
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 December 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1268854
This article is part of the Research TopicCaveolins in Inflammation, Infections, and DiseaseView all 8 articles
 Daniella Cesar-Silva1
Daniella Cesar-Silva1 Filipe S. Pereira-Dutra1Ana Lucia Moraes Giannini2
Filipe S. Pereira-Dutra1Ana Lucia Moraes Giannini2 Clarissa M. Maya-Monteiro1,3
Clarissa M. Maya-Monteiro1,3 Cecília Jacques G. de Almeida1*
Cecília Jacques G. de Almeida1*Lipids perform a series of cellular functions, establishing cell and organelles’ boundaries, organizing signaling platforms, and creating compartments where specific reactions occur. Moreover, lipids store energy and act as secondary messengers whose distribution is tightly regulated. Disruption of lipid metabolism is associated with many diseases, including those caused by viruses. In this scenario, lipids can favor virus replication and are not solely used as pathogens’ energy source. In contrast, cells can counteract viruses using lipids as weapons. In this review, we discuss the available data on how coronaviruses profit from cellular lipid compartments and why targeting lipid metabolism may be a powerful strategy to fight these cellular parasites. We also provide a formidable collection of data on the pharmacological approaches targeting lipid metabolism to impair and treat coronavirus infection.
Lipids serve multiple functions in the cell. They reside mostly in the cell membranes, where they can be distributed distinctly within membrane leaflets and in cholesterol rich nanodomains according to their biophysical and biochemical properties. These lipid nanodomains, (historically called “lipid rafts”), concentrate coronavirus receptors and their disruption affects the expression and proper localization of coronavirus receptors, impacting virus entry (1–7). Virus structural proteins are also found in these lipid domains due to their affinity to cholesterol and this localization seems to be important for the induction of cell-cell fusion observed during coronavirus infections (6, 8, 9).
At the plasma membrane we can also find caveolae, invaginations with lipid nanodomain-like organization. Although caveolae are not directly relevant for coronaviruses entry, one of its main components, caveolin-1, colocalizes with some coronavirus receptors, seems to be involved in virus infectivity and may participate in virus fusion to the plasma membrane in a cholesterol dependent manner (2, 10).
Besides its role in the membrane, cholesterol participates in coronavirus cycle and host response against virus infection in diverse ways. Key components of the cholesterol metabolism are altered and associated with virus infectivity, inflammatory response, and severity of COVID-19 and MERS (8, 11–13). Cholesterol esterification and triacylglyceride synthesis leads to the biogenesis of lipid droplets, organelles where coronavirus replicate and that contribute to the production of inflammatory mediators (14–18). Cholesterol oxidized products also modulate viral infection and host responses (19–31). Fatty acid metabolism is involved in the acylation of the spike protein, contributing to its localization in lipid nanodomains and trafficking, virus entry and assembly (32–36). Other relevant aspects of lipid metabolism for coronaviruses include the role of specific sphingolipids in virus infection and disease severity (37–45). In this review, we discuss some of these roles and their impact on infectivity and host responses. Besides, we present a large collection of results obtained using in vitro and in vivo coronavirus infection models that support the therapeutical benefits of using lipid metabolism drugs as strategies against coronaviruses.
We begin with a brief background on lipid domains and cholesterol metabolism to provide the basis for understanding their involvement in coronavirus infection.
Lipid nanodomains are nanoscale cell membrane domains rich in cholesterol, glycosylated, and saturated lipids, such as sphingolipids, assembled in a liquid-ordered (Lo) compact arrangement (46–48). These domains also segregate glycosylphosphatidyl (GPI)-anchored and lipidated proteins, forming signal transduction platforms (47). The existence of lipid nanodomains has been a matter of intense debate in the past, as their visualization can be challenging due to their transient character in quiescent cells. However, upon stimulation, receptors in these domains cluster, inducing lipid nanodomains coalescence, signaling protein recruitment, and interaction with the cortical actin filaments (49, 50). Solubilization of cell membranes with non-ionic detergents at cold temperatures (usually, Triton X-100 at 4°C) allows isolation of domains, segregating GPI-anchored proteins and Lo domain probes. Thus, this method is often used to separate membrane domains representing lipid nanodomains, although there is controversy about this correspondence. More precisely, this approach helps the isolation of Lo domains and the associated molecules, drawing the inference that these molecules would be associated with lipid nanodomains in the living cell (51). Proteins in these domains display specific characteristics that favor their compartmentalization. Moreover, proteins can be anchored in membranes by different lipid modifications by co- or post-translational modifications, some reversible while others are permanent. Such lipid anchors include glycophosphatidylinositol (GPI) anchors, N-terminal myristic acid tails, cysteine acylation, isoprenylation, and the addition of C-terminal sterol moieties. S-acylation is the reversible and enzyme-mediated attachment of a fatty acid to a cysteine residue via a thioester linkage (called palmitoylation because palmitic acid is frequently the added acyl chain). This process targets proteins to the inner leaflet of the cell membrane, preferentially to lipid nanodomains. Cholesterol-depleting and -sequestering drugs, such as MβCD and nystatin disrupt these lipid nanodomains hampering intracellular signaling triggered by molecules organized in these domains.
Caveolae are specialized lipid nanodomains that form invaginations of 50-200 nm in the plasma membrane. These structures likely serve as safeguards when sudden changes in membrane tension occur, for example, during hypotonic shock or mechanical stretch. In these situations, caveolae flatten, providing membranes required for cellular reshaping (52, 53). Caveolae are also crucial for transcytosis (54, 55) and cell signaling. Significantly, they are also involved in controlling lipid metabolism, the main subject of this review (56). Caveolae bear specific proteins, which are determinants for sculpting these structures and executing caveolar functions. These proteins belong to two prominent families: caveolins and cavins. The caveolin family comprises three members of integral membrane proteins named caveolin-1, caveolin-2, and caveolin-3. Caveolin-1 and -2 are ubiquitous but are enriched in endothelial cells, adipocytes, pneumocytes, and fibroblasts, whereas caveolin-3 is specific to muscle cells (57–61). Caveolin-1 and caveolin-3 share a high degree of identity. They are essential for caveolae formation, whereas caveolin-2, the most divergent member of the family, is not required but favors the formation of deeper caveolae (62, 63). The interaction of signaling molecules with caveolins contributes to their compartmentalization and functions. Although the absence of caveolins is not lethal (64–66), their deficiency is associated with various diseases (67–71). The cavin family comprises four cytosolic proteins: cavin1, cavin2, cavin3, and cavin4; the latter being specific to muscle cells. Cavin1 hetero-trimerizes with the other cavins and is essential for caveolae formation (72–74). The other cavins are not essential, except for cavin 2 in endothelial cells of specific tissues (75), but are important to shape and stabilize caveolae (76–79). Lipids play essential roles in caveolae dynamics and functions. Caveolae are enriched in cholesterol, sphingomyelin, and glycosphingolipids, like GD3 (80). Cholesterol binding to caveolin-1 promotes oligomerization (81) and trafficking from the Golgi to the plasma membrane (82). Cholesterol has a vital role in the caveolae structure, which are also disrupted by treatment with cholesterol-depleting drugs (83–85). Furthermore, not only do lipids affect caveolin expression and caveolae formation and motility, but caveolin expression also regulates lipid homeostasis. In the absence of caveolin-1, alterations in cholesterol metabolism occur, such as reduction of free cholesterol synthesis, enhancement of cholesterol esterification (86), and reduction of cholesterol in lipid droplets (87). Caveolin-1 can also be found in lipid droplets where it is believed to play a role in the stabilization, as well as in the lipolysis of these organelles (82, 88–90). Caveolin itself and caveolae as a whole, traffic to lipid droplets, increasing their size and lipid uptake (91). Taken together, all these data corroborate the relevance of caveolae to lipid metabolism.
The lipid droplet (LD) is an organelle where several hydrophobic reactions involved in lipid, energy, and redox homeostasis take place (92–94). Lipid droplets contain a hydrophobic core composed of neutral lipids, mainly triacylglycerol (TG) and cholesterol ester (CE) and are delimited by a monolayer of phospholipids (95–97) and associated proteins (98–100) Although LDs are ubiquitous, their protein and lipid composition strongly depend on the cell type and cellular metabolic state (101–103). Their numbers and sizes are also variable due to the balance between lipid synthesis and degradation (97, 104, 105). The biogenesis of LDs occurs in the ER (106), where critical enzymes involved in lipid synthesis are located, mainly diacylglycerol O-acyltransferases (DGAT1 and DGAT2) (107, 108) and acyl-CoA: cholesterol O-acyltransferases (ACAT1/SOAT1 and ACAT2/SOAT2) (109–111), responsible for the synthesis of TG and CE, respectively (94, 112). The lipid metabolism pathways leading to lipid uptake, cholesterol efflux, autophagy, β-oxidation, lipophagy, and lipid remodeling (109, 113–118) impact LD accumulation or consumption (119–122). Besides, LDs play a role in lipid availability and membrane biosynthesis (123, 124).
Cellular cholesterol content is regulated by uptake, de novo synthesis, storage, and export. Diet-derived cholesterol is absorbed by the enterocytes, transported to intracellular organelles, and released in the bloodstream (125), where it circulates associated with lipoproteins, like chylomicrons, very low-density proteins (VLDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). LDL is recognized by LDL receptors on cell surface and is endocytosed by clathrin-coated vesicles (126). LDL-bound cholesterol ester (CE) is then hydrolysed by lysosomal acid lipase (LAL) in the lysosomes, from where vesicular and non-vesicular mechanisms transport cholesterol to other organelles (127). The plasma membrane holds 60-90% of this lipid (128). Studies using radiolabeled cholesterol binding probes indicated the existence of three distinct cholesterol pools: one that binds the probe, named the accessible pool, which is sensitive to cholesterol deprivation; the SM-sequestered pool that is not affected by cholesterol deprivation and binds the probe only after SMase treatment; and the essential pool which does not bind to the probe, even after SMase treatment and whose depletion causes rounding and dissociation of the cell from the culture dish. Interestingly, the excess of the accessible pool traffic to the ER, the organelle responsible for cholesterol synthesis, starting with acetyl-CoA and involving more than 20 enzymes (128).
Despite being the center of cholesterol synthesis, the ER disposes of only 1% of the total cellular cholesterol due to the fast transport of newly synthesized cholesterol to cellular membranes or lipid droplets (129). The master regulator of cholesterol synthesis is sterol regulatory element-binding protein 2 (SREBP2), a transcriptional factor synthesized as a precursor protein. In the ER, SREBP2 forms a heterodimer with SCAP (SREBP-cleavage activating protein), a sensor of cholesterol level (130). When ER’s cholesterol level is below 5% of total ER lipids, SCAP binds COPII in transport vesicles and escorts SREBP2 to the Golgi, where it is processed and activated by site 1 (S1P) and site 2 (S2P) proteases (131). The activated soluble N-terminus SREBP2 goes to the nucleus and binds to sterol regulatory elements (SRE) found in genes coding for proteins involved in cholesterol synthesis, like 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and squalene monooxygenase (SQLE). When cholesterol levels increase in the ER, cholesterol binds SCAP, blocking its interaction with COPII, thus inhibiting SREBP2 transport to the Golgi. Besides, cholesterol binding to SCAP induces its interaction with INSIG1 and INSIG2 (insulin-induced gene-1 and -2 proteins), which also interferes with SCAP’s association with COPII (132). Furthermore, the cell manages the cholesterol excess by storing or exporting it. Acyl-coenzyme A: cholesterol acyltransferase (ACAT) esterifies cholesterol that can be stored in LDs. CE from lipid droplets is released by the CEHs associated with the lipid droplet surface and is transported to other organelles or the extracellular milieu (133). ABC-binding cassette (ABC) transporters mediate the efflux of non-esterified apolipoprotein-bound cholesterol originated from diverse organelles (Figure 1) (134).
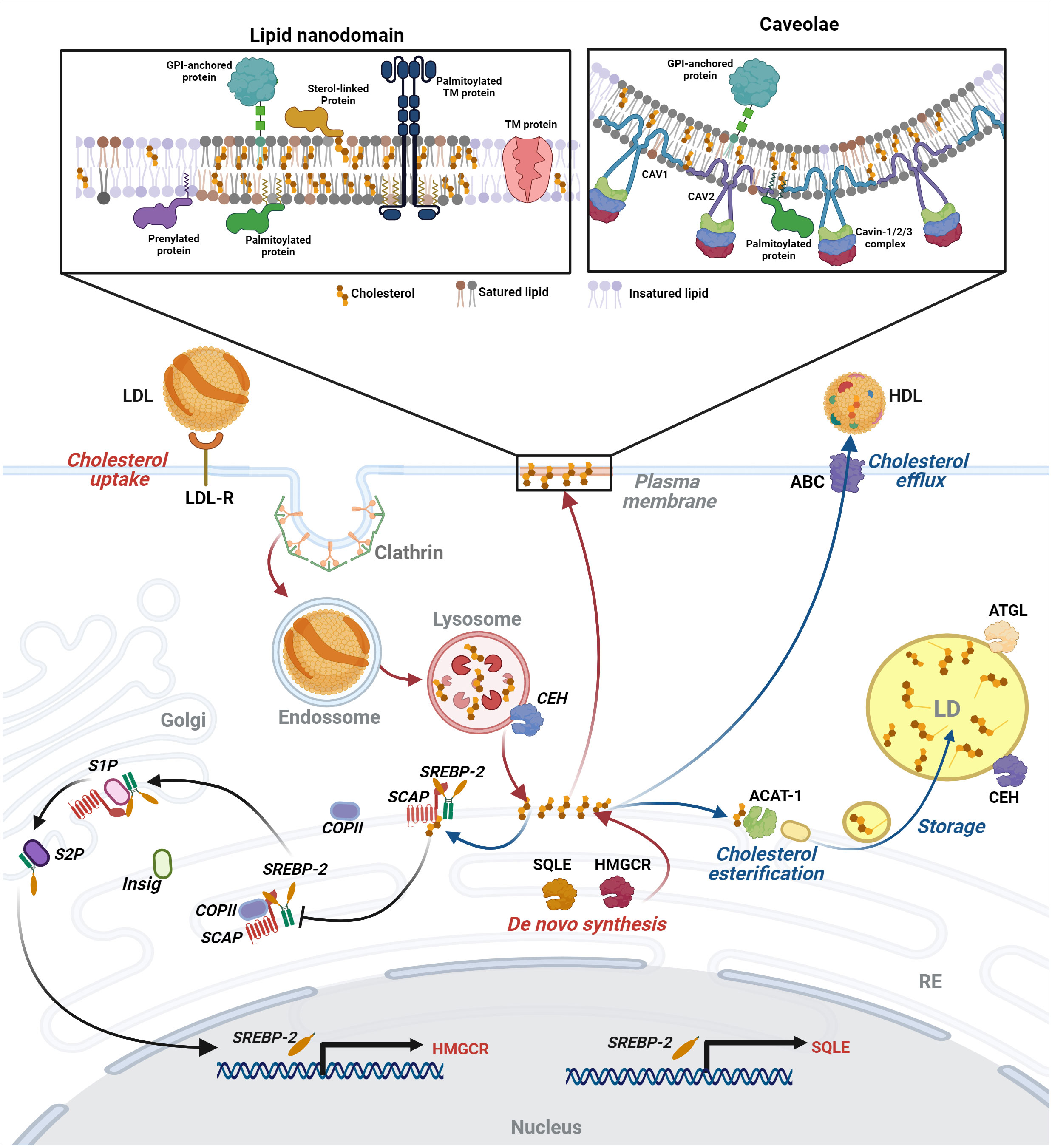
Figure 1 Intracellular lipid compartments and control of cellular cholesterol levels - The cholesterol level in the cell is regulated by uptake, de novo synthesis, storage, and export. LDL-bound cholesterol enters the cell by receptor-mediated endocytosis in clathrin-coated vesicles reaching the lysosomes, from which cholesterol is released and transported by vesicular and non-vesicular mechanisms to cellular organelles and the plasma membrane (PM), where it is more enriched in lipid nanodomains and caveolae. For details of these structures, see the text in “Plasma membrane and cholesterol homeostasis as therapeutic targets in antiviral approaches” section. Excess of the accessible cholesterol pool in PM is transported to the ER, where cholesterol synthesis also occurs. There, SREBP2 forms a heterodimer with SCAP, cholesterol level sensor. Low levels of cholesterol result in cleavage and activation of a soluble N-terminus SREBP2 that goes to the nucleus and activates the transcription of genes involved in cholesterol synthesis, such as HMGCR and SQLE. When cholesterol rises in the ER, it binds SCAP, inhibiting the transport of SREBP2 to the Golgi and its processing, halting its activity as a transcription factor. The excess cholesterol can be stored in lipid droplets or exported through ABC-binding cassette (ABC) transporters. Image created using BioRender.
In the next sections, we will systematize information about how distinct aspects of lipid metabolism favor coronavirus infection or the host response against it. Moreover, we will present results obtained with a wide variety of drugs that aim the lipid metabolism as a strategy to fight coronavirus infection and the harmful consequences of the triggered host inflammatory response. Finally, we will focus on drugs that were tested in in vivo animal models and clinical studies.
Coronaviruses bind to specific plasma membrane proteins, for instance, the angiotensin-converting enzyme-2 (ACE2) in the case of SARS-CoV and SARS-CoV-2 and dipeptidyl peptidase 4 (DPP4) in the case of MERS-CoV. Besides these receptors, other molecules facilitate the attachment of these viruses (Table 1). After binding to cell receptors, coronaviruses can fuse directly to the plasma membrane or enter by endocytosis mediated by different mechanisms, depending on the virus and the cell type, and that involve lipid nanodomains, clathrin-coated vesicles, caveolae, or clathrin- and caveolae-independent pathways. Direct fusion with the plasma membrane relies on the proteolytic processing of Spike by proteases, like TMPRSS2 in the case of SARS-CoV-2. Otherwise, Spike can be processed by cathepsins activated by the lower pH found in endosomes (146). Thus, the cellular repertoire of receptors, proteases, and other cellular factors affect the virus entry route. For instance, in Calu-3 cells that express TMPRSS2, SARS-CoV-2 enters by fusion, whereas in Vero E6 cells, it enters by an endocytic pathway. The inhibition of infection by drugs that raise the endosomal pH, such as chloroquine, hydroxychloroquine, NH4Cl, and bafilomycin, indicate the requirement of endocytosis for successful infection (147–151).
