 Mortimer Svec1†
Mortimer Svec1† Sarah Dötsch1†
Sarah Dötsch1† Linda Warmuth1†
Linda Warmuth1† Manuel Trebo1
Manuel Trebo1 Simon Fräßle1
Simon Fräßle1 Stanley R. Riddell2
Stanley R. Riddell2 Ulrich Jäger3
Ulrich Jäger3 Elvira D’Ippolito1‡Dirk H. Busch1*‡
Elvira D’Ippolito1‡Dirk H. Busch1*‡- 1Institute for Medical Microbiology, Immunology and Hygiene, School of Medicine and Health, Technical University of Munich, Munich, Germany
- 2Translational Sciences and Therapeutics, Fred Hutchinson Cancer Research Center, Seattle, WA, United States
- 3Department of Medicine I, Division of Hematology and Hemostaseology, Medical University of Vienna, Vienna, Austria
Adoptive immunotherapy based on chimeric antigen receptor (CAR)-engineered T cells has exhibited impressive clinical efficacy in treating B-cell malignancies. However, the potency of CAR-T cells carriethe potential for significant on-target/off-tumor toxicities when target antigens are shared with healthy cells, necessitating the development of complementary safety measures. In this context, there is a need to selectively eliminate therapeutically administered CAR-T cells, especially to revert long-term CAR-T cell-related side effects. To address this, we have developed an effective cellular-based safety mechanism to specifically target and eliminate the transferred CAR-T cells. As proof-of-principle, we have designed a secondary CAR (anti-CAR CAR) capable of recognizing a short peptide sequence (Strep-tag II) incorporated into the hinge domain of an anti-CD19 CAR. In in vitro experiments, these anti-CAR CAR-T cells have demonstrated antigen-specific cytokine release and cytotoxicity when co-cultured with anti-CD19 CAR-T cells. Moreover, in both immunocompromised and immunocompetent mice, we observed the successful depletion of anti-CD19 CAR-T cells when administered concurrently with anti-CAR CAR-T cells. We have also demonstrated the efficacy of this safeguard mechanism in a clinically relevant animal model of B-cell aplasia induced by CD19 CAR treatment, where this side effect was reversed upon anti-CAR CAR-T cells infusion. Notably, efficient B-cell recovery occurred even in the absence of any pre-conditioning regimens prior anti-CAR CAR-T cells transfer, thus enhancing its practical applicability. In summary, we developed a robust cellular safeguard system for selective in vivo elimination of engineered T cells, offering a promising solution to address CAR-T cell-related on-target/off-tumor toxicities.
1 Introduction
The use of tumor-specific transgenic receptors to engineer autologous patient-derived T cells and harness them against host cancer cells has opened promising new therapeutic options (1, 2). More importantly, it has allowed the manufacturing of well-characterized T-cell products with highly defined specificity and functionality. Remarkable success has been shown with chimeric antigen receptors (CARs) targeting CD19-expressing refractory and relapsed B-cell malignancies, where the adoptive transfer of CAR-T cells led to durable cancer regression (3–5). Currently, most efforts focus on identifying suitable target/CAR combinations for other malignancies (6) and to increase the specificity, sensitivity and durability of CAR-T cells by a variety of engineering approaches like supporting cytokine production, gene knockouts or smart logic gating (7–9).
However, CAR-T cell therapy also comes with some safety concerns. Short-term side effects like cytokine release syndrome (CRS) (10) and neurotoxicity, termed CAR-T cell-related encephalopathy syndrome (CRES) (11) still occur at high frequencies (5, 12). While these acute toxicities are usually reversible and can be alleviated by immediate immunosuppression by corticosteroids or anti-IL-6 receptor antibodies (13), there is still a need for safeguard mechanisms that are truly specific for the transferred cells and mediate the elimination of the cell product within early and - maybe more relevant - late stages after the transfer. Indeed, relevant on-target/off-tumor long-term side effects experienced by patients receiving anti-CD19 CAR-T cell therapy are persistent B-cell aplasia and consequent hypogammaglobulinemia (14). Substitution with polyclonal antibodies is an established therapy, but there is no consensus on the proper management of this toxicity (15). Moreover, there is still scarce evidence on the consequences of prolonged B-cell aplasia, such as increased risk of infections and mortality. This becomes more important in children and young adults where the development of long-lived plasma cells resistant to anti-CD19 CAR-T cell therapy (16), which could provide a long-lasting humoral response, might still be incomplete.
Besides novel engineering approaches attempting to reduce off-target toxicities by controlling the expression and activity of CARs in terms of timing and dosage (17, 18), there are two main concepts of safety regulation. These are based on the use of suicide genes or cell surface markers that aim at the eradication of the adoptively transferred cells. The predominant suicide genes are the inducible caspase 9 (iCas9) (19) and the herpes simplex virus thymidine kinase (HSV-TK) (20). These suicide genes were sufficient under certain circumstances but showed some drawbacks. In the case of the HSV-TK, the clearance of target cells was incomplete; moreover, immune responses against the transgene - also in immunocompromised patients - limited the in vivo persistence of the therapeutic cells after transfer (21). Immunogenicity was less prevalent for the highly humanized iCasp9 (22). Still, this mechanism relied on a high expression level of the iCasp9, and some degree of spontaneous activation triggering auto-apoptosis could not be avoided entirely (19).
Regarding safeguard mechanisms based on surface molecules, one relevant marker is a truncated version of the human epidermal growth factor receptor (EGFRt) that lacks the intracellular domain and is thus physiologically inert (23). We, among others, have shown in syngeneic preclinical models that transferred EGFRt-expressing CAR-T cells could be efficiently depleted upon administration of the monoclonal anti-EGFR antibody Cetuximab (24) and were long-term maintained (25), hinting against immunogenicity. Nevertheless, this marker also requires a high surface expression to deliver a complete clearance of transferred cells (24). Despite ongoing improvements towards an optimized EGFRt expression (26), one should still be cautious about the on-target side effects of Cetuximab. The other caveat is the dependence of this safety mechanism on the antibody-dependent cellular cytotoxicity of the patient.
In this study, we propose a cellular safeguard mechanism that exploits the highly specific antigen recognition and potency of a CAR-T cell capable of recognizing a tag within a different target CAR-T cell. We could show that these so-called ‘anti-CAR CAR-T cells’ exhibit potent cytotoxicity in vitro as well as in vivo, including a clinically relevant mouse model that mirrors established long-term CAR-T cell-related on-target/off-tumor toxicity. In this context, adoptively transferred anti-CAR CAR-T cells reversed persistent B-cell aplasia induced by anti-CD19 CAR-T cells. Intriguingly, high doses of anti-CAR CAR-T cells could mediate this effect also in the absence of pre-conditioning prior to cell transfer.
2 Materials and methods
2.1 Animal models and cell lines
C57BL/6 mice (female, 8-11 weeks old) were acquired from Envigo. RAG-/- mice and C57BL/6 mice on different congenic backgrounds (CD45.2+/+, CD45.1+/+, CD45.1+/-, CD90.1+/+, CD90.1+/-) were derived from in-house breeding under specific pathogen-free conditions at our mouse facility at the Technical University Munich, Institute for Medical Microbiology, Immunology and Hygiene. The performed animal experiments were approved by the district government of Upper Bavaria (Department 5—Environment, Health and Consumer Protection ROB- 55.2-2532.Vet_02-17-138).
Murine splenocytes were cultured in RMPI (Life Technologies, Cat #31870074) with 10% fetal bovine serum (FCS) (Sigma, Cat #F7524), 0.025% L-glutamine (Sigma, Cat #G8540), 0.1% HEPES (Roth, Cat #HN77.4), 0.001% gentamycin (LifeTechnologies, Cat #15750037), 0.002% penicillin/streptomycin (LifeTechnologies, Cat #10378016) and 15 ng/ml recombinant human (rh) IL-15 (Peprotech, Cat # 200-15). The Platinum-E packaging cell line (PlatE) was cultured in DMEM (Life Technologies, Cat #10938025) supplemented with 10% FCS, 0.025% L-glutamine, 0.1% HEPES, 0.001% gentamycin, 0.002% streptomycin. All cells were grown in a humidified incubator at 37°C and 5% CO2.
2.2 CAR DNA template design
scFvs were generated by fusing the variable regions of the heavy (VH) and light (VL) chains with a short (G4S)3 linker. For CAR constructs, the signal peptide of CD8α was followed by the scFv, a hinge domain (a triple repetitive sequence of Strep-tag II (STII) for anti-CD19 CAR and a CD8a spacer domain for the anti-CAR CAR) and parts of the IgG4-Fc molecule. This extracellular domain was followed by a transmembrane region originated from the CD28 chain and the intracellular signaling domains CD3-zeta. A truncated version of the EGFR (EGFRt) was added at the 5’ and separated from the CAR sequence by a self-splicing viral P2A element. As control, a construct with the same elements as the anti-CD19 CAR but without the scFV (defined as ‘mock’) was generated. DNA templates were designed in silico and synthesized by GeneArt (Thermo Fisher Scientific) or Twist Bioscience into pMP72 vectors. scFv sequence of anti-CAR CAR (clone: 5G2) were kindly provided by the lab of Prof. Riddell.
2.3 Retroviral transduction
For the production of retroviral particles, PlatE cells were transiently transfected with a pMP72 expression vector by calcium phosphate precipitation. For this, 15 µl of a 3.31 M MgCl2 was mixed with 18 µg vector DNA and filled up to 150 µl with ddH2O. This solution was slowly added under vortexing to 150 µl transfection buffer (274 mM NaCl, 9.9 mM KCl, 3.5 mM Na2HPO4 and 41.9 mM HEPES), and the final transfection mix was added to PlatE cells for 6 h at 37°C, followed by a complete medium exchange. The virus-containing supernatant was harvested 48 and 72 h after transfection by filtration (0,45 µm sterile filter) and stored at 4°C for up to one week. Mouse splenocytes were obtained from CD45/CD90 congenic mice by mashing the spleen into single-cell suspensions. After red cell lysis, splenocytes were stimulated at a concentration of 107 cells/mL with purified anti-mouse CD3 (BD Biosciences, clone 145-2C11, 1:1000, Cat #553058) and anti-mouse CD28 (BD Biosciences, clone 37.51, 1:3000, BD Cat #553295) antibodies in presence of 25 U/ml recombinant human interleukin-2 (Peprotech, Cat #200-02). For anti-CAR CAR transduction, splenocytes were previously depleted of B cells by magnetic purification (Miltenyi Biotec, Cat #130-048-701) using an anti-mouse CD19-FITC antibody (BD Biosciences, clone 1D3, 1:100, Cat #553785) according to the manufacturer’s protocol. After 24 h, stimulated splenocytes were collected and transduced via spinoculation. For this, tissue-culture untreated 24-well plates were coated overnight with retronectin (TaKaRa, Cat #T100B), purified anti-mouse CD3 (1:1000) and anti-mouse CD28 (1:3000) antibodies. The virus-containing supernatant was centrifuged onto the plates at 3000 g at 32°C for 2 h. After removal of the supernatant, cells were transferred and centrifuged at 800 g at 32°C for 1.5 h. Cells were then rested for two days before administration to the mice.
2.4 Flow cytometry
For in vitro assays, cells were harvested from the culture and washed twice with FACS buffer (1x PBS, 0.5% (w/v) bovine serum albumin (BSA), pH 7.45) before staining. For blood analysis, blood was sampled from the tail vein and stored in heparin at room temperature. Bone marrow was acquired from the hind legs by removal of muscles and rinsing with complete RPMI medium whereas lymph nodes and spleens were homogenized into single-cell suspension by mashing. Red cell lysis was performed for single-cell suspensions from all organs and blood with RBC lysis buffer (90% 0.17M NH4Cl in Tris-HCl). After lysis, cells were first stained with anti-CD16/32 antibody (Biolegend, clone 93, 1:400, Cat #101301) for 20 min at 4°C (only cells from lymphoid tissues), followed by surface marker staining for 20 min at 4°C in the dark. In this study we used the following antibodies for surface staining: CD3-APC (Biolegend, clone 145-2C11, 1:100, Cat #100312), CD3-FITC (Life Technologies, clone 17A2, 1:100, Cat #11003282), CD8-PacO (Life Technologies, clone 5H10, 1:50, Cat #MCD0830), CD19-ECD (BD Bioscience, clone 1D3, 1:300, Cat #562291), CD45.1-PacBlue (Life Technologies, clone A20, 1:100, Cat #48045382), CD45.1-APC (Life Technologies, clone A20, 1:100, Cat #17045382), CD90.1-PacBlue (Life Technologies, clone HIS51, 1:500, Cat #48-0900-82), CD90.2-APC (Life Technologies, clone 53-2.1, 1:500, Cat #17090283), EGFR-PE (Biolegend, clone AY13, 1:2000, Cat #352904), PD-1-PE-Cy7 (Life Technologies, clone J43, 1:100, Cat #25-9985-80), TIM3-BV421 (Biolegend, clone RMT3-23, 1:100, Cat #119723), CD69-BV510 (Biolegend, clone H1-2F3, 1:100, Cat #104531). Live/dead discrimination was performed either by addition of propidium iodide (LifeTechnologies, 1:100, Cat #P1304MP) 3 min before the end of the staining or with ethidium-monoazide-bromide (LifeTechnologies, 1:1000, Cat #E1374) for 15 min at 4°C under bright light. Absolute quantification was performed by using 123count eBeads (Life Technologies, Cat #01-1234-42) according to the manufacturer’s instructions. Specific information is further provided for each experiment. Samples were acquired on a CytoFLEX S flow cytometer (Beckman Coulter). All flow cytometry data were analyzed with FlowJo v10.
2.5 Antigen-specific in vitro stimulation and intracellular cytokine staining
5x104 effector anti-CAR CAR-T cells (EGFRt+) were co-cultured with target anti-CD19 CAR-T cells (EGFRt+) at different effector-to-target ratios in 96-well plates. After 1 h co-incubation, 1x GolgiPlug was added (BD PharMingen, Cat #555029) and cells were incubated for additional 4 h at 37°C. 25 ng/ml Phorbol-12-myristat-13-acetate (Sigma, Cat #P1585) and 1 μg/ml ionomycin (Sigma, Cat #I9657) were used as positive controls, while untransduced cells were used as negative controls. After co-culture, staining for live/dead discrimination was performed with ethidium-monoazide-bromide as described before, followed by surface marker antibody staining for congenic markers (CD45.1, CD45.2) and EGFRt for 20 min at 4°C. Cells were permeabilized using Cytofix/Cytoperm (BD Biosciences, reference #554714) and stained intracellularly for TNF-α-PE-Cy7 (BD Biosciences, clone MP6-XT22, 1:100, Cat #557644), INF-γ-APC (LifeTech, clone XGM1.2, 1:400, Cat #17-7311-82) and IL-2 (LifeTech, clone JES6-5H4, 1:100, Cat #12-7021-82). As bulk CAR-T cell products were used, the number of transferred cells always refers to the amount of congenic marker+EGFRt+ cells. The total amount of transferred cells was defined by flow cytometry according to the expression of the EGFRt marker.
2.6 Flow cytometry-based cytotoxic T lymphocyte assay
4x104 effector anti-CAR CAR-T cells (EGFRt+) were co-cultured with target anti-CD19 CAR-T cells (EGFRt+) at different effector to target ratios in 96-well U-bottom plate at a concentration of 7.5x105 cells/mL for 48 h. At the beginning of the incubation (0 h), and after 24 and 48 h, 200 µL of the cell suspension was harvested for surface marker antibody staining of congenic markers (CD90.1, CD45.1), CD3, CD8, CD19 and EGFRt. Staining for live/dead discrimination was performed with propidium iodide. As bulk CAR-T cell products were used, the number of transferred cells always refers to the amount of congenic marker+ EGFRt+ cells. The total amount of transferred cells was defined by flow cytometry according to the expression of the EGFRt marker.
2.7 Western blot
Transduced mouse splenocytes were rested for 5 days or activated with anti-CD3 (1:1000) and anti-CD28 (1:3000) antibodies for 24 h prior analyses. Cell lysates were generated by addition of 200 µl 1X SDS sample buffer (62.5 mM Tris pH 6.8, 2% w/v SDS, 10% Glycerol, 0.01% Bromophenol blue, 50 mM DTT) to 2x106 cells, followed by sonication for 10 min and incubation at 95°C for 5 min. The solution was centrifuged for 5 min at 13.000 g, and 15 µl of the supernatant were run on an 8% SDS-PAGE gel (stacking gel: TRIS 0.5 M pH 6.5, 0.4% SDS, 40% Acrylamide, 10% APS TEMED; separating gel: Tris 1.5 M pH 8.8, 0.4% SDS, 40% Acrylamide, 10% APS TEMED) for 1 h at 150 V. The proteins were semi-dry blotted onto a nitrocellulose membrane at 10 V for 30 min. The efficiency of the transfer was evaluated via Ponceau staining (0.5% Ponceau, 1% acetic acid). The membrane was blocked in Tris-buffered saline containing 0.1% Tween (TBS-T buffer) with 5% nonfat dry milk (blocking buffer) for 1 h at room temperature. Afterwards the membrane was stained with anti-pCD3ζ antibody (Life Technologies, clone EM54, 1:3000, Cat #PIMA528538) in TBS-T buffer containing 5% BSA at 4°C. As loading control, GAPDH was stained with anti-GAPDH antibody (Thermo Fisher Scientific, clone 1D4, 1:3000, Cat #MA1-16757). After 12 h, the membrane was washed in TBS-T buffer and incubated with a horseradish peroxidase-conjugated anti-mouse secondary antibody (Cell Signaling, 1:1000, Cat #7076S) in blocking buffer for 1 h at room temperature. After washing, the membrane was incubated with 200 µl enhanced chemiluminescence solution (Biorad, Cat #1705061) for 5 min at room temperature and analyzed with the INTAS chemoluminescence detection system.
2.8 Adoptive T cell transfer
For the co-injection model, Rag-/- or C57BL/6 mice were sublethally irradiated (5 Gy) the day before the simultaneous adoptive transfer of 1x106 anti-CD19 CAR-T cells and 2x106 anti-CAR CAR-T cells by intravenous injection (i.v.). Transferred cells and B cells were followed in the blood at different time points and in bone marrow, spleen and lymph nodes at the endpoint.
For the therapeutic model, 2x106 anti-CD19 CAR-T cells were transferred i.v. into sublethally irradiated mice (5 Gy). After 27 days, mice were either or not irradiated with a 2 Gy dose and treated the day after i.v. with 4x106 or 8x106 anti-CAR CAR-T cells, or 4x106 mock T cells (CAR construct without the scFv). Moreover, one group received the monoclonal antibody Cetuximab (1mg per mouse) (Bristol-Myers Squibb) by intraperitoneal injection. Transferred cells and B cells were followed in the blood at different time points and in bone marrow, spleen and lymph nodes at the endpoint.
As bulk CAR-T cell products were used, the number of transferred cells always refers to the number of congenic marker+EGFRt+ cells. The total amount of transferred cells was defined by flow cytometry according to the expression of the EGFRt marker. The specific congenic marker combinations are indicated for each experiment.
2.9 Visualization and statistical testing
Data were visualized using GraphPad Prism (V9.1). All statistical tests were performed using GraphPad Prism (V9.1). Specific details regarding statistical analyses can be found in the corresponding figure legends. Levels of significance were defined as the following: ns = p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Data are presented as mean ± standard deviation or mean + standard deviation, as indicated in figure legends.
3 Results
3.1 Design and in vitro functionality of anti-CAR CAR-T cells
In this study, we used a fully syngeneic second-generation anti-CD19 CAR as the primary ‘therapeutic’ CAR. We further included in the hinge region a sequence containing a three-time in tandem repetition of an 8 amino acid peptide, named Strep-tag II (27). Previous investigations have demonstrated that the inclusion of this triple Strep-tag II in the CAR construct does not compromise the functionality of the corresponding engineered T cells (28). To target anti-CD19 CAR-T cells, we generated a second CAR construct where the scFv could specifically recognize the Strep-tag II. This CAR, referred to as ‘anti-CAR’ CAR from now on, was additionally equipped with the same CD3zeta/CD28 intracellular domains and truncated human EGFR (23, 24) as the anti-CD19 CAR (Figure 1A). This engineering approach theoretically enables the depletion of fully functional anti-CD19 CAR-T cells by the second anti-CAR CAR-T cells (Figure 1B). For appropriate discrimination, the two CARs were always re-expressed in splenocytes from mice harboring distinct CD45/CD90 congenic marker combinations. Furthermore, B cells were depleted from spleen-derived cell suspensions before viral transduction to avoid stimulation of anti-CD19 CAR-T cells prior any in vitro or in vivo assay. Both receptors could be successfully expressed in primary murine splenocytes through retroviral gene delivery and showed comparable surface expression levels (Figure 1C).
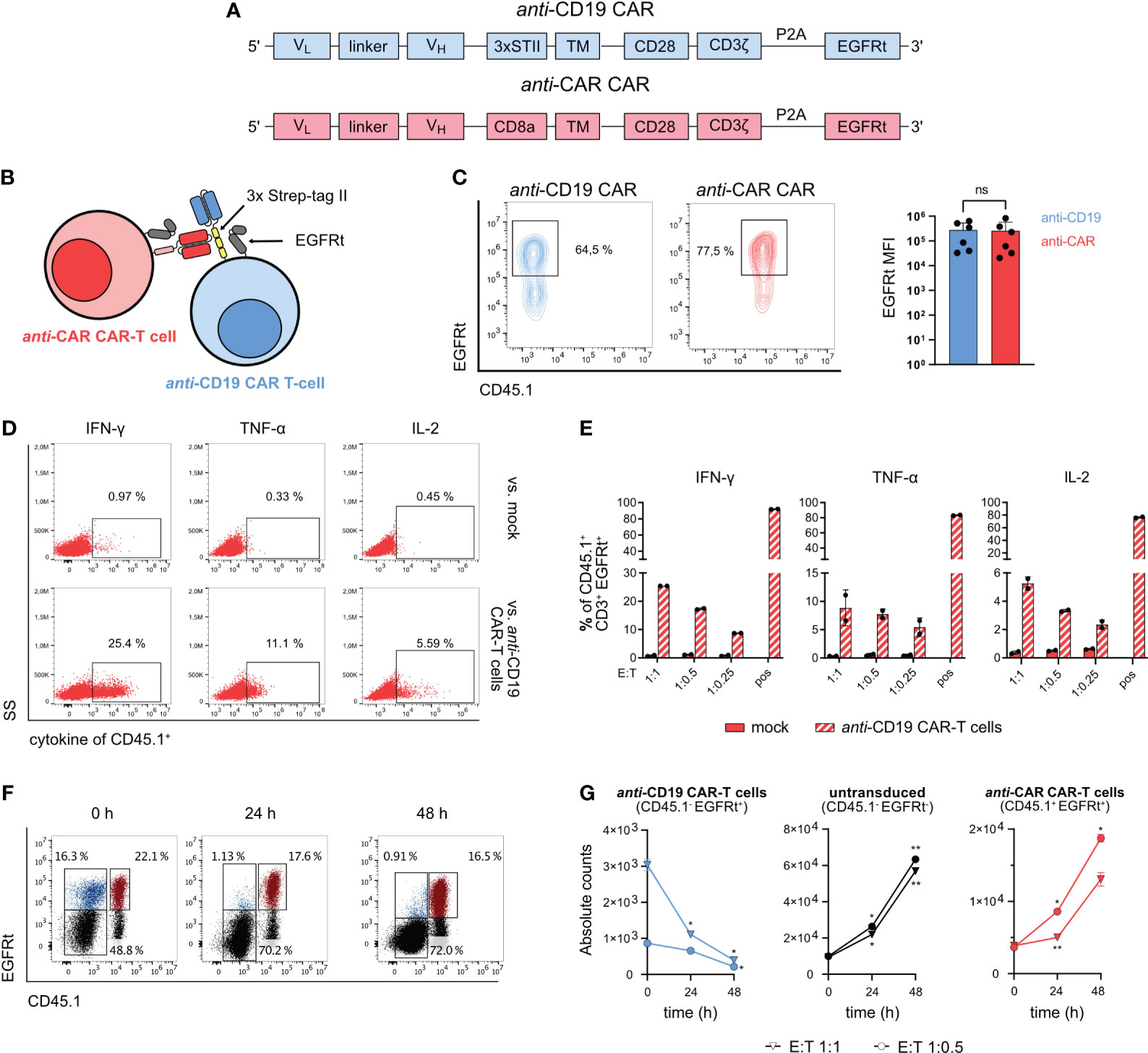
Figure 1 Design and in vitro functionality of anti-CAR CAR-T cells. (A) Schematic of the CAR sequences for retroviral transduction. (B) Schematic of the mechanism of action of anti-CAR CAR-T cells. (C) Cell surface expression of CAR constructs after retroviral transduction measured by EGFRt co-expression. (D, E) 5x104 anti-CAR CAR-T cells were co-cultured with B cell-depleted anti-CD19 CAR-T cells or untransduced cells at the indicated E:T ratios for 4 h. Representative flow cytometry plots (E:T 1:1) (D) and quantification (F) of intracellular staining of IFN-γ, TNF-α and IL-2. Pregated on CD3+ EGFRt+ living lymphocytes. (F, G) 4x104 anti-CAR CAR-T cells (red: CD45.1+ EGFRt+) were co-cultured with B cell-depleted bulk anti-CD19 CAR-T cells (blue: CD45.1- EGFRt+; gray: CD45.1- EGFRt-) at the indicated E:T ratios and analyzed at 0, 24 and 48 h. Representative flow cytometry plots (G) and quantification (H) of the co-culture killing assay. Pregated on CD3+ living lymphocytes. Data are shown as mean + SD in (C) and mean ± SD in (E, G). In (C), statistical analyses have been performed by Mann-Whitney nonparametric test. In (G) statistical analyses have been performed by two-way ANOVA with Dunnett’s multiple comparisons test using 0 h as reference. ns = p > 0.05, *p < 0.05, **p < 0.01.
To demonstrate the in vitro functionality of the newly designed anti-CAR CAR-T cells, we first examined intracellular cytokine release upon target-specific stimulation during co-culture with Strep-tag II-expressing anti-CD19 CAR-T cells. The anti-CAR CAR-T cells showed reliable cytokine production (IFN-γ, TNF-α and IL-2) in a dose-dependent manner in presence of the target cells. Moreover, there was little to no unspecific signaling when co-cultured with untransduced control cells (Figures 1D, E). For evaluating cytotoxic capacity, we again co-cultured anti-CD19 CAR-T cells with anti-CAR CAR-T cells at different effector-to-target (E:T) ratios and monitored the absolute number of each T cell population over 48 hours. By using bulk populations, we could also assess the specificity of killing by tracking the survival of unedited (EGFRt-) cells. Importantly, while the untransduced control cells without antigen expression, as well as the effector cells, proliferated over time, we observed a significant decrease of the target anti-CD19 CAR-T cells (Figures 1F, G). Altogether, these data demonstrate the specific target recognition of our anti-CAR CAR-T cells, which ultimately translates into effective cytotoxic T-cell functions.
Finally, we investigated whether anti-CAR CAR-T cells showed any tonic signaling activity. For this purpose, we firstly analyzed the baseline phosphorylation of CD3zeta in rested splenocytes, using freshly restimulated cells as control. While we observed elevated levels of pCD3z in restimulated anti-CAR CAR-T cells, we did not detect any level exceeding those found in control cells in rested splenocytes (Supplementary Figure 1A). To confirm this phenotype, we also assessed the persistence and activation state of adoptively transferred anti-CAR CAR-T cells under homeostatic conditions (Supplementary Figure 1B). We observed overall similar numbers of circulating anti-CAR CAR-T cells compared to untransduced mock control cells (Supplementary Figure 1C), and, as expected, no influence on the B-cell compartment except for the drop in the first week due to the pre-conditioning regimen prior T-cell transfer (Supplementary Figure 1D). Furthermore, we did not detect neither consistent upregulation of activation markers (CD69 and PD-1) nor exhausted cells (PD-1+TIM3+) (Supplementary Figures 1E, F). These observations were confirmed also in anti-CAR CAR-T cells infiltrating secondary lymphoid organs, except from the detection of a small fraction of CD69+ cells in the bone marrow (Supplementary Figure 1G). Altogether, we could not find robust indications of tonic signaling.
3.2 Anti-CAR CAR-T cells display in vivo killing in immunocompromised mice in a model of acute antigen encounter
Our in vitro data indicated a certain degree of functionality for the designed anti-CAR receptor. However, such evidence could not definitively predict the in vivo functionality, as this relies on additional factors such as the overall antigen load, target cell accessibility and the receptor moiety itself, among others (29, 30).
To assess the in vivo functionality of our anti-CAR construct with minimal confounders, we first conducted a short-term in vivo killing assay in the imunocompromised RAG1-/- mouse model, which lacks mature T cells. As before, we generated anti-CD19 and anti-CAR CAR-T cells from different congenically labelled splenocytes, which were simultaneously injected in pre-conditioned mice. We monitored the adoptively transferred cells in the periphery through blood analysis for 6 days and in tissues on day 10 (Figure 2A). While anti-CD19 CAR-T cells were reliably detected in the blood when transferred alone, we could not detect circulating anti-CD19 CAR-T cells when both T-cell products were co-transferred. This observation was consistent in secondary lymphoid tissues (Figures 2B, C; Supplementary Figure 2), suggesting infiltration properties that are expected from a cell-based mechanism.
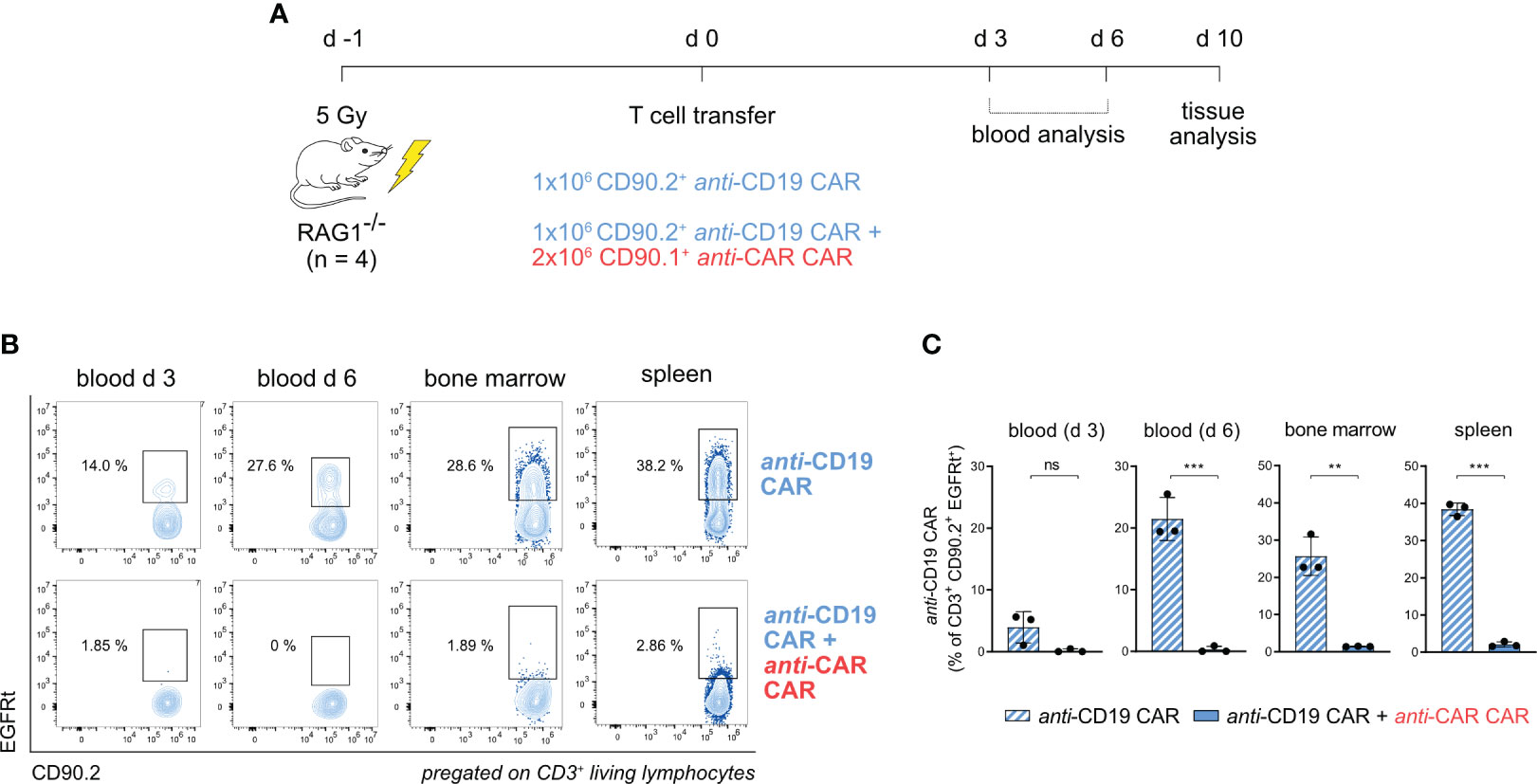
Figure 2 anti-CAR CAR-T cells display in vivo killing in immunocompromised mice in a model of acute antigen encounter. (A) Schematic of the experimental setup. (B, C) Representative flow cytometry plots (B) and quantification (C) of anti-CD19 CAR-T cells (EGFRt+CD90.2+) in the peripheral blood at the indicated time points and in tissues in the presence or not of anti-CAR CAR-T cells. Data are shown as mean ± SD. In (C), statistical analyses were performed by unpaired t-test. ns = p > 0.05, **p < 0.01, ***p < 0.001.
