
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Immunol., 12 January 2024
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1268144
This article is part of the Research TopicInflammatory Response and Immune Disorder in Rheumatic and Musculoskeletal DiseasesView all 10 articles
 Sara Martínez-Ramos1,2
Sara Martínez-Ramos1,2 Carlos Rafael-Vidal1,2
Carlos Rafael-Vidal1,2 Beatriz Malvar-Fernández1,2Nair Pérez1,2Coral Mouriño1,2Sara García Pérez1,2Francisco J. Maceiras Pan1,2
Beatriz Malvar-Fernández1,2Nair Pérez1,2Coral Mouriño1,2Sara García Pérez1,2Francisco J. Maceiras Pan1,2 Carmen Conde3
Carmen Conde3 Jose María Pego-Reigosa1,2
Jose María Pego-Reigosa1,2 Samuel García1,2*
Samuel García1,2*Previous works from our group show that Semaphorin3B (Sema3B) is reduced in RA and plays a protective role in a mouse arthritis model. In turn, MerTK plays a protective function in murine arthritis models, is expressed by synovial tissue macrophages and is linked to remission in patients with RA. In this study, we examined the role of Sema3B in the phenotypic characteristics of RA macrophages and the implication of MerTK. Peripheral blood monocytes from RA patients were differentiated into IFN-γ (RA MØIFN-γ) or M-CSF (RA MØM-CSF) macrophages and stimulated with LPS, Sema3B or their combination. Alternatively, RA fibroblast like synoviocytes (FLS) were stimulated with RA MØIFN-γ and RA MØM-CSF supernatants. Gene expression was determined by qPCR and protein expression and activation by flow cytometry, ELISA and western blot. Sema3B down-regulated the expression of pro-inflammatory mediators, in both RA MØIFN-γ and RA MØM-CSF. We observed a similar reduction in RA FLS stimulated with the supernatant of Sema3B-treated RA MØIFN-γ and RA MØM-CSF. Sema3B also modulated cell surface markers in macrophages towards an anti-inflammatory phenotype. Besides, MerTK expression and activation was up-regulated by Sema3B, just as GAS6 expression, Resolvin D1 secretion and the phagocytic activity of macrophages. Importantly, the inhibition of MerTK and neuropilins 1 and 2 abrogated the anti-inflammatory effect of Sema3B. Our data demonstrate that Sema3B modulates the macrophage characteristics in RA, inducing a skewing towards an anti-inflammatory/pro-resolving phenotype in a MerTK-dependant manner. Therefore, here we identify a new mechanism supporting the protective role of Sema3B in RA pathogenesis.
Rheumatoid arthritis (RA) is a chronic autoimmune rheumatic and musculoskeletal disease (RMD) characterized by articular inflammation, bone erosion and cartilage destruction. Despite the advances in the last decades, current therapies only reach persistent responses in 30% of the patients (1).
Recent studies from our group have reported the relevance of Semaphorin3B (Sema3B) in the pathogenesis of RA. On one hand, Sema3B levels are reduced in the synovial tissue and serum of RA patients compared to arthralgia and undifferentiated arthritis patients, and these levels decrease during the progression of the disease. On the other hand, Sema3B reduces migration, invasion and the secretion of matrix metalloproteases (MMPs) in RA fibroblast-like synoviocytes (FLS). More importantly, Sema3B deficiency enhances the severity of serum-induced arthritis, while Sema3B administration abrogates this effect. This protective role is associated with a reduced mouse FLS migration and the expression of inflammatory mediators in the affected joints. We also found a reduced expression of the macrophage marker CD68, suggesting that Sema3B may modulate this cell population (2–4).
Macrophages are key mediators in RA and are involved in several pathogenic processes, including inflammation, angiogenesis and bone and cartilage destruction (5, 6) importantly, the number of synovial macrophages correlates with the clinical disease activity (7). Historically, macrophages have been classified as pro-inflammatory (M1) or wound healing/anti-inflammatory (M2) macrophages, although these are the extremes of a broad spectrum of intermediated states that depend on the environmental factors and the surrounding cells (5). Recent studies have reported the existence of several macrophage subsets in the synovium of RA patients with different functional roles. Notably, the frequency of these populations is also associated with the clinical status of the patients (8, 9).
Since the effect of Sema3B on RA macrophages is unknown, in the current study we examined the effect of Sema3B on the functional and phenotypic characteristics of this cell population.
Peripheral blood mononuclear cells (PBMCs) (n = 33) and FLS (n = 4) were obtained from blood and inflamed joints of RA patients, respectively. All subjects provided written informed consent, and the protocols were approved by the Ethics Committee of Galicia prior to patient inclusion in this study (studies numbers 2020/159 and 2021/03). RA patients fulfilled the American College of Rheumatology/European Alliance of Associations for Rheumatology 2010 classification criteria for RA (10). Clinical characteristics of patients are detailed in Supplementary Table S1.
PBMCs were obtained by Ficoll gradient (STEMCELL Technologies) and CD14+ monocytes were isolated by using the MagniSort Human pan-Monocyte isolation kit (Thermo Fisher Scientific). Monocytes were differentiated into RA macrophages (RA MØ) by culturing in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% of heat-inactivated fetal bovine serum (FBS, Corning™) and 10000 I.E penicillin-streptomycin (Lonza™ BioWhittaker™), in the presence of IFN-γ (10 ng/mL; R&D Systems; RA MØIFN-γ) or M-CSF (25 ng/mL; PeproTech; RA MØM-CSF) for 7 days. On one side, macrophages were differentiated in the presence or the absence of recombinant human (rh)Sema3B (200 ng/mL; R&D Systems) for 7 days. Conversely, RA MØIFN-γ and RA MØM-CSF were cultured for 7 days and stimulated for 24 h with LPS (10 ng/mL; InvivoGen) in the presence or absence of rhSema3B (200 ng/mL). RA MØIFN-γ and RA MØM-CSF were pre-incubated during 1 hour with neutralizing anti-Neuropilin-1 (α-NRP1) antibody (5µg/mL; R&D Systems), neutralizing anti-Neuropilin-2 (α-NRP2) antibody (5µg/mL; R&D Systems) or their respective isotype controls (sheep and goat IgG; 5µg/mL; R&D Systems), and stimulated with LPS (10 ng/mL) in the presence or absence of rhSema3B (200 ng/mL) for 24 h.
Alternatively, RA MØIFN-γ and RA MØM-CSF were also pre-incubated during 1 hour with DMSO or a specific MerTK inhibitor (100nM; UNC2881, Cayman Chemical) (11, 12) and stimulated with LPS in the presence or absence of rhSema3B for 24 h.
Cells were lysed for mRNA expression analysis or typsinized for Flow Cytometry. Cell-free supernatants were harvested for cytokine analysis.
RA FLS were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Lonza™ BioWhittaker™) containing 10% FBS, 200 mM Glutamine (Lonza™) and 10,000 U/mL penicillin-streptomycin (Thermo Fisher Scientific) and used between passages 6 to 10. Prior to stimulation, RA FLS were cultured overnight in DMEM containing 1% FBS. Afterwards, the cells were stimulated for 4 h with the supernatants (20%, v/v) from RA MØIFN-γ and RA MØM-CSF.
RNA from RA MØ and RA FLS was isolated employing the NucleoSpin RNA/Protein Mini kit (Macherey-Nagel). Total RNA was reverse-transcribed using iScript (Biorad). cDNA was amplified by qPCRs in duplicates using SYBR green (Biorad) and specific primers (Integrated DNA Technologies (IDT); Supplementary Table S2) with a CFX96 Touch Real-Time PCR Detection System (Biorad). Relative levels of gene expression were normalized to the expression of 2 housekeeping genes (GAPDH and B2M). The relative quantity (RQ) of mRNA was calculated by using the formula 2–ΔΔCt.
IL-12p70, IL-6, TNF (R&D Systems) and Resolvin D1 (Cayman Chemical) protein levels were measured by ELISA in cell-free supernatants, according to the manufacturing instructions.
Data were acquired on a CytoFLEX S analyser (Beckman Coulter). 10% of Anti-Hu Fc Receptor (Thermo Fisher Scientific) was used for avoiding non-specific binding. Macrophages were stained with Fixable Viability Dye eFluor for dead cell exclusion (e450; Thermo Fisher Scientific) and antibodies for CD14-PerCPCy5.5, CD64-FITC, CD80-BV510, CD86-PECy7, CD163-APC-Cy7, CD206-APC, HLA-DR-BV605 and MerTK-PE (all Biolegend). In the case of MerTK, Fluorescence Minus One (FMO) enabled the tagging of the positive population. After excluding debris, doublets and dead cells, cell populations were analyzed using CytExpert software and Cytobank platform (Beckman Coulter). Results were expressed as the Median Fluorescence Intensity (MFI).
Macrophage phagocytic activity was determined by the uptake of the fluorogenic substrate DQ Red BSA (FITC; Thermo Fisher Scientific). Stimulated RA MØIFN-γ and RA MØM-CSF were typsinized and plated in conical well plates. DQ Red BSA (20 ng/mL) was added for 0, 30 and 60 minutes. Uptake data were acquired on a CytoFLEX S analyser (Beckman Coulter) and results were expressed as percentage of DQ-BSA uptake versus 30 minutes basal uptake.
Protein from RA MØM-CSF was isolated by Laemmli buffer. Equal quantities of total protein were submitted to electrophoresis on Polyacrylamide gels and transferred to PVDF Transfer membranes (Thermo Fisher Scientific). Membranes were incubated (4°C, overnight) with primary antibodies for (p)hosphoMerTK (FabGennix) and β-actin (R&D Systems) in 4% Milk-TBS/T, washed and incubated in 2% Milk-TBS/T containing HRP-conjugated secondary antibody (anti-mouse IgG, Thermo Fisher Scientific). Protein was developed with ECL Western Blotting Substrate (Thermo Fisher Scientific) employing a ChemiDocTM MP System (Biorad). Densitometry analysis was performed by ImageJ software and relative protein expression was normalized to the values for β-actin.
The gene expression of Sema3B receptors in bone marrow-derived macrophages (BMDM) from wild type (WT) and MerTK deficient (Mertk-/-) mice was retrieved from array profiling data available at the Gene Expression Omnibus (GEO–NCBI; GSE205070) (13).
Statistical analysis was performed by using Windows GraphPad Prism 8 (GraphPad Software, Inc.). Normality was analysed by Shapiro-Wilk and Kolmogorov-Smirnov tests. The potential differences between experimental groups following normal distribution were analysed by One-way ANOVA and Paired t tests, as applicable. Data non-following normal distribution were analysed by Friedman’s test. P values < 0.05 were considered statistically significant.
We firstly analysed the effect of Sema3B in inflammatory macrophages (RA MØIFN-γ). Sema3B alone did not affect the expression of pro-inflammatory mediators, but significantly reduced the LPS-induced expression of IL12B, IL23, CD86, TNF and CCL2 , while moderately IL1B , IL6 and CXCL10 (Figure 1A). In the case of anti-inflammatory mediators, Sema3B induced the expression of STAB1, but did not modulate the expression of IL10, IL13 and TGFB (Supplementary Figure S1A).
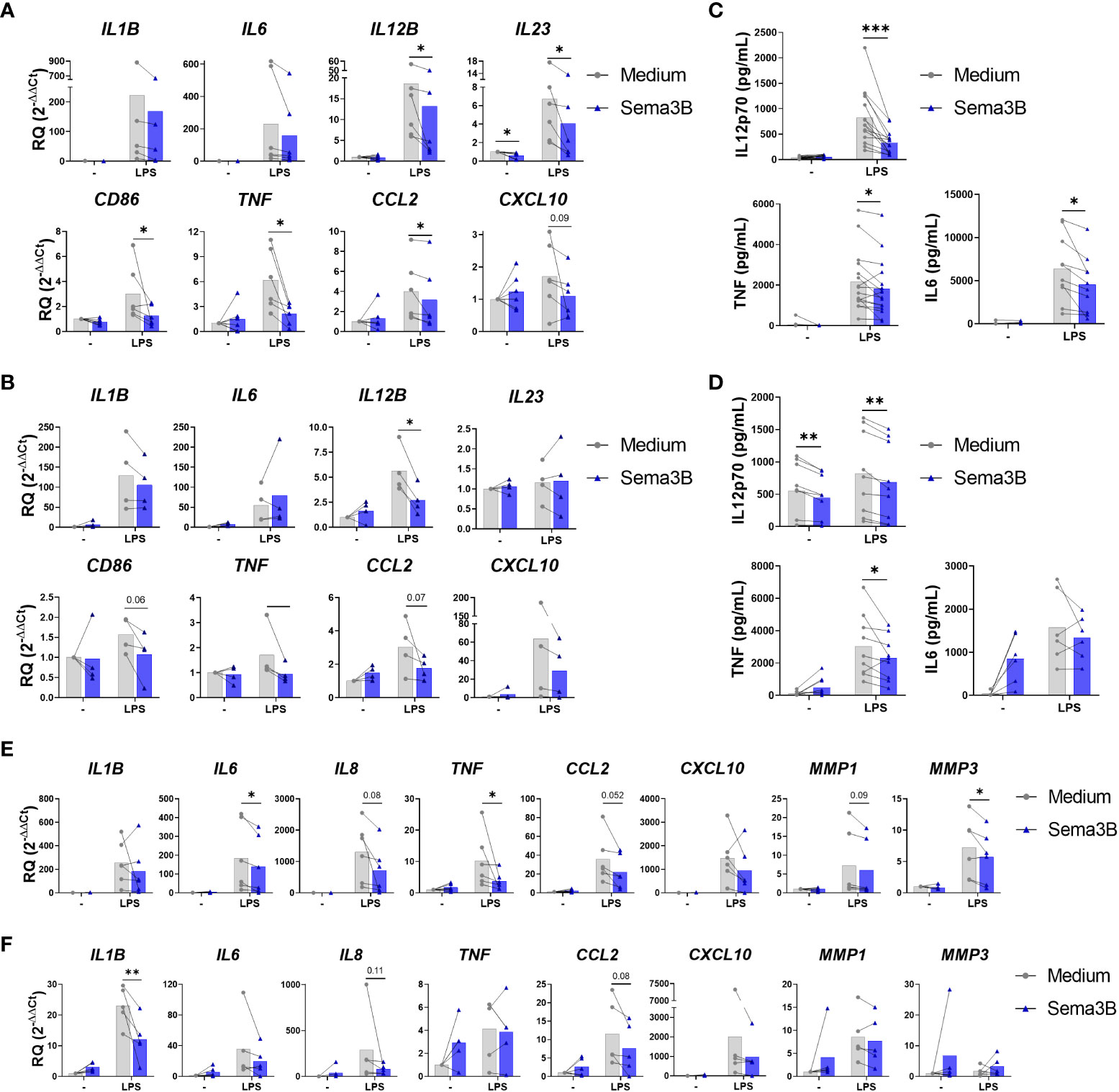
Figure 1 Sema3B reduces the inflammatory phenotype of RA MØ and RA FLS. (A–D) mRNA expression and protein secretion of inflammatory and anti-inflammatory mediators in RA MØIFN-γ [n = 6 – 16] (A, C) and RA MØM-CSF [n = 4 – 9] (B, D) stimulated with rhSema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. (E, F) mRNA expression of inflammatory mediators in RA FLS stimulated with RA MØIFN-γ [n = 6] (E) and RA MØM-CSF [n = 5] (F) supernatants [20%, v/v] for 4 h. Data are shown as RQ (relative quantity) respect to unstimulated cells and serum concentration (pg/mL), and analysed by One-way ANOVA tests. Means and SEM are shown. *P < 0.05 and **P < 0.01 and ***P < 0.001.
The influence of Sema3B was also determined on RA MØ differentiated with M-CSF, which promotes an anti-inflammatory phenotype (14). Similarly to RA MØIFN-γ, Sema3B alone did not affect the expression of pro-inflammatory mediators in RA MØM-CSF, but significantly down-regulated the LPS-induced expression of IL12B , besides reducing CD86, TNF and CCL2 (Figure 1B). In contrast, the anti-inflammatory mediators STAB1 and TGFB were up-regulated by Sema3B, alone and in combination with LPS (Supplementary Figure S1B).
We validated these findings at the protein level in both RA MØIFN and RA MØM-CSF, in which Sema3B reduced the LPS-induced secretion of IL12p70, TNF-α and, in RA MØIFN-γ specifically, IL-6 (Figures 1C, D).
With the aim of determining whether the anti-inflammatory effect of Sema3B could have functional consequences on effector cells, RA FLS were stimulated with cell-free supernatants from RA MØIFN-γ and RA MØM-CSF. Interestingly, supernatants of those macrophages stimulated with LPS in combination with Sema3B reduced the expression of IL1B, IL6, IL8, TNF, CCL2, CXCL10, MMP1 and MMP3 compared to supernatants of LPS-stimulated macrophages, although differences were not significant for all mediators (Figures 1E, F).
The effect of Sema3B on the modulation of M1 (CD80, CD86 and HLA-DR) and M2 (CD163 and CD206) surface markers expressed by RA synovial macrophages was also evaluated (6). In RA MØIFN-γ, Sema3B alone promoted CD64 and CD206 expression and, in LPS-stimulated macrophages, reduced the expression of CD64, HLA-DR and, significatively, CD86 (Figure 2A). In the case of RA MØM-CSF, we found that, in combination with LPS, Sema3B decreased the expression of CD64 and a trend for CD80 (Figure 2B). We also determined whether Sema3B was able to modulate the expression of specific markers during the macrophage differentiation process. Sema3B significantly down-regulated the expression of HLA-DR in IFN-γ-differentiated macrophages, but it did not modulate M2 markers. During the differentiation process with M-CSF, Sema3B reduced the expression of HLA-DR and CD64, while it significantly induced the expression of CD163 (Supplementary Figure S2).
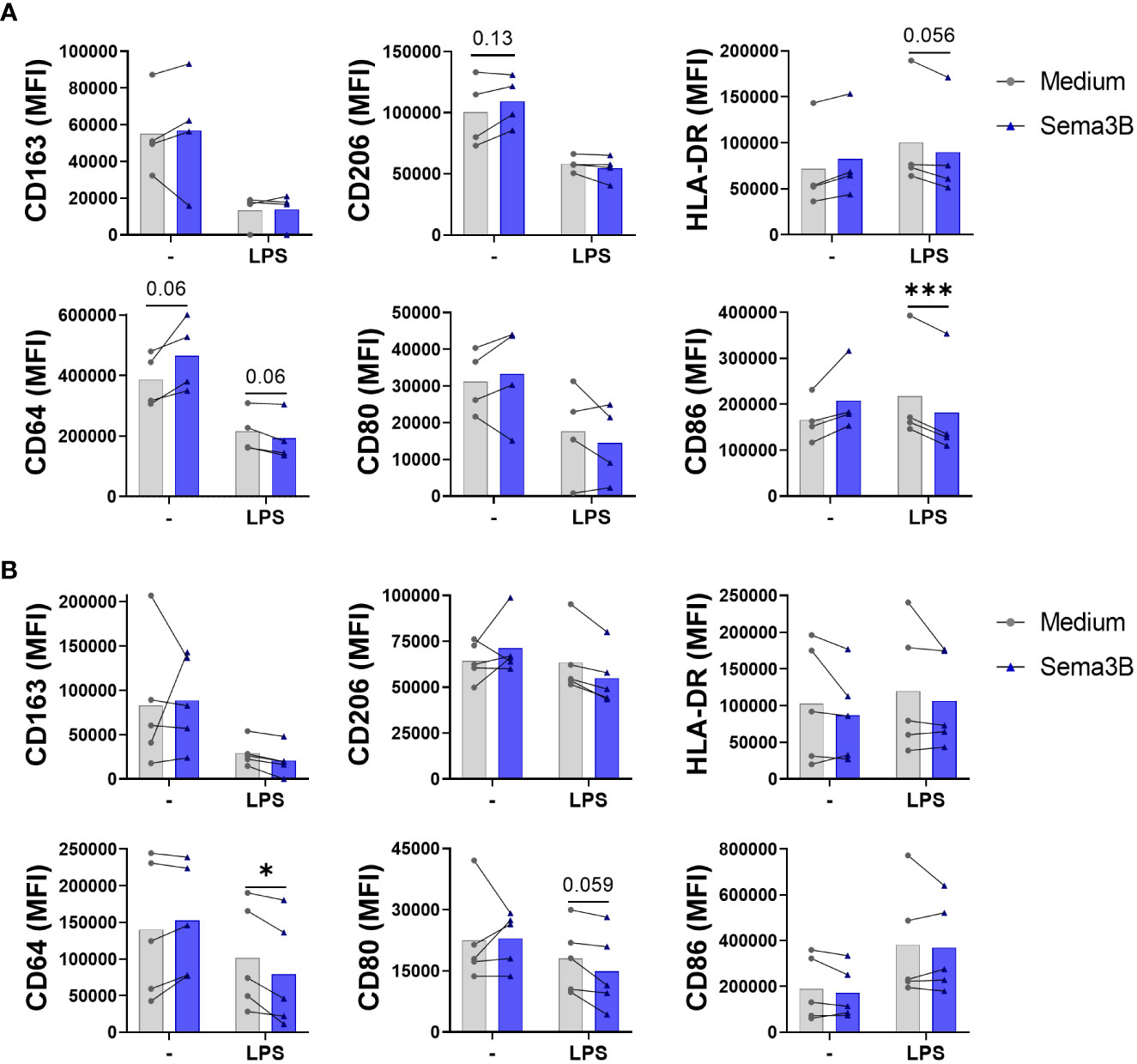
Figure 2 Sema3B promotes an anti-inflammatory phenotype in RA MØ. (A, B) CD163, CD206, HLA-DR, CD64, CD80 and CD86 cell surface marker expression in RA MØIFN-γ [n = 4] (A) and RA MØM-CSF [n = 5] (B) stimulated with rhSema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. Data are shown as MFI (Median Fluorescence Intensity) of cells and analysed by Friedman (A) One-way ANOVA (B) tests. Means and SEM are shown. *P < 0.05, ***P < 0.001.
Altogether, these data demonstrate an anti-inflammatory effect of Sema3B in RA MØ, by reducing the expression of the inflammatory phenotype and modulating the phenotypic characteristics of RA MØ.
We next sought the mechanisms involved in the anti-inflammatory effect of Sema3B. Sema3B did not modulate the expression of its known receptors, PlexinA1 and PlexinA2, and co-receptors, Neuropilin-1 (NRP-1) and Neurolipin-2 (NRP-2) in either RA MØIFN-γ or RA MØM-CSF (15, 16) (Figures 3A, B). However, the neutralization of both NRP-1 and NRP-2 abolished the Sema3B-mediated decrease of IL12p70 in RA MØIFN-γ, demonstrating that both Sema3B co-receptors are involved in the anti-inflammatory role of Sema3B (Figure 3C).
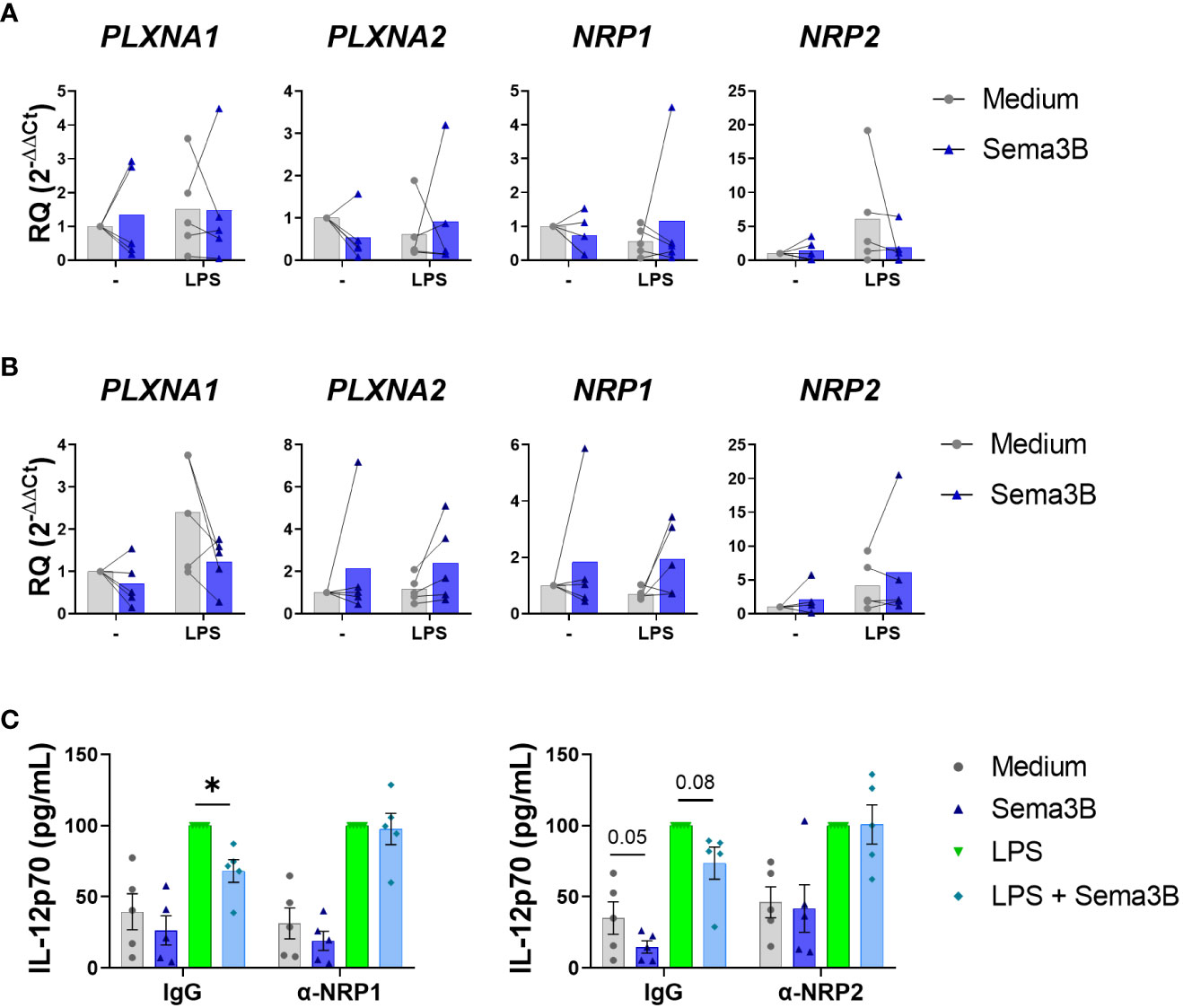
Figure 3 The protective effect of Sema3B in RA MØ is mediated by the co-receptors NRP1 and NRP2. (A, B) mRNA expression of Sema3B receptors in RA MØIFN-γ [n = 5] (A) and RA MØM-CSF [n = 5] (B) stimulated with rhSema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. (C) IL12p70 secretion in RA MØIFN-γ [n = 5] pre-incubated with neutralizing α-NRP1, α-NRP2 antibodies [5µg/mL] or their respective isotype controls, and stimulated with LPS [10 ng/mL] in the presence or absence of rhSema3B [200 ng/mL] for 24 h. Data are shown as RQ (relative quantity) respect to unstimulated cells and serum concentration (pg/mL) and analysed by One-way ANOVA tests. Means and SEM are shown. *P < 0.05.
Due to the crucial role of MerTK in resolving inflammation in RA (9, 17, 18), the involvement of Sema3B on the expression of this tyrosine kinase receptor was also evaluated. Sema3B alone, but not in combination with LPS, up-regulated the mRNA and protein expression of MerTK, and the mRNA expression of the MerTK ligand GAS6 (19–21) in both RA MØIFN-γ (Figure 4A) and RA MØM-CSF (Figure 4B), although differences were more pronounced in the latter. Sema3B also increased MerTK expression during the differentiation process into RA MØM-CSF (Supplementary Figure S2B). Importantly, Sema3B induced the activation of MerTK in RA MØM-CSF at different time points, being this effect significant at 4 hours (Figure 4C).
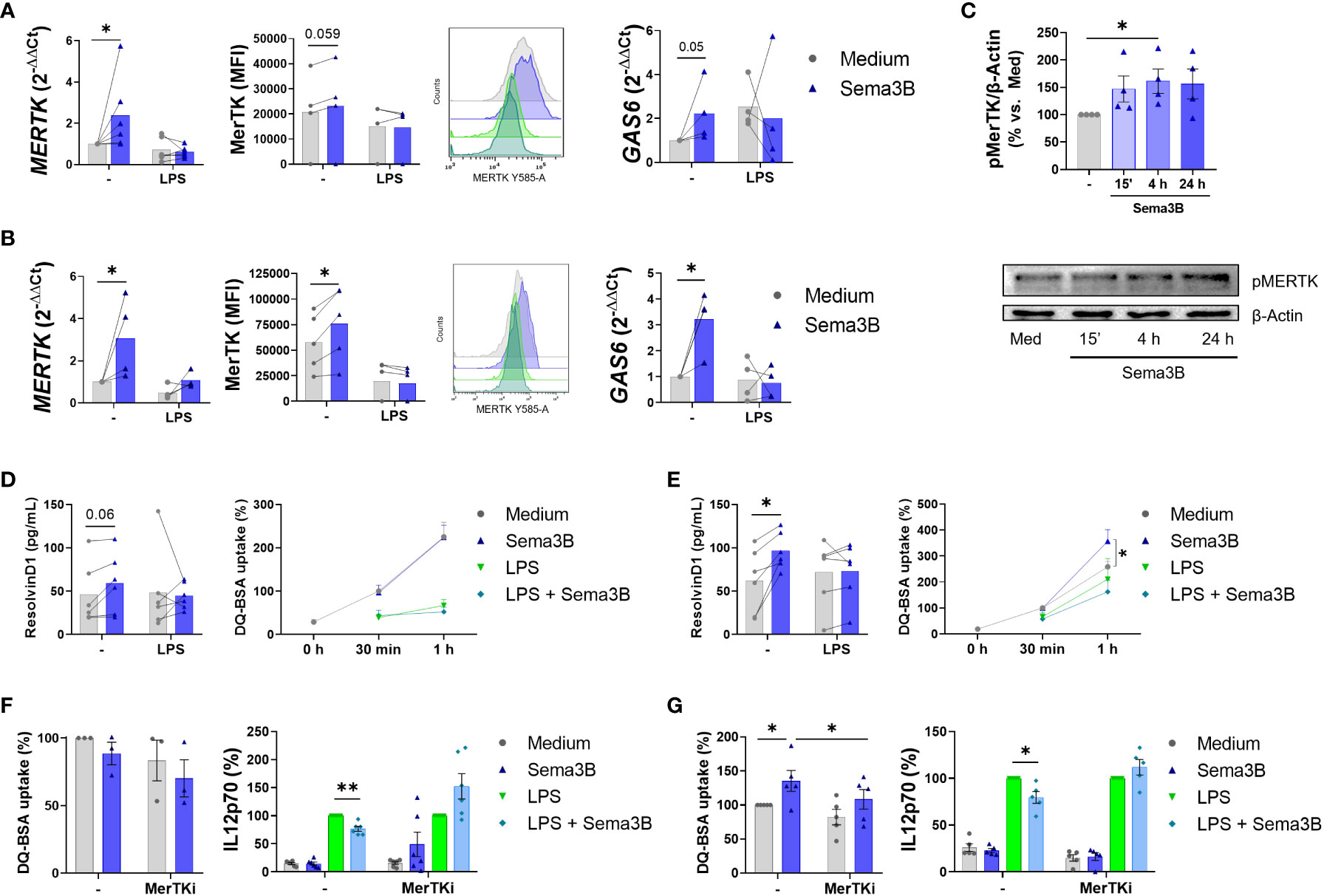
Figure 4 Sema3B induces an anti-inflammatory/pro-resolving phenotype in RA MØ via MerTK. (A, B) MerTK and GAS6 mRNA levels and MerTK cell surface expression in RA MØIFN-γ [n = 4 – 6] (A) and RA MØM-CSF [n = 4 – 5] (B) stimulated with Sema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. (C) Densitometric analysis and representative immunoblot of MertK activation ((p)hosphoMerTK) in RA MØM-CSF [n = 4] stimulated with rhSema3B [200 ng/mL] for 15 minutes, 4 and 24 hours. (D, E) Resolvin D1 secretion and phagocytic activity in RA MØIFN-γ [n = 4 – 6] (D) and RA MØM-CSF [n = 5 – 6] (E) after stimulation with rhSema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. (F, G) Phagocytic activity and IL12p70 secretion in RA MØIFN-γ [n = 3 – 6] (F) and RA MØM-CSF [n = 5] (G) pre-incubated with the MerTKi (i)nhibitor UNC2881 [100 nM] and stimulated with rhSema3B [200 ng/mL] in the presence or absence of LPS [10 ng/mL] for 24 h. Data are shown as RQ (relative quantity) respect to unstimulated cells, MFI (Median Fluorescence Intensity) of cells, % of pMerTK compared to medium, serum concentration (pg/mL), % of DQ-BSA uptake compared to medium and % of IL12p70 secretion compared to LPS. Data are analysed by One-way ANOVA tests. Means and SEM are shown. *P < 0.05, **P < 0.01.
Since MerTK+ RA MØ are involved in the resolution of inflammation, we next analysed the effect of Sema3B in the phagocytic activity of macrophages and the synthesis of Resolvin D1, a lipid induced by macrophage MerTK signalling involved in resolving joint inflammation (22). In RA MØIFN-γ, Sema3B moderately increased Resolvin D1 secretion, although it did not modulate the macrophage phagocytic activity (Figure 4D). On their part, Sema3B-stimulated RA MØM-CSF significantly raised the secretion of Resolvin D1, as well as their phagocytosis capacity, although it resulted insufficient counteracting the effect of LPS (Figure 4E).
Ultimately, we tested if these responses were mediated by MerTK, using a specific MerTK (i)nhibitor. Sema3B reduced the LPS-mediated secretion of IL12p70 in both RA MØIFN-γ and RA MØM-CSF, and this effect was abrogated in the presence of the MerTKi (Figures 4F, G). Also, MerTK inhibition reversed the Sema3B-induced phagocytic activity of RA MØM-CSF (Figure 4G).
Therefore, these results suggest that the anti-inflammatory and pro-resolving responses induced by Sema3B are, at least in part, mediated by MerTK.
In this manuscript we demonstrate that Sema3B modulates the phenotypic characteristics of RA macrophages. More specifically, Sema3B induces a skewing towards an anti-inflammatory/pro-resolving phenotype in a MerTK-dependant manner. Through this work we have found a new protective effect of Sema3B in the pathogenesis of RA. This protective role is in line with previous results from our group, in which we described a reduced expression of inflammatory mediators in both joints and FLS of arthritic mice treated with Sema3B and an impaired invasive phenotype in Sema3B-stimulated RA FLS (2–4).
Firstly, Sema3B reduced the secretion of pro-inflammatory cytokines in monocytes-derived macrophages from RA patients. The expression of surface markers associated to M1 macrophages (CD86, HLA-DR and CD64) was also decreased by Sema3B, while the expression of M2 markers (Stabilin-1 and TGF-β) was raised. Neutralization experiments showed that co-receptors NRP-1 and NRP-2 are involved in the protective signalling induced by Sema3B. Since our previous findings identified NRP-2 and, in lower extent NRP-1, as Sema3B co-receptors essential for reducing the invasive ability of RA FLS (2), we demonstrate that Sema3B signals through these co-receptors in both cell types. Sema3B also increased the phagocytic activity in RA MØM-CSF, which is linked to the resolution of tissue inflammation (6). However, we did not observe this effect in RA MØIFN-γ. This might be due to the IFN-γ-induced M1-like phenotype, which could be modulated to a less inflammatory phenotype by Sema3B, but without reaching pro-resolving characteristics, as it would be the case for Sema3B-modulated RA MØM-CSF. In this regard, the higher phagocytic activity of RA MØM-CSF compared to RA MØIFN-γ (Supplementary Figure S3), and the lack of effect of Sema3B on LPS-stimulated macrophages, support this aim.
Secondly, we proved that the anti-inflammatory/resolving effect of Sema3B in RA MØ was mediated by MerTK. Sema3B has a dual role on MerTK activation. On one hand, Sema3B induced the expression and activation of MerTK. On the other hand, Sema3B up-regulated the macrophage expression of GAS6, which is a ligand that activates MerTK signalling (19–21). MerTK is a crucial tyrosine kinase for the macrophage differentiation towards an anti-inflammatory/resolving phenotype (23, 24) and its deficiency exacerbates the severity of collagen-induced arthritis, while MerTK signalling activation reduces it in this arthritis model (17, 18). Importantly, MerTK+ synovial macrophages are associated with the disease status of RA patients. In fact, the percentage of this macrophage population is reduced in patients with active RA compared to patients in remission and negatively correlates with the disease activity score-28 (DAS28). In contrast, MerTK- synovial MØ express inflammatory mediators, induce the production of inflammatory mediators by RA FLS and participate in both bone and cartilage destruction (9, 17). Therefore, the modulation of MerTK+ macrophages by Sema3B may be a useful approach for the treatment of RA.
We cannot rule out the possibility that MertK modulates Sema3B signalling, since MerTK regulates the expression of the Sema3B receptor PlexinA1 (25). We analysed, using a public dataset (GSE205070), the expression of Sema3B receptors in bone marrow-derived macrophages (BMDM) from WT and Mertk-/- mice. Deficiency of MerTK significantly up-regulated the expression of Plxna1 and Nrp1 (Supplementary Figure 4), suggesting that MerTK signalling might modulate the Sema3B signalling. However, further studies are needed for elucidating this effect.
Sema3B is not the only class 3 semaphorin member able to modulate the macrophage phenotype characteristic. In fact, Sema3A and Sema3E also induce a skewing towards a resolving/anti-inflammatory phenotype in this cell type (26–29). In addition, Sema3A and Sema3F play protective roles in the pathogenesis of RA (2, 4, 29, 30). However, these semaphorins did not modulate the expression of MerTK (data non shown), suggesting that the MerTK regulation is specific for Sema3B.
Lastly, Sema3B also enhanced the secretion of Resolvin D1, a pro-resolving lipid triggered by the MerTK signalling activation, with a protective function in inflammatory arthritis (9, 22). Moreover, Resolvin D1 has been also involved in the modulation of macrophage polarization and in the skewing towards the anti-inflammatory/resolving phenotype (20, 31).
A limitation of this study is that we did not used liquid chromatography–mass spectrometry, the gold standard for the assessment of Resolvin D1 (32). Instead, we employed the well-validated ELISA technique. We neither utilized synovial macrophages from RA patients. We used instead in vitro M-CSF- and IFN-γ- differentiated macrophages from peripheral blood monocytes of RA patients, which possess anti-inflammatory/pro-resolving and pro-inflammatory characteristics, respectively (5, 14). Remarkably, MerTK expression was higher in RA MØM-CSF than in RA MØIFN-γ, therefore these macrophage phenotypes partially mimic the phenotypes of MerTK+ and MerTK- synovial macrophages.
Altogether, our work identifies a new anti-inflammatory mechanism of Sema3B, confirming the protective role of Sema3B in RA pathogenesis and pointing out this semaphorin as a promising therapeutic target.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Ethics Committee of Galicia (studies numbers 2020/159 and 2021/03). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
SM-R: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. CR-V: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. BM-F: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. NP: Data curation, Investigation, Methodology, Writing – review & editing. CM: Data curation, Investigation, Methodology, Writing – review & editing. SGP: Data curation, Investigation, Methodology, Writing – review & editing. FJMP: Data curation, Investigation, Methodology, Writing – review & editing. CC: Conceptualization, Formal analysis, Writing – review & editing. JP-R: Formal analysis, Writing – review & editing. SG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Instituto de Salud Carlos III (ISCIII), #PI20/01472 (SG), #CP19/00005 (SG), #FI2100120 (SM-R) and #IFEQ21/00157, co-funded by the European Union; by Axencia Galega de Innovación, #IN607B-2022-18, #IN606A-2020/043 (CR-V), and by vouchers (#Q222RSV34 and #Q123PV87) from the European Alliance of Associations for Rheumatology (EULAR).
All patients are thanked for their selfless participation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1268144/full#supplementary-material
1. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers (2018) 4:1–23. doi: 10.1038/nrdp.2018.1
2. Tang MW, Malvar Fernández B, Newsom SP, van Buul JD, Radstake TRDJ, Baeten DL, et al. Class 3 semaphorins modulate the invasive capacity of rheumatoid arthritis fibroblast-like synoviocytes. Rheumatol (Oxford) (2018) 57:909–20. doi: 10.1093/rheumatology/kex511
3. Igea A, Carvalheiro T, Malvar-Fernández B, Martinez-Ramos S, Rafael-Vidal C, Niemantsverdriet E, et al. Central role of semaphorin 3B in a serum-induced arthritis model and reduced levels in patients with rheumatoid arthritis. Arthritis Rheumatol (2022) 74:972–83. doi: 10.1002/art.42065
4. Martínez-Ramos S, Rafael-Vidal C, Malvar-Fernández B, Rodriguez-Trillo A, Veale D, Fearon U, et al. HOXA5 is a key regulator of class 3 semaphorins expression in the synovium of rheumatoid arthritis patients. Rheumatology (2022) 62(7):2621–30. doi: 10.1093/rheumatology/keac654
5. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol (2016) 12:472–85. doi: 10.1038/nrrheum.2016.91
6. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol (2022) 13:867260. doi: 10.3389/fimmu.2022.867260
7. Haringman JJ, Gerlag DM, Zwinderman a. H, Smeets TJM, Kraan MC, Baeten D, et al. Synovial tissue macrophages: A sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis (2005) 64:834–8. doi: 10.1136/ard.2004.029751
8. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol (2019) 20:928–42. doi: 10.1038/s41590-019-0378-1
9. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med (2020) 26:1295–306. doi: 10.1038/s41591-020-0939-8
10. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum (2010) 62:2569–81. doi: 10.1002/art.27584
11. Adomati T, Cham LB, Hamdan TA, Bhat H, Duhan V, Li F, et al. Dead Cells Induce Innate Anergy via Mertk after Acute Viral Infection. Cell Rep (2020) 30:3671–3681.e5. doi: 10.1016/j.celrep.2020.02.101
12. Zhang W, McIver AL, Stashko MA, Deryckere D, Branchford BR, Hunter D, et al. Discovery of Mer specific tyrosine kinase inhibitors for the treatment and prevention of thrombosis. J Med Chem (2013) 56:9693–700. doi: 10.1021/jm4013888
13. Akalu YT, Mercau ME, Ansems M, Hughes LD, Nevin J, Alberto EJ, et al. Tissue- specific modifier alleles determine Mertk loss- of- function traits. Elife (2022) 11:e80530. doi: 10.7554/eLife.80530
14. Hamilton TA, Zhao C, Pavicic PG, Datta S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol (2014) 5:554. doi: 10.3389/fimmu.2014.00554
15. Worzfeld T, Offermanns S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discovery (2014) 13:603–21. doi: 10.1038/nrd4337
16. Garcia S. Role of semaphorins in immunopathologies and rheumatic diseases. Int J Mol Sci (2019) 20(2):374. doi: 10.3390/ijms20020374
17. Waterborg CEJ, Beermann S, Broeren MGA, Bennink MB, Koenders MI, van Lent PLEM, et al. Protective role of the MER tyrosine kinase via efferocytosis in rheumatoid arthritis models. Front Immunol (2018) 9:742. doi: 10.3389/fimmu.2018.00742
18. Van Den Brand BT, Abdollahi-Roodsaz S, Vermeij EA, Bennink MB, Arntz OJ, Rothlin CV, et al. Therapeutic efficacy of Tyro3, Axl, and Mer tyrosine kinase agonists in collagen-induced arthritis. Arthritis Rheum (2013) 65:671–80. doi: 10.1002/art.37786
19. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage merTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab (2020) 31:406–421.e7. doi: 10.1016/j.cmet.2019.11.013
20. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal (2018) 11(549):eaar3721. doi: 10.1126/scisignal.aar3721
21. Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene (1997) 14:2033–9. doi: 10.1038/sj.onc.1201039
22. Norling LV, Headland SE, Dalli J, Arnardottir HH, Haworth O, Jones HR, et al. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight (2016) 1(5):e85922. doi: 10.1172/jci.insight.85922
23. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut (2018) 67:333–47. doi: 10.1136/gutjnl-2016-313615
24. Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J, et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflamm (2021) 18:1–20. doi: 10.1186/s12974-020-02041-7
25. Penberthy KK, Rival C, Shankman LS, Raymond MH, Zhang J, Perry JSA, et al. Context-dependent compensation among phosphatidylserine- recognition receptors. Sci Rep (2017) 7(1):14623. doi: 10.1038/s41598-017-15191-1
26. Rienks M, Carai P, Bitsch N, Schellings M, Vanhaverbeke M, Verjans J, et al. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res Cardiol (2017) 112:1–13. doi: 10.1007/s00395-017-0630-5
27. Tian T, Chen L, Wang Z, Zhu M, Xu W, Wu B. Sema3A drives alternative macrophage activation in the resolution of periodontitis via PI3K/AKT/mTOR signaling. Inflammation (2023) 46:876–91. doi: 10.1007/s10753-022-01777-z
28. Mohammed A, Okwor I, Shan L, Onyilagha C, Uzonna JE, Gounni AS. Semaphorin 3E regulates the response of macrophages to lipopolysaccharide-induced systemic inflammation. J Immunol (2020) 204:128–36. doi: 10.4049/jimmunol.1801514
29. Teng Y, Yin Z, Li J, Li K, Li X, Zhang Y. Adenovirus-mediated delivery of Sema3A alleviates rheumatoid arthritis in a serum-transfer induced mouse model. Oncotarget (2017) 8:66270–80. doi: 10.18632/oncotarget.19915
30. Catalano A. The neuroimmune semaphorin-3A reduces inflammation and progression of experimental autoimmune arthritis. J Immunol (2010) 185:6373–83. doi: 10.4049/jimmunol.0903527
31. Schmid M, Gemperle C, Rimann N, Hersberger M. Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32. J Immunol (2016) 196:3429–37. doi: 10.4049/jimmunol.1501701
Keywords: rheumatoid arthritis, Semaphorin3B, macrophages, inflammation, MERTK
Citation: Martínez-Ramos S, Rafael-Vidal C, Malvar-Fernández B, Pérez N, Mouriño C, Pérez SG, Maceiras Pan FJ, Conde C, Pego-Reigosa JM and García S (2024) Semaphorin3B promotes an anti-inflammatory and pro-resolving phenotype in macrophages from rheumatoid arthritis patients in a MerTK-dependent manner. Front. Immunol. 14:1268144. doi: 10.3389/fimmu.2023.1268144
Received: 27 July 2023; Accepted: 26 December 2023;
Published: 12 January 2024.
Edited by:
Chuan-ju Liu, New York University, United StatesReviewed by:
Svetlana P. Chapoval, University of Maryland, United StatesCopyright © 2024 Martínez-Ramos, Rafael-Vidal, Malvar-Fernández, Pérez, Mouriño, Pérez, Maceiras Pan, Conde, Pego-Reigosa and García. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel García, c2FtdWVsLmdhcmNpYUBpaXNnYWxpY2lhc3VyLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.