 Vipin Kumar
Vipin Kumar Marc Hertz
Marc Hertz Albert Agro2
Albert Agro2 Adam J. Byrne
Adam J. Byrne
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 25 September 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1260503
This article is part of the Research TopicCommunity Series in the Role of CD1- and MR1-restricted T cells in Immunity and Disease, volume IIView all 18 articles
Chronic tissue inflammation often results in fibrosis characterized by the accumulation of extracellular matrix components remodeling normal tissue architecture and function. Recent studies have suggested common immune mechanisms despite the complexity of the interactions between tissue-specific fibroblasts, macrophages, and distinct immune cell populations that mediate fibrosis in various tissues. Natural killer T (NKT) cells recognizing lipid antigens bound to CD1d molecules have been shown to play an important role in chronic inflammation and fibrosis. Here we review recent data in both experimental models and in humans that suggest a key role of type 1 invariant NKT (iNKT) cell activation in the progression of inflammatory cascades leading to recruitment of neutrophils and activation of the inflammasome, macrophages, fibroblasts, and, ultimately, fibrosis. Emerging evidence suggests that iNKT-associated mechanisms contribute to type 1, type 2 and type 3 immune pathways mediating tissue fibrosis, including idiopathic pulmonary fibrosis (IPF). Thus, targeting a pathway upstream of these immune mechanisms, such as the inhibition of iNKT activation, may be important in modulating various fibrotic conditions.
Fibrosis is characterized by the deposition of extracellular matrix components, such as collagen and fibronectin. The fibrotic process can affect different tissues and represents an ultimate outcome of chronic inflammatory diseases (1). Although collagen deposition can be a reversible repair process in response to injury, repetitive insult and chronic inflammation can lead to a dysregulation of the wound-healing response and progression to fibrosis (1–4). The fibrotic niche is comprised of several immune cells that drive key steps in the fibrotic process, including neutrophils, mast cells, macrophages, fibroblasts, and NKT cells (5). Here we review some of the recent findings on how a particular NKT cell subset, iNKT cells, play a key role in chronic inflammation and fibrosis in both lung and liver diseases. Although fibrosis is also a major feature of several chronic inflammatory diseases, here we will primarily focus on recent data supporting a key role of iNKT cell activation in the progression of fibrosis in IPF and non-alcoholic steatohepatitis (NASH). IPF is an interstitial lung disease characterized by progressive lung scarring with a median survival of 2-3 years after diagnosis (6). NASH is a leading cause of global liver disease fueled by increasing rates of obesity, diabetes, and metabolic syndrome and is characterized by inflammation, injury, and accumulation of fat in the liver (7). Currently, no therapeutic solution exists that halt disease progression of IPF or NASH and a better understanding of the cellular and molecular mechanisms involved in progressive fibrotic disease is necessary for the development of new therapeutic interventions. We will briefly review different NKT cell subsets, important parameters to consider when characterizing NKT cells in health and disease and highlight some of the key pathways that are impacted by iNKT activation leading to fibrosis. Finally, we will propose a model of complex cellular interactions of different iNKT subsets secreting type 1, type 2 and type 3 cytokines impacting TGF-β-associated macrophage activation, neutrophil infiltration, fibroblast transformation and fibrosis.
NKT cells are a group of innate-like T cells expressing NK cell receptors and antigen T cell receptors (TCR) that recognize lipid antigens presented in the context of a class I MHC-like, non-polymorphic molecule CD1d (8–10). NKT cells become rapidly activated following either TCR recognition of the CD1d-bound lipid antigen or following inflammatory cytokine-associated signaling and produce large amounts of type 1, type 2 or type 3 cytokines. Accordingly, NKT1 predominantly secrete GM-CSF and IFN-γ, NKT2 secrete IL-4, IL-5, and IL-13, and NKT17 secrete IL-17A and IL-22. NKT cells have diverse functions bridging adaptive and innate immune responses. NKT cells are comprised of two main subsets, iNKT cells and type 2 NKT cells (10–12). While both NKT cell subsets are predominantly NK1.1+ (mouse) or CD161+/CD56+ (human) and share common features in mice and in humans (13–15), they can be differentiated by their TCR alpha and beta-chain usage and antigen recognition. Since not all murine strains express the NK1.1 marker, and given its surface expression is modulated following iNKT activation, NK1.1 expression alone is not a good marker for NKT cell identification (see Box 1). iNKT cells mostly express semi-invariant germ line encoded TCR α-chain (75-88%), Vα14/Jα18 in mice and Vα24/Jα18 in humans, paired with a more diverse non-germ line Vβ genes, Vβ8.2, Vβ7 or Vβ2 in mice and Vβ11 in humans, and can be identified using α-galactosyl ceramide (αGalCer)-loaded CD1d-tetramers or a clonotypic TCR-specific mAb. In contrast, type 2 NKT cells express a relatively diverse TCR repertoire. Type 2 NKT cells are not reactive to αGalCer and a major subset recognizes a self-glycolipid 3-sulfated β-galactosyl ceramide (sulfatide) (16, 17). iNKT and type 2 NKT cells also display distinct modes of antigen recognition (18, 19). More importantly, these subsets can have opposite immune functions. iNKT cells promote inflammatory disease while type 2 NKT cells are protective in several chronic inflammatory diseases (20, 21). A protective role of appropriately activated type 2 NKT cells in inflammatory and autoimmune disease has recently been reviewed (22–24) and will not be included here.
Based on the expression of transcription factors and cytokines, mouse and human iNKT cells can be divided into NKT1, NKT2 and NKT17 subsets, similar to CD4+ helper T subsets, and accordingly affect distinct type 1, type 2 and type 3 immune pathways, respectively (25–28). NKT1 express T-bet and predominantly secrete GM-CSF and IFN-γ, NKT2 express GATA3, PLZFhigh and secrete IL-4, IL-5, and IL-13, and NKT17 express RORγt and secrete IL-17A and IL-22 (25–30). It is clear that their innate-like features, including rapid response in hours, place them at the frontline of responses against infection, tumor, and tissue injury. Generally, iNKT cells are tissue-resident and enriched in peripheral tissues, such as lung and liver, and play an important role in tissue homeostasis and immunity (25, 26). Recently, another subset of iNKT cells has been identified in both mouse and in humans referred to as circulating iNKT cells that are distinct developmentally from the tissue-resident iNKT cells (31). These cells are circulating and are CD244+CXCR6+, whereas tissue resident cells are CD244-CXCR6+. It is interesting that tissue-resident iNKT cells are preferentially localized in lung parenchyma, as opposed to the blood vessels where most circulating iNKT cells as well as other lymphocytes typically reside (31). iNKT cell subsets with circulating properties can perhaps sense tissue injuries from chronic inflammation and orchestrate systemic immunity through the activation of a downstream cascade of other immune cells. Thus, circulating and tissue-resident iNKT cell subsets are analogous to the group 1 innate lymphoid cells (ILCs) with tissue resident ILCs and conventional circulating NK cells, and together they provide diverse systemic and tissue-specific immune regulation.
BOX 1 Challenges in characterizing iNKT cells.
•NKT cells can be categorized into two subsets based upon whether they express a semi-invariant or a diverse TCR. Murine and human iNKT cells, but not type 2 NKT cells, recognize the foreign marine sponge-derived glycolipid, α-galactosylceramide (αGalCer), and αGalCer/CD1d-tetramers can be used for the monitoring iNKT cells in both mice and humans (8, 32). Murine type 2 NKT cells, but not iNKT cells, recognize the self-glycolipid, 3-sulfated β-galactosyl ceramide (sulfatide), and the self-lysophospholipid, lysophosphatidylcholine (LPC) (16, 33) and sulfatide/CD1d-tetramers can be used for the monitoring of type 2 NKT cells in mice.
•αGalCer is a high affinity binder for CD1d and TCR and acts like a super antigen for iNKT cells that can activate most iNKT cells in a non-physiological manner. In contrast, self-lipids recognized by iNKT cells, are generally low affinity binders for CD1d or TCR, do not behave like αGalCer (34). αGalCer activation of iNKT cells may skew iNKT responses from a predominantly pathogenic Th1 response to a predominantly protective Th2 repsonse (35–38). Data using αGalCer should be cautiously interpreted regarding the physiological role of iNKT cells.
•A number of studies in humans have used CD3+CD56+ cells as a marker for NKT cells. It is clear from several studies that CD3+CD56+ cells are comprised of population of unconventional T cells, including iNKT cells, MAIT cells, γδ T cells, type 2 NKT cells and a subset of CD8+ T cells (39). Data reporting CD3+CD56+ cells as NKT cells should be interpreted with this in mind.
•It is also important to use tetramer staining in combination with intracytoplasmic cytokine, chemokine staining, or transcription profiling to determine the function of iNKT cells. Absolute iNKT cell number alone, or cell surface expression of TCR, may not provide a complete picture of the specific in vivo role of iNKT cells subsets. At different times during a chronic inflammation different iNKT cell subset may be activated that have distinct roles in the inflammatory cascade (40, 41).
•Chronically activated iNKT cells downregulate their TCR surface expression, thus, reduced cell surface staining may not accurately reflect the absolute number of iNKT cells. A qPCR approach specific for the conserved VJ-region of the invariant TCR, in combination with the tetramer staining, may more accurately reveal their frequency as well as their state of activation (40).
•It should be emphasized that iNKT cells may have a protective role during initial acute inflammation (42–46) whereas chronic activation of iNKT cells may induce a more pathogenic role. Therefore, murine models that are generally acute in nature may not accurately reflect the role of iNKT cells in human chronic conditions such as lung or liver fibrosis.
In several murine models of chronic inflammation and fibrosis, as well as in humans, it has been shown that iNKT cells are selectively activated and play a pathogenic role. For example, in pulmonary fibrosis, carbon tetrachloride (CCL4)-induced fibrosis, ischemia reperfusion injury, Con A-induced hepatitis, primary biliary cirrhosis, Lieber-DeCarli liquid alcohol diet, and choline-deficient amino acid enriched (CDAA) diet, iNKT cells become activated and are pathogenic (18–21, 41, 47, 48). In some tissues, iNKT cells accumulate significantly following injury (41, 47, 49) and the accumulation is dependent on chemokine receptor CXCR6 interactions (50). Thus, in Jα18-/- mice genetically deficient in iNKT cells, but not type 2 NKT cells, both inflammation and fibrosis are significantly ameliorated (41, 47, 51). Consistent with chronic activation, iNKT cells express FAS/FASL and downregulate or internalize their TCR surface expression as shown by reduced staining with CD1d-tetramers (52–54). It is important to mention that the activation of iNKT cells in tissue may be dependent on the presence of CD1d in parenchymal cells in lung, kidney, skin, and heart tissues commonly affected by fibrosis. For example, it has been shown that the CD1d expression in hepatocytes is necessary for the iNKT activation during inflammatory liver injury (55). It is likely that CD1d expression in tissue-resident cells, such as hepatocytes, is needed for either in situ lipid presentation or that CD1d recognition locally is required for full activation or maintenance of iNKT cells in chronic inflammatory conditions. Consistent with the experimental data, iNKT cells are chronically activated and secrete significantly higher levels of proinflammatory cytokines in NASH, severe alcoholic hepatitis, lupus nephritis and IPF patients in comparison to healthy volunteers (23, 40, 56). Importantly, the increase of IFN-γ+iNKT cells correlates with the progression of NAFLD Activity Score (NAS) and fibrosis staging in patients (40, 56). These data are consistent with earlier reports in NASH patients that quantified T cell markers (CD56+CD3+) that are not specific for NKT cells and may include other T cell populations as described in Box 1 (51). Another important finding during fibrosis is a significant increase in adaptive T cells, such as CD8+ T cells in both mice (40, 57) and in humans (58–60). Accordingly, inhibition of iNKT cells results in a decrease of infiltrating CD8+ T cells into liver tissue (40, 41, 57). It is also important to mention that genetic deficiency or inhibition of iNKT cells also leads to significantly reduced macrophages, conventional T cell accumulation and activation, as well as a reduction in the levels of key inflammatory and pro-fibrogenic cytokines including IL-1β, IL-6, and TNFα in fibrotic tissues in mice (23, 40, 41, 47). Among other cells in the fibrotic niche, stellate cell activation has also been shown to be associated with iNKT cell activation (61). Consistent with a critical pathogenic role of iNKT cells, specific inhibition of iNKT cells with a clinically relevant RARβγ agonist, tazarotene, significantly protects mice from hepatic fibrosis (23, 40, 41, 57). One of the key features that provides tazarotene specificity for iNKT cells, and not type 2 NKT cells or conventional T cells, is the significant upregulation of RARγ receptors in iNKT cells compared to these other T cell populations (41).
Similarly, in the bleomycin-induced lung injury model of pulmonary fibrosis, inhibition or skewed activation of iNKT cells by sulfatide or αGalCer, respectively, leads to inhibition of vimentin concentration in lung tissues as well as reduction of fibrosis-promoting cytokines, including TGF-β, IL-5, and IL-13 (48, 49, 62). Inhibition of iNKT cells also results in dampening of arginase+ alveolar macrophages involved in fibrosis (48, 49, 62). Furthermore, manipulation of αGalCer-mediated iNKT-STAAT1-CXCL9 axis has been shown to contribute to vessel fibrosis in pulmonary hypertension caused by lung fibrosis (63). Thus IFN-γ secretion following αGalCer administration inhibits type 2 cytokine-mediated fibrotic responses. The bleomycin-induced model of lung fibrosis used as a model of IPF recapitulates several features but does not accurately replicate the human resolving disease, IPF. For example, the bleomycin model can be induced in recombination-activating gene (RAG) deficient mice that lack mature B and T cells (49) and therefore does not involve a full spectrum of chronic inflammatory components like that observed in the progression of IPF (2, 64–66). Nevertheless, in this model manipulation of the iNKT cytokine response with the super antigen-like ligand αGalCer or following type 2 NKT activation via sulfatide results in the inhibition of TGF-β dependent pathway, blunting fibrosis (48, 49, 67). Consistent with the iNKT involvement in experimental model of lung fibrosis, a significantly increased frequency of IFN-γ+NKT cells in bronchial alveolar lavage (BAL) fluid relative to healthy volunteers also correlates with increased alveolar macrophages, a predictive marker of disease progression in IPF patients. Collectively, data in experimental models as well as in NASH, alcoholic liver disease, and IPF patients implicate iNKT cells in the pathogenesis of fibrosis in humans.
Chronic inflammation plays a key role in promoting tissue fibrosis. Several studies have shown that inflammation associated with fibrosis in both lung and in liver tissues is comprised of a mixture of type 1, type 2 and type 3 cytokine-associated immune pathways (1, 2, 5, 65, 66, 68). Although not clearly understood, several immune cells likely participate or are influenced by the dynamic secretion of these cytokines in the fibrotic tissues eventually leading to macrophage activation and fibroblast transformation. The differential cytokines secreted by iNKT cell subsets can influence type 1 (GM-CSF and IFN-γ), type 2 (IL-4, IL-5, IL-13) and type 3 (IL-17A and IL-22) immune responses (69). The three defined iNKT cell subsets (NKT1, NKT2, NKT17) are present in chronic inflammatory tissues, including the lung and liver. It is likely that with the progression of fibrosis, one or more iNKT cell subset(s) become dominant: for example, NKT17 becomes less prevalent in comparison to NKT1 and NKT 2 subsets with the progression of experimental hepatic fibrosis (40). Thus, cytokines secreted by iNKT subsets can contribute to different pathways of tissue fibrosis at different stages of disease. For example, IFN-γ secreting pulmonary NKT1 cell activation exacerbates experimental lung injury (70). Type 2 cytokines, such as, IL-4, IL-5 and IL-13 are associated with both innate and adaptive immune cells, including NKT2, Th2, ILC2, eosinophils, and IL-13 activated macrophages that are normally involved in tissue repair (2). However, dysregulation or overt activation of the repair process contributes to infection and allergen-driven fibrosis in several organs, including the lung (71–73). In chronic inflammatory lung disease, pathogenic IL-13 producing macrophages are stimulated by a CD1d-dependent iNKT cell interaction both in experimental models as well as in chronic obstructive pulmonary disease (COPD) patients (62). Consistently, in IPF patients, high levels of IL-13 and IL-13R have been found in both blood and in the lungs (2, 74, 75). In a humanized severe combined immunodeficiency (SCID) mouse model of IPF, humanized anti-IL-13 antibody treatment results in a significant reduction in fibrosis and increased epithelial repair (76). Additionally, humanized monoclonal anti-IL-13 antibody treatment has shown some promise in a subset of severe asthma patients (77) but has failed to meet primary efficacy endpoints in IPF clinical trials (78, 79). It is important to emphasize that IL-13-driven fibrosis is both TGF-β-dependent (2, 72, 80) and TGF-β independent (81). Thus, IL-4 and IL-13 cytokines can activate arginase+ macrophages and fibroblasts driving the progression of fibrosis (2). Importantly, type 2 cytokine driven fibroblast activation is not only important but also necessary for the development of hepatic fibrosis (82). Thus, type 2 cytokines in a TGF-β-dependent and independent fashion can have significant impact on two key cell types, fibroblasts, and macrophages, driving the progression of tissue fibrosis. Additionally, chronic activation of iNKT cells secreting IL-4 and IL-5 cytokines results in lung disease where animals develop COPD-like symptoms including increased mucus, fibrosis, and emphysema (83). Notably, patients with COPD also have increased number of iNKT cells in PBMCs (84). Furthermore, OVA-induced airway hypersensitivity is significantly abrogated in iNKT cell deficient Jα18-/- mice with decreases in IL-4, IL-5, and IL-13 cytokines in BAL fluid (85). NKT2-mediated IL-4 signaling has also been shown to promote macrophage activation and fibroblast to myofibroblast transition in renal fibrosis (86). As detailed below, tissue resident NKT17 cells play a key role in neutrophil infiltration into injured tissues, and can subsequently influence other cells, such as macrophages, γδ T cells and conventional Th17 responses. Additionally, IL-17A produced by iNKT cells has been shown to promote liver fibrosis in patients with primary biliary cholangitis (87). Collectively, these data suggest that iNKT cells play an important role in influencing all three cytokine associated pathways involved in tissue fibrosis, including IPF.
One of the hallmarks of inflammatory diseases leading to fibrosis is the infiltration of neutrophils into tissues that are critical regulators of both adaptive and innate immunity (88). We found that neutrophil accumulation into fibrotic liver tissue is dependent on the activation of iNKT cells (40, 41, 47). This is due to the inhibition of the upregulation of several cytokines and chemokines, including MIP-1, MIP-2, IL-6, and osteopontin that are involved in the neutrophil infiltration into tissues following injury. Notably, in non-inflamed lung NKT17 is predominant, whereas NKT1 predominates in the liver of naive mice (25, 89). However, following chronic inflammation and fibrosis, NKT17 replaces NKT1 as the dominant subset, and ultimately as the disease progresses NKT1 and NKT2 subsets dominate in mice as well as in humans (40, 90). In comparison to other lymphocytes, iNKT cells are also abundantly present in the lung vasculature and the interstitial tissue of both mice and humans (25, 91). Similar to circulating iNKT cells, NKT17 predominate within the interstitial tissues, whereas NKT1 and NKT2 are predominantly present in the vasculature. Injury or infections, such as streptococcus preumoniae, has been shown to induce expansion and vascular extravasation of iNKT cells in a CCL2-dependent fashion, and the iNKT cells are intimately connected to neutrophils infiltration (92). Notably, in asthma and airway hypersensitivity models, IL-17A-secreting NKT17 are required for the infiltration of neutrophils into lung tissues (93, 94). Accordingly, iNKT cell activation enhances inflammation in asthma models, and iNKT cell-deficient mice have reduced airway hypersensitivity (25). Recent studies using scRNAseq data of macrophage populations in fibrotic tissues from both liver and lung tissues from NASH and IPF patients, respectively, have clarified the heterogeneity of “scar-associated macrophages” (SAMs) involved in fibrosis (65, 66). These SAMs are a subset of CD9+TREM2+ macrophages that express SPP1, GPNMB, FABP5 and CD63 and can differentiate from monocytes with type 3 cytokines GM-CSF, IL-17A and TGF-β (65). Furthermore, MMP9+ neutrophils that participate in the activation of TGF-β and secrete type 3 cytokines (GM-CSF and IL-17A) are co-clustered with these SAMs in the fibrotic tissue. Accordingly, in murine models, blockade of GM-CSF, IL-17A and TGF-β significantly inhibited the expansion of these FABP5+ SAMs and hepatic and pulmonary fibrosis. These studies indicate that IL-17A, GM-CSF and TGF-β can collaboratively induce monocytes-to-FABP5+ SAM differentiation and promote pathogenic collagen deposition by mesenchymal cells in IPF and NASH patients (65). Collectively, IL-17A signaling associated with NKT17, γδ T cells and neutrophils in the fibrotic niche plays a critical role in fibrosis progression in both IPF and NASH patients.
Several lines of investigation indicate that the NLRP3 inflammasome activation is an important step that drives pro-fibrotic changes in tissue, and that this inflammatory complex could contribute significantly to both IPF and liver fibrosis. Thus, cell specific NLRP3 inflammasome activation in myeloid cells or in neutrophils resulted in extensive hepatic inflammation in parenchyma followed by fibrogenesis and fibrosis in murine NASH models (95–97). These data are consistent with the earlier findings that showed NLRP3 activation blockade leads to inhibition of chronic inflammation and fibrosis (98). Similarly, caspase 1 and NLRP3 knockout mice are protected against hepatitis and associated liver fibrosis (99). Notably, both inflammasome activation and infiltration of immune cells, including neutrophils, is dependent on the presence of activated iNKT cells in experimental models (40, 41, 47). Pro-inflammatory iNKT cells produces TNFα and IL-17A and it is likely that iNKT cell derived cytokines are required for priming of the NLRP3 inflammasome. Consistent with our hypothesis, it has been shown that iNKT cell-derived TNFα was required for the optimal secretion of IL-1α and IL-1β by myeloid cells in response to iNKT cell activation (100, 101). A critical role of TNFα and IL-17A as mediators of tissue fibrosis induced by constitutive NLRP3 activation in myeloid cells has recently been shown (102) (103). NLRP3 activation has also been implicated in interstitial kidney fibrosis involving Smad3 activation that promotes TGF-β signaling (103). Similarly, inflammation and wound healing repair response to injury in lung depend on NLRP3 activation that maintain a balance for MMPs and TIMPS involved in lung fibrosis (4). Collectively, inflammasome activation, an important step in tissue fibrosis, is also critically impacted by the activation of iNKT cells.
Mechanisms driving fibrosis following initial tissue injury are complex and involve key interactions among several immune cells, including macrophages and fibroblasts. As stated in the sections above, recent investigations in experimental models as well as in IPF and NASH patients suggest a key involvement of common or conserved immune pathways in fibrosis in different tissues. Alarmins (IL-25, TSLP) are the earliest cytokines secreted following tissue damage and they contribute to fibrosis indirectly with actions from type 2 cytokine-secreting immune cells, such as NKT2, ILC2, CD4+Th2 cells, etc. Thus IL-25 and IL-13 secreting ILC2 are also found in IPF patients (104). Aside from the important role of TGF-β in fibrosis, type 1, type 2 and type 3 cytokine-associated immune responses are also key inducers of fibrosis (1, 2, 5, 65, 66, 68). iNKT cell derived cytokines, chemokines, and their interactions with other immune cells in the fibrotic milieu play an important role in the progression of tissue inflammation and fibrosis: 1) iNKT subsets, NKT1, NKT2 and NKT17 have major influences on type 1, type 2 and type 3 immunity and are implicated in the progression fibrosis (23, 28, 40, 41, 47, 49, 56, 105); 2) infiltration and the accumulation of neutrophils following tissue injury is inhibited in the absence iNKT cells (21, 23, 40, 41); 3) adaptive Th1/Th2 skewing of adaptive immunity has been shown to be driven by an early activation of iNKT cells; 4) NKT2 interactions with type 2 driven lymphocytes, including eosinophils and ILC2, should play an important role in type 2 cytokine immune responses (25, 27, 28, 30, 34, 106, 107); 5) additionally, iNKT activation also has been shown to promote Hh-Wnt signaling that plays a key role in the transition of epithelial cells into myofibroblasts that deposit matrix proteins (28, 51). Based upon data in experimental models and in humans and the fact that iNKT cell activation is a common upstream pathway in the propagation of chronic inflammation driving fibrosis, we propose a mechanism highlighting key events in the progression of lung fibrosis in IPF (see Figure 1).
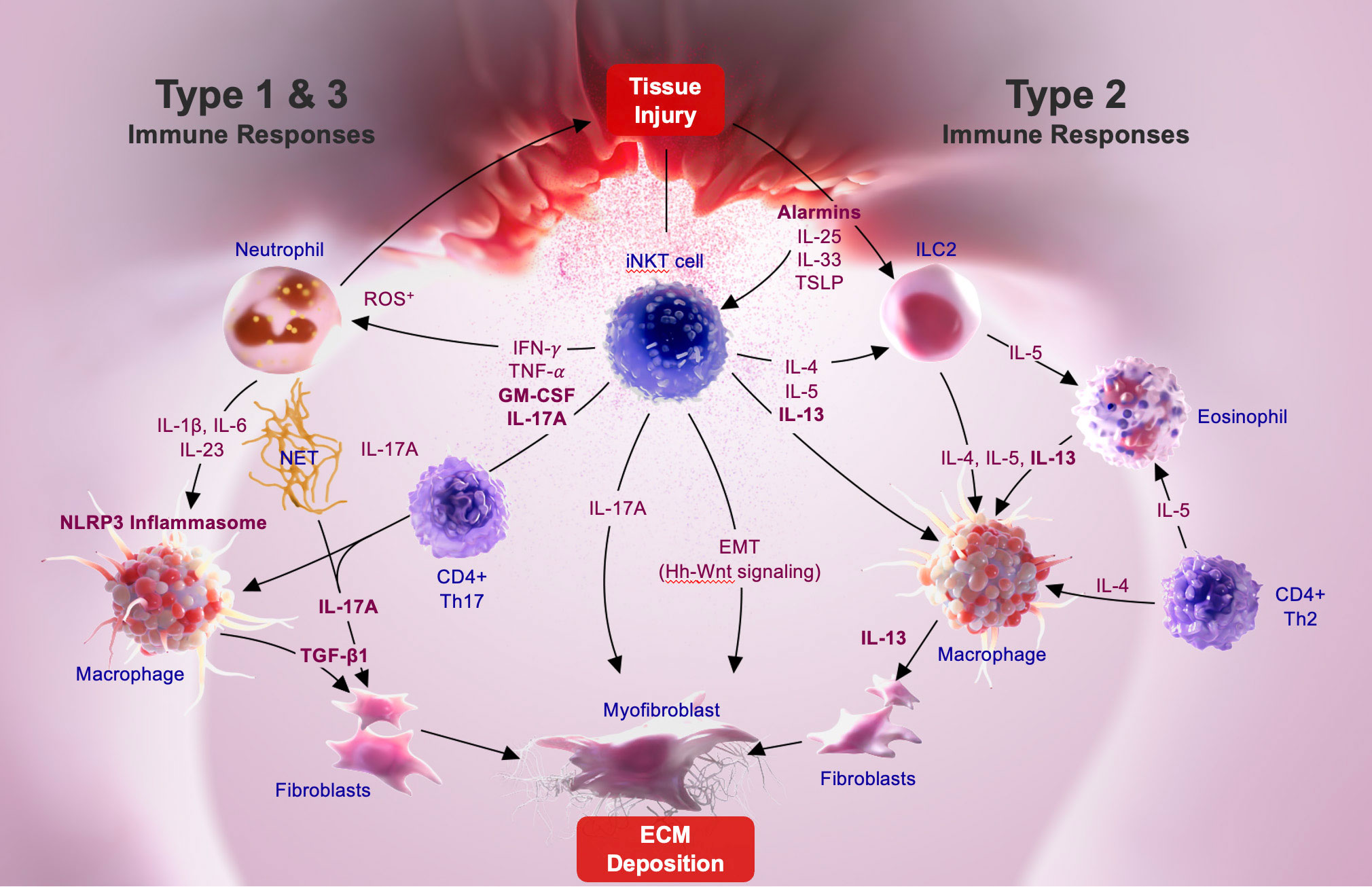
Figure 1 iNKT-mediated pathways driving pulmonary fibrosis.
Alarmins, like IL-33, are constitutively expressed at high levels in epithelial barrier tissues and endothelial barriers and are immediately released following tissue injury. iNKT cells constitutively express the ST2 chain specific of the IL-33 receptor on their surface (108, 109), and IL-33 is a co-stimulatory signal for NKT1, NKT2, and NKT17 activation (108, 110). Following lung epithelial injury, tissue-resident NKT17 cells become activated and secrete IL-17A associated with the recruitment of neutrophils into lung tissue. It is likely that IL-17A secretion from both iNKT17 as well as from γδ T cells become involved in subsequent inflammatory events, including activation of macrophages. As the inflammation progresses other subsets of iNKT cells such as tissue resident NKT1 and NKT2 cells, as well as circulating NKT2 cells that are recruited into lung tissues during chronic immune response, play an important role in activating resident or recruited macrophages. The fibrotic response is either characterized by the predominance of a TGF-β-dependent fibrotic pathway associated with M1-like monocyte/macrophage phenotype driven by type 1 (IL-1β and IL-6) or type 3, (GM-CSF and IL-17) cytokine pathway, or alternatively, by the predominance of TGF-β-independent pathway associated with IL-13-dependent M2-like macrophages driven by the type 2 (IL-4, IL-5, and IL-13) pathway. iNKT cell interactions with macrophages and/or neutrophils also results in inflammasome activation, a key component in fibrosis. Thus, depending on the stimuli or injury, upstream events leading to the activation of either inflammasome-IL-1β-IL-17A-TGF-β axis, or the GM-CSF-IL-17A-TGF-β axis is impacted by iNKT cell subset activation. Acute phase alarmins, such as IL-25, IL-33 and TSLP, function as central initiators of the type 2 immunity driven fibrosis that are triggered by IL-4 and IL-13 produced in association with innate lymphoid cells (ILC2), eosinophils, and Th2 cells in the fibrotic milieu. IL-4, IL-5, and IL-13 cytokine secretion by NKT2 cells, and other type 2 cells, result in IL-13 mediated activation of both macrophages and myofibroblasts that further contribute to fibrosis progression. Thus, iNKT cells are involved in the regulation of all three, type 1, type 2 and type 3 cytokine associated fibrotic pathways. The cross regulation of iNKT cell subsets as well as mechanisms that dictate whether type 1, type 2, or type 3 immunity predominates during the progression of lung fibrosis in IPF are not clear. However, inhibition of iNKT cells, including NKT1, NKT2 and NKT17 subsets, is likely to inhibit type 1, 2 and 3 key cytokine pathways driving fibrosis in IPF.
As investigations continue to unravel the details of the cellular and molecular pathways in fibrosis that are common across tissues, such as the activation of iNKT cell subsets and their role in promoting fibrosis, it may be possible to develop novel biomarkers and therapeutic targets that differentiate stages of fibrosis progression. We have shown that activated iNKT cells in PBMCs from patients strongly correlate with fibrosis and disease progression (40). Development of cellular and molecular markers that predict the dominance of one or more specific pathways at a given time or subset of IPF patients may facilitate patient stratification and/or targeted therapies based upon these biomarkers and may improve outcomes. In complex diseases like IPF and NASH, targeting multiple molecular pathways may be required to address the complex mechanisms driving disease. Some of the disappointing outcomes in clinical trials blocking a single cytokine, for example IL-13 alone, may be a result of: 1) mixed type 1, 2, and 3 driven responses in which a single cytokine response does not dominate; 2) unintended consequences of cross-regulation of cytokine signaling in the fibrotic tissue; and/or 3) disruption of the beneficial or protective aspects of some cytokines. Thus, rather than blocking a particular key cytokine that has many pleiotropic effects, targeting an immune pathway that can facilitate a re-balancing of downstream pathways, and restoring immune homeostasis, may be crucial for an effective treatment strategy for fibrosis. As covered in this brief review, the blocking of an earlier upstream pathway, such as iNKT cell activation, may dampen all three key cytokine-associated pathways and ultimately may lead to the development of novel therapeutic strategies in IPF. As mentioned earlier, it is likely that in patients different NKT cell subsets may promote fibrosis at different timepoints or stages of disease. In this scenario, an agent that blocks all iNKT cell subsets is highly desirable. We have found that an agonist of RARβγ inhibits cytokine secretion from all three iNKT cell subsets and leads to significant inhibition of fibrosis in murine models (40) Future studies are needed to evaluate whether inhibition of iNKT cells in a clinical setting can impact the progression of lung or liver fibrosis. Furthermore, a combination strategy, such as type 2 NKT cell activation leading to a powerful immunoregulatory mechanism along with the inhibition of iNKT cell activation, may be needed as an effective therapeutic approach in tissue fibrosis.
VK: Conceptualization, Writing – original draft. MH: Writing – review & editing. AA: Writing – review & editing. AB: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Work in Kumar laboratory was supported by the National Institutes of Health grants: CA100660 and AA020864 to VK.
VK, MH, and AA are co-founders of GRU Bio and hold equity.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18(7):1028–40. doi: 10.1038/nm.2807
2. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
3. Lupher ML Jr., Gallatin WM. Regulation of fibrosis by the immune system. Adv Immunol (2006) 89:245–88. doi: 10.1016/S0065-2776(05)89006-6
4. Pinar AA, Samuel CS. Immune mechanisms and related targets for the treatment of fibrosis in various organs. Curr Mol Med (2022) 22(3):240–9. doi: 10.2174/1566524022666220114122839
5. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
6. Maher TM, Bendstrup E, Dron L, Langley J, Smith G, Khalid JM, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res (2021) 22(1):197. doi: 10.1186/s12931-021-01791-z
7. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology (2018) 67(1):328–57. doi: 10.1002/hep.29367
8. Bendelac A, Rivera MN, Park SH, Roark JH. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol (1997) 15:535–62. doi: 10.1146/annurev.immunol.15.1.535
9. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi: 10.1038/nri3369
10. Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol (2004) 22:817–90. doi: 10.1146/annurev.immunol.22.012703.104608
11. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
12. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol (2010) 11(3):197–206. doi: 10.1038/ni.1841
13. Exley M, Garcia J, Balk SP, Porcelli S. Requirements for CD1d recognition by human invariant Valpha24+ CD4-CD8- T cells. J Exp Med (1997) 186(1):109–20. doi: 10.1084/jem.186.1.109
14. Exley MA, Bigley NJ, Cheng O, Shaulov A, Tahir SM, Carter QL, et al. Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology (2003) 110(4):519–26. doi: 10.1111/j.1365-2567.2003.01779.x
15. Shaulov A, Yue S, Wang R, Joyce RM, Balk SP, Kim HT, et al. Peripheral blood progenitor cell product contains Th1-biased noninvariant CD1d-reactive natural killer T cells: implications for posttransplant survival. Exp Hematol (2008) 36(4):464–72. doi: 10.1016/j.exphem.2007.12.010
16. Arrenberg P, Halder R, Dai Y, Maricic I, Kumar V. Oligoclonality and innate-like features in the TCR repertoire of type II NKT cells reactive to a beta-linked self-glycolipid. Proc Natl Acad Sci U S A (2010) 107(24):10984–9. doi: 10.1073/pnas.1000576107
17. Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med (2004) 199(7):947–57. doi: 10.1084/jem.20031389
18. Girardi E, Maricic I, Wang J, Mac TT, Iyer P, Kumar V, et al. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat Immunol (2012) 13(9):851–6. doi: 10.1038/ni.2371
19. Patel O, Pellicci DG, Gras S, Sandoval-Romero ML, Uldrich AP, Mallevaey T, et al. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat Immunol (2012) 13(9):857–63. doi: 10.1038/ni.2372
20. Halder RC, Aguilera C, Maricic I, Kumar V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest (2007) 117(8):2302–12. doi: 10.1172/JCI31602
21. Arrenberg P, Maricic I, Kumar V. Sulfatide-mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology (2011) 140(2):646–55. doi: 10.1053/j.gastro.2010.10.003
22. Dhodapkar MV, Kumar V. Type II NKT cells and their emerging role in health and disease. J Immunol (2017) 198(3):1015–21. doi: 10.4049/jimmunol.1601399
23. Marrero I, Maricic I, Feldstein AE, Loomba R, Schnabl B, Rivera-Nieves J, et al. Complex network of NKT cell subsets controls immune homeostasis in liver and gut. Front Immunol (2018) 9:2082. doi: 10.3389/fimmu.2018.02082
24. Marrero I, Ware R, Kumar V. Type II NKT cells in inflammation, autoimmunity, microbial immunity, and cancer. Front Immunol (2015) 6:316. doi: 10.3389/fimmu.2015.00316
25. Crosby CM, Kronenberg M. Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol (2018) 18(9):559–74. doi: 10.1038/s41577-018-0034-2
26. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T cell targets for cancer immunotherapy. Immunity (2018) 48(3):453–73. doi: 10.1016/j.immuni.2018.03.009
27. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. Corrigendum: The burgeoning family of unconventional T cells. Nat Immunol (2016) 17(4):469. doi: 10.1038/ni0416-469a
28. Kumar V. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J Hepatol (2013) 59(3):618–20. doi: 10.1016/j.jhep.2013.02.032
29. Joyce S, Okoye GD, Van Kaer L. Natural killer T lymphocytes integrate innate sensory information and relay context to effector immune responses. Crit Rev Immunol (2021) 41(4):55–88. doi: 10.1615/CritRevImmunol.2021040076
30. Van Kaer L, Postoak JL, Song W, Wu L. Innate and innate-like effector lymphocytes in health and disease. J Immunol (2022) 209(2):199–207. doi: 10.4049/jimmunol.2200074
31. Cui G, Shimba A, Jin J, Ogawa T, Muramoto Y, Miyachi H, et al. A circulating subset of iNKT cells mediates antitumor and antiviral immunity. Sci Immunol (2022) 7(76):eabj8760. doi: 10.1126/sciimmunol.abj8760
32. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med (1998) 188(8):1521–8. doi: 10.1084/jem.188.8.1521
33. Maricic I, Girardi E, Zajonc DM, Kumar V. Recognition of lysophosphatidylcholine by type II NKT cells and protection from an inflammatory liver disease. J Immunol (2014) 193(9):4580–9. doi: 10.4049/jimmunol.1400699
34. Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest (2004) 114(10):1379–88. doi: 10.1172/JCI200423594
35. Cao Z, Yuan Y, Jeyabalan G, Du Q, Tsung A, Geller DA, et al. Preactivation of NKT cells with alpha-GalCer protects against hepatic ischemia-reperfusion injury in mouse by a mechanism involving IL-13 and adenosine A2A receptor. Am J Physiol Gastrointest Liver Physiol (2009) 297(2):G249–58. doi: 10.1152/ajpgi.00041.2009
36. Jahng AW, Maricic I, Pedersen B, Burdin N, Naidenko O, Kronenberg M, et al. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med (2001) 194(12):1789–99. doi: 10.1084/jem.194.12.1789
37. Singh AK, Wilson MT, Hong S, Olivares-Villagomez D, Du C, Stanic AK, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med (2001) 194(12):1801–11. doi: 10.1084/jem.194.12.1801
38. Ferhat MH, Robin A, Barbier L, Thierry A, Gombert JM, Barbarin A, et al. The impact of invariant NKT cells in sterile inflammation: the possible contribution of the alarmin/cytokine IL-33. Front Immunol (2018) 9:2308. doi: 10.3389/fimmu.2018.02308
39. Koay HF, Gherardin NA, Nguyen THO, Zhang W, Habel JR, Seneviratna R, et al. Are NKT cells a useful predictor of COVID-19 severity? Immunity (2022) 55(2):185–7. doi: 10.1016/j.immuni.2022.01.005
40. Maricic I, Marrero I, Eguchi A, Nakamura R, Johnson CD, Dasgupta S, et al. Differential activation of hepatic invariant NKT cell subsets plays a key role in progression of nonalcoholic steatohepatitis. J Immunol (2018) 201(10):3017–35. doi: 10.4049/jimmunol.1800614
41. Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, et al. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology (2015) 61(4):1357–69. doi: 10.1002/hep.27632
42. Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, et al. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology (2009) 49(5):1683–94. doi: 10.1002/hep.22813
43. Lisbonne M, L'Helgoualc'h A, Nauwelaers G, Turlin B, Lucas C, Herbelin A, et al. Invariant natural killer T-cell-deficient mice display increased CCl(4) -induced hepatitis associated with CXCL1 over-expression and neutrophil infiltration. Eur J Immunol (2011) 41(6):1720–32. doi: 10.1002/eji.201041006
44. Duwaerts CC, Sun EP, Cheng CW, van Rooijen N, Gregory SH. Cross-activating invariant NKT cells and kupffer cells suppress cholestatic liver injury in a mouse model of biliary obstruction. PloS One (2013) 8(11):e79702. doi: 10.1371/journal.pone.0079702
45. Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M, Brossay L, et al. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology (2009) 136(3):1048–59. doi: 10.1053/j.gastro.2008.10.027
46. Shiga Y, Sugamata R, Iwamura C, Nagao T, Zao J, Kawakami K, et al. Effect of invariant natural killer T cells with IL-5 and activated IL-6 receptor in ventilator-associated lung injury in mice. Exp Lung Res (2014) 40(1):1–11. doi: 10.3109/01902148.2013.854518
47. Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell Mol Immunol (2016) 13(2):206–16. doi: 10.1038/cmi.2015.06
48. Grabarz F, Aguiar CF, Correa-Costa M, Braga TT, Hyane MI, Andrade-Oliveira V, et al. Protective role of NKT cells and macrophage M2-driven phenotype in bleomycin-induced pulmonary fibrosis. Inflammopharmacology (2018) 26(2):491–504. doi: 10.1007/s10787-017-0383-7
49. Kim JH, Kim HY, Kim S, Chung JH, Park WS, Chung DH. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-gamma. Am J Pathol (2005) 167(5):1231–41. doi: 10.1016/S0002-9440(10)61211-4
50. Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol (2013) 190(10):5226–36. doi: 10.4049/jimmunol.1202909
51. Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut (2012) 61(9):1323–9. doi: 10.1136/gutjnl-2011-301857
52. Bandyopadhyay K, Marrero I, Kumar V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol (2016) 13(3):337–46. doi: 10.1038/cmi.2015.115
53. Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology (2005) 42(4):880–5. doi: 10.1002/hep.20826
54. Valenti L, Fracanzani AL, Fargion S. The immunopathogenesis of alcoholic and nonalcoholic steatohepatitis: two triggers for one disease? Semin immunopathol (2009) 31(3):359–69. doi: 10.1007/s00281-009-0152-9
55. Zeissig S, Peuker K, Iyer S, Gensollen T, Dougan SK, Olszak T, et al. CD1d-Restricted pathways in hepatocytes control local natural killer T cell homeostasis and hepatic inflammation. Proc Natl Acad Sci U S A. (2017) 114(39):10449–54. doi: 10.1073/pnas.1701428114
56. Marrero I, Maricic I, Morgan TR, Stolz AA, Schnabl B, Liu ZX, et al. Differential activation of unconventional T cells, including iNKT cells, in alcohol-related liver disease. Alcohol Clin Exp Res (2020) 44(5):1061–74. doi: 10.1111/acer.14323
57. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P, et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun (2017) 1(4):299–310. doi: 10.1002/hep4.1041
58. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature (2016) 531(7593):253–7. doi: 10.1038/nature16969
59. Sutti S, Bruzzi S, Albano E. The role of immune mechanisms in alcoholic and nonalcoholic steatohepatitis: a 2015 update. Expert Rev Gastroenterol hepatol (2016) 10(2):243–53. doi: 10.1586/17474124.2016.1111758
60. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell (2014) 26(4):549–64. doi: 10.1016/j.ccell.2014.09.003
61. Liu Y, Munker S, Mullenbach R, Weng HL. IL-13 signaling in liver fibrogenesis. Front Immunol (2012) 3:116. doi: 10.3389/fimmu.2012.00116
62. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med (2008) 14(6):633–40. doi: 10.1038/nm1770
63. Jandl K, Marsh LM, Mutgan AC, Crnkovic S, Valzano F, Zabini D, et al. Impairment of the NKT-STAT1-CXCL9 axis contributes to vessel fibrosis in pulmonary hypertension caused by lung fibrosis. Am J Respir Crit Care Med (2022) 206(8):981–98. doi: 10.1164/rccm.202201-0142OC
64. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science (2017) 356(6342):1026–30. doi: 10.1126/science.aam7928
65. Fabre T, Barron AMS, Christensen SM, Asano S, Bound K, Lech MP, et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol (2023) 8(82):eadd8945. doi: 10.1126/sciimmunol.add8945
66. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol (2018) 3(28):eaar7754. doi: 10.1126/sciimmunol.aar7754
67. Kimura T, Ishii Y, Morishima Y, Shibuya A, Shibuya K, Taniguchi M, et al. Treatment with alpha-galactosylceramide attenuates the development of bleomycin-induced pulmonary fibrosis. J Immunol (2004) 172(9):5782–9. doi: 10.4049/jimmunol.172.9.5782
68. Hart KM, Fabre T, Sciurba JC, Gieseck RL 3rd, Borthwick LA, Vannella KM, et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-beta. Sci Transl Med (2017) 9(396):eaal3694. doi: 10.1126/scitranslmed.aal3694
69. Nilsson J, Hornberg M, Schmidt-Christensen A, Linde K, Nilsson M, Carlus M, et al. NKT cells promote both type 1 and type 2 inflammatory responses in a mouse model of liver fibrosis. Sci Rep (2020) 10(1):21778. doi: 10.1038/s41598-020-78688-2
70. Aoyagi T, Yamamoto N, Hatta M, Tanno D, Miyazato A, Ishii K, et al. Activation of pulmonary invariant NKT cells leads to exacerbation of acute lung injury caused by LPS through local production of IFN-gamma and TNF-alpha by Gr-1+ monocytes. Int Immunol (2011) 23(2):97–108. doi: 10.1093/intimm/dxq460
71. Fuschiotti P. Role of IL-13 in systemic sclerosis. Cytokine (2011) 56(3):544–9. doi: 10.1016/j.cyto.2011.08.030
72. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med (2001) 194(6):809–21. doi: 10.1084/jem.194.6.809
73. Weng HL, Liu Y, Chen JL, Huang T, Xu LJ, Godoy P, et al. The etiology of liver damage imparts cytokines transforming growth factor beta1 or interleukin-13 as driving forces in fibrogenesis. Hepatology (2009) 50(1):230–43. doi: 10.1002/hep.22934
74. Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol (2008) 40(10):2174–82. doi: 10.1016/j.biocel.2008.02.016
75. Park SW, Ahn MH, Jang HK, Jang AS, Kim DJ, Koh ES, et al. Interleukin-13 and its receptors in idiopathic interstitial pneumonia: clinical implications for lung function. J Korean Med Sci (2009) 24(4):614–20. doi: 10.3346/jkms.2009.24.4.614
76. Murray LA, Zhang H, Oak SR, Coelho AL, Herath A, Flaherty KR, et al. Targeting interleukin-13 with tralokinumab attenuates lung fibrosis and epithelial damage in a humanized SCID idiopathic pulmonary fibrosis model. Am J Respir Cell Mol Biol (2014) 50(5):985–94. doi: 10.1165/rcmb.2013-0342OC
77. Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med (2011) 365(12):1088–98. doi: 10.1056/NEJMoa1106469
78. Parker JM, Glaspole IN, Lancaster LH, Haddad TJ, She D, Roseti SL, et al. A phase 2 randomized controlled study of tralokinumab in subjects with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2018) 197(1):94–103. doi: 10.1164/rccm.201704-0784OC
79. Wijsenbeek MS, Kool M, Cottin V. Targeting interleukin-13 in idiopathic pulmonary fibrosis: from promising path to dead end. Eur Respir J (2018) 52(6):1802111. doi: 10.1183/13993003.02111-2018
80. Fichtner-, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med (2006) 12(1):99–106. doi: 10.1038/nm1332
81. Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol (2004) 173(6):4020–9. doi: 10.4049/jimmunol.173.6.4020
82. Gieseck RL 3rd, Ramalingam TR, Hart KM, Vannella KM, Cantu DA, Lu WY, et al. Interleukin-13 activates distinct cellular pathways leading to ductular reaction, steatosis, and fibrosis. Immunity (2016) 45(1):145–58. doi: 10.1016/j.immuni.2016.06.009
83. Tsao CC, Tsao PN, Chen YG, Chuang YH. Repeated activation of lung invariant NKT cells results in chronic obstructive pulmonary disease-like symptoms. PloS One (2016) 11(1):e0147710. doi: 10.1371/journal.pone.0147710
84. Pichavant M, Remy G, Bekaert S, Le Rouzic O, Kervoaze G, Vilain E, et al. Oxidative stress-mediated iNKT-cell activation is involved in COPD pathogenesis. Mucosal Immunol (2014) 7(3):568–78. doi: 10.1038/mi.2013.75
85. Akbari O, Faul JL, Hoyte EG, Berry GJ, Wahlstrom J, Kronenberg M, et al. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med (2006) 354(11):1117–29. doi: 10.1056/NEJMoa053614
86. Liu B, Jiang J, Liang H, Xiao P, Lai X, Nie J, et al. Natural killer T cell/IL-4 signaling promotes bone marrow-derived fibroblast activation and M2 macrophage-to-myofibroblast transition in renal fibrosis. Int Immunopharmacol (2021) 98:107907. doi: 10.1016/j.intimp.2021.107907
87. Jia H, Chen J, Zhang X, Bi K, Zhou H, Liu T, et al. IL-17A produced by invariant natural killer T cells and CD3(+) CD56(+) alphaGalcer-CD1d tetramer(-) T cells promote liver fibrosis in patients with primary biliary cholangitis. J Leukoc Biol (2022) 112(5):1079–87. doi: 10.1002/JLB.2A0622-586RRRR
88. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
89. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity (2015) 43(3):566–78. doi: 10.1016/j.immuni.2015.06.025
90. Heinrichs D, Brandt EF, Fischer P, Kohncke J, Wirtz TH, Guldiken N, et al. Unexpected pro-fibrotic effect of MIF in non-alcoholic steatohepatitis is linked to a shift in NKT cell populations. Cells (2021) 10(2):252. doi: 10.3390/cells10020252
91. Scanlon ST, Thomas SY, Ferreira CM, Bai L, Krausz T, Savage PB, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med (2011) 208(10):2113–24. doi: 10.1084/jem.20110522
92. Thanabalasuriar A, Neupane AS, Wang J, Krummel MF, Kubes P. iNKT cell emigration out of the lung vasculature requires neutrophils and monocyte-derived dendritic cells in inflammation. Cell Rep (2016) 16(12):3260–72. doi: 10.1016/j.celrep.2016.07.052
93. Lee KA, Kang MH, Lee YS, Kim YJ, Kim DH, Ko HJ, et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell Immunol (2008) 251(1):50–5. doi: 10.1016/j.cellimm.2008.03.004
94. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med (2007) 204(5):995–1001. doi: 10.1084/jem.20061551
95. Frising UC, Ribo S, Doglio MG, Malissen B, van Loo G, Wullaert A. Nlrp3 inflammasome activation in macrophages suffices for inducing autoinflammation in mice. EMBO Rep (2022) 23(7):e54339. doi: 10.15252/embr.202154339
96. Kaufmann B, Kui L, Reca A, Leszczynska A, Kim AD, Booshehri LM, et al. Cell-specific deletion of NLRP3 inflammasome identifies myeloid cells as key drivers of liver inflammation and fibrosis in murine steatohepatitis. Cell Mol Gastroenterol Hepatol (2022) 14(4):751–67. doi: 10.1016/j.jcmgh.2022.06.007
97. Kaufmann B, Leszczynska A, Reca A, Booshehri LM, Onyuru J, Tan Z, et al. NLRP3 activation in neutrophils induces lethal autoinflammation, liver inflammation, and fibrosis. EMBO Rep (2022) 23(11):e54446. doi: 10.15252/embr.202154446
98. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
99. Dixon LJ, Berk M, Thapaliya S, PapouChado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest (2012) 92(5):713–23. doi: 10.1038/labinvest.2012.45
100. Chow MT, Duret H, Andrews DM, Faveeuw C, Moller A, Smyth MJ, et al. Type I NKT-cell-mediated TNF-alpha is a positive regulator of NLRP3 inflammasome priming. Eur J Immunol (2014) 44(7):2111–20. doi: 10.1002/eji.201344329
101. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol (2009) 183(2):792–6. doi: 10.4049/jimmunol.0900173
102. Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology (2018) 67(2):736–49. doi: 10.1002/hep.29523
103. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J Immunol (2013) 190(3):1239–49. doi: 10.4049/jimmunol.1201959
104. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A. (2014) 111(1):367–72. doi: 10.1073/pnas.1315854111
105. Kim JO, Kim DH, Chang WS, Hong C, Park SH, Kim S, et al. Asthma is induced by intranasal coadministration of allergen and natural killer T-cell ligand in a mouse model. J Allergy Clin Immunol (2004) 114(6):1332–8. doi: 10.1016/j.jaci.2004.09.004
106. Lexmond WS, Neves JF, Nurko S, Olszak T, Exley MA, Blumberg RS, et al. Involvement of the iNKT cell pathway is associated with early-onset eosinophilic esophagitis and response to allergen avoidance therapy. Am J Gastroenterol (2014) 109(5):646–57. doi: 10.1038/ajg.2014.12
107. Jyonouchi S, Smith CL, Saretta F, Abraham V, Ruymann KR, Modayur-Chandramouleeswaran P, et al. Invariant natural killer T cells in children with eosinophilic esophagitis. Clin Exp Allergy (2014) 44(1):58–68. doi: 10.1111/cea.12201
108. Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol (2009) 39(4):1046–55. doi: 10.1002/eji.200838575
109. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol (2008) 20(8):1019–30. doi: 10.1093/intimm/dxn060
Keywords: CD1d, NKT cells, fibrosis, macrophages, fibroblasts, IPF, NASH
Citation: Kumar V, Hertz M, Agro A and Byrne AJ (2023) Type 1 invariant natural killer T cells in chronic inflammation and tissue fibrosis. Front. Immunol. 14:1260503. doi: 10.3389/fimmu.2023.1260503
Received: 17 July 2023; Accepted: 06 September 2023;
Published: 25 September 2023.
Edited by:
Luc Van Kaer, Vanderbilt University Medical Center, United StatesReviewed by:
Chyung-Ru Wang, Northwestern University Feinberg School of Medicine, United StatesCopyright © 2023 Kumar, Hertz, Agro and Byrne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vipin Kumar, dmNrdW1hckBoZWFsdGgudWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.