 Meng Wu1†
Meng Wu1† Guangfeng Long
Guangfeng Long Zhanjun Jia
Zhanjun Jia Weiwei Xia
Weiwei Xia- 1Department of Clinical Laboratory, Children’s Hospital of Nanjing Medical University, Nanjing, China
- 2Department of Nephrology, Children’s Hospital of Nanjing Medical University, Nanjing, China
- 3Jiangsu Key Laboratory of Pediatrics, Nanjing Medical University, Nanjing, China
Mitochondrial antiviral signaling protein (MAVS) is a key innate immune adaptor on the outer mitochondrial membrane that acts as a switch in the immune signal transduction response to viral infections. Some studies have reported that MAVS mediates NF-κB and type I interferon signaling during viral infection and is also required for optimal NLRP3 inflammasome activity. Recent studies have reported that MAVS is involved in various cancers, systemic lupus erythematosus, kidney diseases, and cardiovascular diseases. Herein, we summarize the structure, activation, pathophysiological roles, and MAVS-based therapies for renal diseases. This review provides novel insights into MAVS’s role and therapeutic potential in the pathogenesis of renal diseases.
1 Introduction
The innate immune system plays the essential role in the initial defense mechanism against invading pathogens, serving as the frontline of protection. To mount an effective response, it relies on a sophisticated network of signaling pathways and molecules. Mitochondrial antiviral signaling protein (MAVS) is a key innate immune adaptor on the outer mitochondrial membrane that is required to defend against RNA viral stimulation (1). MAVS is also known as the IFN-β promoter stimulator (IPS-1) (2), caspase activation recruitment domain (CARD) adaptor inducing IFN-β (Cardif) (3), and virus-induced signaling adaptor (VISA) (4). Upon viral recognition, MAVS acts as a signaling hub, orchestrating a series of downstream events that activate the production of antiviral cytokines and interferons. Through its involvement in these intricate signaling cascades, MAVS contributes to the clearance of viral infections and maintenance of immune homeostasis (5). However, increasing evidence indicates that viruses can maintain survival and replication by disturbing multiple points in the MAVS signaling pathway to escape the host antiviral response during long-term coexistence. Furthermore, cells possess a strict regulatory mechanism to prevent spontaneous aggregation of MAVS and maintain immune homeostasis in the resting state (6). Owing to its dual role in antiviral signal transduction and immune homeostasis, the delicate regulation of MAVS has emerged as a central target for cell function and antiviral response. MAVS not only acts as a central hub in orchestrating the innate host response through the induction of antiviral and inflammatory pathways but is also involved in coordinating cellular damage and metabolic functions (7). Recent studies have shown that MAVS is involved in the occurrence and development of many diseases such as cancer (8, 9), systemic lupus erythematosus(SLE) (10), cardiovascular diseases(CVD) (11), acute kidney injury (AKI) (12), and chronic kidney disease(CKD) (13).
An estimated 1.2 million deaths of the global burden of disease can be attributed to kidney failure (14). Notably, patients and clinicians often underrecognize AKI because of limited diagnostic methods and rapid disease progression (15). Worldwide estimates indicate that CKD is the 16th leading cause of years of life lost, directly affecting global morbidity and mortality by increasing the risk of hypertension, diabetes, and CVD (16). The pathophysiological mechanisms underlying AKI and CKD are complex, involving many pathological processes, including glomerular injury, tubular injury, epithelial cell damage, pyroptosis, inflammation, vascular dysfunction, and tubulointerstitial fibrosis. Experimental studies have focused on the pivotal role of MAVS signaling in regulating kidney disease progression (16), indicating the potential of MAVS as a therapeutic approach for managing kidney disease. This review focuses on the role of MAVS in the etiology of renal dysfunction and the pathogenesis of kidney disease.
2 Molecular structure of MAVS
The MAVS gene is located on chromosome 20 in humans and consists of multiple exons and introns. The primary transcript undergoes alternative splicing, generating several isoforms with distinct functional properties. The full-length MAVS protein is a cytosolic protein comprising 540 amino acids. It contains several conserved domains, including an N-terminal caspase activation and recruitment domain(CARD), a central proline-rich region(PRR) and a C-terminal transmembrane domain(TM) (17). The N-terminal CARD domain of MAVS is responsible for protein-protein interactions, enabling MAVS to associate with other signaling molecules in the cytoplasm. This domain serves as a critical platform for the assembly of signaling complexes involved in antiviral signaling. Upon viral infection, the CARD domain of MAVS interacts with the CARD domains of retinoic acid-inducible gene I (RIG-I) or melanoma differentiation associated gene 5 (MDA5), initiating downstream signaling cascades (18). In addition, the MAVS-CARD domain is involved in caspase activation and has six helical structures, including H1–H6. Furthermore, helix1 is divided into two smaller helices, H1a and H1b. H1a, H3, and H4 form hydrophobic positive surfaces, whereas H2 and H6 form hydrophilic negative surfaces on the opposite side (19). The central proline-rich region of MAVS contains multiple conserved motifs, including a TRAF-interacting motif (TIM) and a proline-rich domain (PRD). These motifs enable MAVS to interact with various downstream effectors and adaptors, such as TNF receptor-associated factor (TRAF) proteins, facilitating the transmission of signals to downstream signaling pathways. Additionally, the proline-rich region acts as a scaffold for the recruitment of ubiquitin ligases, which play a crucial role in the activation and regulation of MAVS-mediated immune responses (20). The C-terminal transmembrane domain of MAVS anchors the protein to the outer mitochondrial membrane, allowing it to localize to the mitochondria-associated membranes (MAMs) (21). This localization is essential for MAVS to efficiently transmit signals from viral sensors to downstream effectors. The transmembrane domain also contributes to the formation of large MAVS aggregates, known as “signalosomes,” which serve as platforms for the amplification and propagation of antiviral signaling (20). The structure of the MAVS domain is shown in Figure 1.
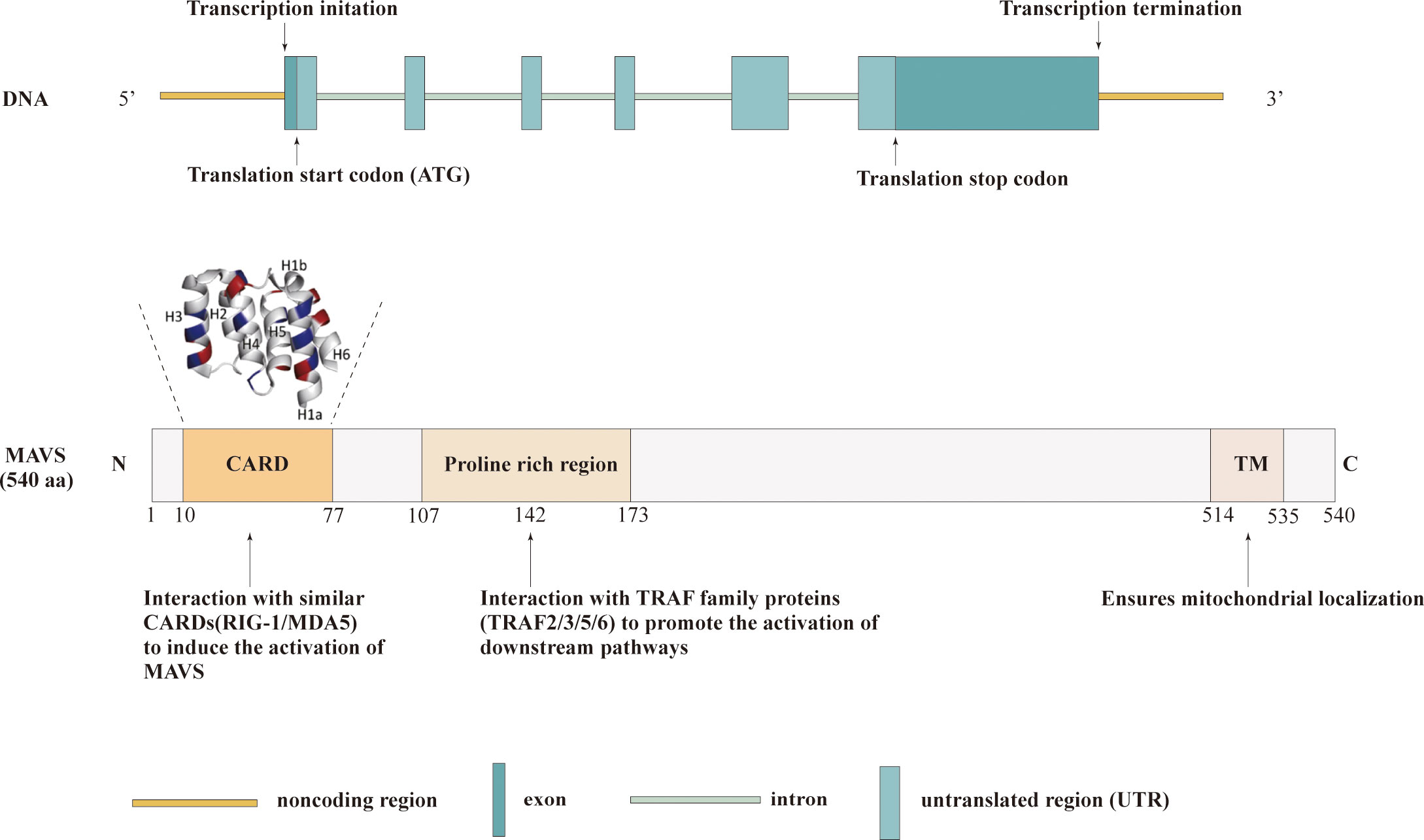
Figure 1 Structures of MAVS. Schematic representation illustrating the three functional domains of MAVS: the CARD domain, proline-rich region, and TM.
