 Dia Roy
Dia Roy Cassandra Gilmour
Cassandra Gilmour Sachin Patnaik
Sachin Patnaik Li Lily Wang
Li Lily Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 October 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1264327
This article is part of the Research Topic Monoclonal Antibodies and Immune Checkpoint Inhibitors in the treatment of Cancer View all 28 articles
The differentiation, survival, and effector function of tumor-specific CD8+ cytotoxic T cells lie at the center of antitumor immunity. Due to the lack of proper costimulation and the abundant immunosuppressive mechanisms, tumor-specific T cells show a lack of persistence and exhausted and dysfunctional phenotypes. Multiple coinhibitory receptors, such as PD-1, CTLA-4, VISTA, TIGIT, TIM-3, and LAG-3, contribute to dysfunctional CTLs and failed antitumor immunity. These coinhibitory receptors are collectively called immune checkpoint receptors (ICRs). Immune checkpoint inhibitors (ICIs) targeting these ICRs have become the cornerstone for cancer immunotherapy as they have established new clinical paradigms for an expanding range of previously untreatable cancers. Given the nonredundant yet convergent molecular pathways mediated by various ICRs, combinatorial immunotherapies are being tested to bring synergistic benefits to patients. In this review, we summarize the mechanisms of several emerging ICRs, including VISTA, TIGIT, TIM-3, and LAG-3, and the preclinical and clinical data supporting combinatorial strategies to improve existing ICI therapies.
The cancer-immunity cycle refers to the process wherein tumor antigen-reactive T cells undergo successful priming and differentiate into cytotoxic killer T cells that infiltrate tumor tissues and eliminate cancer cells (1). The differentiation, expansion, survival, and effector function of these tumor-specific cytotoxic T cells (CTLs) is regulated by the collective signaling effects of the T-cell receptor, costimulatory/coinhibitory receptors, and cytokine receptors, which culminate in transcriptional and epigenetic programs to guide T-cell fate. Unlike in acute viral infections where effector CTLs and memory T-cell responses develop properly, tumor-specific CTLs exhibit dysfunctional states in response to chronic stimulation and a myriad of immunosuppressive factors in the tumor microenvironment (TME). These T cells progressively lose proliferative capacity, memory potential, and effector functions, and enter an “exhausted” state. Exhausted T cells upregulate the expression of multiple ICRs, including PD-1, CTLA-4, VISTA, TIGIT, TIM-3, and LAG-3, which sustain dysfunctional antitumor T-cell responses (2, 3).
Immune checkpoint inhibitors (ICIs) are antibodies or small molecules that bind and block the function of ICRs, thereby reducing tumor-induced T-cell exhaustion and restoring anticancer immunity. Ipilimumab, the monoclonal antibody (mAb) blocking cytotoxic T lymphocyte antigen 4 (CTLA-4), was the first ICI therapy approved by the Food Drug Administration (FDA) in 2011. Currently, several mAbs targeting CTLA-4, PD-1, and PD-L1 have been approved for clinical applications. However, despite revolutionizing the field of oncology, the major challenge of existing ICI therapies is the overall low response rate. Understanding the unique molecular and cellular mechanisms of each ICR may support the development of novel combinatorial therapies that optimally restore antitumor immunity.
This review summarizes updated literature regarding the established and emerging ICRs: PD-1, CTLA-4, VISTA, TIGIT, TIM-3, and LAG-3. Due to the scope limitation, we omit discussions of additional emerging ICRs such as B7-H3, B7-H4, HHLA2, and butyrophilin-like 2 (BTNL2), which have been reviewed elsewhere (4). Herein, we provide an overview of each ICR’s structure, expression, signaling mechanisms, and current preclinical and clinical data. We also elaborate on the concept that multiple ICRs operate concurrently to impair the expansion, survival, and effector functions of tumor-reactive cytotoxic T cells (Figure 1), as well as control the maturation and function of dendritic cells (DCs), macrophages, and myeloid-derived suppressor cells (MDSCs) (Figure 2). Given the frequent coexpression and functional crosstalk of these ICRs, we affirm the concept that combinatorial targeting of ICRs may achieve synergistic therapeutic outcomes compared to monotherapies.

Figure 1 Overview of coinhibitory ICRs and their effects in conventional T cells. T-cell activation requires TCR recognition of cognate antigens presented on APCs and costimulation provided by B7/CD28 or CD115/CD226 interactions. On the other hand, many coinhibitory ligand/receptor pathways are activated to dampen T-cell responses. The B7/CTLA-4 and PD-L1/2/PD-1 pathways are the cornerstones of the immune checkpoint paradigm. Emerging inhibitory ICRs, including TIGIT, LAG-3, TIM-3, and VISTA, each recognized by multiple ligands, play nonredundant yet convergent roles as the “brakes” of T-cell responses.
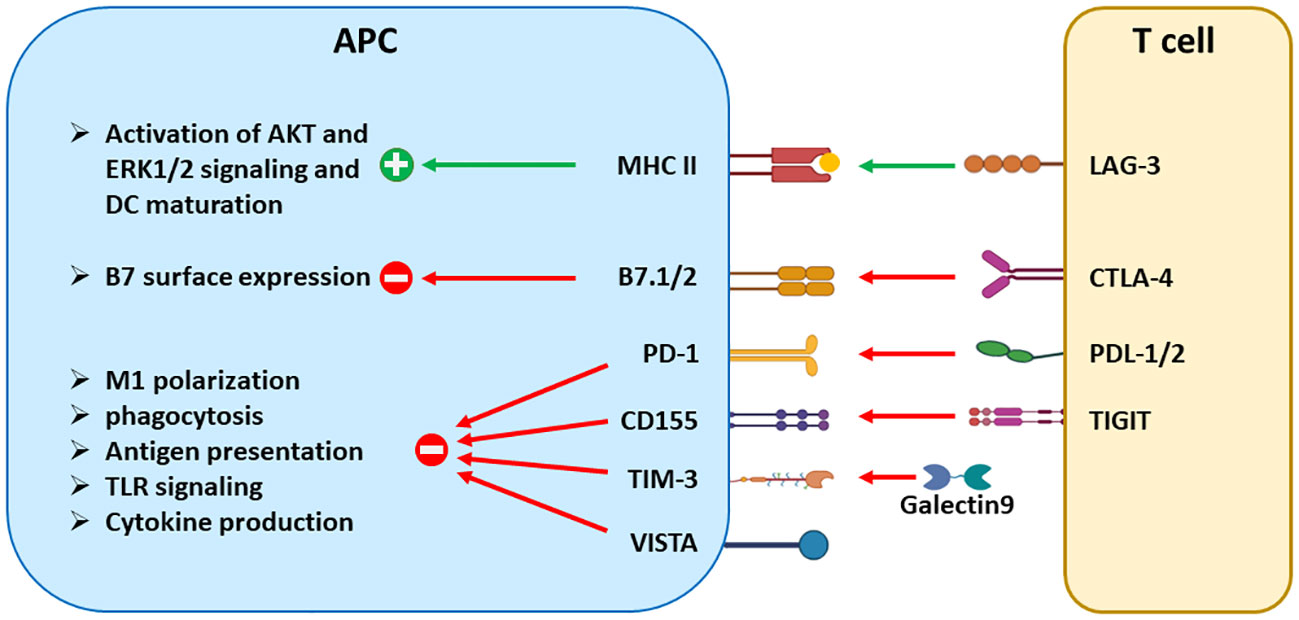
Figure 2 The signaling effects of ICRs in antigen-presenting cells. Aside from suppressing T-cell activation, many ICRs regulate the maturation, antigen presentation, cytokine production, and other effector functions of DCs and tumor-associated macrophages. CTLA-4 reduces the surface expression of B7 molecules through trans-endocytosis. LAG-3 and TIGIT trigger signaling in a reverse direction by engaging their respective binding partners MHCII and CD155. On the other hand, PD-1, TIM-3, and VISTA are expressed in APCs and transmit inhibitory signals to inhibit the effector functions of APCs, including phagocytosis, antigen presentation, and cytokine production. Both PD-1 and VISTA are also expressed in tumor-driven MDSCs and contribute to the differentiation and suppressive function of MDSCs.
Programmed death -1 (PD-1, CD279) belongs to the B7/CD28 family of receptors, which are type-I transmembrane proteins consisting of an immunoglobulin variable (IgV) domain, a transmembrane domain, and a cytoplasmic tail with signaling capacities. PD-1 engages the ligands PD-L1 and PD-L2 and acts as a coinhibitory receptor that regulates both the adaptive and innate arms of the immune system (4, 5).
PD-1 expression is detected in activated T cells, Foxp3+ regulatory T cells (Tregs), natural killer (NK) cells, innate lymphoid cells (ILC2s), B lymphocytes, macrophages, DCs, and monocytes. In T cells, PD-1 gene expression is induced by TCR signaling and positively regulated by multiple transcription factors including AP-1, NFATc1, FoxO1, NF-KB, Notch, STAT, and IRF9 (5). In cancers and chronic viral infections, PD-1 expression in exhausted T cells is significantly higher than in T cells from healthy hosts (3). The expression of PD-1 and its ligand PD-L1 on immune cells and cancer cells may serve as an indicator of disease progression and poor prognosis in a wide range of cancers (6).
The intracellular domain of PD-1 contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) (5). In T cells, the engagement of PD-1 by its ligand PD-L1 results in the recruitment of the tyrosine-protein phosphatases SHP1 and SHP2, which downregulate the phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) pathways. CD28 can be directly dephosphorylated by SHP2 and is the major target of PD-1 inhibitory signaling (7). At the cellular level, the consequences of the PD-1 pathway are multifaceted, resulting in altered T-cell metabolism with impaired glycolysis and augmented fatty acid oxidation, reduced cell expansion and effector cytokine production, and impaired T-cell mobility (3, 4).
In addition to the canonical PD-L1/PD-1 interactions, PD-L1 binds to CD80, which is expressed on antigen-presenting cells (APCs) and activated T cells (8). Trans-interactions of PD-L1 on APCs and CD80 on T cells could transmit inhibitory signaling to T cells and impair antitumor immunity (8, 9). On the other hand, cis-interactions of PD-L1/CD80 on APCs reduced PD-L1/PD-1 interactions and CD80/CTLA4 interactions, without affecting interactions between CD80 on APCs and CD28 on T cells (10–12). Blocking cis-interaction of PD-L1/CD80 reduced CD80 expression on APCs and impaired antitumor immune responses (11). An anti-CD80 antibody blocking PD-L1/CD80 cis-interactions augmented PD-L1/PD-1 interactions and alleviated autoimmune disease (13).
In addition to T cells, PD-1 is expressed in tumor-associated macrophages and inhibits their phagocytic function, which in turn controls antitumor immune responses (14). Furthermore, PD-1 plays a role in regulating tumor-driven emergency myelopoiesis. PD-1 deletion in myeloid progenitors reduced the accumulation of GMPs and MDSCs, which may be the result of elevated ERK1/2 and mTORC1 signaling and metabolic reprogramming (15). In preclinical models and cancer patients, blocking interactions of PD-1 with PD-L1 augments the effector function of PD-1+ exhausted CTLs, and induces the expansion of TCF1+ progenitor-like exhausted T cells with self-renewal capacity (16). On the other hand, blocking PD-1 may trigger hyperproliferation and suppressive function of Tregs and contribute to hyperprogressive diseases (17).
Monoclonal antibodies specific for PD-1 (nivolumab, pembrolizumab), and PD-L1 (durvalumab, atezolizumab, and avelumab) have proven to be clinically effective and gained FDA approval across a wide range of cancers, such as skin cancer, lung cancer, Hodgkin lymphoma, renal cell carcinoma (RCC), head and neck cancer, bladder cancer, colorectal cancer, liver cancer, gastric cancer, triple negative breast cancer, and cervical cancer (18, 19). Additional antibodies blocking PD-1, such as cemiplimab, camrelizumab, sintilimab, toripalimab, tislelizumab, zimberelimab, prolgolimab, and dostarlimab, have been approved for cancer applications worldwide. A meta-analysis of randomized controlled trials has concluded that anti-PD-1/PD-L1 inhibitors are more advantageous for treating advanced and metastatic cancers than conventional therapies, with better overall survival and progression-free survival particularly in male patients with younger age, without central nervous system or liver metastasis, no EGFR mutations, and with higher PD-L1 expression (18).
While PD-L1/PD-1 inhibitors are approved for treating an expanding list of cancers, their use as monotherapies generated an overall low response rate, due to mechanisms of primary and acquired resistance (20, 21). To improve the response rate to ICIs, numerous combination strategies have been studied in preclinical and clinical trials, including combining PD-L1/PD-1 inhibitors with chemotherapeutics such as cyclophosphamide, radiotherapy, targeted therapy, agonistic costimulatory antibodies targeting CD134, CD137 or ICOS, innate immune stimulators such as STING agonists, epigenetic modulators, and cancer vaccines such as oncolytic viruses (19, 22, 23). On the other hand, these combinatorial regimens fail to address the roles of other non-overlapping ICRs that constitute one of the dominant resistance mechanisms to PD-1/PD-L1 inhibitors. In the rest of this review, we will summarize studies of emerging ICRs (i.e., VISTA, TIGIT, TIM-3, and LAG-3) and demonstrate the rationales for combinatorial therapies targeting non-redundant ICRs together with PD-1/PD-L1 inhibitors.
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4, CD152), together with CD28, represents the B7 family of receptors. Similar to PD-1, CTLA-4 contains an extracellular IgV-domain, a transmembrane domain, and a cytoplasmic tail with motifs for intracellular signaling (24, 25). CTLA-4 is constitutively expressed on Foxp3+ regulatory T cells (Tregs) and is inducible upon activation in conventional T cells. In addition, CTLA-4 expression has been detected in natural killer cells, B cells, dendritic cells, and myeloid cells (26–31).
In T cells, CTLA-4 gene expression is induced by Foxp3 and NFAT (32). The stability of CTLA-4 mRNA is regulated post-transcriptionally, by microRNAs such as miR-145 and miR-155 (33, 34). In resting T cells, a majority of CTLA-4 resides intracellularly within endosomes and relocalizes to the cell surface upon TCR stimulation (27, 31, 35–37). CTLA-4 protein localization is dynamically regulated by clathrin-mediated endocytosis and endosomal recycling, which is dependent upon the tyrosine phosphorylation status of its cytoplasmic domain (38).
CTLA-4 inhibits the expansion, cytokine production, and differentiation of conventional T cells and contributes to the development and function of Foxp3+ Tregs. CTLA-4 exerts inhibitory effects by competing against CD28 due to its higher affinity for B7 molecules, as well as by recruiting phosphatases SHP2 and PP2A, which in turn downregulate signaling of TCR and CD28 (39–42). In addition to T-cell intrinsic mechanisms, CTLA-4 indirectly suppresses T-cell responses by modulating dendritic cells: CTLA-4 downregulates the surface expression of B7 molecules through trans-endocytosis (43) or induces the expression of indoleamine 2,3-dioxygenase (IDO), which in turn impairs T-cell proliferation (44). CTLA-4 also reverses the stop signal in activated T cells and reduces the contact time between T cells and APCs, leading to decreased cytokine production and T-cell proliferative responses (45).
The mechanisms of CTLA-4-mediated immunosuppression in cancers are distinct from PD-1 and potentially synergistic with PD-1 (46): although both receptors act on activated conventional T cells, PD-1 controls effector T-cell function at a later stage, mainly within peripheral tissue sites and the tumor microenvironment, while CTLA-4 intercepts T-cell priming in the lymph nodes and governs the function of Tregs (47, 48). CTLA4 is constitutively expressed in Foxp3+ Tregs and CTLA-4-specific antagonistic antibodies not only augment effector T-cell activation but also induce ADCC-mediated depletion of tumor-infiltrating Tregs (49–51). On the other hand, unlike PD-1 and PD-L1, CTLA-4 is not expressed in myeloid cells and does not directly regulate suppressive myeloid cells within the TME. These functional distinctions provide mechanistic rationales for developing combination therapies targeting both axes.
Studies have shown that while CTLA-4 and PD-1 blockade each boosts antitumor T-cell responses, dual blockade results in stronger therapeutic outcomes in preclinical models and human patients (52–54). ICI monotherapies induced the expansion of different tumor-infiltrating T cells (TILs), i.e., PD-1 blockade expanded exhausted-like CD8+ CTLs, whereas CTLA-4 blockade expanded both ICOS+ Th1-like CD4 effectors and exhausted CD8+ CTLs. In contrast, the combined blockade induced the expansion of terminally differentiated effector CD8+ CTLs that are not seen in monotherapies and further increased Th1-like CD4+ effector T cells (52, 53). Similar findings have been shown in human melanoma patients treated with ipilimumab and nivolumab therapy. In addition to melanoma, dual blockade of CTLA-4 and PD-1 was studied in a murine breast cancer model (53). While monotherapies showed modest effects, combination therapy led to complete tumor regression in a majority of mice. The synergistic efficacy was due to the anti-CTLA-4 antibody-induced expansion of the T-cell receptor (TCR) repertoire and augmented functionality of TILs, accompanied by intratumoral Treg depletion. Taken together, these studies have demonstrated the mechanisms of synergy with dual ICI therapy that may guide clinical applications.
Ipilimumab (Yervoy) was the first FDA-approved monoclonal antibody for cancer immunotherapy, owing to robust clinical responses for metastatic melanoma (55, 56). We summarize recent clinical trials that have advanced PD-1 and CTLA-4 combinatorial therapy; comprehensive overviews of other clinical trials involving ipilimumab can be found in other reviews (57, 58). As a monotherapy, the effect of ipilimumab is not as strong as that of the PD-1 antibody nivolumab (Opdivo) for resected stage III or IV melanoma and showed shorter survival and higher toxicity for patients than the PD-1 antibody pembrolizumab (Keytruda) (59, 60). However, when ipilimumab was given concurrently with PD-1 antibody, dual blockade therapy demonstrated significantly improved outcomes in clinical studies. The advantages of dual ICI therapy were first noted in a Phase I dose-escalation study using nivolumab and ipilimumab administered together, which led to better response rates and progression-free survival compared to previously reported results from either monotherapy (61). A subsequent phase III study highlighted better responses and survival with combinatorial therapy when used for metastatic melanoma patients with PD-L1 negative tumors compared to either nivolumab alone or ipilimumab alone, despite the higher occurrence of grade 3 or 4 treatment-related adverse events (62). Follow-up studies showed durable responses and sustained benefits for survival in these patients across multiple years (63–65). Treatment-naive patients with advanced melanoma also benefited from nivolumab-plus-ipilimumab treatment, once again producing higher objective-response rates and progression-free survival with acceptable safety profiles compared to ipilimumab alone (57).
Current research continues to advance PD-1 and CTLA-4 combinatorial immunotherapy in the treatment of other cancers. Beyond melanoma, FDA approval of anti-PD-1 and anti-CTLA-4 dual therapy has expanded to hepatocellular carcinoma (HCC), unresectable pleural mesothelioma, RCC, metastatic non-small cell lung cancer (NSCLC), and advanced or metastatic esophageal squamous cell carcinoma (66–68). Combinatorial ICI therapy in the neoadjuvant setting has also shown promise, with tolerance and strong pathological responses for late-stage melanoma, early-stage colon cancers, and late-stage urothelial cancer (69, 70). Dual blockade of CTLA-4 and PD-1 is currently being evaluated in numerous clinical trials for advanced solid tumors, such as head and neck squamous cell carcinoma (HNSCC) and glioblastomas (NCT04080804, NCT04606316). For testing combined treatment with pembrolizumab (anti-PD-L1), a randomized, double-blind phase III KEYNOTE-598 study (NCT03302234) showed that in patients with metastatic NSCLC, adding ipilimumab to pembrolizumab did not improve efficacy and exhibited greater toxicity than pembrolizumab monotherapy (71). Another phase I expansion trial (NCT02089685) evaluated the efficacy and safety of pembrolizumab combined with a reduced dose of ipilimumab in patients with advanced melanoma and RCC and showed manageable toxicity profile and robust antitumor activity (72).
V-domain immunoglobulin suppressor of T-cell activation (VISTA, alias Gi24, Dies-1, PD-1H, DD1α) is homologous to B7 family receptors and acts as a negative regulator of antitumor immunity and autoimmunity (73–78). VISTA is a type I transmembrane protein containing a single IgV-like extracellular domain (ECD), a transmembrane segment, and a cytoplasmic tail that does not contain ITAM, ITIM, or ITSM motifs. Structural studies have revealed unique features of the VISTA ECD that are distinct from those of other Ig superfamily members, including two additional disulfide bonds, the insertion of an unstructured C-C’ loop, the striking enrichment of histidine residues within the ECD, and an extra H β-strand that forms an intramolecular clamping disulfide bond (79, 80). Mutagenesis studies have demonstrated that these structural features contribute to the surface orientation and suppressive function of VISTA (79, 80).
VISTA expression in mice is largely restricted within the hematopoietic compartment, with the highest expression on CD11b+ myeloid lineages such as monocytes, macrophages, granulocytes, and dendritic cells (73, 74). VISTA is also expressed in lymphocytes including NK cells, TCRγδ T cells, naïve CD4+ and CD8+ TCRαβ T cells, and Foxp3+ Tregs. A similar expression pattern of VISTA is seen in human peripheral blood monocytic cells. VISTA gene expression is positively regulated by the transcription factors P53, HIF1-α, and STAT3 (81–83). However, whether VISTA exerts any impact on the functions of HIF1-α and STAT3 remains unknown. VISTA expression is also regulated by TGF-β/Smad3 signaling in T cells and myeloid cells (84).