3.3 Anti-CAR CAR-T cells can restore B cells in CD19 CAR-treated immunocompetent mice
As the next step, we setup up a similar experiment using an immunocompetent wild type C57BL/6 mouse model as a recipient. We also included an additional control group treated solely with anti-CAR CAR-T cells (Figure 3A; Supplementary Figure 3A). In this model, the administration of anti-CD19 CAR-T cells induces long-lasting B-cell aplasia (31), similar to what reported in patients receiving anti-CD19 CAR therapy (5, 32). Thus, this syngeneic model allowed us to monitor the efficacy of anti-CAR CAR-T cells in terms of depleting the anti-CD19 CAR-T cells as well as reconstituting the B-cell compartment eliminated by the primary therapeutic CAR-T cells (24). Moreover, this model presented challenges more relevant to potential applications in humans, such as the competition with other immune cell populations and, most importantly, a stimulus for the primary anti-CD19 CAR, which was still expected to functionally recognize B cells.
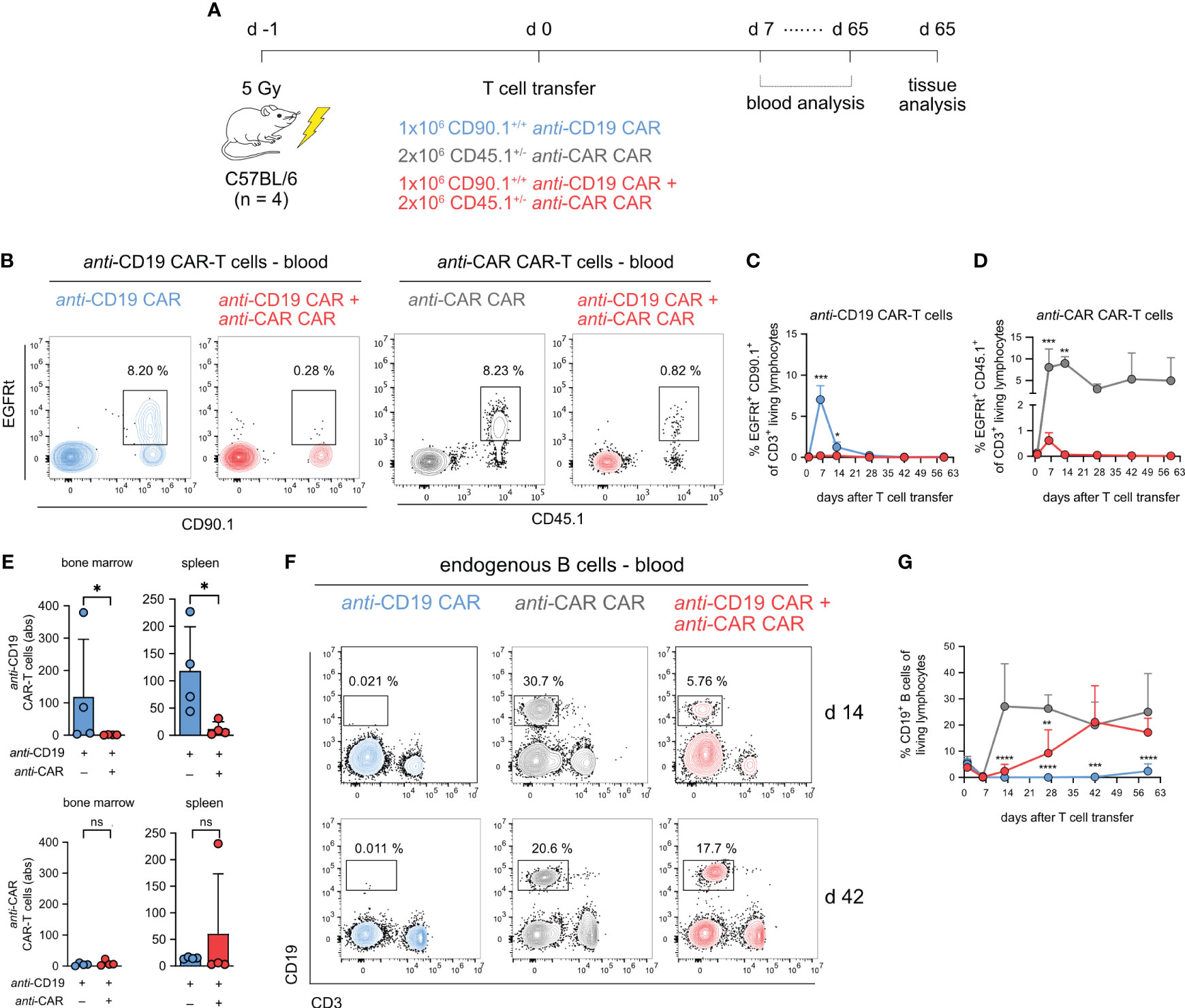
Figure 3 anti-CAR CAR-T cells can restore B cells in CD19 CAR-treated immunocompetent mice. (A) Schematic of the experimental setup. (B) Representative flow cytometry plots of anti-CD19 CAR-T cells (EGFRt+CD90.1+) and anti-CAR CAR-T cells (EGFRt+CD45.1+) at day 6 post infusion according to the different treatment schemes. Cells are pregated on living CD3+ T cells. (C, D) Quantification of the frequencies of anti-CD19 CAR-T cells (C) and anti-CAR CAR-T cells (D) in blood over time. (E) Quantification of tissue-infiltrating anti-CD19 and anti-CAR CAR-T cells. (F) Representative flow cytometry plots of living CD19+ B cells according to the different treatment schemes at the indicated time points. (G) Kinetics of B cells in blood over time. Data are shown as mean + SD. In (C) and (G) statistical analyses were performed by two-way ANOVA with Sidak’s multiple comparisons test (C) and Dunnett’s multiple comparisons test using the anti-CAR group as reference (G). In (E) statistical analyses were performed with non-parametric Mann-Whitney t test. ns = p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p > 0.0001.
Looking at the dynamics of anti-CD19 CAR-T cells in the blood (Supplementary Figure 3B), we observed a well-known pattern of expansion and contraction within the first two weeks in mice treated solely with the primary CAR-T cells (Figures 3B, C; Supplementary Figure 3C). This phenomenon is commonly observed in CD19+ preclinical tumor models as well as in patients receiving anti-CD19 CAR-T cells, and, in our model, was triggered by the endogenous B cells. In sharp contrast, as anticipated from our previous data, this kinetic did not occur in presence of the anti-CAR CAR-T cells. Moreover, we did not detect anti-CD19 CAR-T cells throughout the observation period, further indicating the persistent functionality of the treatment over time (Figures 3B, C; Supplementary Figure 3C). While we observed peripheral expansion and contraction of anti-CAR CAR-T cells in mice receiving both CAR-T cell types, this occurred to a much lesser extent. The high circulating levels of the anti-CAR CAR-T cells when transferred alone, instead, were a sign of T cells with no specificity in the used experimental model (Figures 3B–D; Supplementary Figure 3C). In secondary lymphoid tissues, we found sustained depletion of anti-CD19 CAR-T cells, despite anti-CAR CAR-T cells were barely detectable (Figure 3E).
We finally investigated the endogenous B-cell compartment, which represents the off-tumor target of our primary CAR-T cells. In the anti-CAR-treated control group, B cells recovered within seven days post irradiation, while mice receiving anti-CD19 CAR-T cells experienced long-term aplasia, confirming that the addition of the Strep-tag in the hinge region did not affect functionality. Importantly, in mice receiving the combination of the two CAR-engineered T cells, B cells reconstituted early at lower frequency and eventually reached levels comparable to our control group approximately six weeks after infusion (Figures 3F, G).
In summary, our findings show that anti-CAR CAR-T cells can effectively deplete primary CAR-T cells in an immunocompetent host, leading to the reversal of the unwanted long-term B-cell aplasia induced by anti-CD19 CAR-T cells as a side effect.
