 Alexandra Wallimann
Alexandra Wallimann Mirjam Schenk
Mirjam Schenk- 1Christine Kühne – Center for Allergy Research and Education (CK-CARE), Davos, Switzerland
- 2Institute of Tissue Medicine and Pathology, Experimental Pathology, University of Bern, Bern, Switzerland
IL-32 is a recently described cytokine that performs a variety of functions under inflammatory conditions. Serum IL-32 has been shown to be elevated in several diseases, including type 2 diabetes, cancer, systemic lupus erythematosus, HIV infection, and atopic diseases including atopic dermatitis. There are nine different isoforms of IL-32, with IL-32γ being the most biologically active one. The following review summarizes the different roles of the various IL-32 isoforms in the context of skin inflammation, with a focus on atopic dermatitis.
Take-home messages
● IL-32 levels in skin lesions of atopic dermatitis, hidradenitis suppurativa and alopecia areata are increased compared to healthy skin.
● In atopic dermatitis, IL-32 plays a key role in the interaction between keratinocytes and Langerhans cells and contributes to the inflammatory process, providing a potential therapeutic target.
● Intratumoral IL-32γ promotes an inflammatory tumor microenvironment in melanoma, which is associated with prolonged overall survival and better response to current therapies.
● IL-32 supports T cell mediated host defence in microbial skin infections.
Introduction
Inflammatory skin disorders comprise a wide range of conditions such as atopic dermatitis (AD), psoriasis, hidradenitis suppurativa (HS), alopecia areata (AA), and others. AD is the most prevalent inflammatory skin disease affecting up to 20% of children and 10% of adults (1). AD is characterized by red eczematous patches on the skin and repetitive itch-scratch cycles, leading to a reduced quality of life and an increased risk of developing other atopic diseases such as food allergies, allergic rhinitis, or asthma (2). In addition to an increased risk of developing atopic comorbidities, AD has also been shown to be associated with cardiovascular diseases and mental health comorbidities, including anxiety, depression and attention-deficit hyperactivity disorder (ADHD) (3). Although AD is primarily a childhood disease, it can persist throughout a person’s lifetime and flare up during adulthood. Both, genetic factors, such as Filaggrin mutations, and environmental factors, like urban or rural living environment, pollution, and nutrition, are involved in disease induction and progression. While the basic principles of immune activation in AD are understood, it remains unclear why the disease course is so heterogenous, varying from complete remission to recurrent flares. Several treatment strategies are currently available, including local (topical) treatment with e.g. corticosteroids or calcineurin inhibitors, which can be applied reactively or proactively, systemic treatment with classical immunosuppressants such as cyclosporin, methotrexate or corticosteroids, JAK-inhibitors, or biologicals such as dupilumab and tralokinumab (4). However, treatment responses vary significantly between patients, indicating the existence of different disease endotypes. This underscores the need for a personalized medicine approach to successfully treat AD patients. Novel biomarkers, including cytokines such as IL-32, could become an essential tool for stratifying patients according to disease severity, predicting disease course, and treatment response.
Generally, disruption of the epidermal barrier is considered the origin of AD pathophysiology (5). This disruption can be caused and influenced by genetic mutations of epidermal barrier components, such as the loss-of-function mutation in filaggrin, a dysbalanced skin microbiome, such as a high abundance of Staphylococcus aureus leading to a dysbiosis, and other environmental factors. Consequently, epidermal-derived cytokines, also known as alarmins, including TSLP, IL-33, and IL-25 are secreted, initiating the innate skin-derived immune response. Due to the leaky barrier, microbial and environmental antigens can easily penetrate the epidermis and influence the local immune response. Upon antigen presentation, naïve T cells are primed towards effector Th2 cells. The local immune activation in the skin further develops into systemic immune activation, resulting in a chronic, low-grade inflammatory state and IgE-mediated sensitization. The initial innate immune response plays a pivotal role in the onset of the disease and the progression of local to systemic inflammation. IL-32 is potentially one of the key cytokines in shaping immune activation and inflammation in the early stages of AD development, as it has the capacity to strongly influence dendritic cell (DC) activation (6). Lesional skin of AD patients was found to have increased levels of IL-32 (7). In addition, IL-32 serum levels of AD patients were shown to be increased and correlated with disease severity (7, 8). Treatment with oral cyclosporin for 28 days was found to decrease IL-32 levels in the serum of AD patients, which was accompanied by an alleviation of skin lesions in these patients (9). Besides its role in AD, IL-32 has also been proposed as a biomarker for psoriasis, AA and HS. Therefore, this review focuses on the biological functions of IL-32, particularly in AD and other inflammatory skin disorders (Table 1), and evaluates its potential as a biomarker and therapeutic target in these diseases.
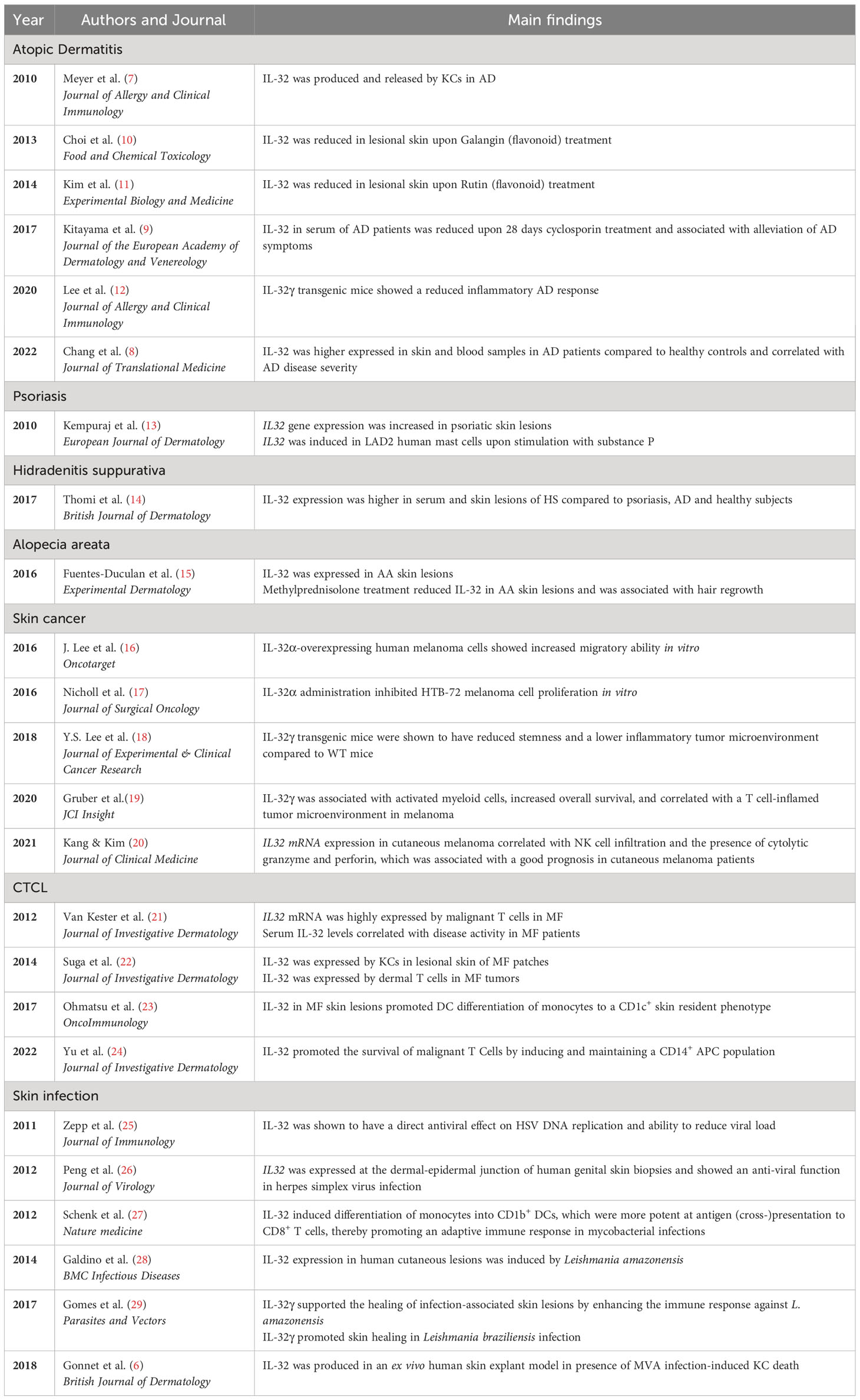
Table 1 Overview of current publications showing roles of IL-32 in skin inflammation.
Biological properties of IL-32
IL-32 is a cytokine initially cloned from human natural killer (NK) cells and its original name was therefore natural killer cell transcript 4 (NK-4) (30). IL-32 is encoded by nine different isoforms through alternative splicing (IL-32α, IL-32β, IL-32γ, IL-32δ, IL-32θ, IL-32ε, IL-32ζ, IL-32η, and IL-32s) (31). The three most extensively studied isoforms are IL-32α, IL-32β and IL-32γ, with IL-32β being the most abundant and IL-32γ being the longest and most biologically active (32, 33). IL-32 induces the production of various cytokines such as TNF-α, IL-8, IL-1β and MIP-2, and exhibits little homology with other cytokines (30). Although the murine orthologue of IL-32 remains unidentified, studies have demonstrated that stimulation of murine macrophages and DCs with human IL-32 leads to activation of similar signalling cascades and biological processes, indicating the existence of the IL-32 receptor and pathway in rodents (19, 27, 30). The role of IL-32 in vivo has been investigated using IL-32 transgenic mice (34–36) and bone marrow transplantation of retroviral constructs containing human IL-32β (37), or application of recombinant human IL-32γ (12, 19).Various immune and non-immune cells including T lymphocytes, NK cells, monocytes/macrophages, DC, and epithelial cells (e.g. keratinocytes) have been shown to express IL-32 (31). A comprehensive comparison showed that lymphocytes and NK cells express the highest levels of IL32, and that IL-32 protein expression was highest in activated effector T cells (19). The presence of a microbial infection, oxidative stress, and pro-inflammatory cytokines (e.g. TNF-α or IL-12) can substantially increase IL-32 expression (31). Depending on the triggering factors, the isoform, and cell type, IL-32 mediates its effect primarily intracellularly, but release of IL-32 by extracellular vesicles of myeloma cells has been described (38). Synovial fibroblasts in rheumatoid arthritis have also been shown to secrete low amounts of IL-32 (39). Furthermore, IL-32 measured in serum or cell culture supernatant suggests the presence of released, extracellular IL-32. To date, the cell surface receptor responsible for binding extracellular IL-32 has not yet been identified. However, extracellular IL-32γ triggers the MAPK/ERK pathway in myeloid cells, thereby inducing their maturation, activation, and chemokine secretion (19). The binding of IL-32α and IL-32β to integrin extracellular domains activates pro-caspase-3 signalling and thus leads to cell death (40). It has also been shown that IL-32 can induce apoptosis in T cells and keratinocytes (KCs) (7, 32).
Furthermore, IL-32 can induce differentiation, maturation and cross-priming in DCs as well as M1 polarization and cytokine/chemokine secretion in macrophages (19, 27). IL-32 can amplify inflammatory cytokines induced by microbial receptors, such as Toll-like receptors (TLR) and intracellular nuclear oligomerization domain (NOD) receptor families and can trigger TLR signalling in the absence of PRR ligands through the protease-activated receptor 2 (PAR2) (41, 42).
Although IL-32 is generally considered pro-inflammatory due to its ability to activate NFκB signalling and p38 MAPKs and enhance tumor immunity, it can exert anti-inflammatory effects under certain circumstances (19, 27, 40). This will be discussed in more detail in the upcoming section, with a focus on skin inflammation. It is noteworthy that the major isoforms of IL-32, namely IL-32α, IL-32β, IL-32γ, and IL-32δ have been detected in both skin and blood samples from individuals with AD, psoriasis, and HS (7, 14).
The role of IL-32 in healthy skin and atopic dermatitis
IL-32γ has been described as key molecular link between KCs and Langerhans cells (LC) under steady-state conditions in healthy skin, where it induces the migration of LC from the epidermis into the dermis before they migrate to the draining lymph node (6). LC and other DC subsets in the skin were shown to express IL-32. The expression of the four main IL-32 isoforms (α, β, γ and δ), particularly IL-32α, was found to be higher in epidermal LCs and dermal CD1adimCD141- DCs compared to dermal CD1adimCD141+ DCs and CD14+ DCs. Furthermore, IL-15 was shown to induce IL-32α expression in these DC subsets. While IL-15 is inducing cytolytic capacity of NK cells, DC-derived IL-32α is antagonizing the IL-15 mediated NK cell effector molecule expression and killing capacity, suggesting a feedback mechanism of IL-32α (43). Importantly, based on expression data of the human protein atlas, T cells appear to be the largest producer of IL-32 in healthy skin (44).
Evidently, IL-32 is upregulated in the skin and serum of AD patients, and serum levels of IL-32 correlate with disease severity, even in patients who respond to treatment. Although IL32 was first identified in activated T and NK cells (45), current literature suggests that KC appear to be an important source of IL-32 in skin inflammation, particularly in AD (6, 7). In addition, IL-32 has been reported to induce apoptosis of KCs. Expression of IL-32 is upregulated in KCs in response to various inflammatory stimuli, such as proinflammatory cytokines, and microbial components (7). Although IL-32 is known to be produced by KCs, it is not actively secreted by them, instead, KC death is required to release IL-32 (Figure 1A). This released IL-32 can promote the activation and migration of immune cells, such as T cells and neutrophils, which further contribute to the inflammatory response in the skin. Since it has been shown that in viral infections of the skin (MVA), KC-released IL-32 can activate LC (6), IL-32 may also play an important role in the activation of LC and the priming of T cells in AD.
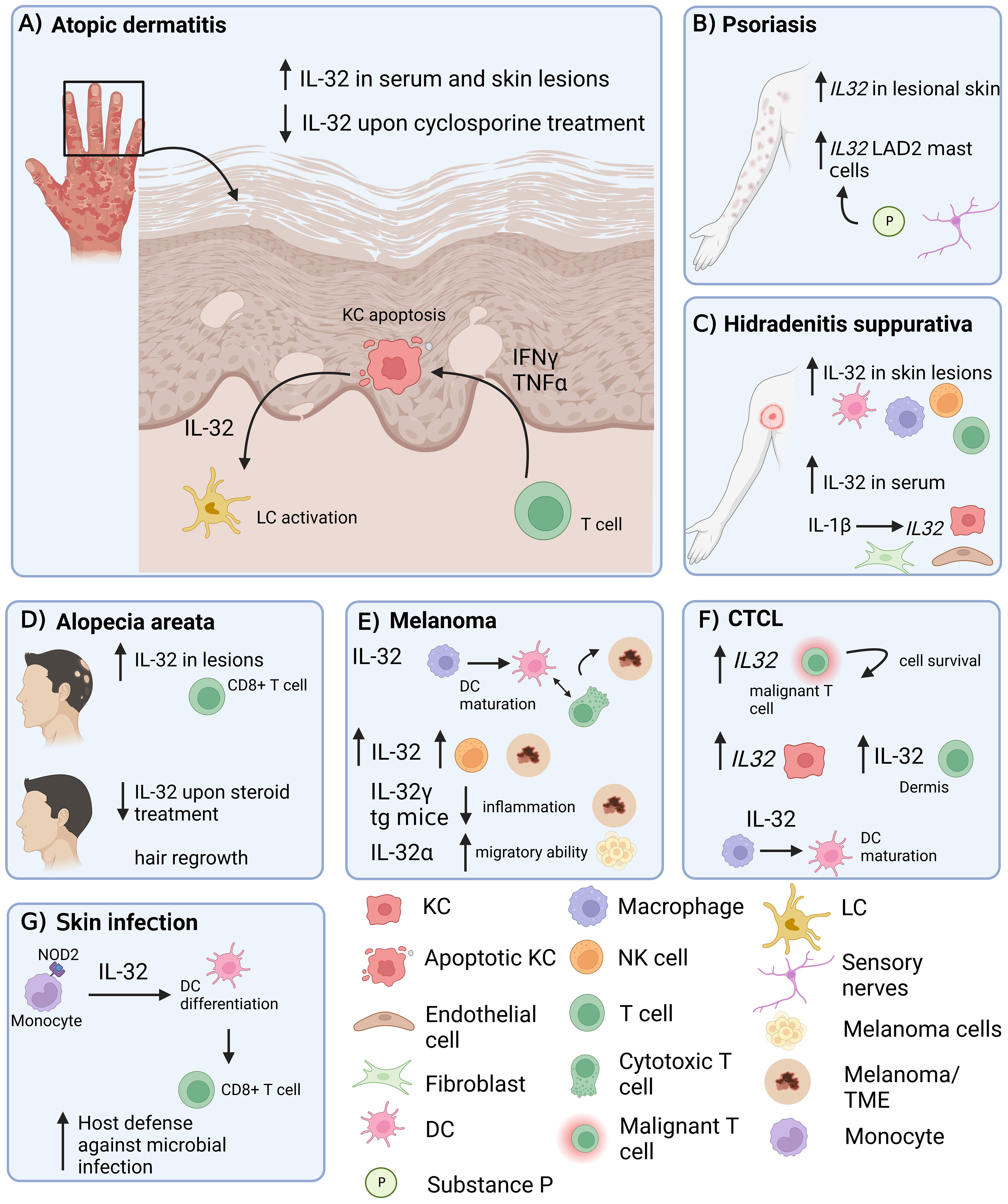
Figure 1 Role of IL-32 in different inflammatory skin diseases. IL-32 has been shown to be expressed in skin lesions in various inflammatory conditions. (A) AD: IL-32 produced by KCs is dependent on T cell-derived IFNγ and TNF-α and drives LC activation. (B) Psoriasis: IL32 gene expression is increased in lesional psoriatic skin. (C) HS: IL-32 is increased in lesional HS skin and in serum. IL-1β induces IL32 expression in KCs, fibroblasts and endothelial cells. (D) IL-32 is elevated in lesional AA skin, produced by CD8+ T cells and decreased upon intra-lesional methylprednisolone treatment. (E) Melanoma: IL-32 is driving DC maturation and supporting a T cell inflamed tumor microenvironment in melanoma. (F) In CTCL, IL32 was expressed by malignant T cells and KCs. IL-32 was driving DC maturation. (G) Skin Infection: IL-32 was driving DC differentiation and antigen cross-presentation to favour T cell mediated host defence in microbial infections. AD, atopic dermatitis; KC, keratinocyte; LC, Langerhans cells; AA, alopecia areata; HS, Hidradenitis suppurativa; DC, dendritic cell; NK cell, natural killer cell; LAD2, laboratory of allergic diseases 2 (human mast cells cell line); CTCL, cutaneous T cell lymphoma; TME, Tumor microenvironment; tg, transgenic. Figure created in biorender.com.
Another study showed that IL-32 promotes AD through JAK-miRNA-155 signalling (8). In this study, the expression of IL-32 in skin and blood samples was higher in AD than in healthy control samples, both in an IL-32 transgenic murine AD model induced by phthalic anhydride (PA) and in human patients. In a DFE/DNCB induced murine dermatitis model, two flavonoids, Galangin and Rutin, were found to suppress IL-32 in lesional skin of these mice, which was associated with reduced clinical scores and lymphocyte proliferation, as well as lower serum IgE levels, highlighting the importance of IL-32 in inflammatory skin lesions (10, 11).
While many studies have emphasized the pro-inflammatory properties of IL-32 and its role in AD progression, Sun Lee et al. found that IL-32γ transgenic mice showed reduced inflammation in PA and MC903 AD models. The reduced manifestations of AD symptoms in IL-32γ transgenic mice was likely a consequence of a lower inflammation in these mice compared to the WT mice, characterized by reduced infiltration of pro-inflammatory immune cells, mast cells, and downregulated Th1, Th2 and Th17 associated cytokines. In contrast, IL-32γ transgenic AD mice showed higher numbers of Foxp3+ Treg cells than WT AD mice (12). These discordant findings could be due to the use of IL-32γ transgenic mice, which could develop an immunosuppressive response due to the continuous expression of IL-32γ. This will be further elaborated in the discussion section of this review.
In summary, IL-32 has pro-inflammatory capabilities and plays a role in the interaction between KCs and LCs in human AD skin. While T cells have been shown to be the main source of IL-32 in other inflammatory conditions, KC-produced IL-32 seems to play and important role in AD. Further studies will elucidate the complex interplay between the cellular sources and targets of IL-32 in AD.
The role of IL-32 in other inflammatory skin conditions: Psoriasis, hidradenitis suppurativa and alopecia areata
In addition to its role in AD, IL-32 has also been shown to be involved in other inflammatory skin diseases such as psoriasis (Figure 1B), HS (Figure 1C) and AA (Figure 1D). Psoriasis is a chronic inflammatory skin disease which is appearing either in separated pustular forms or in chronic plaque forms. IL-17 and IL-23 are key drivers in the pathogenesis of psoriasis and indicate a Th17-dominated inflammation (46). One study showed increased IL32 gene expression in psoriasis lesions compared to healthy controls (13) (Figure 1B). In addition, expression of the IL32 gene in human mast cells (LAD2) was induced upon stimulation with substance P, a mediator released in the skin from sensory nerve endings, indicating potential induction of IL-32 during a neurogenic flare in psoriasis (13). However, IL-32 protein was not detected in the skin of psoriasis patients, only in AD and HS patients (7, 14). A key difference between AD and psoriasis is that KC in psoriasis tend to proliferate rather than undergo apoptosis (47). Since KC apoptosis appears to be required for the release of IL-32 in AD, this would explain the low abundance of IL-32 protein in psoriasis and imply that KC-derived IL-32 may not be involved in the pathogenesis of psoriasis.
Also characterized as a chronic inflammatory skin disease, HS presents with deep-seated nodules, mainly located in the axilla, inguinal and perianal area and sub-mammary folds, that further expand to abscesses (48). Thomi et al. showed that IL-32 protein expression was higher in HS skin lesions as well as in serum compared to skin from AD and psoriasis lesions. Natural killer cells, T cells, macrophages and DCs were found to be the producers of IL-32 in HS skin lesions (Figure 1C). In addition to elevated IL-32 protein levels, total IL32 gene expression was also higher in HS skin. The mRNA expression levels of IL32 positively correlated with the degree of inflammation in HS skin lesions. Although IL32 correlated positively with the numbers of T cells and macrophages, as well as with IFN-γ and IL-17, no correlation was observed with IL-13, an important Th2 cytokine in AD pathogenesis (14). In another study, a highly active IL-1β pathway was detected in HS skin lesions compared to healthy and psoriatic skin, and in a skin explant model, IL-1β was shown to induce IL32 gene expression in endothelial cells, fibroblasts, and KCs (49) (Figure 1C).
AA is an autoimmune inflammatory skin disease characterized by T cell infiltration targeting hair follicles and thus causing patchy hair loss mainly on the scalp (50). It was shown that IL-32 is increased in AA scalp lesions compared to non-lesional regions, and that CD3+ and CD8+ T cells are expressing IL-32. Upon intralesional methylprednisolone treatment, IL-32 expression was decreased and associated with hair regrowth in these regions (Figure 1D). Thus, IL-32 was proposed to be a potential biomarker for disease pathogenesis and hair loss in AA (15). The precise mechanism of how IL-32 promotes the pathogenesis of AA is currently unknown and needs further investigation. Furthermore, it would be interesting to see if blocking IL-32 is sufficient to reduce inflammation and induce hair regrowth.
In summary, IL-32 protein expression seems to be increased in HS and AA lesions compared to healthy skin, whereas only IL32 gene expression is increased in psoriasis. Depending on the inflammatory skin condition, IL-32 appears to originate from different cell types including T cells, KCs, DCs, endothelial cells, and fibroblasts. The use of single cell technologies will help to identify the precise cellular sources and targets of IL-32 in each condition.
IL-32 in skin cancer and cutaneous T-cell lymphoma
Skin cancer is the most common type of cancer worldwide, and its incidence has steadily increased in recent decades. Ultraviolet (UV) exposure is the most important risk factor causing skin cancer by inducing genetic mutations. Three main types of skin cancer exist including basal cell carcinoma, squamous cell carcinoma, and malignant melanoma (51). Different studies have found that IL-32 plays a role in both, the development, and the induction of an immune response in melanoma (Figure 1E) and cutaneous T-cell lymphoma (CTCL, Figure 1F).
IL32 mRNA expression in cutaneous melanoma was correlated with the infiltration of NK cells and the presence of cytolytic granzyme and perforin, which was associated with a good prognosis in cutaneous melanoma patients (20). Another study showed that IL-32γ transgenic mice had reduced carcinogen-induced tumor incidence compared to WT mice, which was associated with a lower inflammatory tumor microenvironment (TME) compared to WT mice. IL-32γ transgenic mice showed reduced gene expression of a variety of pro-inflammatory cytokines including TNF-α, Il-1β, IL-6, as well as Th2-associated IL-4 and IL-13, but increased expression of IL-10 in the skin (18). Eventually, this study showed the ability of IL-32γ to suppress skin carcinogenesis by dampening inflammation within the TME during cancer development. However, Gruber et al. showed that IL-32γ was associated with activated myeloid cells, increased overall survival, and correlated with a T cell-inflamed TME in human melanoma (19). In a murine melanoma model, intratumoral treatment with IL-32γ induced maturation of cross-presenting DC and M1 macrophage polarization. The induced DCs and M1 macrophages act in concert to prime and recruit tumor-specific CD8+ T cells into the TME, leading to an increased number of tumor-specific cytotoxic CD8+ T cells. Therefore, intratumoral IL-32γ is promoting an inflammatory TME, which is associated with increased overall survival and better cancer diagnosis. Intratumoral IL-32γ could function as a novel potent adjuvant in immunotherapy to enhance the efficacy of immune checkpoint blockade.
The effect of IL-32 on melanoma cells has also been studied. While administration of recombinant IL-32γ did not alter the transcriptome of melanoma cells (19) treatment of a human melanoma cell line (HTB-72) with recombinant IL-32α resulted in decreased proliferation of these cells, due to increased expression of the anti-proliferative p21 and p53 in these cells (17). In contrast, IL-32α-overexpressing human melanoma cells showed increased migratory ability in vitro caused by reduced E-cadherin expression favouring F-actin polymerization. The increased migratory capability of IL-32α overexpressing melanoma cells was associated with higher lung metastasis in vivo (16). This discrepancy could be due to the different signalling pathways triggered by intracellularly expressed versus extracellularly administered IL-32α.
CTCL is a type of non-Hodgkin’s lymphoma that affects the skin. It occurs when a mutation or abnormality develops in T-cells, causing them to multiply uncontrollably and accumulate in the skin. The exact cause of CTCL is not fully understood, but it is thought to be related to a combination of genetic and environmental factors. Mycosis fungoides (MF) is the most common type of CTCL, accounting for about 50% of all cases. MF typically starts as a red, scaly rash on the skin, which can be mistaken for other skin conditions such as eczema or psoriasis. Over time, the rash can develop into plaques or tumors and spread to other parts of the body, such as the lymph nodes, blood, and internal organs. The malignant T cells in MF express high and consistent IL32 mRNA and serum IL-32 levels correlated with disease activity in MF patients (21). Consistent with this study, Suga et al. found that IL32 mRNA was higher in lesional skin of MF patches compared to normal skin. KCs were the main producers of IL-32 in the lesional skin of MF patch and plaque, while T cells in the dermis expressed IL-32 in MF tumors (22). Thus, IL-32 might play a role in the formation and persistence of CTCL lesions and could be a potential therapeutic target. On the other hand, IL-32β and IL-32γ have been shown to accelerate the induction of CD1c+ mDCs and CD163+CD68+ macrophages expressing IDO in MF skin lesions and to contribute to a tolerogenic tumor environment (23). In addition, IL-32 was found to be the most abundantly expressed cytokine in MF. Single-cell sequencing revealed that IL32 was mainly expressed by malignant and regulatory T cells, but also by a subset of KCs. Recombinant IL-32β did not affect malignant T cell survival in vitro. However, in the presence of myeloid cells, IL-32β induced and maintained a CD14+ APC population, which was associated with enhanced T cell survival. Thus, IL-32β produced by malignant T cells favours a protective niche for T cell survival in CTCL (24).
IL-32 in skin infections
Amongst its key role in different chronic inflammatory skin conditions, there is evidence that IL-32 is also involved in host defense against infectious pathogens (Figure 1G). IL-32 has been shown to be associated with the control and immunopathology of numerous infectious diseases including tuberculosis, HIV, leprosy, and hepatitis.
In the context of skin infections, IL-32 has been shown to play a supportive role in the healing of infection-related skin lesions (29). Infection with Leishmania amazonensis induced high IL-32 expression which enhanced the immune response against the pathogen and supported healing of skin lesions (28, 29). Similarly, IL-32γ promoted skin healing of skin lesions associated with Leishmania braziliensis infection, indicating its crucial role in controlling parasite load and dissemination (29).
In leprosy, a human mycobacterial infection, IL-32 has been shown to promote the differentiation of monocytes into CD1b+ DCs upon NOD2 activation. These CD1b+ DCs were more efficient in antigen cross-presentation to CD8+ T cells, thereby promoting an adaptive immune response. Accordingly, the expression of NOD2 and IL-32, as well as the number of CD1b+ DCs were higher in tuberculoid than in lepromatous leprosy lesions, indicating a relevant mechanism to control the dissemination of infection (27).
Regarding viral infections, an antiviral function of IL-32 has been demonstrated in herpes simplex virus (HSV) infection, where CD8+ T cells expressed IL32 at the dermal-epidermal junction of human genital skin biopsies (26). IL-32 has previously been shown to have a direct antiviral effect on HSV DNA replication and to reduce viral load (25). In an ex vivo human skin explant model, modified vaccinia virus Ankara (MVA) infection induced KC death, accompanied by production of IL-32 (6). IL-32 induced morphological changes in LCs by shortening dendrites, downregulating adhesion molecules, and inducing release of CXCL10, while upregulating CD80 and HLA-DR, which are hallmarks of LC maturation and migration. Blocking IL-32 with siRNA and an anti-IL-32 antibody significantly inhibited LC activation. Several TLR ligands, including those for TLR-1, 2, 4, and 7 induced IL-32 production by KCs, which subsequently initiated LC activation. This is consistent with the finding that deficiency of MyD88, a universal adaptor molecule for TLRs, in KCs directly impaired LC migration upon antigen exposure (52).