In human cancer tissues, VISTA expression was mostly enriched in tumor-infiltrating myeloid cells and T cells (75, 85). In addition to immune cells, VISTA expression was detected in mesothelioma (86), gastric cancer (87), and AML (83, 88, 89). VISTA expression has been associated with resistance to immunotherapy and poor patient survival in many cancers, including prostate cancer, lymphoma, bladder cancer, melanoma, breast cancer, and AML (88, 90–95),
VISTA impairs antitumor immunity through its ligand activity in myeloid cells and T cell-intrinsic activity. Although it has been speculated that VISTA also acts as an inhibitory receptor (96), the signaling mechanism is unclear and it remains possible that T cell-intrinsic activity may rely on cis interactions with other signaling partners. At the molecular level, several partners, such as PSGL-1, VSIG3, and galectin-9, have been identified to engage VISTA (97–99). While PSGL-1 was suggested as an inhibitory receptor for VISTA, VSIG3 was considered a ligand. Galectin-9 binds VISTA and forms a protein complex that promotes galectin-9-mediated apoptotic signaling. At the cellular level, VISTA regulates the development and function of macrophages, MDSCs, neutrophils, TCRγδ T cells, and CD4+/CD8+ conventional T cells (74, 75, 78, 100, 101). In macrophages, VISTA impairs TLR signaling by regulating the ubiquitination and stability of TRAF6 (102). Blocking VISTA synergizes with a TLR-agonistic vaccine by augmenting the activation of DCs and macrophages, increasing the production of stimulatory cytokines such as IL-12 and IL-27, and promoting the effector function of tumor-specific CTLs. VISTA also contributes to the suppressive function of MDSCs, although the exact molecular mechanisms remain undefined (82, 102).
In preclinical models, genetic deletion of VISTA or treatment with anti-VISTA mAb delayed tumor regression by inducing DC maturation, reducing the abundance of adaptive Foxp3+ Tregs, reducing the abundance of MDSCs, and augmenting the effector function and abundance of CTLs (73, 76).
Studies led by Liu et al. first established the nonredundant and synergistic role of VISTA and PD-1 in mounting immune responses against self and tumor antigens (103). In both B16 melanoma and CT26 colon tumor models, combinatorial treatment with anti-VISTA and anti-PD-L1 mAbs resulted in tumor regression and long-term survival in comparison to monotherapies (103, 104). A separate VISTA-blocking mAb, SG7, suppressed the interaction between VISTA and VSIG3 or PSGL-1 and showed efficacy in combination with PD-1 blockade in the MC38 colon tumor model (105). Finally, a unique role of VISTA in promoting naive T-cell quiescence has been identified (106). Accordingly, a study in a CT26 tumor model showed that a triple blockade of VISTA/PD-1/CTLA-4 could improve the efficacy of PD-1/CTLA-4 dual blockade by promoting antigen-presentation in myeloid cells and reducing the quiescent state of CTLs (107).
Several clinically relevant VISTA-blocking agents have been developed and entered clinical trials. VSTB112 (Janssen Inc) was the first anti-VISTA mAb tested in the clinic (NCT02671955). CA-170 (Curis Inc) is an orally available small molecule that has dual targeting activities against PD-L1/L2 and VISTA. In preclinical models, CA-170 rescued T-cell function similarly to PD-1 antagonists and inhibited the growth of B16 melanoma, CT26, and MC38 murine tumor models (108, 109). CA-170 was tested in a phase I trial (NCT02812875) and a phase II trial (Clinical Trials Registry-India CTRI/2017/12/011026) (110). CA-170 showed an excellent safety profile and encouraging clinical activity in classic Hodgkin lymphoma and advanced NSCLC (109). HMBD-002 (Hummingbird Bioscience) is a human VISTA-specific mAb that binds to the C-C’ loop of VISTA and blocks its interaction with VSIG3 (111). Studies of murine and humanized mouse models showed the effects of HMBD-002 in reducing MDSCs and augmenting T-cell responses. The phase I trial of HMBD-002 is ongoing (NCT05082610). W0180 (Pierre Fabre Inc) is a human VISTA-specific mAb being tested in a phase I trial (NCT04564417). The NCT05082610 and NCT04564417 trials will both test VISTA inhibitors in combination with pembrolizumab. KVA12123 (Kineta Inc) is a third human VISTA-targeting mAb that has recently been granted FDA acceptance for testing in phase I/II trials.
T-cell immunoreceptor with Ig and ITIM domains (TIGIT) is an ICR that contains an IgV-like ECD, a type I transmembrane domain, and a cytoplasmic domain with ITIM and ITT motifs (112). TIGIT is expressed on NK cells, CD4+/CD8+ conventional T cells, and Foxp3+ Tregs. In T cells, TIGIT expression is upregulated following TCR activation and is sustained with increased exhaustion (112).
In human cancers, TIGIT gene expression was found to be upregulated in tumors and correlated with poor prognosis for KIRC, KIRP, LGG, and UVM cancers (113). TIGIT protein expression is abundant in CD4+/CD8+ TILs and Tregs from a wide range of cancer types and is often correlated with poor clinical outcomes or resistance to ICI therapies (114). Coexpression of TIGIT and PD-1 on CD8+ TILs, which is associated with dysfunctional antitumor immune responses, has also been observed in cancers such as HCC, glioblastoma (GBM), acute myeloid leukemia, NSCLC, and melanoma (114).
TIGIT binds to three ligands CD112, CD113, and PVR(CD155), out of which CD155 exhibits the highest affinity (115, 116). The TIGIT/CD155 interaction inhibits the functions of NK cells, T cells, and APCs. Phosphorylation of both the ITT and ITIM domains is required for the inhibitory signaling of TIGIT in NK cells and T cells, partly by recruiting the adaptors Grb2 and SHIP1, which in turn dampen the PI3K, MAPK, and NF-KB signaling pathways (117, 118). TIGIT also outcompetes CD226 for binding to CD155 and disrupts the costimulatory signal from CD226 in T cells (119). In addition to effector T cells, TIGIT is expressed in Foxp3+ Tregs and plays a role in promoting their differentiation, stability, and suppressive function (120–122).
In APCs such as DCs and macrophages, CD155 is phosphorylated upon engaging TIGIT and subsequently inhibits MAPK signaling, resulting in tolerogenic APCs that produce elevated levels of IL-10 but reduced levels of IL-12, and fail to properly stimulate cognate T cells (123). Another recent study demonstrated that leukemia-associated macrophages express TIGIT and that blocking TIGIT drives M1-like phenotypes and increases phagocytosis (124).
The efficacy of the dual blockade of TIGIT and PD-L1 has been demonstrated in murine breast and colon carcinoma models (112). The combination therapy rejuvenated tumor-specific CD8+ CTLs by augmenting their expansion, effector functions, and the development of memory responses (112). A recent study has shown that the PD-1 and TIGIT pathways converge to regulate CD226, as both receptors impair the phosphorylation of CD226 (125). Furthermore, when CD8+ TILs from human liver cancers were treated with TIGIT and PD-1-blocking mAbs, the coblockade of TIGIT and PD1 significantly improved the expansion, cytokine production, and cytotoxicity of CD8+ TILs compared with single PD-1 blockade (126). Similar results were seen in an adoptive T-cell transfer study to treat human lung cancer, where dual blockade of TIGIT/PD-1 or TIM-3/PD-1 resulted in greater tumor control than PD-1 monotherapy (127). Together, these studies provide a strong rationale for blocking both the PD-1 and TIGIT pathways to allow optimal CD226-dependent costimulatory signaling in CD8+ T cells.
Currently, there are approximately > 50 clinical trials in the US testing several TIGIT-targeted mAbs, either as monotherapy or in combination with PD-L1/PD-1 inhibitors (clinicaltrials.gov). Bispecific antibodies targeting both TIGIT and PD-1 are also being tested in these trials. In a phase II clinical trial sponsored by Roche (NCT03563716), anti-TIGIT mAb (Tiragolumab) was granted breakthrough therapy designation and was tested in combination with anti-PD-L1 (atezolizumab) in metastatic NSCLC (128). The combination treatment has improved the overall response rate, progression-free survival, and overall survival, over atezolizumab alone (128). Notably, the benefit of the combination treatment was mainly observed in patients with high PD-L1 expression (> 50%) (128, 129). Another TIGIT antibody Vibostolimab (MK-7684) was evaluated in a phase I trial (NCT02964013) with and without combination with pembrolizumab for advanced solid tumors, including NSCLC, and showed promising antitumor activity (130). Additional TIGIT inhibitors, such as BMS-986207 (NCT04570839) (131), ASP8374 (NCT03260322, NCT04826393) (132), Domvanalimab (AB154) (NCT04262856) (133), BGB-A1217 (NCT04047862) (134), and Etigilimab (OMP-313M32) (NCT04761198) (135) are under investigation as single agents and in combination with PD-1/PD-L1 inhibitors in solid tumors.
T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), along with TIM1 and TIM4, belongs to the TIM family of immunoregulatory proteins. The ECD of TIM-3 contains an immunoglobulin variable domain that binds to several ligands: galectin 9, phosphatidylserine, CEACAM1, and HMGB1 (136). Following the ECD is a mucin domain, a transmembrane domain, and a cytoplasmic domain that does not contain canonical inhibitory signaling motifs such as ITIM or ITSM motifs.
TIM-3 is expressed on subsets of activated CD4+ and CD8+ conventional T cells, Foxp3+ Tregs, NK cells, myeloid cells, and mast cells (136). TIM-3 can be cleaved into a soluble form by ADAM10 and ADAM17 (137). TIM-3 expression in T cells is coregulated with other ICRs including PD-1, TIGIT, and LAG-3 (138). Cytokines such as IL-12, IL-27, and IFN-β can upregulate TIM-3 expression (139, 140). In human cancers, TIM-3 is highly expressed in terminally exhausted CD8+ CTLs, Foxp3+ Tregs, tumor-associated macrophages, and MDSCs. TIM-3 expression levels have been shown to correlate with resistance to immunotherapies and poor prognosis in many cancer types such as melanoma, HCC, prostate cancer, RCC, colon cancer, bladder cancer, cervical cancer, gastric cancer, and esophageal squamous cell carcinoma (122, 141–149).
In conventional T cells, TIM-3 is recruited to the immune synapse upon TCR activation (150). Y256 and Y263, two of the five tyrosines on the cytoplasmic tail of TIM3, bind BAT3, a protein involved in the TCR signaling pathway (151). Bound BAT3 recruits LCK, a major upstream player in the TCR signaling pathway (152). However, engagement with galectin 9 results in the phosphorylation of Y256 and Y263 by interleukin-2-inducible T-cell Kinase (ITK), which releases BAT3 and impairs TCR signaling (153, 154). Another ligand, CEACAM1, binds TIM-3 in cis to promote the stability of TIM-3, while the trans interaction induces similar signaling outcomes as galectin-9 (154). The Galectin 9/TIM-3 axis induces apoptosis of effector Th1 cells and CD8+ CTLs (152, 155, 156). In Foxp3+ Tregs, TIM-3 signaling drives an effector-like phenotype and enhances suppressive function (157).
TIM-3 is also expressed in DCs, where its ligation induces the activation of Bruton’s tyrosine kinase and c-Src, which inhibit NF-kB activation and subsequently reduce DC activation (158). In macrophages, TIM-3 has been reported to promote M2-like polarization by inducing SOCS1 (159). In monocytes and DCs, TIM-3 inhibits the cellular responses to TLR signaling and reduces the production of proinflammatory mediators (160). In a breast cancer model, blocking TIM-3 augmented the production of a key chemokine CXCL9 from CD103+ DCs, thereby improving the antitumor immune responses when combined with chemotherapy (161).
In preclinical models, dual blockade of TIM-3 and PD-1 restored the function of both CD4+ and CD8+ T cells and led to complete tumor regression whereas either monotherapy was not effective (162, 163). A recent study has shown that PD-1 binds galectin-9 and that PD-1/TIM-3/galectin-9 may crosslink and form a lattice. As such, PD-1 functions to attenuate galectin-9/TIM-3-induced apoptosis (164). It should be noted that VISTA also binds to galectin-9 and augments the inhibitory effects of TIM-3 (99). Thus, these findings may provide a rationale for future studies to test the combined blockade of PD-1, TIM-3, and VISTA, to improve the persistence and functions of tumor-reactive PD-1+ TIM-3+ CTLs.
In human cancers, TIM-3 and PD1 are often coexpressed on CD8+ T cells and mark the most dysfunctional T cell subsets. An earlier study of advanced melanoma showed that NY-ESO-1-specific PD-1+CD8+ TILs upregulate TIM-3 expression, which is correlated with dysfunctional phenotypes (165). Blocking TIM-3 augmented cytokine production and proliferation of T cells, while combined blockade of both TIM-3 and PD-1 showed synergistic effects. Similar findings were reported in colorectal cancer, where TIM-3+PD-1+CD8+ TILs represented the predominant fraction of TILs and targeting both TIM-3 and PD-1 enhanced cell expansion, cytokine production, and cytotoxic activity (166). Recent studies of diffuse large B-cell lymphoma found that TIM-3+PD1+ TILs exhibited a transcriptomic signature of T-cell exhaustion, reduced proliferation, and impaired cytokine production, but these dysfunctions were restored by the blockade of PD1 or TIM-3 (167, 168). Although there have not been any FDA-approved therapeutics targeting TIM-3, the pipelines for novel TIM-3 inhibitors are expanding: several TIM-3-specific antibodies (i.e., cobolimab, MBG453, Sym-023, BMS-986258, AZD7789, INCAGN02390, etc.) or TIM-3/PD-1 bispecific antibodies are being tested in clinical trials (169). A phase I/II trial (NCT02608268) evaluated MGB453 (anti-TIM3) in combination with PDR001 (anti-PD-1) in advanced solid cancers such as melanoma and NSCLC and showed excellent safety profile and preliminary antitumor activity (170). Similar encouraging results were shown by trials (NCT02817633 and NCT03680508) that evaluated TSR-022 (anti-TIM3) in combination with PD-1 inhibitors (171, 172). In addition, a phase Ia/b trial evaluated the safety, pharmacokinetics, and efficacy of LY3321367 (anti-TIM3) plus LY3300054 (Anti-PD-L1) and showed modest antitumor activity (173).
Lymphocyte activation gene 3 (LAG-3, CD223) is an Ig superfamily ICR and is homologous to CD4 (174, 175). The ECD of LAG-3 contains four IgV or IgC-like domains that are involved in ligand binding. The cytoplasmic domain of LAG-3 contains a serine phosphorylation site, the conserved KIEELE motif, and the glutamate-proline dipeptide repeat motif that is involved in its inhibitory signaling (176).
LAG-3 is expressed in many immune cell types including activated conventional CD4+/CD8+ T cells, Foxp3+ Tregs, TCRγδ T cells, NK cells, dendritic cells, and B cells (175). In T cells, LAG-3 expression is induced upon TCR stimulation or by cytokines such as IL-12, IL-2, IL-15, IL-7, IL-6, and IL-8 (177–179). LAG3 expression is promoted by transcription factors such as TOX, NFAT, and NR4A, while suppressed by T-bet (180–186). Studies of human cancers have shown that LAG-3 expression is abundant in TILs and associated with T cell dysfunction or insensitivity to PD-1 blockade. These include breast cancer (187), kidney renal clear cell carcinoma (188), melanoma (189), NSCLC (190, 191), HCC (192, 193), and B-cell lymphoma (194). LAG-3 expression in peripheral blood lymphocytes is also associated with resistance to ICI therapies in patients with melanoma and urothelial carcinoma (195). Furthermore, the clinical resistance to PD1 blockade may be correlated with reduced shedding of LAG-3 in CD4+ conventional T cells due to reduced expression of the protease ADAM10 (196).
LAG-3 is recognized by multiple ligands including MHCII (197–199), fibrinogen-like protein 1 (FGL-1) (200), galectin-3 (201), and liver sinusoidal endothelial cell lectin (LSECtin) (202). In conventional T cells, LAG-3 signaling suppresses T cell activation, proliferation, cytokine secretion, and cytotoxic functions (203). LAG-3 interacts in cis with the TCR/CD3 complex and inhibits TCR signaling by promoting local acidification and Lck dissociation (204). LAG-3 and PD1 interact in cis and cluster with pLck at the immunological synapse and recruit SHP1/2, thereby exerting inhibitory effects on T-cell signaling (205). LAG-3 also promotes the activation and suppressive function of Foxp3+ Tregs (206). Soluble LAG-3 acts as an MHCII agonist and induces tyrosine phosphorylation and activation of the AKT and ERK1/2 signaling pathways, thereby inducing DC maturation and improving antitumor T-cell responses (207, 208).
Preclinical studies have established that LAG-3 cooperates with PD-1 in controlling antitumor immunity (175, 209). The striking synergy between LAG-3 and PD-1 has been demonstrated in murine melanoma, colon cancer, and ovarian tumor models, where the dual blockade against LAG-3 and PD-1 effectively controlled tumor progression that was resistant to respective monotherapies (205, 210). A study in the MC38 colon cancer model has shown that PD-L1 blockade elevated the expression of both costimulatory receptors (ICOS) and coinhibitory receptors (LAG3 and PD-1) in TILs, thereby providing a new mechanistic rationale for coblocking LAG3 (211).
In human ovarian cancer, NY-ESO-1-specific CD8+ TILs demonstrated impaired effector function and enriched coexpression of the inhibitory molecules LAG-3 and PD-1. Dual blockade of LAG-3 and PD-1 during T-cell priming efficiently augmented proliferation and cytokine production by NY-ESO-1-specific CD8+ T cells (212).
These preclinical and clinical studies have provided the backbone for combinational treatment strategies. Currently, numerous clinical trials are exploring the therapeutic benefits of simultaneously targeting LAG-3 and PD-1 (209, 213). LAG-3 targeted agents include soluble LAG-3, LAG-3-specific mAbs, or bispecific antibodies recognizing both LAG-3 and PD-1. Relatlimab (anti-LAG-3) in combination with nivolumab received FDA approval in March 2022 for treating unresectable or metastatic melanoma (214). Favezelimab (MK-4280) in combination with pembrolizumab was tested in a phase III trial (NCT02720068) for colorectal cancer and showed promising antitumor activity in PD-L1-positive tumors (213, 215). Ieramilimab (LAG525) was tested in a phase I/II study (NCT02460224) in combination with spartalizumab (PDR001, anti-PD-1) in advanced/metastatic solid tumors such as melanoma and TNBCs, demonstrating a good toxicity profile but moderate antitumor activity (216). Fianlimab (REGN3767, anti-LAG3) is being tested in combination with cemiplimab (anti-PD-1) in a phase I dose-escalation study (NCT03005782) in advanced melanoma patients and showed clinical activities (217). Eftilagimod alpha, a soluble LAG-3 fusion protein, is being tested along with pembrolizumab for treating recurrent or metastatic head and neck squamous cell carcinoma (NCT03625323) (208). An ongoing phase I/II study (NCT04370704) is testing retifanlimab (INCMGA00012, Anti–PD-1), INCAGN02385 (Anti–LAG-3), and INCAGN02390 (Anti–TIM-3) triple combination therapy in patients with advanced solid tumors (218). Multiple trials tested BI-754111 (anti-LAG3) combined with BI-754091 (anti-PD-1) in patients with advanced solid tumors but no significant antitumor activity was reported (219). Lastly, bispecific antibodies targeting PD-1/LAG3, including tebotelimab (MGD013, NCT04212221) and RO7247669 (NCT04140500) are under early-stage clinical investigations (220).
Since the first FDA approval of ICIs in 2011, significant progress has been made toward optimizing existing ICI therapies. Taking the lessons from existing ICIs that target PD-1, PD-L1, and CTLA-4, current efforts in the field focus on identifying and targeting nonredundant ICRs that may potentially synergize with existing therapies. VISTA, TIGIT, TIM-3, and LAG-3 represent such candidates in the pipeline. Recent advances in understanding the converging role of ICRs in driving the dysfunction of both APCs and T cells (Figures 1, 2) have set the conceptual foundation for developing combinatorial therapies targeting these ICRs. Based on the frequent coexpression of ICRs in tumor tissues and the distinct yet convergent mechanisms of action (Table 1), it is expected that combined blockade of these emerging ICRs with PD-L1/PD-1 will result in additive or synergistic outcomes. Indeed, many novel ICI combination therapies are being investigated in early-stage trials (Table 2). To advance this concept into clinical applications, the field still faces some challenges, such as defining the molecular pathways and hierarchy of emerging ICRs, identifying the optimal ICR combinations for distinct cancer types and discrete biomarkers, and developing better preclinical models that present the full extent of immune-related toxicities as seen in human patients. In conclusion, we emphasize that antitumor immunity is controlled by multiple nonredundant ICRs that together maintain immune dysfunction. Recent preclinical and early clinical data strongly support the rational design of novel ICI combinations to achieve synergistic therapeutic efficacies with manageable toxicities.
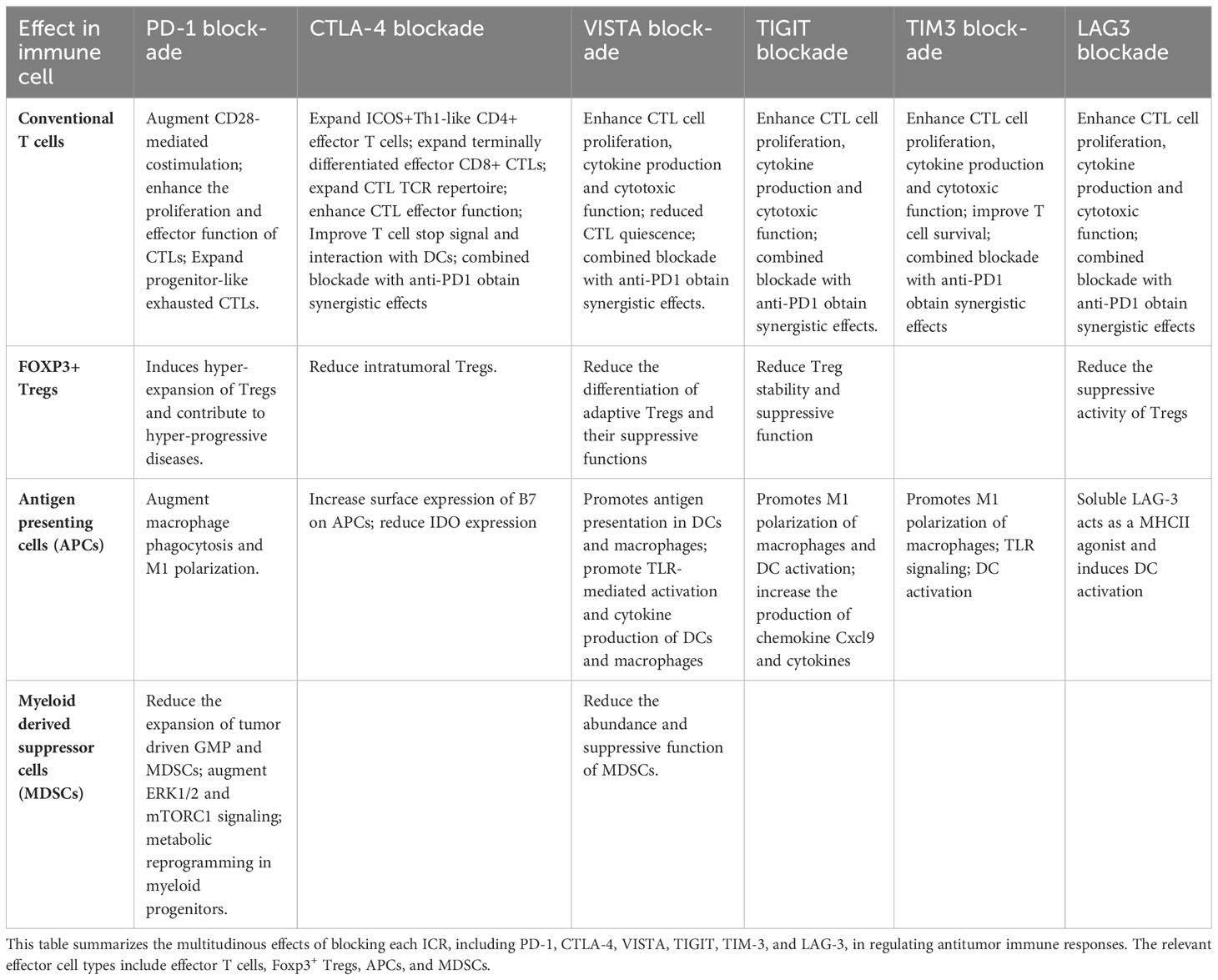
Table 1 Blocking individual ICRs augments antitumor immune responses by convergent cellular and molecular mechanisms.
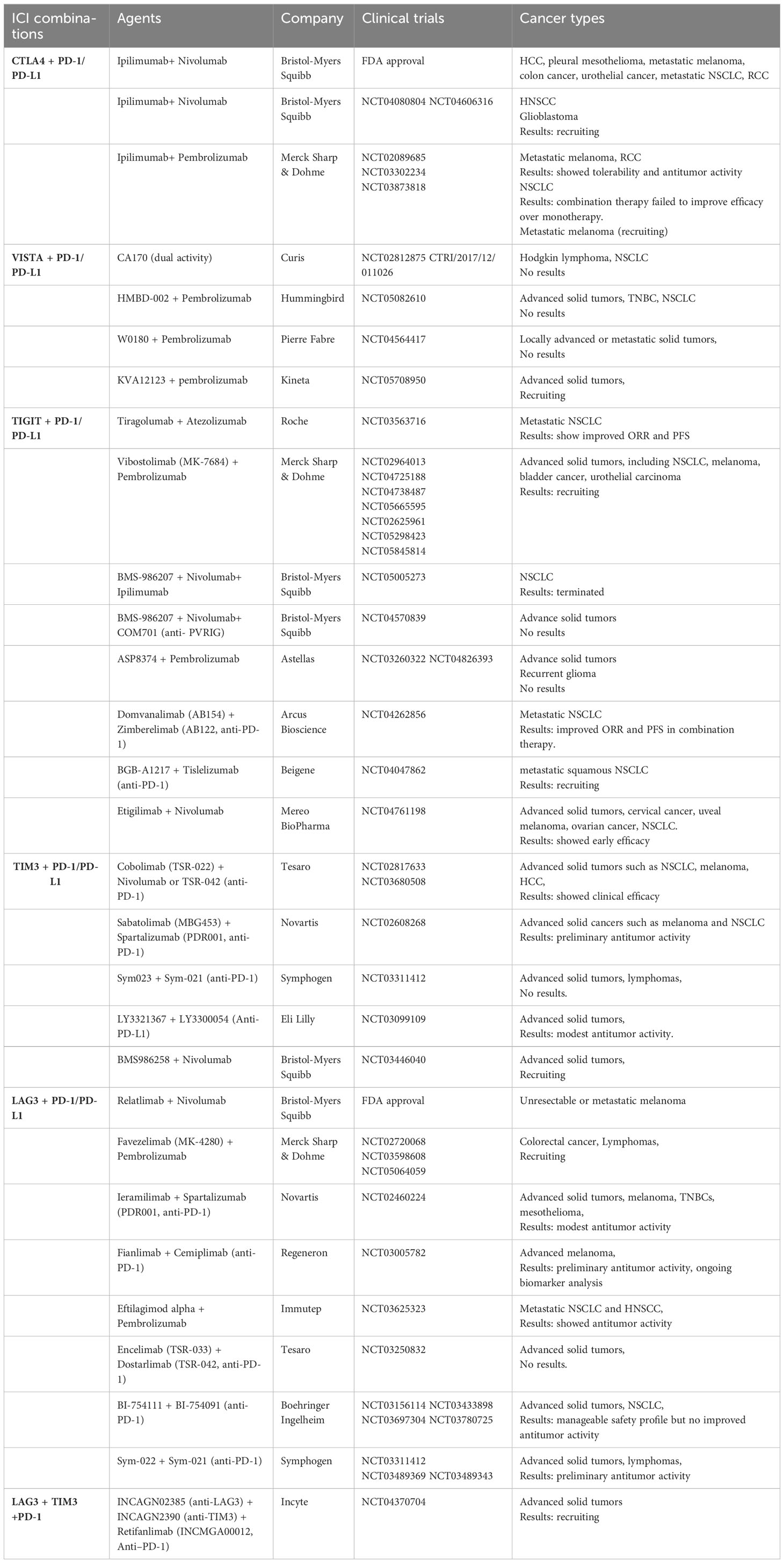
Table 2 Clinical trials testing combined targeting of ICRs.
DR: Writing – original draft, Writing – review & editing. CG: Writing – original draft, Writing – review & editing. SP: Writing – original draft, Writing – review & editing. LW: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Supervision, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. National Institute of Health/National Cancer Institute R01CA164225 (LW), National Institute of Health/National Cancer Institute R01CA223804 (LW), National Institute of Health/National Cancer Institute R21CA258618 (LW), National Institute of Health/National Cancer Institute F31CA257276 (CG). Department of Defense CDMRP W81XWH-21-MRP-MCAA ME210229 (LW), Department of Defense CDMRP W81XWH-21-LCRP-IITRA LC210336 (LW), American Cancer Society RSG-18-045-01-LIB (LW).
LW is an inventor involved with the commercial development of VISTA with ImmuNext Inc Corporation Lebanon, NH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ICR, Immune checkpoint receptor; ICI, Immune checkpoint inhibitor; CTL, Cytotoxic T cell; APC, Antigen presenting cell; MDSC, Myeloid-derived suppressor cell; TCR, T cell receptor; MHCII, Major histocompatibility complex II; TILs, Tumor infiltrating lymphocytes; PD-1, Programmed death-1; CTLA-4, Cytotoxic T lymphocyte-associated protein 4; VISTA, V domain immunoglobulin suppressor of T cell activation; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM-3, T-cell immunoglobulin and mucin domain-containing protein 3; LAG-3, Lymphocyte activation gene 3.
1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
2. Philip M, Schietinger A. CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol (2022) 22:209–23. doi: 10.1038/s41577-021-00574-3
3. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
4. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
5. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv (2020) 6(38):eabd2712. doi: 10.1126/sciadv.abd2712
6. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
7. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33.
8. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
9. Zhang Y, Song Q, Cassady K, Lee M, Tang H, Zheng M, et al. Blockade of trans PD-L1 interaction with CD80 augments antitumor immunity. Proc Natl Acad Sci U.S.A. (2023) 120:e2205085120. doi: 10.1073/pnas.2205085120
10. Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol Res (2018) 6:921–9. doi: 10.1158/2326-6066.CIR-17-0316
11. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity (2019) 51:1059–73 e9. doi: 10.1016/j.immuni.2019.11.003
12. Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science (2019) 364:558–66. doi: 10.1126/science.aav7062
13. Sugiura D, Okazaki IM, Maeda TK, Maruhashi T, Shimizu K, Arakaki R, et al. PD-1 agonism by anti-CD80 inhibits T cell activation and alleviates autoimmunity. Nat Immunol (2022) 23:399–410. doi: 10.1038/s41590-021-01125-7
14. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
15. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863
16. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
17. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U.S.A. (2019) 116:9999–10008. doi: 10.1073/pnas.1822001116
18. Sun L, Zhang L, Yu J, Zhang Y, Pang X, Ma C, et al. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: a systematic review and meta-analysis. Sci Rep (2020) 10:2083. doi: 10.1038/s41598-020-58674-4
19. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer (2022) 21:28. doi: 10.1186/s12943-021-01489-2
20. Karasarides M, Cogdill AP, Robbins PB, Bowden M, Burton EM, Butterfield LH, et al. Hallmarks of resistance to immune-checkpoint inhibitors. Cancer Immunol Res (2022) 10:372–83. doi: 10.1158/2326-6066.CIR-20-0586
21. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2022) 185:576. doi: 10.1016/j.cell.2022.01.008
22. Vafaei S, Zekiy AO, Khanamir RA, Zaman BA, Ghayourvahdat A, Azimizonuzi H, et al. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int (2022) 22:2. doi: 10.1186/s12935-021-02407-8
23. Lu L, Zhan M, Li XY, Zhang H, Dauphars DJ, Jiang J, et al. Clinically approved combination immunotherapy: Current status, limitations, and future perspective. Curr Res Immunol (2022) 3:118–27. doi: 10.1016/j.crimmu.2022.05.003
24. Dariavach P, Mattéi MGG, Golstein P, Lefranc MPP. Human Ig superfamily CTLA-4 gene: chromosomal localization and identity of protein sequence between murine and human CTLA-4 cytoplasmic domains. Eur J Immunol (1988) 18(12):1901–5. doi: 10.1002/eji.1830181206
25. Ramagopal UA, Liu W, Garrett-Thomson SC, Bonanno JB, Yan Q, Srinivasan M, et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc Natl Acad Sci United States America (2017) 114(21):E4223–32. doi: 10.1073/pnas.1617941114
26. Jago CB, Yates J, Saraiva Câmara NO, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol (2004) 136(3):463–71. doi: 10.1111/j.1365-2249.2004.02478.x
27. Linsley PS, Greene JAL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med (1992) 176(6):1595–604. doi: 10.1084/jem.176.6.1595
28. Oyewole-Said D, Konduri V, Vazquez-Perez J, Weldon SA, Levitt JM, Decker WK. Beyond T-cells: functional characterization of CTLA-4 expression in immune and non-immune cell types. Front Immunol (2020) 11:608024. doi: 10.3389/fimmu.2020.608024
29. Sobhani N, Tardiel-Cyril DR, Davtyan A, Generali D, Roudi R, Li Y. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers (2021) 13(6):1440. doi: 10.20944/preprints202102.0150.v1
30. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192(2):303–10. doi: 10.1084/jem.192.2.303
31. Wang XB, Zheng CY, Giscombe R, Lefvert AK. Regulation of surface and intracellular expression of CTLA-4 on human peripheral T cells. Scandinavian J Immunol (2001) 54(5):453–8. doi: 10.1046/j.1365-3083.2001.00985.x
32. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126:375–87. doi: 10.1016/j.cell.2006.05.042
33. Fayyad-Kazan H, Rouas R, Fayyad-Kazan M, Badran R, El Zein N, Lewalle P, et al. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J Biol Chem (2012) 287:9910–22. doi: 10.1074/jbc.M111.337154
34. Vaddi PK, Osborne DG, Nicklawsky A, Williams NK, Menon DR, Smith D, et al. CTLA4 mRNA is downregulated by miR-155 in regulatory T cells, and reduced blood CTLA4 levels are associated with poor prognosis in metastatic melanoma patients. Front Immunol (2023) 14:1173035. doi: 10.3389/fimmu.2023.1173035
35. Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol (Baltimore Md 1950) (1996) 157(11):4762–70. doi: 10.4049/jimmunol.157.11.4762
36. Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol (Baltimore Md 1950) (1996) 156(11):4154–9. doi: 10.4049/jimmunol.156.11.4154
37. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-X
38. Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem (2012) 287:9429–40. doi: 10.1074/jbc.M111.304329
39. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T ceils to stimulation. J Exp Med (1995) 182(2):459–65.
40. Lenschow DJ, Su GHT, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, et al. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci United States America (1993) 90:11054–8. doi: 10.1073/pnas.90.23.11054
41. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA4 is a second receptor for the b cell activation antigen B7. J Exp Med (1991) 174:561–9. doi: 10.1084/jem.174.3.561
42. Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural analysis of CTLA-4 function in vivo. J Immunol (2000) 164:5319–7. doi: 10.4049/jimmunol.164.10.5319
43. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
44. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3:1097–101. doi: 10.1038/ni846
45. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi: 10.1126/science.1131078
46. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
47. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. In: American journal of clinical oncology: cancer clinical trials. (2016). p. 98–106.
48. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev (2008). doi: 10.1111/j.1600-065X.2008.00662.x
49. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271:9543–53. doi: 10.1126/science.271.5256.1734
50. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206:1717–25. doi: 10.1084/jem.20082492
51. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi: 10.1084/jem.20130579
52. Wei SC, Anang NAAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci United States America (2019) 116:22699–709. doi: 10.1073/pnas.1821218116
53. Crosby EJ, Wei J, Yang XY, Lei G, Wang T, Liu CX, et al. Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors. OncoImmunology (2018) 7. doi: 10.1080/2162402X.2017.1421891
54. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci United States America (2010) 107:(9):4275–80. doi: 10.1073/pnas.0915174107
55. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
56. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
57. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol (2015) 4:385. doi: 10.3389/fonc.2014.00385
58. Lipson EJ, Drake CG. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res (2011), 6958–62. doi: 10.1158/1078-0432.CCR-11-1595
59. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. New Engl J Med (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
60. Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. New Engl J Med (2017) 377:1824–35. doi: 10.1056/NEJMoa1709030
61. Callahan MK, Kluger H, Postow MA, Segal NH, Lesokhin A, Atkins MB, et al. Nivolumab plus ipilimumab in patients with advanced melanoma: updated survival, response, and safety data in a phase I dose-escalation study. J Clin Oncol (2018) 36:391–8. doi: 10.1200/JCO.2017.72.2850
62. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
63. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol (2018) 19(11):1480–92. doi: 10.1016/S1470-2045(18)30700-9
64. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. New Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
65. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol (2022) 40(2):127–37. doi: 10.1200/JCO.21.02229
66. Doki Y, Ajani JA, Kato K, Xu J, Wyrwicz L, Motoyama S, et al. Nivolumab combination therapy in advanced esophageal squamous-cell carcinoma. New Engl J Med (2022) 386(5):449–62. doi: 10.1056/NEJMoa2111380
67. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. New Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
68. Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol (2021) 22(2):198–211. doi: 10.1016/S1470-2045(20)30641-0
69. Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med (2020) 26:566–76. doi: 10.1038/s41591-020-0805-8
70. Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol (2019) 20(7):948–60. doi: 10.1016/S1470-2045(19)30151-2
71. Boyer M, Sendur MAN, Rodriguez-Abreu D, Park K, Lee DH, Cicin I, et al. Pembrolizumab plus ipilimumab or placebo for metastatic non-small-cell lung cancer with PD-L1 tumor proportion score >/= 50%: randomized, double-blind phase III KEYNOTE-598 study. J Clin Oncol (2021) 39:2327–38. doi: 10.1200/JCO.20.03579
72. Carlino MS, Atkinson V, Cebon JS, Jameson MB, Fitzharris BM, McNeil CM, et al. KEYNOTE-029: Efficacy and safety of pembrolizumab (pembro) plus ipilimumab (ipi) for advanced melanoma. J Clin Oncol (2017) 35:9545–. doi: 10.1200/JCO.2017.35.15_suppl.9545
73. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
74. Xu W, Hieu T, Malarkannan S, Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol (2018) 15:438–46. doi: 10.1038/cmi.2017.148
75. Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res (2014) 2:510–7. doi: 10.1158/2326-6066.CIR-14-0072
76. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
77. Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci U.S.A. (2014) 111:14846–51. doi: 10.1073/pnas.1407447111
78. Han X, Vesely MD, Yang W, Sanmamed MF, Badri T, Alawa J, et al. PD-1H (VISTA)-mediated suppression of autoimmunity in systemic and cutaneous lupus erythematosus. Sci Transl Med (2019) 11:1–14. doi: 10.1126/scitranslmed.aax1159
79. Mehta N, Maddineni S, Mathews II, Andres Parra Sperberg R, Huang PS, Cochran JR. Structure and functional binding epitope of V-domain ig suppressor of T cell activation. Cell Rep (2019) 28:2509–16 e5. doi: 10.1016/j.celrep.2019.07.073
80. Slater BT, Han X, Chen L, Xiong Y. Structural insight into T cell coinhibition by PD-1H (VISTA). Proc Natl Acad Sci United States America (2020) 117:1648–57. doi: 10.1073/pnas.1908711117
81. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science (2015) 349(6247):1261669. doi: 10.1126/science.1261669
82. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res (2019) 7:1079–90. doi: 10.1158/2326-6066.CIR-18-0507
83. Mo J, Deng L, Peng K, Ouyang S, Ding W, Lou L, et al. Targeting STAT3-VISTA axis to suppress tumor aggression and burden in acute myeloid leukemia. J Hematol Oncol (2023) 16:15. doi: 10.1186/s13045-023-01410-y
84. Schlichtner S, Yasinska IM, Ruggiero S, Berger SM, Aliu N, Prunk M, et al. Expression of the immune checkpoint protein VISTA is differentially regulated by the TGF-beta1 - smad3 signaling pathway in rapidly proliferating human cells and T lymphocytes. Front Med (Lausanne) (2022) 9:790995. doi: 10.3389/fmed.2022.790995
85. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
86. Muller S, Victoria Lai W, Adusumilli PS, Desmeules P, Frosina D, Jungbluth A, et al. V-domain Ig-containing suppressor of T-cell activation (VISTA), a potentially targetable immune checkpoint molecule, is highly expressed in epithelioid Malignant pleural mesothelioma. Mod Pathol (2020) 33:303–11. doi: 10.1038/s41379-019-0364-z
87. Boger C, Behrens HM, Kruger S, Rocken C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology (2017) 6:e1293215. doi: 10.1080/2162402X.2017.1293215
88. Dufva O, Polonen P, Bruck O, Keranen MAI, Klievink J, Mehtonen J, et al. Immunogenomic landscape of hematological Malignancies. Cancer Cell (2020) 38:424–8. doi: 10.1016/j.ccell.2020.08.019
89. Pagliuca S, Gurnari C, Zhang K, Kewan T, Bahaj W, Mori M, et al. Comprehensive transcriptomic analysis of VISTA in acute myeloid leukemia: insights into its prognostic value. Int J Mol Sci (2022) 23(23):14885. doi: 10.3390/ijms232314885
90. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med (2017) 23:551–5. doi: 10.1038/nm.4308
91. He HX, Gao Y, Fu JC, Zhou QH, Wang XX, Bai B, et al. VISTA and PD-L1 synergistically predict poor prognosis in patients with extranodal natural killer/T-cell lymphoma. Oncoimmunology (2021) 10:1907059. doi: 10.1080/2162402X.2021.1907059
92. Seo WI, Lee CH, Jung SJ, Lee DS, Park HY, Jeong DH, et al. Expression of VISTA on tumor-infiltrating immune cells correlated with short intravesical recurrence in non-muscle-invasive bladder cancer. Cancer Immunol Immunother (2021) 70:3113–22. doi: 10.1007/s00262-021-02906-7
93. Kuklinski LF, Yan S, Li Z, Fisher JL, Cheng C, Noelle RJ, et al. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol Immunother (2018) 67:1113–21. doi: 10.1007/s00262-018-2169-1
94. Xie X, Zhang J, Shi Z, Liu W, Hu X, Qie C, et al. The expression pattern and clinical significance of the immune checkpoint regulator VISTA in human breast cancer. Front Immunol (2020) 11:563044. doi: 10.3389/fimmu.2020.563044
95. Choi JW, Kim YJ, Yun KA, Won CH, Lee MW, Choi JH, et al. The prognostic significance of VISTA and CD33-positive myeloid cells in cutaneous melanoma and their relationship with PD-1 expression. Sci Rep (2020) 10:14372. doi: 10.1038/s41598-020-71216-2
96. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest (2014) 124:1966–75. doi: 10.1172/JCI74589
97. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature (2019) 574:565–70. doi: 10.1038/s41586-019-1674-5
98. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology (2019) 156:74–85. doi: 10.1111/imm.13001
99. Yasinska IM, Meyer NH, Schlichtner S, Hussain R, Siligardi G, Casely-Hayford M, et al. Ligand-receptor interactions of galectin-9 and VISTA suppress human T lymphocyte cytotoxic activity. Front Immunol (2020) 11:580557. doi: 10.3389/fimmu.2020.580557
100. Li N, Xu W, Yuan Y, Ayithan N, Imai Y, Wu X, et al. Immune-checkpoint protein VISTA critically regulates the IL-23/IL-17 inflammatory axis. Sci Rep (2017) 7:1485. doi: 10.1038/s41598-017-01411-1
101. Green KA, Wang L, Noelle RJ, Green WR. Selective involvement of the checkpoint regulator VISTA in suppression of B-cell, but not T-cell, responsiveness by monocytic myeloid-derived suppressor cells from mice infected with an immunodeficiency-causing retrovirus. J Virol (2015) 89:9693–8. doi: 10.1128/JVI.00888-15
102. Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell-mediated inflammation and immunosuppression. Cancer Immunol Res (2019) 7:1497–510. doi: 10.1158/2326-6066.CIR-18-0489
103. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci United States America (2015) 112:6682–7. doi: 10.1073/pnas.1420370112
104. Tagliamento M, Agostinetto E, Borea R, Brandão M, Poggio F, Addeo A, et al. VISTA: A promising target for cancer immunotherapy? ImmunoTargets Ther (2021) 10:185–200. doi: 10.2147/ITT.S260429
105. Hernandez-Martinez J-M, Vergara E, Zatarain-Barrón ZL, Barrón-Barrón F, Arrieta O. VISTA/PD-1H: a potential target for non-small cell lung cancer immunotherapy. J Thorac Dis (2018) 10:6378–82. doi: 10.21037/jtd.2018.11.39
106. ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I, et al. VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science (2020) 367(6475):eaay0524. doi: 10.1126/science.aay0524
107. Schaafsma E, Croteau W, ElTanbouly M, Nowak EC, Smits NC, Deng J, et al. VISTA targeting of T-cell quiescence and myeloid suppression overcomes adaptive resistance. Cancer Immunol Res (2023) 11:38–55. doi: 10.1158/2326-6066.CIR-22-0116
108. Musielak B, Kocik J, Skalniak L, Magiera-Mularz K, Sala D, Czub M, et al. CA-170 - A potent small-molecule PD-L1 inhibitor or not? Molecules (Basel Switzerland) (2019) 24:E2804. doi: 10.3390/molecules24152804
109. Sasikumar PG, Sudarshan NS, Adurthi S, Ramachandra RK, Samiulla DS, Lakshminarasimhan A, et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun Biol (2021) 4:699. doi: 10.1038/s42003-021-02191-1
110. Tagliamento M, Bironzo P, Novello S. New emerging targets in cancer immunotherapy: the role of VISTA. ESMO Open (2020) 4(Suppl 3):e000683. doi: 10.1136/esmoopen-2020-000683
111. Thakkar D, Paliwal S, Dharmadhikari B, Guan S, Liu L, Kar S, et al. Rationally targeted anti-VISTA antibody that blockades the C-C' loop region can reverse VISTA immune suppression and remodel the immune microenvironment to potently inhibit tumor growth in an Fc independent manner. J Immunother Cancer (2022) 10(2):e003382. doi: 10.1136/jitc-2021-003382
112. Johnston Robert J, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
113. Wen J, Mao X, Cheng Q, Liu Z, Liu F. A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci Rep (2021) 11(1):22502. doi: 10.1038/s41598-021-01933-9
114. Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer (2022) 10(4):e004711. doi: 10.1136/jitc-2022-004711
115. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
116. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U.S.A. (2012) 109:5399–404. doi: 10.1073/pnas.1120606109
117. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ (2013) 20:456–64. doi: 10.1038/cdd.2012.141
118. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
119. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
120. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest (2015) 125:4053–62. doi: 10.1172/JCI81187
121. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
122. Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight (2018) 3(14):e121157. doi: 10.1172/jci.insight.121157
123. Yu X, Harden K C, Gonzalez L, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
124. Brauneck F, Fischer B, Witt M, Muschhammer J, Oelrich J, da Costa Avelar PH, et al. TIGIT blockade repolarizes AML-associated TIGIT(+) M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J Immunother Cancer (2022) 10(12):e004794. doi: 10.1136/jitc-2022-004794
125. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity (2022) 55:512–26.e9. doi: 10.1016/j.immuni.2022.02.005
126. Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. TIGIT and PD1 Co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2021) 12:443–64. doi: 10.1016/j.jcmgh.2021.03.003
127. Martinez M, Kim S, St Jean N, O'Brien S, Lian L, Sun J, et al. Addition of anti-TIM3 or anti-TIGIT Antibodies to anti-PD1 Blockade Augments Human T cell Adoptive Cell Transfer. Oncoimmunology (2021) 10:1873607. doi: 10.1080/2162402X.2021.1873607
128. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol (2022) 23:781–92. doi: 10.1016/S1470-2045(22)00226-1
129. Recondo G, Mezquita L. Tiragolumab and atezolizumab in patients with PD-L1 positive non-small-cell lung cancer. Lancet Oncol (2022) 23:695–7. doi: 10.1016/S1470-2045(22)00261-3
130. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann Oncol (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
131. Rasco DW, Vaena DA, Fleming GF, Dumbrava EE, Yeku OO, Sharma M, et al. Preliminary antitumor activity of the combination of COM701 + BMS-986207 + nivolumab in patients with recurrent, metastatic MSS endometrial cancer. J Clin Oncol (2023) 41:5595–. doi: 10.1200/JCO.2023.41.16_suppl.5595
132. Shirasuna K, Koelsch G, Seidel-Dugan C, Salmeron A, Steiner P, Winston WM, et al. Characterization of ASP8374, a fully-human, antagonistic anti-TIGIT monoclonal antibody. Cancer Treat Res Commun (2021) 28:100433. doi: 10.1016/j.ctarc.2021.100433
133. Johnson ML, Fox W, Lee Y-G, Lee KH, Ahn HK, Kim Y-C, et al. ARC-7: Randomized phase 2 study of domvanalimab + zimberelimab ± etrumadenant versus zimberelimab in first-line, metastatic, PD-L1-high non-small cell lung cancer (NSCLC). J Clin Oncol (2022) 40:397600–. doi: 10.1200/JCO.2022.40.36_suppl.397600
134. Frentzas S, Meniawy T, Kao SC-H, Wang R, Zuo Y, Zheng H, et al. AdvanTIG-105: Phase 1 dose-escalation study of anti-TIGIT monoclonal antibody ociperlimab (BGB-A1217) in combination with tislelizumab in patients with advanced solid tumors. J Clin Oncol (2021) 39:2583–. doi: 10.1200/JCO.2021.39.15_suppl.2583
135. McKean M, Dumbrava EE, Hamid O, Merriam P, Mettu NB, Call JA, et al. Safety and efficacy of etigilimab in combination with nivolumab in select recurrent/advanced solid tumors. J Clin Oncol (2022) 40:2651–. doi: 10.1200/JCO.2022.40.16_suppl.2651
136. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20:173–85. doi: 10.1038/s41577-019-0224-6
137. Moller-Hackbarth K, Dewitz C, Schweigert O, Trad A, Garbers C, Rose-John S, et al. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3). J Biol Chem (2013) 288:34529–44. doi: 10.1074/jbc.M113.488478
138. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
139. Mujib S, Jones RB, Lo C, Aidarus N, Clayton K, Sakhdari A, et al. Antigen-independent induction of Tim-3 expression on human T cells by the common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 is associated with proliferation and is dependent on the phosphoinositide 3-kinase pathway. J Immunol (2012) 188:3745–56. doi: 10.4049/jimmunol.1102609
140. Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G, et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat Commun (2015) 6:6072. doi: 10.1038/ncomms7072
141. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
142. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PloS One (2013) 8:e58006. doi: 10.1371/journal.pone.0058006
143. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology (2012) 56:1342–51. doi: 10.1002/hep.25777
144. Zang K, Hui L, Wang M, Huang Y, Zhu X, Yao B. TIM-3 as a prognostic marker and a potential immunotherapy target in human Malignant tumors: A meta-analysis and bioinformatics validation. Front Oncol (2021) 11:579351. doi: 10.3389/fonc.2021.579351
145. Cao Y, Zhou X, Huang X, Li Q, Gao L, Jiang L, et al. Tim-3 expression in cervical cancer promotes tumor metastasis. PloS One (2013) 8:e53834. doi: 10.1371/journal.pone.0053834
146. Piao YR, Piao LZ, Zhu LH, Jin ZH, Dong XZ. Prognostic value of T cell immunoglobulin mucin-3 in prostate cancer. Asian Pac J Cancer Prev (2013) 14:3897–901. doi: 10.7314/APJCP.2013.14.6.3897
147. Yuan J, Jiang B, Zhao H, Huang Q. Prognostic implication of TIM-3 in clear cell renal cell carcinoma. Neoplasma (2014) 61:35–40. doi: 10.4149/neo_2014_006
148. Jiang J, Jin MS, Kong F, Cao D, Ma HX, Jia Z, et al. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PloS One (2013) 8(12):e81799. doi: 10.1371/journal.pone.0081799
149. Wang P, Chen Y, Long Q, Li Q, Tian J, Liu T, et al. Increased coexpression of PD-L1 and TIM3/TIGIT is associated with poor overall survival of patients with esophageal squamous cell carcinoma. J Immunother Cancer (2021) 9(10):e002836. doi: 10.1136/jitc-2021-002836
150. Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, et al. T cell ig and mucin domain–containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol (2014) 192:782–91. doi: 10.4049/jimmunol.1302663
151. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of tim-3 to T-cell receptor signaling pathways. Mol Cell Biol (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
152. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med (2012) 18:1394–400. doi: 10.1038/nm.2871
153. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun (2006) 351:571–6. doi: 10.1016/j.bbrc.2006.10.079
154. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi: 10.1038/nature13848
155. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi: 10.1038/ni1271
156. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep (2015) 5:15659. doi: 10.1038/srep15659
157. Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep (2021) 36:109699. doi: 10.1016/j.celrep.2021.109699
158. Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton's tyrosine kinase and c-Src. J Immunol (2014) 193:3417–25. doi: 10.4049/jimmunol.1400395
159. Jiang X, Zhou T, Xiao Y, Yu J, Dou S, Chen G, et al. Tim-3 promotes tumor-promoting M2 macrophage polarization by binding to STAT1 and suppressing the STAT1-miR-155 signaling axis. Oncoimmunology (2016) 5:e1211219. doi: 10.1080/2162402X.2016.1211219
160. Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP, et al. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol (2012) 91:189–96. doi: 10.1189/jlb.1010591
161. de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell (2018) 3360–74:e6. doi: 10.1016/j.ccell.2017.11.019
162. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi: 10.1084/jem.20100643
163. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ–mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096
164. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun (2021) 12:832. doi: 10.1038/s41467-021-21099-2
165. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi: 10.1084/jem.20100637
166. Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, et al. Targeting PD-1 and tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J Immunother (2016) 39:171–80. doi: 10.1097/CJI.0000000000000122
167. Ye X, Wang L, Nie M, Wang Y, Dong S, Ren W, et al. A single-cell atlas of diffuse large B cell lymphoma. Cell Rep (2022) 39:110713. doi: 10.1016/j.celrep.2022.110713
168. Roussel M, Le KS, Granier C, Llamas Gutierrez F, Foucher E, Le Gallou S, et al. Functional characterization of PD1+TIM3+ tumor-infiltrating T cells in DLBCL and effects of PD1 or TIM3 blockade. Blood Adv (2021) 5:1816–29. doi: 10.1182/bloodadvances.2020003080
169. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer (2020) 8(1):e000911. doi: 10.1136/jitc-2020-000911
170. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
171. Falchook GS, Ribas A, Davar D, Eroglu Z, Wang JS, Luke JJ, et al. Phase 1 trial of TIM-3 inhibitor cobolimab monotherapy and in combination with PD-1 inhibitors nivolumab or dostarlimab (AMBER). J Clin Oncol (2022) 40:2504–. doi: 10.1200/JCO.2022.40.16_suppl.2504
172. Acoba JD, Rho Y, Fukaya E. Phase II study of cobolimab in combination with dostarlimab for the treatment of advanced hepatocellular carcinoma. J Clin Oncol (2023) 41:580–. doi: 10.1200/JCO.2023.41.4_suppl.580
173. Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: A phase ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res (2021) 27:2168–78. doi: 10.1158/1078-0432.CCR-20-4405
174. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
175. Andrews LP, Cillo AR, Karapetyan L, Kirkwood JM, Workman CJ, Vignali DAA. Molecular pathways and mechanisms of LAG3 in cancer therapy. Clin Cancer Res (2022) 28:5030–9. doi: 10.1158/1078-0432.CCR-21-2390
176. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol (2002) 169:5392–5. doi: 10.4049/jimmunol.169.10.5392
177. Bruniquel D, Borie N, Hannier S, Triebel F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics (1998) 48:116–24. doi: 10.1007/s002510050411
178. Sun H, Sun C, Xiao W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine (2014) 65:33–41. doi: 10.1016/j.cyto.2013.09.016
179. Somasundaram A, Cillo AR, Lampenfeld C, Workman CJ, Kunning S, Oliveri L, et al. Systemic immune dysfunction in cancer patients driven by IL6 induction of LAG3 in peripheral CD8+ T cells. Cancer Immunol Res (2022) 10:885–99. doi: 10.1158/2326-6066.CIR-20-0736
180. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature (2019) 571:211–8. doi: 10.1038/s41586-019-1325-x
181. Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature (2019) 567:525–9. doi: 10.1038/s41586-019-0979-8
182. Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature (2019) 567:530–4. doi: 10.1038/s41586-019-0985-x
183. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity (2015) 42:265–78. doi: 10.1016/j.immuni.2015.01.006
184. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U.S.A. (2019) 116:12410–5. doi: 10.1073/pnas.1905675116
185. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571:270–4. doi: 10.1038/s41586-019-1324-y
186. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol (2011) 12:663–71. doi: 10.1038/ni.2046
187. Burugu S, Gao D, Leung S, Chia SK, Nielsen TO. LAG-3+ tumor infiltrating lymphocytes in breast cancer: clinical correlates and association with PD-1/PD-L1+ tumors. Ann Oncol (2017) 28:2977–84. doi: 10.1093/annonc/mdx557
188. Zelba H, Bedke J, Hennenlotter J, Mostbock S, Zettl M, Zichner T, et al. PD-1 and LAG-3 dominate checkpoint receptor-mediated T-cell inhibition in renal cell carcinoma. Cancer Immunol Res (2019) 7:1891–9. doi: 10.1158/2326-6066.CIR-19-0146
189. Huuhtanen J, Kasanen H, Peltola K, Lonnberg T, Glumoff V, Bruck O, et al. Single-cell characterization of anti-LAG-3 and anti-PD-1 combination treatment in patients with melanoma. J Clin Invest (2023) 133(6):e164809. doi: 10.1172/JCI164809
190. Thommen DS, Schreiner J, Muller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res (2015) 3:1344–55. doi: 10.1158/2326-6066.CIR-15-0097
191. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
192. Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett (2013) 150:116–22. doi: 10.1016/j.imlet.2012.12.004
193. Cheung CCL, Seah YHJ, Fang J, Orpilla NHC, Lau MC, Lim CJ, et al. Immunohistochemical scoring of LAG-3 in conjunction with CD8 in the tumor microenvironment predicts response to immunotherapy in hepatocellular carcinoma. Front Immunol (2023) 14:1150985. doi: 10.3389/fimmu.2023.1150985
194. Keane C, Law SC, Gould C, Birch S, Sabdia MB, Merida de Long L, et al. LAG3: a novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv (2020) 4:1367–77. doi: 10.1182/bloodadvances.2019001390
195. Shen R, Postow MA, Adamow M, Arora A, Hannum M, Maher C, et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci Transl Med (2021) 13(608):eabf5107. doi: 10.1126/scitranslmed.abf5107
196. Andrews LP, Somasundaram A, Moskovitz JM, Szymczak-Workman AL, Liu C, Cillo AR, et al. Resistance to PD1 blockade in the absence of metalloprotease-mediated LAG3 shedding. Sci Immunol (2020) 5(49):eabc2728. doi: 10.1126/sciimmunol.abc2728
197. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med (1992) 176:327–37. doi: 10.1084/jem.176.2.327
198. Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol (2018) 19:1415–26. doi: 10.1038/s41590-018-0217-9
199. Maruhashi T, Sugiura D, Okazaki IM, Shimizu K, Maeda TK, Ikubo J, et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity (2022) 55:912–24 e8. doi: 10.1016/j.immuni.2022.03.013
200. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176:334–47 e12. doi: 10.1016/j.cell.2018.11.010
201. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
202. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
203. Anderson Ana C, Joller N, Kuchroo Vijay K. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
204. Guy C, Mitrea DM, Chou PC, Temirov J, Vignali KM, Liu X, et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat Immunol (2022) 23:757–67. doi: 10.18632/oncotarget.4751
205. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6:27359–77.
206. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
207. Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood (2003) 102:2130–7. doi: 10.1182/blood-2003-01-0273
208. Atkinson V, Khattak A, Haydon A, Eastgate M, Roy A, Prithviraj P, et al. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J Immunotherapy Cancer (2020) 8:e001681. doi: 10.1136/jitc-2020-001681
209. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9:176–89. doi: 10.18632/genesandcancer.180
210. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
211. Beyrend G, van der Gracht E, Yilmaz A, van Duikeren S, Camps M, Hollt T, et al. PD-L1 blockade engages tumor-infiltrating lymphocytes to co-express targetable activating and inhibitory receptors. J Immunother Cancer (2019) 7:217. doi: 10.1186/s40425-019-0700-3
212. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci United States America (2010) 107:7875–80. doi: 10.1073/pnas.1003345107
213. Chocarro L, Blanco E, Arasanz H, Fernández-Rubio L, Bocanegra A, Echaide M, et al. Clinical landscape of LAG-3-targeted therapy. Immuno-Oncology Technol (2022) 14:100079. doi: 10.1016/j.iotech.2022.100079
214. Lipson EJ, Tawbi HA-H, SChadendorf D, Ascierto PA, Matamala L, Gutiérrez EC, et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J Clin Oncol (2021) 39:9503–. doi: 10.1200/JCO.2021.39.15_suppl.9503
215. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open (2022) 7:100639. doi: 10.1016/j.esmoop.2022.100639
216. Schoffski P, Tan DSW, Martin M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) +/- anti-PD-1 spartalizumab (PDR001) in patients with advanced Malignancies. J Immunother Cancer (2022) 10(2):e003776. doi: 10.1136/jitc-2021-003776
217. Hamid O, Wang D, Kim TM, Kim S-W, Lakhani NJ, Johnson ML, et al. Clinical activity of fianlimab (REGN3767), a human anti-LAG-3 monoclonal antibody, combined with cemiplimab (anti-PD-1) in patients (pts) with advanced melanoma. J Clin Oncol (2021) 39:9515–. doi: 10.1200/JCO.2021.39.15_suppl.9515
218. Hamid O, Gutierrez M, Mehmi I, Dudzisz-Sledz M, Hoyle PE, Wei W, et al. A phase 1/2 study of retifanlimab (INCMGA00012, Anti–PD-1), INCAGN02385 (Anti–LAG-3), and INCAGN02390 (Anti–TIM-3) combination therapy in patients (Pts) with advanced solid tumors. J Clin Oncol (2023) 41:2599–. doi: 10.1200/JCO.2023.41.16_suppl.2599
219. Johnson ML, Patel MR, Cherry M, Kang Y-K, Yamaguchi K, Oh D-Y, et al. Safety of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb, in patients with advanced solid tumors. J Clin Oncol (2020) 38:3063–. doi: 10.1200/JCO.2020.38.15_suppl.3063
220. Ren Z, Guo Y, Bai Y, Ying J, Meng Z, Chen Z, et al. Tebotelimab, a PD-1/LAG-3 bispecific antibody, in patients with advanced hepatocellular carcinoma who had failed prior targeted therapy and/or immunotherapy: An open-label, single-arm, phase 1/2 dose-escalation and expansion study. J Clin Oncol (2023) 41:578–. doi: 10.1200/JCO.2023.41.4_suppl.578
Keywords: immune checkpoint inhibitors, combinatorial immunotherapies, PD-1, CTLA-4, VISTA, TIGIT, TIM3, LAG3
Citation: Roy D, Gilmour C, Patnaik S and Wang L (2023) Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front. Immunol. 14:1264327. doi: 10.3389/fimmu.2023.1264327
Received: 20 July 2023; Accepted: 26 September 2023;
Published: 19 October 2023.
Edited by:
Cory L. Brooks, California State University, United StatesCopyright © 2023 Roy, Gilmour, Patnaik and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Lily Wang, d2FuZ2w5QGNjZi5vcmc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.