 Bryan Gardam
Bryan Gardam Tessa Gargett
Tessa Gargett Michael P. Brown
Michael P. Brown Lisa M. Ebert
Lisa M. Ebert
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 October 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1261257
This article is part of the Research Topic Combination Immunotherapy and Immune Response Assessment of Brain Tumours View all 9 articles
Glioblastoma is an aggressive primary brain tumor that has seen few advances in treatments for over 20 years. In response to this desperate clinical need, multiple immunotherapy strategies are under development, including CAR-T cells, immune checkpoint inhibitors, oncolytic viruses and dendritic cell vaccines, although these approaches are yet to yield significant clinical benefit. Potential reasons for the lack of success so far include the immunosuppressive tumor microenvironment, the blood-brain barrier, and systemic changes to the immune system driven by both the tumor and its treatment. Furthermore, while T cells are essential effector cells for tumor control, dendritic cells play an equally important role in T cell activation, and emerging evidence suggests the dendritic cell compartment may be deeply compromised in glioblastoma patients. In this review, we describe the immunotherapy approaches currently under development for glioblastoma and the challenges faced, with a particular emphasis on the critical role of the dendritic cell-T cell axis. We suggest a number of strategies that could be used to boost dendritic cell number and function and propose that the use of these in combination with T cell-targeting strategies could lead to successful tumor control.
Glioblastoma is the most common and aggressive form of malignant brain tumor (1). Between 2014 and 2018, the Central Brain Tumor Registry of the United States (CBTRUS) reported 133,973 glioblastoma cases; this represented 14.3% of all central nervous system (CNS) tumors. Of these patients, 94% were over the age of 40, while 5% were aged between 15 and 39, and the remaining 1% were less than 14 years old (2). Glioblastoma is considered a rare form of cancer with a diagnosis rate of less than 6 in 100,000 (3); however, the impact of this disease is profound as the prognosis of glioblastoma remains extremely poor, with patient survival being generally 7-17 months post-diagnosis (1, 4–6). In the 5 years covered in the CBTRUS report, glioblastoma patients had a 1-year overall survival rate of 40.9%, with this rate showing a rapid decrease to only 6.8% 5-year overall survival (2). There are few reports of costs in glioblastoma, and there is variance between countries and health care systems; nevertheless, the economic cost is substantial due to both direct healthcare costs and to the years of potential life lost to this disease (7).
Even with the many advances following single-cell sequencing and epigenetic profiling of glioblastoma tumors, there has not yet been translation from scientific discovery to new anti-cancer targets and drugs (8–10). With the exception of tumor treating fields (TTF) administered concurrently with standard post-surgical treatment, there has been no additional survival prolonging treatment in almost 20 years. Glioblastoma remains an incurable disease affecting a broad range of ages with devastating outcomes (1, 11, 12).
In this review, we look at the potential for immune based therapies to treat glioblastoma, and also the challenges facing these immunotherapies. In particular, we explore the benefits that may be achieved by targeting the dendritic cell-T cell axis.
The Stupp protocol, developed almost 20 years ago, remains the standard treatment for newly diagnosed glioblastoma (11); this protocol involves surgery to remove as much of the tumor as is safely possible, followed by radiotherapy and chemotherapy with temozolomide (TMZ) (13). However, following this initial treatment regimen, tumor recurrence is almost inevitable. The importance of removing as much of the tumor as possible was quantified in a study by Stummer et al. that showed that total resection of the tumor was the only factor extending overall survival (14). These findings agreed with a previous study by Lacroix et al. that found that greater than 98% of tumor volume needed to be resected to increase overall survival (15). Even the addition of TMZ in the Stupp protocol only increased the average survival by approximately 2.5 months, and 84% of the patients had died by 2 years post-diagnosis (13).
No standard treatment regimen has been determined for elderly (>60-70 year old) glioblastoma patients who may not withstand the rigors of the Stupp protocol (16). Similarly, treatment of pediatric high grade gliomas is also considered palliative with survival rates of <10% beyond 2 years (17). Likewise, there is no standard second line survival prolonging treatment for recurrent glioblastoma, although bevacizumab may contribute to symptom control (18). Treatment options often depend on the individual and clinician, with survival generally less than six months following recurrence (13, 19, 20). Thus, new therapies, including immunotherapies, need to be developed for glioblastoma patients with the aim of extending the overall survival rates. An overview of the current standard treatments, as well as the immunotherapies discussed in section 5, is shown in Figure 1.
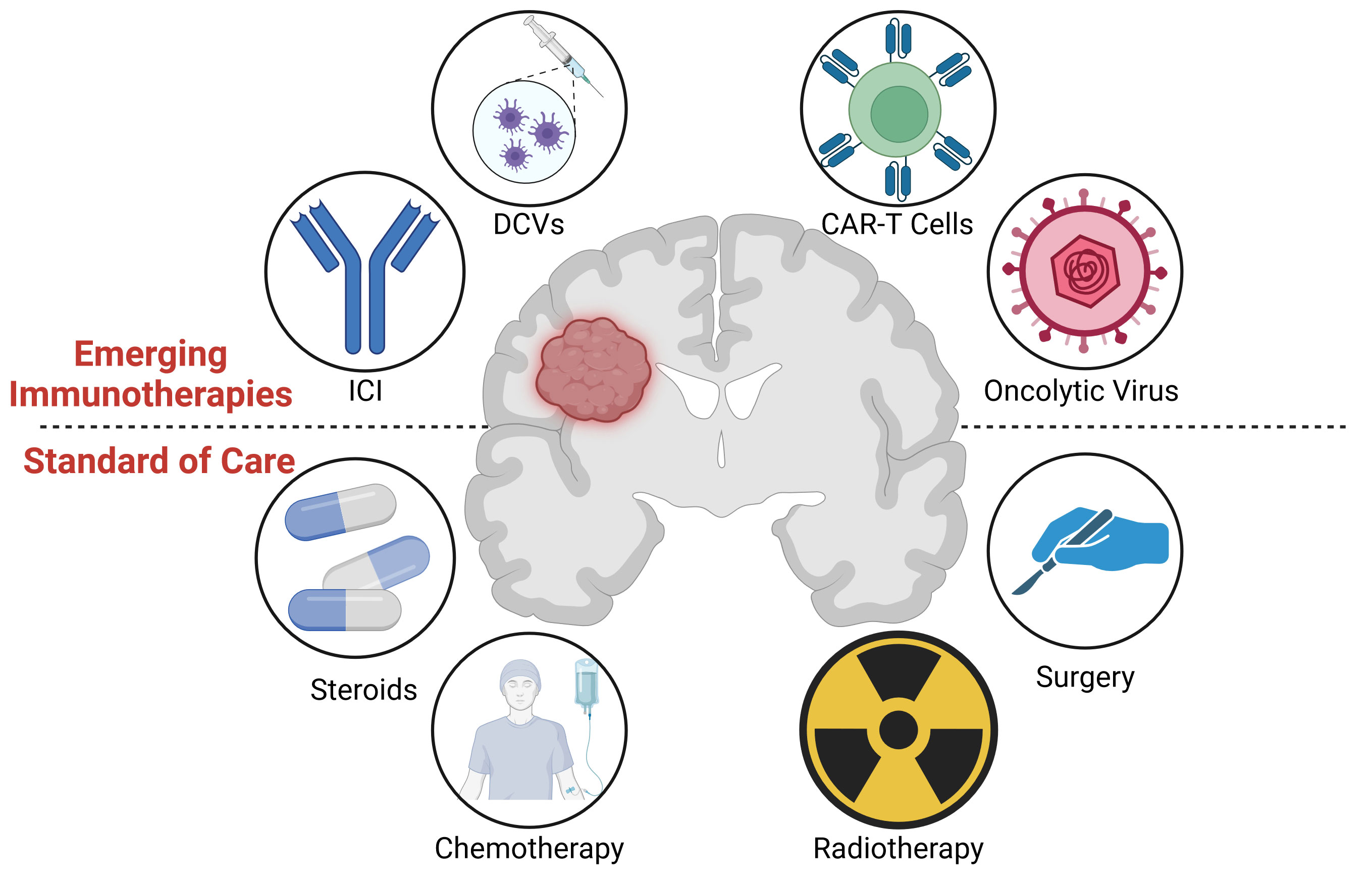
Figure 1 (Lower) Graphical overview of the standard of care treatment for glioblastoma, including surgery, radiotherapy, chemotherapy, and steroids. (Upper) Immunotherapies under investigation, including oncolytic viruses, CAR-T cells, dendritic cell vaccines (DCVs), and Immune Checkpoint Inhibitors (ICIs). Image created with Biorender.com.
From cancer initiation through to progression, interactions between cancer and immune cells proceed through three phases: elimination, equilibrium, and escape (21). Elimination is the phase where immune cells target and kill cancer cells; if successful, all cancerous cells would be removed from the body. However, any surviving cancer cells can persist and compete with the immune system in the equilibrium phase by continuous mutation and adaption. Finally, in the escape phase, the mutated cancer cells either become invisible or impervious to the immune system because of the advantages gained during the equilibrium phase (21), allowing them to grow and spread unopposed.
Immunotherapies aim to shift this balance in favor of the immune system to prevent cancer cell escape. These approaches are seen as a crucial new ‘fourth pillar’ of cancer treatment, in addition to the classical therapies of surgery, radiation and chemotherapy (22). Successful immunotherapy relies on the interaction of key immune cell subsets, including T cells and DCs. Therefore, an understanding of the interplay of these immune cells in the context of the tumor microenvironment is essential for advancing immunotherapy for glioblastoma.
DCs are the most potent professional antigen presenting cells of the immune system. DCs integrate signals of the innate and adaptive components of the immune system and initiate adaptive immunity by activating T cells (23). Most cells of the body are able to express endogenous antigenic peptides on major histocompatibility molecule (MHC) class I molecules, which allows the identification of cancerous or virally infected cells by CD8+ T cells. In addition, all DC subsets have the ability to capture extracellular antigens and present processed peptides on MHC class II molecules to CD4+ T cells, while specialized subsets of DCs are also able to present extracellular antigenic peptides on MHC class I molecules to CD8+ T cells, a process called cross-presentation. Cross-presentation allows the priming of naïve CD8+ T cells specific to antigens external to the DCs, which is crucial to initiating the cytotoxic T cell responses required for anti-tumor immunity (24). Together with antigen presentation, naive T cells require set co-stimulatory signals for activation. DCs provide this additional signal through CD80 or CD86, which are upregulated on the DC surface after antigen capture (25). DCs are thus a critical immune cell type in clearing the body of cancerous cells.
DCs in the tumor microenvironment (TME) are important for the immune system’s anti-tumor effects but can also support tumor growth when present in a tolerogenic form (26). The multiple roles played by DCs in the anti-tumor or pro-tumor response depends on the DC subsets present in the TME. Although only limited data are available on the function of some DC subsets in glioblastoma, other studies in solid tumors may inform the likely DC functions in glioblastoma, and therefore these are also reviewed below. The issue of DC heterogeneity was identified in a detailed review by Pombo Antunes et al. which identified that the DCs characterized in other solid tumors may not translate to glioblastoma and highlighted the need for further analysis of glioblastoma DC subsets (27).
pDCs in humans are identified as human leukocyte antigen-DR (HLA-DR)+ CD123+ CD304+ CD303+ (28). pDCs produce high levels of type 1 interferons (IFN) and play an essential role in viral clearance (29). Further, it is now accepted that mature pDCs can present antigenic peptides and activate naïve CD4+ T cells (30), although the ability of pDCs to cross-present antigens to CD8+ T cells remains unresolved (31). In cancer, pDCs have been reported to induce regulatory T cells (Treg) cells in solid tumors, and have been associated with poor outcomes (32, 33). A study of ovarian cancer patients found a negative correlation between the number of infiltrating pDCs and patient outcomes (32). In keeping with this observation, a study in a humanized murine xenograft model of human melanoma found that pDCs were linked to the generation of T helper Type 2 (Th2) CD4+ T cells, which may promote rather than control tumor growth (33). Conversely, in an immunocompetent murine lymphoma model, pDCs were reported as potent activators of T Helper Type 17 (Th17) cells and cytotoxic T cells leading to an effective anti-tumor response (34). A review by Macri et al. reported that both Th17 and Th2 responses were observed after activation of pDCs (23). A pre-clinical study in a rat model of glioblastoma has identified the recruitment and activation of pDCs; however, the study also concluded that the role of pDCs in an anti-tumor response remained unclear (35).
Thus, the complicated roles of pDCs remain to be fully explored, and it remains unknown whether pDCs support an anti-tumor response or contribute to glioblastoma tumor progression.
Type 1 cDCs (cDC1s) are potent as cytotoxic T cell activators and efficiently present and cross-present antigens on MHC II and MHC I, respectively. In humans, cDC1s are typically identified as HLA-DR+ CD11c+ CD123- CD11b- CD141+ C-type lectin domain family 9 member A (Clec9A)+ (23). cDC1s are excellent antigen cross-presenting cells and lead to high levels of cytotoxic T cell activation in response to tumors (36, 37). Numerous pre-clinical tumor models have identified a role for cDC1 in T cell recruitment to the tumor and in supporting T cell-mediated and NK cell-mediated tumor control (38–40). Further, cDC1s have been identified in studies of murine fibrosarcoma models to be required for successful anti-tumor responses, including the recruitment of effector cells including T cells (41). A study in mice with orthotopic glioblastoma tumors by Bowman-Kirigin et al. demonstrated that cDC1s cross-presented antigens and were required for the priming of CD8+ T cells in the brain and draining lymph nodes, in keeping with other solid tumor studies (42). Likewise, Friedrich et al. found, also in a mouse orthotopic glioblastoma model, that antigen cross-presenting DCs were crucial for the activation of CD8+ T cells; however, they also identified differences in the DC numbers and phenotype depending on whether or not the isocitrate dehydrogenase (IDH) gene was mutated in the tumor cells (43).
Type 2 cDCs (cDC2s) in humans are identified as HLA-DR+ CD11c+ CD141+ CD1low/+ Clec10A+/- CD5+/- (28). Further subdivision of the cDC2 population into CD5+ and CD5- subsets has also recently been reported (28). The CD5+ subset of cDC2 cells was found to have a greater ability to migrate to sites of infection compared to the CD5- subset; however, they produced a suppressive T cell phenotype, whereas CD5- cells elicited a stronger cytotoxic T cell effect (44). A recent study identified cDC2s as the most abundant DC subset in glioblastoma patient tumors, with the majority of these cells being CD5- (26). Although it is widely believed that cDC1s are pivotal for anti-tumor responses, there is an argument for further investigation of CD5- cDC2s in glioblastoma, as the significance of these cells may be under appreciated.
moDCs are viewed as a major subset of DCs of heterogeneous composition that can be generated easily in vitro (45). moDCs are also found in vivo, in both humans, and murine models (46, 47). moDCs in humans are reported as HLA-DR+ CD1c+ CD14+ CD226+ CD163- (28). Conversely, DC3s are a relatively newly defined subset of DCs that resemble moDCs and are thought to derive from moDCs (28, 48). DC3s in humans are identified as HLA-DR+ CD1c+ CD14+ CD163+ CD5- (23, 49). However, a recent study by Bourdely et al. defined DC3s as a discrete subset unrelated to moDCs and found them to be prevalent in solid tumors, where they promote the generation of resident memory T cells (TRM) (49). Ex vivo generation of both moDCs and DC3s can be achieved using the combination of granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin 4 (IL-4) (49–53), suggesting these populations share similarities. DC3 cells have been detected in samples of both lung and breast cancer (54, 55). Although there was no correlation between DC3s and patient outcomes in lung cancer, a positive correlation was observed in triple-negative breast cancer patients (55). Additionally, DC3s have been reported in glioblastoma by single-cell RNA sequencing (scRNAseq) (56). However, data confirming the presence of DC3s using protein markers is yet to be reported.
The true origin of moDCs remains contentious, and whether indeed they are related to cDC2 and DC3 cells is an area identified for continued investigation (28, 48, 57). For the purpose of this review, we will not distinguish between moDCs and DC3s.
The BBB consists of specialized vessels with tight junctions in the CNS that control the transfer of cells and molecules between the blood and the CNS (58) while also preventing toxins and disease- causing agents from entering the CNS (59). When a tumor encroaches on the BBB, it forms a more permeable blood-tumor barrier (BTB) that can permit the entry of small and larger molecules into the main tumor mass. However, the BTB remains heterogeneous, and the passage of molecules and immune cells depends on the location and type of cancer (60). Moreover, the BBB remains a significant obstacle to the efficient delivery of therapeutic agents to the tumor-adjacent normal brain tissue, where invading tumor cells spread, and thus limits the effectiveness of some cytotoxic drug and antibody therapies (61, 62).
The CNS was long considered to be an immune privileged site (63), and the BBB was accordingly thought to limit treatment of glioblastoma with immunotherapies (61). Now, however, evidence that CNS antigens drain through lymphatic vessels to the deep cervical lymph nodes (64, 65) indicates that immune responses can be initiated from antigens within the brain. This highlights the key role that DCs can play in initiating and maintaining T cell responses to brain tumors.
The paucity of DCs in brain tumors and their functional impairment is likely an intrinsic feature of the immunosuppressive TME of glioblastoma, which has been recognized since the early 1970’s (66) and induced by numerous mechanisms, which have been reviewed recently and comprehensively by others (67). Briefly, the glioblastoma TME consists of many different immune cell types, including T regulatory cells (Treg), tumor-associated macrophages (TAMs) and suppressive myeloid cells (68). Remarkably, myeloid cells can make up to 50% of the glioblastoma tumor mass (20), and Friebel et al. reported that up to 80% of all leukocytes in the TME consisted of TAMs and monocytes (9) They have thus been a major focus of investigation (69, 70). The tumor and the TME secrete molecules and cytokines that polarize the TAMs to an immunosuppressive phenotype (71). Further, TAMs can then inhibit T cell functionality via production of immune-suppressive factors such as IL-10 (72). There is increasing evidence of substantial heterogeneity in the glioblastoma TME between patients, including the presence of cDC1s, cDC2s, and pDCs (70, 73), and changes in cell populations have been seen at different stages during a patient’s disease in cDC1s, and pDCs (74).
In addition, recent studies argue that the TME in the brain prevents effective antigen presentation by myeloid lineage cells, including cDC1s, cDC2, and pDCs, and that this is a more important factor leading to ineffective anti-tumor responses than the overall suppressive nature of the TME (75, 76). While the exact mechanism is not fully understood, Simonds et al. reported in a glioblastoma murine model, response to immune checkpoint inhibitors (ICI) relied on T cells and DCs, and mice with subcutaneous tumors could respond to ICI while mice with intracranial tumors could not, indicating the location of the tumor in the brain was a critical factor influencing immune recognition (75). It should be considered that the TME, whether by preventing antigen presentation or its directly suppressive nature, is a significant barrier to treating glioblastoma patients with immunotherapies. There is still extensive work to be done in characterizing the complexity of the immune microenvironment associated with glioblastoma, including a characterization of rarer T cell and DC subsets.
The immune suppression associated with glioblastoma extends beyond the TME and is manifest globally as lymphopenia. The origins of lymphopenia in glioblastoma patients are related both to the disease itself and its treatment. The number of circulating lymphocytes is homeostatically regulated and may reduce with age, or from specific diseases and their treatments (77). Therefore, these effects may have significant implications for the development new immunotherapies for glioblastoma, including those that target the DC-T cell axis.
Chongsathidkiet et al. made the striking observation that lymphopenia resulted in sequestration of T cells in the bone marrow when a brain tumor was also present. A possible mechanism is tumor-mediated downregulation of sphingosine-1 phosphate receptor-1 (S1PR1) on T cells restricting T cells egress from bone marrow (78). Interestingly, this finding was not only found in primary brain tumors but also in other tumors that were xenografted into the brains of mice. This result points to a possible hypothesis that it is an effect specific to the brain, deriving from either from the tumor or TME development, which may be self-protective and prevent inflammation by systemic effects on cells of the immune system.
We found that clinical use of cytotoxic chemotherapy drugs can be associated with an increase in the number of effector cells in recovery phase and that repopulation of the lymphocyte compartment can be associated with a concomitant reduction in suppressive phenotypes in a drug dependent way (79). In glioblastoma patients, lymphopenia often follows treatment with TMZ, especially when combined with radiotherapy, and is associated with a reduction in survival (80). In contrast, another study reports treatment benefits following TMZ treatment-induced lymphopenia (81). A recent study by Ghosh et al. showed that lymphopenia was induced in glioblastoma patients following radiation therapy, which correlated with a reduction in progression-free and overall survival (82). Interestingly, these effects were driven by increased myelopoiesis and the generation of myeloid-derived suppressor cells (MDSC), which in turn were associated with T-cell lymphopenia.