In summary, IL-32 has been shown to support the clearance of viral and bacterial pathogens in the skin, which in turn has been associated with healing of microbial-related skin lesions.
Discussion
In recent years, IL-32 has emerged as a novel pro-inflammatory cytokine that plays a central role in various inflammatory conditions such as rheumatoid arthritis and inflammatory bowel disease, but also in infections and cancer. This review highlights that IL-32, particularly IL-32α, β, and γ, also play a pro-inflammatory role in the pathogenesis of various inflammatory skin disorders. Several studies have shown an association between IL-32 levels and degrees of inflammation in AD, psoriasis, HS, and AA.
Remarkably, studies using IL-32 transgenic mice or IL-32 overexpressing cells in vitro often report an anti-inflammatory effect of IL-32 in various disease models. In AD, IL-32γ transgenic mice showed reduced inflammation compared to WT mice (12). Similarly, IL-32β overexpressing mice showed reduced levels of proinflammatory cytokines TNF-α, IL-6, and IL-1β in a murine model of rheumatoid arthritis, which improved arthritic inflammation in these mice (35). However, exacerbation of collagen-induced arthritis was observed in mice given human IL-32β by bone marrow transplantation (37). Also, application of recombinant IL-32γ in mice resulted in an increase in inflammatory cell infiltration and tissue damage in arthritic mice (53). Furthermore, in the dextran-sodium-sulfate (DSS)-induced colitis model, IL-32γ transgenic mice initially showed a more severe disease course than WT mice. However, after 6 days, the colitis regressed, which was accompanied by an increase in colonic IL-10 levels. (36). Together, these findings indicate that models in which IL-32 is continuously expressed are more likely to elicit immunosuppressive responses in the long-term, which may be a physiological response to the continuous action of IL-32. The use of IL-32 transgenic mice or overexpressing cell lines needs to be carefully evaluated and compared to exogenous administration or bone marrow transplantation of IL-32 to fully elucidate its role.
While KCs have been identified as the major cellular source of IL-32 in AD in an in vitro model with limited cell types (7), a recent paper has shown that IL32 is expressed by COL18A1+ fibroblasts in AD skin (54). Further studies using single cell analysis and spatial resolution will elucidate the precise cellular source and the role of IL-32 during the course of the disease. Inhibition of IL-32 or its deactivation could be a potential approach to reduce skin inflammation. As IL-32 is activated by proteinase-3, Marcondes et al. used the serine protease inhibitor α-1 antitrypsin to suppress IL-32, although α-1 antitrypsin is not a specific inhibitor to IL-32 (55). The suppression of IL-32 by α-1 antitrypsin was associated with reduced T cell proliferation in an allogeneic murine marrow transplantation model. Studies using IL-32 knockdown in human cell lines may also shed light on whether IL-32 can be used as a potential therapeutical target to reduce inflammation.
In general, IL-32 is an interesting target to treat skin inflammation. However, the development of specific IL-32 inhibitors is hampered by the fact that IL-32 can act both intracellularly and extracellularly, that a specific extracellular IL-32 receptor is still elusive, and that the murine orthologue of IL-32 remains unidentified. In addition, the role of the different IL-32 isoforms in skin inflammation remains to be fully elucidated.
In conclusion, IL-32 is elevated locally and systemically in various inflammatory skin disorders and decreases with anti-inflammatory therapies in AD and AA, leading to improvement in disease symptoms. IL-32 could therefore be considered as a novel potential therapeutic target, particularly in AD and AA, to reduce skin inflammation.
Author contributions
AW: Conceptualization, Visualization, Writing – original draft. MS: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
Supported by Christine Kühne -Center for Allergy Research and Education (CK-CARE), Davos, Switzerland.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. (2020) 396(10247):345–60. doi: 10.1016/S0140-6736(20)31286-1
2. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. (2018) 4(1):1. doi: 10.1038/s41572-018-0001-z Atopic dermatitis (2018) 4(1):1. doi: 10.1038/s41572-018-0001-z
3. Paller A, Jaworski JC, Simpson EL, Boguniewicz M, Russell JJ, Block JK, et al. Major comorbidities of atopic dermatitis: beyond allergic disorders. Am J Clin Dermatol (2018) 19:821–38. Springer International Publishing. doi: 10.1007/s40257-018-0383-4
4. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discovery Nat Res (2022) 21:21–40. doi: 10.1038/s41573-021-00266-6
5. Luger T, Amagai M, Dreno B, Dagnelie MA, Liao W, Kabashima K, et al. Atopic dermatitis: Role of the skin barrier, environment, microbiome, and therapeutic agents. J Dermatol Sci (2021) 102 :142–57. doi: 10.1016/j.jdermsci.2021.04.007
6. Gonnet J, Perrin H, Hutton AJ, Boccara D, Bonduelle O, Mimoun M, et al. Interleukin-32 promotes detachment and activation of human Langerhans cells in a human skin explant model. Br J Dermatol (2018) 179(1):145–53. doi: 10.1111/bjd.16721
