 Man Wang
Man Wang Fei Yu
Fei Yu Peifeng Li
Peifeng Li- Institute for Translational Medicine, The Affiliated Hospital of Qingdao University, College of Medicine, Qingdao University, Qingdao, China
The increasing knowledge in the field of oncoimmunology has led to extensive research into tumor immune landscape and a plethora of clinical immunotherapy trials in cancer patients. Immunotherapy has become a clinically beneficial alternative to traditional treatments by enhancing the power of the host immune system against cancer. However, it only works for a minority of cancers. Drug resistance continues to be a major obstacle to the success of immunotherapy in cancer. A fundamental understanding of the detailed mechanisms underlying immunotherapy resistance in cancer patients will provide new potential directions for further investigations of cancer treatment. Noncoding RNAs (ncRNAs) are tightly linked with cancer initiation and development due to their critical roles in gene expression and epigenetic modulation. The clear appreciation of the role of ncRNAs in tumor immunity has opened new frontiers in cancer research and therapy. Furthermore, ncRNAs are increasingly acknowledged as a key factor influencing immunotherapeutic treatment outcomes. Here, we review the available evidence on the roles of ncRNAs in immunotherapy resistance, with an emphasis on the associated mechanisms behind ncRNA-mediated immune resistance. The clinical implications of immune-related ncRNAs are also discussed, shedding light on the potential ncRNA-based therapies to overcome the resistance to immunotherapy.
1 Introduction
Immunotherapy is a type of cancer treatment that harnesses the body’s immune system to target and eliminate cancer cells. Currently, immunotherapy has become a main weapon in fighting cancer (1). Several immunotherapy approaches, such as adoptive cellular immunotherapy and immune checkpoint inhibitor (ICI), have been approved for the treatment of various cancers including melanoma, non-small cell lung cancer (NSCLC) and renal cell carcinoma (RCC) (2). These immunotherapies can achieve long-term tumor regression in subsets of patients. Despite the encouraging improvement in the field of cancer immunotherapy, a significant proportion of cancer patients do not benefit from immunotherapy and some responding patients even undergo relapse after a period of treatment, implying a role of immune resistance (3). The response rates also differ between distinct immunotherapeutic modalities and across cancer types (4). Thus, there is an urgent need to bypass cancer resistance in order to maximize the therapeutic benefits of cancer immunotherapy. Immunotherapy resistance can be generally categorized into intrinsic and acquired resistance (5). Intrinsic resistance, also known as primary resistance, represents a clinical situation where a cancer exhibits low responsiveness to immunotherapy (6). Acquired resistance represents a clinical scenario in which a cancer initially responds to immunotherapy, but later evolves multifarious mechanisms to relapse or progress (6). The mechanisms of cancer immune resistance are very intricate and involve many aspects including genes and metabolism. Particularly, alternation of antitumor immune response pathways and intracellular signaling cascades, abnormal expression of tumor antigens, disruption of antigen presentation processes, formation of an immunosuppressive microniche, functional gene mutations, changes in metabolism in the tumor microenvironment, as well as host-related factors have been associated with immune resistance in cancer (5). An in-depth exploration of immune resistance mechanisms will facilitate the discovery of new therapeutic targets and extend the scope of clinical applications of immunotherapeutic treatments.
Noncoding RNAs (ncRNAs) that account for the vast majority (~98%) of the transcribed genome are a broad and heterogeneous group of RNA molecules in terms of origin, structure, biogenesis and biological function (7, 8). NcRNAs have emerged as ubiquitous players in numerous cellular processes under both the physiological and pathological conditions (9, 10). Based on a length cut-off of 200 nucleotides (nt), ncRNAs are roughly grouped into two classes: short ncRNAs (18-200 nt) and long ncRNAs (lncRNAs, >200 nt) (11, 12). Short ncRNAs include microRNAs (miRNAs), piwi-interacting RNAs (piRNAs) and small nucleolar RNAs (snoRNAs) (13, 14). miRNAs, the most extensively studied type of short ncRNAs, have important roles as regulators of gene expression (15, 16). miRNAs directly bind to the 3’ untranslated region (3’ UTR) of target mRNAs to induce mRNA degradation or in some instances repress protein translation (17). LncRNAs can be further classified into bidirectional transcripts, enhancer RNAs (eRNAs), long intergenic ncRNAs (lincRNAs) and natural antisense transcripts (NATs) (18, 19). Accumulating evidence has demonstrated that lncRNAs act on gene regulation at the epigenetic, transcriptional, post-transcriptional and translational levels via diverse mechanisms (20, 21). Circular RNAs (circRNAs) are recently discovered as a peculiar class of lncRNAs, which form covalently closed continuous loops and are extensively present in mammalian cells (22, 23). CircRNAs can be generally divided into exonic circRNAs (ecircRNAs), circular intronic RNAs (ciRNAs), exon-intron circRNAs (EIciRNAs), and intergenic circRNAs (24–26). CircRNAs act as molecular sponges for miRNAs and proteins, modulators of alternative splicing and transcription and peptide/protein translators (27, 28).
Currently, ncRNAs have become a new research hotspot in the field of cancer immunotherapy. A growing body of evidence has manifested that miRNAs, lncRNAs and circRNAs have a role in regulating the diverse stages of tumor immunity through modulation of the equilibrium between immune activation and immunosuppression (29, 30). These ncRNAs are involved in mediating the communication between cancer cells and immune cells and represent crucial resistance mechanisms that can abrogate the effect of immunotherapy (31, 32). Therefore, ncRNAs may be prospective therapeutic targets to improve the success of immunotherapy in cancer patients. In this review, we present the current state of knowledge implicating miRNAs, lncRNAs and circRNAs in shaping cancer resistance to immunotherapy, with a particular focus on the underlying mechanisms. We also discuss their potential in developing efficacious therapeutic agents for cancer treatment.
2 Cancer immunotherapy
Cancer immunotherapy eliminates cancer cells by various steps in cancer-immunity cycle including antigen presentation, T cell priming and activation and immune cell-mediated cytotoxic effects (33). Tumor-specific T cells can be activated through recognition of a major histocompatibility complex (MHC)-bound cancer epitope by the T cell receptor (TCR) or the interaction between co-stimulatory receptors (e.g., CD28) on T cells and their corresponding ligands (e.g., B7 molecules) on activated antigen-presenting cells (34). Co-stimulatory (e.g., CD27 and OX40) and co-inhibitory molecules (e.g., cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death protein-1 (PD-1)) play a pivotal role in T cell function. The dynamic interaction between stimulatory and inhibitory signals on T cells controls the extent of immune activation to ensure tolerance to self-antigens (inhibitory), while mounting an effective adaptive immune response to exogenous antigens (stimulatory) (35). The engagement of co-inhibitory checkpoint molecules such as CTLA-4 and PD-1 by their respective counterparts can block T cell expansion and activation, culminating in the immune evasion of cancer cells (36) (Figure 1).
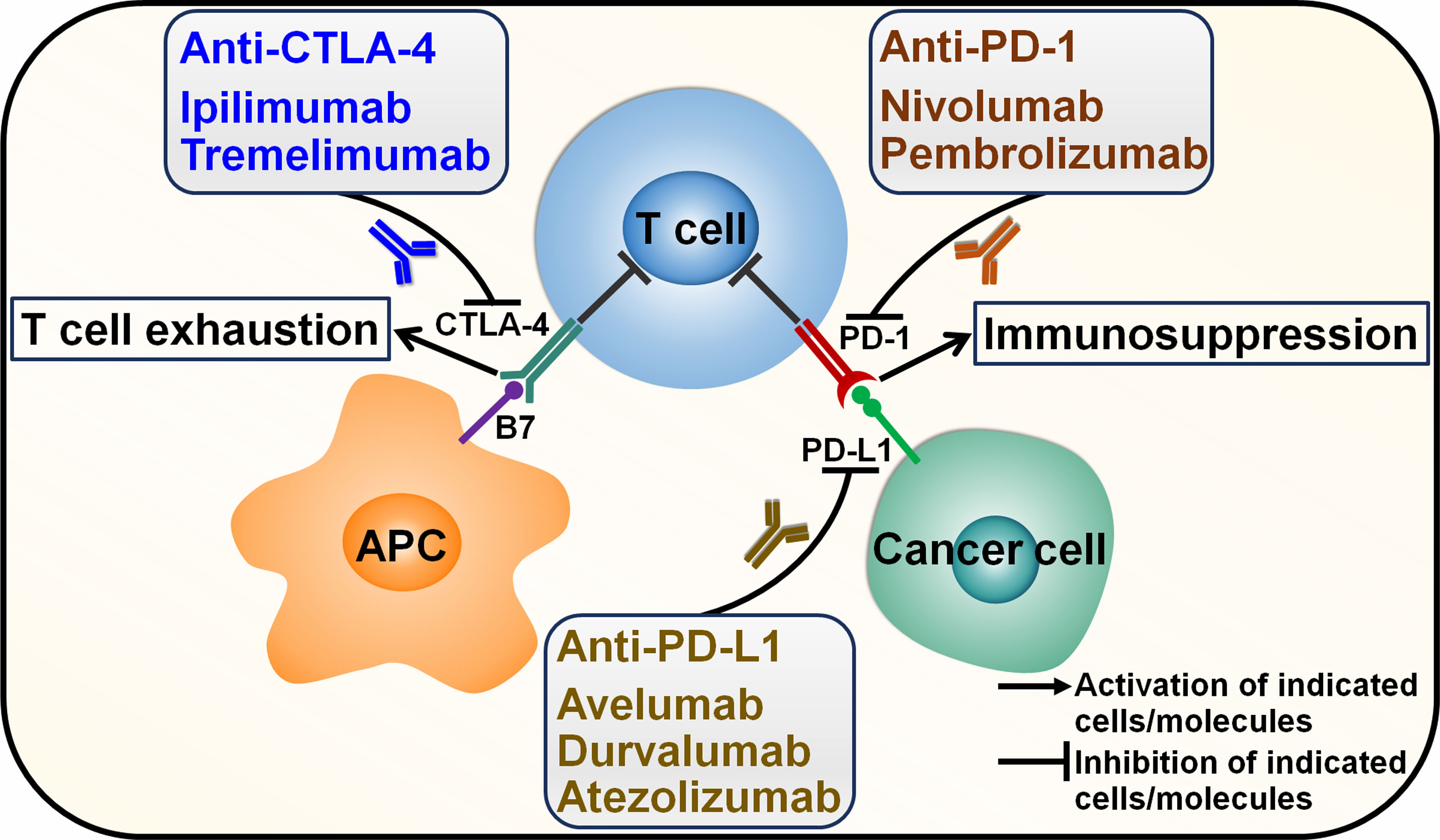
Figure 1 Mechanisms of action of immune checkpoint inhibitors. Cancer cells secrete cancer-associated antigens that are captured by APCs through the MHC-I molecule. APCs then activate T cells, which in turn kill cancer cells. However, cancer cells have evolved multifarious mechanisms to evade immune surveillance. Particularly, immune checkpoints that are expressed on cancer cells and cancer-specific lymphocytes can inhibit T cell activation. The most extensively studied immune checkpoints are CTLA-4, PD-L1 and PD-1. Activation of T cells requires the interaction between CD28 expressed on T cells and B7 on APC. CTLA-4 on T cells binds B7, competing with CD28, and suppresses T cell activation. PD-1 is an inhibitory receptor expressed on T cells. The interaction between PD-1 and its ligand PD-L1 induces T cell exhaustion. Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that specifically target immune checkpoints. ICIs can impede the inhibitory signals and reinvigorate anticancer immune responses. Two monoclonal antibodies ipilimumab and tremelimumab have been developed to inhibit CTLA-4. Nivolumab and pembrolizumab are monoclonal antibodies that target PD-1. Monoclonal antibodies against PD-L1 include avelumab, durvalumab and atezolizumab. CTLA-4, cytotoxic T lymphocyte-associated antigen-4; APC, antigen-presenting cell; PD-1, programmed cell death protein-1; PD-L1, programmed cell death-ligand 1.
ICI therapy functions to impede immune checkpoint-mediated suppression and thus rescues the activity of tumor-specific T cells (37). The introduction and clinical implementation of T cell-directed immunomodulators blocking immune checkpoints CTLA-4, programmed cell death-ligand 1 (PD-L1) and PD-1 have rejuvenated the field of tumor immunology and revolutionized cancer treatment far beyond their impressive clinical activity. ICI immunotherapies alone or in combination with classical therapeutic approaches (e.g., chemotherapy and radiotherapy) are assigned as first-line or second-line treatment for approximately 50 cancer types (37). CTLA-4 inhibitors, ipilimumab and tremelimumab, received approval for use in different cancers, including colorectal cancer (CRC), melanoma, mesothelioma, NSCLC and RCC (38). PD-1/PD-L1-targeting antibodies have become some of the most commonly prescribed cancer treatments. Anti-PD-L1 antibodies avelumab and durvalumab received approval for the treatment of RCC, urothelial carcinoma, advanced bladder cancer and NSCLC (39). Atezolizumab is an anti-PD-L1 antibody used in the treatment of advanced bladder cancer and urothelial cancer (40, 41). The monoclonal antibody targeting PD-1, nivolumab, could be used in the treatment of several advanced cancers, such as Hodgkin’s lymphoma, melanoma, NSCLC, RCC, squamous head and neck cancer and urothelial carcinoma (42). The anti-PD-1 antibody pembrolizumab combined with lenvatinib was approved in treating patients with bladder cancer and endometrial cancer (EC) (43, 44). It is worth noting that a significant proportion of cancer patients receiving immunotherapy do not derive clinical benefit (45). Further development of alternative immune checkpoint-targeting agents is thus critical. The full understanding of the sophisticated mechanisms behind cancer immune escape will hopefully facilitate identification of more effective immunotherapeutic approaches to treat cancer.
3 Mechanisms of action of noncoding RNAs in cancer immune resistance
3.1 MicroRNAs
3.1.1 Coordination of immune cell function
Immunosuppressive cells including myeloid-derived suppressive cells (MDSCs) and tumor-associated macrophages (TAMs) are crucial factors associated with immune resistance (46). Cytokines and signals released by immunosuppressive cells can foster cancer progression and immune evasion. Targeting immunosuppressive cells is a promising therapy to overcome immune resistance. miRNAs may affect the therapeutic effects of immunotherapy by regulating the activity of immunosuppressive cells (Figure 2). The expression level of miR-449c was increased in myeloid progenitor cells of melanoma-bearing mice in response to activation of C-X-C motif chemokine receptor 2 (CXCR2) (47). Overexpressed miR-449c promoted the expansion of monocytic MDSCs (mo-MDSCs) by downregulating signal transducer and activator of transcription 6 (STAT6) (Table 1). Conversely, deficiency of miR-449c repressed differentiation of myeloid progenitor cells into mo-MDSCs. MDSCs are found to play a role in blunting antitumor immunity and enhancing cancer resistance to immunotherapy (58). Thus, it is necessary to verify whether miR-449c can regulate melanoma cell response to immunotherapy.