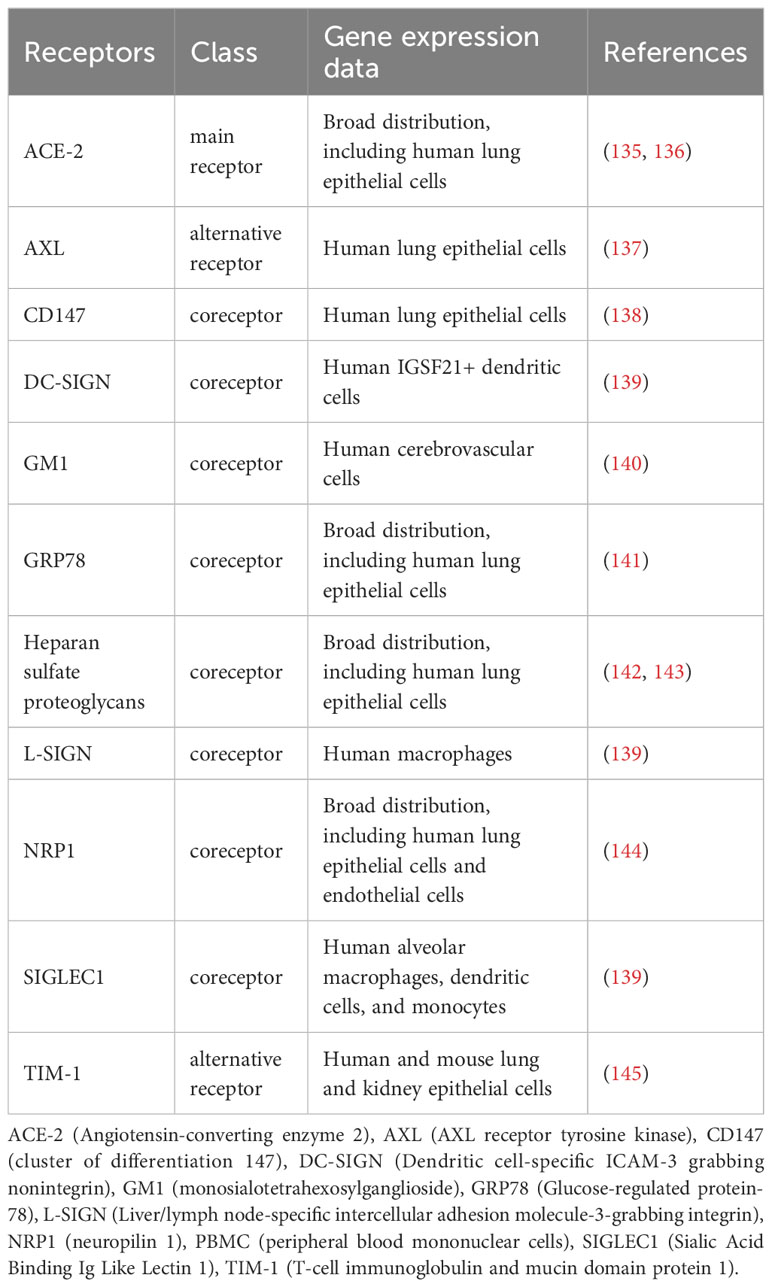
Table 1 Receptors of SARS-COV-2.
In several studies, lipid nanodomains were implicated in coronavirus entry. Disruption of lipid nanodomains by cholesterol-depleting drugs, like MβCD, reduces coronavirus infectivity, whereas cholesterol replenishment restores virus infectivity (1–6). Nevertheless, the timing is crucial for this inhibition as MβCD pretreatment impairs SARS-CoV infection in Vero E6 cells, whereas a 3h post-treatment has no effect, indicating that cholesterol depletion hampers early steps of the virus cycle (3). Similarly, pretreatment, but not post-treatment with simvastatin, a cholesterol-reducing drug, inhibits SARS-CoV-2 replication in Vero E6 and Calu-3 cells. Accordingly, simvastatin impairs binding and entry of SARS-CoV-2 in Calu-3, possibly due to the dislodgment of ACE2 from lipid nanodomains caused by the drug and despite the increase in ACE2 expression that both simvastatin and infection provoke in these cells (7). MβCD treatment also induces relocation of ACE2 from nanodomains to membrane non-raft regions without impacting ACE2 expression levels in Vero E6 or Caco-2 cells (4, 5). Furthermore, a tagged-ectodomain of the SARS-CoV Spike also colocalizes with detergent-resistant membrane domains and with GM1 in fixed cells, which indicates the presence of ACE2 in lipid nanodomains upon Spike binding (4). In contrast to the view that the presence of ACE2 in lipid nanodomains favors SARS-CoV-2 infection, Wing and coworkers (2023) showed that avasimibe - an ACAT inhibitor - increases both free cholesterol in the plasma membrane and the size of GM1-enriched lipid nanodomains, as well as the ACE2 localization in these domains, while reducing SARS-CoV-2 attachment to Vero E6 cells (152) Structural proteins of the avian infectious bronchitis virus (IBV) also localize to detergent-resistant fractions and move to soluble fractions after MβCD treatment. MβCD, as well as mevastatin, another member of the statins’ drug class, also reduces the expression of these proteins (6). In contrast, Li and coworkers (2007) could not detect ACE2 in lipid nanodomains using Vero E6 cells and reported decreased ACE2 expression after MβCD treatment (3). This cholesterol-depleting drug also inhibits mouse hepatitis virus (MHV) replication. Still, the mechanisms cannot be attributed to the relocation of its receptor since CEACAM is detected in detergent-resistant membrane domains when a mild concentration of cold Triton X-100 (0.2%) is used to isolate this receptor in CEACAM-expressing HeLa cells membranes. However, an exogenous GPI-anchored CEACAM expression in detergent-resistant domains (isolated with 1% Triton X-100) fails to enhance MHV infection (1). Nevertheless, Choi and coworkers demonstrated that in the presence of MHV, CEACAM, and Spike redistribute into lipid nanodomains in CEACAM-overexpressing HEK293 cells. These data suggest that cholesterol depletion by MβCD does not affect virus binding but reduces virus entry drastically, possibly because viral proteins redistribute and interact with lipid nanodomains. Spike is detected only in detergent-soluble membranes of the Golgi, meaning that Spike is not associated with lipid nanodomains during virus assembly and budding but is associated with detergent-resistant membrane domains at the plasma membrane, which is important for Spike-induced cell-cell fusion (153). Accordingly, Spike of coronaviruses contains several cholesterol-binding motifs in their sequences (8, 9).
Cholesterol also participates in distinct entry pathways, affecting caveolae- and clathrin-mediated endocytic mechanisms (149, 154). Therefore, excluding alternative entry routes using specific tools is greatly valuable. Wang and coworkers report lipid raft-mediated entry of SARS-CoV in a mechanism independent of caveolae and clathrin in Vero E6 and HEK293-ACE2-GFP cell lines. Clathrin-mediated endocytosis was excluded based on experiments using chlorpromazine (an inhibitor of clathrin-mediated endocytosis), by silencing the clathrin coding gene, or by expressing a dominant-negative Eps15, which participates in clathrin-mediated endocytosis. Caveolae-dependent endocytosis was also excluded by experiments using filipin or nystatin, which inhibited endocytosis of cholera toxin B, used as a control but failed to inhibit SARS-CoV endocytosis. Moreover, caveolin-1 does not colocalize with the SARS-CoV Spike (155).
On the other hand, other studies found that SARS-CoV and SARS-CoV-2 invade HepG2 cells (a hepatocarcinoma cell line) and HEK-293-ACE2 cells, respectively, by clathrin-mediated endocytosis (156, 157). MHV-2 also uses clathrin-mediated endocytosis to enter the mouse astrocytoma DBT cells, which was demonstrated by using chlorpromazine, hypertonic sucrose medium, or by silencing the clathrin heavy-chain gene. Notwithstanding, the clathrin-mediated endocytosis of MHV-2 is Eps15-independent. Besides, the overexpression of a dominant-negative mutant of caveolin-1 did not affect the invasion of MHV-2 in DBT cells, ruling out the participation of this protein or caveolae in virus entry (158). Porcine hemagglutinating encephalomyelitis virus (PHEV) also uses clathrin-mediated endocytosis to invade mouse neuroblastoma cells (Neuro-2a). Entry by endocytosis is evidenced by virus presence in coated pits and EEA1-decorated vesicles and by the inhibition of invasion induced by pH-raising agents, such as chloroquine and NH4Cl. Clathrin’s involvement in PHEV entrance is further demonstrated by PHEV and transferrin coincidental labeling, chlorpromazine sensitivity, and low infectivity detected in a dominant-negative clathrin mutant-expressing or a clathrin heavy chain-silenced cell line. Additionally, silencing of caveolin-1 does not affect PHEV entry in Neuro-2a cells (150).
Other studies propose that caveolin-1, and not necessarily caveolae, are involved in coronavirus infection. The human coronavirus 229E binds to CD13, enriched in membrane detergent-resistant domains where it colocalizes with caveolin-1. MβCD does not hamper virus binding to the cell surface but reduces the colocalization of 229E with caveolin and virus infectivity, while cholesterol replenishment reverses these effects. The silencing of caveolin-1 does not impair 229E binding but reduces its entrance into the cell significantly. 229E particles are detected close to caveolae, but interestingly they are not found in the lumen of caveolae or endosomes. Thus, caveolin-1 may participate in the fusion of the virus particle with the plasma membrane but not in caveolae-mediated endocytosis of the 229E virus (2). Notably, epithelial cells, the primary targets for coronaviruses, do not present caveolae in their apical surfaces, although they express caveolin-1 on these sites (159, 160). The human coronavirus OC43, which uses HLA class I molecule or sialic acid as receptors, colocalizes to caveolin-1 in HCT-8 cells during the early phase of infection. Nystatin, MβCD, or CAV1 silencing inhibits virus invasion and infectivity. The requirement of dynamin was also proved using dynasore and MiTMAB (10). Zhou and colleagues (2022) further showed that SARS-CoV-2 pseudovirus uses CD147 to enter Vero E6 and Huh-7 cells and exploit an Arf-6-dependent caveolar/lipid raft pathway (161).
Enveloped viruses may also present cholesterol in their membranes (162, 163), and cholesterol depletion from the viral envelope of the transmissible gastroenteritis virus (TGEV) by MβCD reduces its infectivity drastically, whereas cholesterol replenishment reverts this effect (164). Curiously, the Spike protein, found in detergent-resistant domains of the TGEV envelope membrane, does not become detergent-soluble after MβCD treatment and does not segregate with the lipid raft marker flotillin-2, indicating that the viral membrane organization is different from that of the plasma membrane (165).
Structural and modeling studies identified that the N-terminal domain of the Spike protein of SARS-CoV-2 interacts with ganglioside sialic acids (166) (see Table 1). SARS-CoV-2 and other betacoronaviruses use these molecules to attach and invade host cells (140, 167). Interestingly, gangliosides are a group of sphingolipids enriched in lipid nanodomains.
In addition to functioning as alternative receptors, sphingolipids metabolism is involved in other aspects of SARS-CoV-2 infection. The central hub of sphingolipid metabolism is ceramide, which can be synthesized by sphingomyelin hydrolysis, de novo synthesis, or recycling from complex sphingolipids (Figure 2). COVID-19 patients’ sera display increased levels of dihydrosphingosine, dihydroceramides, ceramides, sphingosine, and acid sphingomyelinase (ASM) correlating positively with disease severity. In contrast, the level of sphingosine-1-phosphate (S1P) correlates negatively with the disease severity (37). Accordingly, S1P can activate the S1P receptor and induce anti-inflammatory responses. Another study shows that patients with symptomatic COVID-19 have diminished serum sphingosine levels compared to asymptomatic counterparts, who also exhibit elevated acid ceramidase (AC), an enzyme that converts ceramide in sphingosine (38). Accordingly, in vitro treatment of Vero or human primary epithelial cells with exogenous sphingosine protected these cells from infection by pseudoviruses expressing the SARS-CoV-2 Spike protein, possibly due to the binding of the exogenous sphingosine to ACE2, hampering Spike’s interaction with its receptor (39). Notably, in this pseudovirus model, the interaction of ACE2 and Spike activates ASM. This enzyme metabolizes sphingosine into ceramide in acidic endolysosomes and at the plasma membrane at the physiological pH after being released by endolysosomes (168). In the plasma membrane, ASM is important for lipid raft formation in the outer leaflet. In contrast, neutral SM generates ceramide in the inner leaflet, where it interacts with intracellular signaling molecules (169). Increased ceramide in the outer leaflet of the plasma membrane favors SARS-CoV-2 infection. Accordingly, inhibition of ASM by various antidepressant drugs (either selective serotonin reuptake inhibitors (SSRI) or tricyclic class) or silencing of the ASM gene suppresses SARS-CoV-2 infection in Caco-2 cells. Importantly, nasal epithelial cells isolated from amitriptyline-treated (tricyclic class) volunteers were protected from SARS-CoV-2 infection (40). Fluoxetine, an SSRI, also suppresses the replication of different SARS-CoV-2 variants of concern in Vero and A549-hACE2-TMPRSS2 cells, human lung slices (170), and human airway organoid epithelia (41). Geiger and coworkers (2022) showed that fluoxetine and AKS466 - its ASM-independent derivative - target acid ceramidase, leading to the retention of SARS-CoV-2 in lysosomes and inhibiting virus replication in Huh.7, Vero and Calu-3 cells. Ceranib-2, a specific inhibitor of AC, also blocks SARS-CoV-2 replication (42) Further, fluoxetine treatment reduces IL-6 and NF-κB signaling in various cell lines (171). Gene silencing of glucosylceramide synthase (converts ceramide to glucosylceramide), and inhibition by Genz-123346 or Genz-667161, block SARS-CoV-2 early replication steps (43). The same group showed that SARS-CoV-2 infection raises the levels of glycosphingolipids, like dihydrosphingosine, sphingosine, GA1, and GM3, an effect reversed by Genz-123346. This inhibitor does not significantly alter the levels of ceramide, lactosyl ceramide, SM, and GM2 (44). 4-HPR, an inhibitor of dihydroceramide D4-desaturase 1 (DES1- catalyses dihydroceramide in ceramide), inhibits cell-cell fusion and SARS-CoV-2 infection. However, the addition of ceramide fails to revert the effects of 4-HPR. DES1 knockout cells exhibit a similar cell-cell fusion efficiency, despite the enhanced amounts of dihydrosphingosine-derived lipids compared to total sphingolipids. Therefore, the inhibitory effect of 4-HPR on SARS-CoV-2 infection does not rely on DES1, and it is also not related to ACE2 expression or localization. Still, it can be related to the drug effects on decreased membrane fluidity (45).

Figure 2 Coronaviruses infection and sphingolipid metabolism -The central hub of sphingolipid metabolism is ceramide, which can be synthesized by hydrolysis of sphingomyelin, by de novo synthesis, or by recycling from complex sphingolipids. Ceramide has a dual role during SARS-CoV-2 infection, promoting viral entry when it accumulates in the plasma membrane or blocking viral egress of viral particles that accumulate in the endolysosomes. The interaction of ACE2 and Spike activates acid sphingomyelinase (ASM) that metabolizes sphingosine into ceramide, which favors SARS-CoV-2 infection. Inhibition of ASM by various drugs suppresses SARS-CoV-2 infection. Inhibition of acid ceramidase (AC) that metabolizes ceramide into sphingosine also inhibits SARS-CoV-2 infection, as it reduces ceramide levels and increases sphingosine levels that hamper the interaction of Spike with ACE2. Furthermore, sphingosine is metabolized into sphingosine-1-phosphate, which activates an anti-inflammatory response. Drugs inhibiting the activity of enzymes of the sphingolipid metabolism interfering with coronavirus infection are depicted in this figure. AC, acid ceramidase; ASM, acid sphingomyelinase; CS, ceramide synthase; DES1, dihydroceramide D4-desaturase 1; DH-CS, dihydroceramide synthase; GCS, Glucosylceramide Synthase; NSM, neutral sphingomyelinase; SK, sphingosine kinases. Image created using BioRender.
In conclusion, the metabolism of sphingolipids can determine the outcome of SARS-CoV-2 infection. Ceramide has a dual role in SARS-CoV-2 infection, promoting viral entry when it accumulates in the plasma membrane (40) and blocking viral egress of the viral particles that accumulate in the endolysosomes (42). The balance of ceramide and sphingosine seems crucial during the entry stage since engagement of Spike and ACE2 induces the conversion of sphingomyelin to ceramide favoring infection, whereas the conversion of ceramide to sphingosine blocks ACE2 and Spike’s interaction (39, 40). Patients’ shingolipidome shows that the severity of COVID-19 correlates to ceramide accumulation and S1P reduction (37, 38). S1P results from sphingosine phosphorylation and has anti-inflammatory effects, modulating endothelial cell chemotaxis and barrier integrity. While ceramide and sphingosine promote apoptosis, cell cycle arrest, and differentiation, S1P promotes proliferation and survival (172). Thus, S1P reduction may contribute to the exacerbated inflammation observed in severe COVID-19. For this reason, the S1P modulators, fingolimod, and siponimod, used to treat multiple sclerosis, have been proposed as therapy for treating COVID-19 patients (Figure 2) (173, 174).