3.4 Therapeutic anti-CAR CAR-T cell application reconstitutes B cells also in absence of pre-conditioning treatment
Our previous data proved the functionality and reliability of the anti-CAR approach in a model of simultaneous target engagement. Next, we sought to investigate whether it could effectively reverse a long-term side effect caused by a primary therapeutic CAR-T cell product. To address this question, we induced stable B-cell aplasia in wild-type immunocompetent C57BL/6 mice by administering anti-CD19 CAR-T cells, and then transferred anti-CAR CAR-T cells four weeks later (Figure 4A). Achieving successful engraftment of transferred T cells usually requires lymphodepletion, a step commonly used in clinical practice to enhance the effectiveness of CAR-based therapy in patients (33). Nevertheless, consenting patients for a second round of pre-conditioning, in particular in cases of disease remission, can be challenging. Therefore, we compared the efficiency of anti-CAR CAR-T cell transfer with and without pre-conditioning, using different cell doses. Additionally, we included a CAR construct without the scFv (mock) as a negative control, and the monoclonal anti-EGFR Cetuximab as a positive control, known for depleting adoptively transferred EGFRt-expressing CAR-T cells and promoting B-cell reconstitution (24) (Figure 4A; Supplementary Figure 4A).
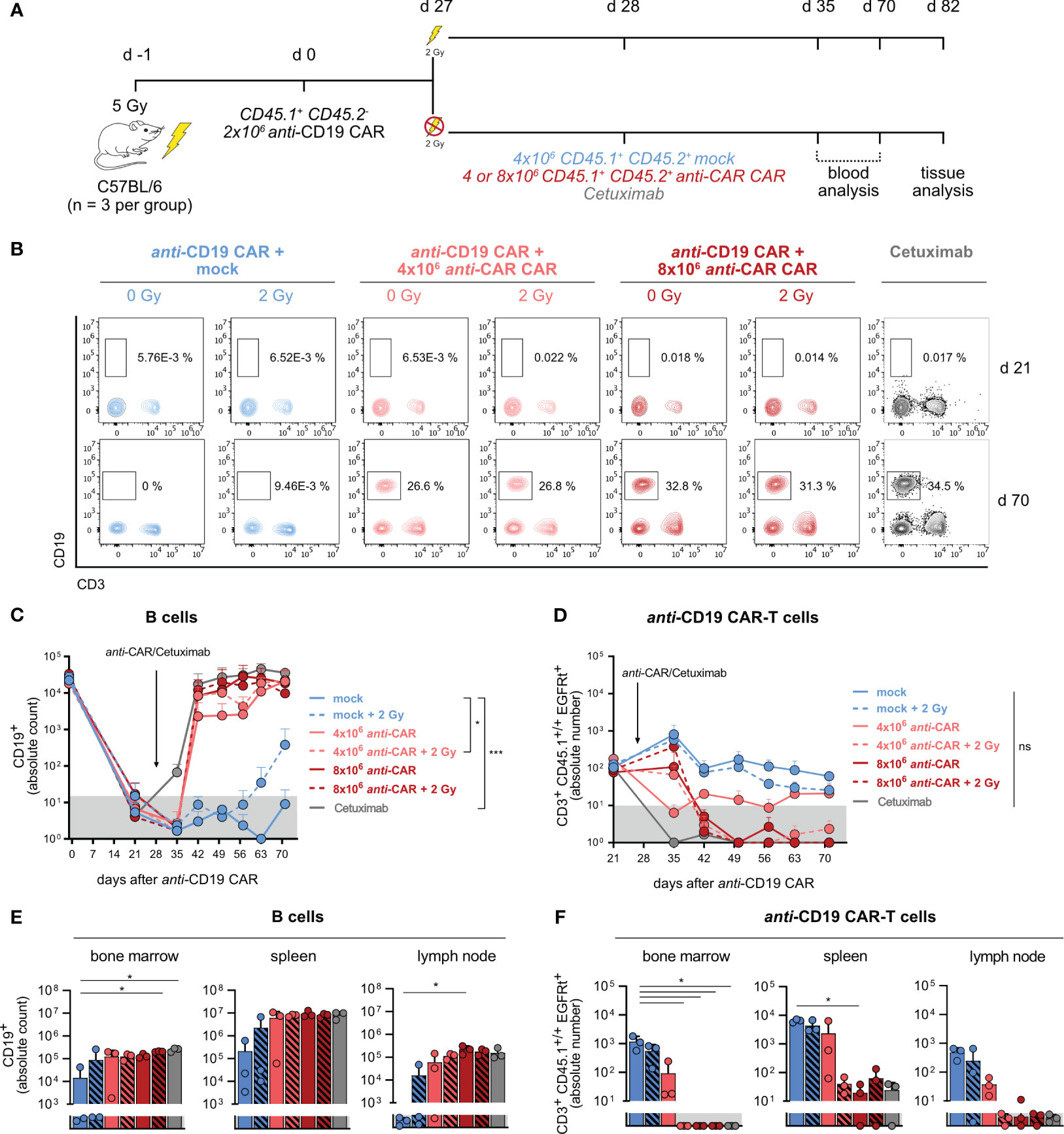
Figure 4 Reliable B-cell reconstitution after therapeutic anti-CAR application also in the absence of pre-conditioning treatment. (A) Schematic of the experimental setup (n=3). (B) Representative flow cytometry plots of B cells one week prior (upper) and six weeks after (lower) anti-CAR CAR-T cell transfer. (C, D) Quantification of B cells (C) and anti-CD19 CAR-T cells (D) in blood. (E, F) Absolute numbers of B cells (E) and anti-CD19 CAR-T cells (F) in secondary lymphoid organs at the endpoint. Data are shown as mean + SD. In (C, D), statistical analyses have been performed by two-way Anova with Dunnett’s multiple comparisons test using the mock group as reference. In (E, F), statistical analyses have been performed by Kruskal-Wallis test with Dunnett’s multiple comparisons test using the mock group as reference. ns = p > 0.05, *p < 0.05, ***p < 0.001.
All mice showed B-cell aplasia until 21 days from anti-CD19 CAR-T cell transfer, which was the last time point of monitoring prior to the second T-cell transfer. Mice receiving anti-CAR CAR-T cells exhibited reliable B-cell reconstitution as early as two weeks post-administration, reaching levels comparable to Cetuximab-treated mice (Figures 4B, C). Noteworthy, there were no significant differences among the four anti-CAR-treated groups, except in mice receiving the lower T-cell dose without pre-conditioning. However, despite a slower reconstitution kinetic, B-cell frequencies in this group eventually reached comparable levels with the other groups at later time points. Importantly, mice receiving the control mock CAR-T cells maintained stable B-cell aplasia throughout the observation period also in combination with the second pre-conditioning, except for one mouse (Figures 4B, C). This observation suggested that anti-CD19 CAR-T cells reliably persisted and remained functional after irradiation. Moreover, it also attributed the B-cell reconstitution to the specific depletion of anti-CD19 CAR-T cells by the secondary safeguard CAR-T cells. This was further supported by the clear presence of anti-CD19 CAR-T cells in mice receiving the mock CAR-T cells at both early (day 21) and late (day 70) time points, regardless of irradiation, while anti-CAR-treated mice showed no detectable anti-CD19 CAR-T cells (Figure 4D; Supplementary Figure 4B).
Similarly to what observed in the peripheral circulation, tissue analysis revealed high B-cell levels and undetectable anti-CD19 CAR-T cells also in the spleen, lymph nodes and bone marrow of anti-CAR CAR-T cell treated mice. Intriguingly, the group receiving low-dose anti-CAR without pre-conditioning showed more variable B-cell levels and persisting infiltrating anti-CD19 CAR-T cells (Figures 4E, F), despite no detectable B cells in the blood.
We finally examined the persistence of the anti-CAR CAR-T cells and observed a clear relationship between the cell dose and pre-conditioning. As expected, irradiation facilitated engraftment of the transferred cells regardless of cell doses. Indeed, circulating anti-CAR CAR-T cells remained detectable only in irradiated mice (Figures 5A, B), with the highest frequencies observed in mice receiving 8x106 cells plus irradiation in both blood and tissues (Figure 5). Intriguingly, the higher cell dose compensated for the absence of pre-conditioning, at least during the initial weeks in the circulation, as indicated by comparable levels between the groups 8x106 cells vs 4x106 cells plus irradiation (Figure 5B). Although the amount of anti-CAR CAR-T cells declined to undeletable levels in the blood of mice receiving this high cell dose at later time points, tissue infiltration remained consistent and comparable to the other groups (Figure 5C). Taking into account the similar kinetics of B-cell reconstitution and complete anti-CD19 CAR-T cell depletion, we could conclude that this condition (8x106 cells, no irradiation) would be still efficient.

