 Johanna Rausch
Johanna Rausch Evelyn Ullrich
Evelyn Ullrich Michael W.M. Kühn
Michael W.M. Kühn
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 22 September 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1269012
This article is part of the Research Topic Understanding Convergent Evasion Mechanisms in Cancer and Chronic Infection: Implications for Immunotherapy View all 14 articles
AML is a malignant disease of hematopoietic progenitor cells with unsatisfactory treatment outcome, especially in patients that are ineligible for intensive chemotherapy. Immunotherapy, comprising checkpoint inhibition, T-cell engaging antibody constructs, and cellular therapies, has dramatically improved the outcome of patients with solid tumors and lymphatic neoplasms. In AML, these approaches have been far less successful. Discussed reasons are the relatively low mutational burden of AML blasts and the difficulty in defining AML-specific antigens not expressed on hematopoietic progenitor cells. On the other hand, epigenetic dysregulation is an essential driver of leukemogenesis, and non-selective hypomethylating agents (HMAs) are the current backbone of non-intensive treatment. The first clinical trials that evaluated whether HMAs may improve immune checkpoint inhibitors’ efficacy showed modest efficacy except for the anti-CD47 antibody that was substantially more efficient against AML when combined with azacitidine. Combining bispecific antibodies or cellular treatments with HMAs is subject to ongoing clinical investigation, and efficacy data are awaited shortly. More selective second-generation inhibitors targeting specific chromatin regulators have demonstrated promising preclinical activity against AML and are currently evaluated in clinical trials. These drugs that commonly cause leukemia cell differentiation potentially sensitize AML to immune-based treatments by co-regulating immune checkpoints, providing a pro-inflammatory environment, and inducing (neo)-antigen expression. Combining selective targeted epigenetic drugs with (cellular) immunotherapy is, therefore, a promising approach to avoid unintended effects and augment efficacy. Future studies will provide detailed information on how these compounds influence specific immune functions that may enable translation into clinical assessment.
Acute myeloid leukemia (AML) is a malignant neoplasm of hematopoietic progenitor cells driven by acquired genetic aberrations that mediate uncontrolled proliferation and a block in differentiation (1, 2).
Novel mechanism-based drugs have improved treatment options in recent years (2), but intensive chemotherapy is still the backbone of curative treatment and induces complete remissions in up to 70% of patients (3). However, relapse is common, and overall survival is generally unsatisfactory and heterogeneous based on two significant factors: the genetic alterations of individual AML blasts and the patient’s age at diagnosis (2, 4). Despite intensive treatment, most elderly patients will ultimately succumb to their disease (2–5). Survival for patients unfit for intensive treatment is dismal, with a 5-year overall survival (OS) below 10% with current standard of care options (3–6) underpinning the need for more efficient and less toxic treatment options.
Epigenetic dysregulation has been recognized as an essential driver for leukemogenesis, thereby providing a therapeutic opportunity. Hypomethylating agents (HMA) are non-selective first-generation epigenetic drugs and are considered a mainstay in treating unfit and elderly patients (7). Several more selective compounds targeting specific epigenetic dependencies have been developed in recent years with promising responses in clinical trials (8–10). Immunotherapy has revolutionized the treatment of solid tumors and lymphatic neoplasms (11–30), but has been far less successful against AML. Mechanisms behind the limited efficacy remain obscure but have been attributed to difficulties in finding a target exclusively expressed on AML blasts, their relatively low mutational burden, and low neo-antigen expression (31–34).
Epigenetic manipulation has been reported to induce immune modulatory effects, including an increased expression of tumor-associated antigens (35, 36) that may sensitize AML blasts for immunotherapy. Here we review the concept of combined epigenetic targeting with immunotherapeutic approaches against AML.
Epigenetic dysregulation has been implicated in the pathogenesis of most cancer types, including AML. Sequencing efforts to characterize the genomic landscape of various cancer types have revealed recurrent mutations in epigenetic regulators, affecting AML in more than 60% of cases (37, 38). Epigenetic regulators determine the chromatin state by controlling regulatory regions and gene expression via chemical modifications, including DNA methylation and histone protein acetylation, methylation, or phosphorylation as reviewed elsewhere (39–41). Therefore, epigenetic regulators were recognized as therapeutic opportunities for many cancers, particularly AML.
First-generation HMAs such as azacitidine and decitabine are non-selective drugs that reduce promotor hypermethylation to restore the expression of tumor suppressor genes (42). These drugs have built the backbone for non-intensive AML treatment (7), and their combination with the BCL2 inhibitor venetoclax is the current standard of care for unfit AML patients resulting in a median overall survival (OS) of 14.7 months (6). Histone deacetylase (HDAC) inhibitors, another class of non-selective epigenetic drugs that initially showed promising activity in preclinical models (43), failed to induce sustainable remissions in clinical trials in monotherapy (44–46). Reasons for the low efficacy in clinical studies are not fully elucidated, however missing predictive biomarkers, the heterogeneous activity of different HDAC inhibitors, and dose-limiting off-target effects of pan-HDAC inhibitors remain an unsolved problem, especially in combination with other anti-neoplastic agents (47–49).
Second-generation epigenetic inhibitors were developed to target specific chromatin modifiers and epigenetic dependencies in various cancers with potentially less off-target toxicity. Research has particularly focused on the development and clinical assessment of drugs targeting the following chromatin modifiers:
Bromodomain-containing transcriptional activators (BRDs) are recruited to histone-acetylated transcription sites to accelerate gene expression. BRD4 is a Bromodomain and extra-terminal (BET) protein, and its function is best characterized in AML (50, 51). Inhibitors of BET proteins, particularly BRD4, have shown promising preclinical activity (52) but demonstrated only modest activity as a single agent against AML with an overall response rate (ORR) of only 6% in relapsed refractory (R/R) AML (53).
The histone methyltransferase Disruptor of Telomeric Silencing 1-like (DOT1L) is the only histone 3 lysine 79 methyltransferase known to date. It maintains leukemic transcription in leukemias with Mixed-Lineage Leukemia (MLL, also known as KMT2A)-rearrangement (MLL-r) or partial tandem duplication and NPM1 mutant (NPM1mut) leukemia (54, 55). Similar to BET inhibitors, the first clinical trials with DOT1L inhibitors demonstrated limited activity with only two complete remissions (CR) in 52 patients in a phase I trial (56) despite promising preclinical activity (54, 55).
Protein Arginine Methyltransferase 5 (PRMT5) regulates gene expression by dimethylation of histone and non-histone proteins (e.g.,RNA splicing factors) (57, 58). Inhibition of PRMT5 has demonstrated anti-leukemic activity and induction of differentiation in preclinical MLL-r and FLT3-ITD AML models (59, 60), and several inhibitors are currently evaluated in early clinical trials for solid tumors, lymphomas, and leukemias, which was reviewed elsewhere (61). In brief, phase I studies have reported limited efficacy, with common adverse effects in solid tumors and primary myelofibrosis (62–64). One phase I study is currently recruiting AML patients (65).
Enhancer of Zeste Homolog 2 (EZH2) is a lysine methyltransferase and the catalytic subunit of Polycomb Repressive Complex 2 (PRC2) that silences its target genes via H3K27 trimethylation (66, 67). EZH2 mutations are found in solid tumors and usually as gain-of-function events in lymphomas (68, 69). The inhibitor tazometestat induced durable and complete responses in Phase I/II trials in sarcomas and lymphomas (70–72). EZH2 has been reported to act context-dependently as a tumor suppressor or sometimes as an oncogene in myeloid malignancies (66, 73). Its loss has been associated with poor prognosis and chemotherapy resistance, and mutations are more common in relapsed AML patients (74–76). EZH1/2 inhibition has demonstrated in vitro and in vivo anti-leukemic activity (77, 78). Clinical outcome data for EZH2 inhibition in AML do not exist, also because a phase I trial was terminated due to insufficient patient recruitment (NCT03110354).
Lysine-Specific Demethylase-1 (LSD1, also known as KDM1A) is a histone 3 demethylase and is believed to participate in the control of leukemic gene expression programs (79). LSD1 inhibition had promising activity in preclinical leukemia models, and preliminary efficacy against AML has been reported from an ongoing clinical phase I/II trial (80, 81). Additional studies are needed to define the clinical activity in specific AML subtypes in detail.
Dramatic clinical responses in AML were observed with specific inhibitors of mutant isocitrate-dehydrogenase (IDH) 1 and 2 enzymes and are also explained by epigenetic mechanisms: Mutations in IDH1 and IDH2 lead to a neo-enzyme activity of both enzymes, accumulating the ordinarily absent oncometabolite 2-hydroxyglutarate (2-HG) (82). 2-HG inhibits ten-eleven translocation (TET) family enzymes responsible for DNA methylation, ultimately resulting in aberrant expression of leukemic genes (83). IDH1/2 inhibition induces cell differentiation of IDH-mutated AML blasts (84). The first phase I trial assessed the IDH2 inhibitor enasidenib as a single agent with an ORR of 40.3% and a median OS rate of 9.3 months in R/R AML patients (85). The combination of the IDH1 inhibitor ivosidenib with azacitidine was recently approved for newly diagnosed IDH1 mutated AML in Europe and the U.S. The approval was based on a randomized, placebo-controlled phase III trial where the combination significantly increased CR rates (47% vs. 15%, p<0.001) and survival (recently updated median OS: 29.3 vs. 7.9 months; HR 0.42, p-value <0.0001) compared to azacitidine plus placebo (9, 86).
A novel epigenetic target and auspicious therapeutic opportunity against specific AML subtypes is the protein interaction of the histone methyltransferase KMT2A (also known as MLL1) with its oncogenic adaptor protein menin (encoded by the MEN1 gene). While it was reported that menin is required for chromatin binding and target gene activation of oncogenic MLL1-fusion proteins in MLL1-rearranged leukemias (87), our group reported that the direct interaction of wildtype MLL with menin is a dependency in the most prevalent NPM1mut AML subtype (55). Characteristic leukemic gene expression programs, including high-level expression of MEIS1, PBX3, and various HOX transcription factor genes, also depend on the protein interaction (55). Pharmacological inhibition of the menin-MLL interaction has demonstrated profound in vitro and in vivo anti-leukemic activity inducing uniform transcriptional repression of MEIS1, PBX3, FLT3, and BCL2, and leading to differentiation and apoptosis in MLL-r and NPM1mut leukemias (55, 87–90). These preclinical data translated into an ongoing clinical assessment of five different menin inhibitors against AML (NCT04067336, NCT04065399, NCT05153330, NCT04811560, NCT04988555) with astonishing first efficacy data from two phase I trials: The oral menin inhibitor revumenib induced complete remissions (combined; CRc) in 38% of heavily pretreated R/R AML with NPM1mut or MLL-r as a single agent, with responding patients exhibiting sustainable responses of more than 9.1 months (8). Ziftomenib also had promising clinical activity in NPM1mut or MLL-r R/R AML, with 35% of patients achieving CR/CRh or CRp rate in a phase I/II study (91). The single-agent evaluation of both drugs is currently ongoing. Combinatorial clinical trial assessment with intensive chemotherapy and specific small molecule inhibitors is also underway, as both inhibitors have exhibited synergistic in vitro and in vivo efficacy with various targeted cancer drugs (92–94).
Within the last decade, similarly great excitement has greeted cancer immunotherapy, revolutionizing the treatment of many cancer types (11–30). Concepts to guide the immune system in recognizing and fighting cancer cells comprise antibody-directed targeting, blockage of immune checkpoints, and adoptive transfer of immune cells. These approaches have led to sustainable responses, prolonged survival, and even cure of previously untreatable malignancies, but single-agent efficacy against AML has been limited.
Immune checkpoint blockade (ICB) with anti-CTLA-4 and anti-PD-L1/PD-1 antibodies dramatically improved overall survival in patients with advanced solid tumors as well as Hodgkin’s lymphoma (13–18) and is now considered the standard of care for the treatment of many other cancer entities.
AML cells also have higher surface expression of inhibitory immune checkpoints (such as PD-L1) compared to normal hematopoietic stem (HSCs) and progenitor cells (HSPCs) and higher expression of PD-1 is observed on T-cells of AML patients compared to healthy donors (95–102). Still, clinical trials assessing therapeutic checkpoint blockade yielded generally discouraging results in myeloid neoplasms. Only 1 out of 9 patients with AML or myelodysplastic syndrome (MDS) responded to the anti-PD-1 antibody pidilizumab in a first phase I trial (103). Also, ORR in studies assessing the anti-PD-1 antibody pembrolizumab and anti-PD-L1 antibody atezolizumab in R/R MDS patients were only 4% and 0%, respectively (104, 105). Responses to the anti-CTLA-4 antibody ipilimumab in early clinical trials assessing selected AML patients that relapsed following allogenic stem cell transplantation (SCT) were more promising, with 23% of patients achieving a CR. However, treatment was commonly associated with severe graft versus host disease (12).
CD47 is a checkpoint of the innate immune system that mediates a “do not eat me “ signal to macrophages (106, 107). Magrolimab, a monoclonal anti-CD47 antibody, demonstrated limited efficacy as a single-agent in AML with no objective responses (stable disease: 73%) (108), but might be more efficacious if added to established combination regimens (discussed below).
Bispecific T-cell engager (BiTE) or dual-affinity retargeting antibodies (DART) are artificial antibody constructs that contain two antigen binding sites, one directed against immune effector cells (mostly CD3 for T-cells) and the other against a specific surface antigen on tumor cells. The convergence leads to T- or NK-cell activation and killing (31). BiTEs targeting CD3 and CD19, such as blinatumomab, are efficient against and approved for treating B-cell neoplasms (28). Defining a unique leukemic target on myeloid blasts has yet limited efforts to extend this concept for successful AML treatment (discussed below), and so far, efficacy has been unsatisfactory. In a phase I trial assessing the anti-CD33xCD3 directed bispecific antibody AMG330 against R/R AML, CR/CRi rates were 17% (109) and 3 and 5% in ongoing phase I studies testing the anti-CD33xCD3 BiTE molecules AMV564 and AMG673 (110, 111). Reported ORR from a phase I/II trial exploring flotetuzumab, an anti-CD123xCD3 DART construct, against R/R AML was 30%. However, treatment was associated with high rates of severe cytokine release syndrome (CRS) (81%, 8% ≥3) (112), which was also commonly observed with the bispecific anti-CD123 antibody XmAb14045 (113). Other CD123-targeting antibodies are under clinical investigation (NCT03647800, NCT02715011).
Several reports suggest that the myeloid antigens WT1, PRAME, and CLL-1 (CLEC12A) are expressed only at low levels on HSCs, which may be associated with less hematologic toxicity if targeted by immunotherapy (32, 114–117). A lower CRS rate was reported from a phase I trial exploring the first CLL-1xCD3-directed bispecific antibody MCLA-117 in R/R AML but with only 15% of patients achieving a partial response (118).
Cellular immunotherapy describes the adoptive transfer of genetically engineered autologous chimeric-antigen receptor (CAR)-T or -Natural Killer (NK) cells. Astonishing successes were reported from treatment of B-cell neoplasms with various CAR-T cell products and have led to their approval in the Europe and the U.S. (19–28). As with BITEs and DARTs, CAR construct development against AML faces similar challenges in defining unique immunotargets on AML blasts. Lineage-specific antigens such as CD33 and CD123 are commonly expressed on AML blasts and evaluated as potential targets. Their expression on hematologic stem cells (HSCs) bears the risk of post-treatment bone marrow failure (32, 119, 120). As CAR-T cells commonly have a “memory effect”, hematologic toxicity might be even more severe compared to BITEs and DARTs.
One strategy to avoid the off-tumor toxicity is the development of AND-gated and NOT-gated CAR-T cells that engage two antigens to increase selectivity (121, 122). Perriello et al. developed cytokine-induced killer (CIK) cells with two CARs directed against CD123 and CD33. In this case, simultaneous binding of both CARs is necessary for a cytotoxic T-cell activation, because the CD33 CAR delivers the essential co-stimulatory signal (122). The authors also demonstrate that reduced binding activity of a CAR may increase selectivity by restricting reactivity to cells with high antigen expression. NOT-gates CARs represent an different approach to avoid off-tumor toxicity: Richards et al. developed CD93-directed CAR T-cells that express a second inhibitory CAR (iCAR) directed against an antigen present on endothelial cells but absent on myeloid blasts. This iCAR contains endodomains from ITIM-containing proteins including PD-1, TIM-3 or TIGIT delivering an inhibitory signal that interferes with the CAR T-cell activation signal (121).