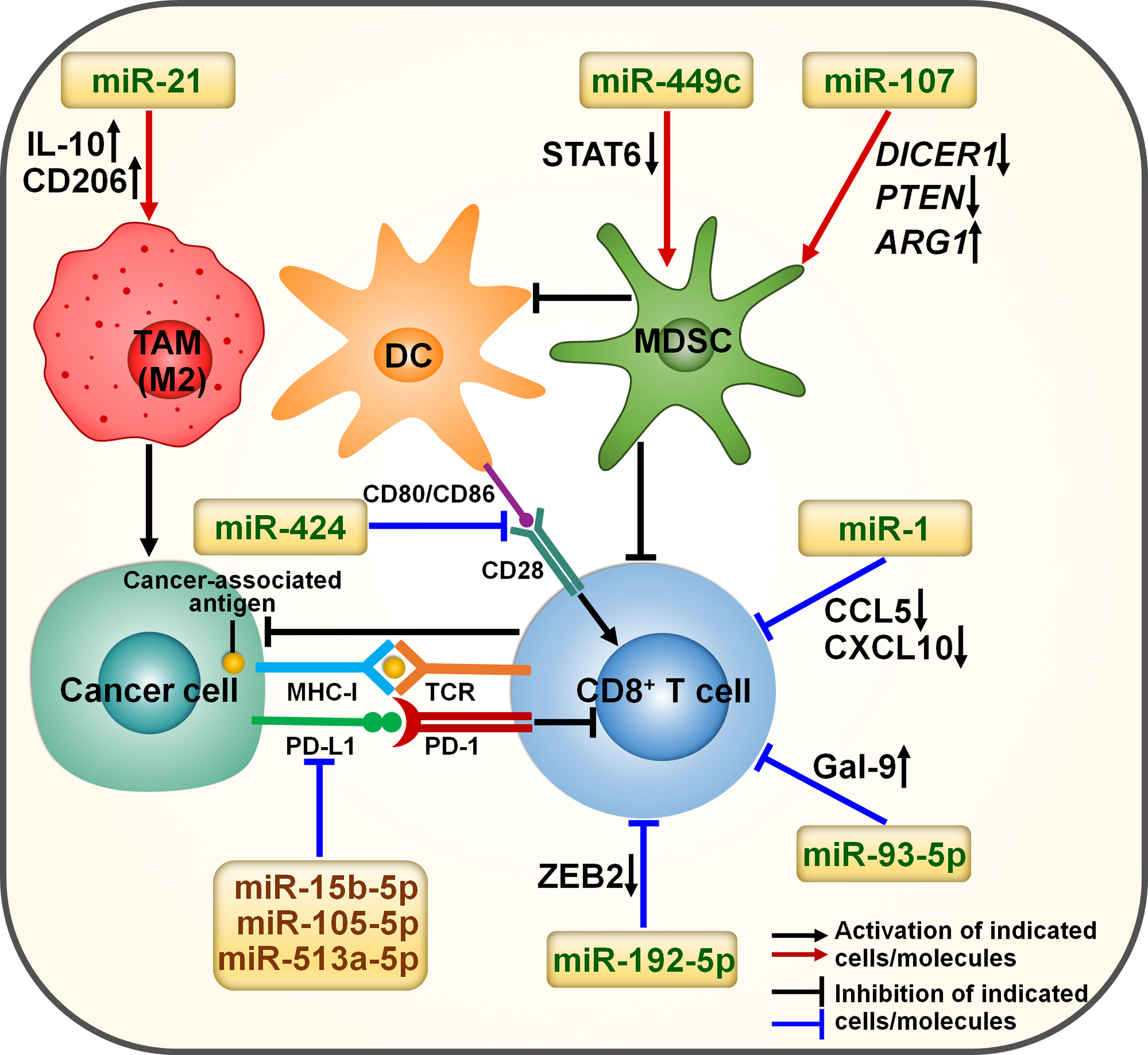
Figure 2 miRNA involvement in immunotherapy resistance in cancer. miRNAs can affect the efficacy of immunotherapy by orchestrating the tumor immune microenvironment. Cancer-derived exosomal miR-21 elevates the expression of IL-10 and CD206, thus promoting M2-like macrophage polarization. Exosomal miR-424 can suppress the CD28-CD80/86 costimulatory signaling, contributing to the impairment of T cell function. miR-449c promotes the expansion of mo-MDSCs by downregulating STAT6. Exosomal miR-107 induces the expansion and activation of MDSCs by regulating DICER1, PTEN and ARG1. miR-1 represses the migration of CD8+ T cells by reducing the expression of CCL5 and CXCL10. miR-15b-5p, miR-105-5p and miR-513a-5p may promote the activation of CD8+ T cells by targeting PD-L1. miR-192-5p restrains the cytotoxic activity of CD8+ T cells by downregulating ZEB2. miR-93-5p induces CD8+ T cell exhaustion by fostering Gal-9 augmentation. IL-10, interleukin-10; STAT6, signal transducer and activator of transcription 6; DICER1, double-stranded RNA-specific endoribonuclease 1; PTEN, phosphatase and tensin homolog; ARG1, arginase 1; TAM, tumor-associated macrophage; DC, dendritic cell; MDSC, myeloid-derived suppressive cell; MHC-I, major histocompatibility complex class I; TCR, T cell receptor; PD-L1, programmed cell death-ligand 1; PD-1, programmed cell death protein-1; CCL5, C-C motif chemokine ligand 5; CXCL10, C-X-C motif chemokine ligand 10; ZEB2, zinc finger E-box binding homeobox 2; Gal-9, galectin-9.
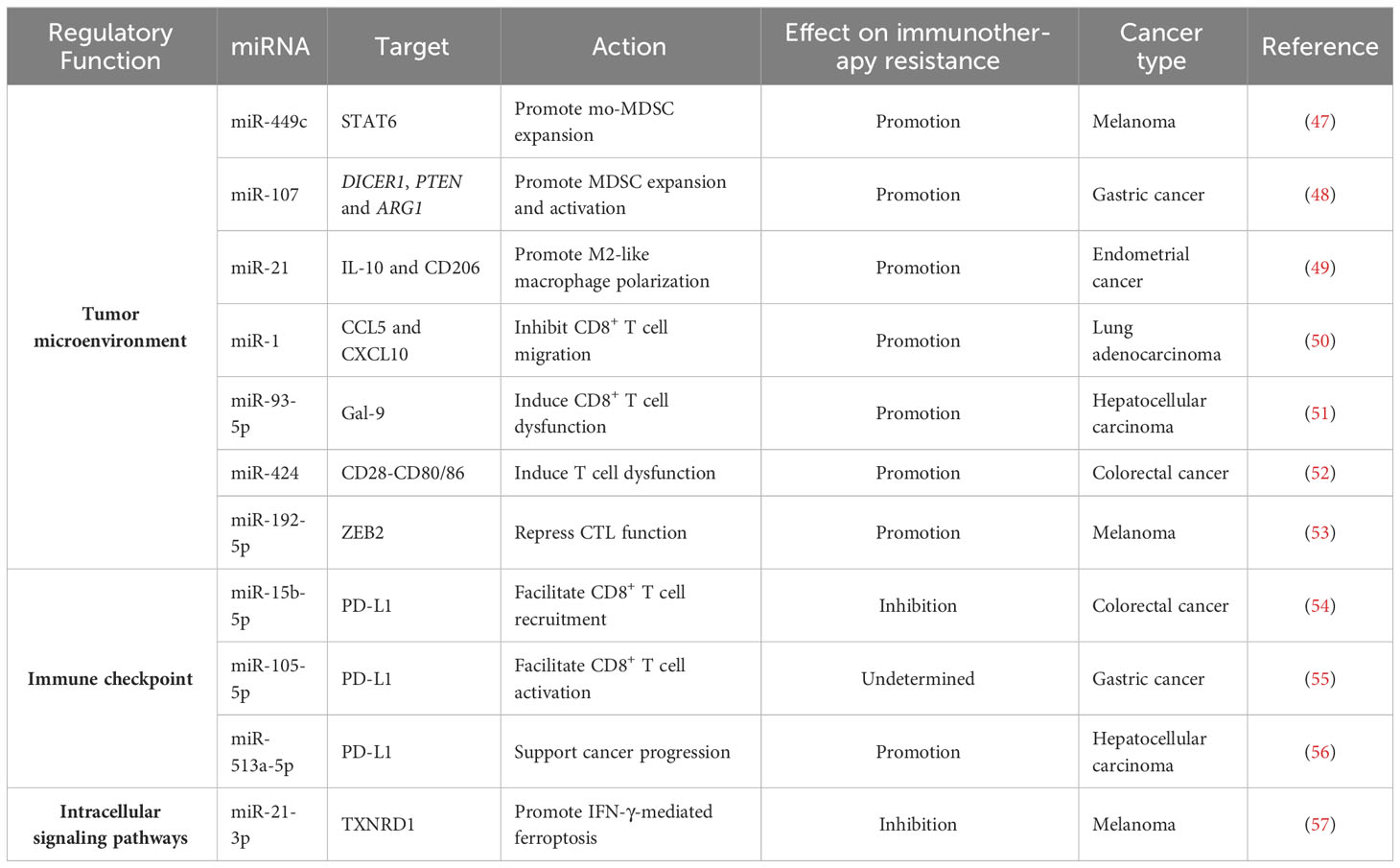
Table 1 Representative miRNAs involved in immunotherapy resistance.
Exosomes are small membrane-enclosed vesicles released by diverse cell types and function as key regulators in cell-to-cell communication via transmitting diverse constituents (e.g., DNAs, lipids, ncRNAs and proteins) (59). Gastric cancer-released exosomes transported miR-107 to MDSCs (48). Exosomal miR-107 promoted the expansion and activation of MDSCs by downregulating tumor suppressors double-stranded RNA-specific endoribonuclease 1 (DICER1) and phosphatase and tensin homolog (PTEN) while upregulating arginase 1 (ARG1). Tumor-derived miR-107 mediated the communication between cancer cells and MDSCs, leading to tumor development and enhancement of immunotherapy resistance. EC cells facilitated monocyte transformation to M2-like TAMs under hypoxic condition (49). Mechanistically, hypoxic EC cells secreted miR-21-containing exosomes, which were taken up by monocytes. Exosome-derived miR-21 increased the expression of interleukin-10 (IL-10) and CD206, hence promoting M2-like macrophage polarization. Exosomal miR-21 reshaped the tumor immune microenvironment, which potentially facilitated EC development. Collectively, miRNAs act as mediators in tumor immune evasion. However, the exact effect of these miRNAs on the anticancer activity of immunotherapy should be further determined.
Cytotoxic CD8+ T cells function to induce apoptosis and efficiently eradicate cancer cells via direct cellular contacts (60, 61). CD8+ T cells have been recognized as the most powerful effectors in antitumor immunity and are the mainstay of successful cancer immunotherapy. Exposure to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) treatment increased miR-1 expression in patients with lung adenocarcinoma (LUAD) (50). The cytokines C-C motif chemokine ligand 5 (CCL5) and C-X-C motif chemokine ligand 10 (CXCL10) can induce migration of intratumoral CD8+ T cells in cancer (62). Upregulated miR-1 inhibited the therapeutic effect of EGFR-TKI and restricted the migration of CD8+ T cells by downregulating CCL5 and CXCL10. miR-1 could be useful in identifying EGFR-TKI-resistant patients who may benefit from immunotherapy. Forced expression of miR-1 may be a mechanism underlying resistance to immunotherapy (e.g., ICI) in LUAD patients. miR-93-5p functioned as a critical initiating oncogene during hepatocarcinogenesis (51). Galectin-9 (Gal-9) causes the inactivation of various immune cells, such as natural killer (NK) cells and T cells, enabling cancer cells to evade from immunosurveillance (63, 64). miR-93-5p induced infiltrated CD8+ T cell dysfunction by favoring Gal-9 augmentation. Anti-Gal-9 antibodies reversed anti-PD-1 therapy resistance in a miR-93-5p-induced hepatocellular carcinoma (HCC) mouse model. The clinical benefit of targeting miR-93-5p and its target Gal-9 in HCC awaits further verification.
Hypoxia caused the enrichment of miR-424 in CRC-derived exosomes, which were subsequently transferred to tumor-infiltrating dendritic cells (DCs) and T cells (52). Exosome-delivered miR-424 impeded the CD28-CD80/86 costimulatory signaling in these immune cells, contributing to the formation of an immunosuppressive tumor microenvironment. Deficiency of tumor-derived miR-424 suppressed CRC development in a murine tumor model. In contrast, intratumoral inoculation of miR-424-containing exosomes accelerated tumor growth by blunting CD28-CD80/86-dependent antitumor immunity. The CD28-CD80/86 costimulatory pathways could repress PD-1-mediated T cell dysfunction and positively correlated with patient response to anti-PD-1 treatment (65–67). It was thus inferred that miR-424 was able to affect immunotherapy response in CRC. Consistently, specifically blocking tumor-derived functional miR-424 restored adaptive antitumor immunity and re-sensitized CRC cells to anti-PD-1/CLTA-4 treatment. Modification of immunosuppressive miRNA-containing exosomes could be developed as an adjuvant treatment for ICI-resistant CRC. Substantial research efforts should be made to comprehensively explore immune-related miRNAs in cancer cells and their derived exosomes.
Gap junctional shuttling of ncRNAs represents a vital mechanism of immunosuppression. miR-192-5p was strikingly enriched in hypoxic melanoma cells (53). The connexin-43 (Cx43)-constituted gap junctions mediated the transfer of miR-192-5p to DCs and melanoma-specific cytotoxic T lymphocytes (CTLs). Moreover, the delivery of miR-192-5p inhibited the cytotoxic activity of CTLs and contributed to cancer immune escape by downregulating zinc finger E-box binding homeobox 2 (ZEB2), a transcription factor that modulated the expression of terminal effector genes in CD8+ T cells. Altogether, these findings may provide new opportunities for the development of anticancer immunotherapies aimed to improve clinical outcomes of cancer patients.
3.1.2 Regulation of inhibitory immune checkpoint-mediated immune evasion
The PD-1/PD-L1-based pathway has become a pivotal immune checkpoint in recent years and is a key target for cancer immunotherapy. However, emerging evidence indicates that the PD-1/PD-L1 pathway has opposite effects on immunotherapy efficacy. On the one hand, reduction of PD-L1 expression via genetic or pharmacological inhibition reinforces the efficacy of anti-PD-1/PD-L1 therapy. The expression level of miR-15b-5p gradually reduced with the development of microsatellite stable (MSS) CRC (54). The protein level of PD-L1 showed an opposite trend. Further study demonstrated that overexpressed miR-15b-5p prevented CRC tumorigenesis and sensitized cancer cells to PD-1 blockade by downregulating PD-L1 at the posttranscriptional level in vivo. Conversely, depletion of miR-15b-5p supported CRC tumorigenesis by upregulating PD-L1 and repressing the recruitment of CD8+ T cells. Increasing miR-15b-5p expression enhanced the efficacy of ICI therapy. The therapeutic effects of combined miR-15b-5p and PD-1 blockade should be verified in further clinical studies. Likewise, miR-105-5p promoted the activation of CD8+ T cells in gastric cancer by directly targeting PD-L1 (55). The expression of miR-105-5p was induced by the hypomethylation of a cancer- and germline-specific promoter of its host gene, GABRA3. miR-105-5p may be a target for gastric cancer immunotherapy, which warrants follow-up studies.
On the other hand, downregulation of PD-L1 mediates the resistant response to ICI (68). miR-513a-5p promoted proliferation, invasion and migration of HCC cells by reducing PD-L1 expression, leading to increased resistance to anti-PD-1 therapy in HCC (56). Specifically, β-glucuronidase-mediated inhibition of miR-513a-5p inhibited HCC progression and improved the efficacy of anti-PD-1 treatment in murine tumor model. Antagonism of miR-513a-5p may be an effective approach for improving the therapeutic potential of anti-PD-1 treatment. The addition of ncRNA-based therapies targeting a large variety of immune checkpoints would pave the way for surmounting the resistance to immunotherapy and enable better clinical outcomes in cancer patients.
3.1.3 Modulation of cell death pathways
Ferroptosis, an iron-dependent form of regulated necrosis, is mainly initiated by the lethal accumulation of lipid peroxidation catalyzed by cellular labile free iron (69). Cancer cell ferroptosis induced by interferon-γ (IFN-γ) released by tumor-infiltrating CD8+ T cells potentiates the efficacy of immunotherapy (70). The involvement of miRNAs in ferroptosis suggests their regulatory effects on the therapeutic effect of immunotherapy. Thioredoxin reductase 1 (TXNRD1) acts as a negative regulator of ferroptosis (71). miR-21-3p promoted IFN-γ-mediated ferroptosis by downregulating TXNRD1 to impel lipid peroxidation (57) (Figure 3). Importantly, miR-21-3p overexpression sensitized melanoma cells to anti-PD-1 therapy through promotion of ferroptosis. Nanoparticle delivery of miR-21-3p was proven to synergize with anti-PD-1 antibody without obvious adverse effects in a preclinical mouse model. Nanoparticle delivery of ferroptosis-related ncRNAs may have translational potential in cancer immunotherapy, which deserves more detailed investigations.