Figure 5 anti-CAR CAR-T cells persist also in absence of pre-conditioning. (A) Representative flow cytometry plots of anti-CAR CAR-T cells one week (upper) and six weeks after (lower) anti-CAR treatment. (B, C) Quantification of anti-CAR CAR-T cells in blood (B) and lymphoid tissues at the endpoint (C). Data are shown as mean+SD. Grey area indicates the detection limit. In (B) statistical analyses have been performed by two-way Anova with Dunnett’s multiple comparisons test using the 4x106 cells group as reference. In (C) statistical analyses have been performed by Kruskal-Wallis test with Dunnett’s multiple comparisons test using the ‘4x106 cells’ group as reference. ns = p > 0.05, **p < 0.01.
In summary, our findings demonstrate that administration of anti-CAR CAR-T cells can reverse established B-cell aplasia in a model simulating a possible therapeutic application, potentially eliminating the need for pre-conditioning before T-cell transfer.
4 Discussion
In this study, we investigated a cellular-based mechanism for the depletion of adoptively transferred CAR-T cells leveraging on the use of a second transgenic receptor-engineered T cell. Our concept featured a CAR targeting a short 8 amino acid peptide sequence (WSHPQFEK) called Strep-tag II, which was located in the hinge region of an anti-CD19 CAR. Targeting this small tag delivered a highly specific cytokine release from the anti-CAR CAR-T cells that ultimately translated into efficient cytotoxicity, restricted only to the Strep-tagged target cells. In two syngeneic mouse models of simultaneous injection of anti-CAR and tagged anti-CD19 CAR-T cells, the safeguard CAR-T cells proved functional, infiltration of secondary lymphoid organs and rapid B-cell reconstitution. More importantly, a later administration of anti-CAR CAR-T cells could reverse established B-cell aplasia induced by anti-CD19 CAR-T cells also in the absence of pre-conditioning regimes.
CAR-T cells against CD19 antigen for relapsed and refractory B-cell malignancies paved the way to cell therapy with CAR-engineered T cells. It should be considered that a significant reason for this success is due to the exclusive expression of the CD19 antigen. Being a unique B-cell lineage marker (34), side effects related to anti-CD19 CAR-T cells have been overall tolerable considering the clinical benefits received by patients with such advanced and aggressive tumors. Indeed, the initially life-threatening acute toxicities (CRS and CRES) (10, 11) that develop within few days after the T-cell transfer have become to a greater extent manageable (13) without any need to deplete the therapeutic living drug. In addition, new short-term manufacturing protocols for the production of CAR-T cells allowed the administration of much lower cell doses while maintaining promising clinical outcomes, presumably due to preserved T cell fitness and functionality during ex vivo manipulation (35, 36). As higher CAR-T cell doses usually mediate more severe side effects (37, 38), these improvements in manufacturing should further lower the risks of acute toxicities.
In contrast, on-target/off-tumor side effects due to shared tumor-associated antigens still pose a major threat, in particular for solid tumors (39, 40). Patients receiving CD19 CAR-T cells showed long-term depletion of healthy B cells and consequent hypogammaglobulinemia (5, 32). While manageable via replacement therapy, these low antibody levels could still potentially predispose CAR-treated patients to severe infections (14). Moreover, the incapacity of building de novo B-cell responses could limit the establishment of functional immunity against new pathogens, as observed during the SARS-CoV-2 pandemic (41). The initial expansion and persistence of CD19 CAR-T cells are crucial for achieving clinical responses (42). Still, their long-term maintenance might not be essential for sustained complete remission. Indeed, a fraction of patients with 5-year complete remission had undetectable CD19 CAR-T cells and recovered B cells already within the first 1-2 years (43). Altogether, while more studies are necessary to clarify the dynamics between CD19 CAR-T cells and endogenous B cells, the existing evidence at least raises the question of whether the maintenance of CD19 CAR-T cells is necessary after years of complete remission at the expense of a functional B-cell compartment.
Precise and complete abrogation of adoptively transferred CAR-T cells might become thus necessary to revert long-term toxicity. Among possible strategies of cell depletion in patients, the use of CAR-T cells engineered with additional suicide genes or that co-express a second targetable marker showed to be efficient but with some limitations, i.e. immunogenicity and incomplete cell eradication (21), high levels of the transgene (19), dependency on the patient’s immune system, tissue penetration and recognition of shared antigens on healthy cells (23). Similarly to others (44), we opted for a T cell-mediated recognition of tagged therapeutic CAR-T cells from a safeguard anti-CAR CAR-T cells, as it harbors the potential of overcoming the limitations mentioned before.
In line with existing evidence (44), we validated the feasibility of the approach in vitro. In addition, we carefully investigated the in vivo efficacy, persistence and tissue infiltration of our anti-CAR CAR-T cells, which would represent a unique benefit of the approach. We could show that anti-CAR CAR-T cells specifically recognize and eliminate anti-CD19 CAR-T cells in the periphery as well as in relevant secondary lymphoid organs. One of the most intriguing observation was that the transfer of anti-CAR CAR-T cells was efficacious also in the absence of lymphodepletion. It is believed that this step of pre-conditioning is crucial for the successful engraftment and maintenance of CAR-T cells (33), and indeed it is routinely performed in the clinic. In our data, we could confirm the beneficial effect of pre-conditioning on T-cell engraftment, as indicated by the higher frequency of circulating anti-CAR CAR-T cells in irradiated mice. Anti-CAR CAR-T cells were barely detectable (low T cell doses) or vanished (high T cell doses) in the periphery without pre-conditioning, but they reliably persisted in lymphoid organs. In line with this, we observed efficient B-cell recovery also in the absence of pre-conditioning, despite the co-existence of anti-CD19 CAR-T and B cells in the spleen and bone marrow of mice treated with the low cell dose. It happened also in patients receiving CD19 CAR-T cells to observe low levels of the transferred cells around the detection limit but still consistent B cell depletion (45), indicating that blood levels do not always reflect the actual persistence of CAR-T cells in disease-relevant organs. Overall, while the long-term maintenance of anti-CAR CAR-T cells in the absence of pre-conditioning may still be questionable, our data indicate that relatively high cell doses could still support sufficient engraftment to efficiently deplete target cells.
One limitation of the proposed approach is the slower rate of anti-CD19 CAR-T cell elimination compared to the use of Cetuximab. This time delay is less significant when such an approach is employed to revert long-term on-target/off-tumor toxicities that do not pose an immediate threat to the patient’s life, such as B-cell aplasia in CD19 CAR-treated patients. In these specific cases, anti-CAR CAR-T cells offer an alternative to mitigate potential safety concerns associated with Cetuximab. Indeed, as EGFR is expressed also in healthy tissues of epithelial, mesenchymal and neuronal origin, Cetuximab poses the risk of relevant toxicities e.g. skin and gastrointestinal toxicities (46). However, in situations involving life-threatening toxicities, more immediate responses are advisable. Another limitation to consider is that the persistence of anti-CAR CAR-T cells after antigen encounter was observed primarily at relatively high doses. Although we did not detect any signs of tonic signaling that might suggest their susceptibility to early dysfunction, further studies are needed to provide more clarity on these observations.
In summary, we demonstrated that it is feasible to generate a safeguard mechanism that exploits the specificity and sensitivity of CAR-T cells to deplete tagged therapeutic CAR-T cells. The proposed approach is generalizable to both other tags and CAR-T cells targeting different tumor-associated antigens, and might therefore be broadly applicable to enhance control over highly potent cell therapies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the district government of Upper Bavaria (Department 5—Environment, Health and Consumer Protection ROB- 55.2-2532.Vet_02-17-138). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LW: Resources, Writing – review & editing. MS: Formal Analysis, Investigation, Methodology, Resources, Visualization, Conceptualization, Writing – original draft. SD: Resources, Writing – review & editing, Formal Analysis, Investigation, Methodology, Visualization. SF: Writing – review & editing, Resources. MT: Resources, Writing – review & editing. SR: Resources, Writing – review & editing. UJ: Resources, Writing – review & editing. ED: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing. DB: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB-TRR 338/1 2021 -452881907 (project A01), NIHCA 18029, and CA114536 (SRR). DB, LW and UJ also received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No. 116026. (T2EVOLVE).
Acknowledgments
We thank our flow cytometry unit for cell sorting, especially I. Andrä, C. Angerpointner, and M. Schiemann. We also thank members of the Dirk H Busch and Veit Buchholz laboratories for experimental help and critical discussions.
Conflict of interest
DB is co-founder of STAGE Cell Therapeutics GmbH now Juno Therapeutics, a Bristol-Myers Squibb Company and T Cell Factory B.V. now Kite/a Gilead Company. DB has a consulting contract with and receives sponsored research support from Juno Therapeutics/BMS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1268698/full#supplementary-material
References
1. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
2. D’Ippolito E, Schober K, Nauerth M, Busch DH. T cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer Immunol Immunother (2019) 68(10):1701–12. doi: 10.1007/s00262-019-02395-9
3. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi: 10.1126/scitranslmed.3005930
4. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
5. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
6. Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic review on CAR-T cell clinical trials up to 2022: academic center input. Cancers (Basel) (2023) 15(4):1003. doi: 10.3390/cancers15041003
7. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-Tumor efficacy of human chimeric antigen receptor T cells. Sci Rep (2017) 7(1):1–11. doi: 10.1038/s41598-017-00462-8
8. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167(2):419–432.e16. doi: 10.1016/j.cell.2016.09.011
9. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell (2019) 35(3):489–503.e8. doi: 10.1016/j.ccell.2019.02.003
10. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
11. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
12. Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol (2021) 22(10):1403–15. doi: 10.1016/S1470-2045(21)00375-2
13. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Locke FL, Lin Y, et al. Toxicity management after chimeric antigen receptor T cell therapy: One size does not fit “ALL ”. Nat Rev Clin Oncol (2018) 15(4):218. doi: 10.1038/nrclinonc.2018.20
14. Wat J, Barmettler S. Hypogammaglobulinemia after chimeric antigen receptor (CAR) T-cell therapy: characteristics, management, and future directions. J Allergy Clin Immunol Pract (2022) 10(2):460–6. doi: 10.1016/j.jaip.2021.10.037
15. Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica (2020) 105:297–316. doi: 10.3324/haematol.2019.229781
16. Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood (2016) 128(3):360–70. doi: 10.1182/blood-2016-01-694356
17. Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol (2019) 12(1):1–10. doi: 10.1186/s13045-019-0813-7
18. Andrea AE, Chiron A, Bessoles S, Hacein-Bey-abina S. Engineering next-generation car-t cells for better toxicity management. Int J Mol Sci (2020) 21(22):1–25. doi: 10.3390/ijms21228620
19. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood (2005) 105(11):4247–54. doi: 10.1182/blood-2004-11-4564
20. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Sci (1997) 276(5319):1719–24. doi: 10.1126/science.276.5319.1719
21. Berger C, Flowers ME, Warren EH, Riddell SR, Berger C, Flowers ME, et al. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK – modified donor T cells after allogeneic hematopoietic cell transplantation. Blood (2012) 107(6):2294–302. doi: 10.1182/blood-2005-08-3503
22. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi: 10.1056/NEJMoa1106152
23. Wang X, Chang WC, Wong CLW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood (2011) 118(5):1255–63. doi: 10.1182/blood-2011-02-337360
24. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest (2016) 126(11):4262–72. doi: 10.1172/JCI84813
25. Dötsch S, Svec M, Schober K, Hammel M, Wanisch A, Gökmen F, et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc Natl Acad Sci (2023) 120(10):2017. doi: 10.1073/pnas.2216830120
26. Shabaneh TB, Moffett HF, Stull SM, Derezes T, Tait LJ, Park S, et al. Safety switch optimization enhances antibody-mediated elimination of CAR T cells. Front Mol Med (2022) 2. doi: 10.3389/fmmed.2022.1026474
27. Skerra A, Schmidt TGM. Use of the Strep-tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol (2000) 326:271–304. doi: 10.1016/S0076-6879(00)26060-6
28. Liu L, Sommermeyer D, Cabanov A, Kosasih P, Hill T, Riddell SR. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat Biotechnol (2016) 34(4):430–4. doi: 10.1038/nbt.3461
29. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res (2015) 3(2):125–35. doi: 10.1158/2326-6066.CIR-14-0127
30. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res (2013) 19(12):3153–64. doi: 10.1158/1078-0432.CCR-13-0330
31. Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PloS One (2013) 8(4):e61338. doi: 10.1371/journal.pone.0061338
32. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
33. Amini L, Silbert SK, Maude SL, Nastoupil LJ, Ramos CA, Brentjens RJ, et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol (2022) 19(5):342–55. doi: 10.1038/s41571-022-00607-3
34. Pontvert-Delucq S, Breton-Gorius J, Schmitt C, Baillou C, Guichard J, Najman A, et al. Characterization and functional analysis of adult human bone marrow cell subsets in relation to B-lymphoid development. Blood (1993) 82(2):417–29. doi: 10.1182/blood.V82.2.417.417
35. Flinn IW, Jaeger U, Shah NN, Blaise D, Briones J, Shune L, et al. A first-in-human study of YTB323, a novel, autologous CD19-directed CAR-T cell therapy manufactured using the novel T-charge TM platform, for the treatment of patients (Pts) with relapsed/refractory (r/r) diffuse large B-cell lymphoma (DLBCL). Blood (2021) 138(Supplement 1). doi: 10.1182/blood-2021-146268
36. Yang J, He J, Zhang X, Li J, Wang Z, Zhang Y, et al. Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J (2022) 12(7):1–9. doi: 10.1038/s41408-022-00694-6
37. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
38. Frigault M, Rotte A, Ansari A, Gliner B, Heery C, Shah B. Dose fractionation of CAR-T cells. A systematic review of clinical outcomes. J Exp Clin Cancer Res (2023) 42(1):1–10. doi: 10.1186/s13046-022-02540-w
39. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
40. Lamers CHJ, Sleijfer S, Van Steenbergen S, Van Elzakker P, Van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. doi: 10.1038/mt.2013.17
41. Gaitzsch E, Passerini V, Khatamzas E, Strobl CD, Muenchhoff M, Scherer C, et al. COVID-19 in patients receiving CD20-depleting immunochemotherapy for B-cell lymphoma. HemaSphere (2021) 5(7):e603. doi: 10.1097/HS9.0000000000000603
42. Faude S, Wei J, Muralidharan K, Xu X, Wertheim G, Paessler M, et al. Absolute lymphocyte count proliferation kinetics after CAR T-cell infusion impact response and relapse. Blood Adv (2021) 5(8):2128–36. doi: 10.1182/bloodadvances.2020004038
43. Chong EA, Ruella M, Schuster SJ. Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N Engl J Med (2021) 384(7):673–4. doi: 10.1056/NEJMc2030164
44. Koristka S, Ziller-Walter P, Bergmann R, Arndt C, Feldmann A, Kegler A, et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol Immunother (2019) 68(9):1401–15. doi: 10.1007/s00262-019-02376-y
45. Peinelt A, Bremm M, Kreyenberg H, Cappel C, Banisharif-Dehkordi J, Erben S, et al. Monitoring of circulating CAR T cells: validation of a flow cytometric assay, cellular kinetics, and phenotype analysis following tisagenlecleucel. Front Immunol (2022) 13(March):1–12. doi: 10.3389/fimmu.2022.830773
Keywords: chimeric antigen receptor, safeguard mechanism, side effects, on-target/off-tumor, B cell aplasia 2
Citation: Svec M, Dötsch S, Warmuth L, Trebo M, Fräßle S, Riddell SR, Jäger U, D’Ippolito E and Busch DH (2024) A chimeric antigen receptor-based cellular safeguard mechanism for selective in vivo depletion of engineered T cells. Front. Immunol. 14:1268698. doi: 10.3389/fimmu.2023.1268698
Received: 28 July 2023; Accepted: 27 November 2023;
Published: 11 January 2024.
Edited by:
Michael Hudecek, University Hospital Würzburg, GermanyReviewed by:
Thomas Nerreter, University Hospital Würzburg, GermanyReona Leo Sakemura, Mayo Clinic, United States
Copyright © 2024 Svec, Dötsch, Warmuth, Trebo, Fräßle, Riddell, Jäger, D’Ippolito and Busch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk H. Busch, ZGlyay5idXNjaEB0dW0uZGU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship