 Feroza Yasinjan
Feroza Yasinjan Yang Xing
Yang Xing Huayue Geng1
Huayue Geng1 Rui Guo
Rui Guo Ziling Liu
Ziling Liu Hong Wang
Hong Wang- 1Cancer Center, The First Hospital of Jilin University, Changchun, China
- 2Department of Neurosurgery, The First Hospital of Jilin University, Changchun, China
- 3Clinical Laboratory, The First Hospital of Jilin University, Jilin University, Changchun, China
Gliomas are the most prevalent primary malignant brain tumors worldwide, with glioblastoma (GBM) being the most common and aggressive type. Despite two decades of relentless pursuit in exploring novel therapeutic approaches for GBM, there is limited progress in improving patients’ survival outcomes. Numerous obstacles impede the effective treatment of GBM, including the immunosuppressive tumor microenvironment (TME), the blood-brain barrier, and extensive heterogeneity. Despite these challenges, immunotherapies are emerging as a promising avenue that may offer new hope for the treatment of gliomas. There are four main types of immunotherapies for gliomas, immune checkpoint blockades, chimeric antigen receptor T-cell therapies, vaccines, and oncolytic viruses. In addition, gene therapy, bispecific antibody therapy, and combine therapy are also briefly introduced in this review. The significant role of TME in the process of immunotherapies has been emphasized in many studies. Although immunotherapy is a promising treatment for gliomas, enormous effort is required to overcome the existing barriers to its success. Owing to the rapid development and increasing attention paid to immunotherapies for gliomas, this article aims to review the recent advances in immunotherapies for gliomas.
1 Introduction
Gliomas are the most prevalent primary malignant brain tumors worldwide. Gliomas often grow in the brain and come from glial tissue, yet they may form elsewhere in the central nervous system (CNS) (1–3). According to the up-to-date WHO classification of CNS tumors, diffuse gliomas in adults have been classified into: astrocytoma IDH-mutant (grade 2, 3, or 4), oligodendroglioma IDH-mutant and 1p/19q co-deleted (grade 2 or 3), and glioblastoma (GBM) IDH-wildtype (grade 4) (4). Moreover, patients diagnosed with GBM usually have a terrible prognosis, with a median overall survival (mOS) of less than two years, and a five-year survival rate of 10% (1–3).
The established treatment paradigm for GBM, known as the Stupp regimen, entails maximal surgical tumor resection followed by a combination of radiotherapy and chemotherapy (5). However, almost all patients show recurrence after receiving standard treatment. Thus, it is necessary to explore novel effective therapies for GBM. In recent years, immunotherapeutic strategies have revolutionized the treatment of various cancers, such as melanoma and lung cancer, and also bring new hope for GBM treatment (6–8).
Currently, more than 88 clinical trials on immunotherapies for GBM are being conducted worldwide (9). Moreover, the efficacy of several immunotherapeutic treatments, including the dendritic cell (DC) vaccine DCVax-L (10, 11) and oncolytic virus (OV) G47Δ (12), has been demonstrated in phase II and III clinical trials. And oncolytic virus G47Δ had also been conditionally approved in Japan for the treatment of malignant gliomas. These events have represented the potential power of immunotherapies in gliomas, and immunotherapies are worthy of our wait to change the bad prognosis of patients with GBM. However, several barriers, including blood-brain barrier (BBB), tumor microenvironment (TME), and substantial heterogeneity, broadly weaken and limit the efficacy of immunotherapies for gliomas. Therefore, novel practical therapeutic approaches are constantly being studied.
2 Immunotherapies in gliomas
Immunotherapies for cancer treatment refer to the engagement of patients’ immune systems to recognize and eliminate cancer. There are several kinds of immunotherapies used in cancer treatment, including immune checkpoint blockades (ICBs), adoptive cell therapies, therapeutic vaccines, OV therapies, etc. (13, 14). And different types of immunotherapies are applicable and suitable for different cancers. There are mainly four types of immunotherapies in treating gliomas: ICBs, Chimeric antigen receptor T (CAR-T) cell therapies, vaccines, and OVs (15, 16). ICB therapy can effectively block immune checkpoints such as PD-1/PD-L1 and thus inhibits the immunosuppressive effect. Peptide and dendritic vaccines are the immune targets of tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). CAR-T cell therapies targeting tumor surface molecules, such as EGFR variant III (EGFRvIII), IL13Rα2, and HER2, have also been explored in GBM treatment. OV therapy is an emerging treatment of GBM that has received widespread attention in recent years, with G47Δ being conditionally approved as a treatment option in Japan, opening the way for further development of immunotherapy (12). Next, we would like to analyze the recent advances in these major immunotherapeutic strategies.
2.1 ICBs
Immune checkpoints are surface molecules on immune cells that can regulate host immunity when they bind to the corresponding ligands or receptors on tumor cells or other cells (9). And tumor cells are proficient at employing this strategy to avoid the lethal effect exerted by immune cells (mainly T cells) (17). Immune checkpoint blockade (ICB) primarily refers to blocking the immunosuppressive immune checkpoints such as PD-1/PD-L1, and CTLA-4, thus inhibiting their corresponding immunosuppressive effects and playing an antitumor effect. And ICB has been proven feasible and effective in treating many cancers (18). However, there are no successful phase III clinical trials or marketing authorizations of ICBs in GBM treatment all over the world. Nevertheless, the exploration of this field is continuous and active, which can be indicated in Table 1. With a deeper understanding of the TME, some co-stimulatory checkpoints and their specific agonists have also been studied. Unlike other cancers, such as melanoma and lung cancer, the application of ICBs seems unfavorable in treating gliomas (19). Nevertheless, the combination therapy of ICB and other treatments, such as standard chemoradiotherapy, targeted therapies, or other different kinds of immunotherapies, may find a way to success (9).
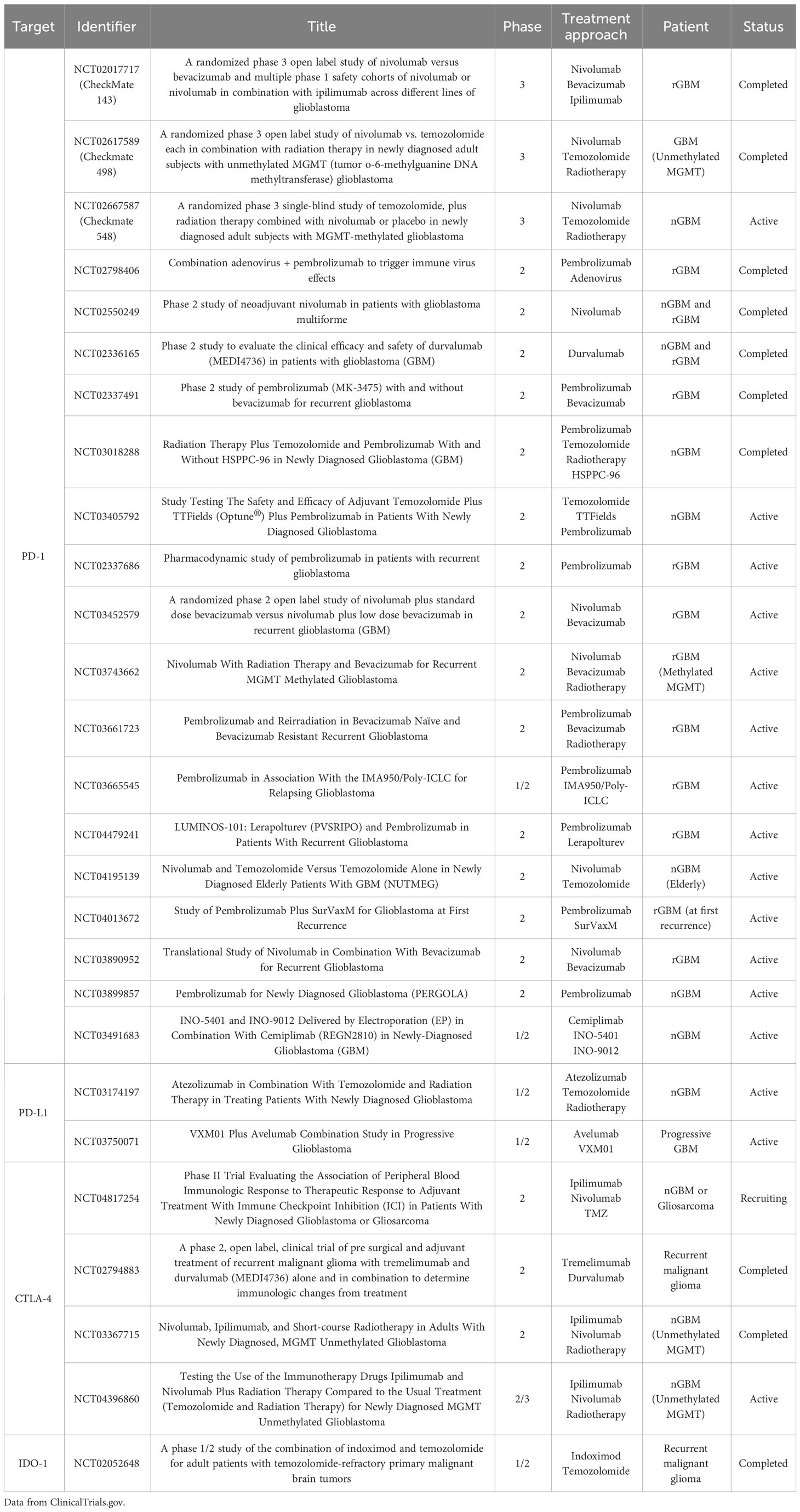
Table 1 Recent phase II/III clinical trials of ICBs for GBM treatment.
There are several completed clinical trials associated with ICBs for GBM treatment. All three clinical trials used anti-PD-1 monoclonal antibodies. PD-1 is a co-repressor molecule of the CD2 family and is expressed on the activated immune cells constitutively (20). After PD-1 binding to its ligands (mainly PD-L1) on tumor cells or antigen-presenting cells (APCs), T cells’ impotence, failure, or even apoptosis can be induced (21). In addition, PD-1 can also stimulate the proliferation of regulatory T cells (Tregs) and reduce the immune responses of natural killer cells and B cells (22).
Pembrolizumab (KEYTRUDA) is a commonly used monoclonal anti-PD-1 antibody. In 2019, satisfactory results of a randomized, multi-institutional trial of neoadjuvant pembrolizumab in patients with recurrent, surgically resectable GBM were published (23). Compared with patients receiving adjuvant, post-surgical administration of pembrolizumab alone, patients receiving neoadjuvant pembrolizumab and sustained adjuvant therapy after surgery had substantially improved OS and progression-free survival (PFS). The median OS of patients in the adjuvant-only group and the neoadjuvant group were 228 days (7.5 months) and 417 days (13.7 months), respectively, with a hazard ratio (neoadjuvant/adjuvant) of 0.39 (P = 0.04). And the median PFS of patients in the adjuvant-only group and the neoadjuvant group were 72.5 days (2.4 months) and 99.5 days (3.3 months), respectively, with a hazard ratio (neoadjuvant/adjuvant) of 0.43 (P = 0.03). The frequency of the focal upregulation of PD-L1 in the TME, the downregulation of PD-1 on T cells in the peripheral blood, and the intensive clonal expansion of T cells was higher in the neoadjuvant group compared with the adjuvant-only group. And the functional activation of tumor-infiltrating lymphocytes (TILs) was induced by neoadjuvant PD-1 blockade, which produced an interferon response within the TME (23).