It has been reported that DCs are lacking in the TME of patients’ glioblastoma tumors (83, 84). However, a recent study by Simonds et al. has shown that DCs are still present in human glioblastoma, although reduced compared to other tumor locations. This study also highlighted that a deficiency in DCs can lead to a delayed immune response through poor priming of T cells in the draining lymph nodes (75). This finding supports a significant role for DCs in immune responses to tumors, including glioblastoma. In addition to the reduced numbers, the functionality of DCs has also been reported to be impaired in glioblastoma patients compared to healthy patient samples (26, 43, 56, 83–89). A summary of the reported changes in DC numbers in glioblastoma patients is presented in Table 1, including analyses of circulating DCs as well as those within tumors. Together, these studies suggest that reduced quantity and quality of DCs in glioblastoma may impair tumor control and the effectiveness of immunotherapies. Although other studies have identified DCs in the tumor and TME of glioblastoma patients from tumor samples (42) or scRNAseq data (43, 56, 91), no quantification compared to healthy patients was reported in these studies.
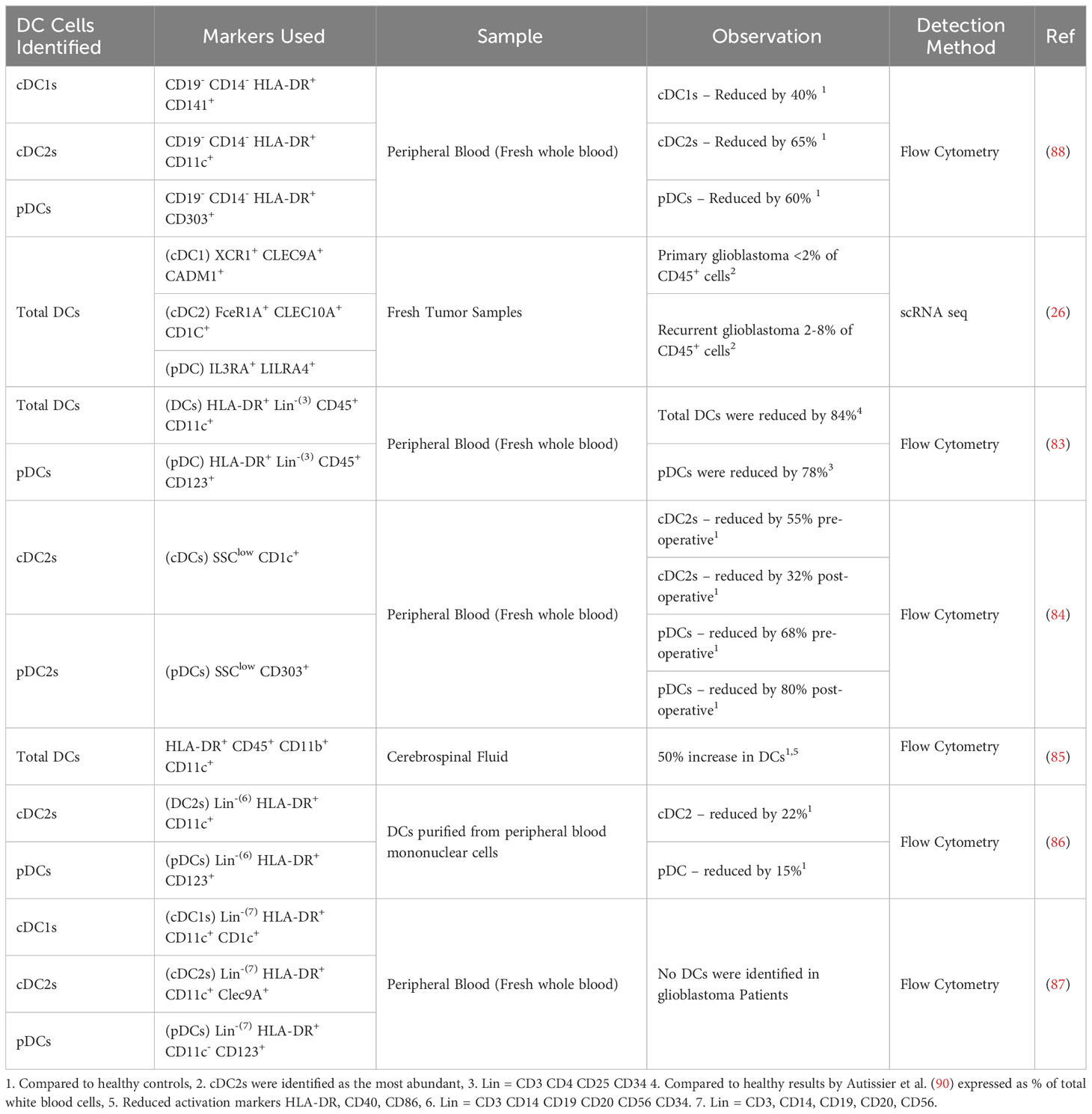
Table 1 Changes in dendritic cells in glioblastoma patients.
Adhikaree et al. reported downregulation of HLA-DR, the T cell costimulatory molecule CD86 and increases programmed death-ligand 1 (PD-L1) when DCs were exposed to glioblastoma tumors or the corticosteroid dexamethasone, which is often given to glioblastoma patients to reduce brain swelling and relieve symptoms (88). This phenomenon has also been reported elsewhere (92). The downregulation of HLA-DR and CD86 indicates a less mature DC phenotype and can lead to the generation of Treg cells (88); thus, different approaches have been used to restore the function of the DCs, including inhibiting p38 (88) and through silencing PD-L1 in DCs (92). Although no studies have reported other effects of p38 inhibition, p38 is responsible for the regulation of many transcription factors, including pro-inflammatory cytokines, growth factors, and adhesion molecules (93), so further studies are required to investigate the biologic specificity of p38 inhibition.
A graphic representation of the changes seen in dendritic cells is shown in Figure 2A.
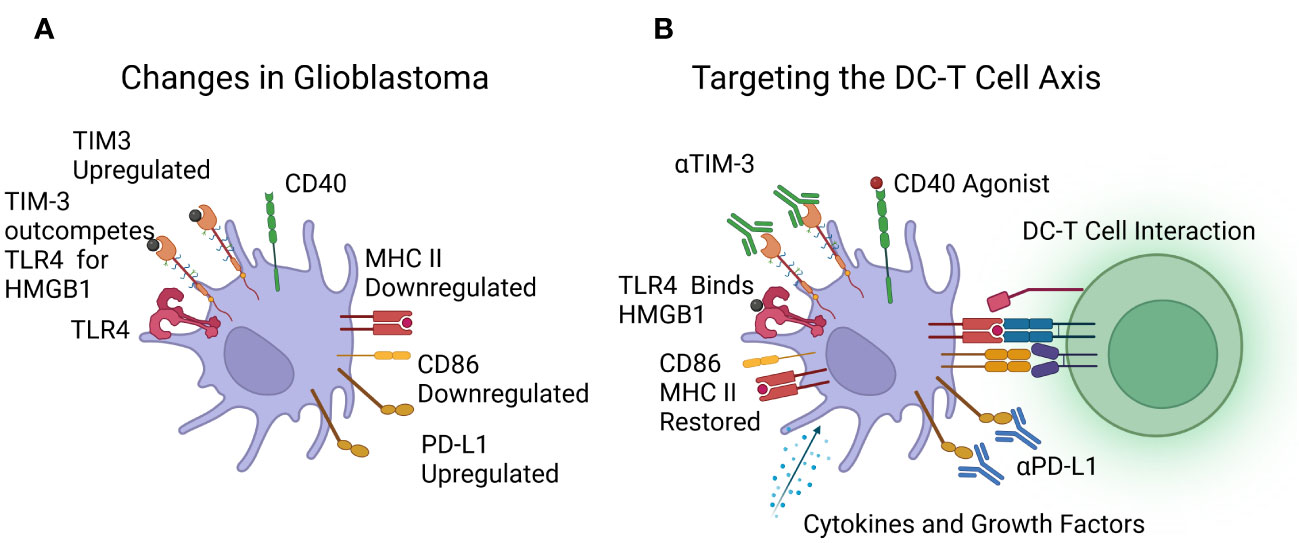
Figure 2 (A) Reported changes in dendritic cells in patients with glioblastoma, including downregulation of MHC class II and CD86, upregulation of PD-L1 and TIM-3, with TLR4 outcompeted. (B) Immunotherapies under investigation to target the DC-T cell axis, including cytokines, growth factors, and CD40 agonists to boost dendritic cell numbers and function, and Immune Checkpoint Inhibitors (ICIs) targeting TIM-3 and PD-L1. Image created with Biorender.com.
CAR-T cell therapy generally involves genetically modifying autologous T cells to express high-affinity chimeric receptors, with the extracellular region specific for tumor antigens and the intracellular portion responsible for T cell activation upon CAR engagement. This receptor facilitates tumor antigen recognition by the engineered T cells and leads to efficient tumor cell killing (94–96). CAR-T cell treatment involves the collection of the patient’s T cells, gene modification for CAR expression, CAR-T cell expansion, and patient infusion with the CAR-T cell product. The first generation of CAR-T cells entered clinical trials in the 1990s’ as a treatment for HIV (97). Second- and third-generation CAR-T cells were developed to include one or multiple costimulatory domains, respectively (98). CAR-T cells have been highly effective in treating B-cell malignancies (99–101); however, the same cannot be said for solid tumors (95). Currently, CAR-T cells targeting multiple antigens are being investigated to treat glioblastoma in preclinical studies and early clinical trials (102, 103). To address the heterogeneity of glioblastoma, multi-antigen targeting by CAR-T cells is being explored in some of these studies (104), but tumor specificity is required to minimize off-tumor, on-target toxicity in normal healthy tissue (105). Notwithstanding the relatively early stage of development of CAR-T cell therapy for glioblastoma, limited effectiveness or resistance to treatment has already been reported, including loss of tumor antigen expression (11, 106) and presumed CAR-T cell induced resistance through the upregulation of programmed death-ligand 1 (PD-L1) and recruitment of Treg cells (107). The likely reasons given for these modest clinical results include the immunosuppressive nature of the TME (96, 108), the high degree of antigen heterogeneity (109), sub-optimal CAR-T cell doses, and route of administration (103).
To address some of the challenges associated with treating solid tumors, including glioblastoma, recent CAR-T cell designs may incorporate additional genes to improve T cell function. For example, cytokine-enabled CAR-T cells, which are sometimes referred to as TRUCKs (T cells redirected for antigen-unrestricted cytokine-initiated killing), secrete T cell supportive cytokines or cytotoxic mediators such as IL-12, IL-15 and IL-18 (110). However, the ability of local, CAR-T-produced cytokines to promote beneficial paracrine changes in the immune microenvironment, including DC stimulation, has also been explored. For example, IL-18-producing TRUCKs reportedly induce changes in the balance of M1 and M2 macrophages and may also alter the phenotype of from suppressive cDC2s, although a change in cDC1s numbers was not observed (111). IL-12 producing CAR-T cells are perhaps the most widely studied TRUCKs to date (112–114); however, there are limited studies of the effects of IL-12 on the TME or DCs. There has been one study of IL-12 enhanced CAR-T cells in an immune-competent murine lymphoma model, which reported evidence of epitope-spreading, implying DC involvement, and another study describing the effect of murine CAR-T cells co-administered with local IL-12 delivery in the murine GL261 glioma model, which has confirmed that in principle IL-12 can re-shape the myeloid compartment in solid tumors (115). Our recent studies of GD2-CAR-T cells expressing transgenic IL-15 also suggest beneficial effects on DCs (116).
While the approaches above may indirectly act on DCs, a promising approach to overcome DC deficits in glioblastoma is to directly enhance intra-tumoral DC number and function via using TRUCKs as a delivery vehicle for DC-stimulating agents. Of particular note, a recent preclinical study by Swan et al. investigated CAR-T cells expressing mouse IL7 and FMS-like tyrosine kinase 3 ligand (Flt3L) in a glioblastoma mouse model. These CAR-T cells increased the number of antigen-presenting cDC1s and increased CAR-T cell survival; further, it was noted that this combination improved the overall survival of the mice (117). Similar results, including an increased number of cDC1s in the tumor, were observed by Lai et al. in other non-glioblastoma cancer models (118). Furthermore, Kuhn et al. found in a mouse B cell lymphoma model that CAR-T cells expressing CD40L stimulated DC1s to activate endogenous T cells and provide greater anti-tumor effects (119). It will be very interesting in future studies to investigate whether similar effects are observed in the context of glioblastoma.
DCV approaches involve the ex vivo culture and maturation of autologous DCs with antigens derived from tumor lysate or specific peptides before the patient is infused with the preparation to activate a T-cell response (120). A comparison of the route of administration was conducted in patients with advanced melanoma and found that intradermal injection resulted in 4% of the DCs migrating to the draining lymph nodes. In contrast, intranodal injection resulted in up to 84% of the DCs migrating to adjacent lymph nodes (121). Like CAR-T cell therapies, the DCV process is personalized and expensive (107). Tumor lysates enable multiple antigens to be loaded onto the DCs; alternatively, specific peptides may be loaded onto the DCs. Most DCV trials use moDCs generated from peripheral blood mononuclear cells (PBMCs) with GM-CSF and IL-4, although the way the DCs are matured prior to infusion differ from one study to the next. A detailed comparison of five different DC phenotype maturation processes used in DCV clinical trials was conducted by Zhang et al., who demonstrated that the protocol utilizing four cytokines produced a more mature DC product (122).
Numerous glioblastoma clinical trials have been conducted, with mixed results, and not all living up to the expectations generated by preclinical results (51, 84, 123–126). For example, a clinical trial by Prins et al. compared tumor lysate and specific peptides as a source of antigens and found dose-limiting toxicity, but no apparent benefit for either approach and no overall patient benefit (125). In the DCV phase II randomized clinical trial in glioblastoma patients by Buchroithner et al., the tumor lysate approach was used and showed no overall increase in patient survival (126). Similarly in a randomized double-blind placebo-controlled phase II DCV trial with six peptides by Wen et al. there was no overall survival benefit, although a prolonged progression-free survival was noted in the treatment group (51). On the other hand, a clinical trial by Wang et al. found that treatment with a tumour-associated antigen (TAA) based personalized DCV showed treatment benefits warranting further exploration in a larger cohort (127).
An important point to note is that the DCV approach relies on the patient having an intact functional immune system. However, as noted earlier, glioblastoma has deleterious effects on circulating T cells and DCs and may impair the effectiveness of these therapies. However, targeting the DC – T cell axis in combination may overcome these challenges.
OV therapy uses viruses that are only weakly pathogenic and have been genetically engineered to maximize anti-cancer effects while minimizing damage to normal cells. Additionally, some OV therapies have further genetic modifications to express immunomodulatory factors (128). Three of the main viruses being studied in glioblastoma include herpes simplex virus (HSV), adenovirus (Adv), and poliovirus; however, the effectiveness of OV in treating glioblastoma has been limited to date despite promising preclinical results (129). Although some of the challenges involve overcoming the suppressive TME, it is suggested that the OV generates its own immune response, and this response neutralizes the virus particles, thus reducing the overall effect (128).
To improve the efficacy of OV therapy, some investigators are engineering viruses to express Flt3L to enhance intra-tumoral DC numbers. For example, King et al. established a rat intracranial glioblastoma model in which Flt3L and herpes simplex virus type 1–thymidine kinase, delivered intratumorally by replication-defective recombinant adenovirus type 5 vectors, was able to control tumors in combination with the antiviral drug Ganciclovir. It was proposed that this was through the recruitment of pDCs (130). However, FLT3L has also been shown to expand cDCs (118, 131). In a similar study, Ali et al. showed a similar response in the same model with Flt3L alone (132). However, it should be noted that in the study by Ali et al., the treatment was administered three days post-cell implantation, which may lack clinical relevance as the tumor may not have established.
Immune checkpoint inhibitors target immune checkpoint pathways with antibodies that disrupt the receptor/ligand interactions to prevent tolerance and restore the function of immune effector cells (133). Programmed cell death protein 1 (PD-1) with its ligand PD-L1, provides an inhibitory mechanism in activated T cells, B cells, monocytes and DCs to prevent the recognition of self-antigens and autoimmunity (134). Many cancers, including glioblastoma, upregulate PD-L1 on DCs, causing tolerogenic cell phenotypes to develop and, in turn, tumor escape (104). Similarly, cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is expressed on T cells and, when engaged by its ligands CD80 or CD86, also suppresses T cell function (104).
Antonios et al. found increased expression of PD-L1 in glioblastoma tumor-infiltrating myeloid cells, and greater efficiency of a DCV was achieved in a preclinical murine glioblastoma model when combined with a PD-1 blocking monoclonal antibody (135, 136). A further study by Aslan et al. in a murine glioblastoma model identified the importance of PD-1, PD-L1 and CTLA-4 in T cell effector function while also acknowledging that other suppressive molecules may be at play on TAMs in the TME (137). In comparison, two reports from the clinical trials of anti-PD-1 agent nivolumab (CheckMate 143) were reported by Omuro et al. in newly diagnosed and recurrent glioblastoma patients. Both reports found that monotherapy treatment was safe, as was combination with CTLA-4 inhibition or standard-of-care treatments; however, there was no increased overall survival. In both cases, it was concluded that further studies and trials were warranted to identify approaches to use these novel agents more effectively, including in combination with other therapies (138, 139).
Although many combination experiments in the setting of glioblastoma are yet to be conducted, findings from other tumor models provide support for novel therapies that enhance DC function while also blocking immune checkpoint function. For example, Salmon et al. showed that treatment with Flt3L resulted in more DCs at the tumor site in a mouse melanoma model. However, DC1s in tumor-draining lymph nodes of these mice upregulated PD-L1, suggesting that a combination of Flt3L and anti-PD-L1 would result in greater T cell activation (131). Indeed, in a separate study, the same approach to expand DCs led to better outcomes following anti-PD-1 therapy in a melanoma mouse model (140).
T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) is another candidate for ICI therapy, although not yet tested clinically in glioblastoma. In addition to its function as a T cell checkpoint receptor, TIM-3 also regulates DC function. In this context, TIM-3 competes as a receptor for the nuclear alarmin protein high-mobility-group box 1 (HMGB1) secreted by dying tumor cells. HMGB1 normally functions as a danger signal, resulting in DC activation following binding to toll-like receptor 4 (TLR4) (141). However, Chiba et al. identified secreted factors in the TME that result in increased expression of TIM-3 on DCs (142). The increased expression of TIM-3 outcompetes TLR4, thus resulting in reduced activation of DCs and, in turn, reduced immune responses. Similarly, another ligand of TIM-3, galectin 9 has been recently identified as upregulated in the TME of melanoma patients (143) and glioblastoma patients (85). The increased presence of galectin 9 in the TME is also associated with a decreased ability of DCs to present tumor antigens.
Several additional studies support the critical role that DCs play in mediating responses to ICI agents. For example, in a subcutaneous mouse tumor model, Peng et al. identified that functional DCs were vital to an effective outcome from anti PD-L1 therapies (144). Similarly, a study in non-small cell lung carcinoma by Cohen et al. found that following PD-1 blockade, DCs are crucial for the reactivation of T cells (145). A recent study by Tomaszewski et al. found in glioblastoma that the upregulation of calmodulin-dependent kinase kinase 2 (CaMKK2) reduced the interaction of DCs with T cells, and reduced the effectiveness of ICI therapies; it was thus suggested that therapies targeting CaMKK2 and DC or T cells would enhance treatment outcomes, although there are no reports of these combination therapies being tested.
In support of these mechanistic studies, a number of reports confirm that combining ICI agents with DC-stimulating agents can promote anti-tumor activity in various cancer types. For example, preclinical mouse breast and pancreatic cancer models demonstrated that ICI therapy combined with a CD40 agonist increased immune cell infiltration and greater tumor control than ICI therapy alone (146). Likewise, in a further preclinical mouse model of pancreatic cancer, ICI therapy and CD40 agonists also showed increased tumor control (147), and an early clinical trial in metastatic colorectal cancer and metastatic pancreatic cancer showed that combining ICI therapy with a toll-like receptor 9 (TLR9) agonist also resulted in an increase in T-cell and DC tumor infiltration (148).
Considering the deficiencies in DC number and function in glioblastoma patients (see above and Table 1), these studies in other tumor types provide a strong rationale to test approaches to support DC maturation and function to improve the efficacy of ICI therapies in treating glioblastoma.
The possible improved outcomes by targeting the DCs with immunotherapies and targeting the DC-T cell axis is shown at Figure 2B.
Despite positive preclinical data, many of the single CAR-T cell, DCV and OV trials have had limited success in treating glioblastoma, and these reports often note that better success may be seen when combined with checkpoint inhibitors or other approaches. This further highlights the need for a holistic approach to immunotherapies for the treatment of glioblastoma; further, given the heterogeneity of glioblastoma tumors, and the immunosuppressive mechanisms operating in the TME, combined immunotherapies are more likely to be effective. However, according to the U.S. National Library of Medicine clinical trials database (https://www.clinicaltrials.gov/) (149), a search for active, recruiting, and not yet recruiting clinical trials testing immunotherapies for glioblastoma returned a total of 43 trials, but of these, only 11 involved a combination of 2 immunotherapies 9 of which are a combination of 2 checkpoint inhibitors, and just 1 trial consisted of a combination of 3 immunotherapies (Table 2). Of particular note, only 1 trial is testing combinations of DC-targeted approaches with any other type of immunotherapy. This is clearly an area that deserves further attention.
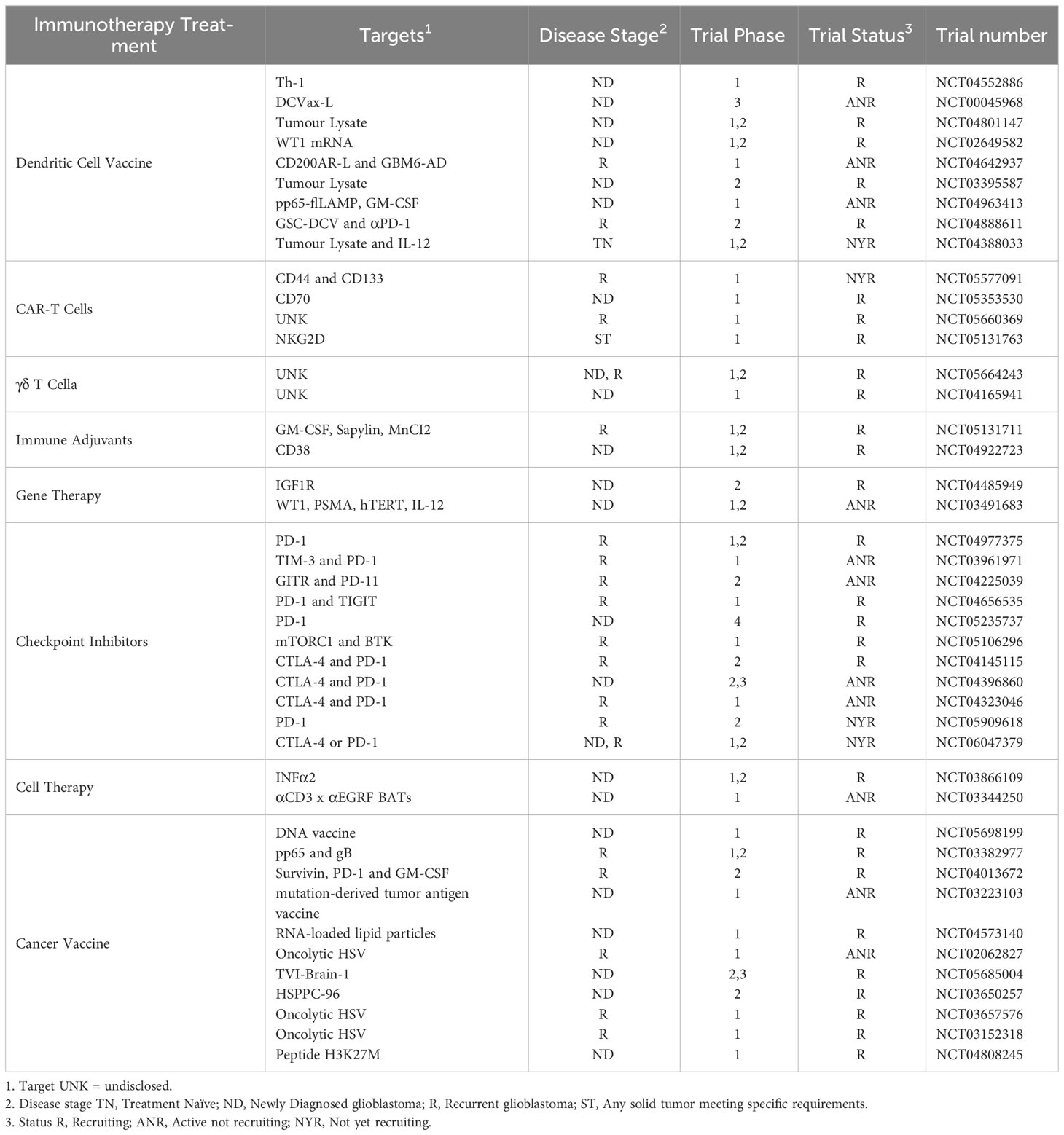
Table 2 Current clinical trials for glioblastoma with Immunotherapies registered on the U.S. National Library of Medicine clinical trials database.
It is recognized that solid tumor control by the immune system requires a multi-step process involving different immune cells, including DCs and T cells (74). However, recent studies suggest that the unique glioblastoma microenvironment, influenced by its intracranial location, results in a particular deficit in DC number and function (75, 85, 86), T cell sequestration in the bone marrow (80), and the suppressive TME (69) all pose hurdles to be overcome in producing effective immunotherapies. Friebel et al. reported interesting links between the immune cells present in the TME of glioblastoma, in that more T cells were associated with more DCs, and conversely, more TAMs were associated with fewer T cells (9). Lee et al. also concluded that the recruitment of T cells and DCs in combination with other immunotherapies would be required to achieve significant results (150). Likewise, Liu et al. conclude that the future of glioblastoma treatments lies in a multifaceted immune response (129). Eiraku et al. had a similar conclusion that combinations of DCV and CAR-T cells with ICI may provide a better outcome (53).
Immunotherapies aim to enhance or enable the patient’s immune system to detect, target and kill cancer cells while protecting normal cells (104, 151). However, the immune system suppression caused by radiotherapy and chemotherapy has raised questions about the effectiveness of using immunotherapies as a treatment for glioblastoma (152). Many of the suppressive attributes of glioblastoma mentioned above likely also contribute to the limited progress in immune-based treatments for glioblastoma (5). Moreover, although corticosteroids such as dexamethasone are given to glioblastoma patients to reduce inflammation in the brain and relieve symptoms, these agents can reduce immune responses, including a loss of antigen presentation by DCs (88, 153–155). Thus, these standard methods to manage glioblastoma symptoms may further limit the effectiveness of immunotherapies. A careful balance between promoting immune function and limiting inflammation in the brain to prevent unwanted side effects must be considered in designing immunotherapeutic approaches (156). As treatments are further developed, the consideration of engaging and supporting all arms of the immune system while targeting the tumor will ultimately lead to better patient outcomes. However, as seen in Table 2, less than 25% of the current glioblastoma trials in Table 2 involve combined immunotherapies. Highlighting the need for combination therapies in glioblastoma is the remarkable report by Zhu et al. that describes a single glioblastoma patient with a progression-free survival of 69 months following treatment with a personalized DCV combined with depletion of Treg cells, ICI with anti-PD-1, and an immune adjuvant Poly I:C (157).
We propose that the approaches most likely to succeed will find a way to enhance the suboptimal T cell and DC populations present in glioblastoma patients, likely through combination treatments targeting both ends of the DC – T cell axis.
BG: Conceptualization, Writing – original draft, Writing – review & editing. TG: Conceptualization, Supervision, Writing – review & editing. MB: Supervision, Writing – review & editing. LE: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant 2020344 awarded through the 2022 Priority-driven Collaborative Cancer Research Scheme and co-funded by Cancer Australia and The Kids’ Cancer Project; additional funding was provided by the Neurosurgical Research Foundation, Tour de Cure, the Ray & Shirl Norman Cancer Research Trust, the Mark Hughes Foundation, and the Health Services Charitable Gifts Board Adelaide.
BG acknowledges the support received through the provision of an Australian Government Research Training Program Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Delgado-Martín B, Medina MÁ. Advances in the knowledge of the molecular biology of glioblastoma and its impact in patient diagnosis, stratification, and treatment. Advanced Sci (2020) 7(9):1902971. doi: 10.1002/advs.201902971
2. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. Cbtrus statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro-Oncology (2021) 23(Supplement_3)::iii1–iii105. doi: 10.1093/neuonc/noab200
3. Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallée V-P, Mendoza A, et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell (2019) 179(4):846–63.e24. doi: 10.1016/j.cell.2019.09.035
4. Nguyen H-M, Guz-montgomery K, Lowe DB, Saha D. Pathogenetic features and current management of glioblastoma. Cancers (2021) 13(4):1–42. doi: 10.3390/cancers13040856
5. Mooney J, Bernstock JD, Ilyas A, Ibrahim A, Yamashita D, Markert JM, et al. Current approaches and challenges in the molecular therapeutic targeting of glioblastoma. World Neurosurg (2019) 129:90–100. doi: 10.1016/j.wneu.2019.05.205
6. Feng E, Sui C, Wang T, Sun G. Temozolomide with or without radiotherapy in patients with newly diagnosed glioblastoma multiforme: A meta-analysis. Eur Neurol (2017) 77(3-4):201–10. doi: 10.1159/000455842
7. De Paepe A, Van den Eede N, Strens D, Specenier P. The economic burden of glioblastoma. Acta clinica belgica (English ed Online) (2015) 70(S3):17.
8. Brennan Cameron W, Verhaak Roel GW, McKenna A, Campos B, Noushmehr H, Salama Sofie R, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
9. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell (2020) 181(7):1626–42.e20. doi: 10.1016/j.cell.2020.04.055
10. Wang L, Jung J, Babikir H, Shamardani K, Jain S, Feng X, et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat Cancer (2022) 3(12):1534–52. doi: 10.1038/s43018-022-00475-x
11. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
12. Janjua TI, Rewatkar P, Ahmed-Cox A, Saeed I, Mansfeld FM, Kulshreshtha R, et al. Frontiers in the treatment of glioblastoma: past, present and emerging. Advanced Drug delivery Rev (2021) 171:108–38. doi: 10.1016/j.addr.2021.01.012
13. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
14. Stummer W, Reulen H-J, Meinel T, Pichlmeier U, Schumacher W, Tonn J-C, et al. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery (2008) 62(3):564–76. doi: 10.1227/01.neu.0000317304.31579.17
15. Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg (2001) 95(2):190–8. doi: 10.3171/jns.2001.95.2.0190
16. Glaser SM, Dohopolski MJ, Balasubramani GK, Flickinger JC, Beriwal S. Glioblastoma multiforme (Gbm) in the elderly: initial treatment strategy and overall survival. J neuro-oncol (2017) 134(1):107–18. doi: 10.1007/s11060-017-2493-x
17. MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro-oncology (2011) 13(10):1049–58. doi: 10.1093/neuonc/nor092
18. Chen W, Wang Y, Zhao B, Liu P, Liu L, Wang Y, et al. Optimal therapies for recurrent glioblastoma: A bayesian network meta-analysis. Front Oncol (2021) 11:641878. doi: 10.3389/fonc.2021.641878
19. Field KM, Simes J, Nowak AK, Cher L, Wheeler H, Hovey EJ, et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro-Oncology (2015) 17(11):1504–13. doi: 10.1093/neuonc/nov104
20. Yeo ECF, Brown MP, Gargett T, Ebert LM. The role of cytokines and chemokines in shaping the immune microenvironment of glioblastoma: implications for immunotherapy. Cells (Basel Switzerland) (2021) 10(3):607. doi: 10.3390/cells10030607
21. Dunn GP, Old LJ, Schreiber RD. The three es of cancer immunoediting. Annu Rev Immunol (2004) 22(1):329–60. doi: 10.1146/annurev.immunol.22.012703.104803
22. Van Gool SW, Makalowski J, Fiore S, Sprenger T, Prix L, Schirrmacher V, et al. Randomized controlled immunotherapy clinical trials for gbm challenged. Cancers (2021) 13(1):1–28. doi: 10.3390/cancers13010032
23. Macri C, Pang ES, Patton T, O’Keeffe M. Dendritic cell subsets. Semin Cell Dev Biol (2018) 84:11–21. doi: 10.1016/j.semcdb.2017.12.009
24. Heath WR, Belz GT, Behrens GMN, Smith CM, Forehan SP, Parish IA, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev (2004) 199(1):9–26. doi: 10.1111/j.0105-2896.2004.00142.x
25. Zammit DJ, Cauley LS, Pham Q-M, Lefrançois L. Dendritic cells maximize the memory cd8 T cell response to infection. Immun (Cambridge Mass) (2005) 22(5):561–70. doi: 10.1016/j.immuni.2005.03.005
26. Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci (2021) 24(4):595–610. doi: 10.1038/s41593-020-00789-y
27. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife (2020) 9:e52176. doi: 10.7554/eLife.52176
28. Villar J, Segura E. Decoding the heterogeneity of human dendritic cell subsets. Trends Immunol (2020) 41(12):1062–71. doi: 10.1016/j.it.2020.10.002
29. Webster B, Assil S, Dreux M. Cell-cell sensing of viral infection by plasmacytoid dendritic cells. J Virol (2016) 90(22):10050–3. doi: 10.1128/JVI.01692-16
30. Liu Y-J. Ipc: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23(1):275–306. doi: 10.1146/annurev.immunol.23.021704.115633
31. Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity (2008) 29(3):352–61. doi: 10.1016/j.immuni.2008.09.002
32. Labidi-Galy SI, Treilleux I, Goddard-Leon S, Combes J-D, Blay J-Y, Ray-Coquard I, et al. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology (2012) 1(3):380–2. doi: 10.4161/onci.18801
33. Aspord C, Leccia M-T, Charles J, Plumas J. Plasmacytoid dendritic cells support melanoma progression by promoting th2 and regulatory immunity through ox40l and icosl. Cancer Immunol Res (2013) 1(6):402–15. doi: 10.1158/2326-6066.CIR-13-0114-T
34. Guéry L, Dubrot J, Lippens C, Brighouse D, Malinge P, Irla M, et al. Ag-presenting cpg-activated pdcs prime th17 cells that induce tumor regression. Cancer Res (Chicago Ill) (2014) 74(22):6430–40. doi: 10.1158/0008-5472.CAN-14-1149
35. Candolfi M, King GD, Yagiz K, Curtin JF, Mineharu Y, Muhammad AKMG, et al. Plasmacytoid dendritic cells in the tumor microenvironment: immune targets for glioma therapeutics. Neoplasia (New York NY) (2012) 14(8):757–IN26. doi: 10.1593/neo.12794
36. Böttcher JP, Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer (2018) 4(11):786–92. doi: 10.1016/j.trecan.2018.09.001
37. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31(5):711–23.e4. doi: 10.1016/j.ccell.2017.04.003
38. Mittal D, Vijayan D, Putz EM, Aguilera AR, Markey KA, Straube J, et al. Interleukin-12 from cd103+ Batf3-dependent dendritic cells required for nk-cell suppression of metastasis. Cancer Immunol Res (2017) 5(12):1098–108. doi: 10.1158/2326-6066.Cir-17-0341
39. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the cxcr3 chemokine system is required for the efficacy of anti-pd-1 therapy. Immunity (2019) 50(6):1498–512.e5. doi: 10.1016/j.immuni.2019.04.010
40. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. Nk cells stimulate recruitment of cdc1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
41. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for cd8α+ Dendritic cells in cytotoxic T cell immunity. Sci (American Assoc Advancement Science) (2008) 322(5904):1097–100. doi: 10.1126/science.1164206
42. Bowman-Kirigin JA, Desai R, Saunders BT, Wang AZ, Schaettler MO, Liu CJ, et al. The conventional dendritic cell 1 subset primes cd8+ T cells and traffics tumor antigen to drive antitumor immunity in the brain. Cancer Immunol Res (2023) 11(1):20–37. doi: 10.1158/2326-6066.Cir-22-0098
43. Friedrich M, Hahn M, Michel J, Sankowski R, Kilian M, Kehl N, et al. Dysfunctional dendritic cells limit antigen-specific T cell response in glioma. Neuro-Oncology (2022) 25(2):263–76. doi: 10.1093/neuonc/noac138
44. Yin X, Yu H, Jin X, Li J, Guo H, Shi Q, et al. Human blood cd1c+ Dendritic cells encompass cd5high and cd5low subsets that differ significantly in phenotype, gene expression, and functions. J Immunol (2017) 198(4):1553–64. doi: 10.4049/jimmunol.1600193
45. Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt A-L, et al. Cellular differentiation of human monocytes is regulated by time-dependent interleukin-4 signaling and the transcriptional regulator ncor2. Immun (Cambridge Mass) (2017) 47(6):1051–66.e12. doi: 10.1016/j.immuni.2017.11.024
46. Rubio MT, Means TK, Chakraverty R, Shaffer J, Fudaba Y, Chittenden M, et al. Maturation of human monocyte-derived dendritic cells (Modcs) in the presence of prostaglandin E2 optimizes cd4 and cd8 T cell-mediated responses to protein antigens: role of pge2 in chemokine and cytokine expression by modcs. Int Immunol (2005) 17(12):1561–72. doi: 10.1093/intimm/dxh335
47. Mansouri S, Katikaneni DS, Gogoi H, Jin L. Monocyte-derived dendritic cells (Modcs) differentiate into bcl6 + Mature modcs to promote cyclic di-gmp vaccine adjuvant-induced memory T H cells in the lung. J Immunol (1950) (2021) 206(9):2233–45. doi: 10.4049/jimmunol.2001347
48. Dutertre C-A, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity (2019) 51(3):573–89.e8. doi: 10.1016/j.immuni.2019.08.008
49. Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, et al. Transcriptional and functional analysis of cd1c+ Human dendritic cells identifies a cd163+ Subset priming cd8+Cd103+ T cells. Immun (Cambridge Mass) (2020) 53(2):335–52.e8. doi: 10.1016/j.immuni.2020.06.002
50. Chometon TQ, Siqueira M, Sant Anna JC, Almeida MR, Gandini M, Martins de Almeida Nogueira AC, et al. A protocol for rapid monocyte isolation and generation of singular human monocyte-derived dendritic cells. PloS One (2020) 15(4):e0231132–e. doi: 10.1371/journal.pone.0231132
51. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A randomized double-blind placebo-controlled phase ii trial of dendritic cell vaccine ict-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res (2019) 25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261
52. Reap EAEA, Suryadevara CMCM, Batich KAKA, Sanchez-Perez LL, Archer GEGE, Schmittling RJRJ, et al. Dendritic cells enhance polyfunctionality of adoptively transferred T cells which target cytomegalovirus in glioblastoma. Cancer Res (Chicago Ill) (2018) 78(1):256–64. doi: 10.1158/0008-5472.CAN-17-0469
53. Eiraku Y, Terunuma H, Yagi M, Deng X, Nicol AJ, Nieda M. Dendritic cells cross-talk with tumour antigen-specific cd8+ T cells, Vγ9γδt cells and Vα24nkt cells in patients with glioblastoma multiforme and in healthy donors. Clin Exp Immunol (2018) 194(1):54–66. doi: 10.1111/cei.13185
54. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immun (Cambridge Mass) (2019) 50(5):1317–34.e10. doi: 10.1016/j.immuni.2019.03.009
55. Michea P, Noël F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol (2018) 19(8):885–97. doi: 10.1038/s41590-018-0145-8
56. Tomaszewski WH, Waibl-Polania J, Chakraborty M, Perera J, Ratiu J, Miggelbrink A, et al. Neuronal camkk2 promotes immunosuppression and checkpoint blockade resistance in glioblastoma. Nat Commun (2022) 13(1):6483–. doi: 10.1038/s41467-022-34175-y
57. Carenza C, Franzese S, Calcaterra F, Mavilio D, Della Bella S. Comprehensive phenotyping of dendritic cells in cancer patients by flow cytometry. Cytometry Part A (2021) 99(3):218–30. doi: 10.1002/cyto.a.24245
58. Daneman R, Prat A. The blood–brain barrier. Cold Spring Harbor Perspect Biol (2015) 7(1):a020412. doi: 10.1101/cshperspect.a020412
59. Srivastava S, Jackson C, Kim T, Choi J, Lim M. A characterization of dendritic cells and their role in immunotherapy in glioblastoma: from preclinical studies to clinical trials. Cancers (2019) 11(4):537. doi: 10.3390/cancers11040537
60. Arvanitis CD, Ferraro GB, Jain RK. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer (2020) 20(1):26–41. doi: 10.1038/s41568-019-0205-x
61. Mangani D, Weller M, Roth P. The network of immunosuppressive pathways in glioblastoma. Biochem Pharmacol (2017) 130:1–9. doi: 10.1016/j.bcp.2016.12.011
62. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology (2017) 20(2):184–91. doi: 10.1093/neuonc/nox175
63. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of cns immune privilege. Trends Immunol (2015) 36(10):569–77. doi: 10.1016/j.it.2015.08.006
64. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
65. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
66. Brooks WH, Netsky MG, Normansell DE, Horwitz DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med (1972) 136(6):1631–47. doi: 10.1084/jem.136.6.1631
67. Sharma P, Aaroe A, Liang J, Puduvalli VK. Tumor microenvironment in glioblastoma: current and emerging concepts. Neurooncol Adv (2023) 5(1):vdad009. doi: 10.1093/noajnl/vdad009
68. Abadi B, Yazdanpanah N, Nokhodchi A, Rezaei N. Smart biomaterials to enhance the efficiency of immunotherapy in glioblastoma: state of the art and future perspectives. Advanced Drug delivery Rev (2021) 179:114035–. doi: 10.1016/j.addr.2021.114035
69. Lin C, Wang N, Xu C. Glioma-associated microglia/macrophages (Gams) in glioblastoma: immune function in the tumor microenvironment and implications for immunotherapy. Front Immunol (2023) 14:1123853. doi: 10.3389/fimmu.2023.1123853
70. Andersen BM, Faust Akl C, Wheeler MA, Chiocca EA, Reardon DA, Quintana FJ. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat Rev Cancer (2021) 21(12):786–802. doi: 10.1038/s41568-021-00397-3
71. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest (2017) 97(5):498–518. doi: 10.1038/labinvest.2017.19
72. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kückelhaus J, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun (2022) 13(1):925. doi: 10.1038/s41467-022-28523-1
73. Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. . Nat Neurosci (2019) 22(12):2098–110. doi: 10.1038/s41593-019-0532-y
74. Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L, et al. Single-cell rna sequencing reveals evolution of immune landscape during glioblastoma progression. Nat Immunol (2022) 23(6):971–84. doi: 10.1038/s41590-022-01215-0
75. Simonds EF, Lu ED, Badillo O, Karimi S, Liu EV, Tamaki W, et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer (2021) 9(6):e002181. doi: 10.1136/jitc-2020-002181
76. Kilian M, Sheinin R, Tan CL, Friedrich M, Krämer C, Kaminitz A, et al. Mhc class ii-restricted antigen presentation is required to prevent dysfunction of cytotoxic T cells by blood-borne myeloids in brain tumors. Cancer Cell (2023) 41(2):235–51.e9. doi: 10.1016/j.ccell.2022.12.007
77. Sheu T-T, Chiang B-L. Lymphopenia, lymphopenia-induced proliferation, and autoimmunity. Int J Mol Sci (2021) 22(8):4152. doi: 10.3390/ijms22084152
78. Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med (2018) 24(9):1459–68. doi: 10.1038/s41591-018-0135-2
79. Truong NTH, Gargett T, Brown MP, Ebert LM. Effects of chemotherapy agents on circulating leukocyte populations: potential implications for the success of car-T cell therapies. Cancers (2021) 13(9):2225. doi: 10.3390/cancers13092225
80. Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res (2011) 17(16):5473–80. doi: 10.1158/1078-0432.Ccr-11-0774
81. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate egfrviii-expressing tumor cells in patients with glioblastoma. Neuro-Oncology (2010) 13(3):324–33. doi: 10.1093/neuonc/noq157
82. Ghosh S, Huang J, Inkman M, Zhang J, Thotala S, Tikhonova E, et al. Radiation-induced circulating myeloid-derived suppressor cells induce systemic lymphopenia after chemoradiotherapy in patients with glioblastoma. Sci Trans Med (2023) 15(680):eabn6758. doi: 10.1126/scitranslmed.abn6758
83. Gousias K, von Ruecker A, Voulgari P, Simon M. Phenotypical analysis, relation to Malignancy and prognostic relevance of icos + T regulatory and dendritic cells in patients with gliomas. J neuroimmunol (2013) 264(1):84–90. doi: 10.1016/j.jneuroim.2013.09.001
84. Löhr M, Freitag B, Technau A, Krauss J, Monoranu C-M, Rachor J, et al. High-grade glioma associated immunosuppression does not prevent immune responses induced by therapeutic vaccines in combination with T reg depletion. Cancer immunol immunother (2018) 10:1545–58. doi: 10.1007/s00262-018-2214-0
85. Wang M, Cai Y, Peng Y, Xu B, Hui W, Jiang Y. Exosomal lgals9 in the cerebrospinal fluid of glioblastoma patients suppressed dendritic cell antigen presentation and cytotoxic T-cell immunity. Cell Death Dis (2020) 11(10):896. doi: 10.1038/s41419-020-03042-3
86. Pinzon-Charry A, Ho CSK, Laherty R, Maxwell T, Walker D, Gardiner RA, et al. A population of hla-dr+ Immature cells accumulates in the blood dendritic cell compartment of patients with different types of cancer. Neoplasia (2005) 7(12):1112–22. doi: 10.1593/neo.05442
87. Holl EK, Frazier VN, Landa K, Beasley GM, Hwang ES, Nair SK. Examining peripheral and tumor cellular immunome in patients with cancer. Front Immunol (2019) 10:1767. doi: 10.3389/fimmu.2019.01767
88. Adhikaree J, Franks HA, Televantos C, Vaghela P, Kaur AP, Walker D, et al. Impaired circulating myeloid cd1c+ Dendritic cell function in human glioblastoma is restored by P38 inhibition - implications for the next generation of dc vaccines. Oncoimmunology (2019) 8(7):1593803–. doi: 10.1080/2162402X.2019.1593803
89. Prins RM, Everson RG. Commentary on "Dysfunctional dendritic cells limit antigen-specific T cell response in glioma". Neuro-oncol (Charlottesville Va) (2023) 25(2):277–8. doi: 10.1093/neuonc/noac256
90. Autissier P, Soulas C, Burdo TH, Williams KC. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry Part A (2010) 77A(5):410–9. doi: 10.1002/cyto.a.20859
91. Karimi E, Yu MW, Maritan SM, Perus LJM, Rezanejad M, Sorin M, et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nat (London) (2023) 614(7948):555–63. doi: 10.1038/s41586-022-05680-3
92. Hassannia H, Ghasemi Chaleshtari M, Atyabi F, Nosouhian M, Masjedi A, Hojjat-Farsangi M, et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology (2020) 159(1):75–87. doi: 10.1111/imm.13126
93. Zhang J, Shen B, Lin A. Novel strategies for inhibition of the P38 mapk pathway. Trends Pharmacol Sci (2007) 28(6):286–95. doi: 10.1016/j.tips.2007.04.008
94. Migliorini D, Dietrich P-Y, Stupp R, Linette GP, Posey AD, June CH. Car T-cell therapies in glioblastoma: A first look. Clin Cancer Res (2018) 24(3):535. doi: 10.1158/1078-0432.CCR-17-2871
95. Chokshi CR, Brakel BA, Tatari N, Savage N, Salim SK, Venugopal C, et al. Advances in immunotherapy for adult glioblastoma. Cancers (2021) 13(14):3400. doi: 10.3390/cancers13143400
96. Dapash M, Castro B, Hou D, Lee-Chang C. Current immunotherapeutic strategies for the treatment of glioblastoma. Cancers (2021) 13(18):4548. doi: 10.3390/cancers13184548
97. Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, et al. T–cell mediated rejection of gene–modified hiv–specific cytotoxic T lymphocytes in hiv–infected patients. Nat Med (1996) 2(2):216–23. doi: 10.1038/nm0296-216
98. Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, et al. Chimeric antigen receptor engineering: A right step in the evolution of adoptive cellular immunotherapy. Int Rev Immunol (2015) 34(2):154–87. doi: 10.3109/08830185.2015.1018419
99. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Sheih A, et al. High rate of durable complete remission in follicular lymphoma after cd19 car-T cell immunotherapy. Hematological Oncol (2019) 37(S2):170–1. doi: 10.1002/hon.127_2629
100. Cao J, Wang G, Cheng H, Wei C, Qi K, Sang W, et al. Potent anti-leukemia activities of humanized cd19-targeted chimeric antigen receptor T (Car-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am J Hematol (2018) 93(7):851–8. doi: 10.1002/ajh.25108
101. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfusion Med hemotheR (2019) 46(1):15–24. doi: 10.1159/000496870
102. Brown MP, Ebert LM, Gargett T. Clinical chimeric antigen receptor-T cell therapy: A new and promising treatment modality for glioblastoma. Clin Trans Immunol (2019) 8(5):e1050. doi: 10.1002/cti2.1050
103. Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, et al. Current progress in chimeric antigen receptor T cell therapy for glioblastoma multiforme. Cancer Med (2021) 10(15):5019–30. doi: 10.1002/cam4.4064
104. Gardeck AM, Sheehan J, Low WC. Immune and viral therapies for Malignant primary brain tumors. Expert Opin Biol Ther (2017) 17(4):457–74. doi: 10.1080/14712598.2017.1296132
105. Ebert LM, Yu W, Gargett T, Brown MP. Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochem Soc Trans (2018) 46(2):391–401. doi: 10.1042/BST20170178
106. Tang X, Wang Y, Huang J, Zhang Z, Liu F, Xu J, et al. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduction Targeted Ther (2021) 6(1):125. doi: 10.1038/s41392-021-00505-7
107. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused egfrviii-directed car T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
108. Elahi R, Khosh E, Tahmasebi S, Esmaeilzadeh A. Immune cell hacking: challenges and clinical approaches to create smarter generations of chimeric antigen receptor T cells. Front Immunol (2018) 9:1717. doi: 10.3389/fimmu.2018.01717
109. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation car T cells against solid cancers. J Hematol Oncol (2020) 13(1):54–. doi: 10.1186/s13045-020-00890-6
110. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation car T cells. Br J Cancer (2019) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
111. Chmielewski M, Abken H. Car T cells releasing il-18 convert to T-bethigh foxo1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
112. Hombach A, Barden M, Hannappel L, Chmielewski M, Rappl G, Sachinidis A, et al. Il12 Integrated into the Car Exodomain Converts Cd8+ T cells to Poly-Functional Nk-Like Cells with Superior Killing of Antigen-Loss Tumors. Mol Ther (2022) 30(2):593–605. doi: 10.1016/j.ymthe.2021.10.011
113. Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human il-12. Cancer Med (2019) 8(10):4753–65. doi: 10.1002/cam4.2361
114. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. Il-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology (2015) 4(3):e994446. doi: 10.4161/2162402X.2014.994446
115. Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Núñez N, Stowe CL, et al. Intratumoral il-12 delivery empowers car-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun (2021) 12(1):444. doi: 10.1038/s41467-020-20599-x
116. Gargett T, Ebert LM, Truong NTH, Kollis PM, Sedivakova K, Yu W, et al. Gd2-targeting car-T cells enhanced by transgenic il-15 expression are an effective and clinically feasible therapy for glioblastoma. J Immunother Cancer (2022) 10(9):e005187. doi: 10.1136/jitc-2022-005187
117. Swan SL, Mehta N, Ilich E, Shen SH, Wilkinson DS, Anderson AR, et al. Il7 and il7 flt3l co-expressing car T cells improve therapeutic efficacy in mouse egfrviii heterogeneous glioblastoma. Front Immunol (2023) 14:1085547. doi: 10.3389/fimmu.2023.1085547
118. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor flt3l drives epitope spreading and antitumor immunity. Nat Immunol (2020) 21(8):914–26. doi: 10.1038/s41590-020-0676-7
119. Kuhn NF, Lopez AV, Li X, Cai W, Daniyan AF, Brentjens RJ. Cd103+ Cdc1 and endogenous cd8+ T cells are necessary for improved cd40l-overexpressing car T cell antitumor function. Nat Commun (2020) 11(1):6171. doi: 10.1038/s41467-020-19833-3
120. Datsi A, Sorg RV. Dendritic cell vaccination of glioblastoma: road to success or dead end. Front Immunol (2021) 12:770390. doi: 10.3389/fimmu.2021.770390
121. Lesterhuis WJ, de Vries IJM, Schreibelt G, Lambeck AJA, Aarntzen EHJG, Jacobs JFM, et al. Route of administration modulates the induction of dendritic cell vaccine–induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res (2011) 17(17):5725–35. doi: 10.1158/1078-0432.Ccr-11-1261
122. Zhang H, Wang Y, Wang Q-T, Sun S-N, Li S-Y, Shang H, et al. Enhanced human T lymphocyte antigen priming by cytokine-matured dendritic cells overexpressing bcl-2 and il-12. Front Cell Dev Biol (2020) 8:205. doi: 10.3389/fcell.2020.00205
123. Batich KA, Mitchell DA, Healy P, Herndon JE, Sampson JH. Once, twice, three times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in glioblastoma. Clin Cancer Res (2020) 26(20):5297–303. doi: 10.1158/1078-0432.CCR-20-1082
124. Akiyama Y, Oshita C, Kume A, Iizuka A, Miyata H, Komiyama M, et al. A-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: A phase I clinical trial. BMC Cancer (2012) 12(1):623. doi: 10.1186/1471-2407-12-623
125. Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in Malignant glioma patients. J Immunother (2013) 36(2):277–8. doi: 10.1097/CJI.0b013e3182811ae4
126. Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: A phase ii randomized trial. Cancers (2018) 10(10):372. doi: 10.3390/cancers10100372
127. Wang Q-T, Nie Y, Sun S-N, Lin T, Han R-J, Jiang J, et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother (2020) 69(7):1375–87. doi: 10.1007/s00262-020-02496-w
128. Zhang Q, Liu F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for Malignant gliomas. Cell Death Dis (2020) 11(6):485. doi: 10.1038/s41419-020-2696-5
129. Liu P, Wang Y, Wang Y, Kong Z, Chen W, Li J, et al. Effects of oncolytic viruses and viral vectors on immunity in glioblastoma. Gene Ther (2020) 29:115–26. doi: 10.1038/s41434-020-00207-9
130. King GD, Muhammad AK, Curtin JF, Barcia C, Puntel M, Liu C, et al. Flt3l and tk gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol (2008) 10(1):19–31. doi: 10.1215/15228517-2007-045
131. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of cd103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic pd-L1 and braf inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
132. Ali S, Curtin JF, Zirger JM, Xiong W, King GD, Barcia C, et al. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble fms-like tyrosine kinase 3 ligand (Hsflt3l): treatment with hsflt3l inhibits intracranial glioma progression. Mol Ther (2004) 10(6):1071–84. doi: 10.1016/j.ymthe.2004.08.025
133. Dapash M, Hou D, Castro B, Lee-Chang C, Lesniak MS. The interplay between glioblastoma and its microenvironment. Cells (2021) 10(9):2257. doi: 10.3390/cells10092257
134. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev (2009) 229(1):126–44. doi: 10.1111/j.1600-065X.2009.00771.x
135. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive tumor-infltrating myeloid cells mediate adaptive immune resistance via a pd-1/pd-L1 mechanism in glioblastoma. Neuro-oncol (Charlottesville Va) (2017) 19(6):796–807. doi: 10.1093/neuonc/now287
136. Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. Pd-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight (2016) 1(10):e87059. doi: 10.1172/jci.insight.87059
137. Aslan K, Turco V, Blobner J, Sonner JK, Liuzzi AR, Núñez NG, et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat Commun (2020) 11(1):931. doi: 10.1038/s41467-020-14642-0
138. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of checkmate 143. Neuro-oncology (2018) 20(5):674–86. doi: 10.1093/neuonc/nox208
139. Omuro A, Reardon DA, Sampson JH, Baehring J, Sahebjam S, Cloughesy TF, et al. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: results from exploratory phase I cohorts of checkmate 143. Neuro-oncol Adv (2022) 4(1):vdac025–vdac. doi: 10.1093/noajnl/vdac025
140. Prokopi A, Tripp CH, Tummers B, Hornsteiner F, Spoeck S, Crawford JC, et al. Skin dendritic cells in melanoma are key for successful checkpoint blockade therapy. J Immunother Cancer (2021) 9(1):e000832. doi: 10.1136/jitc-2020-000832
141. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi: 10.1038/nm1622
142. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating dcs suppress nucleic acid–mediated innate immune responses through interactions between the receptor tim-3 and the alarmin hmgb1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
143. Holderried TAW, de Vos L, Bawden EG, Vogt TJ, Dietrich J, Zarbl R, et al. Molecular and immune correlates of tim-3 (Havcr2) and galectin 9 (Lgals9) mrna expression and DNA methylation in melanoma. Clin Epigenet (2019) 11(1):161. doi: 10.1186/s13148-019-0752-8
144. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. Pd-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun (2020) 11(1):4835–. doi: 10.1038/s41467-020-18570-x
145. Cohen M, Giladi A, Barboy O, Hamon P, Li B, Zada M, et al. The interaction of cd4+ Helper T cells with dendritic cells shapes the tumor microenvironment and immune checkpoint blockade response. Nat Cancer (2022) 3(3):303–17. doi: 10.1038/s43018-022-00338-5
146. Ma HS, Poudel B, Torres ER, Sidhom J-W, Robinson TM, Christmas B, et al. A cd40 agonist and pd-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell–mediated anticancer activity. Cancer Immunol Res (2019) 7(3):428–42. doi: 10.1158/2326-6066.Cir-18-0061
147. Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of cd40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci (2020) 117(14):8022–31. doi: 10.1073/pnas.1918971117
148. Lemech C, Dredge K, Bampton D, Hammond E, Clouston A, Waterhouse NJ, et al. Phase ib open-label, multicenter study of pixatimod, an activator of tlr9, in combination with nivolumab in subjects with microsatellite-stable metastatic colorectal cancer, metastatic pancreatic ductal adenocarcinoma and other solid tumors. J Immunother Cancer (2023) 11(1):e006136. doi: 10.1136/jitc-2022-006136
149. Clinicaltrials.Gov: U.S. National Library of Medicine (2023). Available at: https://www.clinicaltrials.gov/ct2/home.
150. Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant pd-1 blockade induces T cell and cdc1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun (2021) 12(1):6938–. doi: 10.1038/s41467-021-26940-2
151. Putranto TA, Wibisono D, Astoro NW, Yana ML, Rantung Y, Manuaba IBAP. Introduction to dendritic cell vaccines immunotherapy for glioblastoma multiforme : A novel approach. Bali Med J (2019) 8(1):371–5. doi: 10.15562/bmj.v8i1.1500
152. Dutoit V, Philippin G, Widmer V, Marinari E, Vuilleumier A, Migliorini D, et al. Impact of radiochemotherapy on immune cell subtypes in high-grade glioma patients. Front Oncol (2020) 10:89. doi: 10.3389/fonc.2020.00089
153. Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer (2015) 113(2):232–41. doi: 10.1038/bjc.2015.238
154. Hsu AK, Quach H, Tai T, Prince HM, Harrison SJ, Trapani JA, et al. The immunostimulatory effect of lenalidomide on nk-cell function is profoundly inhibited by concurrent dexamethasone therapy. Blood (2011) 117(5):1605–13. doi: 10.1182/blood-2010-04-278432
155. Grabowski MM, Sankey EW, Ryan KJ, Chongsathidkiet P, Lorrey SJ, Wilkinson DS, et al. Immune suppression in gliomas. J Neuro-Oncol (2021) 151(1):3–12. doi: 10.1007/s11060-020-03483-y
156. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest (2010) 120(5):1368–79. doi: 10.1172/JCI41911
Keywords: cancer, immunotherapies, dendritic cells, glioblastoma, T cells, combination therapies, CAR-T cells, brain tumor
Citation: Gardam B, Gargett T, Brown MP and Ebert LM (2023) Targeting the dendritic cell-T cell axis to develop effective immunotherapies for glioblastoma. Front. Immunol. 14:1261257. doi: 10.3389/fimmu.2023.1261257
Received: 19 July 2023; Accepted: 09 October 2023;
Published: 20 October 2023.
Edited by:
You-Wen He, Duke University, United StatesReviewed by:
Marc Garcia-Moure, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Gardam, Gargett, Brown and Ebert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Ebert, bGlzYS5lYmVydEBzYS5nb3YuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.