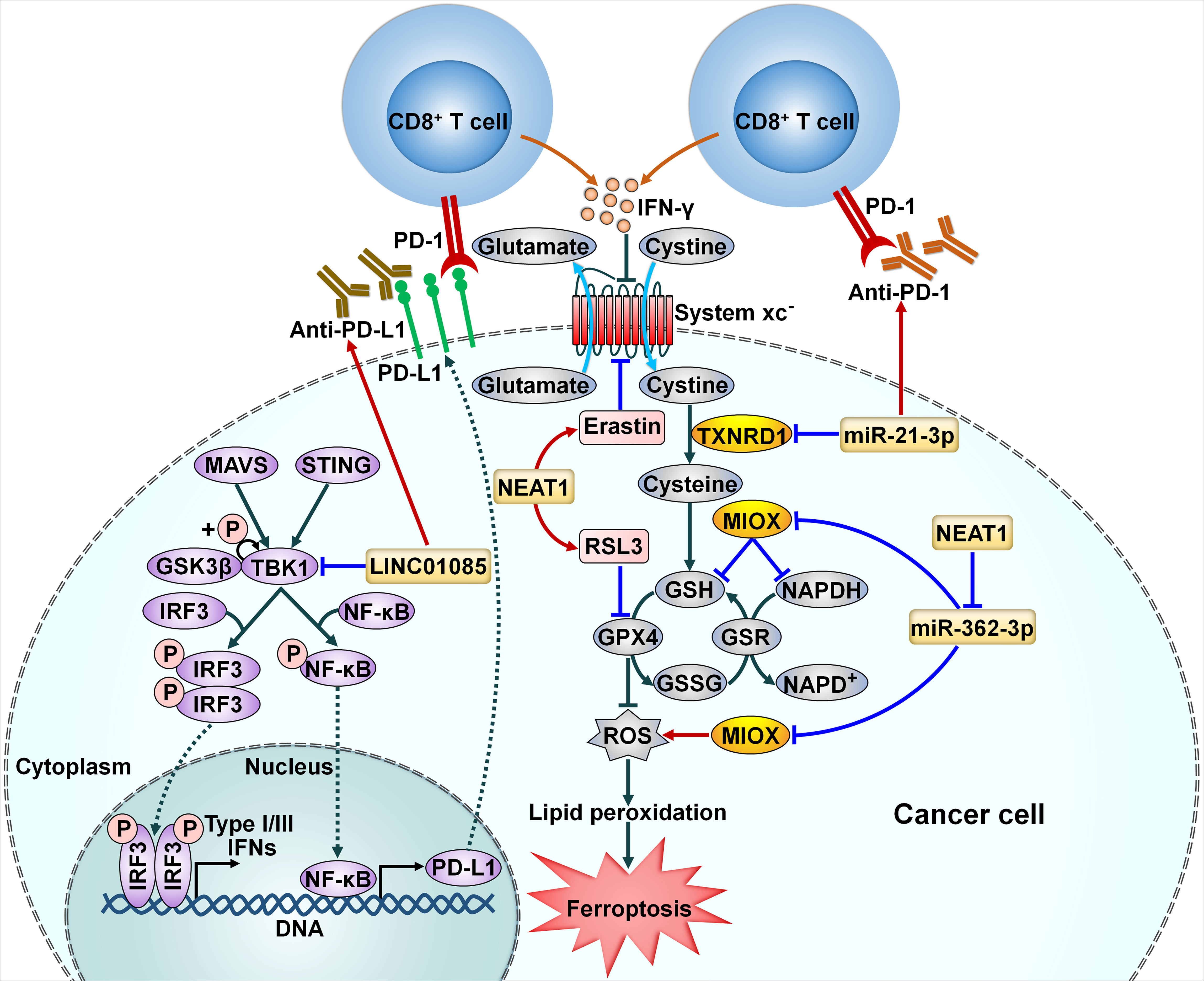
Figure 3 Implication of ncRNAs in cellular signaling pathways related to cancer immune resistance. NcRNAs can affect the efficacy of immunotherapy through modulation of cancer cell ferroptosis. miR-21-3p accelerates IFN-γ-mediated ferroptosis by downregulating TXNRD1 to impel lipid peroxidation. NEAT1 increases MIOX expression by sponging miR-362-3p. This event enhances ROS production and decreases the intracellular levels of NADPH and GSH, eventually leading to cancer cell ferroptosis. Moreover, NEAT1 reinforces the anticancer potency of ferroptosis activators erastin and RSL3. LINC01085 inhibits the phosphorylation of TBK1 by abating its interaction with GSK3β. Accordingly, LINC01085 blocks the activation of the STING/MAVS/IRF3 signaling pathway, resulting in downregulation of NF-κB and PD-L1 and decreased production of type I/III IFNs. LINC01085 can act in synergy with anti-PD-L1 treatment. PD-L1, programmed cell death-ligand 1; PD-1, programmed cell death protein-1; IFN-γ, interferon-γ; MAVS, mitochondrial antiviral signaling protein; STING, stimulator of interferon genes; GSK3β, glycogen synthase kinase 3β; TKB1, TANK-binding kinase 1; IRF3, interferon regulatory factor 3; NF-κB, nuclear factor-κB; TXNRD1, thioredoxin reductase 1; RSL3, RAS-selective lethal 3; MIOX, myo-inositol oxygenase; GSH, glutathione; NAPDH, nicotinamide adenine dinucleotide phosphate; GPX4, glutathione peroxidase 4; GSR, glutathione reductase; GSSG, oxidized glutathione; ROS, reactive oxygen species.
3.2 Long noncoding RNAs
3.2.1 Blockade of antigen presentation
LncRNAs play important roles in cancer immunotherapy resistance. Disruption of the antigen presentation pathway forms a significant mechanism of cancer resistance to immunotherapy. LncRNAs are vital regulators of the antigen presentation process (Figure 4). The lncRNA LINK-A exhibited a tissue-specific expression pattern in mammary glands (72). This lncRNA promoted the interaction between phosphatidylinositol- (3,4,5)-triphosphate (PIP3) and inhibitory G-protein-coupled receptor (GPCR) pathways, resulting in the inactivation of the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling pathway (Table 2). This led to the inhibition of PKA-mediated phosphorylation of an E3 ubiquitin ligase tripartite motif-containing 71 (TRIM71). Hypophosphorylated TRIM71 facilitated K48-linked polyubiquitination and proteasome-mediated degradation of the antigen peptide-loading complex (PLC) components (e.g., tapasin, transporter associated with antigen processing 1 (TAP1), TAP2, and calreticulin (CALR)) and inherent tumor suppressors Rb and p53. Thus, LINK-A acted to attenuate intrinsic tumor suppressor barriers via the GPCR/PKA/TRIM71 signaling cascade, forming a resistance strategy for immunological cancer escape. Knockdown of LINK-A markedly improved the protein stability of the PLC components and MHC class I (MHC-I) complex and enhanced the infiltration of CD8+ T cells into the tumor tissue, hence sensitizing breast cancer cells to ICI.
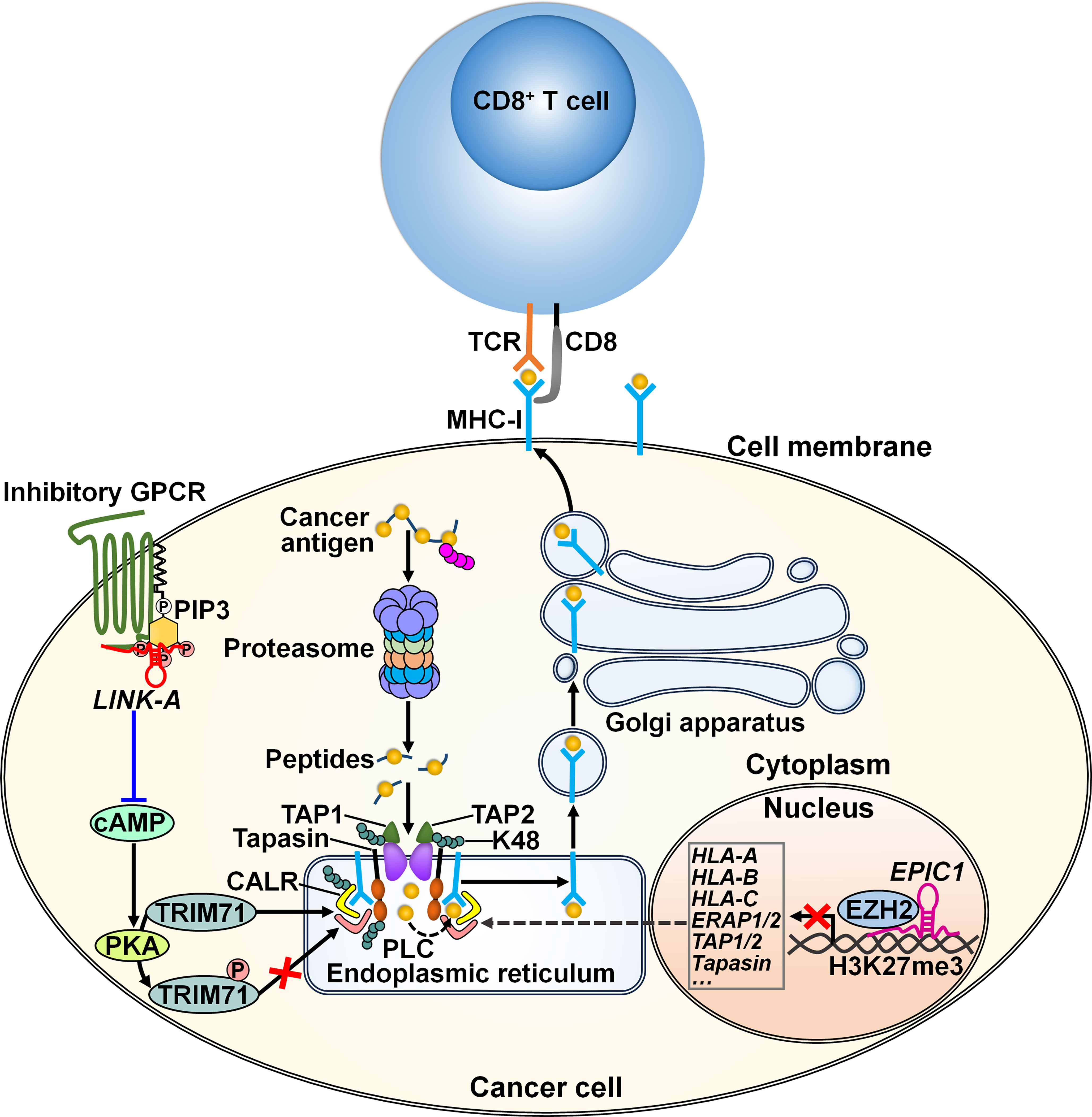
Figure 4 Regulatory mechanisms of lncRNAs in cancer cell antigen presentation. The lncRNA LINK-A supports the interaction between inhibitory GPCR and PIP3 pathways, leading to downregulation of cAMP and consequent inhibition of PKA-mediated TRIM71 phosphorylation. As a result, LINK-A promotes K48-polyubiquitination-mediated degradation of the PLC components including tapasin, TAP1, TAP2, and CALR, constituting a resistance strategy for immunological cancer escape. The linRNA EPIC1 causes the epigenetic silencing of antigen presentation genes (e.g., HLA-A, HLA-B, HLA-C) and antigen processing genes (e.g., ERAP1/2, TAP1/2 and tapasin) through interaction with EZH2. These lncRNAs promotes cancer resistance to immunotherapy. TCR, T cell receptor; MHC-I, major histocompatibility complex class I; GPCR, G-protein-coupled receptor; PIP3, phosphatidylinositol- (3,4,5)-triphosphate; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; TRIM71, tripartite motif-containing 71; TAP1, transporter associated with antigen processing 1; TAP2, transporter associated with antigen processing 2; CALR, calreticulin; PLC, peptide-loading complex; HLA-A, human leukocyte antigen A; HLA-B, human leukocyte antigen B; HLA-C, human leukocyte antigen C; ERAP1/2, endoplasmic reticulum aminopeptidase 1/2; EZH2, zeste homolog 2.
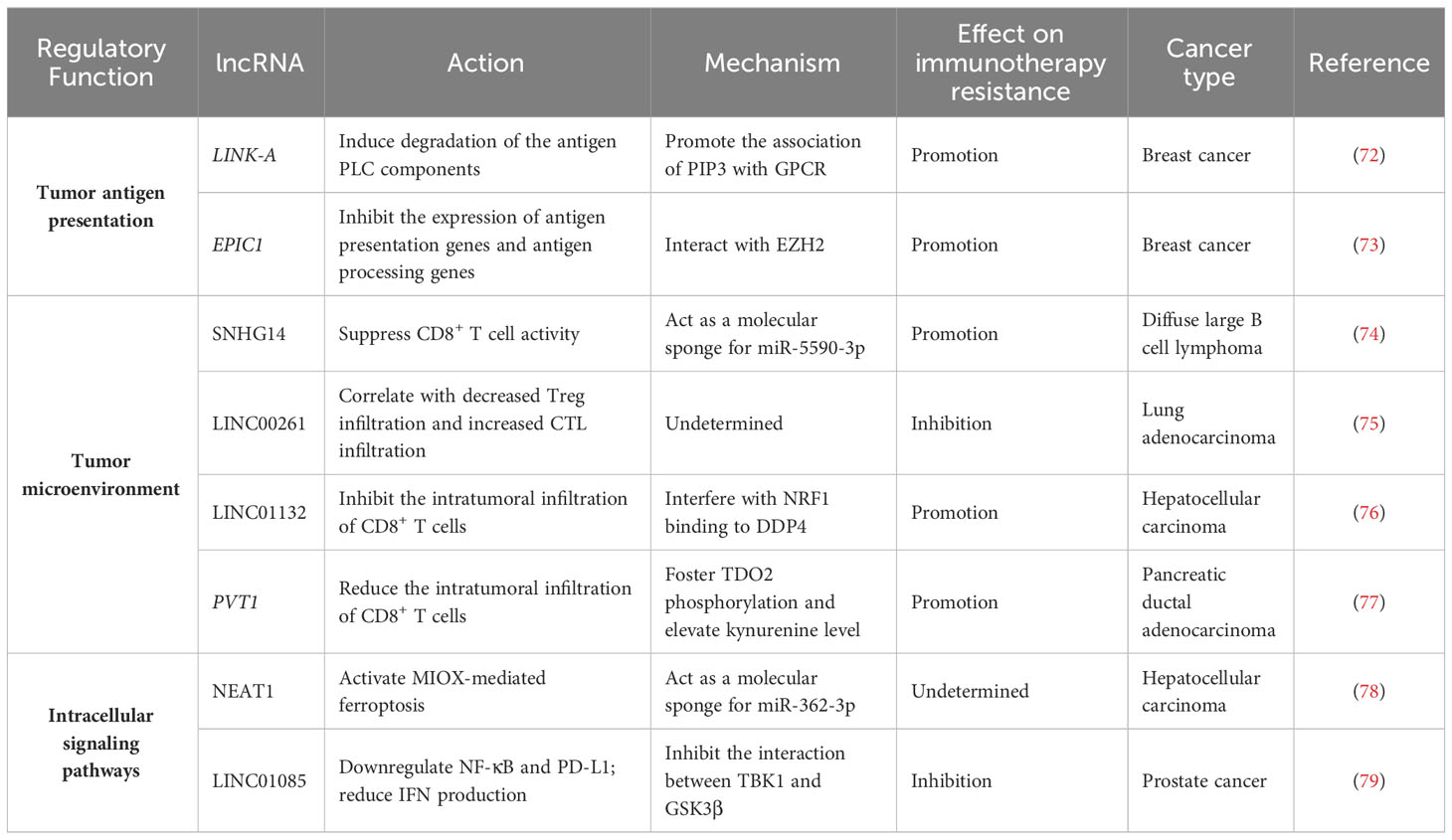
Table 2 Representative lncRNAs associated with cancer immune response.
The long intergenic ncRNA (linRNA) EPIC1 could interact with the histone methyltransferase enhancer of zeste homolog 2 (EZH2), resulting in the epigenetic silencing of antigen presentation genes (e.g., human leukocyte antigen A (HLA-A), HLA-B, HLA-C), antigen processing genes (e.g., endoplasmic reticulum aminopeptidase 1/2 (ERAP1/2), TAP1/2 and tapasin) as well as the IFN-γ receptor IFNGR1 (73). Consequently, EPIC1 induced breast cancer resistance to ICI therapy through inhibition of antigen presentation and the IFN-γ/Janus kinase (JAK)/STAT1 pathways. Oppositely, knockdown of EZH2 counteracted the tumor-promoting effect of EPIC1. The linRNA EPIC1 may serve as a potential target for cancer immunotherapy. The mechanisms by which EPIC1 regulates the interaction between EZH2 and its target genes warrant further study.
3.2.2 Regulation of T cell activity
Cancer immunotherapy predominantly relies on either activating or restoring the function of CD8+ T cells (80). LncRNAs can coordinate the activation of T cells and thus affect the immunotherapy response (Figure 5). LncRNA small nucleolar RNA host gene 14 (SNHG14) served as a tumor promoter in diffuse large B cell lymphoma (DLBCL) (74). SNHG14 increased ZEB1 expression to activate PD-1 by sponging miR-5590-3p. Consequently, SNHG14/miR-5590-3p repressed CD8+ T cell activity and promoted DLBCL progression through ZEB1-mediated activation of PD-1/PD-L1 immune checkpoint. Particularly, the supplementation of anti-PD-1 or anti-PD-L1 reversed the effect of SNHG14 overexpression on CD8+ T cell activity. Depletion of SNHG14 potentially blocks immune evasion and enhances the effectiveness of immunotherapy in DLBCL through regulating the PD-1/PD-L1 axis.
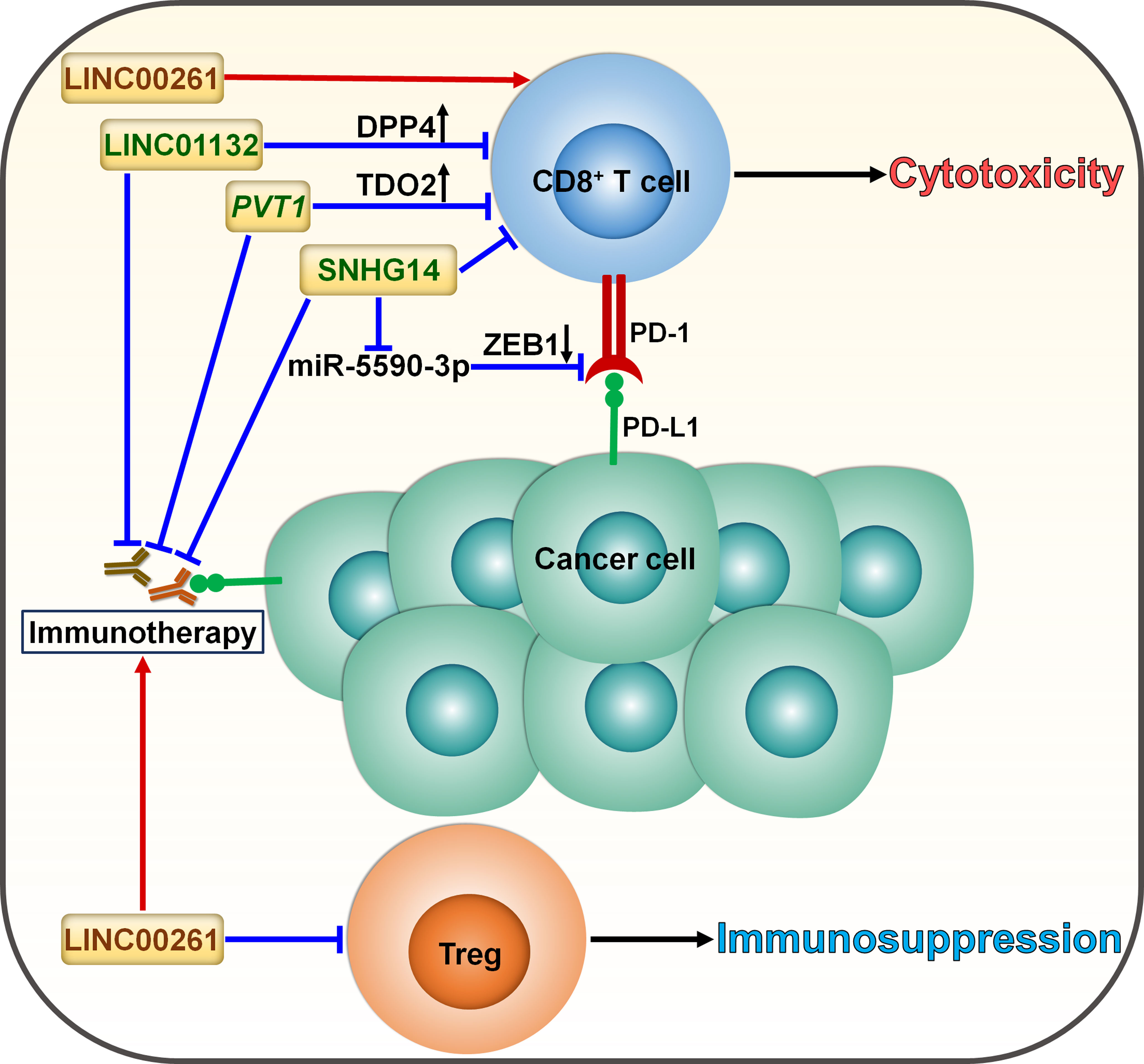
Figure 5 Roles of lncRNAs in skewing T cell balance in cancer. LncRNAs serve as momentous factors influencing the CD8+ T cell/Treg equilibrium. LINC01132 impairs the intratumoral infiltration of CD8+ T cells to compromise antitumor immunity by promoting DPP4 expression. PVT1 inhibits the infiltration and cytotoxicity of CD8+ T cells by provoking TDO2. SNHG14 restricts CD8+ T cell activity and accelerates cancer progression via ZEB1-mediated activation of PD-1/PD-L1 immune checkpoint by sponging miR-5590-3p. These lncRNAs facilitate cancer immune evasion and dampen the effectiveness of immunotherapy through induction of T cell dysfunction. LINC00261 suppresses the expansion of tumor-promoting Tregs and tips the balance toward tumor suppression, potentially strengthening the tumor-killing effect of immunotherapy. DPP4, dipeptidyl peptidase 4; TDO2, tryptophan 2,3-dioxygenase; ZEB1, zinc finger E-box binding homeobox 1; PD-1, programmed cell death protein-1; PD-L1, programmed cell death-ligand 1; Treg, regulatory T cell.
Cyclin-dependent kinase 1 (CDK1)-related lncRNA, LINC00261, was significantly downregulated in LUAD compared with normal tissues and was correlated with poor prognosis in LUAD (75). The level of IL-8, mainly derived from macrophages, dramatically elevated in ICI-resistant patients, which could be ascribed to the downregulation of LINC00261 in LUAD. These results suggested the interrelation between the LINC00261 level and immunotherapy resistance in LUAD. In addition, reduced expression of LINC00261 was likely to correlate with increased infiltration of regulatory T cells (Tregs) and decreased infiltration of cytotoxic T cells, eventually leading to immunosuppression. Enforced expression of LINC00261 may improve the effectiveness of immunotherapy, which is worthy of further study. Concerted research efforts are required to reveal the mechanisms through which LINC00261 affects the responsiveness to immunotherapy in LUAD.
The expression level of LINC01132 was remarkably higher in HCC tissues than normal tissues and correlated with poor prognosis of HCC patients (76). LINC01132 increased the expression of dipeptidyl peptidase 4 (DPP4) by competitively binding to nuclear factor erythroid 2-related factor 1 (NRF1). Depletion of LINC01132 promoted the intratumoral infiltration of CD8+ T cells and potentiated the efficacy of anti-PD-L1 therapy in HCC-bearing mice via DPP4 inhibition.
Pancreatic ductal adenocarcinoma (PDAC)-produced IL-6 induced the expression of the lncRNA plasmacytoma variant translocation 1 (PVT1) in tumor-associated nonmyelinating Schwann cells (TASc) (77). Tryptophan 2,3-dioxygenase (TDO2), an important enzyme in the kynurenine pathway, fostered cancer progression by inducing CD8+ T cell dysfunction (81). PVT1 fostered TDO2 phosphorylation and enhanced its enzymatic activities in the conversion of tryptophan to kynurenine. As a result, PVT1 reduced CD8+ T cell infiltration and promoted PDAC carcinogenesis (77). TASc inhibition significantly suppressed PDAC growth and enhanced the intratumoral infiltration and cytotoxicity of CD8+ T cells. Reportedly, high levels of kynurenine contribute to immunotherapy resistance in cancer patients (82). As expected, TASc inhibition synergized with anti-PD-1 antibody in PDAC-bearing mice, which could be partially attributable to reduced kynurenine production. It will be important to validate the direct effect of PVT1 on the status of the TME and the efficacy of anti-PD-1 antibody in further studies.
3.2.3 Modulation of cellular signaling pathways
LncRNAs have been implicated in the regulation of multiple pathways through diverse genetic or epigenetic mechanisms (83). LncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) increased the expression of myo-inositol oxygenase (MIOX) in HCC by sponging miR-362-3p (78) (Figure 3). MIOX facilitated the production of reactive oxygen species (ROS) and reduced the intracellular levels of nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), thus promoting ferroptosis. In vitro and in vivo experiments manifested that upregulation of NEAT1 strengthened the anticancer potency of ferroptosis activators erastin and RAS-selective lethal 3 (RSL3) through promotion of ferroptosis. Inducing ferroptosis via NEAT1 overexpression could serve as a prospective therapeutic method for HCC immunotherapy.
LINC01085 was deleted in docetaxel-resistant castration resistant prostate cancer (CRPC) and predicted favorable outcomes of CRPC patients (79). It could competitively interact with TANK-binding kinase 1 (TBK1) and glycogen synthase kinase 3β (GSK3β), hence abating the affinity of TBK1 and GSK3β and reducing TBK1 phosphorylation at the Ser-172 site. These events inhibited the activation of the stimulator of interferon genes (STING)/mitochondrial antiviral signaling protein (MAVS)/IFN regulatory factor 3 (IRF3) signaling pathway, contributing to downregulation of nuclear factor-κB (NF-κB) and PD-L1 as well as reduced production of type I/III IFNs. In vivo experiments indicated that overexpression of LINC01085 in combination with anti-PD-L1 treatment produced a significant tumor-antagonizing effect than anti-PD-L1 treatment alone. Overall, this study provided a scientific rationale to combine LINC01085 agonists with ICI therapy to enhance the therapeutic effect for CRPC in the future.
3.3 Circular RNAs
3.3.1 Induction of immune cell dysfunction
CircRNAs have emerged as key factors that act in an entangled network of interactions with diverse miRNAs or proteins to remodel the tumor immune microenvironment (Figure 6). CircUHRF1 was upregulated in HCC tissues compared with matched adjacent normal tissues, and its high expression indicated NK cell exhaustion and poor prognosis in HCC patients (84). CircUHRF1 was also highly expressed in HCC cell-derived exosomes. T cell immunoglobulin and mucin domain-containing protein-3 (TIM-3) is a major inhibitory receptor on NK cells (85), and NK cells with elevated TIM-3 expression displayed a decreased capacity to initiate antitumor immune responses (86). HCC-secreted exosomal circUHRF1 could be delivered into NK cells, where it restricted NK cell ability to secrete IFN-γ and tumor necrosis factor-α (TNF-α) by targeting miR-449c-5p to enhance TIM-3 expression (84) (Table 3). CircUHRF1 supported HCC development in a NK cell- and exosome-dependent manner. CircUHRF1 deficiency increased HCC cell sensitivity to anti-PD-1 treatment and improved the overall survival rate in a xenograft mouse model. Altogether, circUHRF1 mediates the crosstalk between HCC cells and NK cells, leading to the impairment of NK cell function through upregulation of TIM-3 expression. CircUHRF1 functions as a critical factor affecting resistance to anti-PD-1 treatment. Reduction of circUHRF1 expression in HCC may improve patient outcomes by rescuing NK cell function. Tumor-infiltrating CD8+ T cells also had high expression levels of TIM-3. It is intriguing whether exosomal circUHRF1 could induce T cell dysfunction by regulating the miR-449c-5p/TIM-3 pathway. The involvement of circUHRF1 in the communication between cancer cells and tumor microenvironment is worthy of continued study.
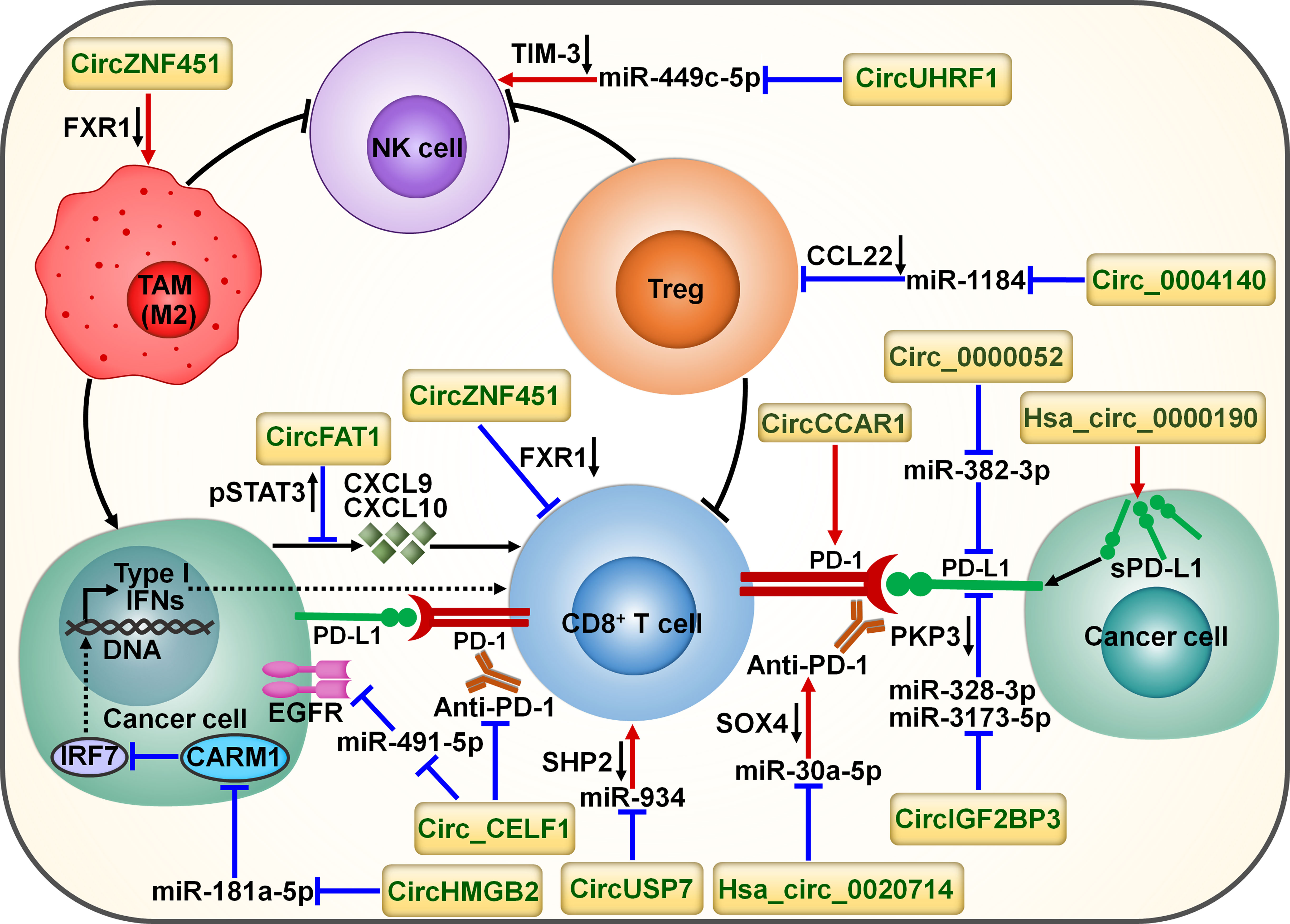
Figure 6 Schematic representation of the interplay between circRNAs and tumor immune microenvironment. Cancer-derived exosomal circZNF451 mediates the degradation of FXR1 in macrophages. It can reshape the immunosuppressive tumor microenvironment by driving macrophage polarization toward the M2-like phenotype as well as dysfunction of cytotoxic CD8+ T cells. Cancer-secreted exosomal circUHRF1 restrains NK cell ability by targeting miR-449c-5p to increase TIM-3 expression. CircUHRF1 attenuates cancer cell sensitivity to anti-PD-1 treatment. Circ_0004140 increases the expression of CCL22 to recruit Tregs in the tumor microenvironment by directly targeting miR-1184. CircFAT1 exerts an inhibitory action on CD8+ T cell activation by stimulating STAT3 to block the secretion of IFN-I-induced chemokines (e.g., CXCL9 and CXCL10). Cancer-secreted exosomal circUSP7 restricted CD8+ T cell function by regulating the miR-934/SHP2 axis. Circ_CELF1 upregulates the oncogene EGFR by acting as a ceRNA for miR-491-5p. Circ_CELF1 can abrogate the anticancer effects of anti-PD-1 therapy. CircHMGB2 increases CARM1 expression by sponging miR-181a-5p, thereby limiting the IFN-I response. Thus, circHMGB2 plays a critical role in the molding of an immunosuppressive tumor microenvironment. CircRNAs are implicated in cancer immune escape by controlling the immune checkpoints. CircCCAR1 contributes to CD8+ T cell dysfunction by stabilizing PD-1. Circ_0000052 directly combines with miR-382-3p to relieve its inhibition of PD-L1. Hsa_circ_0000190 can elevate the expression of sPD-L1 and regulate immunotherapy response. Hsa_circ_0020714 diminishes cancer sensitivity to anti-PD-1 treatment by sponging miR-30a-5p to upregulate SOX4. CircIGF2BP3 prevents proteasome-mediated degradation of PD-L1 by targeting miR-328-3p and miR-3173-5p to upregulate PKP3. FXR1, fragile X-related protein 1; NK cell, natural killer cell; TIM-3, mucin domain-containing protein-3; TAM, tumor-associated macrophage; Treg, regulatory T cell; CCL22, C-C motif chemokine ligand 22; pSTAT3, phosphorylated signal transducer and activator of transcription 3; CXCL9, C-X-C motif chemokine ligand 9; CXCL10, C-X-C motif chemokine ligand 10; IFN, interferon; PD-1, programmed cell death protein-1; PD-L1, programmed cell death-ligand 1; sPD-L1, soluble programmed cell death-ligand 1; PKP3, plakophilin 3; EGFR, epidermal growth factor receptor; SOX4, Sry-related high-mobility group box 4; IRF7, interferon regulatory factor 7; CARM1, coactivator-associated arginine methyltransferase 1; SHP2, Src homology 2 domain-containing phosphatase 2.
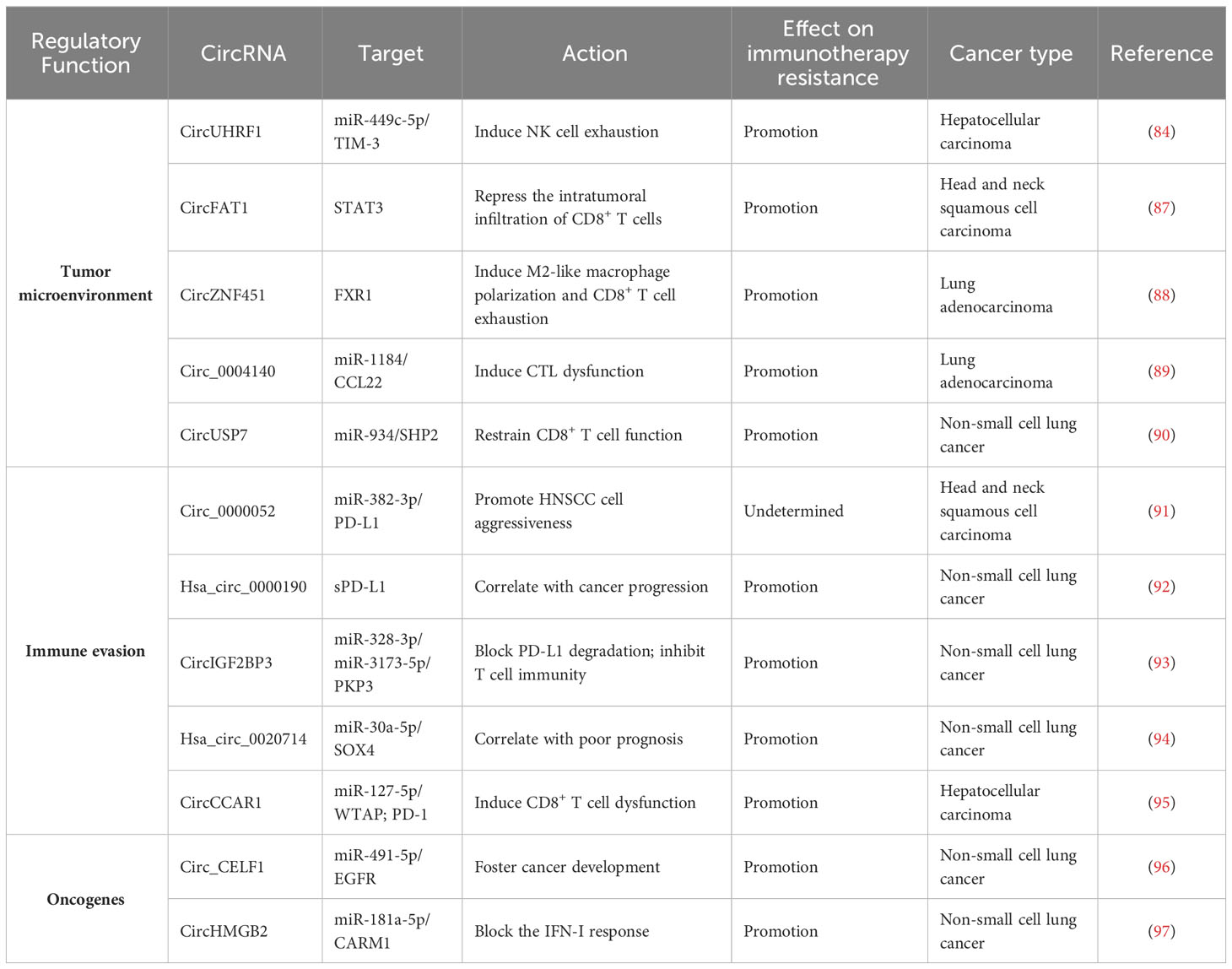
Table 3 Overview of reported immune resistance-related circRNAs in cancer.
CircFAT1 was strikingly upregulated in head and neck squamous cell carcinoma (HNSCC) relative to matched normal tissues and its high expression indicated HNSCC metastasis and poor prognosis (87). CircFAT1 combined with STAT3 and facilitated its activation by reducing Src homology 2 domain-containing phosphatase 1 (SHP1)-mediated dephosphorylation of phosphorylated STAT3 (pSTAT3). pSTAT3 bound to STAT1 to block type I IFN (IFN-I) response (98). As expected, circFAT1 depletion enhanced the production of IFN-I-induced chemokines (e.g., CXCL9 and CXCL10) that were intertwined with recruitment of CD8+ T cells to the tumor site (99). Consistently, circFAT1 knockdown potentiated the efficacy of anti-PD-1 treatment in vivo by promoting CD8+ T cell infiltration into tumor tissues (87). Upregulation of circFAT1 represents a vital molecular mechanism, which contributes to the molding of an immunosuppressive microenvironment in HNSCC.
CircZNF451 was highly expressed in the exosomes of LUAD patients refractory to anti-PD-1 treatment (88). LUAD-derived exosomal circZNF451 could induce the ubiquitination of fragile X-related protein 1 (FXR1) via the E3 ligase TRIM56 in macrophages. CircZNF451-mediated FXR1 degradation abolished the inhibition on the downstream E74-like factor 4 (ELF4)/IRF4 signaling pathway. ELF4 plays an important role in both innate and adaptive immunity by affecting the development and activity of NK and CD8+ T cells (100). Accordingly, exosomal circZNF451 reshaped the immunosuppressive tumor microenvironment by fostering macrophage polarization toward an anti-inflammatory M2-like phenotype as well as dysfunction of cytotoxic CD8+ T cells. Overexpression of circZNF451 attenuated the sensitivity of anti-PD-1 therapy in a xenograft model, while conditional silencing of ELF4 in macrophages reversed this effect. Collectively, exosomal circZNF451 may act as a new indicator for immunotherapy resistance and a target to enhance the efficacy of PD-1 blockade in LUAD. Another LUAD-enriched circ_0004140 could support cancer cell proliferation and migration (89). Circ_0004140 elevated the expression of CCL22 by directly interacting with miR-1184. CCL22 can recruit Tregs in the tumor microenvironment (101). Tregs are able to suppress CTL proliferation and induce immune suppression. Thus, overexpression of circ_0004140 was associated with impairment of CTL function. Combining CCL22/C-C motif chemokine receptor 4 (CCR4) axis inhibitor and anti-PD-1 treatment reversed immune resistance and reduced LUAD progression.
The plasma levels of exosomal circUSP7 were higher in NSCLC patients than healthy controls and correlated with poor clinical prognosis and CD8+ T cell dysfunction in NSCLC (90). CircUSP7 in patient plasma was mainly secreted by NSCLC cells via exosomes. Exosomal circUSP7 suppressed the ability of CD8+ T cells to secrete granzyme B (GzmB), IFN-γ, perforin and TNF-α. SHP2 is a ubiquitous tyrosine phosphatase that affects diverse immune cell signaling cascades (102). Importantly, SHP2 negatively regulated CD8+ T cell proliferation and function. Exosomal circUSP7 restrained CD8+ T cell function by serving as a miR-934 sponge to increase SHP2 expression (90). The in vivo experiment showed that exosomal circUSP7 overexpression enhanced the resistance to anti-PD-1 treatment and promoted tumor progression in NSCLC-bearing humanized NSG mice through induction of CD8+ T cell exhaustion. Furthermore, NSCLC patients with high levels of exosomal circUSP7 showed an apparent phenotype of resistance to anti-PD-1 therapy. Altogether, exosomal circUSP7 played a critical role in promoting tumor immune escape and immunotherapy resistance via the miR-934/SHP2 axis in NSCLC. Peripheral blood exosomal circUSP7 could be used as a novel marker of anti-PD-1 treatment in NSCLC. Reduction of circUSP7 expression may be effective in restoring CD8+ T cell functionality and augmenting immunotherapy efficacy in NSCLC patients.
3.3.2 Promotion of cancer immune evasion
The increased expression of PD-L1 on cancer cells restricts the function of tumor-infiltrating T cells through interaction with PD-1 loaded on T cells and contributes to an immunosuppressive microenvironment (36). Circ_0000052 directly interacted with miR-382-3p to relieve its inhibition of PD-L1 (91). The resultant upregulation of PD-L1 led to HNSCC cell aggressiveness. Hsa_circ_0000190 could enhance the expression of soluble PD-L1 (sPD-L1) in NSCLC (92). The expression level of sPD-L1 showed a positive linkage with disease progression in NSCLC patients following immunotherapy, hinting the crucial role of sPD-L1 in regulating immunotherapy response. Hsa_circ_0000190 might mediate NSCLC immune resistance through upregulation of sPD-L1. The mechanism by which hsa_circ_0000190 coordinates sPD-L1 expression remains to be deciphered.
NSCLC-enriched circIGF2BP3 showed a negative interrelation with the infiltration level of intratumoral CD8+ T cells (93). CircIGF2BP3 acted as a molecular sponge for miR-328-3p and miR-3173-5p to elevate plakophilin 3 (PKP3) expression. PKP3 enhanced the mRNA stability of the deubiquitinase ovarian tumor domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) by binding to FXR1, further protecting PD-L1 from proteasomal degradation. The circIGF2BP3/PKP3 regulatory axis accounted for cancer cell escape from immune surveillance by increasing PD-L1 expression. Accordingly, circIGF2BP3 dampened T cell-mediated antitumor immunity and accelerated NSCLC growth. PKP3 knockdown reinforced the therapeutic effect of PD-1 blockade in tumor-bearing mice.
High expression levels of hsa_circ_0020714 was correlated with resistance to anti-PD-1 immunotherapy and predicted a poor prognosis in NSCLC patients (94). Reportedly, the oncogene Sry-related high-mobility group box 4 (SOX4) favored immune escape and improved resistance to PD-1 blockade in cancer (103). Hsa_circ_0020714 induced NSCLC immune evasion and attenuated sensitivity to anti-PD-1 treatment by sponging miR-30a-5p to upregulate SOX4 (94). Blockade of the hsa_circ_0020714/miR-30a-5p/SOX4-related pathway may result in better immunotherapeutic effects in NSCLC.
CircCCAR1 was enriched in serum exosomes of HCC patients compared with those of healthy controls (95). CircCCAR1 acted as an oncogenic driver in HCC by sponging miR-127-5p to upregulate its target Wilms tumor 1-associated protein (WTAP). Moreover, exosomal circCCAR1 released by HCC cells was ingested by CD8+ T cells and resulted in CD8+ T cell dysfunction by stabilizing PD-1. Exosomal circCCAR1 released by HCC cells enhanced resistance to anti-PD-1 immunotherapy and reduced the survival time of HCC-bearing mice by retarding CD8+ T cell expansion and infiltration into tumor tissues. Modulation of PD-L1/PD-1-regulating circRNAs may represent an effective strategy to reverse T cell exhaustion and maximize immunotherapeutic effect in cancer patients. Nevertheless, clear evidence is needed to better understand the role of these circRNAs in modifying immunotherapy responsiveness.
3.3.3 Upregulation of oncogenes
Oncogenes have been implicated in immune system dysfunction and thus affect the benefits of immune-based cancer treatments (104). CircRNAs counteract the anticancer efficacy of immunotherapy through modulation of oncogenes. The expression level of circ_CELF1 was higher in NSCLC tissues than their adjacent nontumor tissues (96). Circ_CELF1 increased the expression of the oncogene epidermal growth factor receptor (EGFR) by acting as a competitive endogenous RNA (ceRNA) for miR-491-5p. Thus, circ_CELF1 could foster NSCLC development. Moreover, enforced expression of circ_CELF1 diminished the anticancer effects of anti-PD-1 therapy and shortened the survival time of NSCLC-bearing mice. Circ_CELF1 may represent a potential manageable target to overcome therapy resistance in NSCLC. Intensive research efforts should be encouraged to validate and expand this assumption.
The expression level of circHMGB2 was remarkably higher in NSCLC tissues than matched normal tissues (97). High expression of circHMGB2 was associated with poor prognosis of patients with LUAD and lung squamous cell carcinoma (LUSC). CircHMGB2 could upregulate coactivator-associated arginine methyltransferase 1 (CARM1) by sponging miR-181a-5p, thereby blocking the IFN-I response in LUAD and LUSC. These events contributed to formation of an immunosuppressive microenvironment and promotion of tumor growth in NSCLC-bearing mice. As expected, circHMGB2 overexpression profoundly reduced the therapeutic effect of PD-1 blockade in vivo. Pharmacological or genetic inhibition of CARM1 increased the sensitivity of circHMGB2-overexpressing NSCLC cells to anti-PD-1 treatment in murine cancer models. The interaction network between cancer cells and tumor microenvironment is complicated, and elucidating the underlying mechanisms is critically essential to preventing cancer immune evasion. Solid experimental data are still required to establish the role of circHMGB2 in the communication between cancer cells and tumor microenvironment.
4 Noncoding RNA-based anticancer immunotherapy
Due to the interconnection between ncRNAs and immunotherapy resistance, they are considered as prospective therapeutic targets for cancer intervention. Various mimics/antagonists of immunoregulatory miRNAs like miR-26, miR-33a, miR-34, miR-101, miR-125, miR-21, miR-31, miR-32, miR-100, miR-192 and miR-211 are under preclinical and clinical trials against cancer (105–108). However, none has yet reached the therapeutic breakthrough. The physiochemical characteristics of miRNA mimics/antagonists, such as susceptibility to nuclease-mediated degradation, off-target adverse effects and low cellular uptake, hinder their application in cancer treatment (109). Some strategies have been explored to solve the challenges associated with miRNA-targeted therapies. Chemical modifications such as locked nucleic acid (LNA), phosphorothioate-containing oligonucleotides and peptide nucleic acids (PNAs) could heighten the stability, cell targeting and uptake, and delivery efficacy of miRNA-based drugs (110). The engineering of targeted delivery formulations (e.g., liposomal- and polymeric-based delivery platforms) provides an extraordinary opportunity for the application of ncRNA-based therapeutics (111). Further clinical verifications are needed to position immunoregulatory ncRNAs as prospective therapeutic targets for better cancer management.
Several ncRNA-based immunotherapies have been proposed (Figure 7). For instance, a poly(lactic-co-glycolic acid) (PLGA)-based siRNA nanoparticle targeting PD-L1 (siPD-L1@PLGA) was previously generated and could be effectively captured by PDAC cells (112). The siPD-L1@PLGA impeded IFN-γ-mediated PD-L1 induction in recipient cells. Blockade of the PD-1/PD-L1 interaction via siPD-L1@PLGA induced local expansion of tumor-specific CTLs and strengthened the cytotoxicity of CTLs. Accordingly, the siPD-L1@PLGA increased the population of IFN-γ positive CD8+ T cells and markedly reduced tumor growth in a patient-derived xenograft (PDX) mouse model of pancreatic cancer. The upregulation of GzmB in siPD-L1@PLGA-treated pancreatic tumors implied high activity of NK cells or cytotoxic T cells. Given its low toxicity and facile production, the siPD-L1@PLGA could be a promising therapeutic option for PDAC immunotherapy. The extensive evidence outlined in this study warrants the pursuit of future studies to investigate the anticancer effects of this agent in PDAC patients. The co-expression plasmid of CD28-siRNA-PD-1 (pCD28-siRNA-PD-1) delivered by attenuated Salmonella dramatically prolonged the survival of melanoma-bearing mice (113). pCD28-siRNA-PD-1 elevated CD28 expression and suppressed PD-1 expression in tumor tissues. It also promoted cancer cell apoptosis by upregulating cleaved-caspase-3. Mice treated with pCD28-siRNA-PD-1 showed heightened antitumor immune responses, as evidenced by the increased number of NK cells, CD4+ T cells and CD8+ T cells and the decreased number of Tregs. These observations pave the way for developing novel combinatorial therapies that would enhance patient response to anti-PD-1 therapy. CD38 was found to be highly expressed in HCC (114). Extracellular vesicle (EV)-mediated transfer of CD38 siRNA (EVs/siCD38) facilitated macrophage repolarization toward M1-like phenotype and phagocytosis of cancer cells by macrophages through inhibition of CD38-mediated adenosine generation (114). Consequently, EVs/siCD38 exerted an inhibitory effect on HCC growth in vitro and in vivo. EVs/siCD38 also enhanced the sensibility of HCC cells to anti-PD-1/PD-L1 treatment in immunotherapy-resistant mice by attenuating the CD38 level. EVs/siCD38 may hold great promise for the further clinical application in cancer management. It is intriguing whether CD38 can directly modulate the function of immune cells in HCC, such as CD8+ T cells and Tregs. Special attention should be paid to the underlying mechanisms responsible for the anticancer efficacy of EVs/siCD38. A sialic acid-targeted cyclodextrin-based nanoparticle (CD.siRNA.stearic acid-PEG-SA) was previously developed to transport siRNAs to silence colony stimulating factor-1 receptor (CSF-1R) (115). Due to the interaction between sialic acid and its receptor Siglec-1 (CD169), the nanoparticle could specifically transfer CSF-1R siRNAs to M2 macrophages. The targeted formulation exhibited high levels of selective incorporation into M2 macrophages. As expected, the expression level of CSF-1R was profoundly decreased in M2 macrophages. CSF-1R downregulation induced the reprograming of M2 macrophages to M1 phenotype, leading to the apoptosis of prostate cancer cells. Altogether, the targeted cyclodextrin-based siRNA delivery system has potential to be a new immunotherapeutic measure for the intervention of prostate cancer. The tumor suppressor miR-186 was found to be present in NK cell-derived exosomes (116). miR-186 was downregulated in high-risk neuroblastoma patients, and its low expression was connected with poor prognosis of cancer patients. Anti-CD56 coated nanoparticles (CD56-NP) could specifically transfer the mature miR-186 to NK cells. CD56-NP-miR-186 blocked the transforming growth factor-β1 (TGF-β1)-dependent inhibition of NK cytotoxicity in high-risk neuroblastoma by targeting TGF-β receptor 1 (TGFBR1) and TGFBR2. Altogether, miR-186 counteracted TGF-β-dependent immune escape mechanism that hindered the efficiency of antibody-dependent cellular cytotoxicity (ADCC)-based therapies. Modulation of miR-186 abundance may be a feasible strategy to overcome immune evasion in neuroblastoma.
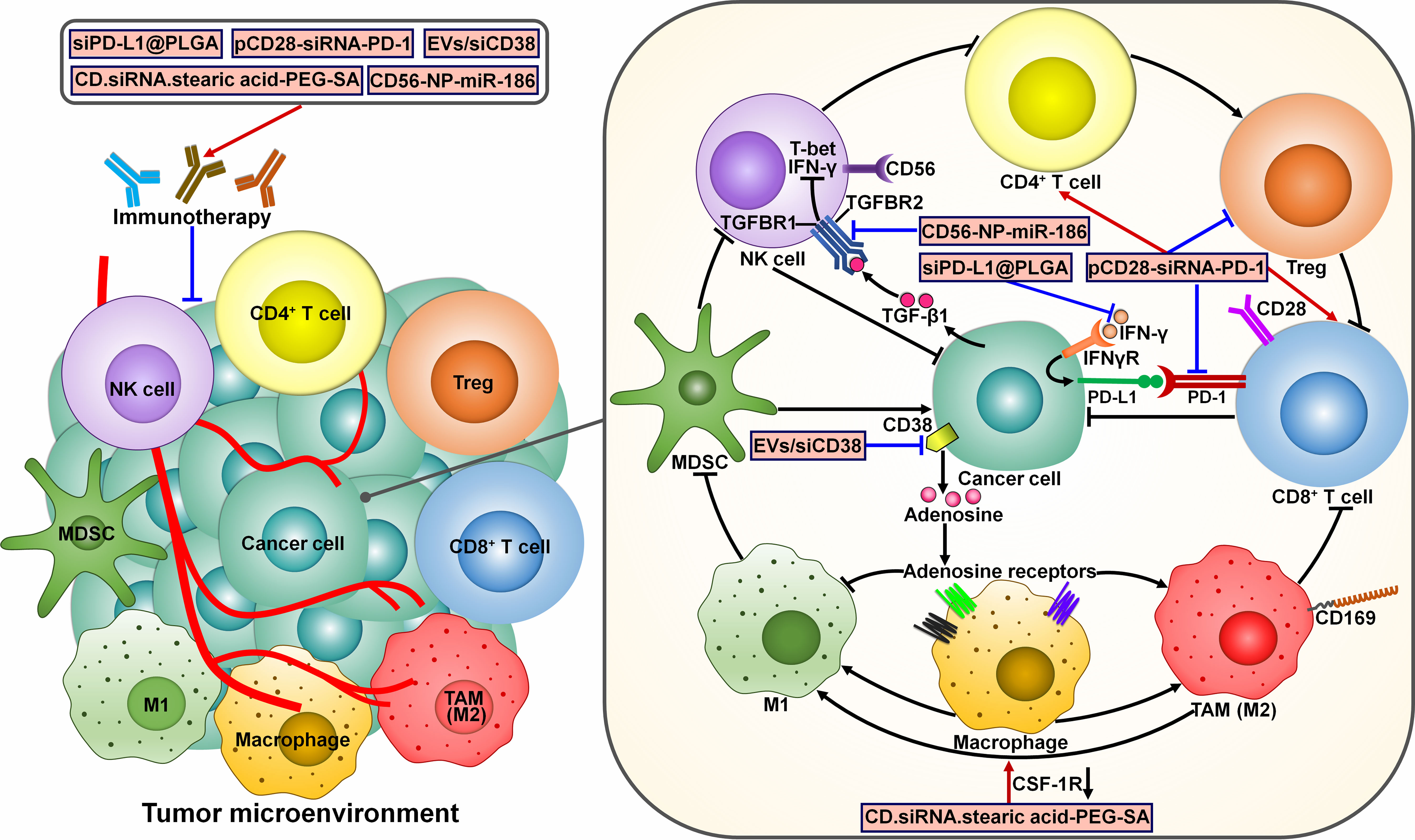
Figure 7 Mechanisms of action of potential ncRNA-based immunotherapeutics. Several ncRNA-centered immunotherapies have been developed. The TGF-β1/SMAD3 signaling pathway can inhibit the expression of T-bet and IFN-γ, thereby blunting NK cell activity. CD56-NP-miR-186 improves NK cytotoxicity by targeting TGFBR1 and TGFBR2. siPD-L1@PLGA suppresses IFN-γ-mediated PD-L1 induction and induces local expansion of CD8+ T cells. pCD28-siRNA-PD-1 can increase CD28 expression and reduce PD-1 expression. It also induces antitumor immune responses by increasing the populations of CD4+ T cells and CD8+ T cells while repressing Treg expansion. EVs/siCD38 impedes CD38-mediated adenosine generation and facilitates macrophage repolarization toward tumor-suppressive M1-like phenotype. CD.siRNA.stearic acid-PEG-SA induces the reprograming of M2 macrophages to M1 phenotype by silencing CSF-1R. These ncRNA-based approaches can reverse cancer resistance to conventional immunotherapies and hold great promise for the clinical use in cancer treatment. IFN-γ, interferon-γ; TGFBR1, transforming growth factor-β receptor 1; TGFBR2, transforming growth factor-β receptor 2; NK cell, natural killer cell; Treg, regulatory T cell; TGF-β1, transforming growth factor-β1; IFNγR, interferon-γ receptor; PD-L1, programmed cell death-ligand 1; PD-1, programmed cell death protein-1; MDSC, myeloid-derived suppressive cell; TAM, tumor-associated macrophage; CSF-1R, colony stimulating factor-1 receptor.
5 Prognostic and predictive value of long noncoding RNAs for immunotherapy in cancer
LncRNAs have been closely associated with both innate and adaptive immunity in cancer. Accordingly, immune-related lncRNAs have a significant impact on response to cancer immunotherapy. Several clinical trials have suggested the correlation between lncRNAs and clinical outcomes of cancer immunotherapy. A phase 2, single arm clinical trial showed that high expression of NF-κB-interacting lncRNA, NKILA, in cancer cells or cancer-specific CTLs predicted a poor prognosis among cancer patients treated with immunotherapy (117). A risk signature comprising seven ferroptosis-related lncRNAs (DUXAP8, LINC00894, LINC01426, LINC02609, MYG1-AS1, PELATON and PVT1) could predict the prognosis of patients with clear cell RCC (ccRCC) (118). Based on this prediction model, ccRCC patients were divided into low- and high-risk groups. Importantly, patients in low-risk group had better immunotherapy response than patients in high-risk group. A prognostic signature was previously constructed based on nineteen pyroptosis-related lncRNAs (e.g., LINC01704, LINC02345 and LINC02560) (119). This signature categorized HNSCC patients into low- and high-risk cohorts. The low-risk cohort was more sensitive to immunotherapeutic agents. A prognostic model consisting of eight lncRNAs AC004080.1, AC078923.1, AC114730.3, AC156455.1, LINC00997, LINC01419, NKILA and U62317.4 was developed (120). Patients with colon cancer were divided into low- and high-risk groups according to the prognostic model. Patients in low-risk group would benefit from immunotherapy. According to a risk signature comprising eight lncRNAs (AC011468.3, AC012306.2, AL441992.1, AP001453.2, AP001922.5, FZD4-DT, RUSC1-AS1 and SOX21-AS1), patients with cervical cancer were classified into different risk groups (121). High-risk patients were less sensitive to ICIs. LncRNA-based risk signatures have great significance to predict immunotherapy response and may aid in clinical decision-making for personalized treatment, but their clinical utility should be further validated.
6 Conclusions and future perspectives
T cell-targeted immunomodulators have been developed and approved for the treatment of various cancer types with an unprecedented speed over the past few decade. Although a tremendous step forward has been achieved, immunotherapy has not overcome existing hurdles of cancer treatment. Remarkably, emergence of resistance is a key factor restricting the efficacy of immunotherapy. NcRNAs, including miRNAs, lncRNAs and circRNAs, have the ability to alter the expression of genes involved in immune activation and suppression, and they also mediate the crosstalk between cancer cells and tumor immune microenvironment. It is not surprising that ncRNAs function as master regulators of immunotherapy resistance. Recent studies have revealed that ncRNAs are involved in cancer cell antigen presentation, immune escape, T cell priming and activation, reshaping of an immunosuppressive tumor microenvironment, cell death pathways during the immunotherapy of cancer. Continual studies are required to discover more ncRNAs that are involved in immune regulation and resistance. Comparison of the expression pattern of ncRNAs between immunotherapy-sensitive and immunotherapy-resistant cancer patient cohorts could be helpful in screening immune-related ncRNAs. The exact mechanisms through which ncRNAs affect immunotherapy response must be systematically defined. NcRNAs can affect both innate and adaptive immune responses. It is important to thoroughly explore the perplexing roles of ncRNAs in tumor immunity. LncRNAs and circRNAs are known to act as miRNA sponges and competitively bind to miRNAs, interfering with their effect on target gene expression. NcRNAs can act alone or crosstalk with each other to remodel the tumor immune microenvironment through different regulatory patterns. The overall impact of complex ncRNA interactions on immunotherapy response is worthy of further study. Notably, in vitro cell-based models or in vivo animal models cannot completely simulate the complicated state of the tumor microenvironment in cancer patients. Thus, the conclusions derived from preclinical studies must be interpreted with caution. Metabolic dysregulation in cancer cells can inhibit the intratumoral infiltration of immune cells and curtail antitumor immunity through the synthesis of immunosuppressive metabolites (122). Thus, aberrant cancer metabolism constitutes a critical mechanism of immunotherapy resistance. The causal link between ncRNA-mediated metabolic alteration in the tumor microenvironment and immunotherapy resistance has yet to be established, sparking continual research efforts in verifying this association. The composition and functionality of the tumor immune microenvironment differ across cancer types. Tumor immune microenvironment predominantly consists of DCs, lymphocytes (e.g., B cells, CD4+ T cells and CD8+ T cells), myeloid cells (e.g., macrophages, MDSCs and neutrophils) and NK cells (123). Tumor immune microenvironment participates in regulating cancer immune evasion and response to immunotherapy. Therefore, more studies investigating the influence of ncRNAs on the functionality of tumor immune microenvironment are required. The effects of ncRNAs on tumor immune microenvironment may be cancer type-specific. It will be important to adequately characterize the roles of ncRNAs in immune regulation across different cancer types. NcRNAs that are related to immunotherapy responses in various types of cancer may have potential to be exploited as future drug targets for effective cancer intervention.