The other two clinical trials applied another anti-PD-1 antibody, nivolumab (OPDIVO). Nivolumab is one of the most studied anti-PD-1 antibodies in treating gliomas. CheckMate 143 was the first phase III randomized clinical trial to test the efficacy of nivolumab in patients with recurrent GBM (rGBM) (24). But this trial’s primary endpoint (median OS, mOS) was not reached. Compared with the control group (bevacizumab) (10.0 months), the mOS was similar in the nivolumab group (9.8 months) (P = 0.76). And the PFS and the ORR (overall response rate) of the bevacizumab group were better than those of the nivolumab group. Besides, the post-subgroup analysis indicated that the potential benefits from nivolumab are more probable to be obtained in patients with methylated MGMT promoters (24). NCT02550249 was another crucial phase II clinical trial of nivolumab for GBM treatment. In this single-arm phase II clinical trial, twenty-seven patients with relapsed GBM and three patients with primary GBM were included to explore the feasibility, safety, and antitumor effects of neoadjuvant nivolumab in patients with resectable GBM (25). The neoadjuvant nivolumab was found to increase the expression of chemokines, infiltration of immune cells, and clonal diversity of T cell receptors in the TME (25).
There were also two completed randomized phase III clinical trials to test the efficacy of nivolumab in patients with newly diagnosed GBM (nGBM) (26, 27). The Checkmate 498 trial aimed to test the effectiveness of nivolumab plus radiotherapy, compared with TMZ plus radiotherapy, in patients with nGBM characterized as unmethylated MGMT promoters. However, its recently published results did not reach the primary endpoint (mOS). The mOSs of the nivolumab plus radiotherapy group and TMZ plus radiotherapy were 13.4 months and 14.9 months, respectively (27). The CheckMate 548 trial was a similar phase III study to evaluate the effectiveness of nivolumab plus the Stupp regime (radiotherapy plus TMZ), compared with the Stupp regime plus placebo, in patients with nGBM with methylated MGMT promoter (26). However, the results also showed a failure in adding nivolumab to the Stupp regime could not improve the OS and PFS of the patients. The mPFSs of nivolumab plus the Stupp regime group and the Stupp regime plus placebo group were 10.6 months and 10.3 months, respectively. And the mOSs were 28.9 months and 32.1 months, respectively.
In addition, some other immune checkpoints in gliomas, such as CTLA-4 (28–30), CD47 (31–33), CD73 (34, 35), TIGIT (36), and CD137 (37), were also studied in either clinical trials and preclinical research. It is worth noting that TILs in GBM are proven to highly express a number of co-inhibitory immune checkpoints, such as TIM-3, PD-1, and LAG-3, as a result of severe exhaustion of T cells (38). And this supports the necessity of exploring other feasible immune checkpoints and combined therapies to increase the effectiveness of the ICB (26, 39).
2.2 Vaccine therapy
Vaccines have a long history of use in cancer treatment (40, 41). Several types of vaccines are used in cancer treatment, and peptide and dendritic cell (DC) vaccines are the main strategies for glioma treatment (42–44). In addition, the TAA and TSA can be used as the immune targets of vaccines to stimulate adaptive immunity (45). Table 2 lists the recent phase II/III clinical trials of vaccine therapy for GBM treatment from ClinicalTrials.gov.
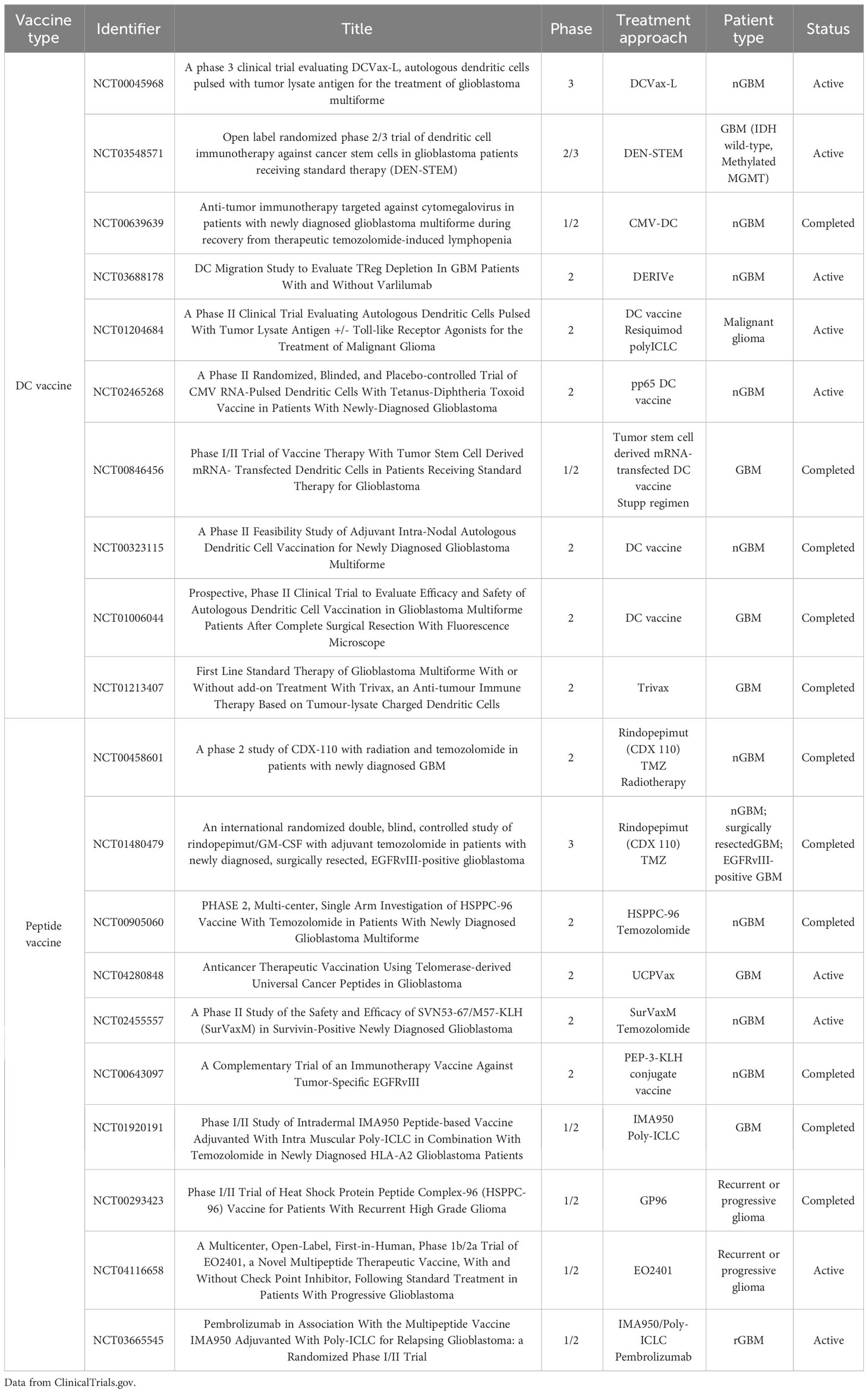
Table 2 Recent phase II/III clinical trials of vaccine therapy for GBM treatment.
Peptide vaccines comprise 8-25 amino acids and have epitopes acting as antigenic targets (43). Moreover, they are usually conjugated to a carrier protein to increase immunogenicity. These vaccines with simple structures are relatively easy to manufacture and store and have relatively lower variability when compared to other vaccines (43, 46). The frequently mutated or highly expressed proteins or antigens in GBM mainly include EGFR, EGFRvIII, NF1, TERT, PDGFRA, PTEN, RB1, IDH1, TP53, PIK3R1, and PIK3CA, some of which are regarded as ideal vaccine targets (43, 47). EGFRvIII is a deletion mutation of EGFR, which can be detected in approximately 20% of GBM (48, 49). And EGFRvIII is proven to enhance tumor growth and chemotherapy resistance. Currently, EGFRvIII is one of the most studied TSAs and an important target in vaccines for GBM treatment (50). There was an important phase III clinical trial that used the peptide vaccine rindopepimut (51). However, the results of this phase III trial of rindopepimut in combination with TMZ in patients with EGFRvIII-positive nGBM did not show a not disappoint. Compared with the TMZ group, the rindopepimut plus TMZ group did not increase the mOS. The mOS of TMZ and rindopepimut plus TMZ groups were 20.1 months and 20.0 months, respectively (P=0.93). Besides, the result also indicated the necessity of multi-peptide vaccines against several targets to overcome the antigenic heterogeneity in GBM (43, 51).
DC vaccine is another studied type of vaccine for glioma treatment (52). Dendritic cells’ function as antigen-presenting cells serves as the foundation for the main mechanism of DC vaccines (53). When immune cells, especially T cells, are activated by DCs, they can cross BBB and enter the brain tumor site to play the antitumor effects (53, 54). And DCs are believed to trigger both innate and adaptive immune responses to facilitate the transformation of immunologically cold gliomas into immunologically hot gliomas (44, 53, 55). Currently, about half of the current phase II and III trials involving vaccines are cell-based strategies, especially DC vaccines (56).
In 2023, a phase III trial of an autologous tumor lysate-loaded DC vaccine (DCVax-L) plus TMZ in patients with nGBM and rGBM reported encouraging findings (10, 11). This study’s primary and secondary endpoints were the mOSs in patients with nGBM and rGBM, respectively (10). In the nGBM part, the mOSs in the DCVax-L plus TMZ group and the TMZ control group were 19.3 and 16.5 months, respectively (P = 0.002). And the survival rates of two and five years in DCVax-L plus TMZ group and TMZ control group were 15.7% vs 9.9% and 13.0% vs 5.7%, respectively (10). In the rGBM part, the mOSs in DCVax-L plus TMZ group and TMZ control group were 13.2 and 7.8 months, respectively (P < 0.001). And the survival rates of two years and 30 months of DCVax-L plus TMZ group and TMZ control group were 15.7% vs. 9.9% and 13.0% vs. 5.7%, respectively (10). Besides, DCVax-L-treated patients with nGBM with methylated MGMT promoter survived longer (21.3 months) than those in the external control group (P = 0.03) (10). Obviously, this phase III trial advances the pace of vaccine therapy for gliomas. However, the individual patient-level data of the external control populations are not accessible. And a more credible and reasonable investigation of DCVax-L in GBM treatment is needed.
2.3 CAR-T cell therapy
CAR-T cell therapy is a typical type of adoptive T-cell therapies (57). This therapeutic approach involves collecting T cells from a patient’s peripheral blood, followed by their modification, amplification, and activation to express CAR molecules on the cell membranes. These genetically engineered T cells are then administered to the patient through injection, allowing them to target specific tumor cell antigens (57). The significance of CAR-T cell therapy in treating gliomas has been identified, although it has yet to exhibit big success (58). Table 3 lists the recent phase II/III clinical trials of CAR-T therapy for GBM treatment from ClinicalTrials.gov.
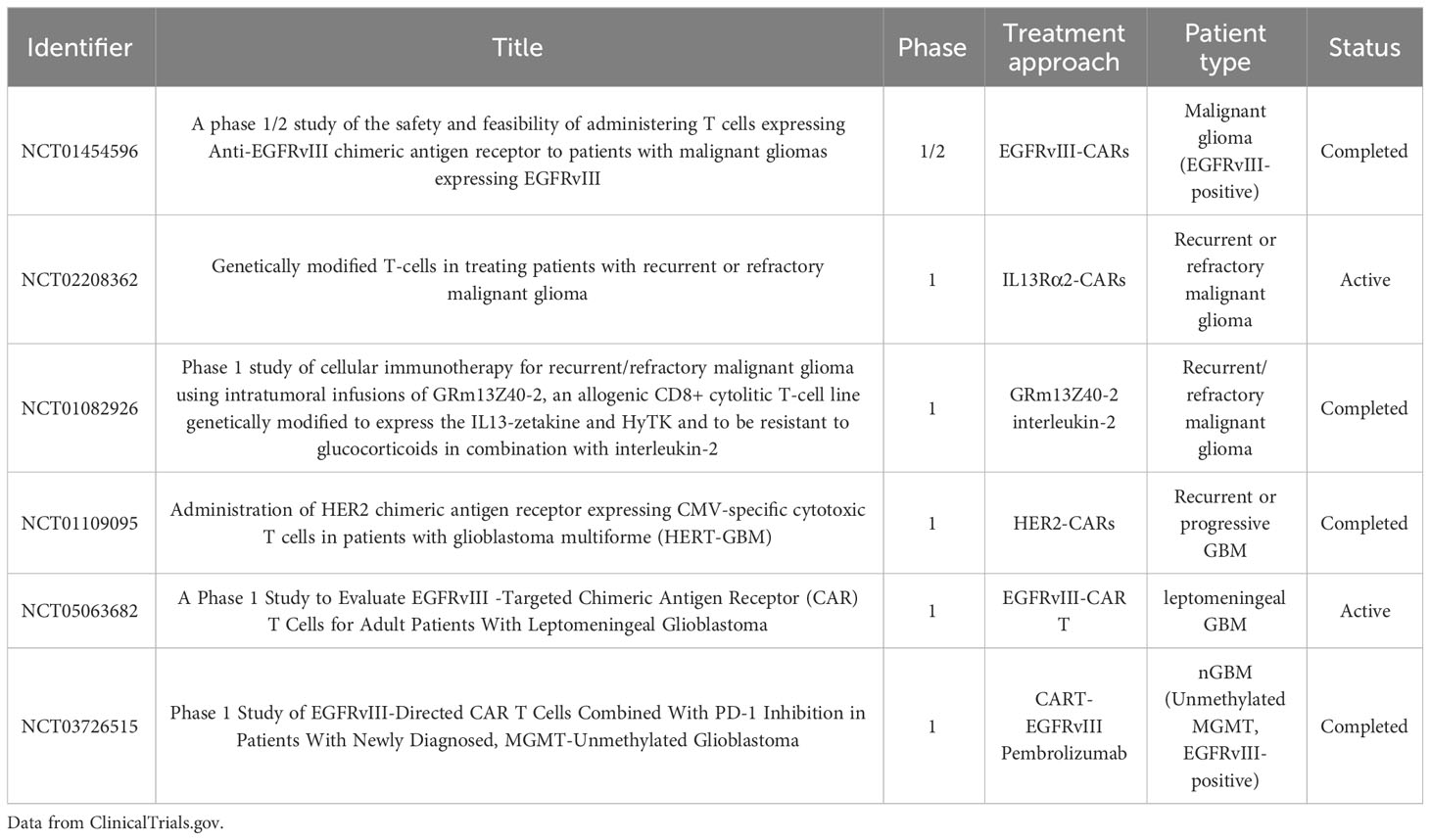
Table 3 Recent clinical trials of CAR-T cell therapy for GBM treatment.
The phase I clinical trials associated with CAR-T cell therapy in GBM treatment shows that scientists are also actively trying in this field. In these clinical trials, several commonly used targeted antigens, including EGFRvIII (59), IL13Rα2 (60), and HER2 (61), had also been indicated. NCT02208362 was a phase I clinical trial of IL13Rα2-targeted CAR-T cell therapy in GBM treatment (60). In this study, two intracranial approaches were applied to deliver CAR-T cells to a patient with rGBM, including infusions into the resected tumor cavity and the ventricular system. And it was found that intraventricular therapy can achieve the wide regression of central nervous system tumors. In comparison, the intracavitary treatment seemed only to control the local tumor recurrence. Moreover, the CAR-T cell therapy provided the patient with a response for 7.5 months. Overall, the result proved that CAR-T cell therapy can show the antitumor activity in GBM treatment, and IL13Rα2 is a valid immunotherapeutic target for CAR-T cell therapy (60). NCT02209376 was another phase I study of CAR-T cell therapy in patients with EGFRvIII-positive rGBM (59). This first-in-human experiment of EGFRvIII-targeting CAR-T cells by intravenous administration proved that CAR-T cells could transfer to the brain tumor site. It is worth noting that the EGFRvIII level on tumor cells was observed to be reduced or eliminated after the CAR-T cell administration. Moreover, the effect of CAR-T cells on the TME in GBM was also emphasized in this study. The immunosuppressive Tregs were the dominant T cell type for TILs in the TME. And many immunosuppressive molecules, including PD-L1, IL-10, and IDO1, were also observed to increase. Besides, the study also indicated the combination therapy of CAR-T cell therapy and the inhibition of immunosuppressive pathways (59). In another phase I study (NCT01109095), researchers investigated the use of HER2-targeting CAR-modified virus-specific T cells (HER2-CAR VSTs) in patients with rGBM (61). Participants in this trial were administered one or more autologous HER2-CAR VST injections across five different dosage levels. Of the 17 patients, eight exhibited clinical benefits, with one showing a partial response and seven maintaining stable disease. The median overall survival (mOS) was 11.1 months following HER2-CAR VSTs administration and 24.5 months from the time of diagnosis (61).
CAR-T cell therapy has made a step forward in both hematologic and solid tumors. However, its use for GBM treatment remains limited due to the BBB, antigen escape, tumor heterogeneity, and TME (62). Though the mentioned three phase I clinical trials indicated firm hopes for GBM treatment, more clinical trials with larger samples and more reliable examinations are needed.
2.4 OV therapy
Oncolytic virus (OV) therapy has emerged as a significant treatment strategy and has become a focus of research in the field of oncology (63). In 2021, the Japanese Ministry of Health, Labor, and Welfare (MHLW) granted conditional and time-limited approval for G47Δ to treat patients with malignant glioma in Japan (12). Table 4 presents a summary of recent phase II/III clinical trials for OV therapy in the treatment of glioblastoma (GBM), as found on ClinicalTrials.gov.
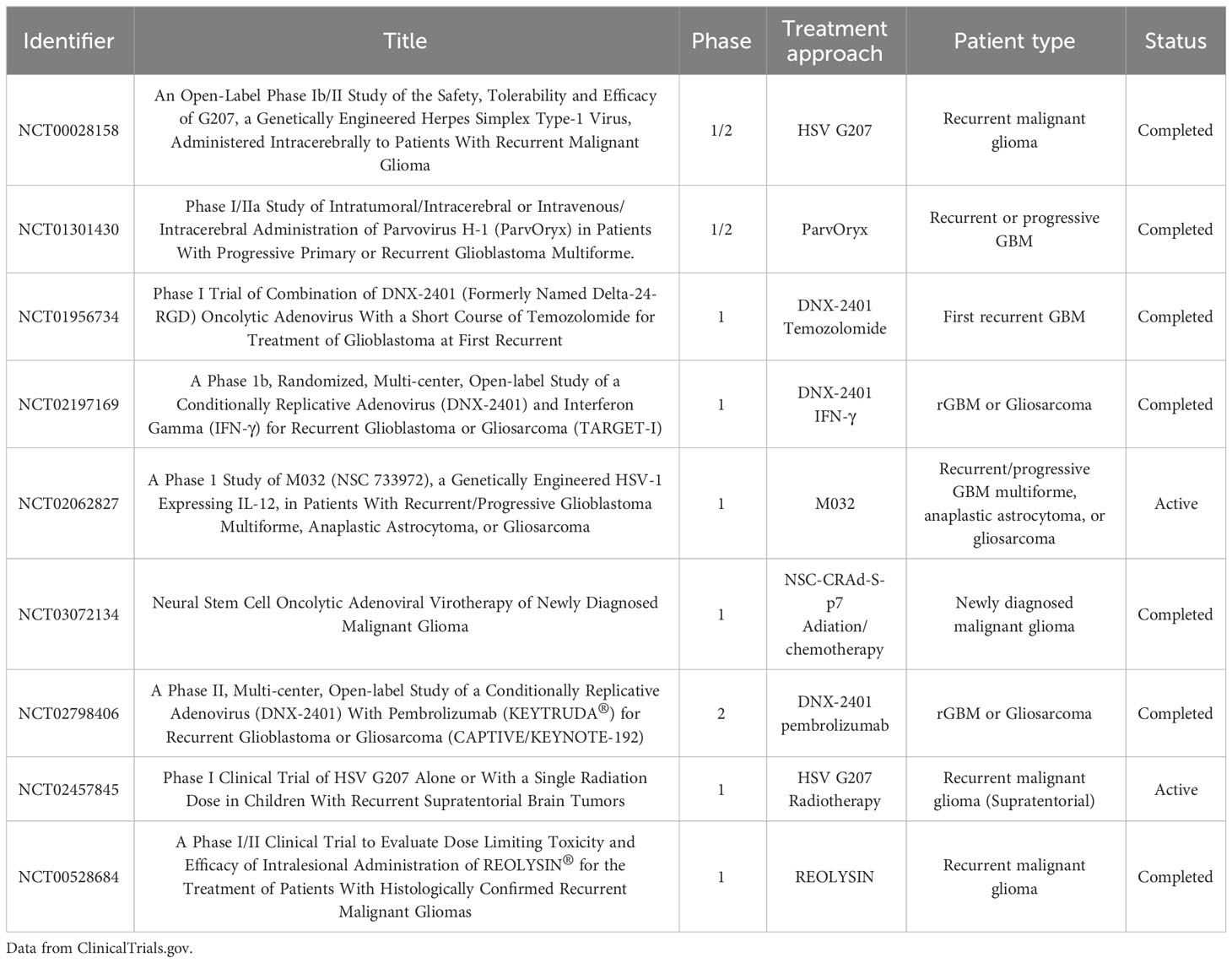
Table 4 Recent phase II/III clinical trials of OV therapy for GBM treatment.
OVs are genetically modified, weakly pathogenic viruses that enhance the antitumor effects without harming normal cells (63). OV therapy hasadvantage in this era of mature genetic engineering. There are three purposes for genetic modification of OVs: 1) delete virulence genes to improve the safety of OVs, 2) enhance the tumor cell tropism and targeting of OVs to tumor cells, and 3) modify OVs with different therapeutic genes to enhance anti-tumor effects (64). On the one hand, OVs can self-replicate in host cancer cells, leading to direct lysis of the cancer cells (63). On the other hand, OVs help activate innate and adaptive immune responses by the releases of damage-related molecular patterns (DAMPs), viral pathogen-associated molecular patterns (PAMPs), and TAAs, to improve the immunosuppressive TME (64–67). Besides, OVs can selectively target and inhibit glioma stem cells (GSCs), which is an essential factor for drug resistance, tumor blood vessel formation, and immunosuppressive glioma microenvironment (68–70).
Since gliomas develop predominantly in the brain and lack distant metastases, which allows for the viruses that need an active cell cycle for reproduction, gliomas are particularly well-suited for OV treatment (71). The viruses used for OV therapy in gliomas include Oncolytic H-1 Parvovirus (72), Oncolytic Reovirus (73, 74), Oncolytic measles virus (75), Newcastle disease virus (74), Oncolytic vaccinia virus (76), Poliovirus (77), Oncolytic adenovirus (78), Oncolytic zika virus (79), Oncolytic herpes simplex virus (64, 80). Moreover, of all the OVs, oHSV progresses furthest in the clinical practice, including G47Δ, G207, HSV1716, and rQNestin-34.5 (80).
UMIN000015995 was a phase II trial of G47Δ in Japan for patients with residual or recurrent GBM (12). The third-generation as well as the triple-mutated oncolytic herpes simplex virus type 1 (HSV-1) G47 was created by deleting the US11 promoter from its parental G207 and overlapping the US11 gene (81). The primary endpoint (1-year survival rate) was achieved ahead of schedule, which was 84.2%. Additionally, the secondary endpoints of OS and PFS were 20.2 months and 4.7 months, respectively, after the G47 initiation (12). Based on the exciting results, G47Δ obtained conditional and time-limited approval in Japan for malignant glioma patients (12).
As a promising cancer treatment approach, OV therapy has a significant effect on gliomas. Moreover, the successful application of G47Δ in Japan also indicates the huge potential of OV therapy. Nevertheless, further research and phase III trials with large samples are needed to verify the real efficacy of this novel treatment approach.
2.5 Other therapies
2.5.1 Gene therapy
Gene therapy, a rapidly evolving field in oncology, encompasses the techniques used to introduce exogenous genes into targeted cells or tissues. These interventions are aimed at correcting or compensating for genetic defects and abnormalities for therapeutic purposes (82). Vectors in gene therapy can deliver entire genes, gene regulatory elements, or oligonucleotides and are categorized into viral and non-viral types (83). Viral vectors, including adenovirus vectors (AdV), adeno-associated viral vectors (AAV), retrovirus vectors (RV), and lentiviral vectors (LV), are non-toxic purified viruses designed to deliver genetic payloads without causing infections (84–88). Recently, non-viral vector-mediated gene therapy, such as nanoparticles with low toxicity and immunogenicity, has emerged as a promising approach in GBM treatment. These non-viral vectors can efficiently traverse the BBB and deliver gene drugs for glioma therapy (89, 90). The primary strategies in gene therapy are suicide gene therapy, tumor suppressor gene therapy, gene target therapy, and immunomodulatory gene therapy (91). Suicide gene therapy involves the delivery of genes coding specific enzymes that convert or activate non-toxic prodrugs into cytotoxic drugs at tumor sites, resulting in oncolytic action and tumor cell apoptosis (92). Tumor suppressor gene therapy focuses on restoring normal tumor suppressor gene function in tumor cells, thereby inhibiting tumor growth through genes like TP53, p16, and PTEN (93). Gene-targeted therapy binds specific tumor antigens and blocks carcinogenic pathways (91), while immunomodulatory gene therapy regulates the immunosuppressive tumor microenvironment (TME) and enhances immune cell effects. This therapeutic approach can be either categorized into gene therapy or immunotherapy. Moreover, with the development of both gene therapy and immunotherapy, the two approaches have become more closely linked. One good example is OV-based immunotherapy. Natural viruses can be genetically modified through genetic engineering into special OVs, which can specifically recognize, infect, and replicate in tumor cells, thereby destroying the tumor cells (71). Besides, tumor vaccines and CAR-T therapies can also be regarded as gene therapies in some situations.