7. Meyer N, Zimmermann M, Bürgler S, Bassin C, Woehrl S, Moritz K, et al. IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol (2010) 125(4):858–65. doi: 10.1016/j.jaci.2010.01.016
8. Chang J, Zhou B, Wei Z, Luo Y. IL-32 promotes the occurrence of atopic dermatitis by activating the JAK1/microRNA-155 axis. J Transl Med (2022) 20(1). doi: 10.1186/s12967-022-03375-x
9. Kitayama N, Otsuka A, Nonomura Y, Nakashima C, Honda T, Kabashima K. Decrease in serum IL-32 level in patients with atopic dermatitis after cyclosporine treatment. J Eur Acad Dermatol Venereol (2017) 31(10):e449–50. doi: 10.1111/jdv.14274
10. Choi JK, Kim SH. Inhibitory effect of galangin on atopic dermatitis-like skin lesions. Food Chem Toxicol (2014) 68:135–41. doi: 10.1016/j.fct.2014.03.021
11. Choi JK, Kim SH. Rutin suppresses atopic dermatitis and allergic contact dermatitis. Exp Biol Med (2013) 238(4):410–7. doi: 10.1177/1535370213477975
12. Lee YS, Han SB, Ham HJ, Park JH, Lee JS, Hwang DY, et al. IL-32γ suppressed atopic dermatitis through inhibition of miR-205 expression via inactivation of nuclear factor-kappa B. J Allergy Clin Immunol (2020) 146(1):156–68. doi: 10.1016/j.jaci.2019.12.905
13. Kempuraj D, Conti P, Vasiadi M, Alysandratos KD, Tagen M, Kalogeromitros D, et al. IL-32 is increased along with tryptase in lesional psoriatic skin and is up-regulated by substance P in human mast cells. Eur J Dermatol (2010) 20(6):865–7. doi: 0.1684/ejd.2010.1110
14. Thomi R, Yerly D, Yawalkar N, Simon D, Schlapbach C, Hunger RE. Interleukin-32 is highly expressed in lesions of hidradenitis suppurativa. Br J Dermatol (2017) 177(5):1358–66. doi: 10.1111/bjd.15458
15. Fuentes-Duculan J, Gulati N, Bonifacio KM, Kunjravia N, Zheng X, Suárez-Fariñas M, et al. Biomarkers of alopecia areata disease activity and response to corticosteroid treatment. Exp Dermatol (2016) 25(4):282–6. doi: 10.1111/exd.12918
16. Lee J, Kim KE, Cheon S, Song JH, Houh Y, Kim TS, et al. Interleukin-32α induces migration of human melanoma cells through downregulation of E-cadherin. Oncotarget (2016) 7(40):65825–36. doi: 10.18632/oncotarget.11669
17. Nicholl MB, Chen X, Qin C, Bai Q, Zhu Z, Davis MR, et al. IL-32α has differential effects on proliferation and apoptosis of human melanoma cell lines. J Surg Oncol (2016) 113(4):364–9. doi: 10.1002/jso.24142
18. Lee YS, Lee CH, Bae JT, Nam KT, Moon DB, Hwang OK, et al. Inhibition of skin carcinogenesis by suppression of NF-κB dependent ITGAV and TIMP-1 expression in IL-32γ overexpressed condition. J Exp Clin Cancer Res (2018) 37(1):293. doi: 10.1186/s13046-018-0943-8
19. Gruber T, Kremenovic M, Sadozai H, Rombini N, Baeriswyl L, Maibach F, et al. IL-32γ potentiates tumor immunity in melanoma. JCI Insight (2020) 5(18). doi: 10.1172/jci.insight.138772
20. Kang JY, Kim KE. Prognostic value of interleukin-32 expression and its correlation with the infiltration of natural killer cells in cutaneous melanoma. J Clin Med (2021) 10(20):4691. doi: 10.3390/jcm10204691
21. van Kester MS, Borg MK, Zoutman WH, Out-Luiting JJ, Jansen PM, Dreef EJ, et al. A meta-analysis of gene expression data identifies a molecular signature characteristic for tumor-stage mycosis fungoides. J Invest Dermatol (2012) 132(8):2050–9. doi: 10.1038/jid.2012.117
22. Suga H, Sugaya M, Miyagaki T, Kawaguchi M, Fujita H, Asano Y, et al. The role of IL-32 in cutaneous T-cell lymphoma. J Invest Dermatol (2014) 134(5):1428–35. doi: 10.1038/jid.2013.488
23. Ohmatsu H, Humme D, Gonzalez J, Gulati N, Möbs M, Sterry W, et al. IL-32 induces indoleamine 2,3-dioxygenase + CD1c + dendritic cells and indoleamine 2,3-dioxygenase + CD163 + macrophages: Relevance to mycosis fungoides progression. Oncoimmunol (2017) 6(2):e1181237. doi: 10.1080/2162402X.2016.1181237
24. Yu KK, Smith NP, Essien SV, Teague JE, Vieyra-Garcia P, Gehad A, et al. IL-32 supports the survival of Malignant T cells in cutaneous T-cell lymphoma. J Invest Dermatol (2022) 142(8):2285–8. doi: 10.1016/j.jid.2022.01.009
25. Zepp JA, Nold-Petry CA, Dinarello CA, Nold MF. Protection from RNA and DNA viruses by IL-32. J Immunol (2011) 186(7):4110–8. doi: 10.4049/jimmunol.1000081
26. Peng T, Zhu J, Phasouk K, Koelle DM, Wald A, Corey L. An Effector Phenotype of CD8 + T Cells at the Junction Epithelium during Clinical Quiescence of Herpes Simplex Virus 2 Infection. J Virol (2012) 86(19):10587–96. doi: 10.1128/JVI.01237-12
27. Schenk M, Krutzik SR, Sieling PA, Lee DJ, Teles RMB, Ochoa MT, et al. NOD2 triggers an interleukin-32-dependent human dendritic cell program in leprosy. Nat Med (2012) 18(4):555–63. doi: 10.1038/nm.2650
28. Galdino H, Maldaner AE, Pessoni LL, Soriani FM, Pereira LIDA, Pinto SA, et al. Interleukin 32γ (IL-32γ) is highly expressed in cutaneous and mucosal lesions of American Tegumentary Leishmaniasis patients: Association with tumor necrosis factor (TNF) and IL-10. BMC Infect Dis (2014) 14(1). doi: 10.1186/1471-2334-14-249
29. Gomes RS, Silva MVT, Dos Santos JC, De Lima Silva LL, Batista AC, MaChado JR, et al. IL-32γ promotes the healing of murine cutaneous lesions caused by Leishmania Braziliensis infection in contrast to Leishmania amazonensis. Parasit Vectors (2017) 10(1). doi: 10.1186/s13071-017-2268-4
30. Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: A cytokine and inducer of TNFα. Immunity (2005) 22:131–42. doi: 10.1016/S1074-7613(04)00380-2