3 The activation of MAVS
MAVS acts as a switch in immune signal transduction during viral infection. In the resting state, MAVS is kept silent. In the viral infection, RIG-I recognizes viral double-stranded RNA to activate RIG-I/MAVS antiviral functions, induce type I interferon production. As known many RNA viruses including dsRNA and ssRNA viruses produced double-stranded RNA (dsRNA) during replication to activate RIG-I/MAVS antiviral signaling. A list of some RNA viruses that have been reported to activate MAVS signaling was shown in Table 1 (22–29). Furthermore, MAVS filament formation reveals structural and functional variability after viral infection (30). Notably, it has been reported that the combination of the MAVS-CARD and RIG-CARD domains results in the formation of MAVS aggregates on the surface of mitochondria, which can efficiently convert endogenous MAVS into functional aggregates. These aggregates activate cytosolic signaling cascades to induce antiviral molecules (31). MAVS TM domain has been shown to be critical to mediate the dimerization of MAVS in the activation of interferon regulatory factor 3/7 (IRF3/7) and NF-κB (32). In addition, the MAVS-PRR domain can recruit proteins to form MAVS aggregates and activate downstream effectors (33).
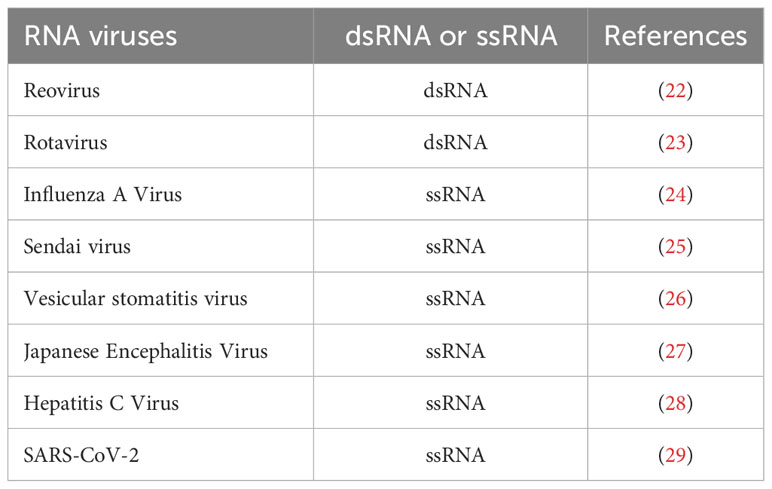
Table 1 List of RNA viruses activated MAVS signaling.
4 The antiviral signaling pathway of MAVS
Several studies have reported activation of the MAVS pathway by its interacting proteins and downstream signaling events. A recent study on the downstream signaling pathways of MAVS revealed that it mediates the activation of NF-κB and IRF3 by viruses (34). In the dormant state, NF-κB is sequestered in the cytoplasm by inhibitory κB (IκB) family members (35). Moreover, under the detection of viral RNA, MAVS then recruits IκB kinases IKKα, IKKβ, and IKKγ (an essential regulatory subunit, also called NF-kappa-B essential modulator (NEMO)) to induce the phosphorylation of IκB.
Phospho-IκB are recognized and ubiquitinated by ubiquitin ligases and subsequently degraded by the proteasome. Then, NF-κB translocates into the nucleus and binds to enhancers/promoters of target genes, activating the proinflammatory response (36). MAVS is a scaffold protein involved in activation upon RIG-I activation, which recognizes MAVS-interacting proteins in the RIG-I-mediated intracellular viral detection pathway. Lee et al. showed that mitochondrial MAVS recruits the signaling molecule TRAF6, activating NF-κB signaling pathways (37). Notably, K48-linked polyubiquitination of TRAF6 and NEMO is critical for NF-κB activation (38). Furthermore, Michallet et al. indicated that TRADD, a crucial adaptor of tumor necrosis factor receptor (TNFRI) and Fas-associated death domain (FADD), together with receptor-interacting protein (RIP) 1, play a crucial role in MAVS -induced NF-kB activation by the E3 ubiquitin ligase RIP1-bound TRAF2 (39). In addition, MAVS interacts with the Toll-interleukin-1 receptor domain-containing adaptor, inducing IFN-β (TRIF) in TLR-independent viral recognition pathways. The N-terminal- and C-terminal regions of TRIF are associated with TRAF6 and RIP1, respectively. RIP1 is further modified by K63 ligand ubiquitination to activate the downstream molecule TAK1. The formation of the TRAF6-RIP1-TAK1 complex plays a positive role in MAVS-induced NF-κB activation (40).
Viral components induce strong IRF3 activation through the Toll-like receptor (TLR) and RIG-I-like receptor (RLR) families. During the induction of IFNs, MAVS recruits TANK-binding kinase 1(TBK1) and inhibitor of kB kinase (IKKϵ), two primary kinases functionally that have been shown to phosphorylate the regulatory factor 3 and 7 (IRF3/IRF7), respectively. Furthermore, phosphorylated factors that translocate into the nucleus are critical for IFN production (41).
Upon RIG-I activation, MAVS activates the downstream pathways by recruiting related proteins. Stimulator of interferon genes (STING) (also known as MITA, MPYS, or ERIS) is an essential adaptor protein in the DNA recognition signaling pathway. The MAVS-STING interaction plays a vital role in mitochondria-mediated antiviral innate immune response (42). Upon viral RNA stimulation, the STING N-terminal fragment binds to the MAVS-CARD domain and interferon regulatory factor 3 (IRF3) to form the MAVS-STING and STING-IRF3 complexes, respectively. STING dimerization leads to the interaction of these two complexes and the activation of the IRF signaling pathway (43).
The Matrix Protein 2 (M2) is expressed by the influenza A virus upon infection, which plays a critical role in the viral replication and transmission cycle by serving as a multifunctional mediator of various processes (44). M2 protein has a proton-selective ion channel activity, which is the target of antiviral drug. M2 protein contains specific structural domains that enable it to interact with MAVS on mitochondria and positively regulating MAVS-mediated innate immunity. Importantly, M2 enhances MAVS signaling via increasing ROS production (45).
WD repeat-containing protein 5 (WDR5), also known as SWD3 and BIG-3, is a member of the WD40 protein family. It has been identified as an important MAVS-associated component in the recruitment of RIG-I/STING to MAVS and the induction of type I IFNs. Further epistatic studies have shown that WDR5 functions upstream of TBK1 and downstream of RIG-I and MAVS (46). Notably, TRAF3 plays a critical role in the TLR-independent viral recognition pathways (47). Upon TLRs stimulation, MAVS activates downstream signaling of the TANK/TBK1/IKKi complex and NEMO the TANK/TBK1 complex by associating with the ubiquitin ligase TRAF3/TRAF6/TRAF5 during IFN induction (48, 49). Two adaptor proteins, SINTBAD and NAP1 and TANK, specifically bind to TBK1/IKKi during virus-induced IRF activation (50, 51). Alternatively, NEMO binds to polyubiquitinated TBK1, and the NEMO/TBK1 complex recruited to MAVS results in the activation of TBK1 and phosphorylation of IRF3 (52). In addition, k63-linked polyubiquitination of NEMO and TRAF3 is important for IRF3 activation in TLR-independent viral recognition pathways (38). All antiviral signaling pathways of MAVS are shown in Figure 2.