So far, CAR-T-cells targeting CD33, CD123, or two antigens at once (e.g., CD33 and CLL-1; CD13 and TIM-3) are currently evaluated in early clinical trials (NCT03971799, NCT03795779, NCT03631576, NCT03190278, NCT03114670, NCT02159495, NCT04272125, NCT03222674, NCT04010877, NCT04097301). Three studies reported activity against heavily pretreated patients (123–125), but longer follow-up efficacy data needed to draw more definitive conclusions are pending. For CD70, another immune target expressed on AML blast and low expression on HSCs, promising activity has been reported in preclinical AML models. Clinical trial evaluation is expected shortly (126, 127).
CAR-engineered NK cells may have potential advantages over CAR-T cells and be a promising alternative for two reasons: a) their HLA-class I independent tumor cell recognition allows maintaining intrinsic anti-tumor activity in case of antigen loss (128), and b) the lack of clonal expansion protects recipients from persistent graft versus host disease (GvHD) or long-term hematologic toxicity, reviewed in (129). First clinical applications have demonstrated encouraging anti-leukemic activity and tolerability with cord-blood-derived CD19-CAR NK cells against chronic lymphatic leukemia (130). CAR-NK cell products are effective against preclinical AML models in vitro and in vivo but clinical activity remains to be demonstrated (131).
While the efficacy of these concepts still needs improvement, the strong graft versus leukemia effect that has been observed over decades following allogenic SCT indicates that AML may still be prone to immunotherapy (132–134). As mentioned above, one potential reason might be the particularly low mutational burden found in AML blasts compared to other cancers, which has been associated with generally lower responses to immune-based treatments (33, 34). Defining an AML-specific immunotarget that is not expressed on HSC is also an ongoing challenge for the development of potent immune-based treatments (32).
Epigenetic mechanisms have been implicated in contributing to the poor responses of AML to immunotherapy. One example is the silencing of HLA class II molecules observed in AML patients that relapsed after allogenic SCT (135–137). This has been attributed to the DNA-hypermethylation of respective promotor regions (96). Therapeutic manipulation with HMAs to reverse promotor-methylation has successfully been used at relapse to boost graft-versus leukemia effects of donor lymphocyte infusions. However, this concept is less efficient with high leukemia burden (138–140). Additional immune modulatory effects of HMA are currently being discussed. These include enhanced expression of tumor-associated antigens such as MAGE-1 and NY-ESO-1 (35, 36). Also, HMA-treatment is associated with tumor re-expression of endogenous retroviruses (ERVs) that is believed to improve T- and NK-cell activation via enhanced IFN-γ expression (141–144), enhances tumor lymphocyte infiltration (145), and impairs expansion of regulatory T-cells (146), (Figure 1). The limited activity of HMAs commonly observed in the clinical setting may partly be explained by the upregulation of the immune inhibitory checkpoints (147, 148).
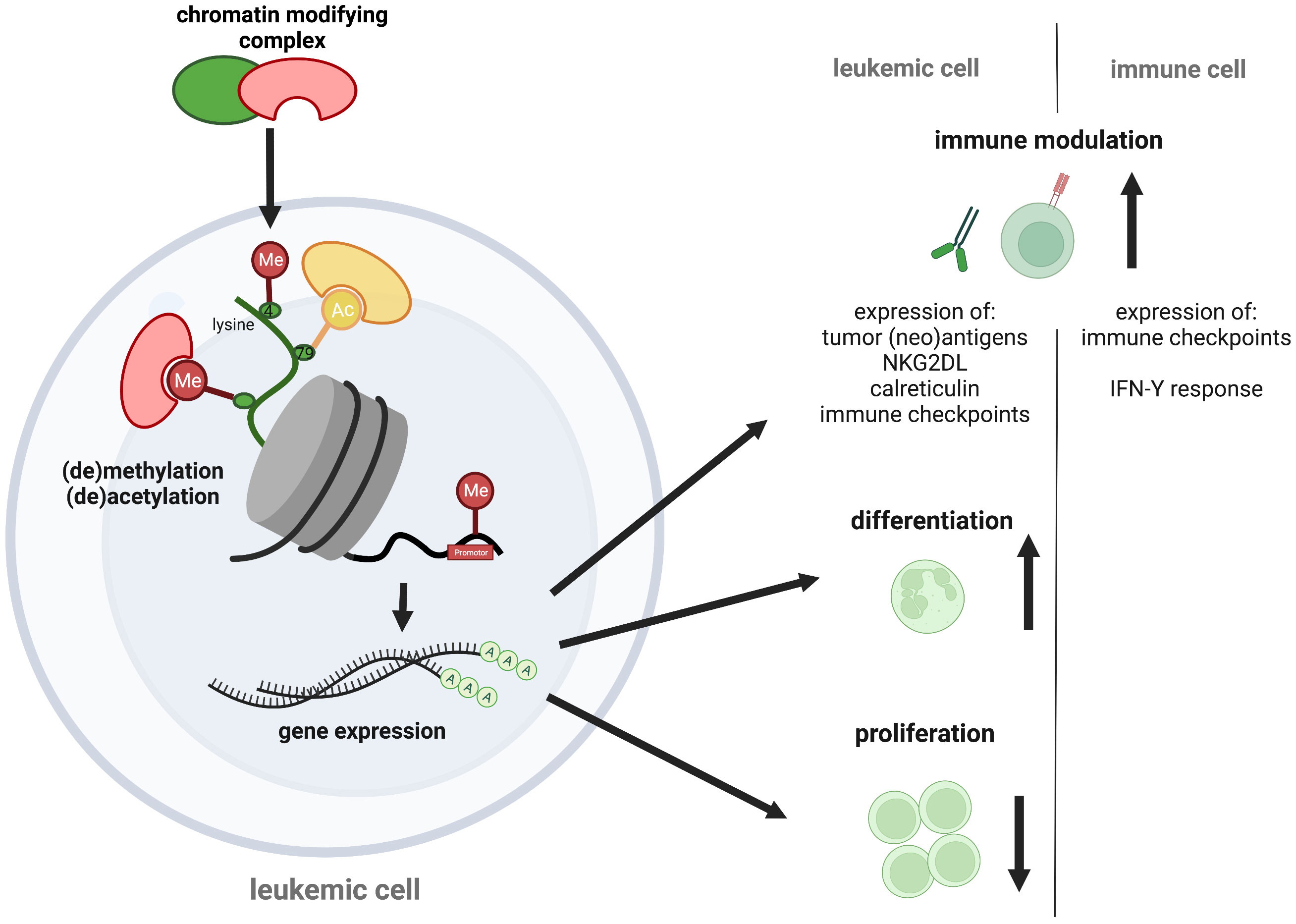
Figure 1 Epigenetic targeting in AML. Epigenetic regulators control transcription via chemical chromatin modifications, including histone protein and DNA (de-)methylation or histone (de-)acetylation that determine chromatin state. As therapeutic opportunities against AML, chromatin modifiers can alter leukemogenic gene expression, causing cell differentiation and proliferation arrest of the malignant blasts. Additional pro-immunogenic effects have recently been discussed, including an increased neoantigen-, immune checkpoint-, NK2GDL- and calreticulin expression on leukemic blasts and an augmented immune checkpoint expression and IFN-Y response of immune cells. The figure was created with BioRender.com.
HMA treatment has also been investigated in combination with immune checkpoint blockade in clinical trials. Encouraging results demonstrated a first phase II trial assessing the combination of PD-1 antibody nivolumab and azacitidine in R/R AML resulting in an ORR of 58% in HMA-naive and 22% in HMA-pretreated patients, respectively (149). Newly diagnosed and R/R patients achieved a CRc in 47% and 14% in a phase II trial assessing the combination of the PD-1 antibody pembrolizumab with azacitidine (150). Azacitidine combined with the anti-TIM-3 monoclonal antibody sabatolimab led to an ORR of 57% and a CRc of 30% in newly diagnosed AML in a phase Ib trial (151). The only randomized data available come from a trial assessing the anti-PD-L1 antibody durvalumab, Here, no significant benefit for the combination of durvalumab and azacitidine was observed over azacitidine alone in MDS/AML patients (152). Consistent with the data above, the authors of a recent meta-analysis concluded that the activity of checkpoint inhibitors is generally low in the relapsed/refractory AML setting (153). Further studies are currently ongoing (Table 1).
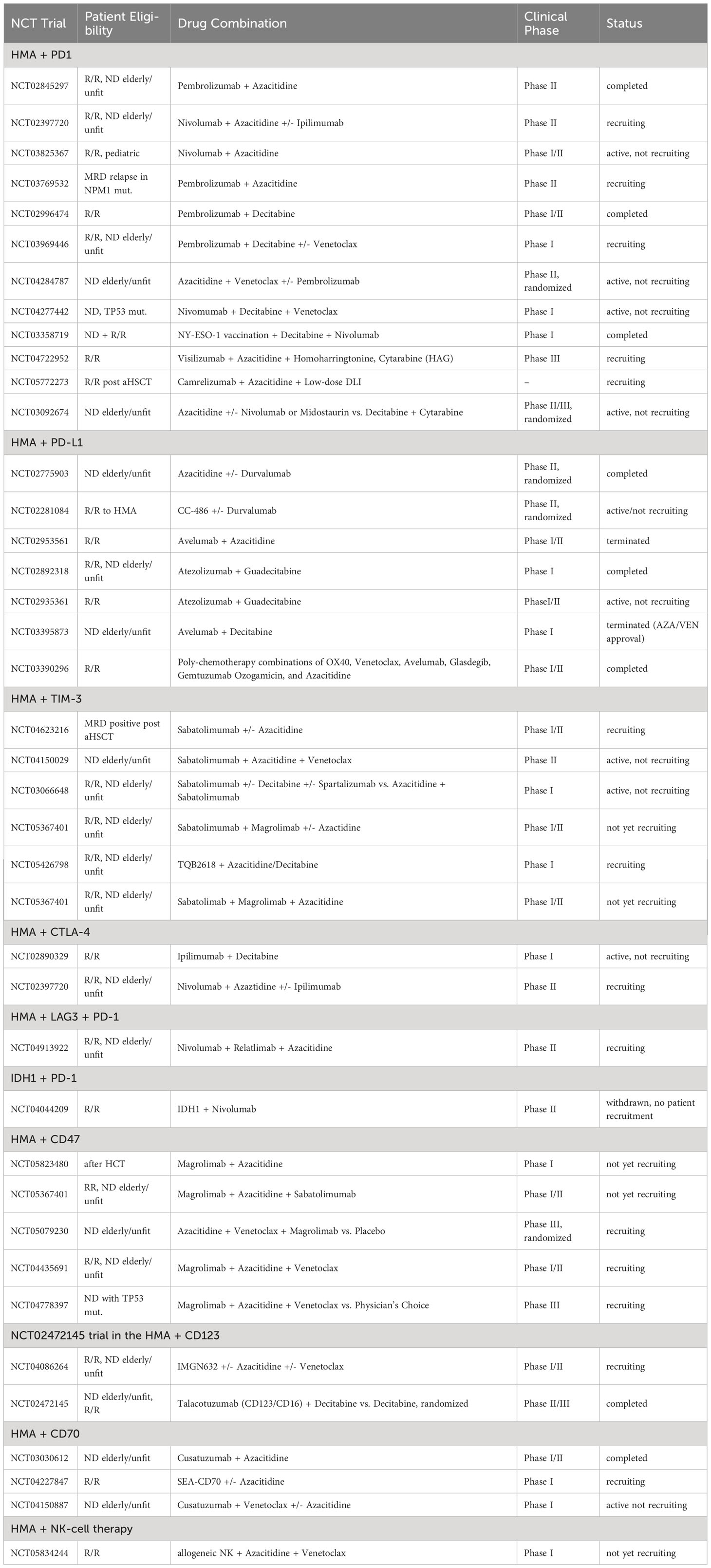
Table 1 Current clinical trials evaluating combinations of epigenetic targeting and immunotherapy in AML.
HMAs in combination with immune checkpoint inhibitors were also assessed in the post-transplant setting, with only a few responses reported and increased immune-related toxicity (12, 154). This was demonstrated by the combination of avelumab and azacitidine, resulting in CR rates of only 10.5% and an increased risk of severe graft versus host disease (155). Several clinical trials are ongoing and will allow more definitive conclusions concerning efficacy and safety.
HDAC inhibitors can also induce tumor-associated antigens, improve antigen presentation, influence T-cell trafficking and activity but also increase PD-1 expression (156–159). Several trials reported responses to HDAC inhibitors in combination with checkpoint blockade in solid tumors (160). However, in R/R MDS/AML patients, no activity of this concept has been reported in a recent phase 1b study assessing pembrolizumab plus entinostat with no responses in any of the patients (161).
In contrast, encouraging activity of combining the anti-CD47 antibody magrolimab with azacitidine and the BCL2-inhibitor venetoclax was reported from a phase I/II trial in the adverse TP53 mutated AML subtype. CRc rates were 63%, with an average one-year overall survival of 53% (162). Two randomized phase III trials are currently ongoing (NCT05079230, NCT04778397, Table 1).
HMAs and HDAC inhibitors were also reported to increase the expression of AML-associated antigens such as CD33 (163) and may therefore be a suitable combination partner for BiTEs, DARTs, and CAR-T, and -NK-cell treatment. Experimental in vitro and in vivo studies indicated improved T-cell activity for combined HMA or HDAC inhibitors with CD33-, CD123-, and CD70-directed CAR-T cells or bispecific antibodies (126, 164–166).
Multiple lines of evidence support the view that epigenetic silencing of NKG2D-ligands (NKG2DL) contributes to impaired NK-cell function, which was reversed with HMA treatment in studies on cultured NK cells (167–169). In preclinical AML models, decitabine enhanced the activity of BI836858, an anti-CD33 antibody that also engages NK cells via CD16 (170). In contrast, combining the NK-cell engaging and CD123 targeting monoclonal talacotuzumab with decitabine could not improve responses over decitabine alone in a phase II/III trial (171). Based on these data, combinations of HMAs with bispecific antibodies or CAR-T/CAR-NK cell treatment may also constitute an attractive combination. A comprehensive assessment of the biological effects of HMAs on cellular treatments is required before these combination treatments can be introduced into clinical testing.
Combining the more selective second-generation targeted epigenetic drugs with cancer immunotherapy appears attractive as it may be associated with fewer unintended effects and more efficacy. However, it also requires detailed studies before those concepts enter clinical trials. In particular, more data are needed on how these individual compounds may modulate effector and regulatory immune cell function in the context of substance-specific effects in leukemia cells. Most selective epigenetic compounds, for example, IDH or menin inhibitors, alter specific gene expression and induce differentiation (54, 55, 84, 93, 94), (Figure 1). These effects may represent a synergistic opportunity for combinatorial approaches as they commonly lead to the induction of surface antigen expression that may be utilized for immunotherapy, as reported with other targeted agents (172). Several other compound-specific effects may confer synergy with immunotherapeutic approaches: BET inhibitors, for instance, have been reported to impair PD-1 expression and T-cell exhaustion in vitro (173). Accordingly, improved T-cell expansion and anti-tumor efficacy have been observed in an adoptive T-cell transfer model upon JQ1 treatment (174). In a landmark study, it was observed that LSD1 inhibition stimulated T-cell-mediated anti-tumor responses by inducing endogenous ERV expression in cancer cells that resulted in type 1 interferon activation (175). Confirmative studies are needed before these approaches can be translated into clinical applications.
As outlined above, immunotherapy has dramatically improved treatment outcomes in patients with many cancers while these approaches have been far less successful in AML.
While the detailed mechanisms behind the relative resistance against immunotherapy remain obscure, the low immunogenicity of myeloid blasts for immune checkpoint blockade (31, 33, 34) and the difficulties in defining AML-specific antigens not expressed on HSCs for immune-directed treatment (32, 119, 120) remains an unsolved challenge. Epigenetic manipulation was shown to improve the responses to immunotherapy by inducing neoantigens, increasing antigen presentation, and co-regulating immune checkpoints (35, 36, 96, 141–144, 146–148). Clinical trials evaluating the combination of non-selective epigenetic drugs (such as HMAs) with checkpoint inhibitors have mainly reported modest activity in the R/R AML setting (12, 153, 176), while approaches combining the anti-CD47 antibody magrolimab with azacitidine with or without venetoclax resulted in very promising response rates in clinical trials (162, 177). Clinical data for the combination of HMAs with cellular immunotherapy is pending, while CAR-NK cell concepts seem auspicious due to their only temporary toxicity for the normal hematopoiesis (129). Promising strategies include the introduction of (second-generation) targeted epigenetic drugs into immunotherapeutic treatment regimens. These drugs commonly have less adverse effects and their common ability to release the differentiation block in AML blasts accompanied by antigen-induction may enhance cellular immunotherapy. Studies that define specific effects of these drugs on various immune cells are underway to enable translation of these concepts into clinical investigation.
JR: Writing – original draft, Writing – review & editing. EU: Writing – review & editing. MK: Writing – review & editing, Writing – original draft.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to MWMK and EU (SFB1292/2/ TP12) and MWMK (KU-2688/2-2).
MWMK receives honoraria and is a consultant for Pfizer, Kura Oncology, Jazz Pharmaceuticals, Bristol-Myers Squibb/Celgene Abbvie, and Servier; is on the speakers bureau of Gilead and receives travel support from Daiichi Sankyo. JR has received travel support from Abbvie. EU has a sponsored research project with Gilead and BMS.
EU declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood (2022) 140(11):1200–28. doi: 10.1182/blood.2022015850
2. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood (2022) 140(12):1345–77. doi: 10.1182/blood.2022016867
3. Röllig C, Kramer M, Schliemann C, Mikesch JH, Steffen B, Krämer A, et al. Does time from diagnosis to treatment affect the prognosis of patients with newly diagnosed acute myeloid leukemia? Blood (2020) 136(7):823–30. doi: 10.1182/blood.2019004583
4. Sasaki K, Ravandi F, Kadia TM, DiNardo CD, Short NJ, Borthakur G, et al. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer (2021) 127(12):2049–61. doi: 10.1002/cncr.33458
5. Rausch C, Rothenberg-Thurley M, Dufour A, Schneider S, Gittinger H, Sauerland C, et al. Validation and refinement of the 2022 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia (2023) 37(6):1234–44. doi: 10.1038/s41375-023-01884-2
6. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New Engl J Med (2020) 383(7):617–29. doi: 10.1056/NEJMoa2012971
7. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood (2015) 126(3):291–9. doi: 10.1182/blood-2015-01-621664
8. Issa GC, Aldoss I, DiPersio J, Cuglievan B, Stone R, Arellano M, et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature (2023) 615(7954):920–4. doi: 10.1038/s41586-023-05812-3
9. Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med (2022) 386(16):1519–31. doi: 10.1056/NEJMoa2117344
10. Dinardo CD, Schuh AC, Stein EM, Montesinos P, Wei A, De Botton S, et al. Effect of enasidenib (ENA) plus azacitidine (AZA) on complete remission and overall response versus AZA monotherapy in mutant-IDH2 (mIDH2) newly diagnosed acute myeloid leukemia (ND-AML). JCO (2020) 38(15_suppl):7501–1. doi: 10.1200/JCO.2020.38.15_suppl.7501
11. Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell–engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol (2023) 41(12):2238–47. doi: 10.1200/JCO.22.01725
12. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med (2016) 375(2):143–53. doi: 10.1056/NEJMoa1601202
13. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372(4):320–30. doi: 10.1056/NEJMoa1412082
14. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
15. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. New Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
16. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. JCO (2014) 53:0105. doi: 10.1200/JCO.2013.53.0105
17. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
18. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Five-year outcomes with pembrolizumab versus chemotherapy for metastatic non–small-cell lung cancer with PD-L1 tumor proportion score ≥ 50%. JCO (2021) 39(21):2339–49. doi: 10.1200/JCO.21.00174
19. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
20. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
21. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
22. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
23. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
24. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet (2021) 398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8
25. Kamdar M, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet (2022) 399(10343):2294–308. doi: 10.1016/S0140-6736(22)00662-6
26. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
27. Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol (2023) 41(6):1265–74. doi: 10.1200/JCO.22.00842
28. Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med (2017) 376(9):836–47. doi: 10.1056/NEJMoa1609783
29. Budde LE, Sehn LH, Matasar M, Schuster SJ, Assouline S, Giri P, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol (2022) 23(8):1055–65. doi: 10.1016/S1470-2045(22)00335-7
30. Dickinson MJ, Carlo-Stella C, Morschhauser F, Bachy E, Corradini P, Iacoboni G, et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med (2022) 387(24):2220–31. doi: 10.1056/NEJMoa2206913
31. Daver N, Alotaibi AS, Bücklein V, Subklewe M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia (2021) 35(7):1843–63. doi: 10.1038/s41375-021-01253-x
32. Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia (2019) 33(1):64–74. doi: 10.1038/s41375-018-0180-3
33. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
34. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
35. Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jäger E, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leukemia Res (2010) 34(7):899–905. doi: 10.1016/j.leukres.2010.02.004
36. Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2’-deoxycytidine. Cancer Res (1994) 54(7):1766–71.
37. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
38. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502(7471):333–9. doi: 10.1038/nature12634
39. Dawson MA, Kouzarides T, Huntly BJP. Targeting epigenetic readers in cancer. N Engl J Med (2012) 367(7):647–57. doi: 10.1056/NEJMra1112635
40. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol (2016) 8(9):a019505. doi: 10.1101/cshperspect.a019505
41. Sasca D, Guezguez B, Kühn MWM. Next generation epigenetic modulators to target myeloid neoplasms. Curr Opin Hematol (2021) 28(5):356–63. doi: 10.1097/MOH.0000000000000673
42. Stomper J, Rotondo JC, Greve G, Lübbert M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: mechanisms of resistance and novel HMA-based therapies. Leukemia (2021) 35(7):1873–89. doi: 10.1038/s41375-021-01218-0
43. San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers (Basel) (2019) 11(11):1794. doi: 10.3390/cancers11111794
44. Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM, et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leuk Lymph. (2014) 55(10):2301–4. doi: 10.3109/10428194.2013.877134
45. Schlenk RF, Krauter J, Raffoux E, Kreuzer KA, Schaich M, Noens L, et al. Panobinostat monotherapy and combination therapy in patients with acute myeloid leukemia: results from two clinical trials. Haematologica (2018) 103(1):e25–8. doi: 10.3324/haematol.2017.172411
46. Abaza YM, Kadia TM, Jabbour EJ, Konopleva MY, Borthakur G, Ferrajoli A, et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic Malignancies. Cancer (2017) 123(24):4851–9. doi: 10.1002/cncr.30949
47. Stahl M, Zeidan AM. Hypomethylating agents in combination with histone deacetylase inhibitors in higher risk myelodysplastic syndromes: Is there a light at the end of the tunnel? Cancer (2017) 123(6):911–4. doi: 10.1002/cncr.30532
48. Issa JP, Garcia-Manero G, Huang X, Cortes J, Ravandi F, Jabbour E, et al. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer (2015) 121(4):556–61. doi: 10.1002/cncr.29085
49. Sekeres MA, Othus M, List AF, Odenike O, Stone RM, Gore SD, et al. Randomized phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American intergroup study SWOG S1117. J Clin Oncol (2017) 35(24):2745–53. doi: 10.1200/JCO.2015.66.2510
50. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell (2005) 19(4):523–34. doi: 10.1016/j.molcel.2005.06.027
51. Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol Cell (2015) 58(6):1028–39. doi: 10.1016/j.molcel.2015.04.011
52. Fiskus W, Cai T, DiNardo CD, Kornblau SM, Borthakur G, Kadia TM, et al. Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells. Blood Cancer J (2019) 9(2):1–13. doi: 10.1038/s41408-018-0165-5
53. Borthakur G, Odenike O, Aldoss I, Rizzieri DA, Prebet T, Chen C, et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer (2021) 127(16):2943–53. doi: 10.1002/cncr.33590
54. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell (2011) 20(1):66–78. doi: 10.1016/j.ccr.2011.06.010
55. Kühn MWM, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discovery (2016) 6(10):1166–81. doi: 10.1158/2159-8290.CD-16-0237
56. Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood (2018) 131(24):2661–9. doi: 10.1182/blood-2017-12-818948
57. Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol (2019) 26(11):999–1012. doi: 10.1038/s41594-019-0313-z
58. Kim H, Ronai ZA. PRMT5 function and targeting in cancer. Cell Stress (2020) 4(8):199–215. doi: 10.15698/cst2020.08.228
59. Kaushik S, Liu F, Veazey KJ, Gao G, Das P, Neves LF, et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia (2018) 32(2):499–509. doi: 10.1038/leu.2017.206
60. Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia (2016) 30(4):789–99. doi: 10.1038/leu.2015.308
61. Feustel K, Falchook GS. Protein arginine methyltransferase 5 (PRMT5) inhibitors in oncology clinical trials: A review. J Immunother Precis Oncol (2022) 5(3):58–67. doi: 10.36401/JIPO-22-1
62. Patel MR, Monga V, Jauhari S, Stevens D, Masarova L, McKean M, et al. A phase 1 dose escalation study of protein arginine methyltransferase 5 (PRMT5) inhibitor PRT543 in patients with myeloid malignancies. Blood (2021) 138(Supplement 1):2609. doi: 10.1182/blood-2021-150938
63. El-Khoueiry AB, Clarke J, Neff T, Crossman T, Ratia N, Rathi C, et al. Phase 1 study of GSK3368715, a type I PRMT inhibitor, in patients with advanced solid tumors. Br J Cancer. (2023) 129(2):309–17. doi: 10.1038/s41416-023-02276-0
64. Monga V, Johanns TM, Stupp R, Chandra S, Falchook GS, Giglio P, et al. A phase 1 study of the protein arginine methyltransferase 5 (PRMT5) brain-penetrant inhibitor PRT811 in patients (pts) with recurrent high-grade glioma or uveal melanoma (UM). JCO (2023) 41(16_suppl):3008–8. doi: 10.1200/JCO.2023.41.16_suppl.3008
65. Watts JM, Bradley TJ, Thomassen A, Brunner AM, Minden MD, Papadantonakis N, et al. A phase I/II study to investigate the safety and clinical activity of the protein arginine methyltransferase 5 inhibitor GSK3326595 in subjects with myelodysplastic syndrome and acute myeloid leukemia. Blood (2019) 134(Supplement_1):2656. doi: 10.1182/blood-2019-130337
66. Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in myeloid Malignancies. Cells (2020) 9(7):1639. doi: 10.3390/cells9071639
67. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature (2011) 469(7330):343–9. doi: 10.1038/nature09784
68. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi: 10.1038/ng.518
69. Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med (2016) 22(2):128–34. doi: 10.1038/nm.4036
70. Gounder M, Schöffski P, Jones RL, Agulnik M, Cote GM, Villalobos VM, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol (2020) 21(11):1423–32. doi: 10.1016/S1470-2045(20)30451-4
71. Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol (2018) 19(5):649–59. doi: 10.1016/S1470-2045(18)30145-1
72. Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol (2020) 21(11):1433–42. doi: 10.1016/S1470-2045(20)30441-1
73. Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med (2019) 216(4):966–81. doi: 10.1084/jem.20181276
74. Göllner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med (2017) 23(1):69–78. doi: 10.1038/nm.4247
75. Greif PA, Hartmann L, Vosberg S, Stief SM, Mattes R, Hellmann I, et al. Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: an exome sequencing study of 50 patients. Clin Cancer Res (2018) 24(7):1716–26. doi: 10.1158/1078-0432.CCR-17-2344
76. Kempf JM, Weser S, Bartoschek MD, Metzeler KH, Vick B, Herold T, et al. Loss-of-function mutations in the histone methyltransferase EZH2 promote chemotherapy resistance in AML. Sci Rep (2021) 11(1):5838. doi: 10.1038/s41598-021-84708-6
77. Porazzi P, Petruk S, Pagliaroli L, De Dominici M, Deming D, Puccetti MV, et al. Targeting chemotherapy to decondensed H3K27me3-marked chromatin of AML cells enhances leukemia suppression. Cancer Res (2022) 82(3):458–71. doi: 10.1158/0008-5472.CAN-21-1297
78. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood (2015) 125(2):346–57. doi: 10.1182/blood-2014-06-581082
79. Cusan M, Cai SF, Mohammad HP, Krivtsov A, Chramiec A, Loizou E, et al. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPα-dependent enhancers in AML. Blood (2018) 131(15):1730–42. doi: 10.1182/blood-2017-09-807024
80. Salamero O, Somervaille TCP, Molero A, Acuña-Cruz E, Perez-Simon JA, Coll R, et al. Iadademstat combination with azacitidine is a safe and effective treatment in first line acute myeloid leukemia. Final results of the alice trial. Blood (2022) 140(Supplement 1):1711–3. doi: 10.1182/blood-2022-168945
81. Maes T, Mascaró C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell (2018) 33(3):495–511.e12. doi: 10.1016/j.ccell.2018.02.002
82. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
83. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
84. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science (2013) 340(6132):622–6. doi: 10.1126/science.1234769
85. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
86. De Botton S, Montesinos P, Vives Polo S, Zarzycka E, Wang J, Riva M, et al. Updated efficacy and safety data from the AGILE study in patients with newly diagnosed acute myeloid leukemia treated with ivosidenib + azacitidine compared to placebo + azacitidine. JCO (2023) 41(16_suppl):7012–2. doi: 10.1200/JCO.2023.41.16_suppl.7012
87. Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell (2015) 27(4):589–602. doi: 10.1016/j.ccell.2015.02.016
88. Klossowski S, Miao H, Kempinska K, Wu T, Purohit T, Kim E, et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest (2020) 130(2):981–97. doi: 10.1172/JCI129126
89. Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell (2019) 36(6):660–673.e11. doi: 10.1016/j.ccell.2019.11.001
90. Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science (2020) 367(6477):586–90. doi: 10.1126/science.aax5863
91. Fathi A, Wang E, Issa G, Altman J, Montesinos P, Botton SD, et al. P504 updated data for ziftomenib in patients with npm1-mutated relapsed or refractory acute myeloid leukemia. Hemasphere (2023) 7(Suppl). doi: 10.1097/01.HS9.0000968924.19161.da
92. Fiskus W, Boettcher S, Daver N, Mill CP, Sasaki K, Birdwell CE, et al. Effective Menin inhibitor-based combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J (2022) 12(1):1–11. doi: 10.1038/s41408-021-00603-3
93. Rausch J, Dzama MM, Dolgikh N, Stiller HL, Bohl SR, Lahrmann C, et al. Menin inhibitor ziftomenib (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax. Haematologica (2023) 27. doi: 10.3324/haematol.2022.282160
94. Dzama MM, Steiner M, Rausch J, Sasca D, Schönfeld J, Kunz K, et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood (2020) 136(21):2442–56. doi: 10.1182/blood.2020005037
95. Daver N, Basu S, Garcia-Manero G, Cortes JE, Ravandi F, Ning J, et al. Defining the immune checkpoint landscape in patients (pts) with acute myeloid leukemia (AML). Blood (2016) 128(22):2900. doi: 10.1182/blood.V128.22.2900.2900
96. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med (2019) 25(4):603–11. doi: 10.1038/s41591-019-0400-z
97. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117(17):4501–10. doi: 10.1182/blood-2010-10-310425
98. Wu Z, Ou J, Liu N, Wang Z, Chen J, Cai Z, et al. Upregulation of Tim-3 is associated with poor prognosis in acute myeloid leukemia. Cancer Med (2023) 12(7):8956–69. doi: 10.1002/cam4.5549
99. Wang M, Bu J, Zhou M, Sido J, Lin Y, Liu G, et al. CD8+T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin Immunol (2018) 190:64–73. doi: 10.1016/j.clim.2017.08.021
100. Radwan SM, Elleboudy NS, Nabih NA, Kamal AM. The immune checkpoints Cytotoxic T lymphocyte antigen-4 and Lymphocyte activation gene-3 expression is up-regulated in acute myeloid leukemia. HLA (2020) 96(1):3–12. doi: 10.1111/tan.13872
101. Chen C, Liang C, Wang S, Chio CL, Zhang Y, Zeng C, et al. Expression patterns of immune checkpoints in acute myeloid leukemia. J Hematol Oncol (2020) 13(1):28. doi: 10.1186/s13045-020-00853-x
102. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer (2019) 125(9):1470–81. doi: 10.1002/cncr.31896
103. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic Malignancies. Clin Cancer Res (2008) 14(10):3044–51. doi: 10.1158/1078-0432.CCR-07-4079
104. Gerds AT, Scott BL, Greenberg PL, Khaled SK, Lin TL, Pollyea DA, et al. PD-L1 blockade with atezolizumab in higher-risk myelodysplastic syndrome: an initial safety and efficacy analysis. Blood (2018) 132(Supplement 1):466. doi: 10.1182/blood-2018-99-118577
105. Garcia-Manero G, Tallman MS, Martinelli G, Ribrag V, Yang H, Balakumaran A, et al. Pembrolizumab, a PD-1 inhibitor, in patients with myelodysplastic syndrome (MDS) after failure of hypomethylating agent treatment. Blood (2016) 128(22):345. doi: 10.1182/blood.V128.22.345.345
106. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138(2):271–85. doi: 10.1016/j.cell.2009.05.046
107. Wang C, Sun C, Li M, Xia B, Wang Y, Zhang L, et al. Novel fully human anti-CD47 antibodies stimulate phagocytosis and promote elimination of AML cells. J Cell Physiol (2021) 236(6):4470–81. doi: 10.1002/jcp.30163
108. Vyas P, Knapper S, Kelly R, Salim R, Lubowiecki M, Royston D, et al. Initial Phase 1 results of the first-in-class anti-CD47 antibody HU5F9- G4 in relapsed/refractory acute myeloid leukemia patients. EHA Library. (2018) 214718:PF232.
109. Ravandi F, Walter RB, Subklewe M, Buecklein V, Jongen-Lavrencic M, Paschka P, et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). JCO (2020) 38(15_suppl):7508–8. doi: 10.1200/JCO.2020.38.15_suppl.7508
110. Subklewe M, Stein A, Walter RB, Bhatia R, Wei AH, Ritchie D, et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a novel half-life extended (HLE) anti-CD33/CD3 biTE® (Bispecific T-cell engager) in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood (2019) 134:833. doi: 10.1182/blood-2019-127977
111. Westervelt P, Roboz GJ, Cortes JE, Kantarjian HM, Lee S, Rettig MP, et al. Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2x2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood (2018) 132(Supplement 1):1455. doi: 10.1182/blood-2018-99-111529
112. Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood (2021) 137(6):751–62. doi: 10.1182/blood.2020007732
113. Ravandi F, Bashey A, Foran JM, Stock W, Mawad R, Blum W, et al. Complete responses in relapsed/refractory acute myeloid leukemia (AML) patients on a weekly dosing schedule of xmAb14045, a CD123 x CD3 T cell-engaging bispecific antibody: initial results of a phase 1 study. Blood (2018) 132(Supplement 1):763. doi: 10.1182/blood-2018-99-119786
114. Laborda E, Mazagova M, Shao S, Wang X, Quirino H, Woods AK, et al. Development of A chimeric antigen receptor targeting C-type lectin-like molecule-1 for human acute myeloid leukemia. Int J Mol Sci (2017) 18(11):2259. doi: 10.3390/ijms18112259
115. Steger B, Floro L, Amberger DC, Kroell T, Tischer J, Kolb HJ, et al. WT1, PRAME, and PR3 mRNA expression in acute myeloid leukemia (AML). J Immunother. (2020) 43(6):204–15. doi: 10.1097/CJI.0000000000000322
116. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and persistence of WT1-specific T-cell receptor gene–transduced lymphocytes in patients with AML and MDS. Blood (2017) 130(18):1985–94. doi: 10.1182/blood-2017-06-791202
117. Rosenfeld C, Cheever MA, Gaiger A. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia (2003) 17(7):1301–12. doi: 10.1038/sj.leu.2402988
118. Mascarenhas J, Cortes J, Huls G, Venditti A, Breems D, Botton SD, et al. Update from the ongoing Phase I multinational study of MCLA-117, a bispecific CLEC12AxCD3 T-cell engager in patients with acute myelogenous leukemia. Vol. 294456. EHA Library Mascarenhas J (2020).
119. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia (2015) 29(8):1637–47. doi: 10.1038/leu.2015.52
120. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood (2014) 123(15):2343–54. doi: 10.1182/blood-2013-09-529537
121. Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discovery (2021) 2(6):648–65. doi: 10.1158/2643-3230.BCD-20-0208
122. Perriello VM, Rotiroti MC, Pisani I, Galimberti S, Alberti G, Pianigiani G, et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv (2023) 7(12):2855–71. doi: 10.1182/bloodadvances.2022008762
123. Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood (2018) 132(Supplement 1):901. doi: 10.1182/blood-2018-99-110579
124. Naik S, Madden RM, Lipsitt A, Lockey T, Bran J, Rubnitz JE, et al. Safety and anti-leukemic activity of CD123-CAR T cells in pediatric patients with AML: preliminary results from a phase 1 trial. Blood (2022) 140(Supplement 1):4584–5. doi: 10.1182/blood-2022-170201
125. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia (2021) 35(11):3282–6. doi: 10.1038/s41375-021-01232-2
126. Cheng J, Ge T, Zhu X, Wang J, Zeng Y, Mu W, et al. Preclinical development and evaluation of nanobody-based CD70-specific CAR T cells for the treatment of acute myeloid leukemia. Cancer Immunol Immunother. (2023) 72(7):2331–46. doi: 10.1007/s00262-023-03422-6
127. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood (2021) 138(4):318–30. doi: 10.1182/blood.2020008221
128. Ljunggren HG, Kärre K. In search of the “missing self”: MHC molecules and NK cell recognition. Immunol Today (1990) 11(7):237–44. doi: 10.1016/0167-5699(90)90097-S
129. Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. (2022) 22(10):557–75. doi: 10.1038/s41568-022-00491-0
130. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
131. Albinger N, Pfeifer R, Nitsche M, Mertlitz S, Campe J, Stein K, et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J (2022) 12(4):61. doi: 10.1055/s-0042-1748710
132. Schmid C, Labopin M, Schaap N, Veelken H, Schleuning M, Stadler M, et al. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation in acute leukaemia - a matched pair analysis by the Acute Leukaemia Working Party of EBMT. Br J Haematol (2019) 184(5):782–7. doi: 10.1111/bjh.15691
133. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood (1990) 75(3):555–62. doi: 10.1182/blood.V75.3.555.555
134. Mathé G, Amiel JL, Schwarzenberg L, Cattan A, Schneider M. Adoptive immunotherapy of acute leukemia: experimental and clinical results. Cancer Res (1965) 25(9):1525–31.
135. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MTL, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. New Engl J Med (2009) 361(5):478–88. doi: 10.1056/NEJMoa0811036
136. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. New Engl J Med (2018) 379(24):2330–41. doi: 10.1056/NEJMoa1808777
137. Jan M, Leventhal MJ, Morgan EA, Wengrod JC, Nag A, Drinan SD, et al. Recurrent genetic HLA loss in AML relapsed after matched unrelated allogeneic hematopoietic cell transplantation. Blood Adv (2019) 3(14):2199–204. doi: 10.1182/bloodadvances.2019000445
138. Steinmann J, Bertz H, Wäsch R, Marks R, Zeiser R, Bogatyreva L, et al. 5-Azacytidine and DLI can induce long-term remissions in AML patients relapsed after allograft. Bone Marrow Transplant. (2015) 50(5):690–5. doi: 10.1038/bmt.2015.10
139. Schroeder C, Gani C, Lamprecht U, von Weyhern CH, Weinmann M, Bamberg M, et al. Pathological complete response and sphincter-sparing surgery after neoadjuvant radiochemotherapy with regional hyperthermia for locally advanced rectal cancer compared with radiochemotherapy alone. Int J Hyperthermia. (2012) 28(8):707–14. doi: 10.3109/02656736.2012.722263
140. Craddock C, Labopin M, Robin M, Finke J, Chevallier P, Yakoub-Agha I, et al. Clinical activity of azacitidine in patients who relapse after allogeneic stem cell transplantation for acute myeloid leukemia. Haematologica (2016) 101(7):879–83. doi: 10.3324/haematol.2015.140996
141. Kübler A, Woiterski J, Witte KE, Bühring HJ, Hartwig UF, Ebinger M, et al. Both mature KIR+ and immature KIR– NK cells control pediatric acute B-cell precursor leukemia in NOD.Cg-Prkdcscid IL2rgtmWjl/Sz mice. Blood (2014) 124(26):3914–23. doi: 10.1182/blood-2014-05-572743
142. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell (2015) 162(5):974–86. doi: 10.1016/j.cell.2015.07.011
143. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell (2015) 162(5):961–73. doi: 10.1016/j.cell.2015.07.056
144. Sohlberg E, Pfefferle A, Andersson S, Baumann BC, Hellström-Lindberg E, Malmberg KJ. Imprint of 5-azacytidine on the natural killer cell repertoire during systemic treatment for high-risk myelodysplastic syndrome. Oncotarget (2015) 6(33):34178–90. doi: 10.18632/oncotarget.6213
145. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
146. Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, et al. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica (2013) 98(8):1196–205. doi: 10.3324/haematol.2012.074823
147. Ørskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget (2015) 6(11):9612–26. doi: 10.18632/oncotarget.3324
148. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28(6):1280–8. doi: 10.1038/leu.2013.355
149. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discovery. (2019) 9(3):370–83. doi: 10.1158/2159-8290.CD-18-0774
150. Gojo I, Stuart RK, Webster J, Blackford A, Varela JC, Morrow J, et al. Multi-center phase 2 study of pembroluzimab (Pembro) and azacitidine (AZA) in patients with relapsed/refractory acute myeloid leukemia (AML) and in newly diagnosed (≥65 years) AML patients. Blood (2019) 134(Supplement_1):832. doi: 10.1182/blood-2019-127345
151. Brunner AM, Esteve J, Porkka K, Knapper S, Traer E, Scholl S, et al. Efficacy and safety of sabatolimab (MBG453) in combination with hypomethylating agents (HMAs) in patients (Pts) with very high/high-risk myelodysplastic syndrome (vHR/HR-MDS) and acute myeloid leukemia (AML): final analysis from a phase ib study. Blood (2021) 138(Supplement 1):244. doi: 10.1182/blood-2021-146039
152. Zeidan AM, Cavenagh J, Voso MT, Taussig D, Tormo M, Boss I, et al. Efficacy and safety of azacitidine (AZA) in combination with the anti-PD-L1 durvalumab (durva) for the front-line treatment of older patients (pts) with acute myeloid leukemia (AML) who are unfit for intensive chemotherapy (IC) and pts with higher-risk myelodysplastic syndromes (HR-MDS): results from a large, international, randomized phase 2 study. Blood (2019) 134(Supplement_1):829. doi: 10.1182/blood-2019-122896
153. Gómez-Llobell M, Raíndo AP, Medina JC, Centurión IG, Orgueira AM. Immune checkpoint inhibitors in acute myeloid leukemia: A meta-analysis. Front Oncol (2022) 12:882531. doi: 10.3389/fonc.2022.882531
154. Gros FX, Cazaubiel T, Forcade E, Lechevalier N, Leguay T, Servant V, et al. Severe acute GvHD following administration of ipilimumab for early relapse of AML after haploidentical stem cell transplantation. Bone Marrow Transplant. (2017) 52(7):1047–8. doi: 10.1038/bmt.2017.78
155. Saxena K, Herbrich SM, Pemmaraju N, Kadia TM, DiNardo CD, Borthakur G, et al. A phase 1b/2 study of azacitidine with PD-L1 antibody avelumab in relapsed/refractory acute myeloid leukemia. Cancer (2021) 127(20):3761–71. doi: 10.1002/cncr.33690
156. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res (2015) 3(12):1375–85. doi: 10.1158/2326-6066.CIR-15-0077-T
157. Wischnewski F, Pantel K, Schwarzenbach H. Promoter demethylation and histone acetylation mediate gene expression of MAGE-A1, -A2, -A3 , and -A12 in human cancer cells. Mol Cancer Res (2006) 4(5):339–49. doi: 10.1158/1541-7786.MCR-05-0229
158. Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol (2000) 165(12):7017–24. doi: 10.4049/jimmunol.165.12.7017
159. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res (2016) 22(16):4119–32. doi: 10.1158/1078-0432.CCR-15-2584
160. Borcoman E, Kamal M, Marret G, Dupain C, Castel-Ajgal Z, Le Tourneau C. HDAC inhibition to prime immune checkpoint inhibitors. Cancers (Basel). (2021) 14(1):66. doi: 10.3390/cancers14010066
161. Bewersdorf JP, Shallis RM, Sharon E, Caldwell A, Wei W, Yacoub A, et al. A multi-center phase ib trial of the histone deactylase inhibitor (HDACi) entinostat in combination with anti-PD1 antibody pembrolizumab in patients with refractory/relapsed myelodysplastic syndromes (RR-MDS) or oligoblastic acute myeloid leukemia (RR-AML) after hypomethylating agent (HMA) failure. Blood (2022) 140(Supplement 1):9084–6. doi: 10.1182/blood-2022-158626
162. Daver N, Senapati J, Maiti A, Loghavi S, Kadia TM, DiNardo CD, et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in patients (pts) with newly diagnosed (ND) older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood (2022) 140(Supplement 1):141–4. doi: 10.1182/blood-2022-170188
163. Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood (2014) 123(4):554–61. doi: 10.1182/blood-2013-09-527044
164. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun (2021) 12(1):6436. doi: 10.1038/s41467-021-26683-0
165. Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med (2020) 26(9):1459–67. doi: 10.1038/s41591-020-0910-8
166. Tang L, Kong Y, Wang H, Zou P, Sun T, Liu Y, et al. Demethylating therapy increases cytotoxicity of CD44v6 CAR-T cells against acute myeloid leukemia. Front Immunol (2023) 14:1145441. doi: 10.3389/fimmu.2023.1145441
167. Baragaño Raneros A, Martín-Palanco V, Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C, et al. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun (2015) 16(1):71–82. doi: 10.1038/gene.2014.58
168. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature (2019) 572(7768):254–9. doi: 10.1038/s41586-019-1410-1
169. Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk Res (2007) 31(10):1393–402. doi: 10.1016/j.leukres.2007.02.020
170. Vasu S, He S, Cheney C, Gopalakrishnan B, Mani R, Lozanski G, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858–mediated natural killer ADCC against AML blasts. Blood (2016) 127(23):2879–89. doi: 10.1182/blood-2015-11-680546
171. Montesinos P, Roboz GJ, Bulabois CE, Subklewe M, Platzbecker U, Ofran Y, et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: results from a multicenter, randomized, phase 2/3 study. Leukemia (2021) 35(1):62–74. doi: 10.1038/s41375-020-0773-5
172. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-cells targeting FLT3 have potent activity against FLT3-ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia (2018) 32(5):1168–79. doi: 10.1038/s41375-018-0009-0
173. Zhong M, Gao R, Zhao R, Huang Y, Chen C, Li K, et al. BET bromodomain inhibition rescues PD-1-mediated T-cell exhaustion in acute myeloid leukemia. Cell Death Dis (2022) 13(8):671. doi: 10.1038/s41419-022-05123-x
174. Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. (2016) 126(9):3479–94. doi: 10.1172/JCI86437
175. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell (2018) 174(3):549–563.e19. doi: 10.1016/j.cell.2018.05.052
176. Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia (2018) 32(5):1094–105. doi: 10.1038/s41375-018-0070-8
177. Daver N, Vyas P, Chao M, Xing G, Renard C, Ramsingh G, et al. A phase 3, randomized, open-label study evaluating the safety and efficacy of magrolimab in combination with azacitidine in previously untreated patients with TP53-mutant acute myeloid leukemia. Blood (2021) 138:3426. doi: 10.1182/blood-2021-145208
Keywords: acute myeloid leukemia, hypomethylating agents, immunotherapy, epigenetics, checkpoint inhibition, cellular therapy, chromatin modifiers, combination therapy
Citation: Rausch J, Ullrich E and Kühn MWM (2023) Epigenetic targeting to enhance acute myeloid leukemia-directed immunotherapy. Front. Immunol. 14:1269012. doi: 10.3389/fimmu.2023.1269012
Received: 28 July 2023; Accepted: 05 September 2023;
Published: 22 September 2023.
Edited by:
Matthieu Perreau, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Vincenzo Maria Perriello, University of Perugia, ItalyCopyright © 2023 Rausch, Ullrich and Kühn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael W.M. Kühn, bWlja3VlaG5AdW5pLW1haW56LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.