Beyond the essential role of cholesterol in structuring lipid nanodomains and involvement in coronavirus entry, its metabolism affects coronavirus infection and host defense mechanisms. Viral infection activates antiviral responses in the host cell, including the transcription of interferon-induced genes (ISGs), such as the cholesterol 25-hydroxylase (CH25H) gene, whose product modifies cholesterol into 25-hydrocholesterol (25HC). 25HC has several antiviral and anti-inflammatory activities in infection by diverse enveloped viruses, including coronaviruses. SARS-CoV-2 infection stimulates the expression of CH25H in Calu-3 and A549-ACE2 cells. The overexpression of CH25H or the treatment with exogenous 25HC abolishes infection by SARS-CoV, MERS-CoV, or SARS-CoV-2 pseudoviruses in Calu-3 cells. Mechanistically, 25HC induces cholesterol trafficking from the plasma membrane to the endoplasmic reticulum, where the ACAT esterifies it, resulting in the formation of lipid droplets and cholesterol depletion in the plasma membrane, restricting the entry of the virus by fusion with the cell surface (19). Heissler and collaborators (2023) report that 25-HC inhibits the common cold coronavirus hCoV-OC43 infection of CHO-K1 cells. They showed that 25HC suppresses SREBP-2 activation, inhibiting cholesterol biosynthesis and activation of ACAT. The latter results in cholesterol sinking into lipid droplets and depletion of the accessible cholesterol from the plasma membrane, which was confirmed by using anthrolysin O (ALOD 4) that binds specifically to this cholesterol pool. Interestingly, 25HC does not alter SM-sequestered cholesterol (20), indicating that 25HC acts independently of lipid nanodomains. In rabbit blood cells that are devoid of internal membranes, the authors show that 25HC is no longer able to induce a reduction of cholesterol at the plasma membrane, suggesting that 25HC effects are dependent on cholesterol transport from the plasma membrane to other organelles (20). Another study indicates that porcine epidemic diarrhea virus (PEDV) downregulates CH25H expression in Vero cells. However, the overexpression of CH25H or 25HC treatment also blocks PEDV infection. Interestingly, CH25H-M, a CH25H mutant lacking hydrolase activity and not producing 25HC, also inhibits PEDV infection, although to a lesser extent. CH25H affects the entry of PEDV and the replication of another porcine coronavirus, the TGEV (21). The infection of IPI-FX cells with porcine deltacoronavirus (PDCoV) upregulates CH25H expression, and 25HC treatment impairs PDCoV infection, not at the adsorption nor at the replication steps (22), but possibly post-entry steps of PDCoV infection such as replication or egress, at least using LLC PK1 cells (23). PDCoV infection induces a transitory increase in IFN-γ and the production of lipid droplets, while 25HC enhances both effects. Furthermore, infection activates the TGF-β signaling pathway, which 25HC reverses. Asiaticoside, an inhibitor of TGF-β, also inhibits PDCoV infection (23). The overexpression of CH25H or 25HC treatment also impairs the infection of HEK293-hACE2 cells by SARS-CoV or SARS-CoV-2 pseudoviruses. The authors show that 25HC accumulates in late endosomes and lysosomes and impairs Spike-mediated membrane fusion (24). The antiviral activity of 25HC was also demonstrated by Yuan and collaborators using the SARS-CoV-2 N antigen expression, viral load reduction, and plaque reduction assays in Vero E6 cells (25). Another relevant aspect of 25HC action on coronavirus infection is the modulation of the inflammatory response since severe cases are characterized by an exacerbated pro-inflammatory response that potentially progresses to organ failure and death. 25HC provides a negative feedback loop of SREBP-dependent activation of pro-inflammatory cytokines (26). Plasma samples of severe COVID-19 patients compared to mild cases show decreased levels of 25HC and increased cytokine levels that correlate with the disease’s severity. Accordingly, CH25H expression is also reduced at mRNA and protein levels in peripheral blood mononuclear cells (PBMC) of severe COVID-19 patients. In this context, treatment of PBMC with 25HC inhibits SREBP activity, the expression of pro-inflammatory genes, such as TNF-α, IL-1β, NOX2, and NLPR3, and the production of inflammatory cytokines, such as IL-6, TNF-α, IL-1b, IFN-γ, IL-8, CCL2, among others (27). In contrast, Zu and collaborators (2020) described a sudden increase of 25HC in the plasma of a severe COVID-19 patient two days before her death, as well as an increase in 25HC levels in serum samples of SARS-CoV-2-infected hACE2 mice compared to the control group. Caco-2 cells also exhibit CH25H upregulation upon SARS-CoV-2 infection, and the overexpression of CH25H inhibits SARS-CoV-2, as well as SARS-CoV-2 pseudovirus particle production in Vero cells, indicating an effect at the entry step of viral infection. 25HC also protects mice from infection by SARS-CoV-2 strain MASCp6.10 (28). Despite the controversial results regarding 25HC levels in the blood upon SARS-CoV-2 infection, both studies reveal a protective role of 25HC against either viral infection or the excessive production of inflammatory mediators caused by infection. In sharp contrast with all these papers reporting a protective role of 25HC, Fessler and collaborators (2022) show that 25HC is effective in controlling the infection caused by the common cold coronavirus (hCoV)-229E, but not the infection and associated damage inflicted by the SARS-CoV-2 (29). Regarding 229E infection, 25HC reduces viral plaque number and size in MRC-5 cells, indicating a post-entry effect. On the contrary, 25HC does not affect SARS-CoV-2 infection in TMPRSS2-expressing VeroE6 cells. Likewise, despite the increase of CH25H expression in K18-hACE2 mice, 25HC treatment does not change the viral load in these mice. Corroborating to the inefficacy of 25HC, CH25H-deficient mice show a similar viral load to wild-type mice. Furthermore, 25HC does not change inflammatory lung indicators to SARS-CoV-2 infection, such as the level of inflammatory mediators and total cell counts in bronchoalveolar lavage fluid (BALF). Of note, 25HC increases inflammatory chemokines in the plasma of infected treated mice and microvascular leakage, and it does not improve lung damage or the survival rate. These observations recommend caution regarding the therapeutic potential of 25HC (29). It is worth mentioning that Marcello and coworkers failed to find any correlation between 25HC in the blood of COVID-19 patients and disease severity. Instead, they observed a 50% decrease in 27HC in COVID-19 patients. In vitro, 27OH inhibits infection of SARS-CoV-2 and HCoV-OC43 (30). The role of other oxysterols in COVID-19 and other coronavirus pathologies still deserves more studies, such as the one by Ohashi and coworkers (2021) describing anti-SARS-CoV-2 activities (31). Finally, we would like to mention that 25HC inhibits caveolin-1 expression and the subsequent Cav-1+-endossome-dependent IFN-γ signaling in the context of Parkinson’s disease (175), an aspect of the regulation of the inflammatory response not investigated so far in coronavirus infection.
Wei and collaborators (2020) correlated SARS-CoV-2 infection with lipoprotein metabolism. First, they demonstrated that SARS-CoV-2 Spike can bind cholesterol and that HDL increases the entry of a pseudovirus expressing SARS-CoV-2 Spike-in Huh-7 cells in a dose-dependent manner. Furthermore, HDL also increases the entry and replication of SARS-CoV-2, a process dependent on the scavenger receptor B type 1 (SR-B1). ACE2 and SR-B1 are co-expressed in pulmonary and extrapulmonary tissue such as the retina, colon, and liver, indicating that SR-B1 facilitates SARS-CoV-2 infection and explaining the viral tropism for these organs (8).
The total cholesterol, HDL, and LDL levels in the blood of COVID-19 patients correlate negatively with infection and disease severity (11). On the other hand, SREBP and NFκB activation are higher in fatal cases compared to survivors. Moreover, the C-terminal fragment of SREBP-2 is present in the blood of COVID-19 patients and correlates to disease severity, cytokine levels, and vascular damage, being higher in septic patients. Treatment of PBMC from COVID-19 patients with fatostatin A (an SREBP-2 processing inhibitor) and SN50 (an NF-κB signaling inhibitor) suppress the production of pro-inflammatory TNF-α and IL-1β. Accordingly, the prototypical pro-inflammatory lipopolysaccharide (LPS) stimulation of HUVEC cells provokes an increase in the C-terminal SREBP content in cell culture lysates and supernatants, whereas the N-terminal SREBP increases only in the cell lysates. These results indicate that the liberation of SREBP C-terminal fragment correlates to inflammatory stimulation and may be a useful indicator of disease severity in COVID-19 patients and a possible target to reduce the exacerbated inflammation response in severe COVID-19 (12). Besides, the same study showed that SREBP-2 silencing blocks the LPS effect. Fatostatin, SN50, and SREBP-2 silencing all diminish sepsis outcomes of the cecal ligation and puncture (CLP) model of sepsis. The authors propose that SARS-CoV-2 infection leads to C-terminal SREBP release, which induces cytokine storm production and the vascular damage observed in COVID-19 septic patients (12). Pharmacological intervention on the transactivation of lipogenic enzymes by the N-terminal SREBP also suppresses MERS-CoV infection, whose primary target is the lipid metabolism pathway, at least in the Calu-3 cells infection model. Using a lipid metabolite library, Yuan and coworkers (2019) identified that AM580, a synthetic analog of retinoic acid receptor alpha (RARα), was protective against MERS-CoV infection in the hepatic cell line Huh7 (13). RARα regulates cholesterol metabolism by stimulating the expression of Apo-A1 in hepatic cells (176), as well as of cholesterol efflux transporters (ABCA1 and ABCG1) in macrophages (177). MERS-CoV infection triggers the expression of genes coding lipogenic enzymes like acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), and hydroxymethylglutaryl-CoA synthase (HMGCS), besides enhancing the production of lipid droplets. Not surprisingly, AM580 suppresses these effects and reduces MERS-CoV replication drastically in various cell lines, such as Calu-3, A549, Vero, THP-1, human primary monocyte-derived macrophages (MDMs), human primary small airway epithelial cells (HSAEC), as well in human intestinal organoids. AM580 binds to the N-terminus forms of SREBP1 and SREBP2, preventing lipogenic genes’ transactivation. The effect of AM580 is counteracted by exogenous palmitate, indicating that MERS-CoV subverts the fatty acid metabolism to its benefit. AM580 also abolishes the inflammatory response in Huh7 cells and MDMs (13). AM580 is further effective in vivo, as described in session 5 of this review. In the following sections of this review, we will contextualize other aspects of coronaviruses’ subversion of cholesterol metabolism.
Drugs addressed to fatty acid metabolism are also effective against coronavirus infection. Orlistat, a drug that inhibits gastric lipases and FAS, and triacsin C, an inhibitor of long-chain acyl CoA synthetase (ACS), both inhibit SARS-CoV-2 infection in VeroE6 cells when added 2h or 4h post-infection, but have minimal effect if cells are treated for 1h before infection, indicating a post-entry action. Orlistat, triacsin C, and other inhibitors that impair de novo fatty acid synthesis, such as TOFA (an ACC inhibitor) and C75 (a FAS inhibitor), also block SARS-CoV-2 infection in Calu-3 cells (14).
Besides their essential role in phospholipids syntheses, fatty acids participate in protein lipidation, energy generation, and lipid droplet formation. The inhibition of β−oxidation by etomoxir (an inhibitor of carnitine palmitoyl-transferase 1A -CPT1A) and trimetazidine (an inhibitor of beta-oxidation of long-chain 3-keoacyl-CoA thiolase) do not impact SARS-CoV-2 replication. On the other hand, the inhibition of palmitoyl acyltransferase (PAT) by 2-bromopalmitate hinders SARS-CoV-2 infection. Accordingly, FASN-deficient cells do not sustain SARS-CoV-2 replication. In contrast, incubation with palmitic acid and oleic acid supports replication in these cells, further demonstrating the central role of fatty acid metabolism during SARS-CoV-2 infection (14). Chu and coworkers (2021) demonstrate the importance of palmitoylation for SARS-CoV-2 using an experimental setting of genome silencing in HEK293T-hACE2 and Caco-2 cells with a library of metabolic shRNAs, followed by infecting these cells with an infectious-clone-derived SARS-CoV-2 carrying a mNeoGreen reporter protein (SARS-CoV-2-mNG virus). This study identified host factors that favor virus replication, such as FASN (which encodes FAS) and ACACA (which encodes ACC1). FASN-KO HEK293T-hACE2 cells are also resistant to virus infection, and eleven out of 22 FAS inhibitors blocked SARS-CoV-2-mNG infection, the most effective ones being TVB-3664, orlistat, TVB-2640, TVB-3166, GSK-214069, FASN-IN-4, and FT113. The first three were effective against the early lineage of SARS-CoV-2 (USA_WA1/2020) and the alpha, beta, and delta variants. BSA-conjugated palmitic acid reverses the inhibition mediated by these inhibitors, indicating that their effect relies on hampering palmitoylation. The orlistat effect is not fully reversed, possibly because this drug also inhibits lipases (178). According to the role of fatty acid metabolism in virus infection, the FASN gene is an interferon-regulated gene suppressed by IFN type I after infection by several viruses, including SARS-CoV-2. The artificial overexpression of FASN increases SARS-CoV-2 load in HeLa-ACE2 and Huh 7.5 cells, whereas knockdown of FASN decreases SARS-CoV-2 replication in HEK293-ACE2 and Huh 7.5 cells. The FASN inhibitors C75, epigallocatechin gallate (EGCG), Cerulenin, and TVB-3166 were also effective in inhibiting SARS-CoV-2 infection in HeLa-ACE2, Huh 7.5 cells, and VeroE6 cells (179). Interestingly, Tang and coworkers (2023) identified that SARS-CoV-2 infection in organoids is regulated by the circadian-associated repressor of transcription (CIART) gene, which regulates fatty acid metabolism by activating the NR4A1 (180).
Palmitoylation is vital for coronavirus infectivity, at least in part because of the S-acylation of Spike (32, 33). This modification has several roles that different studies have demonstrated. For instance, palmitoylation protects SARS-CoV Spike from degradation and confers the ability to partition in detergent-resistant membranes (32, 34, 35). However, others reported that SARS-CoV-2 Spike palmitoylation is unnecessary for its location on lipid nanodomains (33). Palmitoylation of SARS-CoV-2 Spike is also crucial for trafficking to the Golgi and the plasma membrane (36). This section will describe what has been published regarding palmitoylation and coronavirus infection.
Spike proteins from several coronaviruses are heavily palmitoylated in cysteine residues, e.g., SARS-CoV-2 Spike is palmitoylated in 10 cytosolic residues (32, 181–183). Besides, Spike protein with cysteine-to-alanine mutations cannot be palmitoylated, and pseudoviruses bearing these mutated versions of Spike fail to enter cells due to hindered fusion of virus envelope and the plasma membrane (33, 181, 182). Interestingly, the mutation of specific cysteines impacts infectivity at different rates (184).
Palmitoylation is mediated by palmitoyl-transferases (PAT), a family of 23 proteins containing a DHHC motif. Although most PAT proteins reside in the Golgi, ZDHHC5 is enriched in the plasma membrane and somewhat in the endosomal system (185). ZDHHC5 and its accessory protein Golgin A7 (GOLGA7) interact with the SARS-CoV-2 Spike protein (33, 186). Accordingly, ZDHHC5 knockdown impaired SARS-CoV-2 Spike palmitoylation and reduced Spike-mediated cell-cell fusion and pseudovirus entry in HEK293T cells (33). In contrast, Zeng and coworkers (2021) showed that ZDHHC5 and GOLGA7 knockout do not affect SARS-CoV-2 Spike palmitoylation nor its localization but somehow impaired virus entry (187). These results could be explained by the fact that other ZDHHCs also bind SARS-CoV-2 Spike (182). Mesquita and coworkers (2021) claim that ZDHHC20 is the main acetyltransferase that lipidates the SARS-CoV-2 Spike. They knocked down each of the 23 ZDHHC acetyltransferases and reported that silencing of ZDHHC8, ZDHHC9, and ZDHHC20 reduced palmitoylation of ectopically expressed Spike. In their study, ZDHHC5 knockdown did not affect Spike palmitoylation (35). Besides acylation with palmitate, coronavirus Spike can also be modified by myristate and stearate, and inhibition of acylation by the FASN inhibitor TVB3166 reduces 229E spread (188). C75 and 2-bromopalmitate (2-BP), inhibitors of ZDHHCs, decrease the palmitoylation of MHV and SARS-CoV-2 Spike and inhibit the entry of SARS-CoV-2 Spike pseudotyped virus (32, 33). Ramadan and coworkers (2022) used bis-piperazine derivatives, which are more specific palmitoylation inhibitors, and reported inhibition of SARS-CoV-2 infectivity (189).
Besides the role in Spike palmitoylation, silencing of ZDHHCs reduced by 30% the amount of cholesterol in virions, which might be related to reduced virus entry. Furthermore, Mesquita and coworkers (2021) showed that palmitoylation of Spike is responsible for the modification of the lipid environment surrounding it, promoting the formation of detergent-resistant domains both in the virion envelope and in the ERGIC, being crucial for the fusion of SARS-CoV-2 pseudoparticles and viral-like particles with the host cell (35). This observation is interesting since the ERGIC usually has a low cholesterol content. Indeed, in uninfected cells, ERGIC53 (a marker of ERGIC) is detergent-soluble but becomes insoluble after SARS-CoV-2 infection. Moreover, the interaction of the MHV Spike with the M protein and their subsequent incorporation into virions depend on Spike palmitoylation (32). This effect is specific to each coronavirus since palmitoylation is dispensable in the case of SARS-CoV and TGEV Spike proteins (34, 183).
In conclusion, Spike acylation assures coronavirus particles’ entry, assembly, and spreading.
Lipid droplet formation has been detailed in a previous section. Here we will describe the results of several groups that link lipid droplets and coronavirus infections. As mentioned before, fatty acids are used to produce neutral triacylglycerides packaged into lipid droplets. Intriguingly, a consequence of SARS-CoV-2 infection is the accumulation of lipid bodies in monocytes isolated from COVID-19 patients and in SARS-CoV-2 in vitro infected primary monocytes from healthy donors. The infection results in the expression of genes coding proteins involved in lipid uptake and lipid metabolic pathways, like CD36, PPARγ, SREBP-1, and DGAT-1 (15). Accordingly, inhibiting DGAT - involved in lipid droplet formation - with A922500 and Xanthohumol reduces SARS-CoV-2 replication. These treatments also inhibit the production of pro-inflammatory mediators by monocytes. A922500 was also effective in restraining SARS-CoV-2 infection on VeroE6 cells, in which viral replicative RNA and virus particles localize close to lipid droplets (14, 15). Moreover, the knockdown of DGAT1/2 in a single infectious cycle decreases the extracellular, but not the intracellular SARS-CoV-2 load, while it reduces the viral nucleoprotein (NP) production (16). Xanthohumol also inhibits coronaviruses protease Mpro in enzymatic assays impairing infection of SARS-CoV-2 and PEDV in vitro (17).