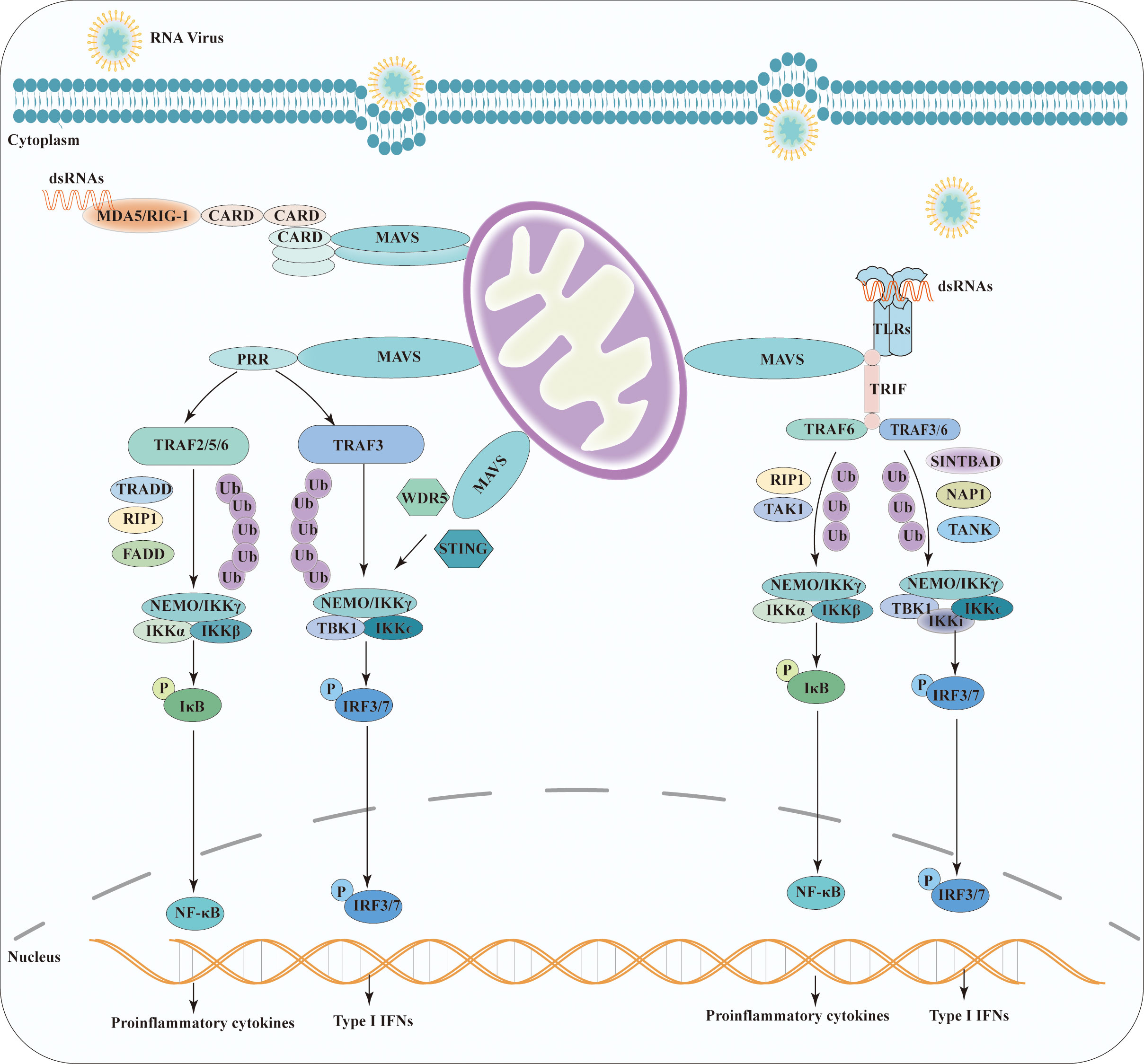
Figure 2 Schematic representation of the antiviral signaling pathway model of MAVS. Viruses induce MAVS activation through TLR and RLR families. Homophilic interactions between CARD domains enable RIG-I and MDA5 to interact with MAVS. Upon TLR stimulation, MAVS triggers downstream signaling cascades by recruiting TRAF and TRIF. MAVS activates two cytosolic protein kinase complexes, leading to the production of immune factors. The TBK1 complex phosphorylates IRF-3/7, promoting the transcription of IFN genes. Simultaneously, the IKK complex activates NF-kB, resulting in the production of proinflammatory cytokines.
5 MAVS in kidney diseases
Accumulating evidence supports that MAVS is participated in the pathophysiology of human diseases. Here, we illustrate recent progress in comprehending the roles of MAVS in AKI, CKD, and other renal diseases, such as diabetic renal disease and hypertensive renal disease.
6 MAVS in AKI
AKI is a heterogeneous, broadly defined clinical syndrome characterized by high incidence and mortality (53). The most common causes of AKI include hypoxia, ischemia-reperfusion injury (IRI), sepsis, and nephrotoxic drugs (i.e., cisplatin and folic acid) (54). Many pathways are involved in AKI pathogenesis, including generating reactive oxygen species (ROS), inflammation, and activating several renal tubular epithelial cell death pathways (apoptosis, necrosis, ferroptosis, and pyroptosis) (55). MAVS, a key trigger in the activation of mitochondrial ROS (mtROS), is essential for optimal NLRP3 inflammasome activity and plays a vital regulatory role in host inflammation (56, 57). Previous studies demonstrated that NLRP3 is a key sensor of cell injury (58) and is expressed in proximal tubular epithelial cells in kidney disease (59). Furthermore, NLRP3 deficiency reduces renal tubular apoptosis in kidney injury and plays a role similar to that of ROS scavenger (59, 60). Moreover, NLRP3 or MAVS deletion attenuates mtROS production and mitochondrial membrane depolarization in renal tubular cells after renal hypoxia injury, indicating that MAVS is an indispensable and independent factor for inflammasome-independent NLRP3 activation (12). Another study indicated that NLRP3 deficiency afforded a significant renal protective effect against IRI in experimental mice, which could be linked with the inflammasome-independent role of NLRP3 in kidney injury (61). A later study revealed that MAVS was overexpressed in trichloroethylene (TCE)-sensitized positive mouse renal tubular cells and, mtROS inhibitor Mito TEMPO significantly decreased the expression of MAVS and the assembly of the NLRP3 inflammasome, suggesting that MAVS associates with NLRP3 and induces the activation of the NLRP3 inflammasome, finally contributing to tubular reabsorption dysfunction and kidney injury (62). Pinto et al. reported that West Nile virus (WNV) infection triggers signaling through MAVS, causing massive proinflammatory cytokine production that can result in sepsis and AKI in mice lacking type I IFN receptor gene (IFNAR) expression only in myeloid cell subsets (63).
Cisplatin, an anti-cancer drug, can cause AKI. However, the direct role of MAVS in cisplatin-induced AKI has not been elucidated. A previous study reported that ovarian cancer (OC) with high MAVS expression was remarkably less responsive to cisplatin and paclitaxel than those with low MAVS expression. Thus, MAVS appears to be a predictive biomarker of poor prognosis in OC (8). Additionally, several studies have indicated that the NLRP3 inflammasome represents a critical player in the pathogenesis of cisplatin-induced AKI (64–67). However, Kim et al. study reported that the kidney injury biomarkers, such as blood urea nitrogen, serum creatinine, tubular apoptosis score, and acute tubular necrosis score, were not statistically different between NLRP3 knockout (NLRP3−/−) mice and wild-type mice with cisplatin-induced AKI, which suggests that the NLRP3 inflammasome is not involved in cisplatin-induced AKI (68). The role of the NLRP3 inflammasome in cisplatin-induced AKI warrants further investigation. These findings suggest that modulating MAVS activity holds therapeutic potential for preventing or treating AKI.
7 MAVS in CKD
CKD is currently defined as a progressive decline in renal function associated with high morbidity and mortality, eventually leading to end-stage renal disease (ESRD) (69). The pathological process of CKD involves various factors, including proteinuria, unilateral ureteral obstruction (UUO), hyperuricemia, aldosterone, and subtotal nephrectomy (SNx) (13, 60, 70). Furthermore, apoptosis of renal tubular cells after AKI is an important initial step in the AKI-to-CKD progression (71). In that regard, a previous study found that MAVS induces cell death in a dose-dependent manner because of its cytotoxic effect; MAVS-induced apoptosis mediates the activation of the caspase-dependent pathway (72). Proteinuria, which is a key driving factor of tubulointerstitial fibrosis, is an independent risk factor for CKD. Moreover, as major targets, mitochondria play a pivotal role in the albumin-induced apoptosis of human proximal tubule cells (73). In the pathology of CKD induced by albumin, the knockdown of MAVS in human renal tubular epithelial cells weakened the colocalization between MAVS and NLRP3, thereby alleviating cell pyroptosis and epithelial-mesenchymal transition (13). Recently, the NLRP3 inflammasome and mitochondria were shown to participate in the development of various types of CKD. In a UUO-treated mouse model, NLRP3 deletion attenuated renal injury by preventing apoptosis and ameliorating tubulointerstitial fibrosis (12). In nephrocalcinosis-related CKD, NLRP3 antagonists decrease proinflammatory and fibrotic macrophage subsets and prevent renal fibrosis (74). Another study showed that NLRP3 knockout mice exhibited reduced renal tubular apoptosis and phenotypic alterations after aldosterone infusion, indicating that the NLRP3 inflammasome plays a vital role in aldosterone-induced renal injury (60). These studies suggest that MAVS and its downstream factor, NLRP3, regulate CKD.
8 MAVS in other kidney diseases
Mounting evidence indicates that NLRP3 has important effects on several kidney diseases, including pyelonephritis (75) and IgA nephropathy (76). These reports indicate that MAVS and its downstream factor, NLRP3, play diverse roles in renal disease by stimulating inflammation, renal tubular epithelial cell injury, apoptosis, epithelial-mesenchymal transition, and tubulointerstitial fibrosis. The function of MAVS signaling in kidney disease is shown in Figure 3A.
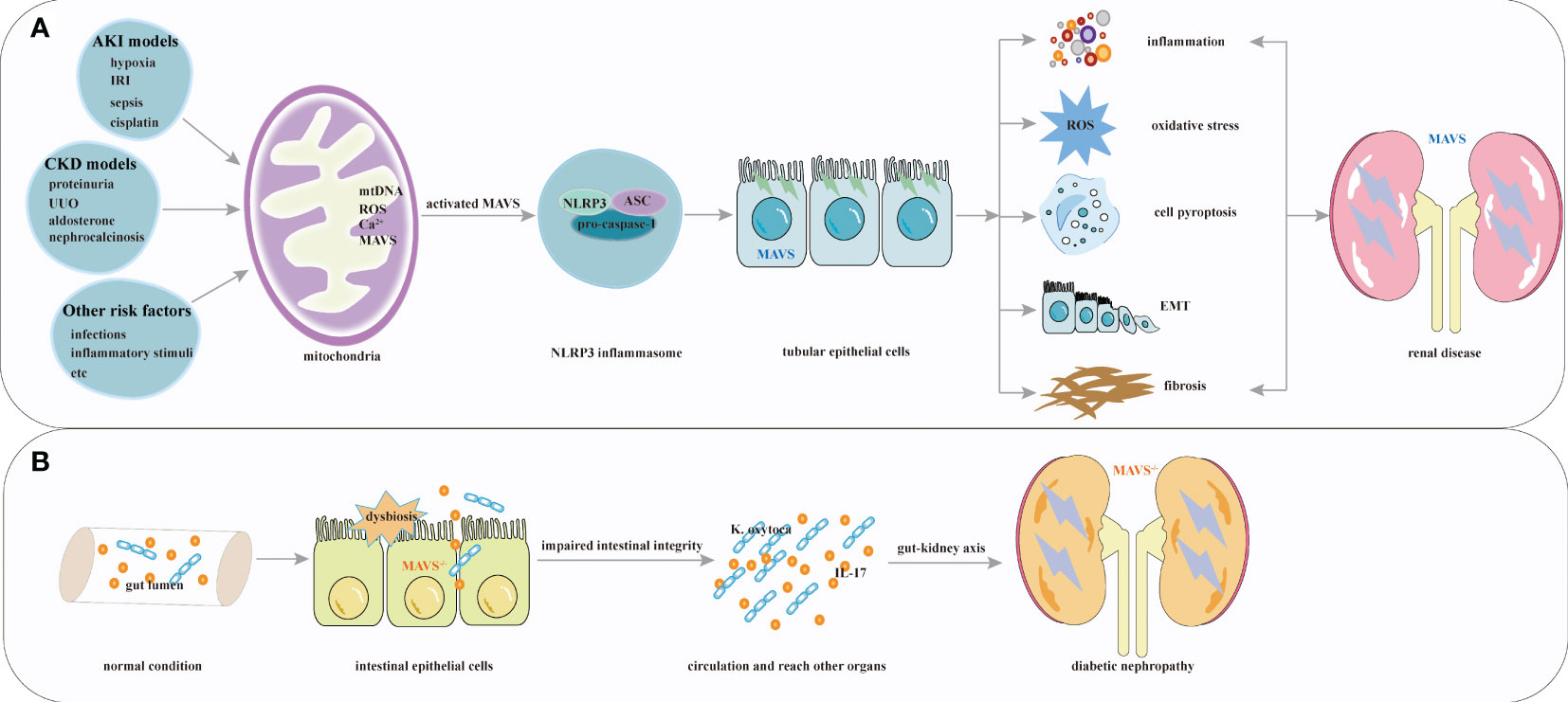
Figure 3 Overview of MAVS roles in renal diseases. (A) Multiple factors including hypoxia, IRI, sepsis, cisplatin, chronic kidney injury associated with proteinuria, UUO, hyperuricemia, aldosterone, and other risk factors activate MAVS and its downstream factor NLRP3 inflammasome. These activations stimulate processes such as inflammation, renal tubular epithelial cell injury, apoptosis, epithelial-mesenchymal transition, and tubulointerstitial fibrosis formation. (B) MAVS knockout contribute to the pathophysiology of diabetic nephropathy.
Diabetic kidney disease (DKD) is the leading cause of ESRD (77). A recent study showed that hyperglycemia might be associated with alterations in the epithelial barrier of the intestinal environment (78). The gut and the kidney are intricately linked through the gut-kidney axis. The gut microbiota and its metabolites affect kidney function and contribute to the development and progression of renal diseases. A previous study demonstrated that MAVS played a pivotal role in regulating and protecting intestinal permeability (79), and Linh et al. revealed elevated MAVS expression in diabetic kidneys. MAVS knockout diabetic mice had more severe glomerular and tubular injuries. In diabetic MAVS-knockout mice kidney, intestine-derived Klebsiella oxytoca and IL-17 production were observed because of impaired intestinal integrity. Furthermore, Klebsiella oxytoca supernatant or IL-17 promoted KIM-1 production in tubular epithelial cells, leading to renal injuries (80). These results suggest that systemic MAVS signaling confers a protective role in DKD. Which is different from the function of MAVS signaling in acute tubular necrosis and renal fibrosis (12, 56). The most straightforward explanation for these results would be that MAVS knockout contribute to the deleterious effects of hyperglycemia (81). Given the above, loss of kidney function can influence the intestinal barrier, and MAVS may play various roles within different microenvironment of the kidney. To date, interactions between intestinal and kidney specific deletion of MAVS have not been validated in kidney disease, and the role of MAVS in kidney tissue await further exploration. These results suggest that MAVS signaling, which is associated with regulating intestinal homeostasis, plays a protective role for DKD. The protective function of MAVS in kidney disease is shown in Figure 3B.
9 MicroRNA-mediated MAVS therapies
MicroRNAs, small non-coding RNA molecules, are pivotal in post-transcriptional gene expression regulation. They exert their influence by binding to mRNA molecules, subsequently guiding the RNA-induced silencing complex (RISC) to either degrade the target mRNA or impede its translation (82). Several miRNAs that potentially regulate the expression or function of MAVS have been identified. These miRNAs may modulate antiviral immune responses by targeting MAVS. The potential use of miRNAs and MAVS as therapeutic agents holds promise for the treatment of viral infections and other diseases.
MicroRNA-125a has been reported to target the MAVS 3’-UTR during viral infection, negatively regulating the antiviral response by inhibiting MAVS expression, leading to reduced production of type I interferons (83). Additionally, miR-125a and -b have recently been proven to bind directly to the endogenous 3’-UTR of MAVS, thereby downregulating the subsequent induction of type I and III IFNs. Furthermore, microRNA-125a identifies potential therapeutic options for the prevention and/or treatment of Influenza A viral infection in chronic obstructive pulmonary disease (84). Hou et al. reported that miR-3470b directly targets MAVS 3’-UTR in Bovine ephemeral fever virus infection. In that study, the authors discovered that the overexpression of miR-3470b resulted in a marked decrease in MAVS at both the transcriptional and protein levels, thereby suppressing the antiviral response (85). Further, miR-3570 negatively regulates rhabdovirus-triggered production of type I interferons by targeting MAVS in miiuy croaker macrophages, thus facilitating viral replication (86). Intriguingly, miR-22 directly targets and represses MAVS expression, which inhibits poly(I:C)-triggered production of type I interferons and inflammatory cytokines (87). Taken together, these findings suggested that specific miRNAs targeting MAVS may be a potential strategy for the management of renal diseases. All the miRNAs are listed in Table 2.
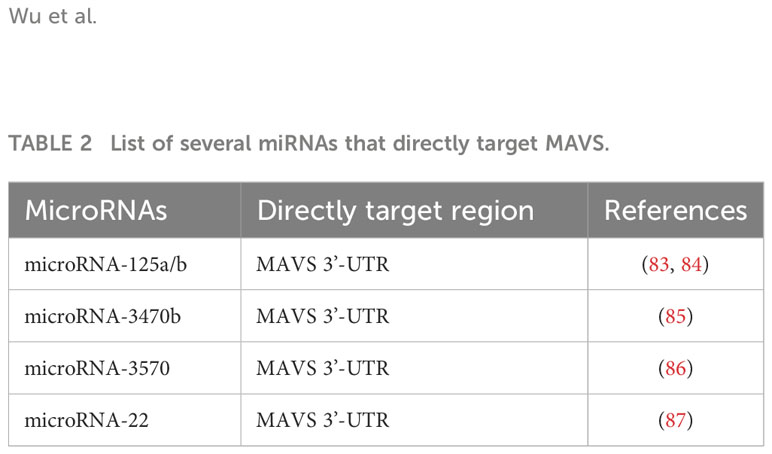
Table 2 List of several miRNAs that directly target MAVS.
10 Inhibitory regulatory of MAVS signaling
MAVS acts as a key protein involved in kidney diseases. Therefore, understanding and inhibiting MAVS and its downstream signaling pathways have significant importance in management of renal diseases.
Recent studies have shown that several E3 ubiquitin ligases, including TRIM25, RNF5, MARCH5, Smurf1, Smurf2, NLRX1, RACK, OTUD1, OTUD4 and AIP4, have been identified to target MAVS for ubiquitination and subsequent degradation through Lys48-linked deubiquitination (88–96). Distinguishing from the above E3s, YOD1 suppress MAVS aggregation through Lys63-linked deubiquitination (97). At the meanwhile, MARCH8 acts as a negative regulator of the MAVS pathway by K27-linked deubiquitination. promoting MAVS degradation (98). The MAVS pathway is also regulated by viral proteases through cleavage of MAVS. For example, NS3/4A, CVB3 2Apro and PB1-F2 cleave MAVS, disrupting the downstream signaling cascade (99–101). RNF34, HFE, and Tetherin degrades MAVS complexes through autophagy (98, 102, 103). PSMA7 serves as a negative regulator of the MAVS for proteasome-dependent degradation (104). The inhibitory regulator of MAVS itself are listed in Table 3.
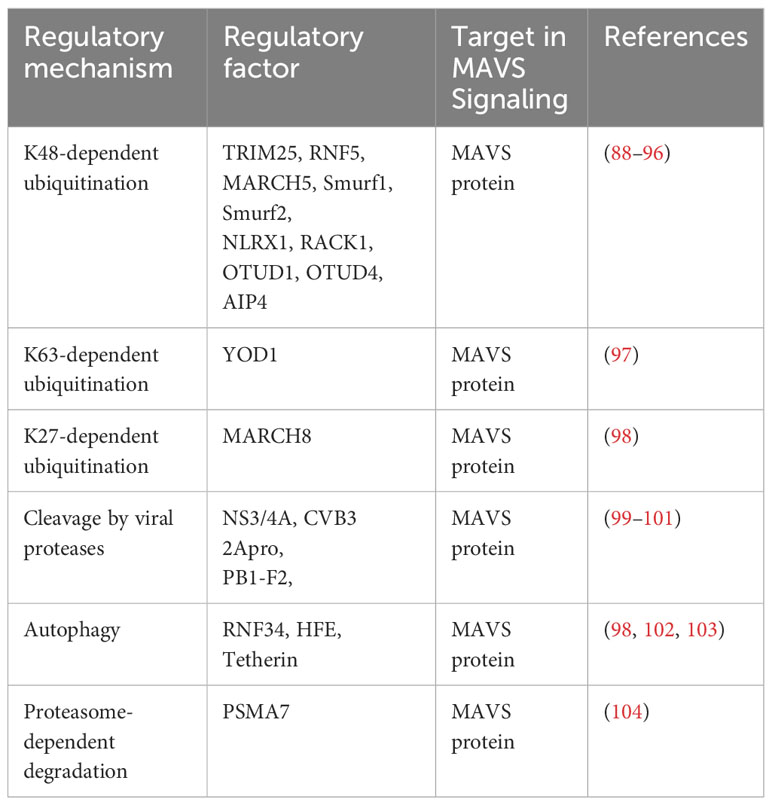
Table 3 List of several proteins that target MAVS protein itself.
Besides above proteins acting on MAVS itself, there are some small molecule inhibitors and natural products acts on MAVS downstream signaling. Several small molecule inhibitors, such as MRT67307, BX795 and BAY11-7082, targets MAVS downstream effector, TBK1, thereby inhibiting MAVS signaling (105–108). Besides, some natural compound (curcumin and resveratrol) derived from turmeric exhibits inhibitory effects on MAVS downstream signalling (109, 110). It has been shown to reduce MAVS-mediated inflammation and immune response in various viral infection models. The inhibitory regulator of MAVS downstream signaling are listed in Table 4.
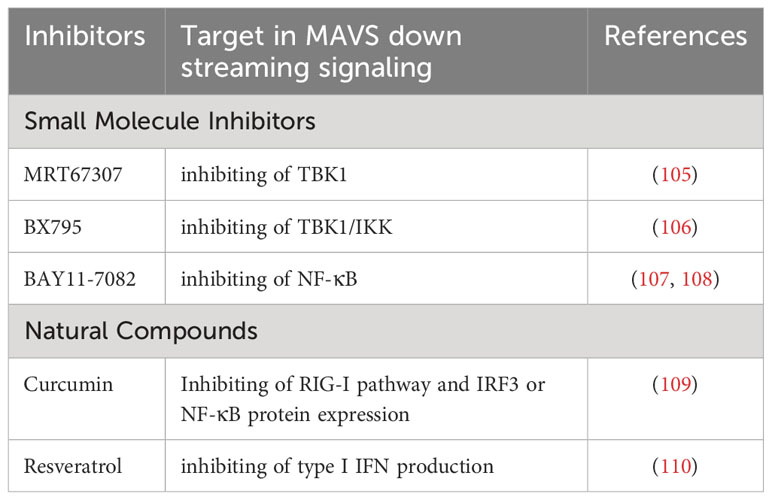
Table 4 List of several inhibitors that target MAVS down streaming signaling.
These negative regulation helps to terminate MAVS signaling and prevent prolonged inflammation immune activation, which could be used for treatment of kidney diseases.
11 Conclusions and perspectives
In conclusion, as a critical adaptor protein, MAVS plays a central role in innate immunity and the pathological processes involved in renal disease. MAVS recruits its interacting proteins and downstream molecules upon viral infection to activate NF-κB and IRF signaling, which plays a crucial role in the mitochondria-mediated antiviral innate immune response. In addition, MAVS is required for optimal NLRP3 inflammasome activity, and the expression of MAVS and NLRP3 is mainly localized in tubular epithelial cells. Several studies have demonstrated the role of MAVS and NLRP3 inflammasomes in various experimental models of renal disease, including IRI, hypoxic injury, sepsis, cisplatin-induced AKI, UUO, albumin overload-induced tubular injury, nephrocalcinosis-related CKD, and DKD. These studies revealed that MAVS and its downstream factor, NLRP3, may provide a promising strategy for treating renal disease.
To date, only phosphorylation and polyubiquitination were verified to regulate the activation of MAVS-mediated signaling, whether MAVS could be modified by other manners such as glycosylation, and the effects of these manners on MAVS activation still require further exploration. However, the existing literature on the regulation of MAVS signaling mainly focuses on animal or cell experimental models of renal disease. Relatively few studies have reported MAVS expression levels in clinical patients. Moreover, direct evidence of the role of MAVS in the pathogenesis of renal disease is lacking. The underlying mechanisms of MAVS in different renal diseases warrant further in-depth investigation.
The current emphasis in MAVS expression research revolves around its potential as a clinical prognostic biomarker and therapeutic target, primarily within the field of cancer. The various applications and development of MAVS agonists and antagonists may provide new therapeutic strategies for future research in renal diseases. It is important to note that miRNA regulation can be complex and that the interactions between miRNAs and their target genes can vary depending on the cellular context and specific conditions. Therefore, further research is required to fully understand the regulatory networks involving miRNAs and MAVS in different biological settings.
Author contributions
MW: Funding acquisition, Writing – original draft, Writing – review & editing. ZP: Writing – original draft, Writing – review & editing. GL: Writing – original draft, Writing – review & editing. HC: Conceptualization, Supervision, Writing – review & editing. ZJ: Conceptualization, Supervision, Writing – review & editing. WX: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant from the National Natural Science Foundation of China (no. 82070760, 82070701), grants from the Nanjing National Commission on Health and Family Planning (no. ZKX19042), grant from the Nanjing Medical Key Science and Technology Development Project Fund (No. YKK19105), and grants from Academic Technology Programof Nanjing Medical University (No. NMUB20210073).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ablasser A, Hur S. Regulation of cgas- and rlr-mediated immunity to nucleic acids. Nat Immunol (2020) 21(1):17–29. doi: 10.1038/s41590-019-0556-1
2. Wang D, Fang L, Li T, Luo R, Xie L, Jiang Y, et al. Molecular cloning and functional characterization of porcine ifn-beta promoter stimulator 1 (Ips-1). Vet Immunol Immunopathol (2008) 125(3-4):344–53. doi: 10.1016/j.vetimm.2008.05.018
3. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the rig-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005) 437(7062):1167–72. doi: 10.1038/nature04193
4. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. Visa is an adapter protein required for virus-triggered ifn-beta signaling. Mol Cell (2005) 19(6):727–40. doi: 10.1016/j.molcel.2005.08.014
5. Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic rig-I and sting sensing pathways. Trends Immunol (2017) 38(3):194–205. doi: 10.1016/j.it.2016.12.004
6. Chen Y, Shi Y, Wu J, Qi N. Mavs: A two-sided card mediating antiviral innate immune signaling and regulating immune homeostasis. Front Microbiol (2021) 12:744348. doi: 10.3389/fmicb.2021.744348
7. Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (Mavs) adapter. Curr Opin Immunol (2011) 23(5):564–72. doi: 10.1016/j.coi.2011.08.001
8. Chen L, Hou J, You B, Song F, Tu X, Cheng X. An analysis regarding the prognostic significance of mavs and its underlying biological mechanism in ovarian cancer. Front Cell Dev Biol (2021) 9:728061. doi: 10.3389/fcell.2021.728061
9. Zhang J, Li S, Zhang J, Zhang W, Jiang J, Wu H, et al. Docetaxel resistance-derived linc01085 contributes to the immunotherapy of hormone-independent prostate cancer by activating the sting/mavs signaling pathway. Cancer Lett (2022) 545:215829. doi: 10.1016/j.canlet.2022.215829
10. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent mavs oligomerization in systemic lupus erythematosus. Sci Signal (2016) 9(456):ra115. doi: 10.1126/scisignal.aaf1933
11. Wang Q, Sun Z, Cao S, Lin X, Wu M, Li Y, et al. Reduced immunity regulator mavs contributes to non-hypertrophic cardiac dysfunction by disturbing energy metabolism and mitochondrial homeostasis. Front Immunol (2022) 13:919038. doi: 10.3389/fimmu.2022.919038
12. Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH, Lee YH, et al. Inflammasome-independent role of nlrp3 mediates mitochondrial regulation in renal injury. Front Immunol (2018) 9:2563. doi: 10.3389/fimmu.2018.02563
13. Zhu Y, Huang G, Yang Y, Yong C, Yu X, Wang G, et al. Chinese herbal medicine suyin detoxification granule inhibits pyroptosis and epithelial-mesenchymal transition by downregulating mavs/nlrp3 to alleviate renal injury. J Inflamm Res (2021) 14:6601–18. doi: 10.2147/JIR.S341598
14. Shabaka A, Cases-Corona C, Fernandez-Juarez G. Therapeutic insights in chronic kidney disease progression. Front Med (Lausanne) (2021) 8:645187. doi: 10.3389/fmed.2021.645187
15. Marx D, Metzger J, Pejchinovski M, Gil RB, Frantzi M, Latosinska A, et al. Proteomics and metabolomics for aki diagnosis. Semin Nephrol (2018) 38(1):63–87. doi: 10.1016/j.semnephrol.2017.09.007
16. Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management: A review. JAMA (2019) 322(13):1294–304. doi: 10.1001/jama.2019.14745
17. De Colibus L, Stunnenberg M, Geijtenbeek TBH. Ddx3x structural analysis: implications in the pharmacology and innate immunity. Curr Res Immunol (2022) 3:100–9. doi: 10.1016/j.crimmu.2022.05.002
18. Koshiba T. Mitochondrial-mediated antiviral immunity. Biochim Biophys Acta (2013) 1833(1):225–32. doi: 10.1016/j.bbamcr.2012.03.005
19. Potter JA, Randall RE, Taylor GL. Crystal structure of human ips-1/mavs/visa/cardif caspase activation recruitment domain. BMC Struct Biol (2008) 8:11. doi: 10.1186/1472-6807-8-11
20. Tang ED, Wang CY. Mavs self-association mediates antiviral innate immune signaling. J Virol (2009) 83(8):3420–8. doi: 10.1128/JVI.02623-08
21. Vazquez C, Horner SM. Mavs coordination of antiviral innate immunity. J Virol (2015) 89(14):6974–7. doi: 10.1128/JVI.01918-14
22. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct rig-I and mda5 signaling by rna viruses in innate immunity. J Virol (2008) 82(1):335–45. doi: 10.1128/JVI.01080-07
23. Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. Rig-I/mda5/mavs are required to signal a protective ifn response in rotavirus-infected intestinal epithelium. J Immunol (2011) 186(3):1618–26. doi: 10.4049/jimmunol.1002862
24. Siren J, Imaizumi T, Sarkar D, Pietila T, Noah DL, Lin R, et al. Retinoic acid inducible gene-I and mda-5 are involved in influenza a virus-induced expression of antiviral cytokines. Microbes Infect (2006) 8(8):2013–20. doi: 10.1016/j.micinf.2006.02.028
25. Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. Activation of the beta interferon promoter by unnatural sendai virus infection requires rig-I and is inhibited by viral C proteins. J Virol (2007) 81(22):12227–37. doi: 10.1128/JVI.01300-07
26. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med (2008) 205(7):1601–10. doi: 10.1084/jem.20080091
27. Chang TH, Liao CL, Lin YL. Flavivirus induces interferon-beta gene expression through a pathway involving rig-I-dependent irf-3 and pi3k-dependent nf-kappab activation. Microbes Infect (2006) 8(1):157–71. doi: 10.1016/j.micinf.2005.06.014
28. Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. Innate immunity induced by composition-dependent rig-I recognition of hepatitis C virus rna. Nature (2008) 454(7203):523–7. doi: 10.1038/nature07106
29. Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, et al. Sars-cov-2 induces double-stranded rna-mediated innate immune responses in respiratory epithelial derived cells and cardiomyocytes. Proc Natl Acad Sci (2021) 118(16):e2022643118. doi: 10.1073/pnas.2022643118
30. Jiang QX. Structural variability in the rlr-mavs pathway and sensitive detection of viral rnas. Med Chem (2019) 15(5):443–58. doi: 10.2174/1573406415666181219101613
31. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. Mavs forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell (2011) 146(3):448–61. doi: 10.1016/j.cell.2011.06.041
32. Baril M, Racine ME, Penin F, Lamarre D. Mavs dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus ns3/4a protease. J Virol (2009) 83(3):1299–311. doi: 10.1128/JVI.01659-08
33. Tanaka Y, Chen ZJ. Sting specifies irf3 phosphorylation by tbk1 in the cytosolic DNA signaling pathway. Sci Signal (2012) 5(214):ra20. doi: 10.1126/scisignal.2002521
34. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates nf-kappab and irf 3. Cell (2005) 122(5):669–82. doi: 10.1016/j.cell.2005.08.012
35. Silverman N, Maniatis T. Nf-kappab signaling pathways in mammalian and insect innate immunity. Genes Dev (2001) 15(18):2321–42. doi: 10.1101/gad.909001
36. Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res (2006) 16(2):141–7. doi: 10.1038/sj.cr.7310019
37. Lee NR, Ban J, Lee NJ, Yi CM, Choi JY, Kim H, et al. Activation of rig-I-mediated antiviral signaling triggers autophagy through the mavs-traf6-beclin-1 signaling axis. Front Immunol (2018) 9:2096. doi: 10.3389/fimmu.2018.02096
38. Liu J, Qian C, Cao X. Post-translational modification control of innate immunity. Immunity (2016) 45(1):15–30. doi: 10.1016/j.immuni.2016.06.020
39. Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, et al. Tradd protein is an essential component of the rig-like helicase antiviral pathway. Immunity (2008) 28(5):651–61. doi: 10.1016/j.immuni.2008.03.013
40. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
41. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. Ikkepsilon and tbk1 are essential components of the irf3 signaling pathway. Nat Immunol (2003) 4(5):491–6. doi: 10.1038/ni921
42. Moriyama M, Koshiba T, Ichinohe T. Influenza a virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat Commun (2019) 10(1):4624. doi: 10.1038/s41467-019-12632-5
43. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins mavs, sting, and trif induces irf3 activation. Science (2015) 347(6227):aaa2630. doi: 10.1126/science.aaa2630
44. Bhowmick S, Chakravarty C, Sellathamby S, Lal SK. The influenza a virus matrix protein 2 undergoes retrograde transport from the endoplasmic reticulum into the cytoplasm and bypasses cytoplasmic proteasomal degradation. Arch Virol (2017) 162(4):919–29. doi: 10.1007/s00705-016-3153-8
45. Wang R, Zhu Y, Lin X, Ren C, Zhao J, Wang F, et al. Influenza M2 protein regulates mavs-mediated signaling pathway through interacting with mavs and increasing ros production. Autophagy (2019) 15(7):1163–81. doi: 10.1080/15548627.2019.1580089
46. Wang YY, Liu LJ, Zhong B, Liu TT, Li Y, Yang Y, et al. Wdr5 is essential for assembly of the visa-associated signaling complex and virus-triggered irf3 and nf-kappab activation. Proc Natl Acad Sci USA (2010) 107(2):815–20. doi: 10.1073/pnas.0908967107
47. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of traf3 in the toll-like receptor-dependent and -independent antiviral response. Nature (2006) 439(7073):208–11. doi: 10.1038/nature04374
48. Guo B, Cheng G. Modulation of the interferon antiviral response by the tbk1/ikki adaptor protein tank. J Biol Chem (2007) 282(16):11817–26. doi: 10.1074/jbc.M700017200
49. Tang ED, Wang CY. Traf5 is a downstream target of mavs in antiviral innate immune signaling. PloS One (2010) 5(2):e9172. doi: 10.1371/journal.pone.0009172
50. Sasai M, Shingai M, Funami K, Yoneyama M, Fujita T, Matsumoto M, et al. Nak-associated protein 1 participates in both the tlr3 and the cytoplasmic pathways in type I ifn induction. J Immunol (2006) 177(12):8676–83. doi: 10.4049/jimmunol.177.12.8676
51. Ryzhakov G, Randow F. Sintbad, a novel component of innate antiviral immunity, shares a tbk1-binding domain with nap1 and tank. EMBO J (2007) 26(13):3180–90. doi: 10.1038/sj.emboj.7601743
52. Wang L, Li S, Dorf ME. Nemo binds ubiquitinated tank-binding kinase 1 (Tbk1) to regulate innate immune responses to rna viruses. PloS One (2012) 7(9):e43756. doi: 10.1371/journal.pone.0043756
53. Pabla N, Bajwa A. Role of mitochondrial therapy for ischemic-reperfusion injury and acute kidney injury. Nephron (2022) 146(3):253–8. doi: 10.1159/000520698
54. So BYF, Yap DYH, Chan TM. Circular rnas in acute kidney injury: roles in pathophysiology and implications for clinical management. Int J Mol Sci (2022) 23(15):8509. doi: 10.3390/ijms23158509
55. Li N, Wang Y, Wang X, Sun N, Gong YH. Pathway network of pyroptosis and its potential inhibitors in acute kidney injury. Pharmacol Res (2022) 175:106033. doi: 10.1016/j.phrs.2021.106033
56. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor mavs promotes nlrp3 mitochondrial localization and inflammasome activation. Cell (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
57. Sandhir R, Halder A, Sunkaria A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim Biophys Acta Mol Basis Dis (2017) 1863(5):1090–7. doi: 10.1016/j.bbadis.2016.10.020
58. Tschopp J, Schroder K. Nlrp3 inflammasome activation: the convergence of multiple signalling pathways on ros production? Nat Rev Immunol (2010) 10(3):210–5. doi: 10.1038/nri2725
59. Liu D, Xu M, Ding LH, Lv LL, Liu H, Ma KL, et al. Activation of the nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int J Biochem Cell Biol (2014) 57:7–19. doi: 10.1016/j.biocel.2014.09.018
60. Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F. Mitochondrial reactive oxygen species-mediated nlrp3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget (2016) 7(14):17479–91. doi: 10.18632/oncotarget.8243
61. Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, et al. An inflammasome-independent role for epithelial-expressed nlrp3 in renal ischemia-reperfusion injury. J Immunol (2010) 185(10):6277–85. doi: 10.4049/jimmunol.1002330
62. Xie H, Peng J, Zhang X, Deng L, Ding Y, Zuo X, et al. Effects of mitochondrial reactive oxygen species-induced nlrp3 inflammasome activation on trichloroethylene-mediated kidney immune injury. Ecotoxicol Environ Saf (2022) 244:114067. doi: 10.1016/j.ecoenv.2022.114067
63. Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, et al. Deficient ifn signaling by myeloid cells leads to mavs-dependent virus-induced sepsis. PloS Pathog (2014) 10(4):e1004086. doi: 10.1371/journal.ppat.1004086
64. Luo S, Yang M, Han Y, Zhao H, Jiang N, Li L, et al. Beta-hydroxybutyrate against cisplatin-induced acute kidney injury via inhibiting nlrp3 inflammasome and oxidative stress. Int Immunopharmacol (2022) 111:109101. doi: 10.1016/j.intimp.2022.109101
65. Zhang Q, Sun Q, Tong Y, Bi X, Chen L, Lu J, et al. Leonurine attenuates cisplatin nephrotoxicity by suppressing the nlrp3 inflammasome, mitochondrial dysfunction, and endoplasmic reticulum stress. Int Urol Nephrol (2022) 54(9):2275–84. doi: 10.1007/s11255-021-03093-1
66. Yang SK, Han YC, He JR, Yang M, Zhang W, Zhan M, et al. Mitochondria targeted peptide ss-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ros-nlrp3 pathway. BioMed Pharmacother (2020) 130:110521. doi: 10.1016/j.biopha.2020.110521
67. Ullah M, Liu DD, Rai S, Concepcion W, Thakor AS. Hsp70-mediated nlrp3 inflammasome suppression underlies reversal of acute kidney injury following extracellular vesicle and focused ultrasound combination therapy. Int J Mol Sci (2020) 21(11):4085. doi: 10.3390/ijms21114085
68. Kim HJ, Lee DW, Ravichandran K, OK D, Akcay A, Nguyen Q, et al. Nlrp3 Inflammasome Knockout Mice Are Protected against Ischemic but Not Cisplatin-Induced Acute Kidney Injury. J Pharmacol Exp Ther (2013) 346(3):465–72. doi: 10.1124/jpet.113.205732
69. Zhu Z, Hu J, Chen Z, Feng J, Yang X, Liang W, et al. Transition of acute kidney injury to chronic kidney disease: role of metabolic reprogramming. Metabolism (2022) 131:155194. doi: 10.1016/j.metabol.2022.155194
70. Zhao YB, Wei W, Lin XX, Chai YF, Jin H. The role of histone H3 methylation in acute kidney injury. Drug Des Devel Ther (2022) 16:2453–61. doi: 10.2147/DDDT.S376673
71. Kim DH, Park JS, Choi HI, Kim CS, Bae EH, Ma SK, et al. The critical role of fxr is associated with the regulation of autophagy and apoptosis in the progression of aki to ckd. Cell Death Dis (2021) 12(4):320. doi: 10.1038/s41419-021-03620-z
72. Lei Y, Moore CB, Liesman RM, O'Connor BP, Bergstralh DT, Chen ZJ, et al. Mavs-mediated apoptosis and its inhibition by viral proteins. PloS One (2009) 4(5):e5466. doi: 10.1371/journal.pone.0005466
73. Erkan E, Devarajan P, Schwartz GJ. Mitochondria are the major targets in albumin-induced apoptosis in proximal tubule cells. J Am Soc Nephrol (2007) 18(4):1199–208. doi: 10.1681/ASN.2006040407
74. Anders HJ, Suarez-Alvarez B, Grigorescu M, Foresto-Neto O, Steiger S, Desai J, et al. The macrophage phenotype and inflammasome component nlrp3 contributes to nephrocalcinosis-related chronic kidney disease independent from il-1-mediated tissue injury. Kidney Int (2018) 93(3):656–69. doi: 10.1016/j.kint.2017.09.022
75. Cheng CH, Lee YS, Chang CJ, Lin JC, Lin TY. Genetic polymorphisms in inflammasome-dependent innate immunity among pediatric patients with severe renal parenchymal infections. PloS One (2015) 10(10):e0140128. doi: 10.1371/journal.pone.0140128
76. Chun J, Chung H, Wang X, Barry R, Taheri ZM, Platnich JM, et al. Nlrp3 localizes to the tubular epithelium in human kidney and correlates with outcome in iga nephropathy. Sci Rep (2016) 6:24667. doi: 10.1038/srep24667
77. Jung CY, Yoo TH. Pathophysiologic mechanisms and potential biomarkers in diabetic kidney disease. Diabetes Metab J (2022) 46(2):181–97. doi: 10.4093/dmj.2021.0329
78. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science (2018) 359(6382):1376–83. doi: 10.1126/science.aar3318
79. Fischer JC, Bscheider M, Eisenkolb G, Lin CC, Wintges A, Otten V, et al. Rig-I/mavs and sting signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med (2017) 9(386):eaag2513. doi: 10.1126/scitranslmed.aag2513
80. Linh HT, Iwata Y, Senda Y, Sakai-Takemori Y, Nakade Y, Oshima M, et al. Intestinal bacterial translocation contributes to diabetic kidney disease. J Am Soc Nephrol (2022) 33(6):1105–19. doi: 10.1681/ASN.2021060843
81. Farre R, Dejongh S, Meijers B. Of mice and mavs-diabetic kidney disease and the leaky gut. J Am Soc Nephrol (2022) 33(6):1053–5. doi: 10.1681/ASN.2022040407
82. Ambros V. The functions of animal micrornas. Nature (2004) 431(7006):350–5. doi: 10.1038/nature02871
83. Yan J, Zhang Y, Su Y, Tian L, Qin P, Xu X, et al. Microrna-125a targets mavs and traf6 to modulate interferon signaling and promote hcv infection. Virus Res (2021) 296:198336. doi: 10.1016/j.virusres.2021.198336
84. Hsu AC, Dua K, Starkey MR, Haw TJ, Nair PM, Nichol K, et al. Microrna-125a and -B inhibit A20 and mavs to promote inflammation and impair antiviral response in copd. JCI Insight (2017) 2(7):e90443. doi: 10.1172/jci.insight.90443
85. Hou P, Wang H, Zhao G, Hu G, Xia X, He H. Mir-3470b promotes bovine ephemeral fever virus replication via directly targeting mitochondrial antiviral signaling protein (Mavs) in baby hamster Syrian kidney cells. BMC Microbiol (2018) 18(1):224. doi: 10.1186/s12866-018-1366-6
86. Xu T, Chu Q, Cui J, Bi D. Inducible microrna-3570 feedback inhibits the rig-I-dependent innate immune response to rhabdovirus in teleost fish by targeting mavs/ips-1. J Virol (2018) 92(2):e01594-17. doi: 10.1128/JVI.01594-17
87. Wan S, Ashraf U, Ye J, Duan X, Zohaib A, Wang W, et al. Microrna-22 negatively regulates poly(I:C)-triggered type I interferon and inflammatory cytokine production via targeting mitochondrial antiviral signaling protein (Mavs). Oncotarget (2016) 7(47):76667–83. doi: 10.18632/oncotarget.12395
88. Castanier C, Zemirli N, Portier A, Garcin D, Bidere N, Vazquez A, et al. Mavs ubiquitination by the E3 ligase trim25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral rig-I-like receptors. BMC Biol (2012) 10:44. doi: 10.1186/1741-7007-10-44
89. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase rnf5 regulates antiviral responses by mediating degradation of the adaptor protein mita. Immunity (2009) 30(3):397–407. doi: 10.1016/j.immuni.2009.01.008
90. Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, et al. The mitochondrial ubiquitin ligase march5 resolves mavs aggregates during antiviral signalling. Nat Commun (2015) 6:7910. doi: 10.1038/ncomms8910
91. Wang Y, Tong X, Ye X. Ndfip1 negatively regulates rig-I-dependent immune signaling by enhancing E3 ligase smurf1-mediated mavs degradation. J Immunol (2012) 189(11):5304–13. doi: 10.4049/jimmunol.1201445
92. Pan Y, Li R, Meng JL, Mao HT, Zhang Y, Zhang J. Smurf2 negatively modulates rig-I-dependent antiviral response by targeting visa/mavs for ubiquitination and degradation. J Immunol (2014) 192(10):4758–64. doi: 10.4049/jimmunol.1302632
93. Qin Y, Xue B, Liu C, Wang X, Tian R, Xie Q, et al. Nlrx1 mediates mavs degradation to attenuate the hepatitis C virus-induced innate immune response through pcbp2. J Virol (2017) 91(23):e01264-17. doi: 10.1128/JVI.01264-17
94. Xie T, Chen T, Li C, Wang W, Cao L, Rao H, et al. Rack1 attenuates rlr antiviral signaling by targeting visa-traf complexes. Biochem Biophys Res Commun (2019) 508(3):667–74. doi: 10.1016/j.bbrc.2018.11.203
95. Zheng Y, Gao C. Fine-tuning of antiviral innate immunity by ubiquitination. Adv Immunol (2020) 145:95–128. doi: 10.1016/bs.ai.2019.11.004
96. You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, et al. Pcbp2 mediates degradation of the adaptor mavs via the hect ubiquitin ligase aip4. Nat Immunol (2009) 10(12):1300–8. doi: 10.1038/ni.1815
97. Liu C, Huang S, Wang X, Wen M, Zheng J, Wang W, et al. The otubain yod1 suppresses aggregation and activation of the signaling adaptor mavs through lys63-linked deubiquitination. J Immunol (2019) 202(10):2957–70. doi: 10.4049/jimmunol.1800656
98. Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, et al. Tetherin suppresses type I interferon signaling by targeting mavs for ndp52-mediated selective autophagic degradation in human cells. Mol Cell (2017) 68(2):308–22 e4. doi: 10.1016/j.molcel.2017.09.005
99. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease ns3/4a cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA (2005) 102(49):17717–22. doi: 10.1073/pnas.0508531102
100. Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, et al. Enterovirus 2apro targets mda5 and mavs in infected cells. J Virol (2014) 88(6):3369–78. doi: 10.1128/JVI.02712-13
101. Varga ZT, Grant A, Manicassamy B, Palese P. Influenza virus protein pb1-F2 inhibits the induction of type I interferon by binding to mavs and decreasing mitochondrial membrane potential. J Virol (2012) 86(16):8359–66. doi: 10.1128/JVI.01122-12
102. He X, Zhu Y, Zhang Y, Geng Y, Gong J, Geng J, et al. Rnf34 functions in immunity and selective mitophagy by targeting mavs for autophagic degradation. EMBO J (2019) 38(14):e100978. doi: 10.15252/embj.2018100978
103. Liu J, Wu X, Wang H, Wei J, Wu Q, Wang X, et al. Hfe inhibits type I ifns signaling by targeting the sqstm1-mediated mavs autophagic degradation. Autophagy (2021) 17(8):1962–77. doi: 10.1080/15548627.2020.1804683
104. Jia Y, Song T, Wei C, Ni C, Zheng Z, Xu Q, et al. Negative regulation of mavs-mediated innate immune response by psma7. J Immunol (2009) 183(7):4241–8. doi: 10.4049/jimmunol.0901646
105. Li C, Liu W, Wang F, Hayashi T, Mizuno K, Hattori S, et al. DNA damage-triggered activation of cgas-sting pathway induces apoptosis in human keratinocyte hacat cells. Mol Immunol (2021) 131:180–90. doi: 10.1016/j.molimm.2020.12.037
106. Sakuragi S, Liao H, Yajima K, Fujiwara S, Nakamura H. Rubella virus triggers type I interferon antiviral response in cultured human neural cells: involvement in the control of viral gene expression and infectious progeny production. Int J Mol Sci (2022) 23(17):9799. doi: 10.3390/ijms23179799
107. Pu J, Chen D, Tian G, He J, Huang Z, Zheng P, et al. All-trans retinoic acid attenuates transmissible gastroenteritis virus-induced inflammation in ipec-J2 cells via suppressing the rlrs/nf-kappab signaling pathway. Front Immunol (2022) 13:734171. doi: 10.3389/fimmu.2022.734171
108. Huang B, Li J, Zhang X, Zhao Q, Lu M, Lv Y. Rig-1 and mda-5 signaling pathways contribute to ifn-beta production and viral replication in porcine circovirus virus type 2-infected pk-15 cells in vitro. Vet Microbiol (2017) 211:36–42. doi: 10.1016/j.vetmic.2017.09.022
109. Wang X, Wang X, Zhang J, Shan Q, Zhu Y, Xu C, et al. Prediction and verification of curcumin as a potential drug for inhibition of pdcov replication in llc-pk1 cells. Int J Mol Sci (2023) 24(6):5870. doi: 10.3390/ijms24065870
Keywords: MAVS, renal disease, innate immune, NLRP3, inflammation
Citation: Wu M, Pei Z, Long G, Chen H, Jia Z and Xia W (2023) Mitochondrial antiviral signaling protein: a potential therapeutic target in renal disease. Front. Immunol. 14:1266461. doi: 10.3389/fimmu.2023.1266461
Received: 25 July 2023; Accepted: 26 September 2023;
Published: 12 October 2023.
Edited by:
Daniela Novick, Weizmann Institute of Science, IsraelReviewed by:
Jiann-Ruey Hong, National Cheng Kung University, TaiwanMoshe Levi, Georgetown University, United States
Copyright © 2023 Wu, Pei, Long, Chen, Jia and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiwei Xia, eGlhd3dwa3VAMTYzLmNvbQ==; Zhanjun Jia, amlhemo3MkBob3RtYWlsLmNvbQ==; Hongbing Chen, Y2hlbmdoYjE5OTlAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship