 Anna Stierschneider
Anna Stierschneider Christoph Wiesner
Christoph Wiesner- Department Science & Technology, Institute Biotechnology, IMC Krems University of Applied Sciences, Krems, Austria
Toll-like receptor 4 (TLR4) are part of the innate immune system. They are capable of recognizing pathogen-associated molecular patterns (PAMPS) of microbes, and damage-associated molecular patterns (DAMPs) of damaged tissues. Activation of TLR4 initiates downstream signaling pathways that trigger the secretion of cytokines, type I interferons, and other pro-inflammatory mediators that are necessary for an immediate immune response. However, the systemic release of pro-inflammatory proteins is a powerful driver of acute and chronic inflammatory responses. Over the past decades, immense progress has been made in clarifying the molecular and regulatory mechanisms of TLR4 signaling in inflammation. However, the most common strategies used to study TLR4 signaling rely on genetic manipulation of the TLR4 or the treatment with agonists such as lipopolysaccharide (LPS) derived from the outer membrane of Gram-negative bacteria, which are often associated with the generation of irreversible phenotypes in the target cells or unintended cytotoxicity and signaling crosstalk due to off-target or pleiotropic effects. Here, optogenetics offers an alternative strategy to control and monitor cellular signaling in an unprecedented spatiotemporally precise, dose-dependent, and non-invasive manner. This review provides an overview of the structure, function and signaling pathways of the TLR4 and its fundamental role in endothelial cells under physiological and inflammatory conditions, as well as the advances in TLR4 modulation strategies.
1 Structure and function of the toll-like receptor 4 and its receptor family
Toll-like receptors (TLRs) are a family of conserved pattern-recognition receptors (PRRs) that detect and bind to evolutionarily conserved molecular motifs in pathogen-associated molecular patterns (PAMPs) of microbes or damage-associated molecular patterns (DAMPs) of injured tissues to induce innate and adaptive immune response (1–3). Other classes of PRRs include Nod-like receptors (NLRs), RIG-I-like receptors (RLRs), AIM2-like receptors (ALRs), C-type lectin receptors (CLRs), and intracellular DNA and RNA sensors such as cyclic GMP-AMP synthase (cGAS) (4, 5). Obviously, TLRs are expressed in innate immune cells such as macrophages, neutrophils, dendritic cells, natural killer cells, mast cells, eosinophils, basophils as well as adaptive immune cells, including B cells and specific T cells. They are also found in and on non-immune cells such as fibroblasts, epithelial and endothelial cells (6–8). TLRs are type I transmembrane proteins consisting of an extracellular domain (ECD) with leucine-rich repeats (LRRs) responsible for the detection of PAMPs and DAMPs, a transmembrane region, and an intracytoplasmic toll/interleukin 1 (IL-1) receptor (TIR) domain necessary for the initiation of the downstream signaling pathways (9) (Figure 1). LRRs exist in different protein combinations depending on the type of receptor and the corresponding ligand recognition and signal transduction (10). This high variety of LRRs is attributable to its consensus sequence motif of L(X2)LXL(X2)NXL(X2)L(X7)L(X2), where X can be any amino acid. Upon interaction of distinct PAMPs or DAMPs with the corresponding LRR, the extracellular domains of the respective TLRs form homo- or heterodimers with co-receptors or accessory molecules, which is essential for the activation of various signaling pathways and thus for an appropriate immune response (11). The LRR carboxy-terminal domain, characterized by the consensus motif CXC(X23)C(X17)C, separates the LRR region from the transmembrane region in TLRs (12). The TIR domain exhibits high homology to that of the IL-1 receptor family (13). Three highly conserved regions in TIR domains have been identified that mediate the protein-protein interactions between TLRs and adaptor proteins necessary for signal transduction. The various TLRs use different sets of adaptor proteins to determine signal transduction, with TLR4 being the only known TLR that uses all TIR domain-containing adaptor proteins, including the myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), also known as MyD88-adaptor like (Mal), TIR domain-containing adaptor inducing interferon beta (TRIF), and TRIF-related adaptor protein (TRAM) (14, 15).
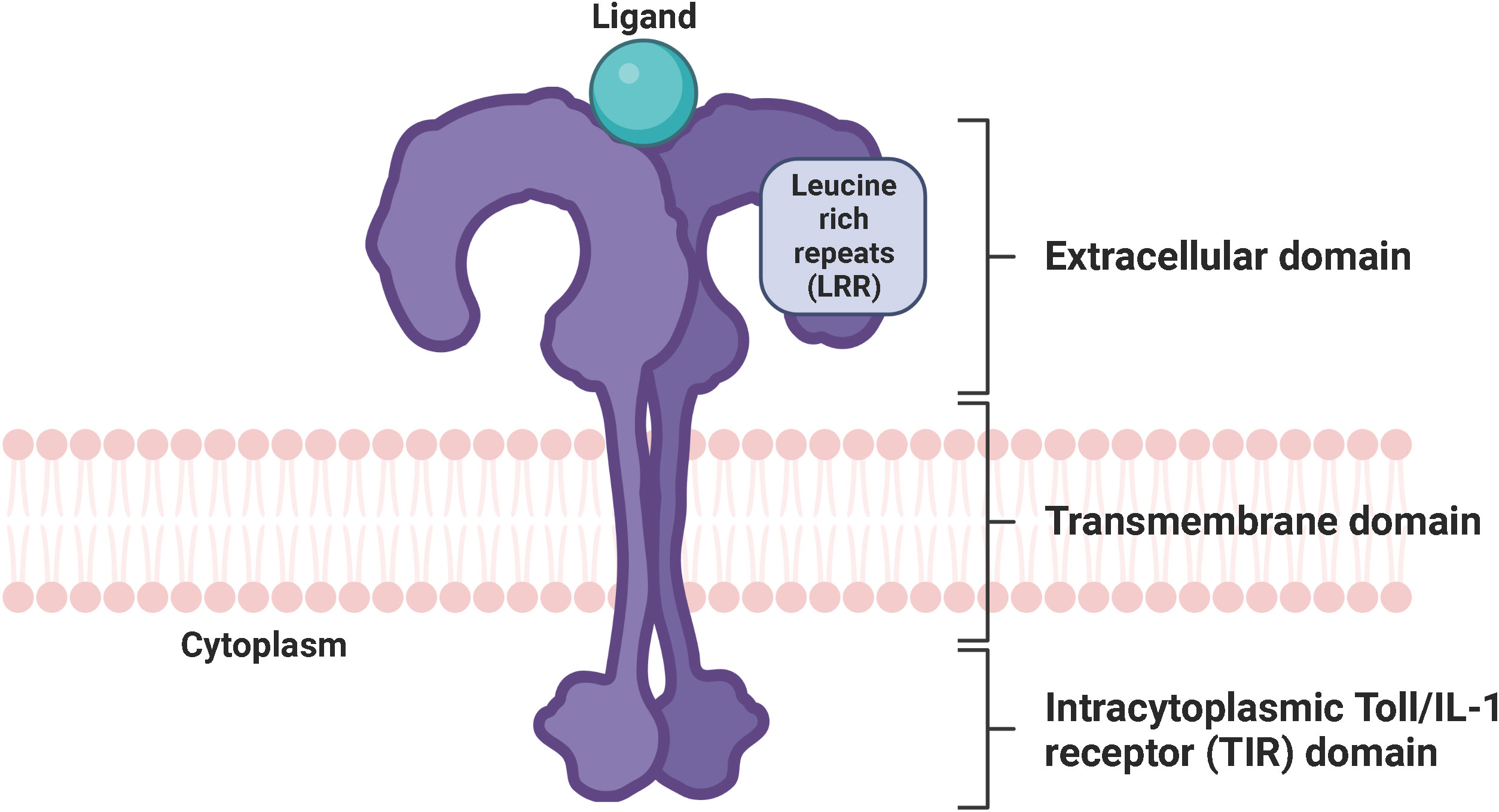
Figure 1 Conserved structure of TLRs. TLRs are characterized by an extracellular domain with leucine-rich repeats responsible for the recognition of PAMPs and DAMPs, a transmembrane domain, and an intracytoplasmic Toll/IL-1 receptor (TIR) domain necessary for signal transduction. TLR, toll-like receptor; LRR, leucine-rich repeats; PAMP, pathogen-associated molecular pattern; DAMP, damage-associated molecular pattern; TIR domain, Toll/interleukin-1 (IL-1) receptor domain. Created with BioRender.com.
In 1985, Anderson et al. originally identified Toll as a gene product essential for the embryonic dorsoventral axis formation in the fruit fly Drosophila melanogaster (16), and 11 years later, its critical antifungal function in this species was described (17). Just one year later, in 1997, the discovery of the mammalian homologue of the toll receptor (now designated TLR4), which plays a major role in the innate immune response by inducing the expression of pro-inflammatory genes, revolutionized our knowledge of the immune system and triggered an irresistible research on PRRs (18). To date, a total of 10 and 12 functional TLRs have been identified in humans and mice, respectively, with TLR1 - TLR9 being conserved in both species. A retroviral insertion into the TLR10 gene in mice impaired its functionality, and the human genome lost TLR11, TLR12, and TLR13 (19). Based on their cellular localization and respective PAMP ligands, human TLRs are classified into cell surface TLRs and intracellular TLRs (Figure 2). TLR2/TLR1 and TLR2/TLR6 heterodimers, TLR4 and TLR5 homodimers, and the orphan TLR10 (incomplete understanding of dimerization) are expressed on the cell surface and primarily recognize microbial membrane components such as lipoteichoic acid (LTA) (TLR2), lipopeptides (LP) (TLR1, 2, 6) and lipopolysaccharide (LPS) (TLR4, 10) as well as flagellin (TLR5, 10) (20, 21). In addition, TLR2 detects zymosan glycan in fungi and like TLR4 glycophosphatidylinositol (GPI) anchors in protists. In contrast, human TLR3, TLR7, TLR8, and TLR9 homodimers are exclusively found in intracellular components such as lysosomes, endosomes, endolysosomes, and the endoplasmic reticulum, where they mainly target microbial nucleic acids (9, 22) (Figure 3).
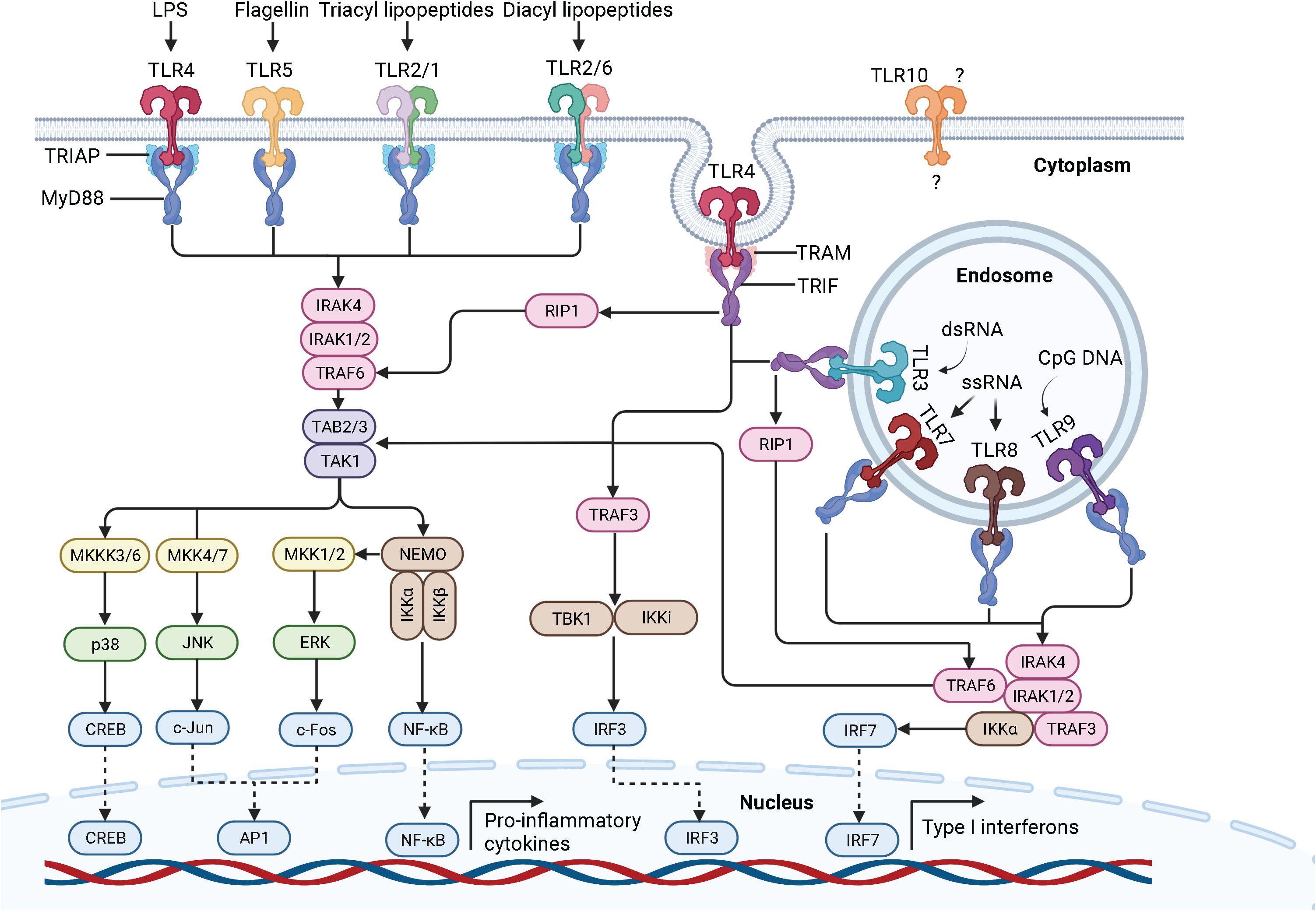
Figure 2 Schematic representation of the TLR signaling pathways. Depending on the type of PAMP and its respective binding affinity to different extracellular domains of the TLRs, TLR signaling is initiated by the dimerization of the extracellular domains of the receptors after recruitment of the adaptor proteins MyD88 and TIRAP (MyD88-dependent pathway) or TRIF and TRAM (TRIF-dependent pathway) through the interactions with the TIR domains of the TLRs. TLR, toll-like receptor; LPS, lipopolysaccharide; dsRNA, double-stranded ribonucleic acid; ssRNA, single-stranded ribonucleic acid; CpG DNA, 5’-cytosine-phosphate-guanine-3’ deoxyribonucleic acid; MyD88, myeloid differentiation factor 88; TRIAP, TIR domain-containing adaptor protein; TRIF, TIR domain-containing adaptor inducing interferon beta; TRAM, TRIF-related adaptor protein; IRAK, interleukin 1 receptor-associated kinase; TRAF, tumor necrosis factor receptor-associated factor; TAK-1, transforming growth factor beta-activated kinase 1; TAB, TAK-1 binding protein; MKK, mitogen-activated protein kinase kinase; JNK, Jun N-terminal kinase; ERK1/2, extracellular signal−regulated protein kinase 1/2; CREB, cyclic adenosine monophosphate-responsive element-binding protein; AP-1, activator protein 1; NF-κB, nuclear factor kappa B; NEMO, NF-κB essential modulator; IKK, IκB kinase; IKKi, inhibitor of NF-κB kinase; RIP1, receptor-interacting protein 1; TBK, TANK-binding kinase; IFN, interferon; IRF, interferon regulatory factor. Created with BioRender.com.