Both NP and Spike associate with ADRP, a lipid droplet structural protein, in lung cells of SARS-CoV-2-infected hamsters. Moreover, silencing ADRP suppressed the SARS-CoV-2 load (16). Whereas the results of Dias and coworkers corroborate the idea that lipid droplets serve as platforms for virus replication (190–192), the study of Yuan and coworkers (2021) suggests they participate in a post-replication effect. The ectopic transfection of NP in Huh7 cells, but not of other viral proteins, activates DGAT-1/2 expression (16). This result contrasts with the study of Wang and collaborators (2023) that shows that among the SARS-CoV-2 proteins (except nsp3), only ORF3a induces lipid droplet accumulation in HeLa cells. The authors also show that ORF3a is necessary for lipid droplet accumulation and virus replication in Caco-2 cells using a trans-complementation system (18). Interestingly, ORF3a also shows other activities that favor SARS-CoV-2 infection, such as inhibition of autophagy (193–195), lysosomal egress (196) and IFN signaling (197). Infection of Vero E6 cells with SARS-CoV-2 resulted in the accumulation of lipid droplets, often connected to mitochondria. In contrast, SARS-CoV-infected Vero E6 cells do not present lipid droplets. Type II pneumocytes of COVID-19 patients also exhibit a large amount of lipid droplets and connections between these organelles and mitochondria (198).
Avasimibe, an inhibitor of ACAT, reduces the production of esterified cholesterol and, consequently, lipid droplets (110). It also augments the free cholesterol content of the plasma membrane in Vero E6 cells, in line with a redistribution of cholesterol from lipid droplets to the plasma membrane. Besides the inhibitory effect on SARS-CoV-2 attachment, avasimibe also reduces the replication of SARS-CoV-2 RNA. Interestingly, supporting evidence for the role of ACAT during SARS-CoV-2 infection comes from studies using a genetic approach of loss- and gain-of-function that indicated ACAT2 plays a preponderant part in inhibiting SARS-CoV-2 infection compared to ACAT1 (152).
Here, it is worth reminding that using exogenous 25HC inhibits coronavirus infection, despite the increase in lipid droplet formation. In this case, the effect of 25CH is attributed to the depletion of the accessible pool of cholesterol in the plasma membrane, affecting virus entry by fusion, which is a preceding event relative to the replication phase, where lipid droplets are necessary. Thus, although coronavirus infection induces CH25H, endogenous 25CH cannot hamper infection effectively as it happens with the addition of exogenous 25CH (19).
The occurrence of lipid droplets in a cell depends on their biogenesis and lipolysis. Viral infection provokes an increase in lipid droplets, followed by the disappearance of these organelles and a concurrent rise of intracellular FFA and glycerol. While the production of lipid droplets favors virus replication in the first hours of infection, lipolysis benefits virus replication and the production of pro-inflammatory cytokines at later time points. Atglistatin and CAY10499 are selective and non-selective lipase inhibitors, respectively. When given at 12 or 18 hpi of SARS-CoV-2 infected-Vero E6 cells, these drugs prevent the lipid droplets’ lipolysis and rising of FFA and glycerol levels. Notably, the reduction of FFA is associated with decreased fatty acid oxidation and Spike palmitoylation, contributing to the inhibition of virus replication and production of TNF-α, IL-6, and MCP-1. Supplementation of FFA reverts the inhibition of virus replication. Thus, lipid droplets are not just platforms for virus replication but are lipid sources for energetic demands and protein modification during viral infection. CAY10499 and Atglistatin also effectively suppress SARS-CoV-2 infection and the associated inflammatory response in vivo, as described in session 5 of this review (199). Interestingly, FFA binds Spike, rendering it unavailable for interaction with ACE2, diminishing SARS-CoV-2 infection (200). The contrasting effects of FFA may be related to the time course of infection.
Coronavirus infections induce extensive membrane remodeling in the host cell, creating double-membrane vesicles (DMVs) and convoluted membranes (CMs), which shelter replication-transcription complexes (RTCs). These complexes harbor viral proteins and host factors, maintaining a lipid microenvironment that protects newly synthesized RNA molecules from nucleases and innate immune sensors (201). The viral non-structural proteins nsp3, nsp4, and nsp6 are involved in the biogenesis of DMVs (the replication platforms of coronaviruses), which are connected and probably originate from the ER (202–207). The ectopic expression of nsp3 and nsp4 induces the formation of DMVs (203, 205, 208, 209), while the nsp6 bridges DMVs to the ER and to lipid droplets, allowing lipids, but not proteins, to flow from the ER to the DMVs (123). The nsp6 of alpha- and beta-coronaviruses also induces the formation of autophagosomes decorated with LC3 in an ATG5- and PI3K-dependent manner (210). Accordingly, treatment with K22, a drug that targets nsp6, inhibits infection of several coronaviruses (123, 211).
DMVs are like the autophagosomes, regarding their double-membrane constitution and the presence of LC3-I, and they emerge from the ER through mechanisms that involve host autophagic and the ERAD machinery (212, 213). The role of autophagy in coronavirus infection is controversial, though, and each coronavirus exploits the autophagic machinery in a particular way (214). Prentice and coworkers (2004) showed that deletion of the ATG5 gene inhibits the replication of murine hepatitis virus (MHV) strain A59 in stem cells (215). However, Zhao and collaborators (2007) reported that ATG5 is dispensable for efficiently replicating the same virus strain in murine bone marrow-derived macrophages or primary murine fibroblasts (216). Thenceforth, extensive literature shows that coronaviruses explore the autophagic process to produce the membrane system that allows its replication while hampering the completion of autophagy, avoiding its degradation in autophagosomes (193–195, 209, 217–222).
During autophagy, autophagosome membranes expand drastically, a process dependent on cellular factors, as will be briefly described. In nutrient-rich conditions, mTOR, a master suppressor of autophagy, interacts with and inhibits the ULK1 complex formed by ULK1, ATG13, FIP200, and ATG101. Stressing conditions relieve mTOR inhibition, and ULK1 translocates to the ER, recruiting the PI3K complex II (PI3KC3-C2), formed by VPS34, Beclin1-1, VPS15, and ATG14. PIP3KC3 originates from PIP3-rich domains in the ER, where the phagophore formation initiates. Assembly of PI3K3 complex depends on the recruitment of WD repeat domain phosphoinositide interacting 2b (WIPI2B) and double FYVE Containing Protein (DFCP1). WIPI2B binds and engages ATG12, ATG5, and ATG16L1 that lipidates microtubule-associated protein light chain 3 (LC3) and γ-aminobutyric acid receptor-associated proteins (GABARAPs) to membrane PE, originating LC3II, a signature of the autophagic process. LC3 II then regulates the elongation and closure of the autophagosomes carrying the material to be degraded after fusion with lysosomes (reviewed by Nakatogawa, 2020) (223).
Autophagosome formation also requires bulk lipid transfer from the ER’s cytosolic leaflet to the phagophore’s cytosolic leaflet. This process is associated with lipid movement between the cytosolic and the luminal leaflets of each organelle to rebalance the amount of lipids between leaflets. These tasks are executed by the ATG2 lipid transfer protein and the scramblases VMP1 and TMEM41B, respectively (224–228). VMP1 also regulates the contact sites of the ER with the phagophore and other organelles, such as mitochondria, endosomes, and lipid droplets (229). Similar to their role in the expansion of autophagosomes, they are also crucial for the formation of DMVs during coronavirus MHV-A59 infection or after ectopic expression of SARS-CoV-2 nsp3 and nsp4 non-structural proteins (230). VMP1 and TMEM41B were identified as important host factors for virus infection in genome-wide screens (231, 232), and genetic analysis revealed that deficiency of each of these proteins blocks DMV formation in distinct ways. In VMP1 KO cells, nsp3/4-bound DMVs are unable to close, while in TMEM41B KO cells, nsp3/4 complexes are unable to form. Interestingly, both proteins function as cholesterol, and PS scramblases (230), but the inhibition of PS synthesis partially rescues the defect of DMV production only in the absence of VMP1 (233). Deficiency in TMEM41B and VPM1 functions leads to the accumulation of accessible cholesterol in the cytosolic leaflet of the plasma membrane (227) and results in the formation of enlarged lipid droplets. In TMEM41B KO cells, this is due to a decrease in the mobilization of fatty acids from lipid droplets to mitochondria and their ß-oxidation (224). The absence of VMP1 also induces large lipid droplets because of the increased lipid flux from the ER to lipid droplets (229).
A genome-wide CRISPR screen of human coronaviruses-infected cells identified genes of the phosphatidylinositol phosphate biosynthesis and the cholesterol homeostasis host pathways, indicating the importance of lipid metabolism in coronavirus infection. Many of these genes were further validated by examining the effectiveness of virus infection in loss-of-function experiments, in which susceptible cells were manipulated to harbor mutant candidate genes. Moreover, several drugs targeting lipid metabolism exhibited inhibitory activity on viral replication: SAR405 (a selective and ATP-competitive inhibitor of class III PI3K - PIK3C3), YM201636 (a PIKfyve inhibitor), PF-429242 (a reversible, competitive aminopyrrolidineamide inhibitor of MBTPS1), 25HC (which promotes ER retention of the SCAP/SREBP complex), Fatostatin (which binds to SCAP and inhibits ER-to-Golgi translocation of SREBPs), Bardoxolone (an activator of the KEAP1-NRF2 complex) (232). Notably, apilimod (a PIKfyve inhibitor) also presents antiviral activity (234–236).
Class III PI3K (also known as VPS34), an essential player in the formation of autophagosomes and vesicular sorting and trafficking, is also central to the formation of DMVs. The VPS34 inhibitors, VPS34-IN1, PIK-III, and SAR405, all inhibit SARS-CoV-2 replication in Calu-3 cells. These drugs may act during early or later steps of the viral infection (14). Other inhibitors of fatty acid metabolism show antiviral activity in post-entry steps: orlistat (inhibits gastric lipases and FASN); triacsin C (targets long chain ACS); TOFA (targets acetyl-CoA) and C75 (inhibits FASN) (14).
Interestingly, both etomoxir (an inhibitor of carnitine palmitoyltransferase 1A -CPT1A), which blocks translocation of fatty acids into the mitochondria, and trimetazidine (inhibits long-chain 3-ketoacyl-CoA thiolase) do not impair SARS-CoV-2 infection, ruling out a role of β-oxidation in viral replication. Furthermore, the inhibitors VPS34-IN1, orlistat, TOFA, or A922500 inhibit the formation of replicative centers based on dsRNA labeling. Furthermore, TOFA and A922500 show much higher inhibition when the viral titers were analyzed, suggesting they have other additional effects besides inhibiting RNA synthesis (14).
The deletion of FASN in Caco-2 cells confirmed the central role of fatty acid metabolism in viral replication, while supplementation with palmitate and oleic acid was partially able to reverse FASN deficiency (14). Yuen and coworkers (2021) also reported an inhibitory effect of VPS34-IN1 in the replication and infectivity of SARS-CoV-2 in VeroE6 cells. VPS34-IN1 and its analog, compound 19, also impair viral infection of ex-vivo human lung tissue (221). They also analyzed the effects of other autophagy inhibitors (targets in parentheses) during viral infection of VeroE6 cells. They found that SBI-0206965 (ULK1), 3-methyladenine – 3-MA (class I PI3K), and hydroxychloroquine – HCQ (acidification of autophagolysosome) impaired viral replication. Interestingly, inhibition of ULK1, which is upstream of VPS34 in the autophagy pathway, stimulated SARS-CoV-2 infection instead of inhibiting it. Moreover, 3-MA showed a minimal inhibitory effect, while HCQ strongly inhibited SARS-CoV-2 replication. Twu and collaborators (2021) also report a central role of the class III PI3K in SARS-CoV-2 replication. The VPS34 inhibitor, VPS34-IN-2 (also known as PIK-III), inhibits nsp3/4-induced DMV formation and SARS-CoV-2 replication in A549/ACE2 and Calu-3 cells. Knockdown of VPS34 and Beclin 1 or the PI3P effector DFCP1 cells have the same effect in the A549 cell model (209). Taken together, these data reinforce the importance of lipid metabolism for SARS-CoV-2 infection and its potential as a therapeutic target.
The dependence of the coronaviruses cycle on host lipids and lipid metabolism motivated the search and assessment of potential antiviral drugs targeting these pathways. In this section, we will focus on drug testing data obtained in vivo using COVID-19 animal models and human clinical trials.
Statins are a class of drugs that target hydroxymethyl-glutaryl Coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol biosynthesis. They are widely used in cardiovascular patients, and since cardiovascular disease is a risk factor for developing severe COVID-19, several studies were conducted to evaluate the effect of statins on COVID-19 patients. Besides their cardiovascular effects, statins protect against SARS-CoV-2 infection and inflammatory response triggered by the virus, both in vitro and in vivo (7, 237, 238). Teixeira and coworkers demonstrated that simvastatin reduces viral replication and lung damage and delays the mortality of SARS-CoV-2-infected K18-hACE2 mice. To our knowledge, this is the only study showing the effectiveness of a statin in a COVID-19 animal model. In vitro, simvastatin effectively reduced the binding and internalization of SARS-CoV-2 due to the dislodgement of ACE2 from lipid nanodomains (7). Other studies using in vitro settings also indicate the role of statins in constraining SARS-CoV-2 infection (237, 238). Besides ACE2 localization in lipid nanodomains, statins may have other effects due to the broad participation of cholesterol in several steps of the viral cycle. For example, statins inhibit the production of lipid droplets (239), viral assembly (240), and protein post-translational modification and trafficking (241, 242). In addition, statins have anti-inflammatory, immunomodulatory, and antithrombotic effects, including during coronavirus infection (7, 241, 243–246). Indeed, numerous studies indicate that the use of statins is associated with reduced admission to the intensive care unit, faster recovery, and lower risk of mortality among patients on statin medication (247–252). However, some studies suggest that statins do not improve COVID-19 outcomes (253) and can even increase the risk of severe COVID-19 development (254). Meta-analysis of retrospective studies shows controversial results about the beneficial effect of statins in reducing the risk of mortality due to COVID-19 (255–260).
Intraperitoneal injection of the retinoid derivative AM580, shown to modulate cholesterol metabolism and SARS-CoV-2 infection in vitro, also rescued body weight loss and death of MERS-CoV-infected hDPP4-transgenic mice. The drug reduced virus load, alveolar damage, and lung inflammatory infiltration (13). Other drugs that affect not only the synthesis but also the traffic of cholesterol through the endosomal pathway, such as itraconazole, fluoxetine, and U18666A, were also tested as adjuvant therapy against coronavirus infection in vitro and in vivo (261–265). Itraconazole - an ergosterol biosynthesis inhibitor and cholesterol traffic inhibitor - showed antiviral effects in vitro against feline coronavirus (FCoV) and SARS-CoV-2 (263, 264). However, it did not suppress the SARS-CoV-2 load in the golden hamster model, which determined the interruption of a clinical study with COVID-19 patients (263, 266, 267). U1866A - an inhibitor of oxidosqualene cyclase, desmosterol reductase, and Niemann-Pick type C1 (NPC1) cholesterol transporter - suppresses intracellular cholesterol biosynthesis and trafficking of cholesterol from lysosomes. In cats, this drug inhibited FCoV replication in vitro in an NPC1-dependent manner (266), but in vivo, testing was inconclusive due to the small number of animals used (265).
The cholesterol metabolism also generates oxysterols, which modulate the immune and inflammatory response. The effects of 25HC have been described in a previous section of this review and will be summarized here. Several studies report the antiviral and anti-inflammatory activities of 25HC against SARS-CoV-2 infection in animal models (19, 24, 27, 28). Some also propose combinations of 25HC with other molecules to improve its delivery to the lungs (27) or enhance its antiviral activity (268). However, there is a divergent study reporting no protective effects of 25HC against infection, and even a possible potentiation of the pro-inflammatory response induced by 25HC (29), as described in section 2.3.
The antidepressant fluoxetine, and other representatives of this class of drugs, modulate the sphingolipid metabolism and their effects against SARS-CoV-2, using animal models or treatment of COVID-19 patients, were promising. In this context, fluoxetine suppresses SARS-CoV-2 replication in K18-hACE2 mice and the production of pro-inflammatory cytokines and chemokines, like IL-6, TNF-α, CCL2, and CXCL10. Differently from the results obtained in in vitro studies, fluoxetine did not alter ceramide and sphingomyelin levels in the lungs but enhanced the levels of hexosylceramide (glucosylceramide) and the ratio HexCer/Cer (41). These effects are encouraging since the metabolomics analysis of COVID-19 patients with pneumonia show increased ceramide levels and decreased long HexCer levels, both associated with disease severity (269). Nasal epithelial cells from healthy volunteers pre-treated with Amitryptiline were resistant to ex-vivo infection by SARS-CoV-2 (40). Moreover, Fluvoxamine reduced clinical signs of severe COVID-19 patients compared to placebo-treated controls (270).
Another study comparing the effects of various antidepressants, including SSRIs and tricyclic ones, in hospitalized COVID-19 patients, reported a lower risk of intubation and death among patients that began the use of antidepressants in the first 48h of hospital admission compared to patients who did not take the drugs (271). Home use of antidepressants is also associated with decreased hospitalization of COVID-19-positive patients (272).
Regarding fatty acid metabolism, orlistat suppressed SARS-CoV-2 infection in two mouse models: K18-hACE2 mice and adenovirus-hACE2 C57BL/6J mice. Orlistat improved survival rates and decreased the viral load and lung inflammation when used simultaneously with the virus inoculation. However, when given the day after infection, it delayed the course of the disease but was unable to prevent death (178).