So far, intensive efforts have been deployed to evaluate the therapeutic potential of ncRNA-centered treatments. Nevertheless, most results are still at the stage of basic or preclinical research. The success of immunotherapy mainly relies on cancer eradication through efficient activation of host immune system. Routine preclinical testing of anticancer agents in cancer cell lines or immune-compromised animals does not adequately take into account the engagement of the immune system. More reliable immune-competent preclinical models are key to understanding the anticancer action and mechanism of immunotherapies. Since ncRNAs play a vital role in various cellular processes under both physiological and pathological conditions, ncRNA-directed therapies, which are based on the functional repression of oncogenic ncRNAs through the application of antisense oligonucleotides and the replacement of tumor-suppressive ncRNA via the introduction of ncRNA mimics, may bring about significant detrimental outcomes. ICI immunotherapies work by reversing the physiological brakes of immune activation, so they may exert off-target effects leading to immune-induced inflammation of multiple organs. Accordingly, more extensive studies are warranted to completely evaluate the anticancer benefits and safe profiles of ncRNA-targeted therapies. Additional research efforts should be directed towards the optimization of immunotherapy regimens, with the aim to ameliorate the immunological-related adverse effects. Given the heterogeneity and complexity of immune states among cancer patients, it is essential to individualize ncRNA-based immunotherapy based on both cancer- and patient-specific characteristics. NcRNA-based immunotherapy could act in synergy with other treatment modalities including chemotherapy, radiotherapy and metabolic checkpoint-targeting therapies. The personalized combinatorial strategy may be further improved by determining the optimal sequencing of different treatments. Clinical translation of ncRNA-based therapeutics can be affected by delivery routes, immunogenicity and specificity. Many delivery vehicles have been utilized to transfer therapeutic ncRNAs, including lipid nanoparticles, metal-based nanoparticles, polymer-based delivery system and viral vectors (124). Exosomes and bacteriophages are considered as ideal and favorable transporters of ncRNA therapeutics in clinical practice due to their poor immunogenicity and high delivery efficiency. NcRNA oligonucleotides are mainly recognized by Toll-like receptors (TLRs), leading to adverse immune reactions. Chemical modification is regarded as a potential approach to reduce the immunogenicity of ncRNA-based therapeutics. However, even after chemical modification in ncRNA oligonucleotides, this issue is not well addressed yet and should be further explored. Increasing knowledge of TLR-associated adverse immune effects will assist in developing novel ncRNA therapies that exhibit low immunogenicity before the initiation of clinical trials. To reduce the detrimental effects of ncRNA mimics or antagonists in undesired cells, expression vectors carrying promoter sequences whose expression is specifically upregulated in target cells could be utilized to express therapeutic ncRNA oligonucleotides. The specificity of ncRNA-based therapeutics may be further improved by targeting the precursors of ncRNAs to inhibit their synthesis and maturation using nucleic acid oligonucleotides or peptides. Despite encouraging preclinical and early clinical evidence, the successful clinical utility of ncRNA-based immunotherapy has not yet been realized. With increasing knowledge of ncRNA involvement in tumor immunity and immunotherapy, there is hope for ncRNA-based therapeutics as novel treatments for cancer.
Author contributions
MW: Conceptualization, Funding acquisition, Investigation, Supervision, Visualization, Writing – original draft. FY: Resources, Writing – review and editing, Visualization. PL: Project administration, Supervision, Writing – review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Shandong Province, China (Grant No. ZR2021MH018).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res (2020) 30(8):660–9. doi: 10.1038/s41422-020-0343-4
2. Clarke JM, George DJ, Lisi S, Salama AKS. Immune checkpoint blockade: the new frontier in cancer treatment. Targeted Oncol (2018) 13(1):1–20. doi: 10.1007/s11523-017-0549-7
3. Cortez MA, Anfossi S, Ramapriyan R, Menon H, Atalar SC, Aliru M, et al. Role of mirnas in immune responses and immunotherapy in cancer. Genes Chromosomes Cancer (2019) 58(4):244–53. doi: 10.1002/gcc.22725
4. Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol (2014) 8(6):1132–9. doi: 10.1016/j.molonc.2014.07.011
5. Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of cancer resistance to immunotherapy. Front Oncol (2020) 10:1290. doi: 10.3389/fonc.2020.01290
6. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
7. Diamantopoulos MA, Tsiakanikas P, Scorilas A. Non-coding rnas: the riddle of the transcriptome and their perspectives in cancer. Ann Trans Med (2018) 6(12):241. doi: 10.21037/atm.2018.06.10
8. Liu Y, Ding W, Wang J, Ao X, Xue J. Non-coding rna-mediated modulation of ferroptosis in cardiovascular diseases. Biomed pharmacotheR (2023) 164:114993. doi: 10.1016/j.biopha.2023.114993
9. Slack FJ, Chinnaiyan AM. The role of non-coding rnas in oncology. Cell (2019) 179(5):1033–55. doi: 10.1016/j.cell.2019.10.017
10. Micheel J, Safrastyan A, Wollny D. Advances in non-coding Rna sequencing. Non-coding RNA (2021) 7(4):70. doi: 10.3390/ncrna7040070
11. Carninci P. Molecular biology: the long and short of rnas. Nature (2009) 457(7232):974–5. doi: 10.1038/457974b
12. Borga C, Meeran SM, Fassan M. Non-coding rnas, a real next-gen class of biomarkers? Non-coding RNA Res (2019) 4(3):80–1. doi: 10.1016/j.ncrna.2019.10.001
13. Choudhuri S. Small noncoding rnas: biogenesis, function, and emerging significance in toxicology. J Biochem Mol Toxicol (2010) 24(3):195–216. doi: 10.1002/jbt.20325
14. Ao X, Ding W, Li X, Xu Q, Chen X, Zhou X, et al. Non-coding rnas regulating mitochondrial function in cardiovascular diseases. J Mol Med (2023) 101(5):501–26. doi: 10.1007/s00109-023-02305-8
15. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microrna biogenesis, mechanisms of actions, and circulation. Front Endocrinol (2018) 9:402. doi: 10.3389/fendo.2018.00402
16. Liu Y, Ao X, Zhou X, Du C, Kuang S. The regulation of pbxs and their emerging role in cancer. J Cell Mol Med (2022) 26(5):1363–79. doi: 10.1111/jcmm.17196
17. Wilczynska A, Bushell M. The complexity of mirna-mediated repression. Cell Death Differ (2015) 22(1):22–33. doi: 10.1038/cdd.2014.112
18. Fernandes JCR, Acuna SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long non-coding rnas in the regulation of gene expression: physiology and disease. Non-coding RNA (2019) 5(1):17. doi: 10.3390/ncrna5010017
19. Liu Y, Wang Y, Li X, Jia Y, Wang J, Ao X. Foxo3a in cancer drug resistance. Cancer Lett (2022) 540:215724. doi: 10.1016/j.canlet.2022.215724
20. Sun W, Yang Y, Xu C, Guo J. Regulatory mechanisms of long noncoding Rnas on gene expression in cancers. Cancer Genet (2017) 216-217:105–10. doi: 10.1016/j.cancergen.2017.06.003
21. Liu Y, Ao X, Wang Y, Li X, Wang J. Long non-coding Rna in gastric cancer: mechanisms and clinical implications for drug resistance. Front Oncol (2022) 12:841411. doi: 10.3389/fonc.2022.841411
22. Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, et al. Circular rna: A new star of noncoding Rnas. Cancer Lett (2015) 365(2):141–8. doi: 10.1016/j.canlet.2015.06.003
23. Liu Y, Ao X, Jia Y, Li X, Wang Y, Wang J. The foxo family of transcription factors: key molecular players in gastric cancer. J Mol Med (2022) 100(7):997–1015. doi: 10.1007/s00109-022-02219-x
24. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular rnas are a large class of animal Rnas with regulatory potency. Nature (2013) 495(7441):333–8. doi: 10.1038/nature11928
25. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular Rna expression. PloS Genet (2013) 9(9):e1003777. doi: 10.1371/journal.pgen.1003777
26. Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, et al. Circular intronic long noncoding Rnas. Mol Cell (2013) 51(6):792–806. doi: 10.1016/j.molcel.2013.08.017
27. Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of circrnas. Mol Cell (2017) 66(1):9–21 e7. doi: 10.1016/j.molcel.2017.02.021
28. Chen LL. The biogenesis and emerging roles of circular Rnas. Nat Rev Mol Cell Biol (2016) 17(4):205–11. doi: 10.1038/nrm.2015.32
29. Vishnubalaji R, Shaath H, Elango R, Alajez NM. Noncoding Rnas as potential mediators of resistance to cancer immunotherapy. Semin Cancer Biol (2020) 65:65–79. doi: 10.1016/j.semcancer.2019.11.006
30. Zhou X, Ao X, Jia Z, Li Y, Kuang S, Du C, et al. Non-coding Rna in cancer drug resistance: underlying mechanisms and clinical applications. Front Oncol (2022) 12:951864. doi: 10.3389/fonc.2022.951864
31. Benedetti A, Turco C, Fontemaggi G, Fazi F. Non-coding Rnas in the crosstalk between breast cancer cells and tumor-associated macrophages. Non-coding RNA (2022) 8(1):16. doi: 10.3390/ncrna8010016
32. Su T, Zhang P, Zhao F, Zhang S. Exosomal micrornas mediating crosstalk between cancer cells with cancer-associated fibroblasts and tumor-associated macrophages in the tumor microenvironment. Front Oncol (2021) 11:631703. doi: 10.3389/fonc.2021.631703
33. Li A, Yi M, Qin S, Song Y, Chu Q, Wu K. Activating cgas-sting pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol (2019) 12(1):35. doi: 10.1186/s13045-019-0721-x
34. O’Neill RE, Cao X. Co-stimulatory and co-inhibitory pathways in cancer immunotherapy. Adv Cancer Res (2019) 143:145–94. doi: 10.1016/bs.acr.2019.03.003
35. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not all immune checkpoints are created equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
36. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
37. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun (2020) 11(1):3801. doi: 10.1038/s41467-020-17670-y
38. Kooshkaki O, Derakhshani A, Hosseinkhani N, Torabi M, Safaei S, Brunetti O, et al. Combination of ipilimumab and nivolumab in cancers: from clinical practice to ongoing clinical trials. Int J Mol Sci (2020) 21(12):4427. doi: 10.3390/ijms21124427
39. Twomey JD, Zhang B. Cancer immunotherapy update: fda-approved checkpoint inhibitors and companion diagnostics. AAPS J (2021) 23(2):39. doi: 10.1208/s12248-021-00574-0
40. Chen R, Tao Y, Xu X, Shan L, Jiang H, Yin Q, et al. The efficacy and safety of nivolumab, pembrolizumab, and atezolizumab in treatment of advanced non-small cell lung cancer. Discov Med (2018) 26(143):155–66.
41. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (Poplar): A multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0
42. Haanen JB, Robert C. Immune checkpoint inhibitors. Prog tumor Res (2015) 42:55–66. doi: 10.1159/000437178
43. Collin M. Immune checkpoint inhibitors: A patent review (2010-2015). Expert Opin Ther patents (2016) 26(5):555–64. doi: 10.1080/13543776.2016.1176150
44. Ott PA, Bang YJ, Berton-Rigaud D, Elez E, Pishvaian MJ, Rugo HS, et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: results from the keynote-028 study. J Clin Oncol (2017) 35(22):2535–41. doi: 10.1200/JCO.2017.72.5952
45. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
46. Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol (2022) 15(1):61. doi: 10.1186/s13045-022-01282-8
47. Han X, Luan T, Sun Y, Yan W, Wang D, Zeng X. Microrna 449c mediates the generation of monocytic myeloid-derived suppressor cells by targeting stat6. Molecules Cells (2020) 43(9):793–803. doi: 10.14348/molcells.2020.2307
48. Ren W, Zhang X, Li W, Feng Q, Feng H, Tong Y, et al. Exosomal mirna-107 induces myeloid-derived suppressor cell expansion in gastric cancer. Cancer Manage Res (2019) 11:4023–40. doi: 10.2147/CMAR.S198886
49. Xiao L, He Y, Peng F, Yang J, Yuan C. Endometrial cancer cells promote M2-like macrophage polarization by delivering exosomal mirna-21 under hypoxia condition. J Immunol Res (2020) 2020:9731049. doi: 10.1155/2020/9731049
50. Kawana S, Saito R, Miki Y, Kimura Y, Abe J, Sato I, et al. Suppression of tumor immune microenvironment via microrna-1 after epidermal growth factor receptor-tyrosine kinase inhibitor resistance acquirement in lung adenocarcinoma. Cancer Med (2021) 10(2):718–27. doi: 10.1002/cam4.3639
51. Dong ZR, Cai JB, Shi GM, Yang YF, Huang XY, Zhang C, et al. Oncogenic mir-93-5p/gal-9 axis drives cd8 (+) T-cell inactivation and is a therapeutic target for hepatocellular carcinoma immunotherapy. Cancer Lett (2023) 564:216186. doi: 10.1016/j.canlet.2023.216186
52. Zhao X, Yuan C, Wangmo D, Subramanian S. Tumor-secreted extracellular vesicles regulate T-cell costimulation and can be manipulated to induce tumor-specific T-cell responses. Gastroenterology (2021) 161(2):560–74.e11. doi: 10.1053/j.gastro.2021.04.036
53. Tittarelli A, Navarrete M, Lizana M, Hofmann-Vega F, Salazar-Onfray F. Hypoxic melanoma cells deliver micrornas to dendritic cells and cytotoxic T lymphocytes through connexin-43 channels. Int J Mol Sci (2020) 21(20):7567. doi: 10.3390/ijms21207567
54. Liu C, Liu R, Wang B, Lian J, Yao Y, Sun H, et al. Blocking il-17a enhances tumor response to anti-pd-1 immunotherapy in microsatellite stable colorectal cancer. J immunothe Cancer (2021) 9(1):e001895. doi: 10.1136/jitc-2020-001895
55. Miliotis C, Slack FJ. Mir-105-5p regulates pd-L1 expression and tumor immunogenicity in gastric cancer. Cancer Lett (2021) 518:115–26. doi: 10.1016/j.canlet.2021.05.037
56. Kong X, Zheng Z, Song G, Zhang Z, Liu H, Kang J, et al. Over-expression of gusb leads to primary resistance of anti-pd1 therapy in hepatocellular carcinoma. Front Immunol (2022) 13:876048. doi: 10.3389/fimmu.2022.876048
57. Guo W, Wu Z, Chen J, Guo S, You W, Wang S, et al. Nanoparticle delivery of mir-21-3p sensitizes melanoma to anti-pd-1 immunotherapy by promoting ferroptosis. J immunothe Cancer (2022) 10(6):e004381. doi: 10.1136/jitc-2021-004381
58. Sui H, Dongye S, Liu X, Xu X, Wang L, Jin CQ, et al. Immunotherapy of targeting mdscs in tumor microenvironment. Front Immunol (2022) 13:990463. doi: 10.3389/fimmu.2022.990463
59. Koosha F, Alimohammadi N, Rafeian-Kopaei M. The exosomes: staring biomarkers and novel therapeutic strategies. Curr Pharm design (2021) 27(35):3714–21. doi: 10.2174/1381612827666210614102340