31. Aass KR, Kastnes MH, Standal T. Molecular interactions and functions of IL-32. J Leukoc Biol (2021) 109(1):143–59. doi: 10.1002/JLB.3MR0620-550R
32. Goda C, Kanaji T, Kanaji S, Tanaka G, Arima K, Ohno S, et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol (2006) 18(2):233–40. doi: 10.1093/intimm/dxh339
33. Da Choi J, Bae SY, Hong JW, Azam T, Dinarello CA, Her E, et al. Identification of the most active interleukin-32 isoform. Immunol (2009) 126(4):535–42. doi: 10.1111/j.1365-2567.2008.02917.x
34. Neupane S, Srivastav S, Bhurtel S, Katila N, Shadfar S, Park PH, et al. Enhanced neuroinflammatory responses after systemic LPS injection in IL-32β transgenic mice. J Chem Neuroanat (2018) 94:173–82. doi: 10.1016/j.jchemneu.2018.07.002
35. Park MH, Yoon DY, Ban JO, Kim DH, Lee DH, Song S, et al. Decreased severity of collagen antibody and lipopolysaccharide-induced arthritis in human IL-32β overexpressed transgenic mice. Oncotarget (2015) 6(36):38566–77. doi: 10.18632/oncotarget.6160
36. Choi J, Bae S, Hong J, Ryoo S, Jhun H, Hong K, et al. Paradoxical effects of constitutive human IL-32γ in transgenic mice during experimental colitis. Proc Natl Acad Sci USA (2010) 107(49):21082–6. doi: 10.1073/pnas.1015418107
37. Shoda H, Fujio K, Yamaguchi Y, Okamoto A, Sawada T, Kochi Y, et al. Interactions between IL-32 and tumor necrosis factor alpha contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther (2006) 8(6). doi: 10.1186/ar2074
38. Zahoor M, Westhrin M, Aass KR, Moen SH, Misund K, Psonka-Antonczyk KM, et al. Hypoxia promotes IL-32 expression in myeloma cells, and high expression is associated with poor survival and bone loss. Blood Adv (2017) 1(27):2656–66. doi: 10.1182/bloodadvances.2017010801
39. Heinhuis B, Koenders MI, Van De Loo FA, Netea MG, Van Den Berg WB, Joosten LAB. Inflammation-dependent secretion and splicing of IL-32γ in rheumatoid arthritis. Proc Natl Acad Sci U S A (2011) 108(12):4962–7. doi: 10.1073/pnas.1016005108
40. Boreika R, Sitkauskiene B. Interleukin-32 in pathogenesis of atopic diseases: proinflammatory or anti-inflammatory role? J Interferon Cytokine Res (2021) 41:235–42. doi: 10.1089/jir.2020.0230
41. Netea MG, Azam T, Ferwerda G, Girardin SE, Walsh M, Park JS, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1β and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci (2005) 102(45):16309–14. doi: 10.1073/pnas.0508237102
42. Nakayama M, Niki Y, Kawasaki T, Takeda Y, Ikegami H, Toyama Y, et al. IL-32-PAR2 axis is an innate immunity sensor providing alternative signaling for LPS-TRIF axis. Sci Rep (2013) 3(1):2960. doi: 10.1038/srep02960
43. Gorvel L, Korenfeld D, Tung T, Klechevsky E. Dendritic cell–derived IL-32α: A novel inhibitory cytokine of NK cell function. J Immunol (2017) 199(4):1290–300. doi: 10.4049/jimmunol.1601477
44. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Sci (1979) (2015) 347(6220). doi: 10.1126/science.1260419
45. Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol (1992) 148:597–603. doi: 10.4049/jimmunol.148.2.597
46. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker JNWN. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
47. Nestle FO, Kaplan DH, Barker JNWN. Psoriasis. N Engl J Med. (2009) 361(5):496–509. doi: 10.1056/NEJMra0804595
48. Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa. J Am Acad Dermatol (2020) 82(5):1045–58. doi: 10.1016/j.jaad.2019.08.090
49. Witte-Händel E, Wolk K, Tsaousi A, Irmer ML, Mößner R, Shomroni O, et al. The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J Invest Dermatol (2019) 139(6):1294–305. doi: 10.1016/j.jid.2018.11.018
50. Fukuyama M, Ito T, Ohyama M. Alopecia areata: Current understanding of the pathophysiology and update on therapeutic approaches, featuring the Japanese Dermatological Association guidelines. J Dermatol (2022) 49(1):19–36. doi: 10.1111/1346-8138.16207
51. Asadi LK, Khalili A, Wang SQ. The sociological basis of the skin cancer epidemic. Int J Dermatol (2023) 62(2):169–76. doi: 10.1111/ijd.15987
52. Didovic S, Opitz FV, Holzmann B, Förster I, Weighardt H. Requirement of MyD88 signaling in keratinocytes for Langerhans cell migration and initiation of atopic dermatitis-like symptoms in mice. Eur J Immunol (2016) 46(4):981–92. doi: 10.1002/eji.201545710
53. Joosten LAB, Heinhuis B, Netea MG, Dinarello CA. Novel insights into the biology of interleukin-32. Cell Mol Life Sci (2013) 70:3883–92. doi: 10.1007/s00018-013-1301-9
54. Mitamura Y, Reiger M, Kim J, Xiao Y, Zhakparov D, Tan G, et al. Spatial transcriptomics combined with single-cell RNA -sequencing unravels the complex inflammatory cell network in atopic dermatitis. Allergy (2023) 78:2215–31. doi: 10.1016/j.jaci.2022.12.790
Keywords: IL-32, atopic dermatitis, skin inflammation, melanoma, infection, CTCL
Citation: Wallimann A and Schenk M (2023) IL-32 as a potential biomarker and therapeutic target in skin inflammation. Front. Immunol. 14:1264236. doi: 10.3389/fimmu.2023.1264236
Received: 20 July 2023; Accepted: 15 August 2023;
Published: 01 September 2023.
Edited by:
Joanna Cichy, Jagiellonian University, PolandReviewed by:
Nicholas Gulati, Icahn School of Medicine at Mount Sinai, United StatesGabriela Fonseca-Camarillo, Instituto Nacional de Cardiología Ignacio Chávez, Mexico
Copyright © 2023 Wallimann and Schenk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Schenk, TWlyamFtLlNjaGVua0Bjay1jYXJlLmNo