 Weihan Wang1†
Weihan Wang1† Cong Pang
Cong Pang Xianghua Zhang
Xianghua Zhang Lin Shi
Lin Shi- 1Department of Neurosurgery, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 2Department of Neurosurgery, The Affiliated Huai’an No.1 People’s Hospital of Nanjing Medical University, Huai’an, China
- 3Graduate School of Capital Medical University, Beijing, China
Transforming growth factor-β-activated kinase 1 (TAK1) positively regulates oxidative stress and inflammation in different diseases. Takinib, a novel and specific TAK1 inhibitor, has beneficial effects in a variety of disorders. However, the effects of takinib on early brain injury (EBI) after subarachnoid hemorrhage (SAH) and the underlying molecular mechanisms remain unknown. Our study showed that takinib administration significantly inhibited phosphorylated TAK1 expression after SAH. In addition, takinib suppressed M1 microglial polarization and promoted M2 microglial polarization. Furthermore, blockade of TAK1 by takinib reduced neuroinflammation, oxidative damage, brain edema, and neuronal apoptosis, and improved neurological behavior after SAH. Mechanistically, we revealed that TAK1 inhibition by takinib mitigated reactive oxygen species (ROS) production and ROS-mediated nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome activation. In contrast, NLRP3 activation by nigericin abated the neuroprotective effects of takinib against EBI after SAH. In general, our study demonstrated that takinib could protect against EBI by targeting TAK1-ROS-NLRP3 inflammasome signaling. Inhibition of TAK1 might be a promising option in the management of SAH.
1 Introduction
Subarachnoid hemorrhage (SAH) remains a life-threatening disease with poor prognosis. How to improve the clinical outcome after SAH is still a tough challenge. Multiple pathophysiological processes occurring after SAH, including early brain injury (EBI), cerebral vasospasm, and delayed cerebral ischaemia (1). Till now, mounting evidence has demonstrated that the neurological outcome after SAH is seriously influenced by early brain injury (EBI) (2–4). Multiple cellular mechanisms are involved in the EBI pathophysiology after SAH. Among them, activation of inflammatory response and oxidative damage contribute greatly to the development of EBI (5–7). Thus, a potential therapy for improving the prognosis after SAH is intervening neuroinflammation and oxidative stress.
Transforming growth factor-β-activated kinase 1 (TAK1), a member of the mitogen‐activated protein kinase (MAPK) family, has a wide range of biological functions (8–10). It has demonstrated that TAK1 activation could strongly elicit pro-inflammatory cytokines release and drive microglial toward a proinflammatory phenotype (11–14). In addition, TAK1 mediates reactive oxygen species (ROS) generation to aggravate oxidative damage in different diseases models (15, 16). In central nervous system (CNS), TAK1 is mainly expressed in neurons and could regulate a variety of intracellular signaling pathways, including nuclear factor‐κB (NF‐κB), AMP-activated protein kinase (AMPK), and nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome (17–19). Since these molecular mechanisms contribute to EBI pathophysiology, targeting TAK1 might be an effective treatment for SAH.
Takinib, a novel and specific TAK1 inhibitor, has been recently evaluated in a variety of disease models (20–22). Previous studies have demonstrated that takinib could inhibit inflammation and apoptosis by suppression of TAK1-mediated signaling pathway (20, 21). However, whether takinib could protect against EBI after SAH and the potential molecular mechanisms remain unclear. Hence, this study aimed to investigate the possible role of takinib in EBI after SAH and its underlying mechanisms.
2 Materials and methods
All the experimental procedures were approved by the Animal Ethics Review Committee of Beijing Friendship Hospital and carried out in accordance with the National Institutes of Health guidelines. A total of 190 rats (weighing 250 to 300 g) were used in our study. Among them, 154 rats underwent SAH insults and 8 rats were excluded due to mild SAH grading score (less than 8) and intracerebral hematoma. All rats were numbered consecutively and randomization was conducted by using the website Randomization (http://www.randomization.com).
2.1 SAH model
The SAH model was performed according to our previous studies (23, 24). Briefly, rats were anesthetized with avertin (200 mg/kg). After anesthetization, rats were operated in a stereotactic frame. A hole was drilled into the skull in the midline 7.5 mm anterior to the bregma. SAH animals were injected with 0.35 ml of nonheparinized fresh autologous arterial blood from the femoral artery through the burr hole under aseptic conditions. Sham group rats underwent the same procedures, but were injected with 0.35 ml physiological saline. The basal temporal lobe adjacent to the clotted blood were collected for evaluation. After recovering from anesthesia, rats were housed in their cages and were free to food and water.
2.2 Experiment design
In the first set of experiments, the dose-dependent effects of takinib on TAK1 activation were examined. Total of 70 rats were randomly assigned into 5 groups, including sham + vehicle (n = 12), SAH + vehicle (n = 15, 3 rats died), SAH + 0.1 mM takinib (n = 15, 3 rats died), SAH + 0.3 mM takinib (n = 14, 2 rats died), and SAH + 0.9 mM takinib (n = 14, 2 rats died). Western blotting and double immunofluorescence staining were conducted to show the protein expression and cellular distribution of TAK1 in the brain cortex.
In the second set of experiments, we further explored the effects of takinib on microglial activation, ROS overproduction, and the subsequent brain damage after SAH. A total of 83 rats were randomly assigned into 4 groups, including sham + vehicle (n = 18), SAH + vehicle (n = 22, 4 rats died), SAH + 0.9 mM takinib (n = 21, 3 rats died), and SAH + 0.9 mM takinib + nigericin (n = 22, 4 rats died). Western blotting, double immunofluorescence staining, biochemical estimation, brain edema, and neurological behavior were conducted to determine the effects of takinib on EBI after SAH and the possible mechanisms.
In the third set of experiments, we investigated the effects of takinib on histopathological change and neurological behavior at day 3 after SAH. A total of 29 rats were randomly assigned into 4 groups, including sham + vehicle (n = 6), SAH + vehicle (n = 8, 1 rats died), SAH + 0.9 mM takinib (n = 7, 1 rats died), and SAH + 0.9 mM takinib + nigericin (n = 8, 1 rats died). Nissl staining and neurological behavior were performed. The experiment design and timeline were shown in Supplementary Figure 1.
2.3 Drug administration
Takinib (Selleck) was dissolved in dimethyl sulfoxide (DMSO) and diluted in physiologic saline at concentrations of 0.1, 0.3, and 0.9 mM. Various doses of takinib (10 μl) or vehicle was administered into the left lateral ventricle at 30 min post SAH insults. Nigericin (Selleck, 2 μg) was dissolved in 2 μl ethanol and physiologic saline and was administered to rats by intracerebroventricular route 2 h before SAH construction. For the intracerebroventricular administration, rats were put on a stereotactic frame. Coordinates for the intracerebroventricular injection were 1.5 mm posterior and 1.0 mm lateral to bregma, and 4.5 mm below the dural layer. The doses of takinib and nigericin were selected according to previous studies (20, 25).
2.4 Neurological scoring
Neurological function was evaluated by using a neurological severity scoring system as previously reported (26). Six test sub-scores were included in this scoring system. The high scores represented a relative normal neurological function. In addition, the rotarod test was conducted to measure motor function. The duration on the rotarod was recorded for statistical analysis (1, 27).
2.5 Brain water content
Brain water content was determined by using a wet/dry method (28). At 24 h following SAH, rats were deeply anesthetized with an overdose of avertin and the brains were separated into three parts, including cerebrum, cerebellum and brain stem. The wet weight was recorded. Each part of brain was dried at 80°C for 72 h and weighed again (dry weight). Brain water content was calculated by [(wet weight − dry weight)/wet weight] × 100%.
2.6 Western blotting
Western blotting was conducted according to previous studies (23, 29). In brief, the protein samples were separated by SDS-PAGE gel. And then they were transferred to nitrocellulose membrane. Afterward, the membranes were incubated with primary antibodies against TAK1 (1:1000, Cell Signaling), p-TAK1 (1:1000, Cell Signaling), NLRP3 (1:200, Santa Cruz Biotechnology), ASC (1:200, Santa Cruz Biotechnology), caspase-1 (1:200, Santa Cruz Biotechnology), cleaved caspase-1 (1:200, Santa Cruz Biotechnology), 3-nitrotyrosine (1:2000, Abcam), and β-actin (1:3000, Bioworld Technology) overnight at 4°C. After that, appropriate secondary antibodies were incubated at room temperature. Membranes were then exposed by ECL reagent.
2.7 Immunofluorescence
Immunofluorescence staining was conducted according to previous studies (23, 24). In brief, brain sections were blocked with 5% donkey serum. And then they were incubated overnight at 4°C with primary antibodies against p-TAK1 (1:100, Cell Signaling), CD16/32 (1:100, BD Biosciences), CD206 (1:100, Invitrogen), NLRP3 (1:50, Santa Cruz Biotechnology), 8-hydroxydeoxyguanosine (8-OhdG) (1:100, Abcam), IL-1β (1:100, Abcam), NeuN (1:200, EMD Millipore), and Iba-1 (1:50, Santa Cruz Biotechnology). Next, the brain sections were incubated with appropriate secondary antibodies. The fluorescently stained cells were visualized and photographed under a fluorescence microscope.
2.8 TUNEL staining
TUNEL staining (Beyotime) was conducted in line with the operation instructions. In brief, the brain sections were incubated with primary antibody against NeuN overnight. Afterward, the slides were incubated with TUNEL reaction mixture. Sections were visualized and photographed under a fluorescence microscope.
2.9 Malondialdehyde detection
The level of malondialdehyde (MDA) in brain samples were examined according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute). MDA was determined at the wavelength of 535 nm using a spectrophotometer.
2.10 Statistical analysis
All values were expressed as means ± s.d. GraphPad Prism 8.02 was used to conduct statistical analysis. All data were tested for normality via Shapiro–Wilk test. Measurements were analyzed with one-way ANOVA followed by Tukey’s post-hoc test. P <0.05 was verified as statistically different.
3 Results
3.1 Effects of takinib on TAK1 activation after SAH
Takinib is a novel and highly selective TAK1 inhibitor. However, the influence of takinib on TAK1 activation after SAH remains obscure. Western blotting was conducted to detect the expression of p-TAK1 and TAK-1 after SAH. The results showed that doses of 0.3 mM and 0.9 mM takinib, but not 0.1 mM takinib, markedly inhibited p-TAK1 expression as compared with SAH + vehicle group (Figures 1A, B). There was no significant difference among all experimental groups in total TAK1 expression (Figure 1C). It has been demonstrated that TAK1 is mainly expressed in neurons. Consistent with previous studies, double immunofluorescence revealed that TAK1 activation was mainly distributed in neurons after SAH. In contrast, takinib treatment at 0.3 mM and 0.9 mM could significantly suppress TAK1 activation in neurons (Figures 1D, E). Since 0.9 mM takinib had the maximal effects, we used this dose in the subsequent experiments.
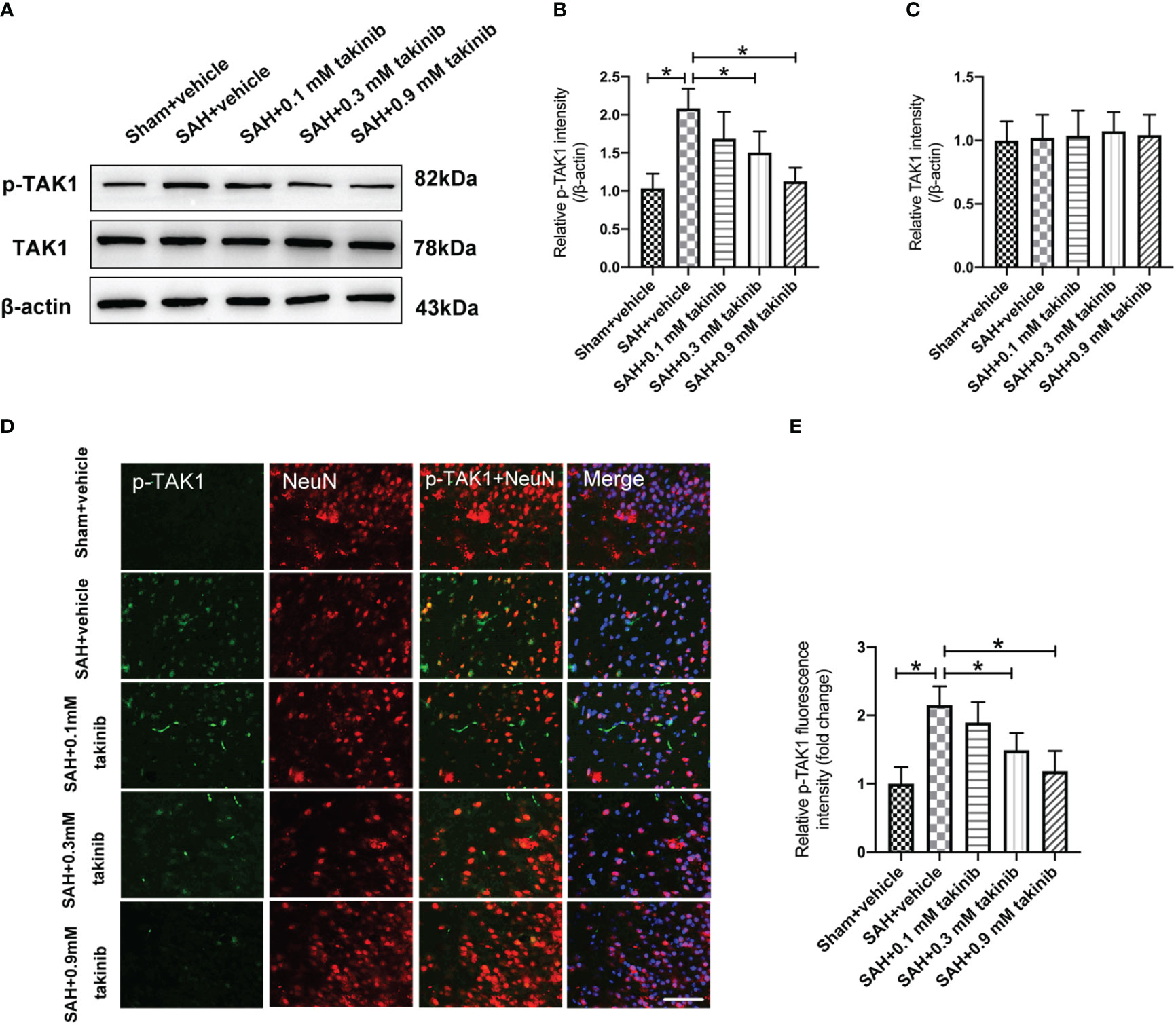
Figure 1 Dose-response effects of takinib on TAK1 activation after SAH. (A) Representative western blot bands and quantitative analyses of p-TAK1 (B) and TAK1 (C) after SAH (n = 6 each group). (D) Representative photomicrographs and quantification (E) of p-TAK1 immunofluorescence staining in the basal cortex at 24 h after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
3.2 Influence of takinib on NLRP3 inflammasome signaling after SAH
TAK1 has been verified as a key regulator of NLRP3 inflammasome activation. However, whether takinib could modulate NLRP3 inflammasome signaling after SAH remains unknown. In this experiment, we evaluated the effects of takinib on NLRP3 inflammasome signaling after SAH. As shown, western blotting data revealed that SAH insults significantly induced the protein levels of p-TAK1, NLRP3, ASC, cleaved caspase1, IL-1β, and IL-18, all of which were reversed by takinib treatment (Figures 2A–H). However, in addition to p-TAK1, other molecular targets alterations by takinib were counteracted by nigericin administration (Figures 2A–H). No significant differences in the expressions of TAK1 (Figure 2C) and caspase1 (Figure 2E) were detected among all experimental groups.
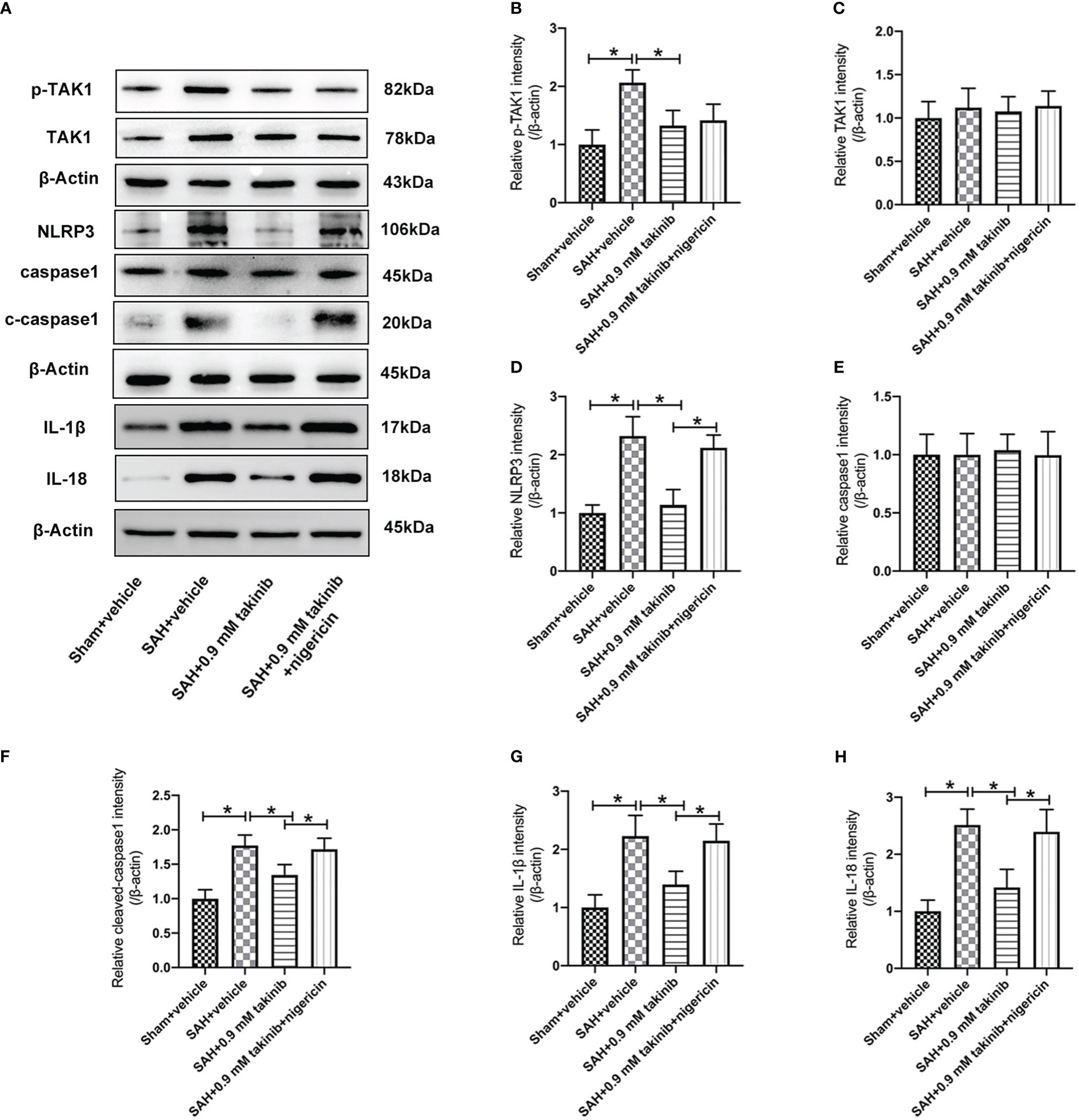
Figure 2 Effects of takinib on TAK1-NLRP3 inflammasome activation after SAH. (A) Representative western blot bands and quantitative analyses of p-TAK1 (B), TAK1 (C), NLRP3 (D), caspase1 (E), cleaved caspase1 (F), IL-1β (G), and IL-18 (H) after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
3.3 Influence of takinib on M1/M2 microglial polarization after SAH
Microglial polarization plays a critical role in neuroinflammation after SAH. Numerous studies have demonstrated that TAK1 could induce microglial activation. However, whether TAK1 could modulate microglial polarization after SAH remains elusive. We then evaluated the effects of takinib on M1/M2 microglial polarization after SAH. The double immunofluorescence showed that takinib treatment significantly reduced the proportion of M1 microglial (CD16/32+) and enhanced the quantity of M2 microglial (CD206+) following SAH (Figures 3A–F). In contrast, NLRP3 activator nigericin abated the effects of takinib on M1/M2 microglial polarization (Figures 3A–F).
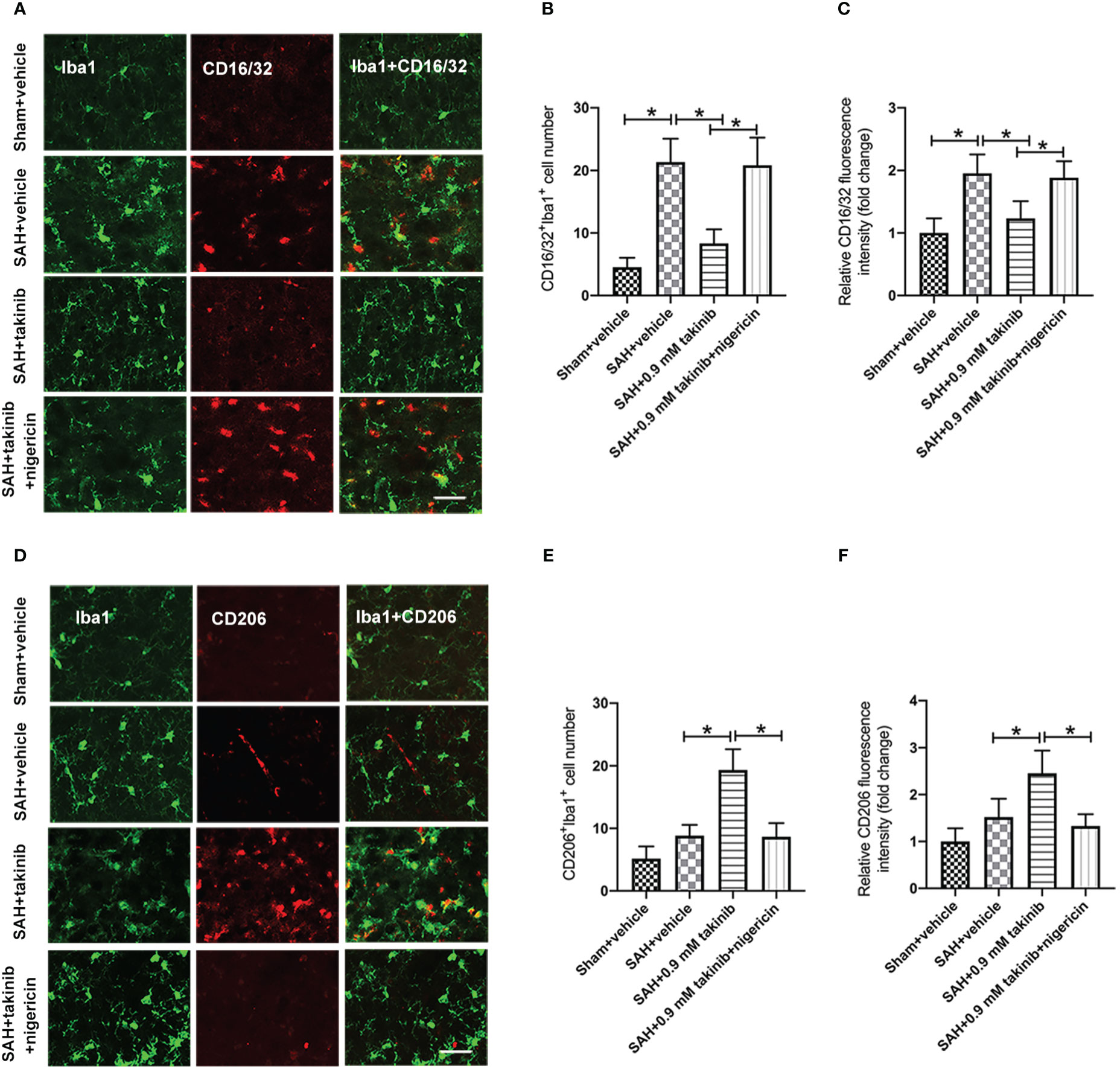
Figure 3 Effects of takinib on microglial phenotypic transformation after SAH. (A) Double immunofluorescence staining for CD16/32 in microglial in the basal cortex after SAH. (B, C) Quantification of CD16/32 immunofluorescence staining in the basal cortex at 24 h after SAH (n = 6 each group). (D) Double immunofluorescence staining for CD206 in microglial in the basal cortex after SAH. (E, F) Quantification of CD206 immunofluorescence staining in the basal cortex at 24 h after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
3.4 Influence of takinib on ROS production, and oxidative damage after SAH
ROS overproduction plays a key role in oxidative damage and contributes greatly to the development of EBI after SAH. Previous studies have demonstrated that TAK1 activation could aggravate ROS overproduction and oxidative damage. However, the influence of takinib on ROS production and oxidative damage following SAH remains unknown. We further evaluated the effects of takinib on ROS production and oxidative damage after SAH. 3-nitrotyrosine, a major product of tyrosine oxidation, is an important biomarker for ROS production. MDA is a biological marker for oxidative damage and lipid peroxidation. Our data revealed that SAH insults significantly induced ROS overproduction, lipid peroxidation, and oxidative damage, all of which were ameliorated by takinib treatment (Figures 4A–E). In contrast, the anti-oxidative effects of takinib could be partly reversed by nigericin treatment (Figures 4A–E).
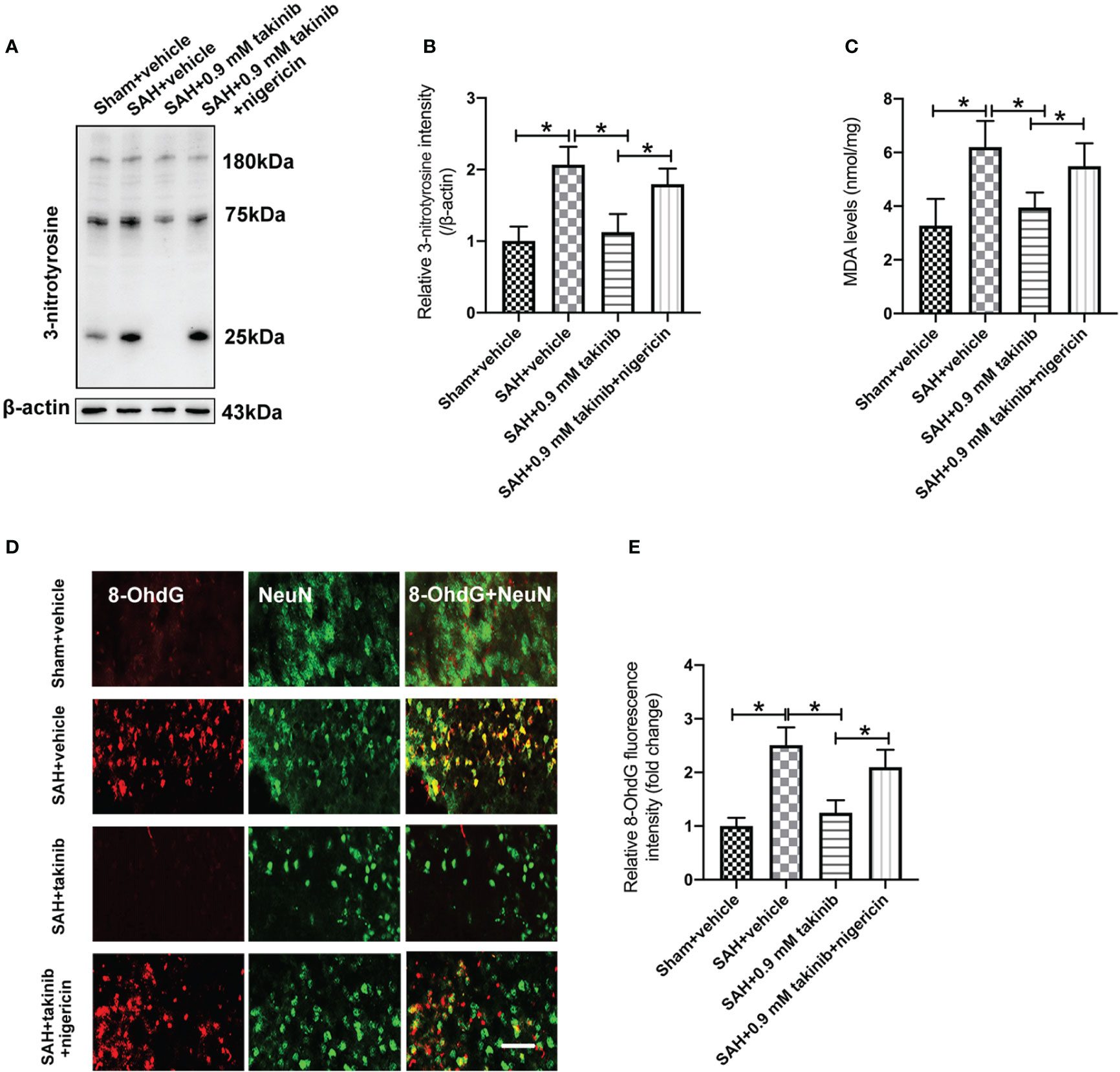
Figure 4 Effects of takinib on ROS and oxidative damage after SAH. Western blot assay (A) and quantification (B) for expression of 3-nitrotyrosine in different groups (n = 6 each group). (C) Quantification of MDA level in brain tissue at 24 h post-SAH (n = 6 each group). (D) Representative photomicrographs and quantification (E) of 8-OhdG immunofluorescence staining in the basal cortex after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
3.5 Influence of takinib on neurological function, brain edema, and neuronal apoptosis at day 1 after SAH
We then evaluated the effects of takinib on neurological outcomes, brain edema, and neuronal apoptosis after SAH. Consistent with the reduced neuroinflammation and oxidative damage, takinib treatment significantly improved neurological scores and motor functions, ameliorated brain edema, and reduced neuronal apoptosis after SAH (Figures 5A–E). In contrast, the pretreatment of nigericin statistically deteriorated the beneficial effects of takinib on neurological behavior, brain edema, and neuronal apoptosis after SAH (Figures 5A–E).
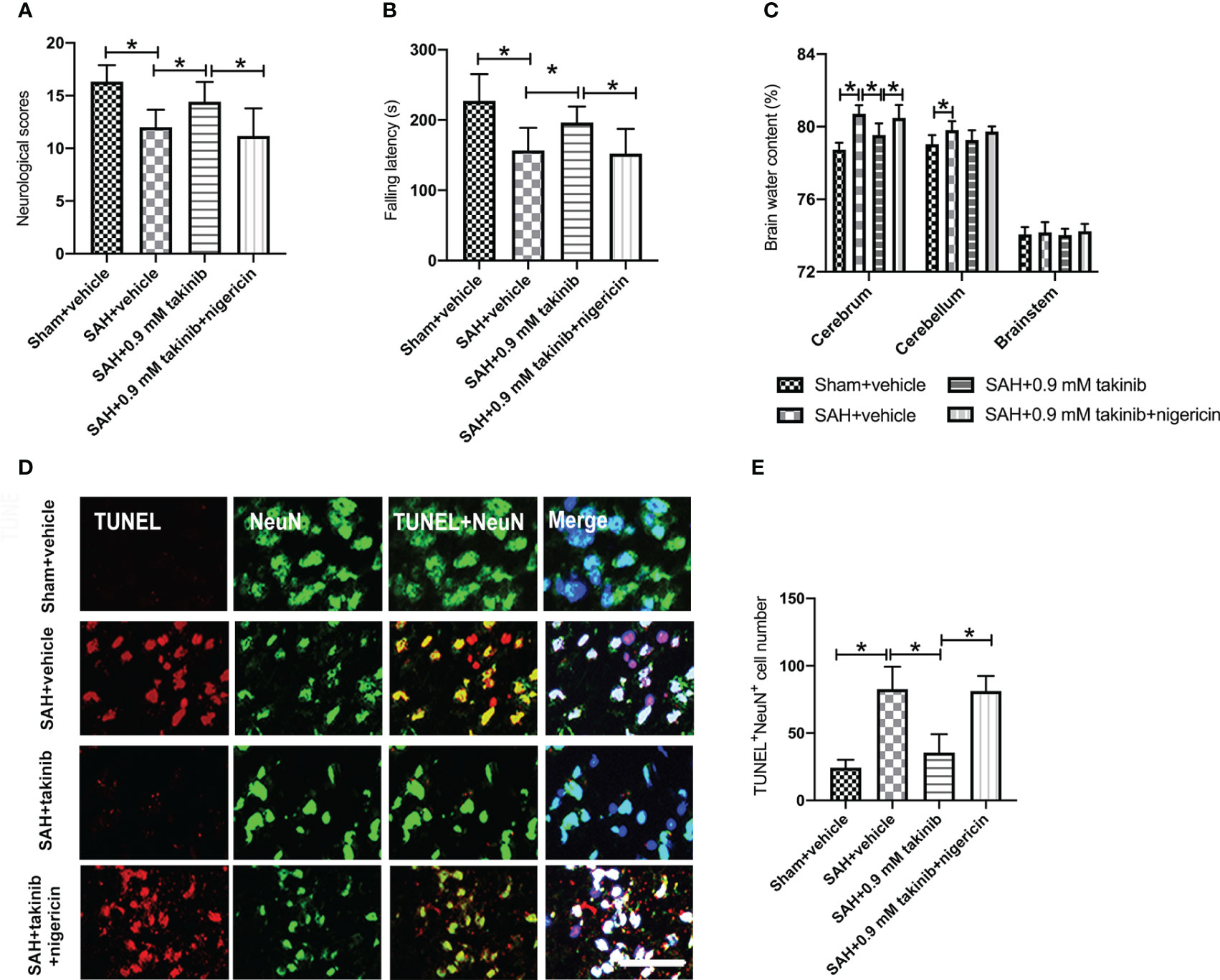
Figure 5 Effects of takinib on neurological behavior, brain water content, and neuronal survival after SAH. Quantification of (A) neurological deficits scores, (B) rotarod performance, and (C) brain water content in different groups after SAH (n = 10-12 each group). (D) Representative photomicrographs and quantification (E) of TUNEL staining in the basal cortex after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
3.6 Influence of takinib on histopathological change and neurological behavior at day 3 after SAH
The first 72 h following SAH play a vital role in determining overall outcome. We then investigated the effects of takinib on histopathological change and neurological behavior at day 3 after SAH. It indicated that SAH insults induced an evident histopathological damage in brain cortex as evidenced by sparse cell arrangements and integrity loss. In contrast, takinib significantly improved histopathological change and reduced neuronal degeneration. In addition, takinib also improved motor function in the early period after SAH. However, all these cerebroprotective effects were abated by nigericin (Figures 6A–D).
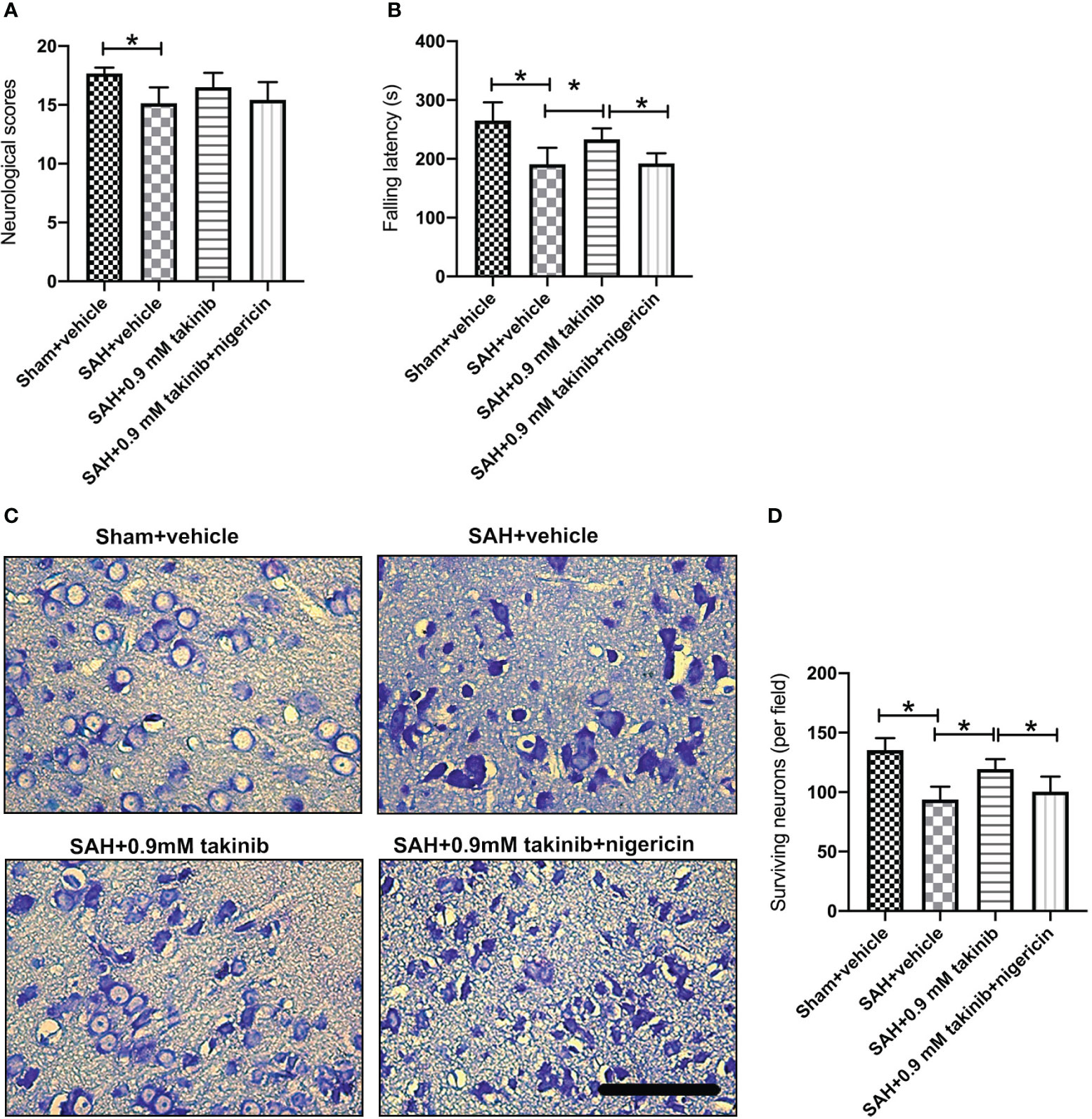
Figure 6 Effects of takinib on histopathological change and neurological behavior at day 3 after SAH. Quantification of (A) neurological deficits scores and (B) rotarod performance in different groups after SAH (n = 6 - 7 each group). (C) Representative photomicrographs and quantification (D) of survival neurons in the basal cortex after SAH (n = 6 each group). One-way ANOVA with Tukey, bars represent the mean ± SD. *P < 0.05.
4 Discussion
In this study, we verified the beneficial effects of takinib on EBI after SAH. Our data indicated that takinib could ameliorate SAH-induced inflammatory response by inhibiting M1-microglial phenotype polarization and promoting microglial polarization to M2 phenotype. In addition, takinib reduced ROS generation and suppressed oxidative damage after SAH. Concomitant with the reduced neuroinflammation and oxidative stress, takinib improved functional outcome and neuronal survival after SAH. Mechanistically, takinib inhibited TAK1 activation and the subsequent ROS-NLRP3 inflammasome signaling pathway following SAH. In contrast, NLRP3 activation by nigericin abated the neuroprotective effects of takinib against EBI after SAH (Figure 7).
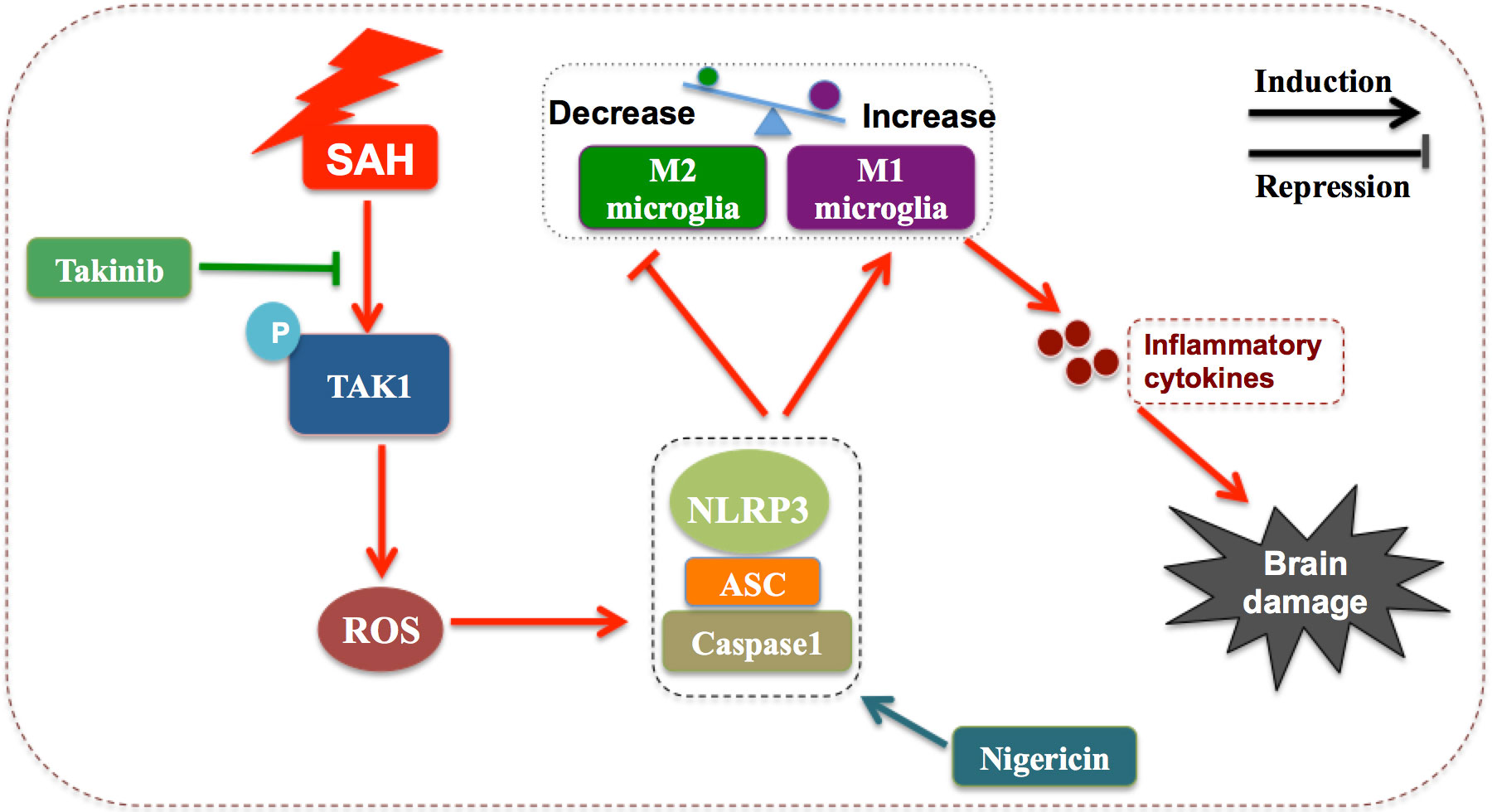
Figure 7 Schematic illustrating the possible mechanisms of takinib action after SAH. As illustrated, SAH significantly increases the expression level of p-TAK1(Thr187) indicating that TAK1 is activated after SAH. TAK1 activation triggers reactive oxygen species (ROS) generation. In response to ROS accumulation after SAH, NLRP3 recruits the adaptor apoptosis-related speck-like protein (ASC) and pro-caspase-1 to form a large multiprotein complex. NLRP3 inflammasome activation promotes microglial phenotype toward M1 and inhibits M2 microglial polarization after SAH, subsequently aggravating neuroinflammation and brain damage. Takinib, a novel and highly selective TAK1 inhibitor, could suppress TAK1 activation and TAK1-mediaed ROS-NLRP3 inflammasome signaling after SAH. In contrast, NLRP3 inflammasome activator nigericin reverses the beneficial effects of takinib against SAH, eventually aggravating SAH-induced brain damage.
microglial polarization plays a critical role in the pathogenesis of neuroinflammation after SAH (29–31). Activated microglial exhibit different phenotypes under different stimulants and have distinct functions. M1-polarized microglial could increase proinflammatory cytokines release and aggravate ROS production. In contrast, M2-polarized microglial produce anti-inflammatory cytokines to keep the immune balance (29, 32). It has demonstrated that inhibition of M1 microglial polarization and induction microglial polarization into M2 could alleviate neuroinflammation and improve neurological outcomes after SAH (29). In addition, oxidative stress also participates in the development of EBI after SAH. Numerous studies have reported that ROS overproduction could induce neuroinflammation to exacerbate brain damage after SAH (1, 24, 33). Therefore, inhibition of M1-like microglial polarization and oxidative damage might be a successful strategy to reduce EBI after SAH.
Multiple studies have proved that TAK1 inhibition is a promising therapeutic application for various CNS diseases including traumatic brain injury (TBI), ischemic stroke, and SAH (34–36). For example, Shen et al. demonstrated that microglial-selective deletion of Tak1 inhibited IL-18 production and ameliorated ischemic stroke injury in prolonged obesity (37). Shi et al. reported that pharmacological inhibition of TAK1 with 5Z-7-oxozeaenol provided long-lasting improvement of stroke outcomes (38). In SAH area, TAK1 inhibition also attenuated EBI by reducing neuroinflammation and neuronal death (34). These indicated that TAK1 might be a promising target for treating SAH. However, the effects of TAK1 inhibition on microglial polarization and oxidative damage after SAH remain unclear.
It has been reported that 5Z-7-oxozeaenol is a potent selective inhibitor of TAK1 and vascular endothelial growth factor receptor 2 (39). Different with 5Z-7-oxozeaenol, takinib is a novel and highly selective TAK1 inhibitor. Recent studies have showed that takinib could effectively inhibit TAK1 activation-mediated inflammatory and apoptotic signaling cascades in different research fields (20, 21). However, to date, no study investigated the potential role of takinib on EBI after SAH. In our study, we first evaluated the effects of takinib on TAK1 expression after SAH. In accordance with previous studies, our data revealed that p-TAK1 (Thr 187) was significantly increased in neurons after SAH. In contrast, takinib treatment significantly decreased p-TAK1 expression. We then evaluated the possible influence of takinib on microglial polarization. Intriguingly, it showed that SAH insults significantly induced M1 microglial polarization, which could be inhibited by takinib treatment. Moreover, TAK1 inhibition by takinib increased M2 phenotype microglial. These suggested that TAK1 inhibition by takinib might have the potential to inhibit M1 microglial polarization and promote the microglial phenotype toward M2.
ROS overproduction plays an important role in the development of EBI after SAH. It can disrupt cellular functions by damaging nucleic acids, proteins, and lipids (1). Moreover, ROS production can aggravate neuroinflammation by activation of NLRP3 inflammasome signaling (25, 40). NLRP3 inflammasome has been demonstrated to participate in microglial polarization after SAH. Previous studies have demonstrated that inhibition of ROS/NLRP3 inflammasome signaling could decrease microglial M1 polarization and promote microglial polarization to M2 phenotype (29, 41). Our experiments revealed that TAK1 inhibition by takinib regulated microglial M1-M2 phenotype transition after SAH. However, the possible mechanisms remain elusive. Mounting evidence has showed that TAK1 activation could induce the aggravation of oxidative stress by promoting ROS production (16, 42). Inhibition of TAK1 is able to attenuate ROS overproduction and might be a potential therapeutic target for oxidative stress-related injuries. Interestingly, TAK1 has been verified as a key regulator of NLRP3 inflammasome activation (11, 34, 43). In our experiments, our data showed that takinib successfully inhibited the ROS overproduction and the subsequent activation of NLRP3 inflammasome signaling after SAH. Moreover, nigericin, a NLRP3 activator, abated the protective effects of takinib against EBI after SAH, validating the interaction between TAK1 and NLRP3 inflammasome. Concomitant with the reduced oxidative damage and neuroinflammation, takinib treatment significantly reduced neuronal apoptosis and improved functional behavior after SAH. Together with our experimental results, we provided the evidence that TAK1-ROS-NLRP3 inflammasome axis involved in the development of EBI after SAH. By intervening with TAK1-ROS-NLRP3 inflammasome axis, takinib modulated microglial polarization and inhibited oxidative damage.
It should be noted that NLRP3 inflammasome could modulate microglial polarization in a variety of disorders, including intracerebral hemorrhage, depression, ischemic stroke, white matter injury, as well as Alzheimer’s disease (44–46). These suggested that targeting NLRP3 inflammasome might be a feasible method to relieve neuroinflammation. In SAH area, Xu et al. reported that TAK1 inhibition by siRNA could reduce NLRP3 inflammasome-mediated neuronal pyroptosis (34). However, the previous studies did not investigate whether TAK1 inhibition affected microglial polarization. In the present study, we demonstrated that TAK1 could affect microglial polarization by modulating NLRP3 inflammasome. Inhibition TAK1 by takinib suppressed NLRP3 inflammasome and decreased M1 microglial polarization. However, how TAK1 regulates NLRP3 inflammasome is not fully investigated. In addition to ROS, K+ efflux, endosomal rupture, and mitochondrial dysfunction could trigger NLRP3 inflammasome activation (47). Hindi et al. indicated that TAK1 plays a critical role in regulating skeletal muscle mass and oxidative metabolism. TAK1 activation could induce an accumulation of dysfunctional mitochondria as well as oxidative damage in skeletal muscle (19). We suspected that TAK1 might also affect mitochondrial dysfunction to trigger NLRP3 inflammation. But further studies are still needed to decipher this question and whether other molecular targets are involved in this modulation.
Our study has several limitations. Firstly, the long-term effects of TAK1 inhibition in the delayed phase of SAH remains unclear. Meanwhile, no studies have performed to investigate the influence of TAK1 on cerebral vasospasm and delayed ischemic neurologic deficits after SAH. A recent study by Shen et al. indicated that prolonged high-fat diet (for 32 weeks) feeding-induced obesity has a significant cerebrovascular dysfunction, which is closely associated with microglial TAK1 activation. They showed that microglial TAK1 activation significantly aggravated basilar artery abnormalities. Both pharmacological suppression and genetic microglial-selective TAK1 deletion relieved basilar artery dysfunction and improved the outcome of ischemic stroke (37). On this background, we suspected that TAK1 inhibition might ameliorate cerebral vasospasm and relieve delayed ischemic neurologic deficits in the late phase of SAH. Secondly, the intracerebroventricular administration limits the clinical utility of takinib. Other administration routes instead of ventricle injections should be conducted to validate its clinical translatability. Thirdly, our data indicated that takinib could protect against M1 microglial polarization and oxidative damage after SAH by modulation of TAK1-ROS-NLRP3 inflammasome axis. However, whether these effects are mediated by NF‐κB, AMPK, and p38 should be further determined (48, 49). Lastly, in addition to activate NLRP3, nigericin might modulate other signaling targets to aggravate neuroinflammation. Thus, more experiments are needed to solve these questions.
5 Conclusion
In conclusion, we provided the first evidence that takinib could modulate microglial polarization and inhibit oxidative damage after SAH primarily by targeting the TAK1-ROS-NLRP3 inflammasome axis. These findings implied that takinib might be a potential new therapeutic candidate for the treatment of SAH.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by the Animal Ethics Review Committee of Beijing Friendship Hospital. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
WW: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. CP: Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft. JZ: Investigation, Methodology, Validation, Writing – review & editing. LP: Methodology, Writing – review & editing. XZ: Methodology, Supervision, Writing – review & editing. LS: Conceptualization, Methodology, Project administration, Writing – original draft, Writing – review & editing. HZ: Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from Beijing Friendship Hospital (No. PYZ21061).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1266315/full#supplementary-material
Supplementary Figure 1 | Schematic illustration of experiment design.
Abbreviations
8-OhdG, 8-hydroxydeoxyguanosine; AMPK, AMP-activated protein kinase; CNS, central nervous system; EBI, early brain injury; MAPK, mitogen‐activated protein kinase; MDA, malondialdehyde; NF-‐κB, nuclear factor‐κB; NLRP3, nod-like receptor pyrin domain-containing protein 3; ROS, reactive oxygen species; SAH, subarachnoid hemorrhage; TAK1, Transforming growth factor-β-activated kinase 1.
References
1. Zhang XS, Lu Y, Li W, Tao T, Wang WH, Gao S, et al. Cerebroprotection by dioscin after experimental subarachnoid haemorrhage via inhibiting NLRP3 inflammasome through SIRT1-dependent pathway. Br J Pharmacol (2021) 178(18):3648–66. doi: 10.1111/bph.15507
2. Schleicher RL, Bevers MB, Rubin DB, Koch MJ, Bache S, Lissak IA, et al. Early brain injury and soluble ST2 after nontraumatic subarachnoid hemorrhage. Stroke (2021) 52(8):e494–6. doi: 10.1161/STROKEAHA.121.035372
3. Al-Mufti F, Amuluru K, Smith B, Damodara N, El-Ghanem M, Singh IP, et al. Emerging markers of early brain injury and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. World Neurosurg (2017) 107:148–59. doi: 10.1016/j.wneu.2017.07.114
4. Lauzier DC, Jayaraman K, Yuan JY, Diwan D, Vellimana AK, Osbun JW, et al. Early brain injury after subarachnoid hemorrhage: incidence and mechanisms. Stroke (2023) 54(5):1426–40. doi: 10.1161/STROKEAHA.122.040072
5. Topkoru B, Egemen E, Solaroglu I, Zhang JH. Early brain injury or vasospasm? An overview of common mechanisms. Curr Drug Targets (2017) 18(12):1424–9. doi: 10.2174/1389450117666160905112923
6. Geraghty JR, Davis JL, Testai FD. Neuroinflammation and microvascular dysfunction after experimental subarachnoid hemorrhage: emerging components of early brain injury related to outcome. Neurocrit Care (2019) 31(2):373–89. doi: 10.1007/s12028-019-00710-x
7. Krenzlin H, Wesp D, Schmitt J, Frenz C, Kurz E, Masomi-Bornwasser J, et al. Decreased superoxide dismutase concentrations (SOD) in plasma and CSF and increased circulating total antioxidant capacity (TAC) are associated with unfavorable neurological outcome after aneurysmal subarachnoid hemorrhage. J Clin Med (2021) 10(6):1188. doi: 10.3390/jcm10061188
8. Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell Death Differ (2014) 21(11):1667–76. doi: 10.1038/cdd.2014.123
9. Totzke J, Scarneo SA, Yang KW, Haystead TAJ. TAK1: a potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol (2020) 10(9):200099. doi: 10.1098/rsob.200099
10. Tan WQ, Fang QQ, Shen XZ, Giani JF, Zhao TV, Shi P, et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-beta-activated kinase 1 (TAK1) pathways in mice. Br J Pharmacol (2018) 175(22):4239–52. doi: 10.1111/bph.14489
11. Heinisch O, Zeyen T, Goldmann T, Prinz M, Huber M, Jung J, et al. Erythropoietin abrogates post-ischemic activation of the NLRP3, NLRC4, and AIM2 inflammasomes in microglial/macrophages in a TAK1-dependent manner. Transl Stroke Res (2022) 13(3):462–82. doi: 10.1007/s12975-021-00948-8
12. Wang R, Pu H, Ye Q, Jiang M, Chen J, Zhao J, et al. Transforming growth factor beta-activated kinase 1-dependent microglial and macrophage responses aggravate long-term outcomes after ischemic stroke. Stroke (2020) 51(3):975–85. doi: 10.1161/STROKEAHA.119.028398
13. Iwamoto M, Nakamura Y, Takemura M, Hisaoka-Nakashima K, Morioka N. TLR4-TAK1-p38 MAPK pathway and HDAC6 regulate the expression of sigma-1 receptors in rat primary cultured microglial. J Pharmacol Sci (2020) 144(1):23–9. doi: 10.1016/j.jphs.2020.06.007
14. Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, et al. A new type of microglial gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci (2013) 16(11):1618–26. doi: 10.1038/nn.3531
15. Tang P, Chen WX, Gao HL, Dai JY, Gu Y, Xie ZA, et al. Small molecule inhibitor of TAK1 ameliorates rat cartilaginous endplate degeneration induced by oxidative stress in vitro and in vivo. Free Radic Biol Med (2020) 148:140–50. doi: 10.1016/j.freeradbiomed.2020.01.002
16. Suzuki M, Asai Y, Kagi T, Noguchi T, Yamada M, Hirata Y, et al. TAK1 mediates ROS generation triggered by the specific cephalosporins through noncanonical mechanisms. Int J Mol Sci. (2020) 14;21(24):9497. doi: 10.3390/ijms21249497
17. Bosman MC, Schepers H, Jaques J, Brouwers-Vos AZ, Quax WJ, Schuringa JJ, et al. The TAK1-NF-kappaB axis as therapeutic target for AML. Blood (2014) 124(20):3130–40. doi: 10.1182/blood-2014-04-569780
18. Mangan MS, Latz E. TAK1ng control: TAK1 restrains NLRP3 activation. J Exp Med (2018) 215(4):1007–8. doi: 10.1084/jem.20180282
19. Hindi SM, Sato S, Xiong G, Bohnert KR, Gibb AA, Gallot YS, et al. TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight (2018) 3(3):e98441. doi: 10.1172/jci.insight.98441
20. Scarneo SA, Eibschutz LS, Bendele PJ, Yang KW, Totzke J, Hughes P, et al. Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res Ther (2019) 21(1):292. doi: 10.1186/s13075-019-2073-x
21. Totzke J, Gurbani D, Raphemot R, Hughes PF, Bodoor K, Carlson DA, et al. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF-alpha inhibition for cancer and autoimmune disease. Cell Chem Biol (2017) 24(8):1029–1039 e7. doi: 10.1016/j.chembiol.2017.07.011
22. Scarneo SA, Hughes PF, Yang KW, Carlson DA, Gurbani D, Westover KD, et al. A highly selective inhibitor of interleukin-1 receptor-associated kinases 1/4 (IRAK-1/4) delineates the distinct signaling roles of IRAK-1/4 and the TAK1 kinase. J Biol Chem (2020) 295(6):1565–74. doi: 10.1074/jbc.RA119.011857
23. Zhang XS, Wu Q, Wu LY, Ye ZN, Jiang TW, Li W, et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis (2016) 7(10):e2416. doi: 10.1038/cddis.2016.292
24. Zhang X, Wu Q, Lu Y, Wan J, Dai H, Zhou X, et al. Cerebroprotection by salvianolic acid B after experimental subarachnoid hemorrhage occurs via Nrf2- and SIRT1-dependent pathways. Free Radic Biol Med (2018) 124:504–16. doi: 10.1016/j.freeradbiomed.2018.06.035
25. Shao A, Gao S, Wu H, Xu W, Pan Y, Fang Y, et al. Melatonin Ameliorates Hemorrhagic Transformation via Suppression of ROS-Induced NLRP3 Activation after Cerebral Ischemia in Hyperglycemic Rats. Oxid Med Cell Longev (2021) 2021:6659282. doi: 10.1155/2021/6659282
26. Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods (2008) 167(2):327–34. doi: 10.1016/j.jneumeth.2007.08.004
27. Sreenivasmurthy SG, Iyaswamy A, Krishnamoorthi S, Senapati S, Malampati S, Zhu Z, et al. Protopine promotes the proteasomal degradation of pathological tau in Alzheimer's disease models via HDAC6 inhibition. Phytomedicine (2022) 96:153887. doi: 10.1016/j.phymed.2021.153887
28. Zhang XS, Lu Y, Li W, Tao T, Peng L, Wang WH, et al. Astaxanthin ameliorates oxidative stress and neuronal apoptosis via SIRT1/NRF2/Prx2/ASK1/p38 after traumatic brain injury in mice. Br J Pharmacol (2021) 178(5):1114–32. doi: 10.1111/bph.15346
29. Xia DY, Yuan JL, Jiang XC, Qi M, Lai NS, Wu LY, et al. SIRT1 promotes M2 microglial polarization via reducing ROS-mediated NLRP3 inflammasome signaling after subarachnoid hemorrhage. Front Immunol (2021) 12:770744. doi: 10.3389/fimmu.2021.770744
30. Akamatsu Y, Pagan VA, Hanafy KA. The role of TLR4 and HO-1 in neuroinflammation after subarachnoid hemorrhage. J Neurosci Res (2020) 98(3):549–56. doi: 10.1002/jnr.24515
31. Heinz R, Brandenburg S, Nieminen-Kelha M, Kremenetskaia I, Boehm-Sturm P, Vajkoczy P, et al. microglial as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (SAH). J Neuroinflamm (2021) 18(1):36. doi: 10.1186/s12974-021-02085-3
32. Orihuela R, McPherson CA, Harry GJ. microglial M1/M2 polarization and metabolic states. Br J Pharmacol (2016) 173(4):649–65. doi: 10.1111/bph.13139
33. Peng J, Pang J, Huang L, Enkhjargal B, Zhang T, Mo J, et al. LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol (2019) 21:101121. doi: 10.1016/j.redox.2019.101121
34. Xu P, Tao C, Zhu Y, Wang G, Kong L, Li W, et al. TAK1 mediates neuronal pyroptosis in early brain injury after subarachnoid hemorrhage. J Neuroinflamm (2021) 18(1):188. doi: 10.1186/s12974-021-02226-8
35. Naito MG, Xu D, Amin P, Lee J, Wang H, Li W, et al. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc Natl Acad Sci U.S.A. (2020) 117(9):4959–70. doi: 10.1073/pnas.1916427117
36. Ridder DA, Schwaninger M. TAK1 inhibition for treatment of cerebral ischemia. Exp Neurol (2013) 239:68–72. doi: 10.1016/j.expneurol.2012.09.010
37. Shen Q, Chen Z, Zhao F, Pan S, Zhang T, Cheng X, et al. Reversal of prolonged obesity-associated cerebrovascular dysfunction by inhibiting microglial Tak1. Nat Neurosci (2020) 23(7):832–41. doi: 10.1038/s41593-020-0642-6
38. Liu Y, Li S, Wang R, Pu H, Zhao Y, Ye Q, et al. Inhibition of TGFbeta-activated kinase 1 promotes inflammation-resolving microglial/macrophage responses and recovery after stroke in ovariectomized female mice. Neurobiol Dis (2021) 151:105257. doi: 10.1016/j.nbd.2021.105257
39. Dakas PY, Barluenga S, Totzke F, Zirrgiebel U, Winssinger N. Modular synthesis of radicicol A and related resorcylic acid lactones, potent kinase inhibitors. Angew Chem Int Ed Engl (2007) 46(36):6899–902. doi: 10.1002/anie.200702406
40. Zhang ZH, Liu JQ, Hu CD, Zhao XT, Qin FY, Zhuang Z, et al. Luteolin Confers Cerebroprotection after Subarachnoid Hemorrhage by Suppression of NLPR3 Inflammasome Activation through Nrf2-Dependent Pathway. Oxid Med Cell Longev (2021) 2021:5838101. doi: 10.1155/2021/5838101
41. Wisitpongpun P, Potup P, Usuwanthim K. Oleamide-mediated polarization of M1 macrophages and IL-1beta production by regulating NLRP3-inflammasome activation in primary human monocyte-derived macrophages. Front Immunol (2022) 13:856296. doi: 10.3389/fimmu.2022.856296
42. Omori E, Inagaki M, Mishina Y, Matsumoto K, Ninomiya-Tsuji J. Epithelial transforming growth factor beta-activated kinase 1 (TAK1) is activated through two independent mechanisms and regulates reactive oxygen species. Proc Natl Acad Sci U.S.A. (2012) 109(9):3365–70. doi: 10.1073/pnas.1116188109
43. Tenshin H, Teramachi J, Ashtar M, Hiasa M, Inoue Y, Oda A, et al. TGF-beta-activated kinase-1 inhibitor LL-Z1640-2 reduces joint inflammation and bone destruction in mouse models of rheumatoid arthritis by inhibiting NLRP3 inflammasome, TACE, TNF-alpha and RANKL expression. Clin Transl Immunol (2022) 11(1):e1371. doi: 10.1002/cti2.1371
44. Tao W, Hu Y, Chen Z, Dai Y, Hu Y, Qi M. Magnolol attenuates depressive-like behaviors by polarizing microglial towards the M2 phenotype through the regulation of Nrf2/HO-1/NLRP3 signaling pathway. Phytomedicine (2021) 91:153692. doi: 10.1016/j.phymed.2021.153692
45. Slusarczyk J, Trojan E, Glombik K, Piotrowska A, Budziszewska B, Kubera M, et al. Targeting the NLRP3 inflammasome-related pathways via tianeptine treatment-suppressed microglial polarization to the M1 phenotype in lipopolysaccharide-stimulated cultures. Int J Mol Sci (2018) 19(7):1965. doi: 10.3390/ijms19071965
46. Nasoohi S, Ismael S, Ishrat T. Thioredoxin-interacting protein (TXNIP) in cerebrovascular and neurodegenerative diseases: regulation and implication. Mol Neurobiol (2018) 55(10):7900–20. doi: 10.1007/s12035-018-0917-z
47. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci (2014) 15(2):84–97. doi: 10.1038/nrn3638
48. Panipinto PM, Singh AK, Shaikh FS, Siegel RJ, Chourasia M, Ahmed S. Takinib inhibits inflammation in human rheumatoid arthritis synovial fibroblasts by targeting the janus kinase-signal transducer and activator of transcription 3 (JAK/STAT3) pathway. Int J Mol Sci (2021) 22(22):12580. doi: 10.3390/ijms222212580
Keywords: subarachnoid hemorrhage, early brain injury, takinib, TAK1, NLRP3
Citation: Wang W, Pang C, Zhang J, Peng L, Zhang X, Shi L and Zhang H (2023) Takinib inhibits microglial M1 polarization and oxidative damage after subarachnoid hemorrhage by targeting TAK1-dependent NLRP3 inflammasome signaling pathway. Front. Immunol. 14:1266315. doi: 10.3389/fimmu.2023.1266315
Received: 24 July 2023; Accepted: 27 October 2023;
Published: 14 November 2023.
Edited by:
Anwen Shao, Zhejiang University, ChinaReviewed by:
Zongqi You, Fudan University, ChinaJiang Shao, First Affiliated Hospital of Harbin Medical University, China
Niansheng Lai, The First Affiliated Hospital of Wannan Medical College, China
Copyright © 2023 Wang, Pang, Zhang, Peng, Zhang, Shi and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zhang, emhhbmdoYW9heWZ5NTIzQDE2My5jb20=; Lin Shi, c2hpbGluX2NhcGl0YWxAMTYzLmNvbQ==
†These authors have contributed equally to this work