60. Weigelin B, Friedl P. T cell-mediated additive cytotoxicity - death by multiple bullets. Trends Cancer (2022) 8(12):980–7. doi: 10.1016/j.trecan.2022.07.007
61. Iwahori K. Cytotoxic cd8(+) lymphocytes in the tumor microenvironment. Adv Exp Med Biol (2020) 1224:53–62. doi: 10.1007/978-3-030-35723-8_4
62. Liu J, Li F, Ping Y, Wang L, Chen X, Wang D, et al. Local production of the chemokines ccl5 and cxcl10 attracts cd8+ T lymphocytes into esophageal squamous cell carcinoma. Oncotarget (2015) 6(28):24978–89. doi: 10.18632/oncotarget.4617
63. Lhuillier C, Barjon C, Baloche V, Niki T, Gelin A, Mustapha R, et al. Characterization of neutralizing antibodies reacting with the 213-224 amino-acid segment of human galectin-9. PloS One (2018) 13(9):e0202512. doi: 10.1371/journal.pone.0202512
64. Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature (2018) 562(7725):69–75. doi: 10.1038/s41586-018-0519-y
65. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted cd8 T cells by pd-1-targeted therapies is cd28-dependent. Science (2017) 355(6332):1423–7. doi: 10.1126/science.aaf0683
66. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, et al. Pd-1 primarily targets tcr signal in the inhibition of functional T cell activation. Front Immunol (2019) 10:630. doi: 10.3389/fimmu.2019.00630
67. Fenwick C, Loredo-Varela JL, Joo V, Pellaton C, Farina A, Rajah N, et al. Tumor suppression of novel anti-pd-1 antibodies mediated through cd28 costimulatory pathway. J Exp Med (2019) 216(7):1525–41. doi: 10.1084/jem.20182359
68. Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, Zhang Y. Pd-L1 promoter methylation mediates the resistance response to anti-pd-1 therapy in nsclc patients with egfr-tki resistance. Oncotarget (2017) 8(60):101535–44. doi: 10.18632/oncotarget.21328
69. Wu H, Wang F, Ta N, Zhang T, Gao W. The multifaceted regulation of mitochondria in ferroptosis. Life (2021) 11(3):222. doi: 10.3390/life11030222
70. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. Cd8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
71. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
72. Hu Q, Ye Y, Chan LC, Li Y, Liang K, Lin A, et al. Oncogenic lncrna downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat Immunol (2019) 20(7):835–51. doi: 10.1038/s41590-019-0400-7
73. Guo W, Wang Y, Yang M, Wang Z, Wang Y, Chaurasia S, et al. Lincrna-immunity landscape analysis identifies epic1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci Adv (2021) 7(7):eabb3555. doi: 10.1126/sciadv.abb3555
74. Zhao L, Liu Y, Zhang J, Liu Y, Qi Q. Lncrna snhg14/mir-5590-3p/zeb1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating pd-1/pd-L1 checkpoint. Cell Death Dis (2019) 10(10):731. doi: 10.1038/s41419-019-1886-5
75. Xue J, Song Y, Xu W, Zhu Y. The cdk1-related lncrna and cxcl8 mediated immune resistance in lung adenocarcinoma. Cells (2022) 11(17):2688. doi: 10.3390/cells11172688
76. Zhang J, Pan T, Zhou W, Zhang Y, Xu G, Xu Q, et al. Long noncoding Rna linc01132 enhances immunosuppression and therapy resistance via nrf1/dpp4 axis in hepatocellular carcinoma. J Exp Clin Cancer Res (2022) 41(1):270. doi: 10.1186/s13046-022-02478-z
77. Sun C, Ye Y, Tan Z, Liu Y, Li Y, Hu W, et al. Tumor-associated nonmyelinating schwann cell-expressed pvt1 promotes pancreatic cancer kynurenine pathway and tumor immune exclusion. Sci Adv (2023) 9(5):eadd6995. doi: 10.1126/sciadv.add6995
78. Zhang Y, Luo M, Cui X, O’Connell D, Yang Y. Long noncoding rna neat1 promotes ferroptosis by modulating the mir-362-3p/miox axis as a cerna. Cell Death Differ (2022) 29(9):1850–63. doi: 10.1038/s41418-022-00970-9
79. Zhang J, Li S, Zhang J, Zhang W, Jiang J, Wu H, et al. Docetaxel resistance-derived linc01085 contributes to the immunotherapy of hormone-independent prostate cancer by activating the sting/mavs signaling pathway. Cancer Lett (2022) 545:215829. doi: 10.1016/j.canlet.2022.215829
80. Cane S, Ugel S, Trovato R, Marigo I, De Sanctis F, Sartoris S, et al. The endless saga of monocyte diversity. Front Immunol (2019) 10:1786. doi: 10.3389/fimmu.2019.01786
81. Greene LI, Bruno TC, Christenson JL, D’Alessandro A, Culp-Hill R, Torkko K, et al. A role for tryptophan-2,3-dioxygenase in cd8 T-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res (2019) 17(1):131–9. doi: 10.1158/1541-7786.MCR-18-0362
82. Sordillo PP, Sordillo LA, Helson L. The kynurenine pathway: A primary resistance mechanism in patients with glioblastoma. Anticancer Res (2017) 37(5):2159–71. doi: 10.21873/anticanres.11551
83. Nadhan R, Isidoro C, Song YS, Dhanasekaran DN. Signaling by lncrnas: structure, cellular homeostasis, and disease pathology. Cells (2022) 11(16):2517. doi: 10.3390/cells11162517
84. Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM, et al. Cancer cell-derived exosomal circuhrf1 induces natural killer cell exhaustion and may cause resistance to anti-pd1 therapy in hepatocellular carcinoma. Mol Cancer (2020) 19(1):110. doi: 10.1186/s12943-020-01222-5
85. Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol (2010) 52(3):322–9. doi: 10.1016/j.jhep.2009.12.005
86. Sanchez-Correa B, Lopez-Sejas N, Duran E, Labella F, Alonso C, Solana R, et al. Modulation of nk cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer immunol immunothe (2019) 68(5):861–70. doi: 10.1007/s00262-019-02336-6
87. Jia L, Wang Y, Wang CY. Circfat1 promotes cancer stemness and immune evasion by promoting stat3 activation. Adv Sci (2021) 8(13):2003376. doi: 10.1002/advs.202003376
88. Gao J, Ao YQ, Zhang LX, Deng J, Wang S, Wang HK, et al. Exosomal circznf451 restrains anti-pd1 treatment in lung adenocarcinoma via polarizing macrophages by complexing with trim56 and fxr1. J Exp Clin Cancer Res (2022) 41(1):295. doi: 10.1186/s13046-022-02505-z
89. Liu Y, Zhang H, Zhang W, Xiang L, Yin Z, Xu H, et al. Circ_0004140 promotes luad tumor progression and immune resistance through circ_0004140/mir-1184/ccl22 axis. Cell Death Discov (2022) 8(1):181. doi: 10.1038/s41420-022-00983-w
90. Chen SW, Zhu SQ, Pei X, Qiu BQ, Xiong D, Long X, et al. Cancer cell-derived exosomal circusp7 induces cd8(+) T cell dysfunction and anti-pd1 resistance by regulating the mir-934/shp2 axis in nsclc. Mol Cancer (2021) 20(1):144. doi: 10.1186/s12943-021-01448-x
91. Zhang DJ, Fu ZM, Guo YY, Guo F, Wan YN, Guan GF. Circ_0000052/mir-382-3p axis induces pd-L1 expression and regulates cell proliferation and immune evasion in head and neck squamous cell carcinoma. J Cell Mol Med (2023) 27(1):113–26. doi: 10.1111/jcmm.17643
92. Luo YH, Yang YP, Chien CS, Yarmishyn AA, Adekunle Ishola A, Chien Y, et al. Circular rna hsa_Circ_0000190 facilitates the tumorigenesis and immune evasion by upregulating the expression of soluble pd-L1 in non-small-cell lung cancer. Int J Mol Sci (2021) 23(1):64. doi: 10.3390/ijms23010064
93. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N(6)-methyladenosine-modified circigf2bp3 inhibits cd8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of pd-L1 in non-small cell lung cancer. Mol Cancer (2021) 20(1):105. doi: 10.1186/s12943-021-01398-4
94. Wu J, Zhu MX, Li KS, Peng L, Zhang PF. Circular rna drives resistance to anti-pd-1 immunotherapy by regulating the mir-30a-5p/sox4 axis in non-small cell lung cancer. Cancer Drug resistance (2022) 5(2):261–70. doi: 10.20517/cdr.2021.100
95. Hu Z, Chen G, Zhao Y, Gao H, Li L, Yin Y, et al. Exosome-derived circccar1 promotes cd8 + T-cell dysfunction and anti-pd1 resistance in hepatocellular carcinoma. Mol Cancer (2023) 22(1):55. doi: 10.1186/s12943-023-01759-1
96. Ge W, Chi H, Tang H, Xu J, Wang J, Cai W, et al. Circular rna celf1 drives immunosuppression and anti-pd1 therapy resistance in non-small cell lung cancer via the mir-491-5p/egfr axis. Aging (2021) 13(22):24560–79. doi: 10.18632/aging.203576
97. Zhang LX, Gao J, Long X, Zhang PF, Yang X, Zhu SQ, et al. The circular rna circhmgb2 drives immunosuppression and anti-pd-1 resistance in lung adenocarcinomas and squamous cell carcinomas via the mir-181a-5p/carm1 axis. Mol Cancer (2022) 21(1):110. doi: 10.1186/s12943-022-01586-w
98. Tsai MH, Pai LM, Lee CK. Fine-tuning of type I interferon response by stat3. Front Immunol (2019) 10:1448. doi: 10.3389/fimmu.2019.01448
99. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of th1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
100. Suico MA, Shuto T, Kai H. Roles and regulations of the ets transcription factor elf4/mef. J Mol Cell Biol (2017) 9(3):168–77. doi: 10.1093/jmcb/mjw051
101. Martinenaite E, Munir Ahmad S, Hansen M, Met O, Westergaard MW, Larsen SK, et al. Ccl22-specific T cells: modulating the immunosuppressive tumor microenvironment. Oncoimmunology (2016) 5(11):e1238541. doi: 10.1080/2162402X.2016.1238541
102. Wang Y, Mohseni M, Grauel A, Diez JE, Guan W, Liang S, et al. Shp2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci Rep (2021) 11(1):1399. doi: 10.1038/s41598-021-80999-x
103. Bagati A, Kumar S, Jiang P, Pyrdol J, Zou AE, Godicelj A, et al. Integrin alphavbeta6-tgfbeta-sox4 pathway drives immune evasion in triple-negative breast cancer. Cancer Cell (2021) 39(1):54–67 e9. doi: 10.1016/j.ccell.2020.12.001
104. Zakiryanova GK, Wheeler S, Shurin MR. Oncogenes in immune cells as potential therapeutic targets. ImmunoTargets Ther (2018) 7:21–8. doi: 10.2147/ITT.S150586
105. Abd-Aziz N, Kamaruzman NI, Poh CL. Development of micrornas as potential therapeutics against cancer. J Oncol (2020) 2020:8029721. doi: 10.1155/2020/8029721
106. Mondal P, Natesh J, Penta D, Meeran SM. Progress and promises of epigenetic drugs and epigenetic diets in cancer prevention and therapy: A clinical update. Semin Cancer Biol (2022) 83:503–22. doi: 10.1016/j.semcancer.2020.12.006
107. Ishida M, Selaru FM. Mirna-based therapeutic strategies. Curr anesthesiol Rep (2013) 1(1):63–70. doi: 10.1007/s40139-012-0004-5
108. Bonneau E, Neveu B, Kostantin E, Tsongalis GJ, De Guire V. How close are mirnas from clinical practice? A perspective on the diagnostic and therapeutic market. Ejifcc (2019) 30(2):114–27.
109. Segal M, Slack FJ. Challenges identifying efficacious mirna therapeutics for cancer. Expert Opin Drug Discov (2020) 15(9):987–92. doi: 10.1080/17460441.2020.1765770
110. Baumann V, Winkler J. Mirna-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future medicinal Chem (2014) 6(17):1967–84. doi: 10.4155/fmc.14.116
111. Mondal P, Kaur B, Natesh J, Meeran SM. The emerging role of mirna in the perturbation of tumor immune microenvironment in chemoresistance: therapeutic implications. Semin Cell Dev Biol (2022) 124:99–113. doi: 10.1016/j.semcdb.2021.04.001
112. Jung JY, Ryu HJ, Lee SH, Kim DY, Kim MJ, Lee EJ, et al. Sirna nanoparticle targeting pd-L1 activates tumor immunity and abrogates pancreatic cancer growth in humanized preclinical model. Cells (2021) 10(10):2734. doi: 10.3390/cells10102734
113. Jia H, Guo J, Liu Z, Chen P, Li Y, Li R, et al. High expression of cd28 enhanced the anti-cancer effect of sirna-pd-1 through prompting the immune response of melanoma-bearing mice. Int Immunopharmacol (2022) 105:108572. doi: 10.1016/j.intimp.2022.108572
114. Deng J, Ke H. Overcoming the resistance of hepatocellular carcinoma to pd-1/pd-L1 inhibitor and the resultant immunosuppression by cd38 sirna-loaded extracellular vesicles. Oncoimmunology (2023) 12(1):2152635. doi: 10.1080/2162402X.2022.2152635
115. Sun Y, Cronin MF, Mendonca MCP, Guo J, O’Driscoll CM. Sialic acid-targeted cyclodextrin-based nanoparticles deliver csf-1r sirna and reprogram tumour-associated macrophages for immunotherapy of prostate cancer. Eur J Pharm Sci (2023) 185:106427. doi: 10.1016/j.ejps.2023.106427
116. Neviani P, Wise PM, Murtadha M, Liu CW, Wu CH, Jong AY, et al. Natural killer-derived exosomal mir-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res (2019) 79(6):1151–64. doi: 10.1158/0008-5472.CAN-18-0779
117. Yu Y, Zhang W, Li A, Chen Y, Ou Q, He Z, et al. Association of long noncoding rna biomarkers with clinical immune subtype and prediction of immunotherapy response in patients with cancer. JAMA Netw Open (2020) 3(4):e202149. doi: 10.1001/jamanetworkopen.2020.2149
118. Ju L, Shi Y, Liu G. Identification and validation of a ferroptosis-related lncrna signature to robustly predict the prognosis, immune microenvironment, and immunotherapy efficiency in patients with clear cell renal cell carcinoma. PeerJ (2022) 10:e14506. doi: 10.7717/peerj.14506
119. Zhou C, Shen Y, Jin Y, Shen Z, Ye D, Shen Y, et al. A novel pyroptosis-related long non-coding rna signature for predicting the prognosis and immune landscape of head and neck squamous cell carcinoma. Cancer Med (2022) 11(24):5097–112. doi: 10.1002/cam4.4819
120. Chen Y, Zhang Y, Lu J, Liu Z, Zhao S, Zhang M, et al. Characteristics of prognostic programmed cell death-related long noncoding rnas associated with immune infiltration and therapeutic responses to colon cancer. Front Immunol (2022) 13:828243. doi: 10.3389/fimmu.2022.828243
121. Kong X, Xiong Y, Xue M, He J, Lu Q, Chen M, et al. Identification of cuproptosis-related lncrna for predicting prognosis and immunotherapeutic response in cervical cancer. Sci Rep (2023) 13(1):10697. doi: 10.1038/s41598-023-37898-0
122. Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, et al. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther (2019) 195:162–71. doi: 10.1016/j.pharmthera.2018.11.004
123. Kaur M, Kaur B, Konar M, Sharma S. Noncoding rnas as novel immunotherapeutic tools against cancer. Adv Protein Chem Struct Biol (2022) 129:135–61. doi: 10.1016/bs.apcsb.2021.11.011
Keywords: cancer, tumor microenvironment, immunotherapy resistance, ncRNAs, tumor immunity, ncRNA-based therapies
Citation: Wang M, Yu F and Li P (2023) Noncoding RNAs as an emerging resistance mechanism to immunotherapies in cancer: basic evidence and therapeutic implications. Front. Immunol. 14:1268745. doi: 10.3389/fimmu.2023.1268745
Received: 28 July 2023; Accepted: 30 August 2023;
Published: 12 September 2023.
Edited by:
Kevin Charles Conlon, Clinical Center (NIH), United StatesReviewed by:
Mei Luo, Huazhong University of Science and Technology, ChinaNick Tschernia, National Cancer Institute (NIH), United States
Copyright © 2023 Wang, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Wang, d2FuZ21hbkBxZHUuZWR1LmNu; Peifeng Li, cGVpZmxpQHFkdS5lZHUuY24=