Pharmacological inhibition of enzymes involved in lipid droplet metabolism showed promising results as therapeutic strategies to fight SARS-CoV-2 infection and the associated inflammatory response in the golden-hamster model (16, 152, 199). The use of Xanthohumol – a DGAT-1/2 inhibitor – or atglistatin (inhibitor of LD-associated lipases) diminished SARS-CoV-2 replication and inhibited the production of IFN-γ, IL-6, and TNF-α. The former also reduced inflammatory infiltration and consolidation of the lung parenchyma in a golden hamster model (16, 199). The combination of atglistatin and remdesivir showed synergistic effects reducing the lung lesions provoked by three different SARS-CoV-2 strains.
The success of coronavirus infection relies on the usage of cell lipids. Coronaviruses dock at the plasma membrane and invade cells either by fusing their envelope directly to the cell membrane or by endocytosis mediated by distinct pathways, including lipid nanodomains/caveolae. After entry, they reach the cytosol and induce the synthesis and extensive remodeling of internal membranes that form the replicative organelles of coronaviruses (273). Besides, coronaviruses elicit the formation of lipid droplets, which serve as replication platforms and an energy source (15, 123). Lipid droplets also contribute to the production of inflammatory mediators induced by the infection (15). In order to support a successful infection, coronaviruses prevent the completion of the autophagic process (193–195, 274), hampering the maturation of the autophagosome and use the exocytic route to exit the cell (218).
In conclusion, coronaviruses can modulate the cell lipid metabolism in many aspects that favor viral infection and spread. Hence, a broad spectrum of drugs targeting lipid metabolism inhibits coronavirus infection and the subsequent inflammatory and immune responses (Figure 3 and Table 2). Among the drugs described here, some are already FDA-approved; others still require safety and efficacy tests before being released for human treatment. Drugs targeting the host instead of virus factors may be a preferable strategy to prevent the selection of drug-resistance variants (277). Additionally, treatments combining drugs (25, 262, 278) aimed at distinct targets are also a promising therapeutic strategy since this reduces the odds of drug resistance variants and allows the use of lower drug doses. On the other hand, some drug combinations are harmful, such as the combined use of simvastatin and Paxlovid, as the latter inhibits simvastatin metabolization, increasing the risk of cardiovascular problems among statin users. In this case, simvastatin can be substituted by fluvastatin or pravastatin, which can be safely used with Paxlovid (279, 280). A in silico study indicated that fluvastatin binds the SARS-CoV-2 Mpro protein, which is the target of Paxlovid. Thus, the combined use of these drugs may potentially provide a dual inhibition of Mpro, which may be particularly relevant since Paxlovid is used for just five days, and infection rebounds in some patients after stopping the use of this drug (280). Another aspect of the pharmacology against coronaviruses that should be appreciated is the discovery that SARS-CoV-2 infection is affected by circadian regulators, such as Bmal1 and CIART (180, 281), the latter acting on the fatty acid metabolism. So far, drug therapy against coronaviruses has not considered this information (282, 283).
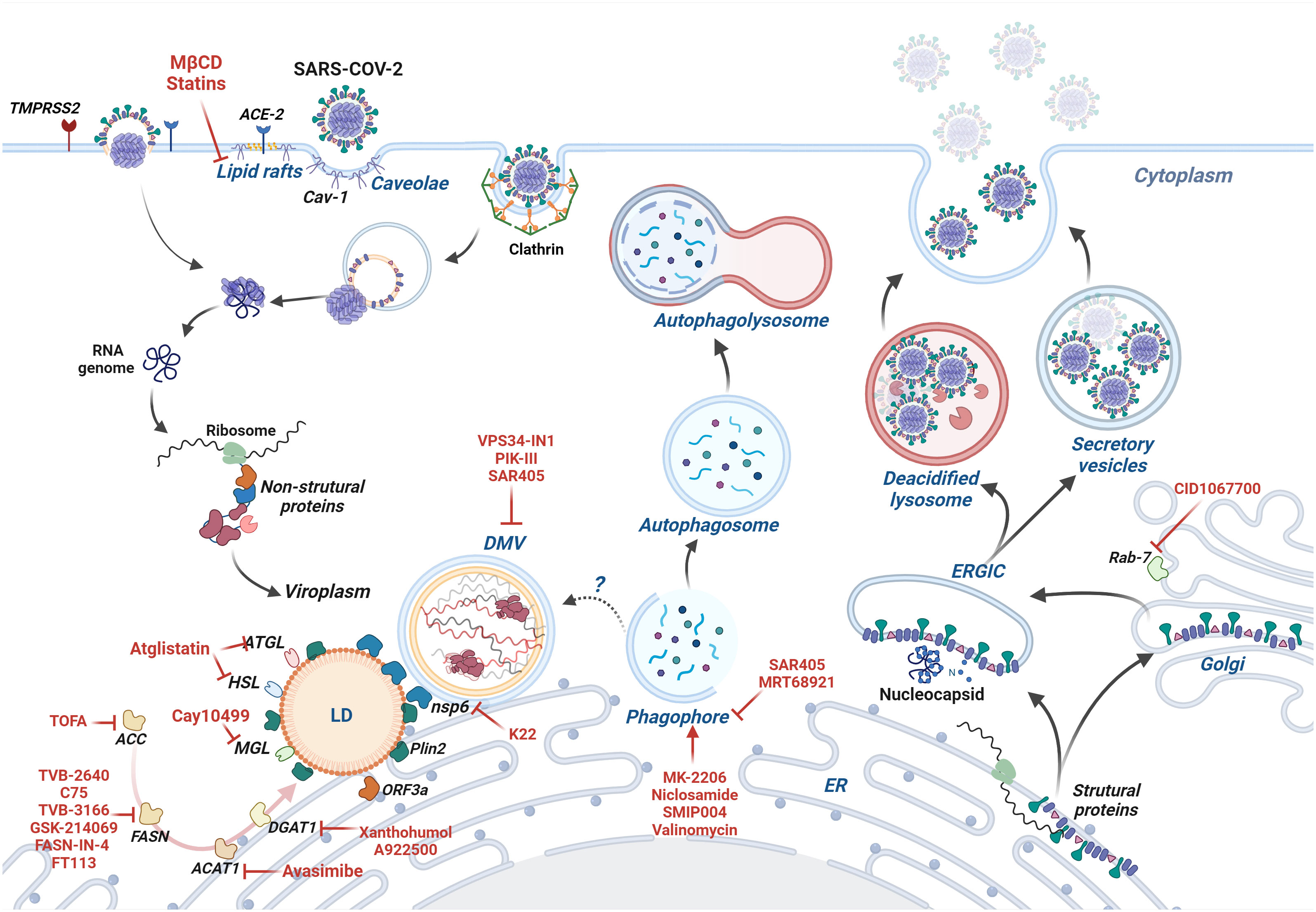
Figure 3 The coronavirus infection and cell lipid metabolism - SARS-CoV-2 cycle is depicted as a model of the coronavirus cycle. SARS-CoV-2 enters the cell by direct fusion with the plasma membrane or clathrin-mediated endocytosis. Caveolae-dependent-endocytosis is not involved in SARS-CoV-2 but is used by other coronaviruses. Once in the cell, the viral genome is transcribed, and non-structural proteins, such as nsp3 and nsp4, induce the formation of double-membrane vesicles, which provide a protected niche for genome replication. Nsp6 bridges DMVs to the ER and lipid droplets, allowing lipids to flow to DMVs. ORF3a, a viral accessory protein, induces the formation of lipid droplets that serve as platforms for virus replication and post-replication events. Drugs affecting cholesterol and fatty acid metabolism inhibit the formation of lipid droplets and viral infection. DMVs share many characteristics with autophagosomes, and there is controversy about whether coronaviruses sequester the autophagic machinery to induce DMVs. Late steps of autophagy are blocked by coronavirus proteins, such as ORF3a, hampering the formation of autolysosomes and virus degradation. The virus particles are assembled during their traffic through the ER, ERGIC, and Golgi until they leave the cell. The egress involves deacidified lysosomes, recycling endosomes and other virus-containing vesicles from the secretory pathway. Drugs targeting molecules of lipid metabolism are written in red, and their mode of action is described in the review and summarized in Table 2. Image created using BioRender.

Table 2 Effects of modulation of lipid metabolism on coronavirus infection.
DC-S: Writing – original draft. FP-D: Writing – original draft, Software. AG: Writing – review & editing. CM-M: Writing – review & editing, Writing – original draft. CA: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Oswaldo Cruz Foundation/FIOCRUZ Inova Program, by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Thorp EB, Gallagher TM. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J Virol (2004) 78:2682–92. doi: 10.1128/jvi.78.6.2682-2692.2004
2. Nomura R, Kiyota A, Suzaki E, Kataoka K, Ohe Y, Miyamoto K, et al. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol (2004) 78:8701–8. doi: 10.1128/jvi.78.16.8701-8708.2004
3. Li GM, Li YG, Yamate M, Li SM, Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect (2007) 9:96–102. doi: 10.1016/j.micinf.2006.10.015
4. Lu Y, Liu DX, Tam JP. Lipid rafts are involved in SARS-coV entry into vero E6 cells. Biochem Biophys Res Commun (2008) 369:344–9. doi: 10.1016/j.bbrc.2008.02.023
5. Glende J, Schwegmann-Wessels C, Al-Falah M, Pfefferle S, Qu X, Deng H, et al. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology (2008) 381:215–21. doi: 10.1016/j.virol.2008.08.026
6. Guo H, Huang M, Yuan Q, Wei Y, Gao Y, Mao L, et al. The important role of lipid raft-mediated attachment in the infection of cultured cells by coronavirus infectious bronchitis virus beaudette strain. PloS One (2017) 12:1–12. doi: 10.1371/journal.pone.0170123
7. Teixeira L, Temerozo JR, Pereira-Dutra FS, Ferreira AC, Mattos M, Gonçalves BS, et al. Simvastatin downregulates the SARS-coV-2-induced inflammatory response and impairs viral infection through disruption of lipid rafts. Front Immunol (2022) 13:1–16. doi: 10.3389/fimmu.2022.820131
8. Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X, et al. HDL-scavenger receptor B type 1 facilitates SARS-coV-2 entry. Nat Metab (2020) 2:1391–400. doi: 10.1038/s42255-020-00324-0
9. Baier CJ, Barrantes FJ. Role of cholesterol-recognition motifs in the infectivity of SARS-coV-2 variants. Colloids Surf B Biointerfaces (2023) 222:113090. doi: 10.1016/j.colsurfb.2022.113090
10. Owczarek K, Szczepanski A, Milewska A, Baster Z, Rajfur Z, Sarna M, et al. Early events during human coronavirus OC43 entry to the cell. Sci Rep (2018) 8:1–12. doi: 10.1038/s41598-018-25640-0
11. Feingold KR. The bidirectional link between HDL and COVID-19 infections. J Lipid Res (2021) 62:100067. doi: 10.1016/J.JLR.2021.100067
12. Lee W, Ahn JH, Park HH, Kim HN, Kim H, Yoo Y, et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct Target Ther (2020) 5. doi: 10.1038/s41392-020-00292-7
13. Yuan S, Chu H, Chan JFW, Ye ZW, Wen L, Yan B, et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat Commun (2019) 10:120. doi: 10.1038/s41467-018-08015-x
14. Williams CG, Jureka AS, Silvas JA, Nicolini AM, Chvatal SA, Carlson-Stevermer J, et al. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-coV-2 replication. Cell Rep (2021) 36:109479. doi: 10.1016/j.celrep.2021.109479
15. da Silva Gomes Dias S, Soares VC, Ferreira AC, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, et al. Lipid droplets fuel SARS-coV-2 replication and production of inflammatory mediators. PloS Pathog (2020) 16:1–19. doi: 10.1371/journal.ppat.1009127
16. Yuan S, Yan B, Cao J, Ye ZW, Liang R, Tang K, et al. SARS-coV-2 exploits host DGAT and ADRP for efficient replication. Cell Discovery (2021) 7. doi: 10.1038/s41421-021-00338-2
17. Lin Y, Zang R, Ma Y, Wang Z, Li L, Ding S, et al. Xanthohumol is a potent pan-inhibitor of coronaviruses targeting main protease. Int J Mol Sci (2021) 22. doi: 10.3390/ijms222212134
18. Wang W, Qu Y, Wang X, Xiao MZX, Fu J, Chen L, et al. Genetic variety of ORF3a shapes SARS-coV-2 fitness through modulation of lipid droplet. J Med Virol (2023) 95. doi: 10.1002/jmv.28630
19. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, et al. Cholesterol 25-hydroxylase inhibits SARS -coV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J (2020) 39:1–13. doi: 10.15252/embj.2020106057
20. Heisler DB, Johnson KA, Ma D, Ohlson MB, Zhang L, Tran M, et al. A concerted mechanism involving ACAT and SREBPs by which oxysterols deplete accessible cholesterol to restrict microbial infection. Elife (2023) 12. doi: 10.7554/eLife.83534
21. Zhang Y, Song Z, Wang M, Lan M, Zhang K, Jiang P, et al. Cholesterol 25-hydroxylase negatively regulates porcine intestinal coronavirus replication by the production of 25-hydroxycholesterol. Vet Microbiol (2019) 231:129–38. doi: 10.1016/j.vetmic.2019.03.004
22. Ke W, Wu X, Fang P, Zhou Y, Fang L, Xiao S. Cholesterol 25-hydroxylase suppresses porcine deltacoronavirus infection by inhibiting viral entry. Virus Res (2021) 295:198306. doi: 10.1016/j.virusres.2021.198306
23. Zhang J, Yang G, Wang X, Zhu Y, Wang J. 25-hydroxycholesterol mediates cholesterol metabolism to restrict porcine deltacoronavirus infection via suppression of transforming growth factor B1. Microbiol Spectr (2022) 10. doi: 10.1128/spectrum.02198-22
24. Zang R, Case JB, Yutuc E, Ma X, Shen S, Castro MFG, et al. Cholesterol 25-hydroxylase suppresses SARS-coV-2 replication by blocking membrane fusion. Proc Natl Acad Sci U.S.A. (2020) 117:32105–13. doi: 10.1073/pnas.2012197117
25. Yuan S, Chan CCY, Chik KKH, Tsang JOL, Liang R, Cao J, et al. Broad-spectrum host-based antivirals targeting the interferon and lipogenesis pathways as potential treatment options for the pandemic coronavirus disease 2019 (COVID-19). Viruses (2020) 12:1–16. doi: 10.3390/v12060628
26. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. 25-hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Sci (1979) (2014) 345:679–84. doi: 10.1126/science.1254790
27. Kim H, Lee HS, Ahn JH, Hong KS, Jang JG, An J, et al. Lung-selective 25-hydroxycholesterol nanotherapeutics as a suppressor of COVID-19-associated cytokine storm. Nano Today (2021) 38:101149. doi: 10.1016/j.nantod.2021.101149
28. Zu S, Deng YQ, Zhou C, Li J, Li L, Chen Q, et al. 25-hydroxycholesterol is a potent SARS-coV-2 inhibitor. Cell Res (2020) 30:1043–5. doi: 10.1038/s41422-020-00398-1
29. Fessler MB, Madenspacher J, Baker PJ, Hilligan KL, Castro E, Chen S, et al. Evaluation of endogenous and therapeutic 25-hydroxycholesterols in murine models of pulmonary SARS-coV-2. Infection (2022). doi: 10.1101/2022.09.12.507671
30. Marcello A, Civra A, Milan Bonotto R, Nascimento Alves L, Rajasekharan S, Giacobone C, et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-coV-2 and is markedly decreased in COVID-19 patients. Redox Biol (2020) 36:101682. doi: 10.1016/j.redox.2020.101682
31. Ohashi H, Wang F, Stappenbeck F, Tsuchimoto K, Kobayashi C, Saso W, et al. Identification of anti-severe acute respiratory syndrome-related coronavirus 2 (Sars-cov-2) oxysterol derivatives in vitro. Int J Mol Sci (2021) 22:1–14. doi: 10.3390/ijms22063163
32. Thorp EB, Boscarino JA, Logan HL, Goletz JT, Gallagher TM. Palmitoylations on murine coronavirus spike proteins are essential for virion assembly and infectivity. J Virol (2006) 80:1280–9. doi: 10.1128/jvi.80.3.1280-1289.2006
33. Wu Z, Zhang Z, Wang X, Zhang J, Ren C, Li Y, et al. Palmitoylation of SARS-coV-2 S protein is essential for viral infectivity. Signal Transduct Target Ther (2021) 6. doi: 10.1038/s41392-021-00651-y
34. McBride CE, Machamer CE. Palmitoylation of SARS-CoV S Protein Is Necessary for Partitioning into Detergent-Resistant Membranes and Cell-Cell Fusion but Not Interaction with M Protein. Virology (2010) 405:139–48. doi: 10.1016/j.virol.2010.05.031
35. Mesquita FS, Abrami L, Sergeeva O, Turelli P, Qing E, Kunz B, et al. S-acylation controls SARS-coV-2 membrane lipid organization and enhances infectivity. Dev Cell (2021) 56:2790–2807.e8. doi: 10.1016/j.devcel.2021.09.016
36. Tien C, Tsai W, Chen CH, Chou H-J, Zhang MM, Lin J-J, et al. Glycosylation and S-palmitoylation regulate SARS-coV-2 spike protein intracellular trafficking. iScience (2022) 25:104709. doi: 10.1016/j.isci.2022.104709
37. Torretta E, Garziano M, Poliseno M, Capitanio D, Biasin M, Santantonio TA, et al. Severity of covid-19 patients predicted by serum sphingolipids signature. Int J Mol Sci (2021) 22. doi: 10.3390/ijms221910198
38. Janneh AH, Kassir MF, Dwyer CJ, Chakraborty P, Pierce JS, Flume PA, et al. Alterations of lipid metabolism provide serologic biomarkers for the detection of asymptomatic versus symptomatic COVID-19 patients. Sci Rep (2021) 11:1–10. doi: 10.1038/s41598-021-93857-7
39. Edwards MJ, Becker KA, Gripp B, Hoffmann M, Keitsch S, Wilker B, et al. Sphingosine prevents binding of SARS-coV-2 spike to its cellular receptor ACE2. J Biol Chem (2020) 295:15174–82. doi: 10.1074/jbc.RA120.015249
40. Carpinteiro A, Edwards MJ, Hoffmann M, Kochs G, Gripp B, Weigang S, et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-coV-2 by epithelial cells. Cell Rep Med (2020) 1. doi: 10.1016/j.xcrm.2020.100142
41. Péricat D, Leon-Icaza SA, Sanchez Rico M, Mühle C, Zoicas I, Schumacher F, et al. Antiviral and anti-inflammatory activities of fluoxetine in a SARS-coV-2 infection mouse model. Int J Mol Sci (2022) 23. doi: 10.3390/ijms232113623
42. Geiger N, Kersting L, Schlegel J, Stelz L, Fähr S, Diesendorf V, et al. The acid ceramidase is a SARS-coV-2 host factor. Cells (2022) 11:1–16. doi: 10.3390/cells11162532
43. Vitner EB, Achdout H, Avraham R, Politi B, Cherry L, Tamir H, et al. Glucosylceramide synthase inhibitors prevent replication of SARS-coV-2 and influenza virus. J Biol Chem (2021) 296:100470. doi: 10.1016/j.jbc.2021.100470
44. Vitner EB, Avraham R, Politi B, Melamed S, Israely T. Elevation in sphingolipid upon SARS-coV-2 infection: possible implications for COVID-19 pathology. Life Sci Alliance (2022) 5:1–9. doi: 10.26508/lsa.202101168
45. Hayashi Y, Tsuchiya K, Yamamoto M, Nemoto-Sasaki Y, Tanigawa K, Hama K, et al. N -(4-hydroxyphenyl) retinamide suppresses SARS-coV-2 spike protein-mediated cell-cell fusion by a dihydroceramide Δ4-desaturase 1-independent mechanism. J Virol (2021) 95. doi: 10.1128/jvi.00807-21
46. Simons K, Van Meer G. Lipid sorting in epithelial cells. Biochemistry (1988) 27:6197–202. doi: 10.1021/bi00417a001
47. Simons K, Ikonen E. Functional rafts in cell membranes. Nature (1997) 387:569–72. doi: 10.1038/42408
48. Kusumi A, Fujiwara TK, Tsunoyama TA, Kasai RS, Liu AA, Hirosawa KM, et al. Defining raft domains in the plasma membrane. Traffic (2020) 21:106–37. doi: 10.1111/tra.12718
49. Kusumi A, Koyama-Honda I, Suzuki K. Molecular dynamics and interactions for creation of stimulation-induced stabilized rafts from small unstable steady-state rafts. Traffic (2004) 5:213–30. doi: 10.1111/j.1600-0854.2004.0178.x
50. Kusumi A, Fujiwara TK, Morone N, Yoshida KJ, Chadda R, Xie M, et al. Membrane mechanisms for signal transduction: the coupling of the meso-scale raft domains to membrane-skeleton-induced compartments and dynamic protein complexes. Semin Cell Dev Biol (2012) 23:126–44. doi: 10.1016/j.semcdb.2012.01.018
51. Brown DA. Analysis of raft affinity of membrane proteins by detergent-insolubility. Lipid Rafts. Methods Mol Biol (2007) 398:9–20. doi: 10.1007/978-1-59745-513-8_2
52. Gervásio OL, Phillips WD, Cole L, Allen DG. Caveolae respond to cell stretch and contribute to stretch-induced signaling. J Cell Sci (2011) 124:3581–90. doi: 10.1242/jcs.084376
53. Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell (2011) 144:402–13. doi: 10.1016/j.cell.2010.12.031
54. Frank PG, Pavlides S, Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res (2009) 335:41–7. doi: 10.1007/s00441-008-0659-8
55. Zhou M, Shi SX, Liu N, Jiang Y, Karim MS, Vodovoz SJ, et al. Caveolae-mediated endothelial transcytosis across the blood-brain barrier in acute ischemic stroke. J Clin Med (2021) 10. doi: 10.3390/jcm10173795
56. Pilch PF, Liu L. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol Metab (2011) 22:318–24. doi: 10.1016/j.tem.2011.04.001
57. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev (2002) 54:431–67. doi: 10.1124/pr.54.3.431
58. Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U.S.A. (1996) 93:131–5. doi: 10.1073/pnas.93.1.131
59. Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells: caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem (1996) 271:15160–5. doi: 10.1074/jbc.271.25.15160
60. Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem (1996) 271:2255–61. doi: 10.1074/jbc.271.4.2255
61. Way M. Parton, R.G. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett (1996) 378:108–12. doi: 10.1016/0014-5793(96)82884-5
62. Fujimoto T, Kogo H, Nomura R, Une T. Isoforms of caveolin-1 and caveolar structure. J Cell Sci (2000) 113 Pt 19:3509–17. doi: 10.1242/jcs.113.19.3509
63. Sowa G, Pypaert M, Fulton D, Sessa WC. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc Natl Acad Sci U.S.A. (2003) 100:6511–6. doi: 10.1073/pnas.1031672100
64. Drab M. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Sci (1979) (2001) 293:2449–52. doi: 10.1126/science.1062688
65. Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol (2002) 160:2207–17. doi: 10.1016/S0002-9440(10)61168-6
66. Razani B, Wang XB, Engelman JA, Lagaud G, Zhang XL, Kneitz B, et al. Caveolin-2-Deficient Mice Show Evidence of Severe Pulmonary Dysfunction without Disruption of Caveolae Caveolin-2-Deficient Mice Show Evidence of Severe Pulmonary Dysfunction without Disruption of Caveolae. Mol Cell Biol (2002) 22:2329–44. doi: 10.1128/MCB.22.7.2329
67. Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathology: Mech Dis (2012) 7:423–67. doi: 10.1146/annurev-pathol-011811-120856
68. Haddad D, Al Madhoun A, Nizam R, Al-Mulla F. Role of caveolin-1 in diabetes and its complications. Oxid Med Cell Longev (2020) 2020:9761539. doi: 10.1155/2020/9761539
69. Mathew R. Medical sciences critical role of caveolin-1 loss/dysfunction in pulmonary hypertension. Med Sci (2021) 9:1–15. doi: 10.3390/medsci9040058
70. Takamura N, Yamaguchi Y. Involvement of caveolin-1 in skin diseases. Front Immunol (2022) 13:1035451. doi: 10.3389/fimmu.2022.1035451
71. Tourkina E. Caveolin-1 signaling in lung fibrosis. Open Rheumatol J (2012) 6:116–22. doi: 10.2174/1874312901206010116
72. Hayer A, Stoeber M, Bissig C, Helenius A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic (2010) 11:361–82. doi: 10.1111/j.1600-0854.2009.01023.x
73. Liu L, Pilch PFA. Critical role of cavin (Polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem (2008) 283:4314–22. doi: 10.1074/jbc.M707890200
74. Parton RG, Mcmahon K, Wu Y. ScienceDirect caveolae : formation, dynamics, and function. (Frances M. Brodsky and Jennifer L. Stow) (2020). pp. 8–16.
75. Hansen CG, Shvets E, Howard G, Riento K, Nichols BJ. Deletion of cavin genes reveals tissue-specific mechanisms for morphogenesis of endothelial caveolae. Nat Commun (2013) 4. doi: 10.1038/ncomms2808
76. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, et al. PTRF-cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell (2008) 132:113–24. doi: 10.1016/j.cell.2007.11.042
77. Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol (2009) 11:807–14. doi: 10.1038/ncb1887
78. McMahon KA, Zajicek H, Li WP, Peyton MJ, Minna JD, Hernandez VJ, et al. SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J (2009) 28:1001–15. doi: 10.1038/emboj.2009.46
79. Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP, et al. MURC/cavin-4 and cavin family members form tissue-specific caveolar complexes. J Cell Biol (2009) 185:1259–73. doi: 10.1083/jcb.200903053
80. Örtegren U, Karlsson M, Blazic N, Blomqvist M, Nystrom FH, Gustavsson J, et al. Lipids and glycosphingolipids in caveolae and surrounding plasma membrane of primary rat adipocytes. Eur J Biochem (2004) 271:2028–36. doi: 10.1111/j.1432-1033.2004.04117.x
81. Murata M, Peranen J, Schreinert R, Wielandt F, Kurzchalia TV, Simons KAI. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA (1995) 92:10339–43. doi: 10.1073/pnas.92.22.10339
82. Pol A, Martin S, Fernández MA, Ingelmo-Torres M, Ferguson C, Enrich C, et al. Cholesterol and fatty acids regulate dynamic caveolin trafficking through the golgi complex and between the cell surface and lipid bodies. Mol Biol Cell (2005) 16:2091–105. doi: 10.1091/mbc.e04-08-0737
83. Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RGW. Caveolin, a protein component of caveolae membrane coats. Cell (1992) 68:673–82. doi: 10.1016/0092-8674(92)90143-Z
84. Westermann M, Steiniger F, Richter W. Belt-like localisation of caveolin in deep caveolae and its re-distribution after cholesterol depletion. Histochem Cell Biol (2005) 123:613–20. doi: 10.1007/s00418-004-0750-5
85. Breen MR, Camps M, Carvalho-Simoes F, Zorzano A, Pilch PF. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PloS One (2012) 7:2–9. doi: 10.1371/journal.pone.0034516
86. Frank PG, Cheung MWC, Pavlides S, Llaverias G, Park DS, Lisanti MP. Caveolin-1 and regulation of cellular cholesterol homeostasis. Am J Physiol Heart Circ Physiol (2006) 291. doi: 10.1152/ajpheart.01092.2005
87. Le Lay S, Hajduch E, Lindsay MR, Le Lièpvre X, Thiele C, Ferré P, et al. Cholesterol-induced caveolin targeting to lipid droplets in adipocytes: A role for caveolar endocytosis. Traffic (2006) 7:549–61. doi: 10.1111/j.1600-0854.2006.00406.x
88. Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem (2004) 279:46835–42. doi: 10.1074/jbc.M409340200
89. Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, et al. Lipid droplet formation. Diabetes (2004) 53:21–5. doi: 10.2337/diabetes.53.5.1261
90. Alejandro Fernández-Rojo M, Restall C, Ferguson C, Martel N, Martin S, Bosch M, et al. Caveolin-1 orchestrates the balance between glucose and lipid-dependent energy metabolism: implications for liver regeneration. Hepatology (2012) 55:1574–84. doi: 10.1002/hep.24810
91. Matthaeus C, Lahmann I, Kunz S, Jonas W, Melo AA, Lehmann M, et al. EHD2-mediated restriction of caveolar dynamics regulates cellular fatty acid uptake. Proc Natl Acad Sci U.S.A. (2020) 117:7471–81. doi: 10.1073/pnas.1918415117
92. Bosch M, Parton RG, Pol A. Lipid droplets, bioenergetic fluxes, and metabolic flexibility. Semin Cell Dev Biol (2020) 108:33–46. doi: 10.1016/J.SEMCDB.2020.02.010
93. Pereira-Dutra FS, Bozza PT. Lipid droplets diversity and functions in inflammation and immune response. Expert Rev Proteomics (2021) 18:809–25. doi: 10.1080/14789450.2021.1995356
94. Farese RV, Walther TC. Glycerolipid synthesis and lipid droplet formation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol (2023) 15. doi: 10.1101/cshperspect.a041246
95. Walther TC, Farese RV. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem (2012) 81:687–714. doi: 10.1146/annurev-biochem-061009-102430
96. Bozza P, D’Avila H, Almeida P, Magalhães K, Almeida C, Maya-Monteiro C. Lipid droplets in host–pathogen interactions. Clin Lipidol (2009) 4:791–807. doi: 10.2217/clp.09.63
97. Pol A, Gross SP, Parton RG. Biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol (2014) 204:635–46. doi: 10.1083/jcb.201311051
98. Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res (2007) 48:2547–59. doi: 10.1194/jlr.R700014-JLR200
99. Itabe H, Yamaguchi T, Nimura S, Sasabe N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis (2017) 16:83. doi: 10.1186/s12944-017-0473-y
100. Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem (1991) 266:11341–6. doi: 10.1016/S0021-9258(18)99168-4
101. Thiam AR, Beller M. The why, when and how of lipid droplet diversity. J Cell Sci (2017) 130:315–24. doi: 10.1242/jcs.192021
102. Onal G, Kutlu O, Gozuacik D, Dokmeci Emre S. Lipid droplets in health and disease. Lipids Health Dis (2017) 16:128. doi: 10.1186/s12944-017-0521-7
103. Cruz ALS, Barreto EdeA, Fazolini NPB, Viola JPB, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis (2020) 11:105. doi: 10.1038/s41419-020-2297-3
104. Welte MA. Expanding roles for lipid droplets. Curr Biol (2015) 25:R470–81. doi: 10.1016/j.cub.2015.04.004
105. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol (2019) 20:137–55. doi: 10.1038/s41580-018-0085-z
106. Walther TC, Chung J, Farese RV. Lipid droplet biogenesis. Annu Rev Cell Dev Biol (2017) 33:491–510. doi: 10.1146/annurev-cellbio-100616-060608
107. Chitraju C, Walther TC, Farese RV. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J Lipid Res (2019) 60:1112–20. doi: 10.1194/jlr.M093112
108. McFie PJ, Banman SL, Stone SJ. Diacylglycerol acyltransferase-2 contains a c-terminal sequence that interacts with lipid droplets. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2018) 1863:1068–81. doi: 10.1016/j.bbalip.2018.06.008
109. Dove DE, Su YR, Zhang W, Jerome WG, Swift LL, Linton MF, et al. ACAT1 deficiency disrupts cholesterol efflux and alters cellular morphology in macrophages. Arterioscler Thromb Vasc Biol (2005) 25:128–34. doi: 10.1161/01.ATV.0000148323.94021.e5
110. Zhu Y, Chen C-Y, Li J, Cheng J-X, Jang M, Kim K-H. In vitro exploration of ACAT contributions to lipid droplet formation during adipogenesis. J Lipid Res (2018) 59:820–9. doi: 10.1194/jlr.M081745
111. Xu Y, Du X, Turner N, Brown AJ, Yang H. Enhanced acyl-coA : cholesterol acyltransferase activity increases cholesterol levels on the lipid droplet surface and impairs adipocyte function. J Biol Chem (2019) 294:19306–21. doi: 10.1074/jbc.RA119.011160
112. Santinho A, Salo VT, Chorlay A, Li S, Zhou X, Omrane M, et al. Membrane curvature catalyzes lipid droplet assembly. Curr Biol (2020) 30:2481–2494.e6. doi: 10.1016/j.cub.2020.04.066
113. Szatmari I, Töröcsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARγ Regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood (2007) 110:3271–80. doi: 10.1182/blood-2007-06-096222
114. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα Action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol (2015) 62:720–33. doi: 10.1016/j.jhep.2014.10.039
115. Silva AR, Pacheco P, Vieira-de-Abreu A, Maya-Monteiro CM, D’Alegria B, Magalhães KG, et al. Lipid bodies in oxidized LDL-induced foam cells are leukotriene-synthesizing organelles: A MCP-1/CCL2 regulated phenomenon. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2009) 1791:1066–75. doi: 10.1016/j.bbalip.2009.06.004
116. Jarc E, Kump A, Malavašič P, Eichmann TO, Zimmermann R, Petan T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2018) 1863:247–65. doi: 10.1016/j.bbalip.2017.12.006
117. Mashek DG. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol Metab (2020) 50:101115. doi: 10.1016/j.molmet.2020.101115
118. Kiarely Souza E, Pereira-Dutra FS, Rajão MA, Ferraro-Moreira F, Goltara-Gomes TC, Cunha-Fernandes T, et al. Lipid droplet accumulation occurs early following salmonella infection and contributes to intracellular bacterial survival and replication. Mol Microbiol (2022) 117:293–306. doi: 10.1111/mmi.14844
119. Bosch M, Sánchez-Álvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Sci (1979) (2020) 370. doi: 10.1126/science.aay8085
120. Vieyres G, Reichert I, Carpentier A, Vondran FWR, Pietschmann T. The ATGL lipase cooperates with ABHD5 to mobilize lipids for hepatitis C virus assembly. (San Diego, United States: Aleem Siddiqui, University of California), Vol. 16. (2020).
121. Padmanabha Das KM, Wechselberger L, Liziczai M, de la Rosa Rodriguez M, Grabner GF, Heier C, et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J Lipid Res (2018) 59:531–41. doi: 10.1194/jlr.M082388
122. Proikas-Cezanne T, Simos G, Liu X, Kounakis K, Chaniotakis M, Markaki M, et al. Emerging roles of lipophagy in health and disease. Front Cell Dev Biol (2019) 7:185. doi: 10.3389/fcell.2019.00185
123. Ricciardi S, Guarino AM, Giaquinto L, Polishchuk EV, Santoro M, Di Tullio G, et al. The role of NSP6 in the biogenesis of the SARS-coV-2 replication organelle. Nature (2022) 606:761–8. doi: 10.1038/s41586-022-04835-6
124. Viktorova EG, Nchoutmboube JA, Ford-Siltz LA, Iverson E, Belov GA. Phospholipid synthesis fueled by lipid droplets drives the structural development of poliovirus replication organelles. PloS Pathog (2018) 14:e1007280. doi: 10.1371/journal.ppat.1007280
125. Altmann SW, Davis HR, Zhu LJ, Yao X, Hoos LM, Tetzloff G, et al. Niemann-pick C1 like 1 protein is critical for intestinal cholesterol absorption. Sci (1979) (2004) 303:1201–4. doi: 10.1126/science.1093131
126. Brown MS, Goldstein JLA. Receptor-mediated pathway for cholesterol homeostasis. Sci (1979) (1986) 232:34–47. doi: 10.1126/science.3513311
127. Luo J, Jiang LY, Yang H, Song BL. Intracellular cholesterol transport by sterol transfer proteins at membrane contact sites. Trends Biochem Sci (2019) 44:273–92. doi: 10.1016/j.tibs.2018.10.001
128. Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife (2014) 2014:1–16. doi: 10.7554/eLife.02882
129. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9:112–24. doi: 10.1038/nrm2330
130. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology-divergent pathophysiology. Nat Rev Endocrinol (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
131. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab (2008) 8:512–21. doi: 10.1016/j.cmet.2008.10.008
132. Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell (2002) 110:489–500. doi: 10.1016/S0092-8674(02)00872-3
133. Korber M, Klein I, Daum G. Steryl ester synthesis, storage and hydrolysis: A contribution to sterol homeostasis. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862:1534–45. doi: 10.1016/j.bbalip.2017.09.002
134. Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang NHDL, Transporters ABC. and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab (2008) 7:365–75. doi: 10.1016/j.cmet.2008.03.001
135. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-coV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty (2020) 9:1–7. doi: 10.1186/s40249-020-00662-x
136. Zhou P, Yang X, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
137. Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T, et al. AXL is a candidate receptor for SARS-coV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res (2021) 31:126–40. doi: 10.1038/s41422-020-00460-y
138. Wang K, Chen W, Zhou Y, Lian JQ, Zhang Z, Du P, et al. SARS-coV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv (2020) 5:283. doi: 10.1101/2020.03.14.988345
139. Lempp FA, Soriaga LB, Montiel-Ruiz M, Benigni F, Noack J, Park YJ, et al. Lectins enhance SARS-coV-2 infection and influence neutralizing antibodies. Nature (2021) 598:342–7. doi: 10.1038/s41586-021-03925-1
140. McQuaid C, Solorzano A, Dickerson I, Deane R. Uptake of severe acute respiratory syndrome coronavirus 2 spike protein mediated by angiotensin converting enzyme 2 and ganglioside in human cerebrovascular cells. Front Neurosci (2023) 17:1117845. doi: 10.3389/fnins.2023.1117845
141. Carlos AJ, Ha DP, Yeh DW, Van Krieken R, Tseng CC, Zhang P, et al. The chaperone GRP78 is a host auxiliary factor for SARS-coV- 2 and GRP78 depleting antibody blocks viral entry and infection. J Biol Chem (2021) 296:100759. doi: 10.1016/J.JBC.2021.100759
142. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-coV-2 infection depends on cellular heparan sulfate and ACE2. Cell (2020) 183:1043–1057.e15. doi: 10.1016/j.cell.2020.09.033
143. Liu L, Chopra P, Li X, Bouwman KM, Tompkins SM, Wolfert MA, et al. Heparan sulfate proteoglycans as attachment factor for SARS-coV-2. ACS Cent Sci (2021) 7:1009–18. doi: 10.1021/acscentsci.1c00010
144. Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-coV-2 infection. Sci (1979) (2020) 370:861–5. doi: 10.1126/science.abd3072
145. Ichimura T, Mori Y, Aschauer P, Padmanabha Das KM, Padera RF, Weins A, et al. KIM-1/TIM-1 is a receptor for SARS-coV-2 in lung and kidney. medRxiv (2020). doi: 10.1101/2020.09.16.20190694
146. Thunders M, Delahunt B. Gene of the month: TMPRSS2 (Transmembrane serine protease 2). J Clin Pathol (2020) 73:773–6. doi: 10.1136/jclinpath-2020-206987
147. Yang Z-Y, Huang Y, Ganesh L, Leung K, Kong W-P, Schwartz O, et al. PH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol (2004) 78:5642–50. doi: 10.1128/jvi.78.11.5642-5650.2004
148. Wang S, Guo F, Liu K, Wang H, Rao S, Yang P, et al. Endocytosis of the receptor-binding domain of SARS-coV spike protein together with virus receptor ACE2. Virus Res (2008) 136:8–15. doi: 10.1016/j.virusres.2008.03.004
149. Shi B-J, Liu C-C, Zhou J, Wang S-Q, Gao Z-C, Zhang X-M, et al. Entry of Classical Swine Fever Virus into PK-15 Cells via a PH-, Dynamin-, and Cholesterol-Dependent, Clathrin-Mediated Endocytic Pathway That Requires Rab5 and Rab7. J Virol (2016) 90:9194–208. doi: 10.1128/jvi.00688-16
150. Li Z, Zhao K, Lan Y, Lv X, Hu S, Guan J, et al. Porcine hemagglutinating encephalomyelitis virus enters neuro-2a cells via clathrin-mediated endocytosis in a rab5-, cholesterol-, and PH-dependent manner. J Virol (2017) 91:1–22. doi: 10.1128/JVI.01083-17
151. Li X, Zhu W, Fan M, Zhang J, Peng Y, Huang F, et al. Dependence of SARS-coV-2 infection on cholesterol-rich lipid raft and endosomal acidification. Comput Struct Biotechnol J (2021) 19:1933–43. doi: 10.1016/j.csbj.2021.04.001
152. Wing PAC, Schmidt NM, Peters R, Erdmann M, Brown R, Wang H, et al. An ACAT inhibitor suppresses SARS-coV-2 replication and boosts antiviral T cell activity. PloS Pathog (2023) 19:1–26. doi: 10.1371/journal.ppat.1011323
153. Choi KS, Aizaki H, Lai MMC. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J Virol (2005) 79:9862–71. doi: 10.1128/jvi.79.15.9862-9871.2005
154. Pichler H, Riezman H. Where sterols are required for endocytosis. Biochim Biophys Acta Biomembr (2004) 1666:51–61. doi: 10.1016/j.bbamem.2004.05.011
155. Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, et al. SARS Coronavirus Entry into Host Cells through a Novel Clathrin- and Caveolae-Independent Endocytic Pathway. Cell Res (2008) 18:290–301. doi: 10.1038/cr.2008.15
156. Inoue Y, Tanaka N, Tanaka Y, Inoue S, Morita K, Zhuang M, et al. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J Virol (2007) 81:8722–9. doi: 10.1128/jvi.00253-07
157. Bayati A, Kumar R, Francis V, McPherson PS. SARS-CoV-2 Infects Cells after Viral Entry via Clathrin-Mediated Endocytosis. J Biol Chem (2021) 296:100306. doi: 10.1016/j.jbc.2021.100306
158. Pu Y, Zhang X. Mouse hepatitis virus type 2 enters cells through a clathrin-mediated endocytic pathway independent of eps15. J Virol (2008) 82:8112–23. doi: 10.1128/jvi.00837-08
159. Vogel U, Sandvig K, Van Deurs B. 1998-vogel-expression of caveoline 1 and polarized formation of invaginated caveolae in caco2 and mdck ii. Cells (1998) 832:825–32. doi: 10.1242/jcs.111.6.825
160. Lahtinen U, Honsho M, Parton RG, Simons K, Verkade P. Involvement of caveolin-2 in caveolar biogenesis in MDCK cells. FEBS Lett (2003) 538:85–8. doi: 10.1016/S0014-5793(03)00135-2
161. Zhou YQ, Wang K, Wang XY, Cui HY, Zhao Y, Zhu P, et al. SARS-coV-2 pseudovirus enters the host cells through spike protein-CD147 in an arf6-dependent manner. Emerg Microbes Infect (2022) 11:1135–44. doi: 10.1080/22221751.2022.2059403
162. Sun X, Whittaker GR. Role for influenza virus envelope cholesterol in virus entry and infection. J Virol (2003) 77:12543–51. doi: 10.1128/jvi.77.23.12543-12551.2003
163. Imhoff H, von Messling V, Herrler G, Haas L. Canine distemper virus infection requires cholesterol in the viral envelope. J Virol (2007) 81:4158–65. doi: 10.1128/jvi.02647-06
164. Ren X, Glende J, Yin J, Schwegmann-Wessels C, Herrler G. Importance of cholesterol for infection of cells by transmissible gastroenteritis virus. Virus Res (2008) 137:220–4. doi: 10.1016/j.virusres.2008.07.023
165. Yin J, Glende J, Schwegmann-Wessels C, Enjuanes L, Herrler G, Ren X. Cholesterol is important for a post-adsorption step in the entry process of transmissible gastroenteritis virus. Antiviral Res (2010) 88:311–6. doi: 10.1016/j.antiviral.2010.10.002
166. Fantini J, Di Scala C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-coV-2 infection. Int J Antimicrob Agents (2020) 55:105960. doi: 10.1016/j.ijantimicag.2020.105960
167. Matrosovich M, Herrler G, Klenk HD. Sialic acid receptors of viruses. In: Peptide-based materials. (Cham: Springer), vol. 310. (2013). p. 1–28.
168. Schissel SL, Jiang XC, Tweedie-Hardman J, Jeong TS, Camejo EH, Najib J, et al. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral PH. Implications for athereosclerotic lesion development. J Biol Chem (1998) 273:2738–46. doi: 10.1074/jbc.273.5.2738
169. Hollmann C, Werner S, Avota E, Reuter D, Japtok L, Kleuser B, et al. Inhibition of acid sphingomyelinase allows for selective targeting of CD4+ Conventional versus foxp3+ Regulatory T cells. J Immunol (2016) 197:3130–41. doi: 10.4049/jimmunol.1600691
170. Zimniak M, Kirschner L, Hilpert H, Geiger N, Danov O, Oberwinkler H, et al. The serotonin reuptake inhibitor fluoxetine inhibits SARS-coV-2 in human lung tissue. Sci Rep (2021) 11:1–5. doi: 10.1038/s41598-021-85049-0
171. Creeden JF, Imami AS, Eby HM, Gillman C, Becker KN, Reigle J, et al. Fluoxetine as an anti-inflammatory therapy in SARS-coV-2 infection. Biomedicine Pharmacotherapy (2021) 138:111437. doi: 10.1016/j.biopha.2021.111437
172. Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol (2003) 4:397–407. doi: 10.1038/nrm1103
173. Teymouri S, Pourbayram Kaleybar S, Hejazian SS, Hejazian SM, Ansarin K, Ardalan M, et al. The effect of fingolimod on patients with moderate to severe COVID-19. Pharmacol Res Perspect (2023) 11:1–9. doi: 10.1002/prp2.1039
174. Hach T, Shakeri-Nejad K, Bigaud M, Dahlke F, de Micco M, Petricoul O, et al. Rationale for use of sphingosine-1-phosphate receptor modulators in COVID-19 patients: overview of scientific evidence. J Interferon Cytokine Res (2023) 43:246–56. doi: 10.1089/jir.2022.0078
175. Lee JH, Han J, Woo JH, Jou I. 25-hydroxycholesterol suppress IFN-γ-induced inflammation in microglia by disrupting lipid raft formation and caveolin-mediated signaling endosomes. Free Radic Biol Med (2022) 179:252–65. doi: 10.1016/j.freeradbiomed.2021.11.017
176. Rottman JN, Widom RL, Nadal-Ginard B, Mahdavi V, Karathanasis SK. NOTES: A retinoic acid-responsive element in the apolipoprotein AI gene distinguishes between two different retinoic acid response pathways. Mol Cell Biol (1991) 11:3814–20. doi: 10.1128/mcb.11.7.3814-3820.1991
177. Costet P, Lalanne F, Gerbod-Giannone MC, Molina JR, Fu X, Lund EG, et al. Retinoic acid receptor-mediated induction of ABCA1 in macrophages. Mol Cell Biol (2003) 23:7756–66. doi: 10.1128/mcb.23.21.7756-7766.2003
178. Chu J, Xing C, Du Y, Duan T, Liu S, Zhang P, et al. Pharmacological inhibition of fatty acid synthesis blocks SARS-coV-2 replication. Nat Metab (2021) 3:1466–75. doi: 10.1038/s42255-021-00479-4
179. Aliyari SR, Ghaffari AA, Pernet O, Parvatiyar K, Wang Y, Gerami H, et al. Suppressing fatty acid synthase by type I interferon and chemical inhibitors as a broad spectrum anti-viral strategy against SARS-coV-2. Acta Pharm Sin B (2022) 12:1624–35. doi: 10.1016/j.apsb.2022.02.019
180. Tang X, Xue D, Zhang T, Nilsson-Payant BE, Carrau L, Duan X, et al. A multi-organoid platform identifies CIART as a key factor for SARS-coV-2 infection. Nat Cell Biol (2023) 25:381–9. doi: 10.1038/s41556-023-01095-y
181. Petit CM, Chouljenko VN, Iyer A, Colgrove R, Farzan M, Knipe DM, et al. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology (2007) 360:264–74. doi: 10.1016/j.virol.2006.10.034
182. Li D, Liu Y, Lu Y, Gao S, Zhang L. Palmitoylation of SARS-coV-2 S protein is critical for S-mediated syncytia formation and virus entry. J Med Virol (2022) 94:342–8. doi: 10.1002/jmv.27339
183. Gelhaus S, Thaa B, Eschke K, Veit M, Schwegmann-Weßels C. Palmitoylation of the Alphacoronavirus TGEV Spike Protein S Is Essential for Incorporation into Virus-like Particles but Dispensable for S-M Interaction. Virology (2014) 464–465:397–405. doi: 10.1016/j.virol.2014.07.035
184. Puthenveetil R, Lun CM, Murphy RE, Healy LB, Vilmen G, Christenson ET, et al. Banerjee, A. S-acylation of SARS-coV-2 spike protein: mechanistic dissection, in vitro reconstitution and role in viral infectivity. J Biol Chem (2021) 279:101112. doi: 10.1016/j.jbc.2021.101112
185. Woodley KT, Collins MO. Regulation and function of the palmitoyl-acyltransferase ZDHHC5. FEBS J (2021) 288:6623–34. doi: 10.1111/febs.15709
186. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-coV-2 protein interaction map reveals targets for drug repurposing. Nature (2020) 583:459–68. doi: 10.1038/s41586-020-2286-9
187. Zeng XT, Yu XT, Cheng W. The interactions of ZDHHC5/GOLGA7 with SARS-coV-2 spike (S) protein and their effects on S protein’s subcellular localization, palmitoylation and pseudovirus entry. Virol J (2021) 18:1–16. doi: 10.1186/s12985-021-01722-w
188. Mekhail K, Lee M, Sugiyama M, Astori A, St-Germain J, Latreille E, et al. FASN inhibitor TVB-3166 prevents S-acylation of the spike protein of human coronaviruses. J Lipid Res (2022) 63:100256. doi: 10.1016/j.jlr.2022.100256
189. Ramadan AA, Mayilsamy K, McGill AR, Ghosh A, Giulianotti MA, Donow HM, et al. Identification of SARS-coV-2 spike palmitoylation inhibitors that results in release of attenuated virus with reduced infectivity. Viruses (2022) 14. doi: 10.3390/v14030531
190. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol (2007) 9:1089–97. doi: 10.1038/ncb1631
191. Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PloS Pathog (2009) 5. doi: 10.1371/journal.ppat.1000632
192. Cheung W, Gill M, Esposito A, Kaminski CF, Courousse N, Chwetzoff S, et al. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J Virol (2010) 84:6782–98. doi: 10.1128/jvi.01757-09
193. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, et al. ORF3a of the COVID-19 virus SARS-coV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell (2021) 56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010
194. Qu Y, Wang X, Zhu Y, Wang W, Wang Y, Hu G, et al. ORF3a-mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication. Front Cell Dev Biol (2021) 9:716208. doi: 10.3389/fcell.2021.716208
195. Zhang Y, Sun H, Pei R, Mao B, Zhao Z, Li H, et al. The SARS-coV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discovery (2021) 7. doi: 10.1038/s41421-021-00268-z
196. Chen D, Zheng Q, Sun L, Ji M, Li Y, Deng H, et al. ORF3a of SARS-coV-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell (2021) 56:3250–3263.e5. doi: 10.1016/j.devcel.2021.10.006
197. Wang R, Yang X, Chang M, Xue Z, Wang W, Bai L, et al. ORF3a protein of severe acute respiratory syndrome coronavirus 2 inhibits interferon-activated janus kinase/signal transducer and activator of transcription signaling via elevating suppressor of cytokine signaling 1. Front Microbiol (2021) 12:752597. doi: 10.3389/fmicb.2021.752597
198. Nardacci R, Colavita F, Castilletti C, Lapa D, Matusali G, Meschi S, et al. Evidences for lipid involvement in SARS-coV-2 cytopathogenesis. Cell Death Dis (2021) 12. doi: 10.1038/s41419-021-03527-9
199. Baek Y-B, Kwon H-J, Sharif M, Lim J, Lee I-C, Ryu YB, et al. Therapeutic strategy targeting host lipolysis limits infection by SARS-coV-2 and influenza A virus. Signal Transduct Target Ther (2022) 7:367. doi: 10.1038/s41392-022-01223-4
200. Toelzer C, Gupta K, Yadav SKN, Borucu U, Davidson AD, Williamson MK, et al. Free fatty acid binding pocket in the locked structure of SARS-coV-2 spike protein. Sci (1979) (2020) 370:725–30. doi: 10.1126/science.abd3255
201. Roingeard P, Eymieux S, Burlaud-Gaillard J, Hourioux C, Patient R, Blanchard E. The double-membrane vesicle (DMV): A virus-induced organelle dedicated to the replication of SARS-coV-2 and other positive-sense single-stranded RNA viruses. Cell Mol Life Sci (2022) 79:1–9. doi: 10.1007/s00018-022-04469-x
202. Knoops K, Kikkert M, Van Den Worm SHE, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PloS Biol (2008) 6:1957–74. doi: 10.1371/journal.pbio.0060226
203. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio (2013) 4:4. doi: 10.1128/mBio.00524-13
204. Snijder EJ, Limpens RWAL, de Wilde AH, de Jong AWM, Zevenhoven-Dobbe JC, Maier HJ, et al. Unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PloS Biol (2020) 18:1–25. doi: 10.1371/journal.pbio.3000715
205. Wolff G, Limpens RWAL, Zevenhoven-Dobbe JC, Laugks U, Zheng S, de Jong AWM, et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Sci (1979) (2020) 369:1395–8. doi: 10.1126/SCIENCE.ABD3629
206. Mihelc EM, Baker SC, Lanman JK. Coronavirus infection induces progressive restructuring of the endoplasmic reticulum involving the formation and degradation of double membrane vesicles. Virology (2021) 556:9–22. doi: 10.1016/j.virol.2020.12.007
207. Cortese M, Lee J-Y, Cerikan B, Neufeldt CJ, Oorschot VMJ, Köhrer S, et al. Integrative imaging reveals SARS-coV-2-induced reshaping of subcellular morphologies. Cell Host Microbe (2020) 28:853–866.e5. doi: 10.1016/j.chom.2020.11.003
208. Oudshoorn D, Rijs K, Limpens RWAL, Groen K, Koster AJ, Snijder EJ, et al. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio (2017) 8. doi: 10.1128/mBio.01658-17
209. Twu WI, Lee JY, Kim H, Prasad V, Cerikan B, Haselmann U, et al. Contribution of autophagy machinery factors to HCV and SARS-coV-2 replication organelle formation. Cell Rep (2021) 37. doi: 10.1016/j.celrep.2021.110049
210. Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy (2011) 7:1335–47. doi: 10.4161/auto.7.11.16642
211. Lundin A, Dijkman R, Bergström T, Kann N, Adamiak B, Hannoun C, et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle east respiratory syndrome virus. PloS Pathog (2014) 10. doi: 10.1371/journal.ppat.1004166
212. Liang H, Luo D, Liao H, Li S. Coronavirus usurps the autophagy-lysosome pathway and induces membranes rearrangement for infection and pathogenesis. Front Microbiol (2022) 13:846543. doi: 10.3389/fmicb.2022.846543
213. Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, et al. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe (2010) 7:500–8. doi: 10.1016/j.chom.2010.05.013
214. Miller K, McGrath ME, Hu Z, Ariannejad S, Weston S, Frieman M, et al. Coronavirus interactions with the cellular autophagy machinery. Autophagy (2020) 16:2131–9. doi: 10.1080/15548627.2020.1817280
215. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem (2004) 279:10136–41. doi: 10.1074/jbc.M306124200
216. Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy (2007) 3:581–5. doi: 10.4161/auto.4782
217. Zou D, Xu J, Duan X, Xu X, Li P, Cheng L, et al. Porcine epidemic diarrhea virus ORF3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet Microbiol (2019) 235:209–19. doi: 10.1016/j.vetmic.2019.07.005
218. Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, et al. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell (2020) 183:1520–1535.e14. doi: 10.1016/j.cell.2020.10.039
219. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-coV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep (2021) 35. doi: 10.1016/j.celrep.2021.109126
220. Singh K, Chen YC, Hassanzadeh S, Han K, Judy JT, Seifuddin F, et al. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-coV-2 infection. Front Genet (2021) 12:599261. doi: 10.3389/fgene.2021.599261
221. Yuen CK, Wong WM, Mak LF, Wang X, Chu H, Yuen KY, et al. Suppression of SARS-coV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J Med Virol (2021) 93:2076–83. doi: 10.1002/jmv.26583
222. Gassen NC, Niemeyer D, Muth D, Corman VM, Martinelli S, Gassen A, et al. SKP2 attenuates autophagy through beclin1-ubiquitination and its inhibition reduces MERS-coronavirus infection. Nat Commun (2019) 10:2–4. doi: 10.1038/s41467-019-13659-4
223. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol (2020) 21:439–58. doi: 10.1038/s41580-020-0241-0
224. Moretti F, Bergman P, Dodgson S, Marcellin D, Claerr I, Goodwin JM, et al. TMEM 41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep (2018) 19:1–14. doi: 10.15252/embr.201845889
225. Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol (2019) 218:1787–98. doi: 10.1083/JCB.201811139
226. Ghanbarpour A, Valverde DP, Melia TJ, Reinisch KMA. Model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc Natl Acad Sci (2021) 118. doi: 10.1073/pnas.2101562118
227. Li YE, Wang Y, Du X, Zhang T, Mak H, Hancock SE, et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J Cell Biol (2021) 220. doi: 10.1083/jcb.202103105
228. Huang D, Xu B, Liu L, Wu L, Zhu Y, Ghanbarpour A, et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab (2021) 33:1655–1670.e8. doi: 10.1016/j.cmet.2021.05.006
229. Zhao YG, Chen Y, Miao G, Zhao H, Qu W, Li D, et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell (2017) 67:974–989.e6. doi: 10.1016/j.molcel.2017.08.005
230. Ji M, Li M, Sun L, Zhao H, Li Y, Zhou L, et al. VMP1 and TMEM41B are essential for DMV formation during β-coronavirus infection. J Cell Biol (2022) 221. doi: 10.1083/jcb.202112081
231. Schneider WM, Luna JM, Hoffmann HH, Sánchez-Rivera FJ, Leal AA, Ashbrook AW, et al. Genome-scale identification of SARS-coV-2 and pan-coronavirus host factor networks. Cell (2021) 184:120–132.e14. doi: 10.1016/j.cell.2020.12.006
232. Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, et al. Genetic screens identify host factors for SARS-coV-2 and common cold coronaviruses. Cell (2021) 184:106–119.e14. doi: 10.1016/j.cell.2020.12.004
233. Ji M, Li M, Sun L, Deng H, Zhao YG. DMV biogenesis during β-coronavirus infection requires autophagy proteins VMP1 and TMEM41B. Autophagy (2022) 00:1–2. doi: 10.1080/15548627.2022.2103783
234. Kang YL, Chou YY, Rothlauf PW, Liu Z, Soh TK, Cureton D, et al. Inhibition of PIKfyve kinase prevents infection by zaire ebolavirus and SARS-coV-2. Proc Natl Acad Sci U.S.A. (2020) 117:20803–13. doi: 10.1073/pnas.2007837117
235. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-coV-2 on virus entry and its immune cross-reactivity with SARS-coV. Nat Commun (2020) 11. doi: 10.1038/s41467-020-15562-9
236. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. The global phosphorylation landscape of SARS-coV-2 infection. Cell (2020) 182:685–712.e19. doi: 10.1016/j.cell.2020.06.034
237. Zapatero-Belinchón FJ, Moeller R, Lasswitz L, van Ham M, Becker M, Brogden G, et al. Fluvastatin mitigates SARS-coV-2 infection in human lung cells. iScience (2021) 24. doi: 10.1016/j.isci.2021.103469
238. Zapata-Cardona MI, Flórez-Álvarez L, Zapata-Builes W, Guerra-Sandoval AL, Guerra-Almonacid CM, Hincapié-García J, et al. Atorvastatin effectively inhibits ancestral and two emerging variants of SARS-coV-2 in vitro. Front Microbiol (2022) 13:721103. doi: 10.3389/fmicb.2022.721103
239. Episcopio D, Aminov S, Benjamin S, Germain G, Datan E, Landazuri J, et al. Atorvastatin restricts the ability of influenza virus to generate lipid droplets and severely suppresses the replication of the virus. FASEB J (2019) 33:9516–25. doi: 10.1096/fj.201900428RR
240. Martínez-Gutierrez M, Castellanos JE, Gallego-Gómez JC. Statins reduce dengue virus production via decreased virion assembly. Intervirology (2011) 54:202–16. doi: 10.1159/000321892
241. Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol (2006) 6:358–70. doi: 10.1038/nri1839
242. Kumar GA, Chattopadhyay A. Statin-induced chronic cholesterol depletion switches GPCR endocytosis and trafficking: insights from the serotonin 1A receptor. ACS Chem Neurosci (2020) 11:453–65. doi: 10.1021/acschemneuro.9b00659
243. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discovery (2005) 4:977–87. doi: 10.1038/nrd1901
244. Violi F, Calvieri C, Ferro D, Pignatelli P. Statins as antithrombotic drugs. Circulation (2013) 127:251–7. doi: 10.1161/CIRCULATIONAHA.112.145334
245. Schönbeck U, Libby P. Inflammation, immunity, and HMG-coA reductase inhibitors: statins as antiinflammatory agents? Circulation (2004) 109:II18–26. doi: 10.1161/01.CIR.0000129505.34151.23
246. Zeiser R. Immune modulatory effects of statins. Immunology (2018) 154:69–75. doi: 10.1111/imm.12902
247. Tan WYT, Young BE, Lye DC, Chew DEK, Dalan R. Statin use is associated with lower disease severity in COVID-19 infection. Sci Rep (2020) 10:17458. doi: 10.1038/s41598-020-74492-0
248. Daniels LB, Sitapati AM, Zhang J, Zou J, Bui QM, Ren J, et al. Relation of statin use prior to admission to severity and recovery among COVID-19 inpatients. Am J Cardiol (2020) 136:149–55. doi: 10.1016/j.amjcard.2020.09.012
249. Zhang X-J, Qin J-J, Cheng X, Shen L, Zhao Y-C, Yuan Y, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab (2020) 32:176–187.e4. doi: 10.1016/j.cmet.2020.06.015
250. Masana L, Correig E, Rodríguez-Borjabad C, Anoro E, Arroyo JA, Jericó C, et al. EFFECT oF STATIN THERAPY oN SARS-CoV-2 INFECTION-RELATED. Eur Heart J Cardiovasc Pharmacother (2020) 8:pvaa128. doi: 10.1093/ehjcvp/pvaa128
251. Diaz-Arocutipa C, Melgar-Talavera B, Alvarado-Yarasca Á, Saravia-Bartra MM, Cazorla P, Belzusarri I, et al. Statins reduce mortality in patients with COVID-19: an updated meta-analysis of 147 824 patients. Int J Infect Dis (2021) 110:374–81. doi: 10.1016/j.ijid.2021.08.004
252. Liu C, Yan W, Shi J, Wang S, Peng A, Chen Y, et al. Biological actions, implications, and cautions of statins therapy in COVID-19. Front Nutr (2022) 9:927092. doi: 10.3389/fnut.2022.927092
253. Mitacchione G, Schiavone M, Curnis A, Arca M, Antinori S, Gasperetti A, et al. Impact of prior statin use on clinical outcomes in COVID-19 patients: data from tertiary referral hospitals during COVID-19 pandemic in Italy. J Clin Lipidol (2021) 15:68–78. doi: 10.1016/j.jacl.2020.12.008
254. Ayeh SK, Abbey EJ, Khalifa BAA, Nudotor RD, Osei AD, Chidambaram V, et al. Statins use and COVID-19 outcomes in hospitalized patients. PloS One (2021) 16:1–11. doi: 10.1371/journal.pone.0256899
255. Khalaji A, Behnoush AH, Alilou S, Rezaee M, Peiman S. Adjunctive therapy with lipid - Lowering agents in COVID - 19 : A systematic review and meta - Analysis of randomized controlled trials. Lipids Health Dis (2023) 22:1–14. doi: 10.1186/s12944-023-01828-w
256. Xavier DP, Cavalcante G, Chagas L, Gisele L, Gomes F, Ferri-guerra J, et al. Effects of statin therapy in hospitalized adult COVID-19 patients : A systematic review and meta-analysis of randomized controlled trials. einstein (São Paulo) (2023) 21:1–7. doi: 10.31744/einstein
257. Zein AFMZ, Sulistiyana CS, Khasanah U, Wibowo A, Lim MA, Pranata R. Statin and mortality in COVID-19: A systematic review and meta-analysis of pooled adjusted effect estimates from propensity-matched cohorts. Postgrad Med J (2022) 98:503–8. doi: 10.1136/postgradmedj-2021-140409
258. Kow CS, Hasan SS. The association between the use of statins and clinical outcomes in patients with COVID-19: A systematic review and meta-analysis. Am J Cardiovasc Drugs (2022) 22:167–81. doi: 10.1007/s40256-021-00490-w
259. Lao US, Law CF, Baptista-Hon DT, Tomlinson B. Systematic review and meta-analysis of statin use and mortality, intensive care unit admission and requirement for mechanical ventilation in COVID-19 patients. J Clin Med (2022) 11. doi: 10.3390/jcm11185454
260. Alhallak I, Paydak H, Mehta JL. Prior statin vs in-hospital statin usage in severe COVID-19: review and meta-analysis. Curr Probl Cardiol (2023) 48. doi: 10.1016/j.cpcardiol.2023.101810
261. Schloer S, Brunotte L, Goretzko J, Mecate-Zambrano A, Korthals N, Gerke V, et al. Targeting the endolysosomal host-SARS-coV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg Microbes Infect (2020) 9:2245–55. doi: 10.1080/22221751.2020.1829082
262. Schloer S, Brunotte L, Mecate-Zambrano A, Zheng S, Tang J, Ludwig S, et al. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-coV-2 infection in vitro. Br J Pharmacol (2021) 178. doi: 10.1111/bph.15418
263. Van Damme E, De Meyer S, Bojkova D, Ciesek S, Cinatl J, De Jonghe S, et al. In vitro activity of itraconazole against SARS-coV-2. J Med Virol (2021) 93:4454–60. doi: 10.1002/jmv.26917
264. Takano T, Akiyama M, Doki T, Hohdatsu T. Antiviral Activity of Itraconazole against Type i Feline Coronavirus Infection. Vet Res (2019) 50:1–6. doi: 10.1186/s13567-019-0625-3
265. Doki T, Tarusawa T, Hohdatsu T, Takano T. In vivo antiviral effects of U18666A against type I feline infectious peritonitis virus. Pathogens (2020) 9:67. doi: 10.3390/pathogens9010067
266. Takano T, Wakayama Y, Doki T. Endocytic pathway of feline coronavirus for cell entry: differences in serotype-dependent viral entry pathway. Pathogens (2019) 8. doi: 10.3390/pathogens8040300
267. Liesenborghs L, Spriet I, Jochmans D, Belmans A, Gyselinck I, Teuwen LA, et al. Itraconazole for COVID-19: preclinical studies and a proof-of-concept randomized clinical trial. EBioMedicine (2021) 66. doi: 10.1016/j.ebiom.2021.103288
268. Lan Q, Wang C, Zhou J, Wang L, Jiao F, Zhang Y, et al. 25-hydroxycholesterol-conjugated EK1 peptide with potent and broad-spectrum inhibitory activity against SARS-coV-2, its variants of concern and other human coronaviruses. Int J Mol Sci (2021) 22:1–10. doi: 10.3390/ijms222111869
269. Marín-Corral J, Rodríguez-Morató J, Gomez-Gomez A, Pascual-Guardia S, Muñoz-Bermúdez R, Salazar-Degracia A, et al. Metabolic signatures associated with severity in hospitalized covid-19 patients. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22094794
270. Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, et al. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: A randomized clinical trial. JAMA - J Am Med Assoc (2020) 324:2292–300. doi: 10.1001/jama.2020.22760
271. Hoertel N, Sánchez-Rico M, Vernet R, Beeker N, Jannot AS, Neuraz A, et al. Association between antidepressant use and reduced risk of intubation or death in hospitalized patients with COVID-19: results from an observational study. Mol Psychiatry (2021) 26:5199–212. doi: 10.1038/s41380-021-01021-4
272. Fritz BA, Hoertel N, Lenze EJ, Jalali F, Reiersen AM. Association between antidepressant use and ED or hospital visits in outpatients with SARS-coV-2. Transl Psychiatry (2022) 12:1–9. doi: 10.1038/s41398-022-02109-3
273. Neuman BW, Angelini MM, Buchmeier MJ. Does form meet function in the coronavirus replicative organelle? Trends Microbiol (2014) 22:642–7. doi: 10.1016/j.tim.2014.06.003
274. Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy (2014) 10:1426–41. doi: 10.4161/auto.29309
275. Takano T, Endoh M, Fukatsu H, Sakurada H, Doki T, Hohdatsu T. The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antiviral Res (2017) 145:96–102. doi: 10.1016/j.antiviral.2017.07.022
276. Gassen NC, Papies J, Bajaj T, Emanuel J, Dethloff F, Chua RL, et al. SARS-coV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-24007-w
277. Cesar-Silva D, Pereira-Dutra FS, Giannini ALM, de Almeida CJG. The endolysosomal system: the acid test for SARS-coV-2. Int J Mol Sci (2022) 23:1–26. doi: 10.3390/ijms23094576
278. Kreutzberger AJB, Sanyal A, Ojha R, Pyle JD, Vapalahti O, Balistreri G, et al. Synergistic block of SARS-coV-2 infection by combined drug inhibition of the host entry factors PIKfyve kinase and TMPRSS2 protease. J Virol (2021) 95. doi: 10.1128/jvi.00975-21
279. Spencer FA, Fonarow GC, Frederick PD, Wright RS, Every N, Goldberg RJ, et al. Early withdrawal of statin therapy in patients with non-ST-segment elevation myocardial infarction: national registry of myocardial infarction. Arch Intern Med (2004) 164:2162–8. doi: 10.1001/archinte.164.19.2162
280. Vuorio A, Kovanen PT, Raal F. Cholesterol-lowering drugs for high-risk hypercholesterolemia patients with COVID-19 while on paxlovid™ Therapy. Future Virol (2022) 17:761–5. doi: 10.2217/fvl-2022-0060
281. Zhuang X, Tsukuda S, Wrensch F, Wing PAC, Borrmann H, Harris JM, et al. Circadian regulation of SARS-coV-2 infection in lung epithelial cells. bioRxiv (2021) 24:33758862. doi: 10.1101/2021.03.20.436163
282. Yu F, Liu Y, Zhang R, Zhu L, Zhang T, Shi Y. Recent advances in circadian-regulated pharmacokinetics and its implications for chronotherapy. Biochem Pharmacol (2022) 203. doi: 10.1016/j.bcp.2022.115185
Keywords: lipid nanodomains, caveolae, caveolin, lipid droplets, coronavirus, SARS-CoV-2, lipid metabolism, inflammation
Citation: Cesar-Silva D, Pereira-Dutra FS, Giannini ALM, Maya-Monteiro CM and de Almeida CJG (2023) Lipid compartments and lipid metabolism as therapeutic targets against coronavirus. Front. Immunol. 14:1268854. doi: 10.3389/fimmu.2023.1268854
Received: 28 July 2023; Accepted: 24 October 2023;
Published: 01 December 2023.
Edited by:
Gan Zhao, University of Pennsylvania, United StatesReviewed by:
Miguel A. Martín-Acebes, Instituto Nacional de Investigación y Tecnología Agroalimentaria (INIA), SpainCopyright © 2023 Cesar-Silva, Pereira-Dutra, Giannini, Maya-Monteiro and de Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecília Jacques G. de Almeida, Y2FsbWVpZGFAaW9jLmZpb2NydXouYnI=; Y2FsbWVpZGFpb2NAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.