 Olivia C. Ihedioha
Olivia C. Ihedioha Anutr Sivakoses
Anutr Sivakoses Stephen M. Beverley
Stephen M. Beverley Diane McMahon-Pratt
Diane McMahon-Pratt Alfred L. M. Bothwell
Alfred L. M. Bothwell- 11Department of Immunobiology, College of Medicine, University of Arizona, Tucson, AZ, United States
- 2Department of Molecular Microbiology, Washington University School of Medicine in St Louis, St. Louis, MI, United States
- 3Department of Epidemiology of Microbial Diseases, Yale School of Public Health, New Haven, CT, United States
Background: Platelets are rapidly deployed to infection sites and respond to pathogenic molecules via pattern recognition receptors (TLR, NLRP). Dickkopf1 (DKK1) is a quintessential Wnt antagonist produced by a variety of cell types including platelets, endothelial cells, and is known to modulate pro-inflammatory responses in infectious diseases and cancer. Moreover, DKK1 is critical for forming leukocyte-platelet aggregates and induction of type 2 cell-mediated immune responses. Our previous publication showed activated platelets release DKK1 following Leishmania major recognition.
Results: Here we probed the role of the key surface virulence glycoconjugate lipophosphoglycan (LPG), on DKK1 production using null mutants deficient in LPG synthesis (Δlpg1- and Δlpg2-). Leishmania-induced DKK1 production was reduced to control levels in the absence of LPG in both mutants and was restored upon re-expression of the cognate LPG1 or LPG2 genes. Furthermore, the formation of leukocyte-platelet aggregates was dependent on LPG. LPG mediated platelet activation and DKK1 production occurs through TLR1/2.
Conclusion: Thus, LPG is a key virulence factor that induces DKK1 production from activated platelets, and the circulating DKK1 promotes Th2 cell polarization. This suggests that LPG-activated platelets can drive innate and adaptive immune responses to Leishmania infection.
Introduction
Cutaneous leishmaniasis (CL) is a zoonotic protozoan disease caused by over 15 different parasite species of Leishmania (1). Current data suggest that more than 1 billion people are at risk of cutaneous leishmaniasis, and more than 1 million new cases occur each year in approximately 90 countries worldwide (2). Cutaneous leishmaniasis is endemic in many regions of the world, including the Mediterranean, North Africa, Central America, the Middle East, and northern parts of South America (3). Most of the detected cases of leishmaniasis in the United States are amongst travellers to endemic regions (4). Therapeutic treatment for managing cutaneous leishmaniasis involves the use of a limited number of drugs, known to cause serious side effects (5). Thus, therapeutic approaches that promote healing and reduce drug toxicity are highly desired. Combinatorial therapy involving a combination of immunomodulatory molecules with current therapies has the potential of limiting drug toxicity (6). Therefore, there is a need to identify immunomodulatory mechanisms and compounds that will serve as a therapeutic target for controlling leishmaniasis. Identifying immunomodulatory molecules for controlling leishmaniasis requires understanding Leishmania surface component interaction with host immune cells.
Platelets are known to play a role in hemostasis and for their essential contribution to protection against infectious pathogens (7). By interaction with macrophage cells, monocytes, neutrophils, lymphocytes, and the endothelium, platelets are therefore important executors during inflammatory and immune responses (8, 9). The membrane glycoprotein CD62P, a member of the selectin family, is expressed on activated platelets (10) and is redistributed from the secretory α-granules (11). PSGL-1, one of the best-characterized selectin ligands, is mainly expressed in leukocytes (12). Leukocyte platelet aggregation formed by adhesion between activated platelet and mature leukocyte is mediated by CD62P and PSGL-1 (13–15). The formation of leukocyte platelet aggregation (LPA) is necessary for effective migration to the infection site (16), which might be induced by L. major surface membrane components.
Surface membrane components of pathogens (virus, bacteria, parasite) are important in initiation of infection and interaction with the host. The Leishmania parasite has a surface coat (glycocalyx) that is composed of glycosylated proteins and lipids that are crucial for the parasite’s survival, replication and pathogenesis in the insect vector and mammalian hosts (17, 18). These glycoconjugates on the parasite coat are differentially expressed in the different developmental stages of the Leishmania parasite (18). Of these molecules, lipophosphoglycan (LPG) and proteophosphoglycan (PPG) show structural similarities through the presence of the canonical phosphoglycan repeating units (Gal-Man-PO4) (19, 20). Thus, there is a strong potential for cross-activity of these shared motifs in biochemical or cell biological tests of purified molecules. This limitation can be overcome using Leishmania major rendered genetically deficient in either or both LPG or PPG. Parasites specifically lacking LPG1 were obtained through homologous gene replacement, deleting the LPG1 galactofuranosyl transferase required for synthesis of a critical linkage within the LPG glycan core domain (21). These mutants otherwise express normal levels of other surface virulence glycoconjugates including GPI anchored proteins, PPG and glycoinositolphospholipids (GIPLS) (22). Similarly, homologous gene replacement of LPG2 encoding the Golgi GDP-mannose nucleotide sugar transporter completely ablate PG repeat synthesis, rendering parasites deficient in both intact LPG and PPG (23). Extensive studies using these mutants have shown that LPG plays critical roles in pathogenesis and parasite survival, through the evasion of complement-mediate lysis, preventing natural killer T cells from recognizing the Leishmania-infected macrophage, suppression of oxidative burst response, macrophage-mediated killing and modulation of host immune responses (24–26). Further, LPG has the ability to alter dendritic cell function, which affects the host’s immunological responses by blocking antigen presentation and promoting an early IL-4 response (27).
We demonstrated activated platelets release of DKK1 following recognition of L. major (28). In that report, we found that DKK1 is maintained at a high concentration in L. major infected mice, and that depletion of platelets resulted in the complete loss of the DKK1 response to infection (28). Further inhibition of DKK1 activity resulted in a significant reduction in cells recruited to the site of infection. The DKK1 response was shown to be critical to the development of the Th2 response to infection through the induction of MAPK and mTOR signaling pathways. It has been reported that platelet-derived DKK1 is elevated in different diseases following the recognition of pathogen-associated molecular patterns through receptors other than TLR activation. However, no one so far has related DKK1 elevation to platelet-TLR activation or has identified leishmanial-associated molecules involved in DKK1 production.
In the current study, we expand upon our initial observations to demonstrate that Leishmania major-mediated platelet activation is initiated via the TLR1/2 signalling pathway, by the specific engagement of the leishmanial virulence factor, lipophosphoglycan (LPG). Overall, our studies demonstrate that LPG plays a critical role in Th2 induction, polymorphonuclear neutrophil (PMN) recruitment and the establishment of infection, through engagement and activation of platelets.
Experimental procedures
Mice
BALB/c mice (6 weeks old) were purchased from the Jackson Laboratory. All mice were housed at the University of Arizona Animal Care Facilities. All mouse protocols were approved by the Arizona University Institutional Animal Care and Use Committee (IACUC) in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Parasite strains and infection protocol
The L. major mutants (Δlpg1- and Δlpg2-) and add-backs (Δlpg1-/+LPG1 and Δlpg2-/+LPG2) were derivatives of WT L. major LV39 clone 5 background. They were made by homologous gene targeting and maintained in selective media as described previously (22, 23, 29, 30). Leishmania major parasites were maintained at 26°C in M199 culture medium (Thermo Fisher Scientific) supplemented with 20% heat-inactivated FBS (Thermo Fisher Scientific)), 20 mM HEPES (Sigma-Aldrich) and 50 ug/ml gentamycin (Thermo Fisher Scientific). Prior to using parasites for infection, metacyclic promastigotes were isolated from stationary-phase cultures using density gradient centrifugation (31, 32). Metacyclic promastigotes were washed three times in cold phosphate-buffered saline (PBS) (Thermo Fisher Scientific) by centrifugation, resuspended in PBS at 2X108/ml and 10 μl containing 2x106 metacyclic promastigotes were injected into the top of the right hind footpad.
Measurement of lesion size and estimation of parasite burden
Lesion size was measured weekly with Vernier calipers and determined by subtracting the size of the uninfected from that of the infected footpad. Parasite burden in the infected footpad was estimated by limiting dilution analysis as previously described (33).
Platelet preparation and P-selectin expression
Blood was drawn by retro-orbital bleeding under isoflurane anesthesia (MWI Animal Health) into tubes containing 3.2% citrate buffer (G-Biosciences) from naïve mice or at days 3, 14 and 42 PI. Platelets were prepared as previously described (34). In brief, blood was diluted with 250 μl of modified Tyrode’s-HEPES buffer (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, and 1 mM MgCl2, pH 7.3) and centrifuged at 250 x g for 15 minutes at room temperature. Platelet-rich plasma was removed, and platelets were then isolated by centrifugation at 900 × g for 30 min. Plasma was collected for DKK1 ELISA.
In vitro stimulation of platelets was done using various stock concentrations of soluble leishmania antigen (SLAG) (1:25, 1:50, 1:200, 1:400), Pam2CSK4, Pam3CSK4 (InvivoGen), LPS (Escherichia coli O111:B4-Sigma) and control isotype antibodies (IgG1, IgG2a and IgG2b) (Invitrogen). DKK1 inhibition was done using anti-TLR4, anti-TLR2, anti-TLR1/2, anti-TLR2/6, anti-TLR1/2/6 antibody (Invitrogen), and Go 6976 PKC-alpha inhibitor (Abcam) before incubation for 1 hr at room temperature. Platelets were isolated by centrifugation at 550 x g for 10 minutes, and the supernatant was collected for DKK1 ELISA. For SLAG preparation, L. major parasites were prepared at 5 × 108 parasites/ml in M199 medium (without FCS). SLAG was prepared by repeated five freeze and thaw cycles at -80°C and room temperature, respectively. SLAG supernatant was collected after centrifugation at 1500rpm for 20 minutes and stored at -80°C. For detection of LPG expression, Western Blot of SLAG (7 µg) was resolved by SDS-PAGE electrophoresis using 10% PAGE; molecules were transferred to PVDF membrane using the iBlot 2 semi-dry blotting system (Thermofisher Scientific Inc.). The membrane was blocked with 5% powdered milk in Tris Buffered Saline (TBS) and incubated for 1 hour at room temperature. Blots were probed with WIC 79.3 antibody (1:1000). After washing in TBS supplemented with 0.05% Tween 20 (TBST), the membrane was incubated for 1 h with anti-mouse IgG conjugated with Horseradish peroxidase (1:7500-Invitrogen) and the reaction was visualized using Pico PLUS Chemiluminescent substrate kit (Thermo Fisher Scientific). Signals were acquired using ChemiDoc Imaging System (Bio-Rad).