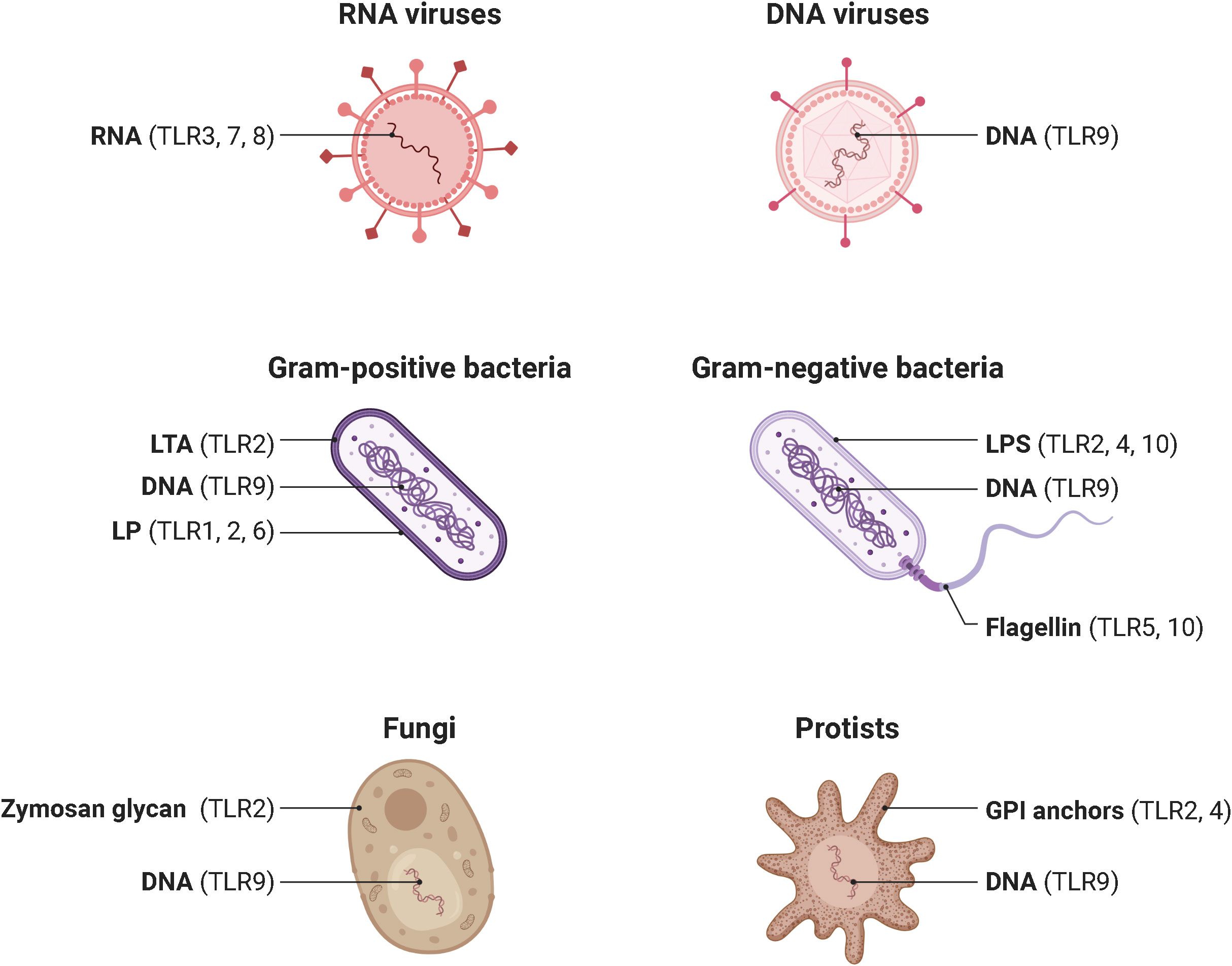
Figure 3 Pathogens and their ligand targets for different human TLRs. Cell surface TLRs primarily recognize components of microbial membranes such as lipoteichoic acid (TLR2), lipopeptides (TLR1, 2, 6) and lipopolysaccharide (TLR4, 10) as well as flagellin (TLR5, 10). Additionally, TLR2 detects zymosan glycan of fungi and, like TLR4, protist GPI anchors. In contrast, cytosolic TLR3, TLR7, TLR8, and TLR9 mainly recognize microbial nucleic acids. TLR, toll-like receptor; DNA, deoxyribonucleic acid; RNA, ribonucleic acid; LTA, lipoteichoic acid. LP, lipopeptides. LPS, lipopolysaccharide. GPI, glycosylphosphatidylinositol. Adapted from “Pathogen Ligand Targets for Different TLRs”, by BioRender.com (2023).
2 TLR4 signaling
One of the best characterized PAMPs to date is the endotoxin LPS, which is derived from the outer membrane of Gram-negative bacteria. The general structure of LPS is divided into lipid A, a core oligosaccharide, and an O side chain, with lipid A being the major PAMP of LPS (23, 24). LPS stimulation of human cells triggers a series of interactions involving the soluble LPS binding protein (LBP), the soluble glycosylphosphatidylinositol-anchored protein cluster of differentiation 14 (CD14), the soluble myeloid differentiation factor 2 (MD-2), and TLR4 (25, 26). LBP extracts LPS from the outer membrane of Gram-negative bacteria and transports it to CD14, which then transfers LPS to the TLR4/MD-2 receptor complex (14, 27). Interestingly, LPS can bind to MD-2 even in the absence of TLR4, which does not seem to be possible the other way around, as there is only evidence that TLR4 can enhance the binding of LPS to MD-2 (28, 29). Upon LPS recognition, the extracellular domains of TLR4 dimerize and adaptor proteins recruit through interactions with the TIR domains of TLR4. Initial recruitment of the adaptor proteins MyD88 and TRIAP induces the MyD88-dependent signaling pathway, which triggers an early activation of the nuclear factor kappa B (NF-κB) and the mitogen-activated protein kinases (MAPK) (30, 31). Sequential binding of the adaptor proteins TRIF and TRAM and subsequent dynamin-driven endosomal translocation of TLR4 initiates the MyD88-independent pathway, also known as the TRIF-dependent pathway, which culminates in the late-phase activation of NF-κB and the IFN-inducing transcription factor IFN regulatory factor 3 (IRF3). The MyD88-dependent pathway leads to the production of pro-inflammatory cytokines. In contrast, the TRIF-dependent pathway additionally results in the secretion of type I interferons (IFN) (32). The signal transduction of the MyD88-dependent and TRIF-dependent pathway is described in detail in the following sections and is graphically displayed in Figure 2.
The MyD88-dependent pathway is characterized by the formation of the Myddosome consisting of MyD88, TRIAP, IRAK4, IRAK1, and IRAK2. MyD88 recruits IL-1 receptor-associated kinase-4 (IRAK4) through homotypic interaction of their death domains (14). IRAK4 is a member of the IRAK family, which shares a death domain and a kinase domain (33). IRAK4 knock-in mutation studies in mice revealed that the kinase activity of IRAK4 is crucial for TLR signaling (34, 35). Of note, IRAK4 also activates NF-κB and MAPK (36). IRAK4 sequentially engages and activates IRAK1 and IRAK2, which are then autophosphorylated at multiple sites (37), leading to the binding of tumor necrosis factor receptor (TNFR)-associated factor-6 (TRAF6) (38). IRAK1 activates the E3 ubiquitin ligase TRAF6, which together with the E2 ubiquitin-conjugating enzymes Ubc13 and Uev1A, induces the synthesis of Lys63 (K63)-linked polyubiquitin to degrade both TRAF6 and IRAK1 and to recruit the transforming growth factor beta (TGF-β)-activated kinase 1 (TAK1) and the adaptor molecules TAK-1 binding protein 2 and 3 (TAB2, TAB3) (11). Subsequent phosphorylation of TAK1 simultaneously induces the IκB kinase (IKK)-NF-κB and MAPK signaling cascades. The IKK complex consists of two catalytic subunits, IKKα and IKKβ, and the regulatory subunit NF-κB essential modulator (NEMO), also called IKKγ. Upon its association with TAK1 through ubiquitin chains, the IKK complex is phosphorylated, leading to the activation of IKKβ (9, 19). Following phosphorylation and proteasomal degradation of the NF-κB inhibitory protein, IκBα releases NF-κB. Released NF-κB then translocates into the nucleus to initiate transcription of pro-inflammatory genes (39). Activated TAK1 simultaneously phosphorylates the MAPK kinases (MMK) 3/6, 4/7, and 1/2, thereby initiating the activation of p38, Jun N-terminal kinases (JNKs), and extracellular signal−regulated protein kinase 1/2 (ERK1/2). Once activated, they phosphorylate the cyclic adenosine monophosphate (cAMP)-responsive element-binding protein (CREB) and activator protein 1 (AP-1) transcription factors, which consist of a heterodimer of c-Fos and c-Jun subunits. The final interaction of CREB and AP-1 with NF-κB facilitates the transcription of pro-inflammatory genes (9, 40).
The TRIF-dependent pathway is characterized by the recruitment of TRAM and TRIF to form the so-called Triffosome (41). TRIF associates with TRAF6 and TRAF3. Activated TRAF6 recruits the kinase receptor-interacting protein 1 (RIP1), which subsequently engages the TAK1 and IKK complex, leading to the activation of NF-κB and MAPKs. Activated TRAF3 recruits the IKK-related kinase TANK-binding kinase 1 (TBK1) and the inhibitor of NF-κB kinase (IKKi), which in turn phosphorylates and activates IRF3, ultimately leading to the induction of IFN (42, 43).
Importantly, once activated, TLR4 is the only member of the TLR family that induces both the MyD88-dependent and TRIF-dependent signaling pathways. TLR2/TLR1, TLR2/TLR6, TLR5, TLR7, TLR8, and TLR9 initiate the MyD88-dependent pathway, whereas TLR3 induces the TRIF-dependent pathway (9, 11). Of note, upon Myddosome formation in response to TLR7, TLR8, or TLR9 activation, IRF7 binds and is directly activated by IRAK1 and IKKα. Like IRF3, IRF7 leads to the induction of IFN (44, 45). We already have a quite clear understanding of the ligand recognition, dimerization, signaling, and biological functions of human TLR1-9, but not of the orphan TLR10. While we know that TLR10 is localized on the cell surface, its ligands, homo- or heterodimerization, TIR domains and adaptor molecules, signal transduction and biological function are still unclear (21).
3 TLR4 signaling in endothelial cells under physiological and inflammatory conditions
Endothelial cells (ECs) form the innermost layer of blood vessels, which include arteries, veins, and capillaries, and separate the circulating blood from the surrounding tissues (46) (Figure 4). Since the blood pressure is much higher in the arteries than in veins, arteries require more layers of smooth muscle cells than veins, making them the thickest blood vessels. Capillaries consist of just one single layer of ECs and no layers of smooth muscle cells because they do not have to withstand any pressure (47).
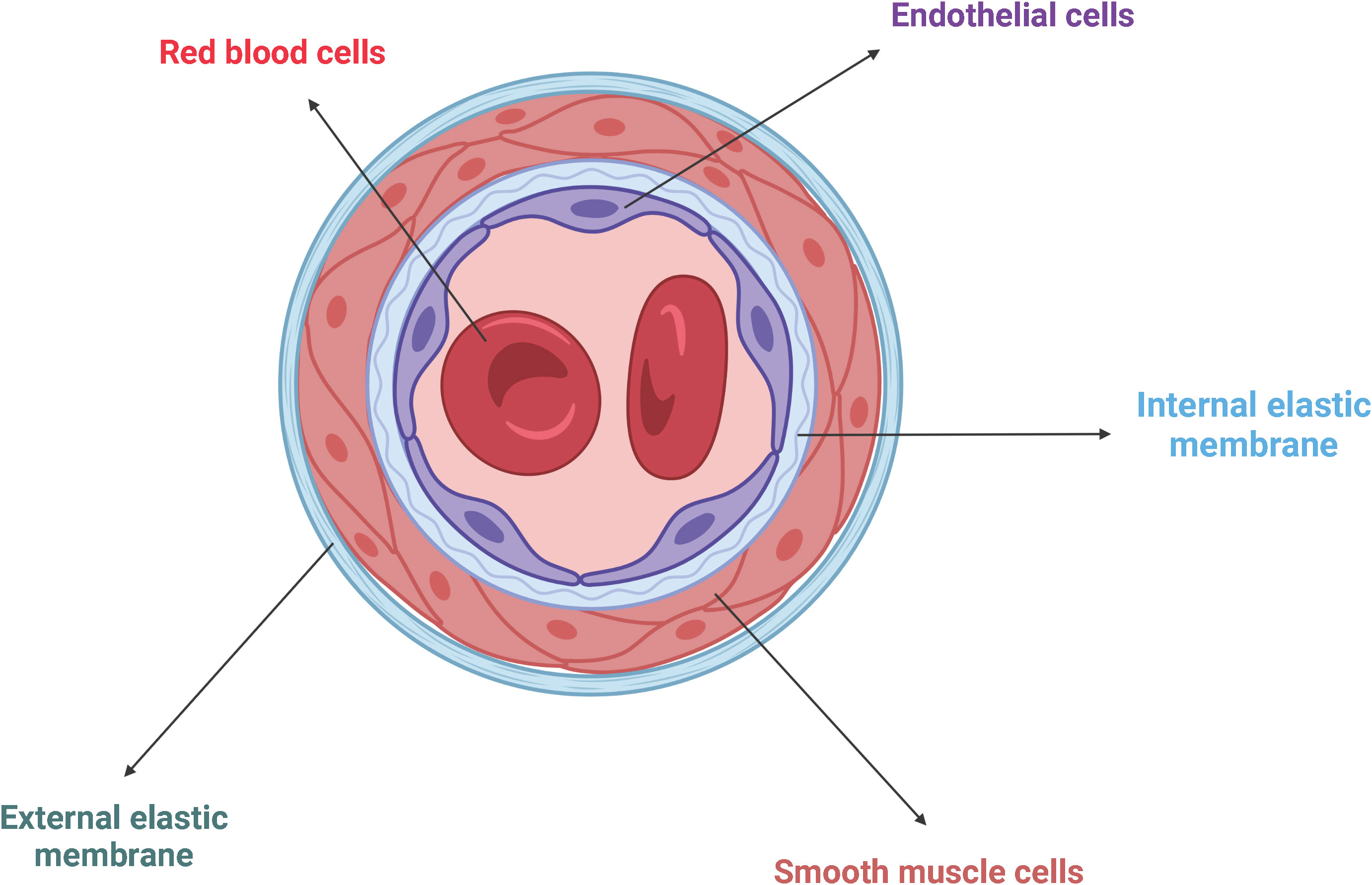
Figure 4 Schematic representation of the general architecture of blood vessels. ECs line the innermost wall of blood vessels, separating the circulating blood from the surrounding tissue. Adjacent smooth muscle cells are trapped between the internal and the external elastic membranes. Created with BioRender.com.
Due to their unique position, they are the first cells exposed to circulating substances in the blood, under healthy and pathological conditions, and thus act as gatekeepers of vascular health and function (48). Under physiological conditions, the endothelium perform several functions to maintain organ homeostasis, including vasoregulation, selective vascular permeability, and provision of an anti-coagulant environment (49). Upon recognition of PAMPs and DAMPs by PRRs, including TLR1-10, NOD1, and RIG-1, ECs secrete various cytokines, chemokines, growth factors, and adhesion molecules to facilitate vasodilation, promote neutrophil trafficking to the endothelium, activate coagulation pathways, and increase vascular permeability (48, 50–52) (Table 1). Of note, studies have highlighted that TLR2, TLR4, and TLR9 are the main drivers of the endothelial immune response (50, 59, 77, 78). In addition to that, by analyzing the baseline mRNA transcription levels of TLR1-10 in different endothelial cell lines, including human umbilical vein endothelial cells (HUVECs), human coronary artery endothelial cells (HCAECs), human microvascular endothelial cells (HMVECs) derived from the brain, liver, and lung, Khakpour et al. clearly demonstrated that TLR4 is the most highly expressed of all TLRs, suggesting that TLR4 is the central PRR in the acute and chronic endothelial immune response (50).
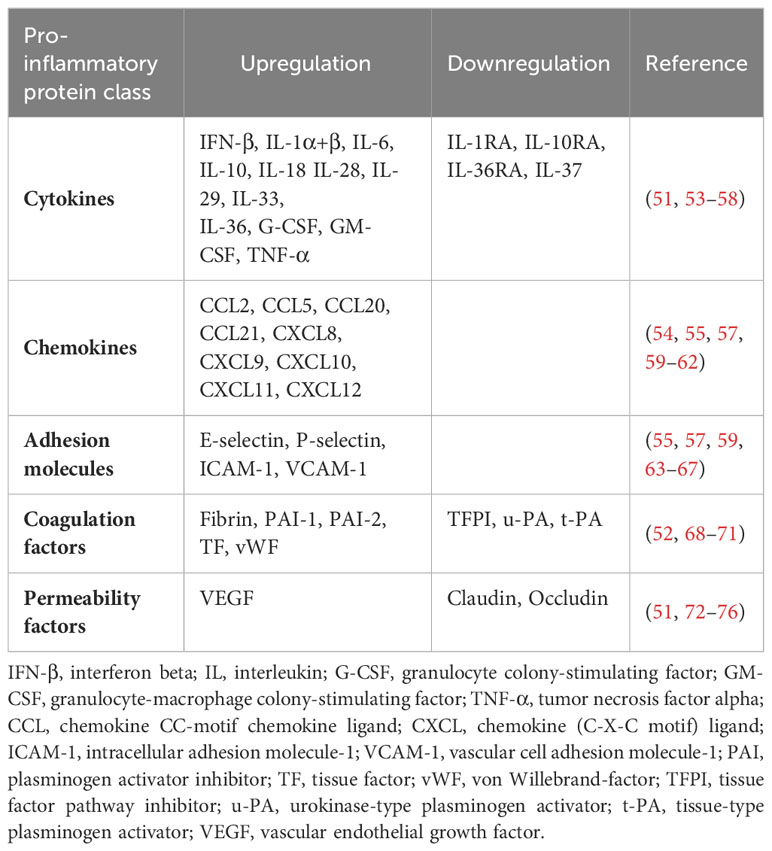
Table 1 PRR, especially TLR4-induced responses in ECs.
3.1 Vasoregulation
To control vascular tone, ECs secrete various paracrine substances. Vasodilators, including nitric oxide (NO), prostacyclin (PGI2), and the endothelium-derived hyperpolarizing factor (EDHF), increase the intravascular diameter by relaxing the adjacent smooth muscle cells, whereas vasoconstrictors such as angiotensin, thromboxane (TXA2), and endothelin-1 (ET-1), counteract the function of vasodilators to keep a balance in vascular resistance. It is noteworthy that activation of endothelial PRRs, especially TLR4, enhances the expression of vasodilators, leading to decreased blood pressure and increased blood flow, which facilitates endothelial permeability (47, 49, 79).
3.2 Leukocyte extravasation/diapedesis/transendothelial migration
PRR, especially TLR4-induced secreted chemokines, such as CXCL8 and CCL2, initiate the expression of E- and P-selectins at the site of EC activation, which interact with sialyl Lewis X glycan epitopes expressed on leukocytes to tether and roll them (51, 63, 80). Sialyl Lewis X is a tetrasaccharide composed of N-acetylneuramic acid (Neu5Ac) α2-3 galactose (Gal) β1-4 [fucose (Fuc) α1-3] N-acetylglucosamine and is attached to glycoproteins or glycolipids of cell surface proteins (81–83). Notably, lymphocytes express L-selectin and interact with sialyl Lewis X glycan epitopes expressed on ECs (84). Subsequent deceleration and firm adhesion of rolling leukocytes to the ECs is mediated by β2- and α4-containing integrins expressed on the surface of leukocytes and the intracellular and vascular adhesion molecules (ICAM-1, VCAM-1) upregulated on the surface of ECs in response to the PRR, especially TLR4-induced endothelial cytokine production, such as IL-1 and IL-6 (85–87). Leukocytes then migrate from the luminal to the abluminal side of the vascular barrier either via the vesicle-based transcellular route through the EC body or via the junctional disruptive paracellular route between adjacent ECs (85, 88) (Figure 5). Upon arrival at the site of the injured or infected tissue, leukocytes eliminate the necrotic cell debris or pathogens by phagocytosis (50). Of note, neutrophils are the first leukocytes to appear at the site of inflammation and form neutrophil extracellular traps (NETs) to bind, eradicate, and inhibit the distribution of invading pathogens (89).
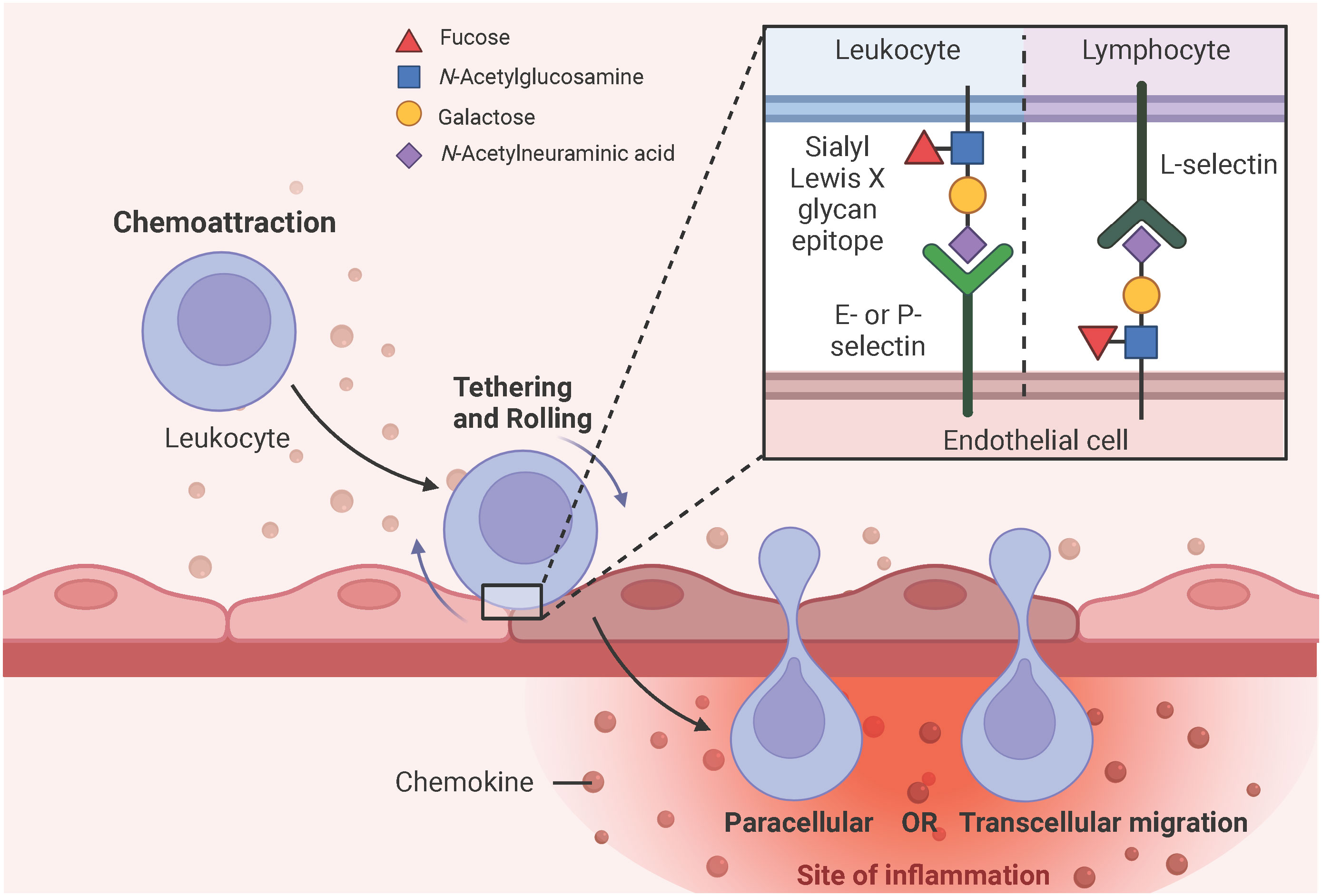
Figure 5 Simplified schematic representation of leukocyte extravasation. PRR, particularly TLR4-induced chemokine secretion initiates the expression of E- and P-selectins on endothelial surfaces, which have a high affinity for sialyl Lewis X glycan epitopes expressed on leukocytes. These selectin-glycan interactions facilitate leukocyte tethering, rolling, and final endothelial transmigration to the site of inflammation. Leukocytes can migrate from the luminal to the abluminal side of the vascular barrier either by the vesicle-based transcellular route through the EC body or by the junctional paracellular route between adjacent ECs. Lymphocytes express L-selectins on their surface and interact with sialyl Lewis X glycan epitopes expressed on ECs to facilitate diapedesis into the subendothelial compartment. Adapted from “Leukocyte Extravasation - The Role of Glycans in Inflammation”, by BioRender.com (2023).
3.3 Coagulation and fibrinolysis
Generally, the coagulation system can be divided into the fast extrinsic and the slower intrinsic pathway, both of which converge on a common pathway initiated by the activation of factor X to Xa (Figure 6A). Tissue factor (TF) and the plasma factor VII are the major initiators of the extrinsic coagulation pathway (90). TF is a membrane-bound glycoprotein that is latent under physiological conditions and released into the blood upon injury. Notably, TF can also be expressed by monocytes and ECs in response to pathogen invasion (91). Factor VII associates with TF to convert to its active form, VIIa. The TF/VIIa complex, in turn, converts circulating factor X to its active serine protease, factor Xa. In contrast, the intrinsic coagulation pathway is initiated by the auto-activation of factor XII to the serine protease factor XIIa upon interaction with negatively charged surfaces such as polyphosphates and phospholipids on activated platelets. This results in the sequential activation of factors XI and IX (92, 93). Thus, both coagulation pathways culminate in the generation of the active factor Xa, which is the starting point of the common pathway. Xa interacts with the cofactor Va to convert prothrombin (factor II) to thrombin (factor IIa). Finally, thrombin cleaves fibrinogen (factor I) to fibrin (factor Ia). The subsequent polymerization of fibrin and aggregation of platelets leads to the formation of a blood clot (94). During the healing process, tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA) convert the fibrin-bound plasminogen to the active enzyme plasmin, which in turn lyses the fibrin network into a degradable form. Freely circulating t-PA and u-PA are inactivated by the plasminogen activator inhibitor (PAI) 1 and 2, and plasmin is blocked by α2 antiplasmin (Figure 6B) (95, 96).
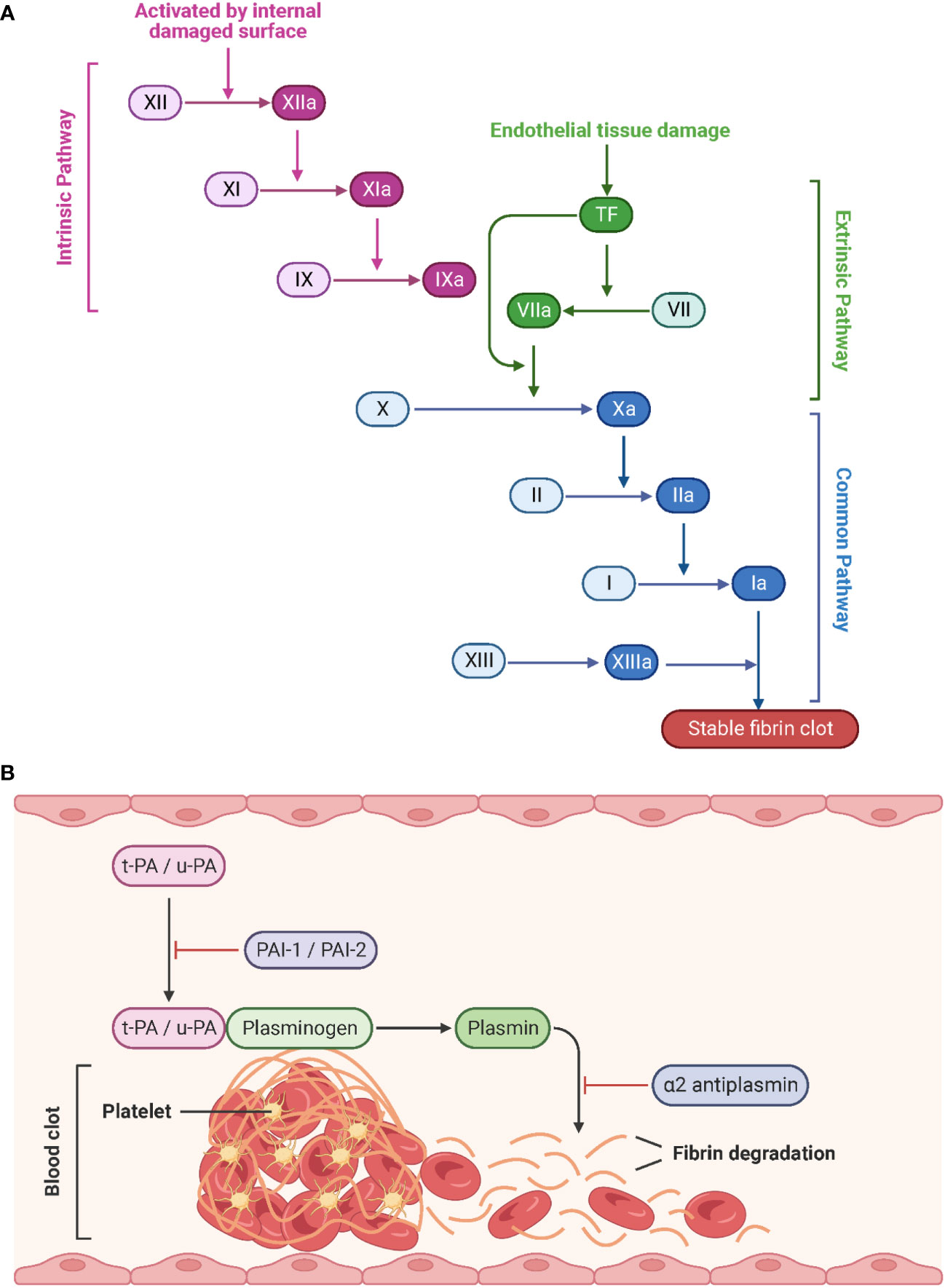
Figure 6 Schematic representation of the coagulation pathways and fibrinolytic pathways. (A) Tissue factor (initiation of the extrinsic pathway) and contact activation (initiation of the intrinsic pathway) lead to a common pathway that generates thrombin. Created with BioRender.com. (B) During fibrinolysis, t-PA and u-PA convert plasminogen to plasmin, which degrades the fibrin network. Adapted from “Process of Blood Clot Formation”, by BioRender.com (2023).
Since platelets, leukocytes, and erythrocytes circulate continuously in the blood vessels until they are needed, the endothelium must provide an anti-coagulant and anti-thrombogenic environment to prevent platelet adhesion under physiological conditions (97) (Figure 7). Therefore, ECs express nitric oxide (NO), prostacyclin (PGI2), and adenosine diphosphatase (ADPase) to prevent platelet adhesion and aggregation (93). The glycocalyx is a layer of proteoglycans (PGs) (syndecans and glypicans) and glycosaminoglycan chains (GAGs) (heparan sulfate (HS), chondroitin sulfate (CS) that covers the vascular lumen. Due to its negatively charged composition, the glycocalyx repels platelets and leukocytes from contacting with the ECs (98–101). In particular, heparan sulfate proteoglycans cooperate with antithrombin (AT) to interfere with several coagulation factors, including thrombin, IXa, Xa, XIa, and XIIa (102). Tissue factor pathway inhibitors (TFPIs), as the name suggests, inhibit the coagulation-activating TF, VII, and X. Thrombomodulin (TM) binds to thrombin, which associates with the endothelial cell protein C receptor (EPCR). The subsequently released activated protein C (APC) interacts with protein S (PS) to block coagulation factors Va and VIIIa (103). Notably, Nur77 and Nor1, as well as the inflammatory stimuli C-reactive protein (CRP) and oxidized low-density lipoprotein (oxLDL), have been identified as potential regulators of TM expression and were found to be downregulated in activated ECs, thus exerting a pro-thrombotic effect (104).
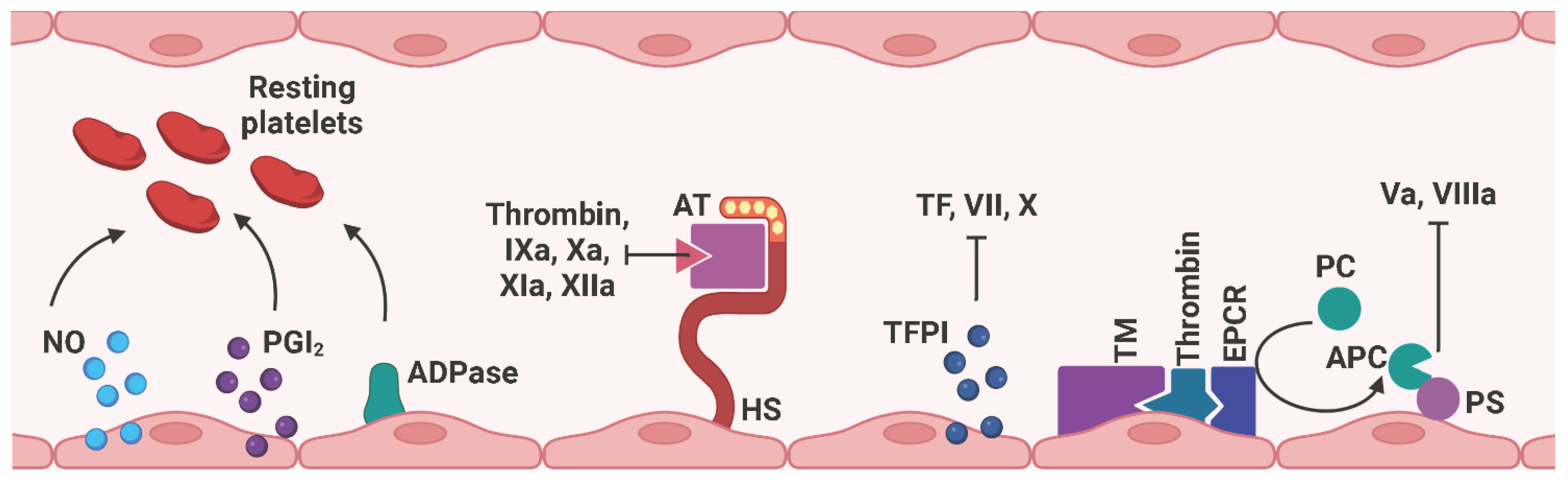
Figure 7 Schematic representation of anti-coagulant mediators expressed on ECs. Under physiological conditions, the endothelium provides an anti-coagulant/anti-thrombotic environment to prevent thrombus formation by free circulating platelets and red blood cells within the blood vessel. NO, PGI2, and ADPase prevent platelet adhesion and aggregation. HS cooperates with AT to interfere with thrombin, factors IXa, Xa, XIa, and XIIa. TFPI binds to thrombin, which associates with EPCR, and subsequently released APC interacts with PS to block factors Va and VIIIa. EC, endothelial cell; NO, nitric oxide; PGI2, prostacyclin; ADPase, adenosine diphosphatase; AT, antithrombin; HS, heparan sulfate proteoglycan; TM, thrombomodulin; EPCR, endothelial cell protein C receptor; PC, protein C; APC, activated protein C; PS, protein S. Created with BioRender.com.
Upon vascular injury or pathogen invasion, the endothelium shifts from the physiological anti-coagulant/anti-thrombotic environment to a pro-coagulant/pro-thrombotic state that promotes fibrin clot formation and reduces clot lysis to prevent blood loss and trap pathogens, respectively (103). This alteration in endothelial function is initiated by sustained activation of ECs by inflammatory stimuli, including circulating PAMPs as described in 1, DAMPs (high-mobility group box 1 (HGMB1), heat shock proteins, heme), cytokines (IL-1β, IL-6, IL-17, IL-19, IFN-γ, TGF-β, TNF-α), chemokines (CXCL1, CXCL8, CCL2), complement system-derived receptors (C1q, C3a or C5a) and proteins (C1, C3, C5, factor B), and reactive oxygen species (ROS) (superoxide anion (O2−)) (105–111). Weibel-Palade bodies (WPBs) synthesize and store von Willebrand factor (vWF), P-selectin, and different pro-coagulant and pro-inflammatory proteins. PRR, especially TLR4-induced, exocytosis of WPBs and liberation of their storage components from inflamed ECs leads to the expression of vWF and P-selectin, recruiting platelets and leukocytes such as neutrophils and monocytes, respectively (110, 112, 113). Leukocytes expressing P-selectin glycoprotein ligand-1 (PSGL-1) are recruited upon interaction with endothelial P-selectin, and platelets adhere upon the interaction of endothelial vWF and platelet-derived glycoprotein Ib-alpha (GPIbα) (114). Activated platelets recognize pathogens and further secrete pro-inflammatory and pro-coagulant proteins, including platelet factor 4 (PF4), CXCL4, CXCL5, CXCL8, CCL3, CCL5, and CCL7, which facilitate neutrophil recruitment, tethering, and NET formation. Neutrophils release NETs to capture pathogens, facilitate thrombus formation, and activate platelets. Active TF from the surface of monocytes and microvesicles further enhances the propagation of thrombosis by inducing fibrin formation and trapping red blood cells (Figure 8) (68, 115, 116).
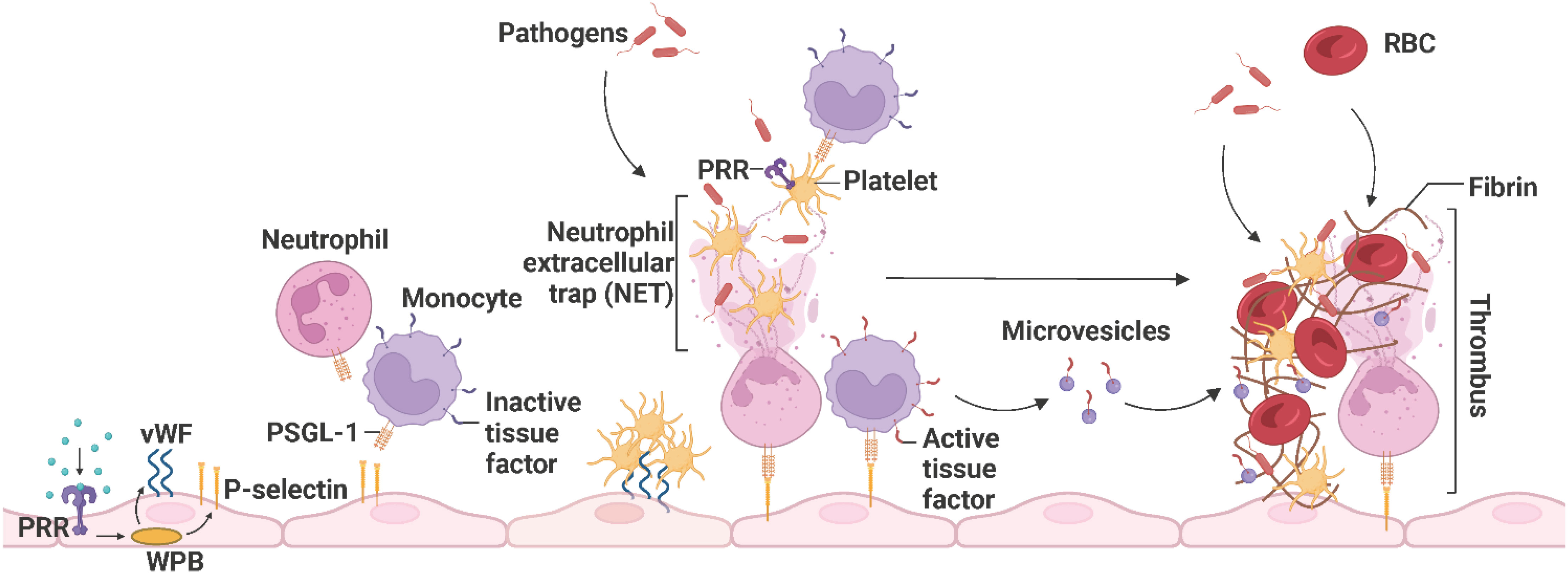
Figure 8 The propagation of immunothrombosis by leukocytes and platelets. During inflammation, PRRs, particularly TLR4, initiate the exocytosis of WPB from ECs and subsequently the expression of P-selectins and vWFs. Leukocytes expressing PSGL-1 are recruited upon interaction with endothelial P-selectin and platelets adhere upon the interaction of endothelial vWF and platelet-derived GPIbα. Neutrophils release NETs that trap pathogens, facilitate thrombus formation, and activate platelets. The activated platelets, recruit leukocytes and recognize pathogens. Active TFs on the surface of monocytes and microvesicles further enhance thrombus propagation by inducing fibrin formation and trapping RBCs. The resulting thrombus promotes pathogen capture. PRR, pathogen recognition receptor; WPB, Weibel-Palade bodies; vWF, von Willebrand factor; PSGL-1, P-selectin glycoprotein ligand-1; NET, neutrophil extracellular trap; TF, tissue factor; RBC, red blood cell. Adapted from “Propagation of Immunothrombosis by Leukocytes and Platelets”, by BioRender.com (2023).
3.4 Permeability of the endothelial barrier
Endothelial integrity and permeability are determined by intracellular junctions between adjacent ECs to regulate the extravasation of water, plasma proteins (e.g., albumin, globulins, fibrinogen, hormone-transporting plasma proteins, cytokines, chemokines), nutrients (e.g., glucose, amino acids, fatty acids, vitamins, minerals), metabolic waste products (e.g., urea, creatinine, carbon dioxide), electrolytes (e.g., sodium, potassium, calcium, magnesium, chloride, bicarbonate, phosphate, sulfate, organic acids), and immune cells (e.g., lymphocytes, monocytes, natural killer cells, erythrocytes, platelets, eosinophils, basophils, neutrophils) (117–119). Inter-EC junctions include tight junctions (claudin, occludin, junction adhesion molecule (JAM) A, B, and C), adherens junctions (VE-cadherin, nectin), gap junctions (connexin 32, 37, 40, and 43), and the platelet-EC adhesion molecule-1 (PECAM-1) (120, 121) (Figure 9). Gap junctions are channels that directly link the cytoplasm of adjacent ECs and allow the transmission of electrical impulses, ions, and small molecules to pass between cells (122). Endothelial tight junctions mediate the diffusion of polar solutes and ions and prevent the penetration of macromolecules across the ECs (123). Interestingly, occludins and claudins are indirectly linked to adherens junctions through zonula occludens (ZO) -1, -2, and -3 and further via the actin cytoskeleton (124). Adherens junctions are particularly important in the endothelium, where they stabilize endothelial cell-cell contact and regulate the expression and organization of tight junctions (125, 126). Adherens junctions found in ECs have VE-cadherin as the key transmembrane component and link adjacent cells through its extracellular domain. The cytoplasmic tail associates with p120-catenin through its juxtamembrane domain (JMD) and with β-catenin and plakoglobin through its C-terminal domain (CTD). Plakoglobin or β-catenin are connected to α-catenin, thereby indirectly linking VE-cadherin to the actin cytoskeleton (119, 127). Nectin, a specific member of the endothelial adherens junction family, is a transmembrane protein of the IgG superfamily and enhances homophilic cell-cell adhesion. Nectin sequentially binds afadin, ponsin, α-catenin and vinculin, and finally actin (128). PECAM-1 is a type I transmembrane glycoprotein of the immunoglobulin (Ig) superfamily of cell adhesion molecules and has been found to be highly expressed at inter-EC junctions to maintain barrier integrity by interacting with tight and adherens junctions and acting as a scaffold by engaging β- and γ-catenins (129, 130). PRRs, especially TLR4-induced inflammatory proteins (vascular endothelial growth factor (VEGF), histamine, thrombin, and IL-6) enhance endothelial permeability by activating vasodilators, kinases, and phosphatases to induce augmented actin-myosin contractility, destabilization of the inter-EC junctions and the formation of focal gaps between adjacent ECs (72, 131). Primarily, VEGF, histamine, and thrombin trigger the activation of Src-family tyrosine kinases, which are responsible for the phosphorylation of VE-cadherin, mainly at the tyrosine residue Y685, and its subsequent internalization, thereby strongly promoting endothelial permeability (132). Interestingly, Alsaffar et al. recently demonstrated that the IL-6/Janus kinase (JAK) signaling induces an initial and short-term (2 h) loss of barrier function dependent on Src and MEK/ERK activation, and a sustained permeability requiring signal transducer and activator of transcription 3 (STAT3) phosphorylation at Y705 (131). Transiently increased endothelial permeability in the acute inflammatory response is crucial for tissue repair or pathogen clearance. However, prolonged hyperpermeability leads to pathological conditions such as edema, hypotension, and impaired vascular perfusion and oxygenation of adjacent tissues (127).
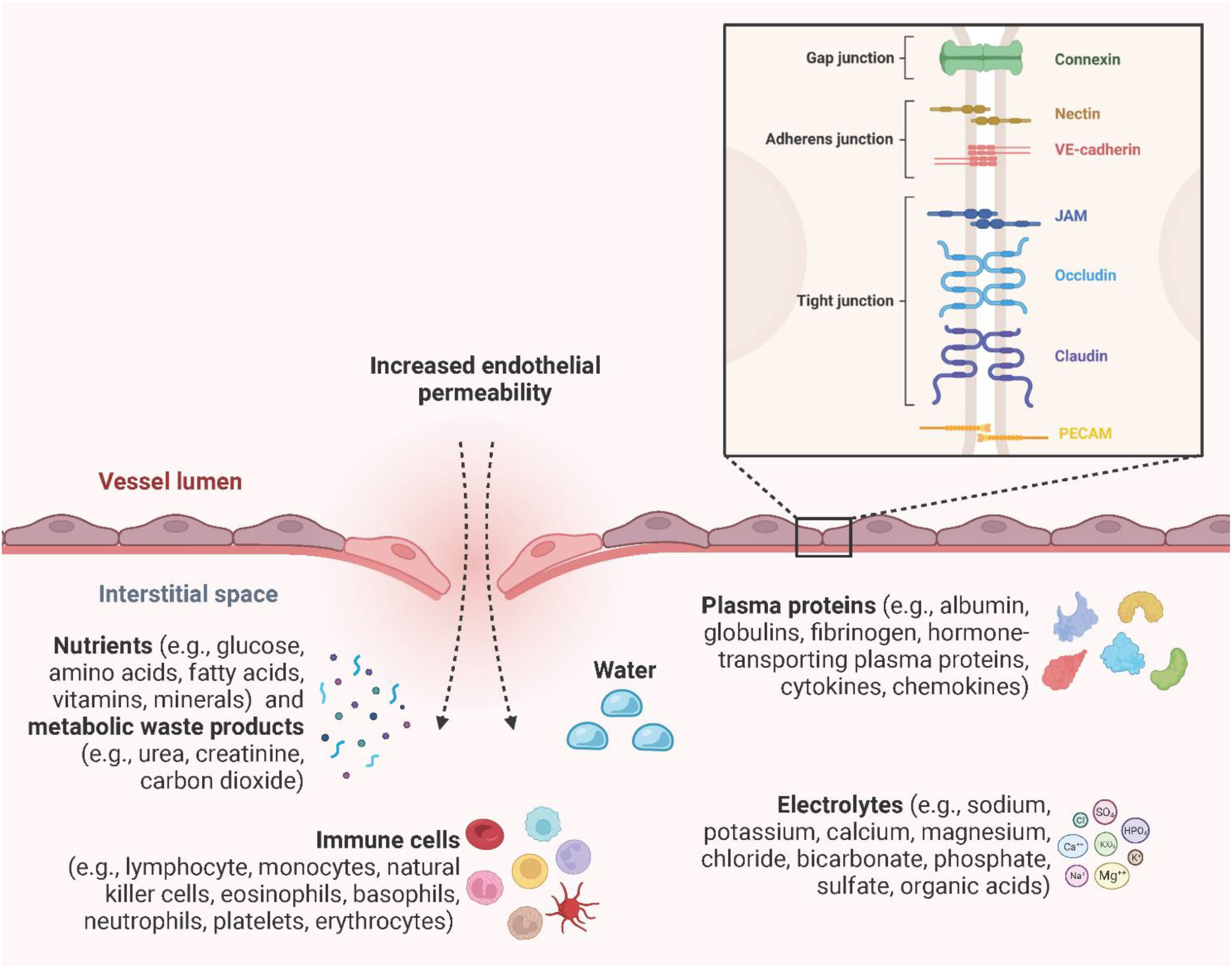
Figure 9 Endothelial permeability. Endothelial integrity and permeability are determined by intracellular junctions, including gap junctions (connexin 32, 37, 40, and 43), adherens junctions (VE-cadherin, nectin), tight junctions (claudin, occludin, JAM A, B, and C), and PECAM-1 to regulate the extravasation of water, plasma proteins, electrolytes, nutrients, metabolic waste products, and immune cells. Created with BioRender.com.
4 Optogenetics: the art of studying the TLR4 signaling pathway
TLR4 signaling is a complex, highly dynamic, and tightly regulated network of two distinct pathways that initiate the innate immune response. Persistent TLR4 signaling is responsible for chronic and acute inflammatory disorders, including sepsis (133), atherosclerosis (134, 135), rheumatoid arthritis (136), acute and chronic lung injury (137), sickle cell disease (63, 110, 138), neurodegenerative diseases (139, 140), and cancer (141, 142). For instance, sickle cell disease is a chronic inflammatory condition with hemolysis, vaso-occlusion, and ischemia-reperfusion due to the heme-induced MD-2/TLR4 activation leading to the production of pro-inflammatory mediators and a persistent activation of leukocytes, platelets, and endothelial cells (63, 110, 111, 138). Therefore, modulation of the TLR4 signaling pathway is a promising strategy to specifically target these pathologies. Common approaches used to study TLR4 signaling are primarily based on genetic manipulation through gain or loss-of-function mutations of the TLR4 or treatment with the bacterial endotoxin LPS. However, these strategies are often associated with the generation of irreversible phenotypes in the target cells or unintended cytotoxicity and signaling crosstalk due to off-target or pleiotropic effects. Furthermore, ligands are often unable to penetrate complex tissues, spheroids, or organoids, resulting in surface activation only. On top of that, the use of reagents is associated with complex operational design, high costs, and sources or errors (isolation impurities, batch variations, pipetting errors, instability upon solvation, etc.) (143–145). Here, optogenetics offers an alternative strategy to control and monitor cellular signaling in an unprecedented spatiotemporally precise, dose-dependent, and non-invasive manner (Figure 10). It is based on utilizing light-sensitive protein domains of microbial or plant photoreceptors, integrated into effector proteins, to direct them with light stimuli. Hence, light induction allows activation, inactivation, stabilization, destabilization, or localization of signaling pathways depending on the protein type and setup (146–148). Initial optogenetic applications used naturally derived photosensitive opsins to investigate and control neuronal activity and later to study brain circuits. This allowed to replace conventional strategies, most of which were highly invasive, slow in kinetics, and imprecise in targeting specific neurons (149–152). Since then, optogenetics has revolutionized the study of cell biological processes, including signaling pathways, protein movement, or metabolic processes, and was even voted for the Method of the Year 2010 (153, 154). The present available repertoire of light-sensitive domains allows for the formation of protein complexes in response to blue (155–157), red (158, 159), or green (160) light. An important system involves the light oxygen voltage (LOV) domain isolated from the yellow-green algae Vaucheria frigida. This xanthophyte contains two distinct aureochromes, aureochrome 1 (AUREO1) and aureochrome 2 (AUREO2), each consisting of a LOV domain and a basic leucine zipper (bZIP) domain. AUREO1 controls blue light-induced cell branching, whereas AUREO2 mediates the development of a sex organ (161, 162). LOV domains are a subfamily of the Per-ARNT-Sim (PAS) family and shear a common PAS domain fold comprising of a five-stranded antiparallel β-sheet and four α-helices (163). The LOV domain derived from the AUREO1 of Vaucheria frigida (VfAU1-LOV), noncovalently binds a flavin chromophore that, upon blue light (λmax ≈ 470 nm) absorption, induces a photochemical reaction that leads to the formation of a covalent adduct between the conserved cysteine and the flavin ring. By fusing this blue light-sensing protein domain to the TLR4 and stably incorporating it into endothelial and pancreatic adenocarcinoma cells, Stierschneider et al. developed two physiologically relevant in vitro cell culture models in which the TLR4 can be turned on with blue light (470 nm) and turned off in the dark. These newly established optogenetic endothelial and pancreatic adenocarcinoma cell lines allow TLR4-specific studies of the underlying molecular and regulatory mechanisms in inflammation and cancer as well as high content screening for compounds that block TLR4 signaling with spatiotemporal precision (148, 164).
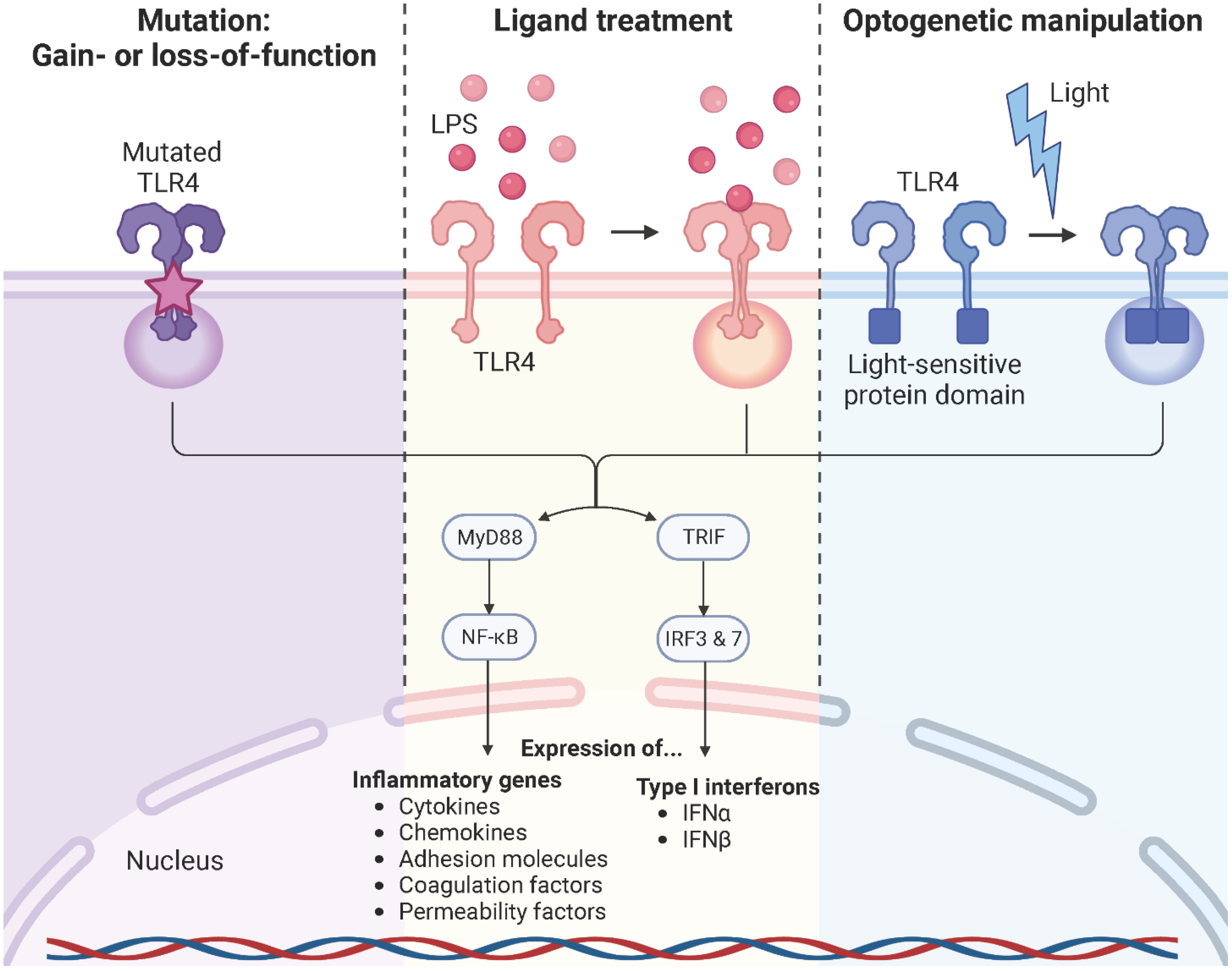
Figure 10 TLR4 modulation strategies. Common approaches used to study TLR4 signaling rely on genetic manipulation through gain- or loss-of-function mutations of the TLR4 or downstream signaling molecules, treatment with its naturally occurring ligand LPS, or optogenetic manipulation. Created with BioRender.com.
5 Conclusion and perspectives
In ECs, activation of TLR4 by LPS ultimately leads to the release of cytokines, chemokines, adhesion molecules, coagulation and permeability factors, that are necessary for an immediate immune response to invading pathogens and tissue injury (12, 165). However, their systemic secretion is a major driver of autoimmune, acute and chronic inflammatory diseases (19). Therefore, negative regulation of TLR4 signaling pathways is a promising strategy to specifically target these pathologies (166). The search for such modulation options requires cell culture models with fast and unambiguous TLR4 signaling. Compared to available standard cell culture models that rely on genetic manipulation of TLR4 or the treatment with agonists such as LPS, optogenetic cell lines with light-inducible TLR4 provide these requirements. Hence, they are predestined for receptor-specific fundamental studies as well as for high content screening of biological active agents that negatively interfere with the TLR4 signaling pathway in inflammation.
Author contributions
CW: Funding acquisition, Project administration, Supervision, Writing – review & editing. AS: Conceptualization, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Lower Austrian FTI Program, grant number K3-F-744/005-2019 and the FFG Austria, grant number 35467434.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature (2000) 406(6797):782–7. doi: 10.1038/35021228
2. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer (2009) 9(1):57–63. doi: 10.1038/nrc2541
3. Santoni M, Andrikou K, Sotte V, Bittoni A, Lanese A, Pellei C, et al. Toll like receptors and pancreatic diseases: From a pathogenetic mechanism to a therapeutic target. Cancer Treat Rev (2015) 41(7):569–76. doi: 10.1016/j.ctrv.2015.04.004
4. Cai X, Chiu Y-H, Zhijian J, Chen. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54(2):289–96. doi: 10.1016/j.molcel.2014.03.040
5. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
6. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
7. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
8. McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol (2014) 5:386. doi: 10.3389/fimmu.2014.00386
9. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
10. Kobe B, Deisenhofer J. Proteins with leucine-rich repeats. Curr Opin Struct Biol (1995) 5(3):409–16. doi: 10.1016/0959-440X(95)80105-7
11. Duan T, Du Y, Xing C, Wang HY, Wang R-F. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
12. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol (2001) 1(2):135–45. doi: 10.1038/35100529
13. Sameer AS, Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. BioMed Res Int (2021) 2021:1157023. doi: 10.1155/2021/1157023
14. Lu Y-C, Yeh W-C, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine (2008) 42(2):145–51. doi: 10.1016/j.cyto.2008.01.006
15. O'Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
16. Anderson KV, Bokla L, Nüsslein-Volhard C. Establishment of dorsal-ventral polarity in the drosophila embryo: The induction of polarity by the Toll gene product. Cell (1985) 42(3):791–8. doi: 10.1016/0092-8674(85)90275-2
17. Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/toll/cactus controls the potent antifungal response in drosophila adults. Cell (1996) 86(6):973–83. doi: 10.1016/S0092-8674(00)80172-5
18. Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi: 10.1038/41131
19. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
20. Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure (2011) 19(4):447–59. doi: 10.1016/j.str.2011.02.004
21. Su S-B, Tao L, Deng Z-P, Chen W, Qin S-Y, Jiang H-X. TLR10: Insights, controversies and potential utility as a therapeutic target. Scandinavian J Immunol (2021) 93(4):e12988. doi: 10.1111/sji.12988
22. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi: 10.1128/CMR.00046-08
23. Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol (2005) 3(1):36–46. doi: 10.1038/nrmicro1068
24. Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem (2002) 71(1):635–700. doi: 10.1146/annurev.biochem.71.110601.135414
25. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci (2021) 78(4):1233–61. doi: 10.1007/s00018-020-03656-y
26. Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol (2007) 19(1):3–10. doi: 10.1016/j.smim.2006.12.002
27. Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science (1990) 249(4975):1431–3. doi: 10.1126/science.1698311
28. Mitsuzawa H, Nishitani C, Hyakushima N, Shimizu T, Sano H, Matsushima N, et al. Recombinant soluble forms of extracellular TLR4 domain and MD-2 inhibit lipopolysaccharide binding on cell surface and dampen lipopolysaccharide-induced pulmonary inflammation in mice. J Immunol (2006) 177(11):8133–9. doi: 10.4049/jimmunol.177.11.8133
29. Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol (2002) 3(7):667–72. doi: 10.1038/ni809
30. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature (2001) 413(6851):78–83. doi: 10.1038/35092578
31. Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature (2002) 420(6913):324–9. doi: 10.1038/nature01182
32. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol (2008) 9(4):361–8. doi: 10.1038/ni1569
33. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature (2002) 416(6882):750–4. doi: 10.1038/nature736
34. Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, et al. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J Exp Med (2007) 204(5):1013–24. doi: 10.1084/jem.20061523
35. Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh K-H, et al. A critical role for IRAK4 kinase activity in Toll-like receptor–mediated innate immunity. J Exp Med (2007) 204(5):1025–36. doi: 10.1084/jem.20061825
36. Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci (2002) 99(8):5567–72. doi: 10.1073/pnas.082100399
37. Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh W-C. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling*. J Biol Chem (2004) 279(39):40653–8. doi: 10.1074/jbc.M402666200
38. Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature (1996) 383(6599):443–6. doi: 10.1038/383443a0
39. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev (2004) 18(18):2195–224. doi: 10.1101/gad.1228704
40. Clark K, Nanda S, Cohen P. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol (2013) 14(10):673–85. doi: 10.1038/nrm3644
41. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198(7):1043–55. doi: 10.1084/jem.20031023
42. Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, et al. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol (2003) 4(12):1223–9. doi: 10.1038/ni1010
43. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the myD88-independent toll-like receptor signaling pathway. Science (2003) 301(5633):640–3. doi: 10.1126/science.1087262
44. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction. Nature (2005) 434(7036):1035–40. doi: 10.1038/nature03547
45. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi: 10.1038/nature03464
46. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7(10):803–15. doi: 10.1038/nri2171
47. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J (2010) 4:302–12. doi: 10.2174/1874192401004010302
48. Al-Soudi A, Kaaij MH, Tas SW. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun Rev (2017) 16(9):951–62. doi: 10.1016/j.autrev.2017.07.008
49. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest (2006) 86(1):9–22. doi: 10.1038/labinvest.3700366
50. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun (2015) 21(8):827–46. doi: 10.1177/1753425915606525
51. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
52. Ren M, Li R, Luo M, Chen N, Deng X, Yan K, et al. Endothelial cells but not platelets are the major source of Toll-like receptor 4 in the arterial thrombosis and tissue factor expression in mice. Am J Physiology-Regulatory Integr Comp Physiol (2014) 307(7):R901–7. doi: 10.1152/ajpregu.00324.2014
53. Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev (2009) 227(1):248–63. doi: 10.1111/j.1600-065X.2008.00733.x
54. Gatheral T, Reed DM, Moreno L, Gough PJ, Votta BJ, Sehon CA, et al. A key role for the endothelium in NOD1 mediated vascular inflammation: comparison to TLR4 responses. PloS One (2012) 7(8):e42386. doi: 10.1371/journal.pone.0042386
55. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells — partnering up with the immune system? Nat Rev Immunol (2022) 22(9):576–88. doi: 10.1038/s41577-022-00694-4
56. Fahey E, Doyle SL. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol (2019) 10:1426. doi: 10.3389/fimmu.2019.01426
57. Menghini R, Campia U, Tesauro M, Marino A, Rovella V, Rodia G, et al. Toll-like receptor 4 mediates endothelial cell activation through NF-κB but is not associated with endothelial dysfunction in patients with rheumatoid arthritis. PloS One (2014) 9(6):e99053. doi: 10.1371/journal.pone.0099053
58. Faure E, Thomas L, Xu H, Medvedev AE, Equils O, Arditi M. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol (2001) 166(3):2018–24. doi: 10.4049/jimmunol.166.3.2018
59. Zeuke S, Ulmer AJ, Kusumoto S, Katus HA, Heine H. TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc Res (2002) 56(1):126–34. doi: 10.1016/S0008-6363(02)00512-6
60. Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol (2008) 15(1):138–46. doi: 10.1128/CVI.00257-07
61. Pegu A, Qin S, Fallert Junecko BA, Nisato RE, Pepper MS, Reinhart TA. Human lymphatic endothelial cells express multiple functional TLRs1. J Immunol (2008) 180(5):3399–405. doi: 10.4049/jimmunol.180.5.3399
62. Kang S, Lee S-P, Kim KE, Kim H-Z, Mémet S, Koh GY. Toll-like receptor 4 in lymphatic endothelial cells contributes to LPS-induced lymphangiogenesis by chemotactic recruitment of macrophages. Blood (2009) 113(11):2605–13. doi: 10.1182/blood-2008-07-166934
63. Beckman JD, Abdullah F, Chen C, Kirchner R, Rivera-Rodriguez D, Kiser ZM, et al. Endothelial TLR4 expression mediates vaso-occlusive crisis in sickle cell disease. Front Immunol (2020) 11:613278. doi: 10.3389/fimmu.2020.613278
64. Chiu J-J, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, et al. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-α in endothelial cells. Arteriosclerosis Thrombosis Vasc Biol (2004) 24(1):73–9. doi: 10.1161/01.ATV.0000106321.63667.24
65. Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arteriosclerosis Thrombosis Vasc Biol (1995) 15(1):2–10. doi: 10.1161/01.ATV.15.1.2
66. Sheikh S, Rainger GE, Gale Z, Rahman M, Nash GB. Exposure to fluid shear stress modulates the ability of endothelial cells to recruit neutrophils in response to tumor necrosis factor-α: a basis for local variations in vascular sensitivity to inflammation. Blood (2003) 102(8):2828–34. doi: 10.1182/blood-2003-01-0080
67. van Hinsbergh VW. Endothelium–role in regulation of coagulation and inflammation. Semin Immunopathol (2012) 34(1):93–106. doi: 10.1007/s00281-011-0285-5
68. Pilard M, Ollivier EL, Gourdou-Latyszenok V, Couturaud F, Lemarié CA. Endothelial cell phenotype, a major determinant of venous thrombo-inflammation. Front Cardiovasc Med (2022) 9:864735. doi: 10.3389/fcvm.2022.864735
69. Muth H, Maus U, Wygrecka M, Lohmeyer J, Grimminger F, Seeger W, et al. Pro- and antifibrinolytic properties of human pulmonary microvascular versus artery endothelial cells: Impact of endotoxin and tumor necrosis factor-α. Crit Care Med (2004) 32(1):217–26. doi: 10.1097/01.CCM.0000104941.89570.5F
70. Roth RI. Hemoglobin enhances the production of tissue factor by endothelial cells in response to bacterial endotoxin. Blood (1994) 83(10):2860–5. doi: 10.1182/blood.V83.10.2860.2860
71. Colucci M, Balconi G, Lorenzet R, Pietra A, Locati D, Donati MB, et al. Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest (1983) 71(6):1893–6. doi: 10.1172/JCI110945
72. Claesson-Welsh L, Dejana E, McDonald DM. Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol Med (2021) 27(4):314–31. doi: 10.1016/j.molmed.2020.11.006
73. Nagyőszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NTK, Haskó J, et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochemistry Int (2010) 57(5):556–64. doi: 10.1016/j.neuint.2010.07.002
74. Hamada K, Kakigawa N, Sekine S, Shitara Y, Horie T. Disruption of ZO-1/claudin-4 interaction in relation to inflammatory responses in methotrexate-induced intestinal mucositis. Cancer Chemotherapy Pharmacol (2013) 72(4):757–65. doi: 10.1007/s00280-013-2238-2
75. Liu L, Jiang Y, Steinle JJ. Toll-Like Receptor 4 Reduces Occludin and Zonula Occludens 1 to Increase Retinal Permeability Both in vitro and in vivo. J Vasc Res (2017) 54(6):367–75. doi: 10.1159/000480455
76. Riddell JR, Maier P, Sass SN, Moser MT, Foster BA, Gollnick SO. Peroxiredoxin 1 stimulates endothelial cell expression of VEGF via TLR4 dependent activation of HIF-1α. PloS One (2012) 7(11):e50394. doi: 10.1371/journal.pone.0050394
77. El Kebir D, Jo´zsef L, Pan W, Wang L, Filep JG, et al. Bacterial DNA activates endothelial cells and promotes neutrophil adherence through TLR9 signaling. J Immunol (2009) 182(7):4386–94. doi: 10.4049/jimmunol.0803044
78. Shin HS, Xu F, Bagchi A, Herrup E, Prakash A, Valentine C, et al. Bacterial lipoprotein TLR2 agonists broadly modulate endothelial function and coagulation pathways in vitro and in vivo. J Immunol (2011) 186(2):1119–30. doi: 10.4049/jimmunol.1001647
79. Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesthesia (2004) 93(1):105–13. doi: 10.1093/bja/aeh163
80. Kubes P. Introduction: The complexities of leukocyte recruitment. Semin Immunol (2002) 14(2):65–72. doi: 10.1006/smim.2001.0343
81. Zandberg WF, Kumarasamy J, Pinto BM, Vocadlo DJ. Metabolic inhibition of sialyl-Lewis X biosynthesis by 5-thiofucose remodels the cell surface and impairs selectin-mediated cell adhesion. J Biol Chem (2012) 287(47):40021–30. doi: 10.1074/jbc.M112.403568
82. Taylor ME, Drickamer K. Mammalian sugar-binding receptors: known functions and unexplored roles. FEBS J (2019) 286(10):1800–14. doi: 10.1111/febs.14759
83. Chen Q, Liu H, Li X. Essential functions, syntheses and detection of sialyl Lewis X on glycoproteins. Explor Drug Sci (2023) 1(1):31–54. doi: 10.37349/eds.2023.00004
84. Mitoma J, Bao X, Petryanik B, Schaerli P, Gauguet J-M, Yu S-Y, et al. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol (2007) 8(4):409–18. doi: 10.1038/ni1442
85. Salminen AT, Allahyari Z, Gholizadeh S, McCloskey MC, Ajalik R, Cottle RN, et al. In vitro studies of transendothelial migration for biological and drug discovery. Front Med Technol (2020) 2:600616. doi: 10.3389/fmedt.2020.600616
86. Ulbrich H, Eriksson EE, Lindbom L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol Sci (2003) 24(12):640–7. doi: 10.1016/j.tips.2003.10.004
87. Xu K, Saaoud F, Yu S, Drummer C 4th, Shao Y, Sun Y, et al. Monocyte AdhesionAdhesion Assays for Detecting Endothelial Cell Activation in Vascular InflammationInflammation and AtherosclerosisAtherosclerosis. In: Ramji D, editor. Atherosclerosis: Methods and Protocols. New York, NY: Springer US (2022). p. 169–82.
88. Schimmel L, Heemskerk N, van Buul JD. Leukocyte transendothelial migration: A local affair. Small GTPases (2017) 8(1):1–15. doi: 10.1080/21541248.2016.1197872
89. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12(3):324–33. doi: 10.1016/j.chom.2012.06.011
90. Stallone G, Paola P, Federica R, Giuseppe C, Loreto G, Giuseppe G. Coagulation and fibrinolysis in kidney graft rejection. Front Immunol (2020) 11:1807. doi: 10.3389/fimmu.2020.01807
91. Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood (2014) 123(17):2605–13. doi: 10.1182/blood-2013-09-526277
92. Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII contact activation. Semin Thromb Hemost (2017) 43(08):814–26. doi: 10.1055/s-0036-1598003
93. Neubauer K, Zieger B. Endothelial cells and coagulation. Cell Tissue Res (2022) 387(3):391–8. doi: 10.1007/s00441-021-03471-2
94. Crawley JTB, Zanardelli S, Chion CKNK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemostasis (2007) 5(s1):95–101. doi: 10.1111/j.1538-7836.2007.02500.x
95. Kaviarasi S, Yuba E, Harada A, Krishnan UM. Emerging paradigms in nanotechnology for imaging and treatment of cerebral ischemia. J Controlled Release (2019) 300:22–45. doi: 10.1016/j.jconrel.2019.02.031
96. Yaron JR, Zhang L, Guo Q, Haydel SE, Lucas AR. Fibrinolytic serine proteases, therapeutic serpins and inflammation: fire dancers and firestorms. Front Cardiovasc Med (2021) 8:648947. doi: 10.3389/fcvm.2021.648947
97. Bouvy C, Gheldof D, Chatelain C, Mullier F, Dogné J-M. Contributing role of extracellular vesicles on vascular endothelium haemostatic balance in cancer. J Extracellular Vesicles (2014) 3. doi: 10.3402/jev.v3.24400
98. Alphonsus CS, Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia (2014) 69(7):777–84. doi: 10.1111/anae.12661
99. Jin J, Fang F, Gao W, Chen H, Wen J, Wen X, et al. The structure and function of the glycocalyx and its connection with blood-brain barrier. Front Cell Neurosci (2021) 15:739699. doi: 10.3389/fncel.2021.739699
100. Mende M, Bednarek C, Wawryszyn M, Sauter P, Biskup MB, Schepers U, et al. Chemical synthesis of glycosaminoglycans. Chem Rev (2016) 116(14):8193–255. doi: 10.1021/acs.chemrev.6b00010
101. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflügers Archiv (2000) 440(5):653–66. doi: 10.1007/s004240000307
102. Mayfosh AJ, Baschuk N, Hulett MD. Leukocyte heparanase: A double-edged sword in tumor progression. Front Oncol (2019) 9:331. doi: 10.3389/fonc.2019.00331
103. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
104. Theofilis P, Sagris M, Oikonomou E, Antonopoulos AS, Siasos G, Tsioufis C, et al. Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines (2021) 9(7):1–21. doi: 10.3390/biomedicines9070781
105. Fischetti F, Tedesco F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity (2006) 39(5):417–28. doi: 10.1080/08916930600739712
106. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
107. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc Pharmacol (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
108. Najem MY, Couturaud F, Lemarié CA. Cytokine and chemokine regulation of venous thromboembolism. J Thromb Haemostasis (2020) 18(5):1009–19. doi: 10.1111/jth.14759
109. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathology: Mech Dis (2020) 15(1):493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
110. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood (2014) 123(3):377–90. doi: 10.1182/blood-2013-04-495887
111. Belcher JD, Zhang P, Nguyen J, Kiser ZM, Nath KA, Hu J, et al. Identification of a heme activation site on the MD-2/TLR4 complex. Front Immunol (2020) 11:1370. doi: 10.3389/fimmu.2020.01370
112. El-Mansi S, Nightingale TD. Emerging mechanisms to modulate VWF release from endothelial cells. Int J Biochem Cell Biol (2021) 131:105900. doi: 10.1016/j.biocel.2020.105900
113. McCormack JJ, Silva Lopes M, Ferraro F, Patella F, Cutler DF, et al. Weibel–Palade bodies at a glance. J Cell Sci (2017) 130(21):3611–7. doi: 10.1242/jcs.208033
114. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemostasis (2018) 16(2):231–41. doi: 10.1111/jth.13911
115. Kaiser R, Escaig R, Erber J, Nicolai L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc Med (2021) 8:824112. doi: 10.3389/fcvm.2021.824112
116. Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, et al. Platelets and immune responses during thromboinflammation. Front Immunol (2019) 10:1731. doi: 10.3389/fimmu.2019.01731
117. Mathew J Fau - Sankar P, Sankar P, Varacallo M. Physiology, blood plasma. BTI - StatPearls (2023).
118. Ono S, Egawa G, Kabashima K. Regulation of blood vascular permeability in the skin. Inflammation Regeneration (2017) 37(1):11. doi: 10.1186/s41232-017-0042-9
119. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev (2006) 86(1):279–367. doi: 10.1152/physrev.00012.2005
120. Okamoto T, Suzuki K. The role of gap junction-mediated endothelial cell-cell interaction in the crosstalk between inflammation and blood coagulation. Int J Mol Sci (2017) 18(11):1–17. doi: 10.3390/ijms18112254
121. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta (BBA) - Biomembranes (2008) 1778(3):794–809. doi: 10.1016/j.bbamem.2007.09.003
122. Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res (2017) 120(1):179–206. doi: 10.1161/CIRCRESAHA.116.306534
123. Cerutti C, Ridley AJ. Endothelial cell-cell adhesion and signaling. Exp Cell Res (2017) 358(1):31–8. doi: 10.1016/j.yexcr.2017.06.003
124. Balda MS, Matter K. Tight junctions as regulators of tissue remodelling. Curr Opin Cell Biol (2016) 42:94–101. doi: 10.1016/j.ceb.2016.05.006
125. Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol (2012) 19(3):218–23. doi: 10.1097/MOH.0b013e3283523e1c
126. Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM, et al. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res (2009) 335(1):17–25. doi: 10.1007/s00441-008-0694-5
127. Hellenthal KEM, Brabenec L, Wagner NM. Regulation and dysregulation of endothelial permeability during systemic inflammation. Cells (2022) 11(12):1–21. doi: 10.3390/cells11121935
128. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev (2004) 84(3):869–901. doi: 10.1152/physrev.00035.2003
129. Ilan N, Cheung L, Pinter E, Madri JA. Platelet-endothelial cell adhesion molecule-1 (CD31), a scaffolding molecule for selected catenin family members whose binding is mediated by different tyrosine and serine/threonine phosphorylation *. J Biol Chem (2000) 275(28):21435–43. doi: 10.1074/jbc.M001857200
130. Wimmer I, Silvia T, Hideaki N, Urban D, Federica S, Fabien G, et al. PECAM-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front Immunol (2019) 10:711. doi: 10.3389/fimmu.2019.00711
131. Alsaffar H, Martino N, Garrett JP, Adam AP. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am J Physiology-Cell Physiol (2018) 314(5):C589–602. doi: 10.1152/ajpcell.00235.2017
132. Gavard J. Endothelial permeability and VE-cadherin. Cell Adhesion Migration (2014) 8(2):158–64. doi: 10.4161/cam.29026
133. Zhang Y-y, Ning B-t. Signaling pathways and intervention therapies in sepsis. Signal Transduction Targeted Ther (2021) 6(1):407. doi: 10.1038/s41392-021-00471-0
134. Li H, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med (2007) 11(1):88–95. doi: 10.1111/j.1582-4934.2007.00011.x
135. Jin M, Fang J, Wang J-j, Shao X, Xu S-w, Liu P-q, et al. Regulation of toll-like receptor (TLR) signaling pathways in atherosclerosis: from mechanisms to targeted therapeutics. Acta Pharmacologica Sin (2023). doi: 10.1038/s41401-023-01123-5
136. Pierer M, Wagner U, Rossol M, Ibrahim S. Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PloS One (2011) 6(8):e23539. doi: 10.1371/journal.pone.0023539
137. Lafferty EI, Qureshi ST, Schnare M. The role of toll-like receptors in acute and chronic lung inflammation. J Inflammation (2010) 7(1):57. doi: 10.1186/1476-9255-7-57
138. Zhang P, Nguyen J, Abdulla F, Nelson AT, Beckman JD, Vercellotti GM, et al. Soluble MD-2 and heme in sickle cell disease plasma promote pro-inflammatory signaling in endothelial cells. Front Immunol (2021) 12:632709. doi: 10.3389/fimmu.2021.632709
139. Conte C, Ingrassia A, Breve J, Bol JJ, Timmermans-Huisman E, van Dam A-M, et al. Toll-like receptor 4 is upregulated in parkinson’s disease patients and co-localizes with pSer129αSyn: A possible link with the pathology. Cells (2023) 12:1–12. doi: 10.3390/cells12101368
140. Zhou Y, Chen Y, Xu C, Zhang H, Lin C, et al. TLR4 targeting as a promising therapeutic strategy for alzheimer disease treatment. Front Neurosci (2020) 14:602508. doi: 10.3389/fnins.2020.602508
141. Wang L, Liu Q, Sun Q, Zhang C, Chen T, Cao X, et al. TLR4 signaling in cancer cells promotes chemoattraction of immature dendritic cells via autocrine CCL20. Biochem Biophys Res Commun (2008) 366(3):852–6. doi: 10.1016/j.bbrc.2007.12.030
142. Giallongo C, Tibullo D, Camiolo G, Parrinello NL, Romano A, Puglisi F, et al. TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis (2019) 10(10):704. doi: 10.1038/s41419-019-1959-5
143. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med (2007) 13(5):552–9. doi: 10.1038/nm1589
144. Lee H-K, Dunzendorfer S, Tobias PS. Cytoplasmic domain-mediated dimerizations of toll-like receptor 4 observed by beta-lactamase enzyme fragment complementation *. J Biol Chem (2004) 279(11):10564–74. doi: 10.1074/jbc.M311564200
145. Oosenbrug T, van de Graaff MJ, Ressing ME, van Kasteren SI. Chemical tools for studying TLR signaling dynamics. Cell Chem Biol (2017) 24(7):801–12. doi: 10.1016/j.chembiol.2017.05.022
147. Repina NA, Rosenbloom A, Mukherjee A, Schaffer DV, Kane RS, et al. At light speed: advances in optogenetic systems for regulating cell signaling and behavior. Annu Rev Chem Biomolecular Eng (2017) 8(1):13–39. doi: 10.1146/annurev-chembioeng-060816-101254
148. Stierschneider A, Zhang F, Bamberg E, Nagel G, Deisseroth K, et al. Light-inducible spatio-temporal control of TLR4 and NF-kappaB-gluc reporter in human pancreatic cell line. Int J Mol Sci (2021) 22(17):1–20. doi: 10.3390/ijms22179232
149. Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K, et al. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci (2005) 8(9):1263–8. doi: 10.1038/nn1525
150. Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci (2015) 18(9):1213–25. doi: 10.1038/nn.4091
151. Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, Lecea L, et al. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc (2010) 5(3):439–56. doi: 10.1038/nprot.2009.226
152. Zhang F, Wang L-P, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods (2006) 3(10):785–92. doi: 10.1038/nmeth936
153. Shikata H, Denninger P. Plant optogenetics: Applications and perspectives. Curr Opin Plant Biol (2022) 68:102256. doi: 10.1016/j.pbi.2022.102256
155. Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W, et al. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J (2014) 33(15):1713–26. doi: 10.15252/embj.201387695
156. Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL, et al. Rapid blue-light–mediated induction of protein interactions in living cells. Nat Methods (2010) 7(12):973–5. doi: 10.1038/nmeth.1524
157. Wang X, Chen X, Yang Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat Methods (2012) 9(3):266–9. doi: 10.1038/nmeth.1892
158. Reichhart E, Ingles-Prieto A, Tichy A-M, McKenzie C, Janovjak H, et al. A phytochrome sensory domain permits receptor activation by red light. Angewandte Chemie Int Edition (2016) 55(21):6339–42. doi: 10.1002/anie.201601736
159. Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature (2009) 461(7266):997–1001. doi: 10.1038/nature08446
160. Kainrath S, Stadler M, Reichhart E, Distel M, Janovjak H, et al. Green-light-induced inactivation of receptor signaling using cobalamin-binding domains. Angewandte Chemie Int Edition (2017) 56(16):4608–11. doi: 10.1002/anie.201611998
161. Takahashi F, Hishinuma T, Kataoka H. Blue light-induced branching in vaucheria. Requirement of nuclear accumulation in the irradiated region. Plant Cell Physiol (2001) 42(3):274–85. doi: 10.1093/pcp/pce033V
162. Takahashi F, Yamagata D, Ishikawa M, Fukamatsu Y, Ogura Y, Kasahara M, et al. AUREOCHROME, a photoreceptor required for photomorphogenesis in stramenopiles. Proc Natl Acad Sci (2007) 104(49):19625–30. doi: 10.1073/pnas.0707692104
163. Kerruth S, Ataka K, Frey D, Schlichting I, Heberle J, et al. Aureochrome 1 illuminated: structural changes of a transcription factor probed by molecular spectroscopy. PloS One (2014) 9(7):e103307. doi: 10.1371/journal.pone.0103307
164. Stierschneider A, Neuditschko B, Colleselli K, Hundsberger H, Herzog F, Wiesner C, et al. Comparative and temporal characterization of LPS and blue-light-induced TLR4 signal transduction and gene expression in optogenetically manipulated endothelial cells. Cells (2023) 12(5):1–21. doi: 10.3390/cells12050697
165. Molteni M, Gemma S, Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm (2016) 2016:6978936. doi: 10.1155/2016/6978936
Keywords: toll-like receptor 4 signaling, endothelium, pro-inflammatory response, lipopolysaccharide, optogenetic control
Citation: Stierschneider A and Wiesner C (2023) Shedding light on the molecular and regulatory mechanisms of TLR4 signaling in endothelial cells under physiological and inflamed conditions. Front. Immunol. 14:1264889. doi: 10.3389/fimmu.2023.1264889
Received: 21 July 2023; Accepted: 08 November 2023;
Published: 24 November 2023.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Gregory M. Vercellotti, University of Minnesota Twin Cities, United StatesToshiyuki Murai, Osaka University, Japan
Copyright © 2023 Stierschneider and Wiesner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Wiesner, Y2hyaXN0b3BoLndpZXNuZXJAaW1jLWtyZW1zLmFjLmF0