epi4.12409
121. Takemiya T, Matsumura K, Sugiura H, Yasuda S, Uematsu S, Akira S, et al. Endothelial microsomal prostaglandin E synthase-1 facilitates neurotoxicity by elevating astrocytic Ca2+ levels. Neurochem Int (2011) 58:489–96. doi: 10.1016/j.neuint.2011.01.003
122. Schlichtiger J, Pekcec A, Bartmann H, Winter P, Fuest C, Soerensen J, et al. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br J Pharmacol (2010) 160:1062–71. doi: 10.1111/j.1476-5381.2010.00765.x
123. Soldner ELB, Hartz AMS, Akanuma SI, Pekcec A, Doods H, Kryscio RJ, et al. Inhibition of human microsomal PGE2 synthase-1 reduces seizure-induced increases of P-glycoprotein expression and activity at the blood-brain barrier. FASEB J (2019) 33:13966–81. doi: 10.1096/fj.201901460RR
124. Feldmann M, Asselin MC, Liu J, Wang S, McMahon A, Anton-Rodriguez J, et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: a case-control study. Lancet Neurol (2013) 12:777–85. doi: 10.1016/S1474-4422(13)70109-1
125. Shimada T, Takemiya T, Sugiura H, Yamagata K. Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflammation (2014) 2014:901902. doi: 10.1155/2014/901902
126. Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat (2010) 91:104–12. doi: 10.1016/j.prostaglandins.2009.04.003
127. Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, et al. Prostaglandin E2 modulates Na+,K+-ATPase activity in rat hippocampus: implications for neurological diseases. J Neurochem (2009) 109:416–26. doi: 10.1111/j.1471-4159.2009.05961.x
128. Reschke CR, Poersch AB, Masson CJ, Jesse AC, Marafiga JR, Lenz QF, et al. Systemic delivery of selective EP1 and EP3 receptor antagonists attenuates pentylenetetrazole-induced seizures in mice. Int J Physiol Pathophysiol Pharmacol (2018) 10:47–59.
129. Collins SA, Huff C, Chiaia N, Gudelsky GA, Yamamoto BK. 3,4-methylenedioxymethamphetamine increases excitability in the dentate gyrus: role of 5HT2A receptor-induced PGE2 signaling. J Neurochem (2016) 136:1074–84. doi: 10.1111/jnc.13493
130. Rojas A, Gueorguieva P, Lelutiu N, Quan Y, Shaw R, Dingledine R. The prostaglandin EP1 receptor potentiates kainate receptor activation via a protein kinase C pathway and exacerbates status epilepticus. Neurobiol Dis (2014) 70:74–89. doi: 10.1016/j.nbd.2014.06.004
131. Marchi N. Experimental status epilepticus, COX-2 and BDNF: Connecting the dots. Epilepsia Open (2021) 6:466–7. doi: 10.1002/epi4.12501
132. Kang X, Qiu J, Li Q, Bell KA, Du Y, Jung DW, et al. Cyclooxygenase-2 contributes to oxidopamine-mediated neuronal inflammation and injury via the prostaglandin E2 receptor EP2 subtype. Sci Rep (2017) 7:9459. doi: 10.1038/s41598-017-09528-z
133. Liu Q, Liang X, Wang Q, Wilson EN, Lam R, Wang J, et al. PGE(2) signaling via the neuronal EP2 receptor increases injury in a model of cerebral ischemia. Proc Natl Acad Sci U.S.A. (2019) 116:10019–24. doi: 10.1073/pnas.1818544116
134. Hou R, Yu Y, Jiang J. PGE2 receptors in detrusor muscle: Drugging the undruggable for urgency. Biochem Pharmacol (2021) 184:114363. doi: 10.1016/j.bcp.2020.114363
135. Rojas A, Ganesh T, Wang W, Wang J, Dingledine R. A rat model of organophosphate-induced status epilepticus and the beneficial effects of EP2 receptor inhibition. Neurobiol Dis (2020) 133:104399. doi: 10.1016/j.nbd.2019.02.010
136. Jiang J, Yu Y, Kinjo ER, Du Y, Nguyen HP, Dingledine R. Suppressing pro-inflammatory prostaglandin signaling attenuates excitotoxicity-associated neuronal inflammation and injury. Neuropharmacology (2019) 149:149–60. doi: 10.1016/j.neuropharm.2019.02.011
137. Varvel NH, Espinosa-Garcia C, Hunter-Chang S, Chen D, Biegel A, Hsieh A, et al. Peripheral myeloid cell EP2 activation contributes to the deleterious consequences of status epilepticus. J Neurosci (2021) 41:1105–17. doi: 10.1523/JNEUROSCI.2040-20.2020
138. Dhir A. An update of cyclooxygenase (COX)-inhibitors in epilepsy disorders. Expert Opin Investig Drugs (2019) 28:191–205. doi: 10.1080/13543784.2019.1557147
139. Claycomb RJ, Hewett SJ, Hewett JA. Prophylactic, prandial rofecoxib treatment lacks efficacy against acute PTZ-induced seizure generation and kindling acquisition. Epilepsia (2011) 52:273–83. doi: 10.1111/j.1528-1167.2010.02889.x
140. Polascheck N, Bankstahl M, Loscher W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp Neurol (2010) 224:219–33. doi: 10.1016/j.expneurol.2010.03.014
141. Holtman L, van Vliet EA, Edelbroek PM, Aronica E, Gorter JA. Cox-2 inhibition can lead to adverse effects in a rat model for temporal lobe epilepsy. Epilepsy Res (2010) 91:49–56. doi: 10.1016/j.eplepsyres.2010.06.011
142. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
143. Bordon Y. Infectious disease: hushing mTOR boosts immunity to pathogens. Nat Rev Immunol (2013) 13:847. doi: 10.1038/nri3562
144. Nowak W, Grendas LN, Sanmarco LM, Estecho IG, Arena AR, Eberhardt N, et al. Pro-inflammatory monocyte profile in patients with major depressive disorder and suicide behaviour and how ketamine induces anti-inflammatory M2 macrophages by NMDAR and mTOR. EBioMedicine (2019) 50:290–305. doi: 10.1016/j.ebiom.2019.10.063
145. Zhao Y, Shen X, Na N, Chu Z, Su H, Chao S, et al. mTOR masters monocyte development in bone marrow by decreasing the inhibition of STAT5 on IRF8. Blood (2018) 131:1587–99. doi: 10.1182/blood-2017-04-777128
146. Yang K, Blanco DB, Chen X, Dash P, Neale G, Rosencrance C, et al. Metabolic signaling directs the reciprocal lineage decisions of alphabeta and gammadelta T cells. Sci Immunol (2018) 3(25):eaas9818. doi: 10.1126/sciimmunol.aas9818
147. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
148. Heikamp EB, Patel CH, Collins S, Waickman A, Oh MH, Sun IH, et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol (2014) 15:457–64. doi: 10.1038/ni.2867
149. Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol (2018) 314:H693–703. doi: 10.1152/ajpheart.00570.2017
150. Zhang L, Huang T, Teaw S, Bordey A. Hypervascularization in mTOR-dependent focal and global cortical malformations displays differential rapamycin sensitivity. Epilepsia (2019) 60:1255–65. doi: 10.1111/epi.15969
151. Hu Y, Mai W, Chen L, Cao K, Zhang B, Zhang Z, et al. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia (2020) 68:1031–45. doi: 10.1002/glia.23760
152. Xiao Z, Peng J, Gan N, Arafat A, Yin F. Interleukin-1beta plays a pivotal role via the PI3K/akt/mTOR signaling pathway in the chronicity of mesial temporal lobe epilepsy. Neuroimmunomodulation (2016) 23:332–44. doi: 10.1159/000460254
153. Deason K, Troutman TD, Jain A, Challa DK, Mandraju R, Brewer T, et al. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J Exp Med (2018) 215:2413–28. doi: 10.1084/jem.20171810
154. Ren W, Yin J, Duan J, Liu G, Tan B, Yang G, et al. mTORC1 signaling and IL-17 expression: Defining pathways and possible therapeutic targets. Eur J Immunol (2016) 46:291–9. doi: 10.1002/eji.201545886
155. Wyszynski RW, Gibbs BF, Varani L, Iannotta D, Sumbayev VV. Interleukin-1 beta induces the expression and production of stem cell factor by epithelial cells: crucial involvement of the PI-3K/mTOR pathway and HIF-1 transcription complex. Cell Mol Immunol (2016) 13:47–56. doi: 10.1038/cmi.2014.113
156. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet (2016) 388:2153–63. doi: 10.1016/S0140-6736(16)31419-2
157. Abdelsalam M, Abd Elmagid DS, Magdy H, El-Sabbagh AM, Mostafa M. The association between toll-like receptor 4 (TLR4) genotyping and the risk of epilepsy in children. Egyptian J Med Hum Genet (2020) 21:61. doi: 10.1186/s43042-020-00102-3
158. Hsieh MY, Lin JJ, Hsia SH, Huang JL, Yeh KW, Chang KW, et al. Diminished toll-like receptor response in febrile infection-related epilepsy syndrome (FIRES). BioMed J (2020) 43:293–304. doi: 10.1016/j.bj.2020.05.007
159. Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol (2012) 12:168–79. doi: 10.1038/nri3151
160. Gross A, Benninger F, Madar R, Illouz T, Griffioen K, Steiner I, et al. Toll-like receptor 3 deficiency decreases epileptogenesis in a pilocarpine model of SE-induced epilepsy in mice. Epilepsia (2017) 58:586–96. doi: 10.1111/epi.13688
161. Wang FX, Xiong XY, Zhong Q, Meng ZY, Yang H, Yang QW. Foxp3 exhibits antiepileptic effects in ictogenesis involved in TLR4 signaling. FASEB J (2017) 31:2948–62. doi: 10.1096/fj.201600989R
162. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
163. Zhan X, Li Q, Xu G, Xiao X, Bai Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front Immunol (2022) 13:1109938. doi: 10.3389/fimmu.2022.1109938
164. Jin X, Liu MY, Zhang DF, Zhong X, Du K, Qian P, et al. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-kappaB signaling pathway. CNS Neurosci Ther (2019) 25:575–90. doi: 10.1111/cns.13086
165. Rong S, Wan D, Fan Y, Liu S, Sun K, Huo J, et al. Amentoflavone affects epileptogenesis and exerts neuroprotective effects by inhibiting NLRP3 inflammasome. Front Pharmacol (2019) 10:856. doi: 10.3389/fphar.2019.00856
166. Shen K, Jiang W, Zhang C, Cai L, Wang Q, Yu H, et al. Molecular mechanism of a specific NLRP3 inhibitor to alleviate seizure severity induced by pentylenetetrazole. Curr Mol Pharmacol (2021) 14:579–86. doi: 10.2174/1874467213666200810140749
167. Yue J, Wei YJ, Yang XL, Liu SY, Yang H, Zhang CQ. NLRP3 inflammasome and endoplasmic reticulum stress in the epileptogenic zone in temporal lobe epilepsy: molecular insights into their interdependence. Neuropathol Appl Neurobiol (2020) 46:770–85. doi: 10.1111/nan.12621
168. Cristina de Brito Toscano E, Leandro Marciano Vieira E, Boni Rocha Dias B, Vidigal Caliari M, Paula Goncalves A, Varela Giannetti A, et al. NLRP3 and NLRP1 inflammasomes are up-regulated in patients with mesial temporal lobe epilepsy and may contribute to overexpression of caspase-1 and IL-beta in sclerotic hippocampi. Brain Res (2021) 1752:147230. doi: 10.1016/j.brainres.2020.147230
169. Pohlentz MS, Muller P, Cases-Cunillera S, Opitz T, Surges R, Hamed M, et al. Characterisation of NLRP3 pathway-related neuroinflammation in temporal lobe epilepsy. PloS One (2022) 17:e0271995. doi: 10.1371/journal.pone.0271995
170. Meng XF, Tan L, Tan MS, Jiang T, Tan CC, Li MM, et al. Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J Neuroinflamm (2014) 11:212. doi: 10.1186/s12974-014-0212-5
171. Qin Z, Song J, Lin A, Yang W, Zhang W, Zhong F, et al. GPR120 modulates epileptic seizure and neuroinflammation mediated by NLRP3 inflammasome. J Neuroinflamm (2022) 19:121. doi: 10.1186/s12974-022-02482-2
Keywords: epileptogenesis, seizures, neuroinflammation, microglia, astrocytes
Citation: Li W, Wu J, Zeng Y and Zheng W (2023) Neuroinflammation in epileptogenesis: from pathophysiology to therapeutic strategies. Front. Immunol. 14:1269241. doi: 10.3389/fimmu.2023.1269241
Received: 29 July 2023; Accepted: 07 December 2023;
Published: 22 December 2023.
Edited by:
Claudia Espinosa-Garcia, Yale University, United StatesReviewed by:
Alberto Javier Ramos, National Scientific and Technical Research Council (CONICET), ArgentinaMarco I. Gonzalez, University of California, Davis, United States
Copyright © 2023 Li, Wu, Zeng and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zheng, emhlbmd3ZW41MzA5QDE2My5jb20=
†ORCID: Wen Zheng, orcid.org/0000-0002-2047-8496