For FACS analyses, isolated platelets (10x106/ml) were washed at 550 x g for 10 minutes in the presence of PGE1 (140 nM; Sigma-Aldrich) and indomethacin (10 μM; Thermo Fisher Scientific). Platelets were resuspended to the required density in modified Tyrode’s-HEPES buffer (Sigma-Aldrich) and rested for 30 minutes at 37°C in the presence of 10 μM indomethacin prior to staining. Staining for P-selectin expression was done using PE-conjugated CD41 (BioLegend) (to determine platelet purity) and APC-conjugated CD62P (Invitrogen; a marker of P- selectin). Stained platelets (1 × 106/ml in Tyrode’s-HEPES buffer; 100 μl) were analyzed using a BD FACSCanto flow cytometer; analysis was done using FlowJo software. A total of 10,000 events per sample were collected. The mean fluorescent intensity of P-selectin was compared among the different groups in the overlay.
Leukocyte-platelet aggregation
Leukocyte-platelet aggregation assessment was performed as described previously with minor modifications (28). Briefly, 100 μl blood was collected via the retro-orbital sinus into tubes containing 3.2% citrate buffer. Peripheral blood (10 µl) was stained with PE-conjugated anti-mouse CD41 antibody and Pacific blue conjugated CD45 antibody (BioLegend) for 10 mins in the dark at room temperature. Fix/red blood cell lysis solution (eBiosciences) was added and incubated for 15 min in the dark at room temperature. Samples were analyzed by flow cytometry within 4 to 6 hours. Live gating was performed on leukocyte-sized events to exclude single platelets. Leukocytes were identified by their forward and side scatter characteristics and CD45 expression. The CD41+ subpopulation identified leukocyte-platelet aggregates.
DKK1 ELISA
Enzyme-linked immunosorbent assays were performed using a mouse DKK1 ELISA kit (Thermo Fisher Scientific) to determine the concentration of DKK1 in plasma and cell culture supernatants according to the manufacturer’s protocol.
Statistical analyses
The in vivo expression of P-selectin and DKK1 production was analyzed using one-way ANOVA with Bonferroni’s post hoc test. Also, in vitro DKK1 production and P-selectin expression following SLAG stimulation was analyzed using one-way ANOVA with Bonferroni’s post hoc test. Comparison of neutralizing antibody inhibition of platelet DKK1 induced by SLAG obtained from WT, addback and mutant parasites was done statistically using Student’s t-test. Likewise, a comparison of PKC-alpha inhibition of DKK1 induced by SLAG obtained from WT, addback and mutant parasites was done statistically using one-way ANOVA with Bonferroni’s post hoc test. In addition, lesion size, parasitic load and percentage of LPA was analyzed using one-way ANOVA with Bonferroni’s post hoc test. Data presented as means ± standard errors were performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA).
Results
Δlpg1- and Δlpg2- parasites induce minimal expression of P-selectin in platelets
Platelets are rapidly deployed to sites of infection where they can modulate immune/inflammatory processes by secreting cytokines, chemokines, and other inflammatory mediators (35). P-selectin is an adhesion receptor for leukocytes expressed by activated platelets (36). To investigate the possible effects of LPG1- and or LPG2-dependent molecules on platelet activation in vivo, expression of P-selectin was determined from platelets obtained from mice that had been infected by WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasites for varying periods of time. Relative to the WT controls, P-selectin expression was significantly suppressed in platelets obtained from Δlpg1- and Δlpg2- mutant infected mice on days 3, 14 and 42 PI. As expected, mice infected with “add-back’ control parasites (Δlpg1-/+LPG1 and Δlpg2-/+LPG2) manifested restoration of P-selectin expression similar to those infected with WT parasites (Figures 1A–F).
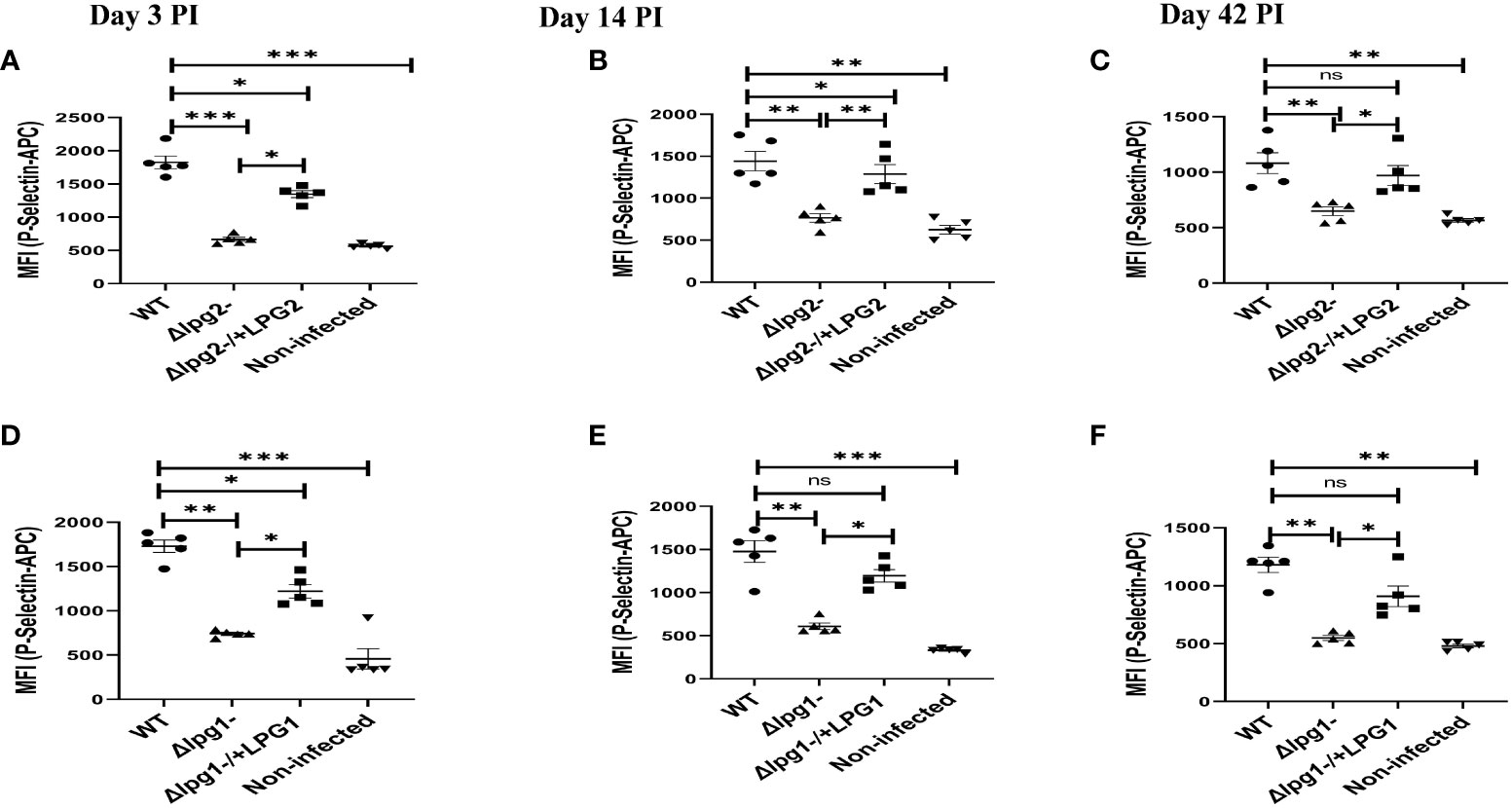
Figure 1 Δlpg1- and Δlpg2- induce minimal P-selectin expression in activated platelets. BALB/c mice were challenged with infective metacyclic promastigote (2 x 106 parasites, n = 5) of WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains via the footpad. Control mice (n = 5) were given 0.9% NaCl saline. Blood was collected via retro-orbital sinus at days 3, 14 and 42 PI. Isolated platelet samples were analyzed by flow cytometry for P-selectin expression. In all the experiments, WT-infected and non-infected mice served as positive and negative controls, respectively. Each dot indicates the expression of P-selectin by CD41+ cells (A–F). Results are presented as mean (± SEM) and are representative of 3 independent experiments. One-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05).
We also characterized P-selectin expression in vitro using SLAG-stimulated platelets obtained from naïve mice. Stimulation of platelets obtained from naive mice with SLAG derived from Δlpg1- and Δlpg2- parasites induced poor P-selectin expression. In contrast, platelets stimulated with SLAG derived from WT, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasites induced robust P-selectin expression (Figures 2A, B). Taken together, these data suggest that the inability of Δlpg1- and Δlpg2- parasites to induce P-selectin expression arises from a lack of LPG1 or LPG2 dependent products. As Δlpg2- parasites lack all phosphoglycan-containing glycans (LPG, PPG and others) while Δlpg1- lacks only LPG, the lack of a significant difference between Δlpg1- and Δlpg2- mutants indicates that platelet activation and P-selectin expression are primarily regulated by LPG.
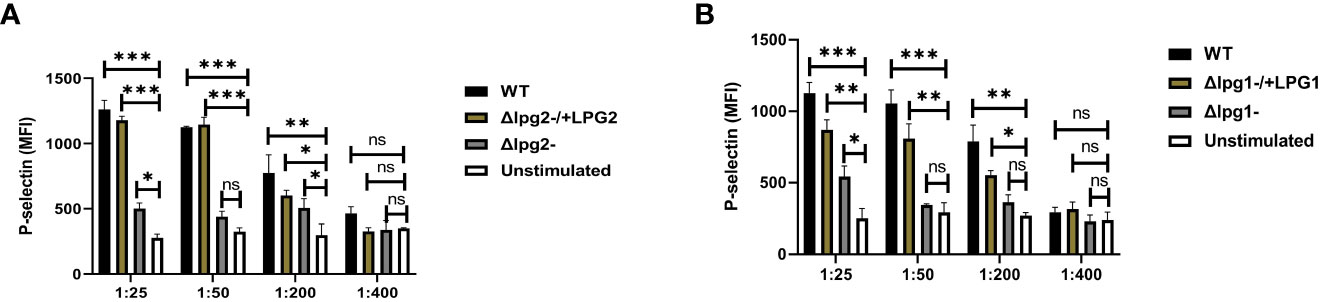
Figure 2 Less efficient expression of P-selectin from Δlpg1- and Δlpg2- SLAG-activated platelets. Platelets isolated from naïve mice were incubated with various concentrations of SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) for 1hr. Platelet samples were analyzed by Flow cytometry for P-selectin expression. In all the experiments, WT-SLAG activated and non-activated platelets served as a positive and negative control, respectively. Column graphs (A, B) indicate the expression of P-selectin by CD41+ cells. Results are presented as mean +/- SEM of replicate wells and represent 3 independent experiments. Data are presented by comparing DKK1 production from the non-SLAG and SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) activated platelets. One-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05).
Δlpg1- and Δlpg2- parasites are less efficient in inducing DKK1 production in platelets
We previously showed that activated platelets release DKK1 following recognition of L. major (28). Furthermore, DKK1 is maintained at a high concentration in L. major infected mice; depletion of platelets resulted in the complete loss of DKK1 (28). To identify the L. major ligand involved in DKK1 production, we first compared the effect of LPG1 and LPG2 gene-dependent molecules in plasma DKK1 production at days 3, 14 and 42 PI. We confirmed a significantly decreased production of plasma DKK1 in Δlpg1- and Δlpg2- mutant infected mice compared to WT controls, but in Δlpg1-/+LPG1 and Δlpg2-/+LPG2 infected mice, DKK1 production was restored (Figures 3A–F). Compared to days 3 and 14 PI, there was a reduction in the concentration of DKK1 produced by activated platelets on day 42 PI.
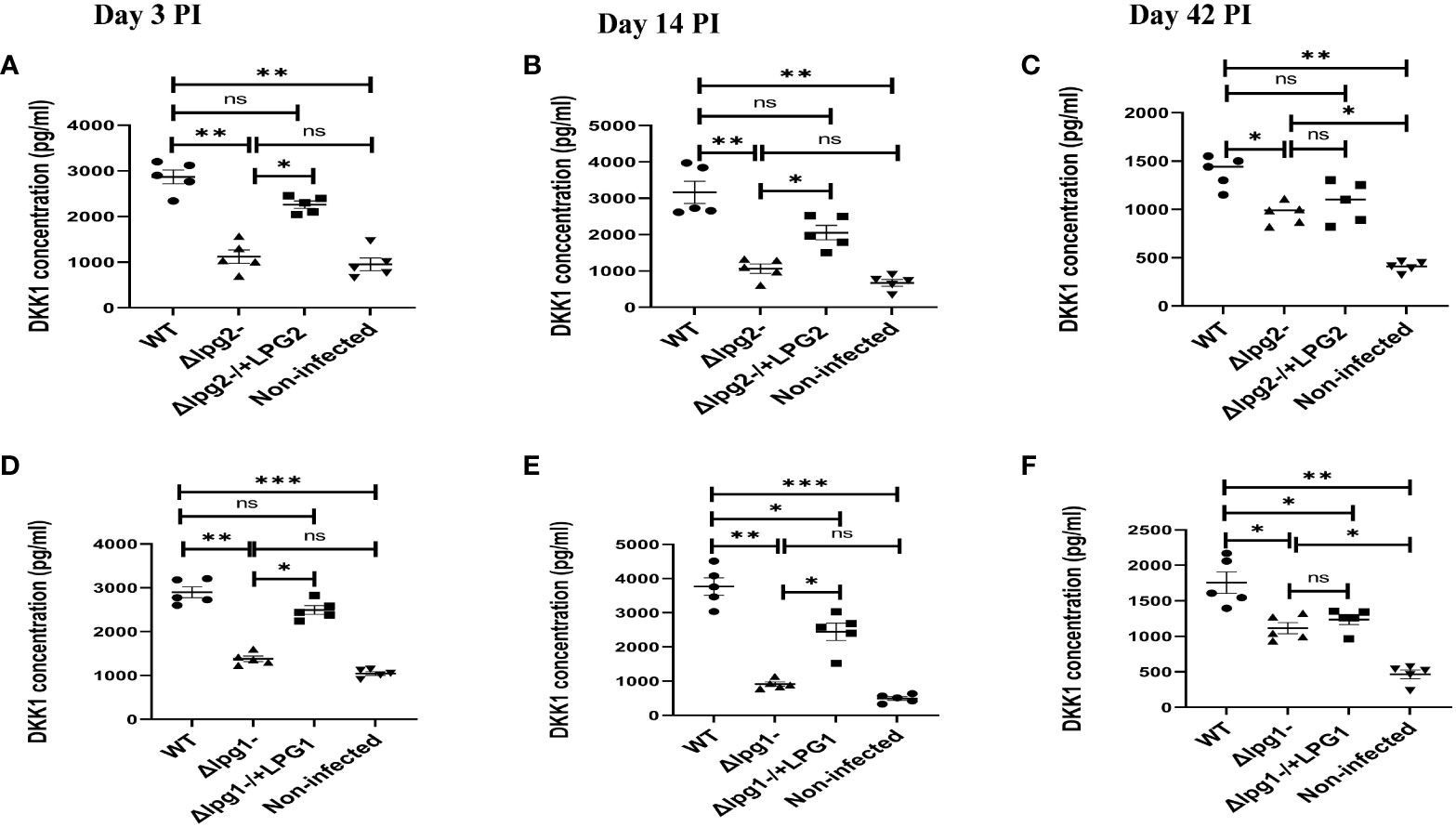
Figure 3 Δlpg1- and Δlpg2- parasites induce less plasma DKK1 production from the platelets. Six-week-old female BALB/c mice were challenged with infective metacyclic promastigote (2 x 106 parasites, n = 5) of WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains via the footpad. Control mice (n = 5) were given 0.9% NaCl Saline. Blood was collected via retro-orbital sinus at days 3, 14 and 42 PI. Plasma samples were analyzed by ELISA For DKK1 production (A–F). In all experiments, WT-infected and non-infected mice served as positive and negative controls, respectively. Results are presented as mean +/- SEM of replicate wells and are representative of 3 independent experiments. One-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05).
Impaired DKK1 production from the Δlpg1- and Δlpg2- were further confirmed via an in vitro stimulation of platelets with SLAG obtained from Δlpg1- and Δlpg2- parasites. Consistent with the plasma DKK1, in vitro stimulation of naïve platelets with SLAG obtained from Δlpg1- and Δlpg2- parasites failed to induce significant production of DKK1, in contrast a significant level of DKK1 was released by platelets stimulated with SLAG derived from WT L. major (Figures 4A, B) or the genetically reconstituted organisms. Since the levels of DKK1 produced in Δlpg1- and Δlpg2- infected mice are comparable, these data suggest that activated platelets release DKK1, a process which depends on the leishmania-derived LPG virulence factor; PPG does not appear to additionally contribute to platelet activation.
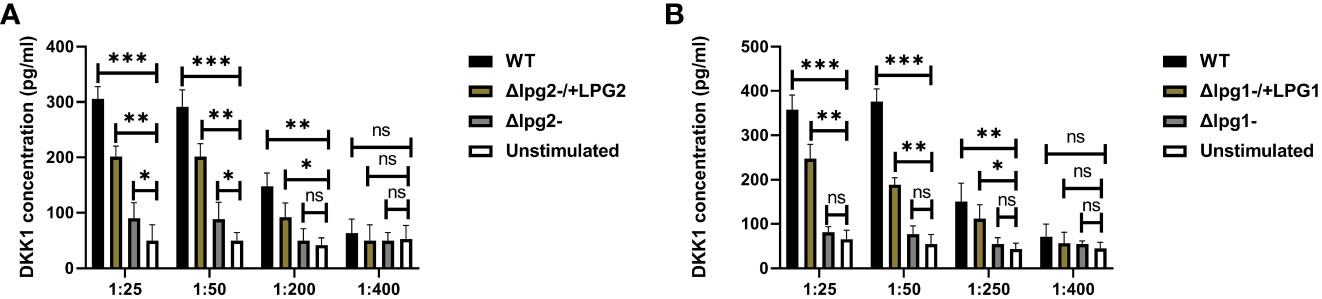
Figure 4 Poor induction of DKK1 from Δlpg1- and Δlpg2- SLAG-activated platelets. Platelets from naïve mice were incubated with various concentrations of SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) for 1hr. Cell culture supernatant samples were analyzed by ELISA for DKK1 production as shown in the column graphs (A, B). In all experiments, WT-SLAG activated, and non-activated platelets served as positive and negative controls, respectively. Results are presented as mean +/- SEM of replicate wells and represent 3 independent experiments. Data are presented by comparing the DKK1 production from non-SLAG and SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) activated platelets. One-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05).
Significant reduction in lesion size and parasitic burden observed in Δlpg1- and Δlpg2- infected mice was restored in mice infected with Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasites
The Δlpg1- and Δlpg2- parasites had a greatly delayed formation of lesions compared with WT controls, whereas lesions produced by the Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasites showed minimal delay and resembled WT controls (Figures 5A, B). In addition, the parasitic burden was significantly decreased in Δlpg1- and Δlpg2- infected mice compared to WT controls, and restoration of the LPG1 and LPG2 genes resulted in parasite survival, which is comparable with the WT controls (Figures 5C, D). The pattern of lesion size in the footpads and parasitic burdens of mice infected with genetically deficient Leishmania (LPG1 or LPG2 gene) were comparable (Figures 5A–D). These data are consistent with previous publications concerning the virulence of these lpg mutant parasites (25, 29, 37). Repetition of this experiment serves as a confirmation of these results for the mutant lines used in the study.
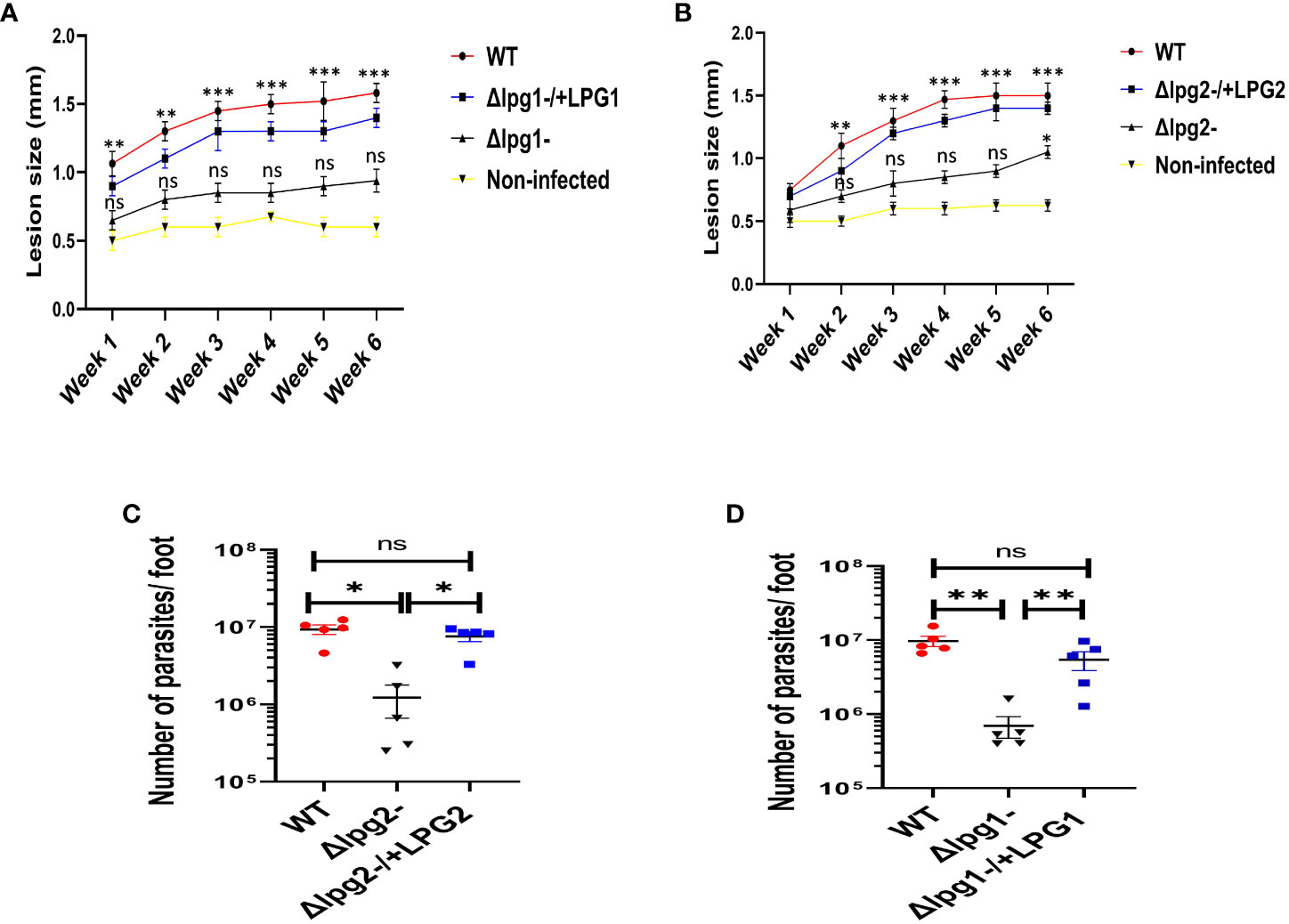
Figure 5 Δlpg1- and Δlpg2- parasites differ from specific add-backs and WT parasites in their capacity to induce lesions and increase the parasite burdens. The infected foot from each mouse in WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasite-infected groups were measured for lesion size weekly using a vernier caliper (A, B), and parasite burden (at day 42 PI) was determined by limiting dilution assay (C, D). Results are presented as mean +/- SEM. For Figures (A, B), mice in all the infected groups were compared with the non-infected group and data analysis was done using one-way ANOVA with Bonferroni’s post hoc test *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05).
Decrease production of DKK1 in SLAG-stimulated platelets in the presence of PKC-alpha inhibitor and anti-TLR1/2 antibody
Platelets express functional TLR2/4 and MyD88, which participate in platelet responsiveness to infection (38). We previously showed that activated platelets release DKK1 following recognition of L. major and PKC-alpha inhibitor or neutralization of TLR2 blocks secretion of DKK1 from SLAG-stimulated human platelets (28). PKC-alpha inhibitor is known to block the TLR2/MyD88 signalling pathway (39). An important feature of TLR2 is the ability to form heterodimers with its co-receptors (TLR1 and TLR6). Therefore, to confirm that TLR1/2/6 recognizes Leishmania-derived LPG and initiates the production of DKK1, neutralizing antibodies were used to inhibit the function of these specific TLRs in the presence of SLAG. Exposure of Δlpg1- and Δlpg2- SLAG-stimulated platelets to blocking anti-TLR1/2/6 antibodies showed no significant effect, but significant inhibition of DKK1 production by anti TLR1/2/6 antibodies was observed in WT, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 SLAG treated platelets (Figures 6A, B). To determine the level of DKK1 inhibition by the neutralizing antibodies, platelets were treated with neutralizing antibodies (anti-TLR2, TLR1/2 and TLR2/6) or control isotype antibodies (IgG2a and IgG1) in the presence of WT-derived SLAG. Results showed that neutralization of TLR1/2/6 with an anti-TLR1/2/6 mAb markedly reduced SLAG-induced DKK1 production in platelets compared with control isotype antibodies. Relative to DKK1 inhibition by anti-TLR2 blocking antibody, addition of anti-TLR1 (TLR1/2) antibody significantly deceased DKK1 production, while addition of anti-TLR6 antibody had no significant effect (Figure 6C). This suggests that only TLR1/2 played a primary role in LPG induced DKK1 production. Previous studies reported that inhibition of TLR2 and 4 attenuates inflammatory response and parasite burden in cutaneous leishmaniasis (40). Therefore, to determine whether TLR4 contributes in DKK1 production after LPG recognition by platelets, platelets were treated with anti-TLR4 neutralizing antibody or control isotype antibody (IgG2b) in the presence of WT-derived SLAG. Results showed that neutralization of TLR4, with an anti-TLR4 mAb failed to reduce SLAG-induced DKK1 production. Also, there was no change in DKK1 production in platelets treated with the control isotype antibody. This suggests that TLR4 played no role in LPG induced DKK1 production (Figure 6C). To confirm the specificity of this data, we assessed the possibility of other PAMPs (not directly related to LPG) to induce DKK1 release. Platelets treated with LPS (TLR4) and Pam2CSK4 (TLR2/6) failed to elicit DKK1 production. However, Pam3CSK4 (TLR1/2) slightly induced DKK1. This suggests that LPS and Pam2CSK4 lack the ability to induce DKK1 production (Figure S3). To verify that SLAG preparations of the mutant lines lack LPG, Western blot assay of SLAG preparations confirms the absence of LPG in Δlpg1- derived SLAG and the presence of LPG in the SLAG of the WT and lpg1 add-back lines (Figure S1).
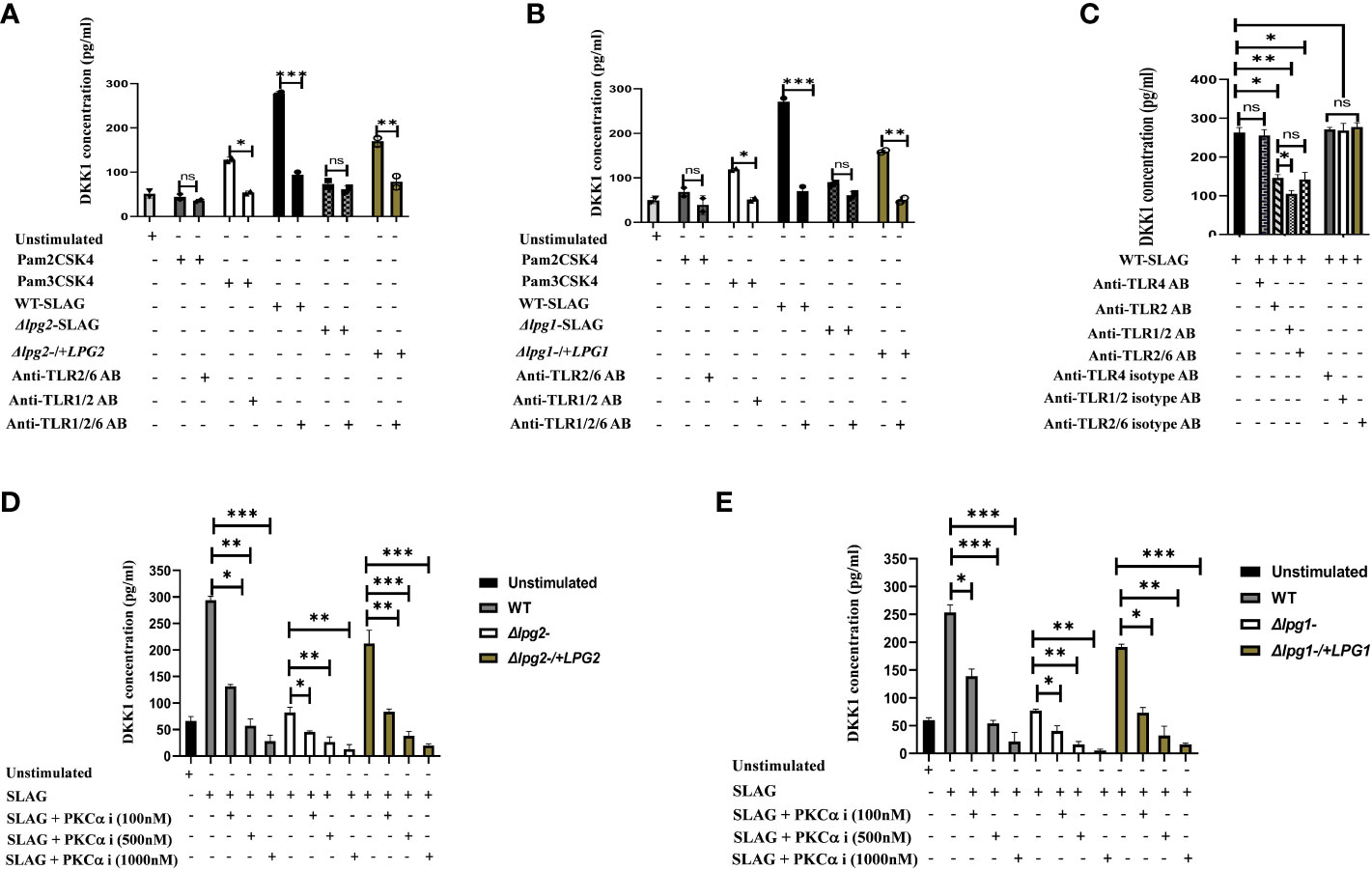
Figure 6 Anti-TLR1/2 antibody and PKC-alpha inhibitor significantly decreased DKK1 production in SLAG-activated platelets. Inhibition of DKK1 by neutralizing antibodies (10 μg/ml) following 1hr incubation with SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) activated platelets. SLAG concentration is 1:50. In all experiments, Pam2CSK4 (10 μg/ml), Pam3CSK4 (10 μg/ml) and unstimulated samples served as a positive and negative control, respectively (A, B). Inhibition of DKK1 by neutralizing antibodies (10 μg/ml) following 1hr incubation with WT SLAG (1:50) activated platelets (C). Inhibition of DKK1 by PKC-alpha inhibitor (100nM, 500nM and 1000nM) following 1hr incubation with SLAG (derived from WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains) activated platelets (D, E). Results are presented as mean +/- SEM of replicate wells and represent 3 independent experiments. For Figures (A, B), Student’s t-test was performed, while one-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data in Figures (C–E). *p < 0.05, **p < 0.01, ***p < 0.001, ‘ns’ indicates not significant (p > 0.05). Note that the DKK1 production from Δlpg1- and Δlpg2- SLAG-stimulated platelets was found to be background platelet DKK1, as non-stimulated platelets showed comparable levels of DKK1 release/PKC-alpha inhibition (Figure S2).
In addition, when platelets were incubated with SLAG obtained from WT, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 parasites in the presence of varying concentrations of PKC-alpha inhibitor, a significant reduction in secretion of DKK1 was observed at higher dose concentrations of PKC-alpha inhibitor. In contrast, DKK1 production from Δlpg1- and Δlpg2- SLAG-stimulated platelets exhibited some degree of inhibition by the PKC-alpha inhibitor at the higher concentrations (Figures 6D, E), but this was confirmed to be background platelet DKK1 that is being inhibited, as the non-stimulated platelets show a comparable level of inhibition (Figure S2). This suggests that the recognition of leishmania-derived LPG and induction of DKK1 from activated platelets may occur via the TLR1/2-MyD88 pathway.
The lack of LPA formation induced by Δlpg1- and Δlpg2- parasites is restored in addback parasite (Δlpg1-/+LPG1 and Δlpg2-/+LPG2) infected mice
Platelet-leukocyte interactions are indispensable events in hemostasis and inflammation (41, 42). Under inflammatory conditions, platelets interact with leukocytes, thus promoting their recruitment by the formation of platelet leukocyte aggregates via the interaction of PSGL-1 and P-selectin (28, 43). The consequence of this interaction enables leukocytes to fulfill their multiple cell-intrinsic functions and immunological tasks. We showed previously that pre-treatment of mice with DKK1 inhibitor 24 hours prior to infection with L. major reduced the elevation of LPA formation at 4 h post-infection (28). Furthermore, repeated administration of DKK1 inhibitor led to significant reduction in cellular recruitment at the site of infection and to the draining lymph node. These findings suggest that leukocyte platelet aggregation and infiltration of leukocytes to the infection site is driven by DKK1 production.
Given that DKK1 production in mice infected with Δlpg1- and Δlpg2- parasites was impaired, we considered the possibility that the loss of LPG1 and LPG2 gene-dependent molecules might decrease LPA formation in blood obtained from Δlpg1- and Δlpg2- mutant infected mice (Figures 7). Relative to the WT-infected mice, LPA circulating in the blood was comparable to those observed in Δlpg1-/+LPG1 and Δlpg2-/+LPG2 infected mice, but this aggregate was significantly impaired in Δlpg1- and Δlpg2- infected groups at day 3 PI. In addition, the LPA level formed in response to infection with Δlpg1- parasites is comparable to those formed in response to infection with Δlpg2- parasites (Figures 7A–E). This suggests that LPG-activated platelets enhance P-selectin expression, resulting in platelet leukocyte aggregation.
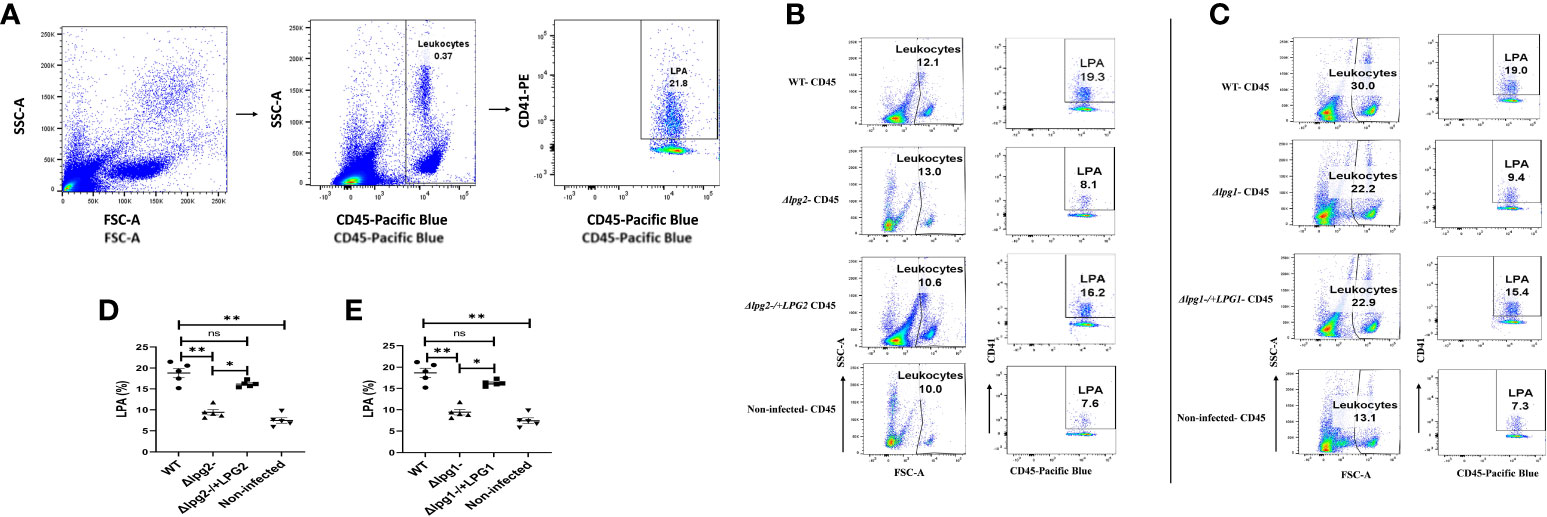
Figure 7 Δlpg1- and Δlpg2- parasites are ineffectual in inducing LPA formation. BALB/c mice were challenged with infective metacyclic promastigote (2 x 106 parasites, n = 5) of WT, Δlpg1-, Δlpg2-, Δlpg1-/+LPG1 and Δlpg2-/+LPG2 strains via the footpad. Control mice (n = 5) were given 0.9% NaCl saline. Blood was collected via retro-orbital sinus at day 3 PI. Blood samples were analyzed by flow cytometry for LPA. Representative flow cytometry dot plots showing the analyses of LPA performed on day 3 PI (A). Representative dot plots shown in (B, C) are from concatenated samples of each experimental group, and the corresponding graphs (D, E) indicate the percentage of LPA molecules by CD45+ cells. A dot plot of each sample in all the experimental groups is presented in Figure S4 (A, B). In all the experiments, WT-infected and non-infected mice served as a positive and negative control, respectively. Results are presented as mean (± SEM). One-way ANOVA with Bonferroni’s post hoc test was performed to analyze the data *p < 0.05, **p < 0.01, ‘ns’ indicates not significant (p > 0.05).
Discussion
The interactions between Leishmania and host cells have fundamental effects on the disease outcome (44). Leishmaniasis is thought to be initiated by direct parasitization of neutrophils and monocytic cells following parasite deposition into the skin (45–47). However, it has been demonstrated that platelets can be stimultated by Leishmania through activation of complement to produce PDGF, which is a potent inducer of CCL2 (MCP1). MCP-1 promotes the recruitment of a subpopulation of effector monocytes to the infection site. Complement was found to be essential for the generation of platelet PDGF, which was found for the first 30-60 minutes at the site of L. major infection (48). Complement activation is also known to generate chemotactic peptides (C5a, C3a), recruiting polymorphonuclear cells (PMN), which are critical for the establishment of infection. Further, C5a has been shown recently to be important for PMN trafficking to the draining lymph node (49). We have previously shown that DKK1 released from activated platelets in response to L. major infection promoted a Th2 inflammatory response and leukocyte-platelet aggregation (28); this is important biologically for the recruitment or migration of PMNs across the vasculature (36, 50, 51). Further, inhibition of DKK1 in vivo or platelet depletion resulted in reduction in IL-4, IL-10 and L. major parasite burden as well as cellular recruitment to the draining lymph node and site of infection. These studies suggest that platelets are likely one of the first cells to initiate innate immune responses to Leishmania, and are important in determining the outcome of infection (48, 52, 53).
Therefore, the primary aim of this study was to determine the Leishmania surface molecules involved in platelet activation and in the subsequent DKK1 production from activated platelets that could help account for the pathological type 2 inflammation observed in mice infected with L. major (28). We focused initially on the glycoconjugates LPG and PPG and compared the contribution of L. major Δlpg1- and Δlpg2- parasites on platelet activation and subsequent DKK1 production in infected mice. Data show that the absence of PPG and LPG in the Δlpg2- mutant and deletion of LPG alone in the Δlpg1- mutant significantly influence the activation of platelets by impairing the expression of P-selectin and DKK1 production compared to the responses obtained from WT and add-back L. major infected mice. In addition, the percentage of LPA at day 3 PI in both Δlpg1- and Δlpg2- mutant infected mice was significantly lower than those obtained from WT controls. In contrast, mice infected with Δlpg1- and Δlpg2- parasites complemented with the LPG1 and LPG2 genes (Δlpg1-/+LPG1 and Δlpg2-/+LPG2) manifested a phenotype similar to those infected with WT parasites. As Δlpg1- and Δlpg2- parasite infections result in relatively similar poor pathogenicity, implying that the deletion of PPG glycoconjugates did not influence the activation of platelets, instead LPG is the key molecule responsible for platelet activation.
Leishmania LPG has been reported to induce inflammation through TLR2 and 4 in macrophages and other cells (54–58). Notably, although there is considerable structural variation in leishmanial LPG between species (59), activation of TLR2 by LPG appears to be found across the genus (L. (V.). braziliensis, L. mexicana, L.infantum, L. major) (24, 57, 60–62). Although TLR4 activation does not lead to DKK1 release from platelets,the finding that platelet TLR2 mediates the induction of DKK1 suggests that this mechanism may play a role in the initiation of infection and pathogenesis of other Leishmania species (63–65).
However, previous studies have demonstrated that the LPG virulence factor is expressed and promotes the establishment of infection by metacyclic promastigotes in host cells; LPG expression significantly diminishes in amastigote stage (66–69). This suggests, as level of LPG expression declines in the mammalian host, it would have little/no critical function as the disease progresses. Since the expression of LPG decreases in the amastigote phase, it is unclear how P-selectin and DKKI observed in the WT and add-back infected mice on days 14 and 42 PI remained elevated. This may be associated with multiple factors. One possibility is the presence of residual membrane-anchored LPG, which might prolong platelet activation and DKK1 production. Previous studies showed that the down-regulation of the promastigote-specific virulence factor LPG varies in different host cell environments (68). Thus, the elevated DKK1 and P-selectin expression observed on days 14 and 42 PI could also be associated with prolonged retention of LPG virulence factor in platelets, or macrophages or possibly dendritic cells. Alternatively, studies demonstrate that TNF-alpha exerts its effects through TNF-RI expressed on megakaryocytes to activate platelets (70–74). Thus, the continuity of platelet activation and DKK1 production observed at later phase of infection may be associated with TNF-alpha activated platelets. Further, presence of Leishmania antigens and induction of immunoglobulins have been shown to induce the formation of circulating immune complexes (75). Antigen-antibody complexes are capable of activating platelets via interaction with FcγRIIa (76–78). Hence, as infection progresses the immune complexes formed could lead to sustained platelet activation and DKK1 release. Upon activation, platelets release certain complement components including C1q, C3, C4, and C5b-9 (79, 80), and they also express receptor for C3a, C5a, C1q and C4 (81–84). Previous studies reported that C3a/C5a and C1q binds to their respective ligands and enhance platelet activation (85, 86). This suggests that C3a and C1q may prolong platelet activation and DKK1 production. Inflammatory responses induced could also provide alternate modes for platelet activation. Collagen is a unique agonist and a potent activator for platelets (87). Glycoproteins Ia/IIa, glycoprotein VI and integrin α2β1, has been identified as the major platelet receptor for collagen (87–89). This indicates that collagen-activated platelets may as well provide a possible mechanism of sustained platelet activation. The WT, addback and mutant infected mice released DKK1 at day 42 PI, but the level of P-selectin expression was impaired in the mutant infected mice. Thus, DKK1 prodution and platelet activation dissociate at the later phase of infection. This suggests that by day 42 PI, the surviving amastigotes could be inducing the DKK1 via an alternate pathway (which may or may not be platelet-mediated). The mechanism of the sustained DKK1 release is not clear but of interest and requires further investigation.
Leishmania infection triggers cascades of events in macrophages that influence the ensuing immune response. One of the most crucial initial signaling events is interleukin-12 production by the infected macrophage, resulting in subsequent induction of Th1 response and production of interferon-gamma (IFN-γ) required to kill the pathogen (90–92). Unlike most microbial pathogens, Leishmania promastigotes have developed immune evasion strategies that prevent immediate “classical” macrophage proinflammatory activation (93). These strategies include engaging suppression-associated macrophage surface receptors such as complement receptor 3 (CR3) (94). Earlier studies of macrophages from CR3-deficient mice demonstrated that CR3 engagement is required for IL-12 suppression during Leishmania infection (95). In addition, Leishmania is known to delay antimicrobial activity in infected macrophages by inhibiting host defence mechanisms such as protein kinase C activation (96), and upregulation of inducible nitric oxide synthase following IFN-γ stimulation (92, 97). Thus, infected macrophages response to IFN-γ is repressed. Furthermore, it has been shown that Leishmania donovani-infected U937 cells exhibit decreased activation of the IFN-γ receptor (98, 99), and L. donovani amastigotes negatively influence the expression of major histocompatibility complex class II (MHC-II) following IFN-γ stimulation (99). Most importantly, Leishmania has been demonstrated to suppress TLR4-mediated proinflammatory cytokine production in BMDMs (100). Thus, these studies show that Leishmania evades the host immune response by inhibiting macrophage activation. Although Leishmania parasites interact and infect various host cell types- macrophages, neutrophils and dendritic cells are arguably the most important cells that regulate the outcome of infection (44, 101). However, there is limited information on the recruitment of these cells to the site of infection. Our study highlighted that DKK1 released by LPG-activated platelets regulates leukocyte platelet aggregation required for infiltration of leukocytes to the infection site. This is consistent with our earlier study that showed that inhibition of DKK1 diminished macrophage accumulation at the infection site, impaired Th2 polarization, and dampened parasitic load (28). This suggests that LPG-activated platelets might be the primary cells that significantly influence the initiation and outcome of infection.
In conclusion, our studies establish the importance of L. major-derived LPG in the induction of DKK1, which may serve as a novel immunomodulatory molecule regulating LPA formation (PMN recruitment) and chronic Th2 inflammatory response. This observation stresses the need to evaluate further the mechanism through which DKK1 facilitates leukocyte migration and LPA formation at the infection site.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Arizona University Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
OI: Conceptualization, Data curation, Methodology, Validation, Writing – original draft, Writing – review & editing. AS: Validation, Writing – review & editing, Data curation, Formal Analysis, Methodology. SB: Validation, Writing – review & editing, Funding acquisition, Resources. DM-P: Conceptualization, Supervision, Validation, Writing – review & editing, Methodology. AB: Conceptualization, Funding acquisition, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by AI-137060 (awarded to AB) and AI-31078 (awarded to SB).
Acknowledgments
We thank John Fitch for assisting with data acquisition. Also, we appreciate Dr. Martha Dua-Awereh, Haley Quinn Marcarian, Anika Arias, Dr. Deborah E. Dobson, Dr. Justin E. Wilson, Dakota M. Reinartz, Dr. Sambamurthy Chandrasekaran and Dr. Rithy Meas for their excellent suggestions and helpful advice.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1257046/full#supplementary-material
References
1. Castro M, Rode J, MaChado PR, Llanos-Cuentas A, Hueb M, Cota G, et al. Cutaneous leishmaniasis treatment and therapeutic outcomes in special populations: A collaborative retrospective study. PloS Negl Trop Dis (2023) 17(1):e0011029. doi: 10.1371/journal.pntd.0011029
2. Sheet WF. WHO Fact Sheet on Leishmaniasis . Geneva, Switzerland: World Health Organization (Accessed July. 2021;10:2021).
3. Cowan R, Varadarajan S, Wei A, Salim T, DallaPiazza M. Microbial perils of the tropics: A case of cutaneous leishmaniasis in an immigrant from South America. IDCases (2023) 31:e01669. doi: 10.1016/j.idcr.2022.e01669
4. Barry MA, Koshelev MV, Sun GS, Grekin SJ, Stager CE, Diwan AH, et al. Case report: cutaneous leishmaniasis in Cuban immigrants to Texas who traveled through the Darien Jungle, Panama. Am J Trop Med Hyg (2014) 91(2):345. doi: 10.4269/ajtmh.14-0124
5. Horev A, Sagi O, Zur E, Ben-Shimol S. Topical liposomal amphotericin B gel treatment for cutaneous leishmaniasis caused by Leishmania major: a double-blind, randomized, placebo-controlled, pilot study. Wiley Online Library; (2023) 62:40–7. doi: 10.1111/ijd.16407
6. Alcântara LM, Ferreira TC, Gadelha FR, Miguel DC. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. Int J Parasitol: Drugs Drug Resist (2018) 8(3):430–9. doi: 10.1016/j.ijpddr.2018.09.006
7. Gautam I, Huss CW, Storad ZA, Krebs M, Bassiouni O, Ramesh R, et al. Activated Platelets mediate monocyte killing of klebsiella pneumoniae. Infect Immun (2023) 91(3):e00556–22. doi: 10.1128/iai.00556-22
8. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb haemostasis (2015) 114(09):449–58. doi: 10.1160/TH14-12-1067
9. Jenne CN, Kubes P. Platelets in inflammation and infection. Platelets (2015) 26(4):286–92. doi: 10.3109/09537104.2015.1010441
10. Ley K. The role of selectins in inflammation and disease. Trends Mol Med (2003) 9(6):263–8. doi: 10.1016/s1471-4914(03)00071-6
11. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res (2007) 100(1):27–40. doi: 10.1161/01.RES.0000252802.25497.b7
12. Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ. PSGL-1 function in immunity and steady state homeostasis. Immunol Rev (2009) 230(1):75–96. doi: 10.1111/j.1600-065X.2009.00797.x
13. Yu M, Xiao G, Han L, Peng L, Wang H, He S, et al. QiShen YiQi and its components attenuate acute thromboembolic stroke and carotid thrombosis by inhibition of CD62P/PSGL-1-mediated platelet-leukocyte aggregate formation. Biomed Pharmacother (2023) 160:114323. doi: 10.1016/j.biopha.2023.114323
14. León B, Ardavín C. Monocyte migration to inflamed skin and lymph nodes is differentially controlled by L-selectin and PSGL-1. Blood J Am Soc Hematol (2008) 111(6):3126–30. doi: 10.1182/blood-2007-07-100610
15. McEver RP. P-selectin/PSGL-1 and other interactions between platelets, leukocytes, and endothelium. Platelets (2007) 2:231–49. doi: 10.1016/B978-012369367-9/50774-6
16. Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood (1990) 75(3):550–4. doi: 10.1182/blood.V75.3.550.550
17. Pimenta PF, Saraiva EM, Sacks DL. The comparative fine structure and surface glycoconjugate expression of three life stages of Leishmania major. Exp parasitol (1991) 72(2):191–204. doi: 10.1016/0014-4894(91)90137-L
18. Ilgoutz SC, McConville MJ. Function and assembly of the Leishmania surface coat. Int J parasitol (2001) 31(9):899–908. doi: 10.1016/S0020-7519(01)00197-7
19. Beverley SM, Turco SJ. Lipophosphoglycan (LPG) and the identification of virulence genes in the protozoan parasite Leishmania. Trends Microbiol (1998) 6(1):35–40. doi: 10.1016/S0966-842X(97)01180-3
20. Llg T, Handman E, Stierhof Y-D. Proteophosphoglycans from Leishmania promastigotes and amastigotes. Biochem Soc Trans (1999) 27(4):518–25. doi: 10.1042/bst0270518
21. Zhang K, Barron T, Turco SJ, Beverley SM. The LPG1 gene family of Leishmania major. Mol Biochem parasitol (2004) 136(1):11–23. doi: 10.1016/j.molbiopara.2004.02.012
22. Späth GF, Epstein L, Leader B, Singer SM, Avila HA, Turco SJ, et al. Lipophosphoglycan is a virulence factor distinct from related glycoconjugates in the protozoan parasite Leishmania major. Proc Natl Acad Sci (2000) 97(16):9258–63. doi: 10.1073/pnas.160257897
23. Capul AA, Barron T, Dobson DE, Turco SJ, Beverley SM. Two functionally divergent UDP-Gal nucleotide sugar transporters participate in phosphoglycan synthesis in Leishmania major. J Biol Chem (2007) 282(19):14006–17. doi: 10.1074/jbc.M610869200
24. Rojas-Bernabe A, Garcia-Hernandez O, Maldonado-Bernal C, Delegado-Dominguez J, Ortega E, Gutierrez-Kobeh L, et al. Leishmania mexicana lipophosphoglycan activates ERK and p38 MAP kinase and induces production of proinflammatory cytokines in human macrophages through TLR2 and TLR4. Parasitology (2014) 141(6):788–800. doi: 10.1017/S0031182013002187
25. Späth GF, Garraway L, Turco SJ, Beverley SM. The role (s) of lipophosphoglycan (LPG) in the establishment of Leishmania major infections in mammalian hosts. Proc Natl Acad Sci (2003) 100(16):9536–41. doi: 10.1073/pnas.153060410
26. Svárovská A, Ant TH, Seblová V, Jecná L, Beverley SM, Volf P. Leishmania major glycosylation mutants require phosphoglycans (lpg2–) but not lipophosphoglycan (lpg1–) for survival in permissive sand fly vectors. PloS Negl Trop Dis (2010) 4(1):e580. doi: 10.1371/journal.pntd.0000580
27. Liu D, Kebaier C, Pakpour N, Capul AA, Beverley SM, Scott P, et al. Leishmania major phosphoglycans influence the host early immune response by modulating dendritic cell functions. Infect Immun (2009) 77(8):3272–83. doi: 10.1128/IAI.01447-08
28. Chae W-J, Ehrlich AK, Chan PY, Teixeira AM, Henegariu O, Hao L, et al. The Wnt antagonist Dickkopf-1 promotes pathological type 2 cell-mediated inflammation. Immunity (2016) 44(2):246–58. doi: 10.1016/j.immuni.2016.01.008
29. Spath GF, Lye L-F, Segawa H, Sacks DL, Turco SJ, Beverley SM. Persistence without pathology in phosphoglycan-deficient Leishmania major. Science (2003) 301(5637):1241–3. doi: 10.1126/science.1087499
30. Capul AA, Hickerson S, Barron T, Turco SJ, Beverley SM. Comparisons of mutants lacking the Golgi UDP-galactose or GDP-mannose transporters establish that phosphoglycans are important for promastigote but not amastigote virulence in Leishmania major. Infect Immun (2007) 75(9):4629–37. doi: 10.1128/IAI.00735-07
31. Späth GF, Beverley SM. A lipophosphoglycan-independent method for isolation of infective Leishmania metacyclic promastigotes by density gradient centrifugation. Exp parasitol (2001) 99(2):97–103. doi: 10.1006/expr.2001.4656
32. Jara M, Barrett M, Maes I, Regnault C, Imamura H, Domagalska MA, et al. Transcriptional shift and metabolic adaptations during Leishmania quiescence using stationary phase and drug pressure as models. Microorganisms (2022) 10(1):97. doi: 10.3390/microorganisms10010097
33. Titus RG, Marchand M, Boon T, Louis J. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol (1985) 7(5):545–55. doi: 10.1111/j.1365-3024.1985.tb00098.x
34. Pula G, Schuh K, Nakayama K, Nakayama KI, Walter U, Poole AW. PKCδ regulates collagen-induced platelet aggregation through inhibition of VASP-mediated filopodia formation. Blood (2006) 108(13):4035–44. doi: 10.1182/blood-2006-05-023739
35. Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci (2010) 67:499–511. doi: 10.1007/s00018-009-0205-1
36. Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol (2018) 9:2712. doi: 10.3389/fimmu.2018.02712
37. Spaüth GF, Lye L-F, Segawa H, SJ T, Beverley SM. Identification of a compensatory mutant (lpg2– Rev) of Leishmania major able to survive as amastigotes within macrophages without LPG2-dependent glycoconjugates and its significance to virulence and immunization strategies. Infect Immun (2004) 72(6):3622–7. doi: 10.1128/IAI.72.6.3622-3627.2004
38. De Stoppelaar S, Claushuis T, Jansen M, Hou B, Roelofs JJ, Van’t Veer C, et al. The role of platelet MyD88 in host response during gram-negative sepsis. J Thromb Haemostasis (2015) 13(9):1709–20. doi: 10.1111/jth.13048
39. Loegering DJ, Lennartz MR. Protein kinase C and toll-like receptor signaling. Enzyme Res (2011) 2011:537821. doi: 10.4061/2011/537821
40. Carneiro PP, Dórea AS, Oliveira WN, Guimarães LH, Brodskyn C, Carvalho EM, et al. Blockade of TLR2 and TLR4 attenuates inflammatory response and parasite load in cutaneous leishmaniasis. Front Immunol (2021) 12:706510. doi: 10.3389/fimmu.2021.706510
41. Franks ZG, Campbell RA, Weyrich AS, Rondina MT. Platelet–leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann N Y Acad Sci (2010) 1207(1):11–7. doi: 10.1111/j.1749-6632.2010.05733.x
42. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
43. Zhang T, Liu L, Huang X, Gao X, Chen D, Huan X, et al. Application of microfluidic chip technology to study the inhibitory effect of tetramethylpyrazine on platelet aggregation, activation, and phosphatidylserine exposure mediated by pathological high shear rate. Blood Coagul Fibrinolysiss (2023) 34(1):47–60. doi: 10.1097/MBC.0000000000001179
44. Liu D, Uzonna JE. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol (2012) 2:83. doi: 10.3389/fcimb.2012.00083
45. Charmoy M, Auderset F, Allenbach C, Tacchini-Cottier F. The prominent role of neutrophils during the initial phase of infection by Leishmania parasites. J Biomed Biotechnol (2009) 2010:719361. doi: 10.1155/2010/719361
46. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (2008) 321(5891):970–4. doi: 10.1126/science.1159194
47. Passelli K, Billion O, Tacchini-Cottier F. The impact of neutrophil recruitment to the skin on the pathology induced by Leishmania infection. Front Immunol (2021) 12:649348. doi: 10.3389/fimmu.2021.649348
48. Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of Leishmania major infection. J Exp Med (2011) 208(6):1253–65. doi: 10.1084/jem.20101751
49. Prat-Luri B, Neal C, Passelli K, Ganga E, Amore J, Firmino-Cruz L, et al. The C5a-C5aR1 complement axis is essential for neutrophil recruitment to draining lymph nodes via high endothelial venules in cutaneous leishmaniasis. Cell Rep (2022) 39(5):110777. doi: 10.1016/j.celrep.2022.110777
50. Liu X, Gorzelanny C, Schneider SW. Platelets in skin autoimmune diseases. Front Immunol (2019) 10:1453. doi: 10.3389/fimmu.2019.01453
51. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PloS Biol (2016) 14(5):e1002459. doi: 10.1371/journal.pbio.1002459
52. Mosser D, Brittingham A. Leishmania, macrophages and complement: a tale of subversion and exploitation. Parasitology (1997) 115(7):9–23. doi: 10.1017/s0031182097001789
53. Puentes SM, Sacks DL, da Silva RP, Joiner KA. Complement binding by two developmental stages of Leishmania major promastigotes varying in expression of a surface lipophosphoglycan. J Exp Med (1988) 167(3):887–902. doi: 10.1084/jem.167.3.887
54. Faria MS, Reis FC, Lima APC. Toll-like receptors in leishmania infections: guardians or promoters? J Parasitol Res (2012) 2012:930257. doi: 10.1155/2012/930257
55. Mukherjee S, Karmakar S, Babu SPS. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis (2016) 20:193–204. doi: 10.1016/j.bjid.2015.10.011
56. Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem parasitol (2003) 130(2):65–74. doi: 10.1016/s0166-6851(03)00160-9
57. de Veer MJ, Curtis JM, Baldwin TM, DiDonato JA, Sexton A, McConville MJ, et al. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol (2003) 33(10):2822–31. doi: 10.1002/eji.200324128
58. Kavoosi G, Ardestani S, Kariminia A. The involvement of TLR2 in cytokine and reactive oxygen species (ROS) production by PBMCs in response to Leishmania major phosphoglycans (PGs). Parasitology (2009) 136(10):1193–9. doi: 10.1017/S0031182009990473
59. Forestier C-L, Gao Q, Boons G-J. Leishmania lipophosphoglycan: how to establish structure-activity relationships for this highly complex and multifunctional glycoconjugate? Front Cell Infect Microbiol (2015) 4:193. doi: 10.3389/fcimb.2014.00193
60. Zamora-Chimal J, Fernández-Figueroa EA, Ruiz-Remigio A, Wilkins-Rodríguez AA, Delgado-Domínguez J, Salaiza-Suazo N, et al. NKT cell activation by Leishmania mexicana LPG: Description of a novel pathway. Immunobiology (2017) 222(2):454–62. doi: 10.1016/j.imbio.2016.08.003
61. Sacramento LA, Da Costa JL, De Lima MH, Sampaio PA, Almeida RP, Cunha FQ, et al. Toll-like receptor 2 is required for inflammatory process development during Leishmania infantum infection. Front Microbiol (2017) 8:262. doi: 10.3389/fmicb.2017.00262
62. Polari LP, Carneiro PP, Macedo M, MaChado PR, Scott P, Carvalho EM, et al. Leishmania Braziliensis infection enhances toll-like receptors 2 and 4 expression and triggers TNF-α and IL-10 production in human cutaneous leishmaniasis. Front Cell Infect Microbiol (2019) 9:120. doi: 10.3389/fcimb.2019.00120
63. Ibraim IC, de Assis RR, Pessoa NL, Campos MA, Melo MN, Turco SJ, et al. Two biochemically distinct lipophosphoglycans from Leishmania Braziliensis and Leishmania infantum trigger different innate immune responses in murine macrophages. Parasites vectors (2013) 6(1):1–11. doi: 10.1186/1756-3305-6-54
64. Ray S, Maunsell JH. Different origins of gamma rhythm and high-gamma activity in macaque visual cortex. PloS Biol (2011) 9(4):e1000610. doi: 10.1371/journal.pbio.1000610
65. Soto-Olguín N, Zamora-Chimal J, Delgado-Domínguez J, Becker I. Leishmania mexicana lipophosphoglycan activates dermal γδ T cells with participation of TLR2. Acta Parasitologica (2022) 68(1):122–9. doi: 10.1007/s11686-022-00639-w
66. Salako E. The role of alkyldihydroxyacetone phosphate synthase (ADS) on host immune response and virulence of leishmania major. (2023). Available at: http://hdl.handle.net/1993/37258.
67. de Carvalho RV, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep (2019) 26(2):429–37. e5. doi: 10.1016/j.celrep.2018.12.047
68. Mandell MA, Beatty WL, Beverley SM. Quantitative single-cell analysis of Leishmania major amastigote differentiation demonstrates variably extended expression of the lipophosphoglycan (LPG) virulence factor in different host cell types. PloS Negl Trop Dis (2022) 16(10):e0010893. doi: 10.1371/journal.pntd.0010893
69. Moody SF, Handman E, McConville M, Bacic A. The structure of Leishmania major amastigote lipophosphoglycan. J Biol Chem (1993) 268(25):18457–66. doi: 10.1016/S0021-9258(17)46648-8
70. Philippe C, Philippe B, Fouqueray B, Perez J, Lebret M, Baud L. Protection from tumor necrosis factor-mediated cytolysis by platelets. Am J pathol (1993) 143(6):1713. doi: 10.1016/S0021-9258(18)54804-3
71. Tacchini-Cottier F, Vesin C, Redard M, Buurman W, Piguet PF. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J Immunol (1998) 160(12):6182–6. doi: 10.1055/s-0038-1657724
72. Bar J, Zosmer A, Hod M, Elder M, Sullivan M. The regulation of platelet aggregation in vitro by interleukin-1 β and tumor necrosis factor-α: changes in pregnancy and in pre-eclampsia. Thromb haemostasis (1997) 78(10):1255–61. doi: 10.1055/s-0038-1657724
73. Pignatelli P, De Biase L, Lenti L, Tocci G, Brunelli A, Cangemi R, et al. Tumor necrosis factor-α as trigger of platelet activation in patients with heart failure. Blood (2005) 106(6):1992–4. doi: 10.1182/blood-2005-03-1247
74. Baj-Krzyworzeka M, Majka M, Pratico D, Ratajczak J, Vilaire G, Kijowski J, et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp hematol (2002) 30(5):450–9. doi: 10.1016/s0301-472x(02)00791-9
75. Parody N, Cacheiro-Llaguno C, Osuna C, Renshaw-Calderón A, Alonso C, Carnés J. Circulating immune complexes levels correlate with the progression of canine leishmaniosis in naturally infected dogs. Vet parasitol (2019) 274:108921. doi: 10.1016/j.vetpar.2019.108921
76. Jevtic SD, Nazy I. The COVID complex: a review of platelet activation and immune complexes in COVID-19. Front Immunol (2022) 13:807934. doi: 10.3389/fimmu.2022.807934
77. Nazy I, Jevtic SD, Moore JC, Huynh A, Smith JW, Kelton JG, et al. Platelet-activating immune complexes identified in critically ill COVID-19 patients suspected of heparin-induced thrombocytopenia. J Thromb Haemostasis (2021) 19(5):1342–7. doi: 10.1111/jth.15283
78. McKenzie SE, Taylor SM, Malladi P, Yuhan H, Cassel DL, Chien P, et al. The role of the human Fc receptor FcγRIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol (1999) 162(7):4311–8. doi: 10.4049/jimmunol.162.7.4311
79. Kim H, Conway EM. Platelets and complement cross-talk in early atherogenesis. Front Cardiovasc Med (2019) 6:131. doi: 10.3389/fcvm.2019.00131
80. Speth C, Rambach G, Würzner R, Lass-Flörl C, Kozarcanin H, Hamad OA, et al. Complement and platelets: mutual interference in the immune network. Mol Immunol (2015) 67(1):108–18. doi: 10.1016/j.molimm.2015.03.244
81. Peerschke EI, Murphy TK, Ghebrehiwet B. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb haemostasis (2003) 89(02):331–9. doi: 10.1055/s-0037-1613450
82. Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhöch S, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation (2018) 138(16):1720–35. doi: 10.1161/CIRCULATIONAHA.118.034600
83. Vik DP, Fearon D. Cellular distribution of complement receptor type 4 (CR4): expression on human platelets. J Immunol (Baltimore Md: 1950) (1987) 138(1):254–8. doi: 10.4049/jimmunol.138.1.254
84. Patzelt J, Müller KA-L, Breuning S, Karathanos A, Schleicher R, Seizer P, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis (2015) 238(2):289–95. doi: 10.1016/j.atherosclerosis.2014.12.002
85. Polley M, Nachman R. Human platelet activation by C3a and C3a des-arg. J Exp Med (1983) 158(2):603–15. doi: 10.1084/jem.158.2.603
86. Peerschke E, Reid K, Ghebrehiwet B. Platelet activation by C1q results in the induction of alpha IIb/beta 3 integrins (GPIIb-IIIa) and the expression of P-selectin and procoagulant activity. J Exp Med (1993) 178(2):579–87. doi: 10.1084/jem.178.2.579
87. Siljander P, Lassila R. Studies of adhesion-dependent platelet activation: distinct roles for different participating receptors can be dissociated by proteolysis of collagen. Arteriosclerosis thrombosis Vasc Biol (1999) 19(12):3033–43. doi: 10.1161/01.atv.19.12.3033
88. Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, et al. Glycoprotein VI but not α2β1 integrin is essential for platelet interaction with collagen. EMBO J (2001) 20(9):2120–30. doi: 10.1093/emboj/20.9.2120
89. Kunicki TJ, Nugent D, Staats S, Orchekowski R, Wayner E, Carter W. The human fibroblast class II extracellular matrix receptor mediates platelet adhesion to collagen and is identical to the platelet glycoprotein Ia-IIa complex. J Biol Chem (1988) 263(10):4516–9. doi: 10.1016/S0021-9258(18)68811-8
90. Boehm U, Klamp T, Groot M, Howard J. Cellular responses to interferon-γ. Annu Rev Immunol (1997) 15(1):749–95. doi: 10.1146/annurev.immunol.15.1.749
91. Heinzel F, Schoenhaut DS, Rerko R, Rosser L, Gately M. Recombinant interleukin 12 cures mice infected with Leishmania major. J Exp Med (1993) 177(5):1505–9. doi: 10.1084/jem.177.5.1505
92. Peters N, Sacks D. Immune privilege in sites of chronic infection: Leishmania and regulatory T cells. Immunol Rev (2006) 213(1):159–79. doi: 10.1111/j.1600-065X.2006.00432.x
93. Sacks D, Sher A. Evasion of innate immunity by parasitic protozoa. Nat Immunol (2002) 3(11):1041–7. doi: 10.1038/ni1102-1041
94. Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol (2002) 20(1):825–52. doi: 10.1146/annurev.immunol.20.103001.114744
95. Carter CR, Whitcomb JP, Campbell JA, Mukbel RM, McDowell MA. Complement receptor 3 deficiency influences lesion progression during Leishmania major infection in BALB/c mice. Infect Immun (2009) 77(12):5668–75. doi: 10.1128/IAI.00802-08
96. Vannier-Santos M, Martiny A, Souza WD. Cell biology of Leishmania spp.: invading and evading. Curr Pharm design (2002) 8(4):297–318. doi: 10.2174/1381612023396230
97. Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev (2005) 18(2):293–305. doi: 10.1128/CMR.18.2.293-305.2005
98. Ray M, Gam AA, Boykins RA, Kenney RT. Inhibition of interferon-γ signaling by Leishmania donovani. J Infect Dis (2000) 181(3):1121–8. doi: 10.1086/315330
99. Reiner NE, Ng W, Ma T, McMaster WR. Kinetics of gamma interferon binding and induction of major histocompatibility complex class II mRNA in Leishmania-infected macrophages. Proc Natl Acad Sci (1988) 85(12):4330–4. doi: 10.1073/pnas.85.12.433
100. Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK. Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol (2012) 189(2):924–34. doi: 10.4049/jimmunol.1102845
Keywords: Leishmaniasis, P-selectin, innate response, leukocyte-platelet aggregates, platelet
Citation: Ihedioha OC, Sivakoses A, Beverley SM, McMahon-Pratt D and Bothwell ALM (2023) Leishmania major-derived lipophosphoglycan influences the host’s early immune response by inducing platelet activation and DKK1 production via TLR1/2. Front. Immunol. 14:1257046. doi: 10.3389/fimmu.2023.1257046
Received: 11 July 2023; Accepted: 25 September 2023;
Published: 11 October 2023.
Edited by:
Neetu Singh, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Tiago Rodrigues Ferreira, National Institute of Allergy and Infectious Diseases (NIH), United StatesDjalma Souza Lima-Junior, National Institutes of Health (NIH), United States
Diego Luis Costa, University of São Paulo, Brazil
Copyright © 2023 Ihedioha, Sivakoses, Beverley, McMahon-Pratt and Bothwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfred L. M. Bothwell, YWJvdGh3ZWxsQGFyaXpvbmEuZWR1