2.5.2 Bispecific antibody therapy
In recent years, bispecific antibodies (BsAbs) have emerged as a promising therapeutic approach in oncology, with notable applications in cancer treatment (94, 95). Unlike conventional antibodies, BsAbs possess two distinct antigen-binding sites, allowing them to function through various mechanisms, including immune cell activation, co-inhibitory receptor blockade, co-stimulatory molecule triggering, signaling pathway suppression, and collaborative targeting of cancer-related antigens (96). Several BsAbs, such as blinatumomab, mosunetuzumab, teclistamab, epcoritamab, and glofitamab, have gained approval for cancer treatment (95). However, the development of BsAbs for glioma therapy has been relatively slow, with most investigations confined to preclinical stages (97–101). One particular focus of this review is AG596, a bispecific T-cell engager (BiTE) that has entered phase I clinical trials, offering insight into the mechanism of BsAbs in oncological applications (102, 103). BiTEs represent a distinct class of BsAbs, comprised of two single-chain variable fragments (scFv) connected by a short peptide linker. Each scFv serves a specific function: one targets a tumor-associated antigen on tumor cells, while the other binds to CD3 expressed on T cells. The design of BiTEs in glioma treatment often involves EGFRvIII, similar to vaccine and CAR-T therapies (98–101). AG596, for instance, can simultaneously engage the tumor-specific antigen (EGFRvIII) on glioblastoma (GBM) cells and CD3 on T cells, thereby activating T cell proliferation and cytotoxic secretion. This process leads to T cell-mediated GBM cell destruction. Preclinical studies have demonstrated that AG596 effectively mediates the lysis of EGFRvIII-positive GBM cell lines, improving overall survival (OS) rates in mice bearing EGFRvIII-expressing GBM cells (100). Moreover, the selectivity of AMG 596 has been confirmed through testing. Its binding was specific to EGFRvIII-positive GBM cells, with no observed T cell activity in EGFRvIII-negative cells and no toxicity detected in normal tissues (EGFRvIII-negative) in cynomolgus monkeys (100). A phase I, first-in-human, sequential dose-escalation/expansion study of AMG 596 in patients with EGFRvIII-positive GBM or malignant glioma (either recurrent or newly diagnosed) (NCT03296696) provided preliminary evidence for its safety, tolerability, and anti-tumor activity in recurrent GBM (104). However, further research is required to fully understand and harness the potential of AMG 596 in GBM therapy. Other ongoing clinical trials of BiTEs for glioma treatment include NCT04903795 and NCT03344250 (phase I).
2.5.3 Combined therapies
GBM is recognized as a highly malignant brain tumor, characterized by pronounced heterogeneity, an immunosuppressive TME, and a propensity for recurrence (105, 106). This complexity has necessitated the adoption of combined therapies, a strategy that neurosurgeons have embraced early on. The Stupp regimen, combining surgery with chemotherapy and radiotherapy, is a prototypical example (5). The emergence of novel treatments, including immunotherapy, targeted therapy, and tumor treatment fields, has given rise to innovative combinations, showing promise in both preclinical and clinical settings. Several tables from sections 2.1 to 2.4, along with most of our discussions, touch upon these combined therapies. This section would like to briefly introduce several prevalent combined therapeutic modes that incorporate immunotherapies in both clinical practice and trials.
Immunotherapy combined with chemotherapy (especially temozolomide (TMZ)). This approach is commonly used in clinical trials, and immunotherapy combined with current standard therapy (Stupp Regimen) can also be categorized into this category. Examples of clinical trials are Checkmate 498 and Checkmate 548 (using nivolumab, TMZ, and radiotherapy), NCT03018288 (Pembrolizumab, Temozolomide Radiotherapy, and HSPPC-96), NCT03548571, NCT00458601 (Rindopepimut, TMZ, and radiotherapy), NCT01480479 (Rindopepimut and TMZ), NCT02455557 (SurVaxM and TMZ), NCT01956734 (DNX-2401 and TMZ), etc.
Immunotherapy combined with targeted therapy (frequently paired with bevacizumab). This combined approach has also been explored in clinical trials such as CheckMate 143 (nivolumab and bevacizumab), NCT02337491 (Pembrolizumab and Bevacizumab), and NCT03452579 (Nivolumab and Bevacizumab), etc.
Combination of different types of immunotherapies or immunotherapeutic drugs (in the same type). Examples of combined immunotherapies in the same categories mainly include NCT02794883 (Tremelimumab and Durvalumab), NCT04396860 (Ipilimumab, Nivolumab, and Radiotherapy), etc. And examples of combined immunotherapies in different categories include NCT02798406 (Pembrolizumab and Adenovirus), NCT03750071 (Avelumab and VXM01), NCT01082926 (GRm13Z40-2 and IL-2), NCT03726515 (CART-EGFRvIII and Pembrolizumab), NCT02798406 (DNX-2401 and pembrolizumab), etc.
Given the current landscape of immunotherapies in gliomas, it is anticipated that more combined therapies yielding convincing results will emerge in the near future. These combined modalities hold the potential to address the multifaceted challenges posed by GBM, offering a more comprehensive approach to treatment.
3 TME in gliomas
As introduced above, immunotherapies have demonstrated promising outcomes in preclinical and clinical studies, especially the successful phase III trial of DCVax-L and the approval of G47Δ in Japan. However, there are also many obstructions on the way to success. The tumor microenvironment (TME) is regarded as one of the most critical factors in various treatments, including immunotherapy. On the one hand, the immunosuppressive TME in gliomas can result in drug resistance and tumor recurrence. On the other hand, understanding deeply and making great use of the TME can also promote the progress of immunotherapy in gliomas.
The TME of gliomas is complex and heterogeneous, consisting of various components, including astrocytes, pericytes, endothelial cells, GSCs, blood vessels, glioma-associated stromal cells, immune cells including myeloid-derived suppressor cells (MDSCs), glioma-associated microglia/macrophages (GAMs), CD4+ T cells, Tregs and NK cells, and extracellular matrix (ECM) (107–109) (Figure 1). These elements may interact with one another to promote the spread and proliferation of glioma cells. It is widely acknowledged that TME is one of the main reasons for the unsatisfactory immunotherapeutic effect of gliomas. Additionally, more and more immunotherapy-related research pays much attention to TME in gliomas (23, 25, 59).
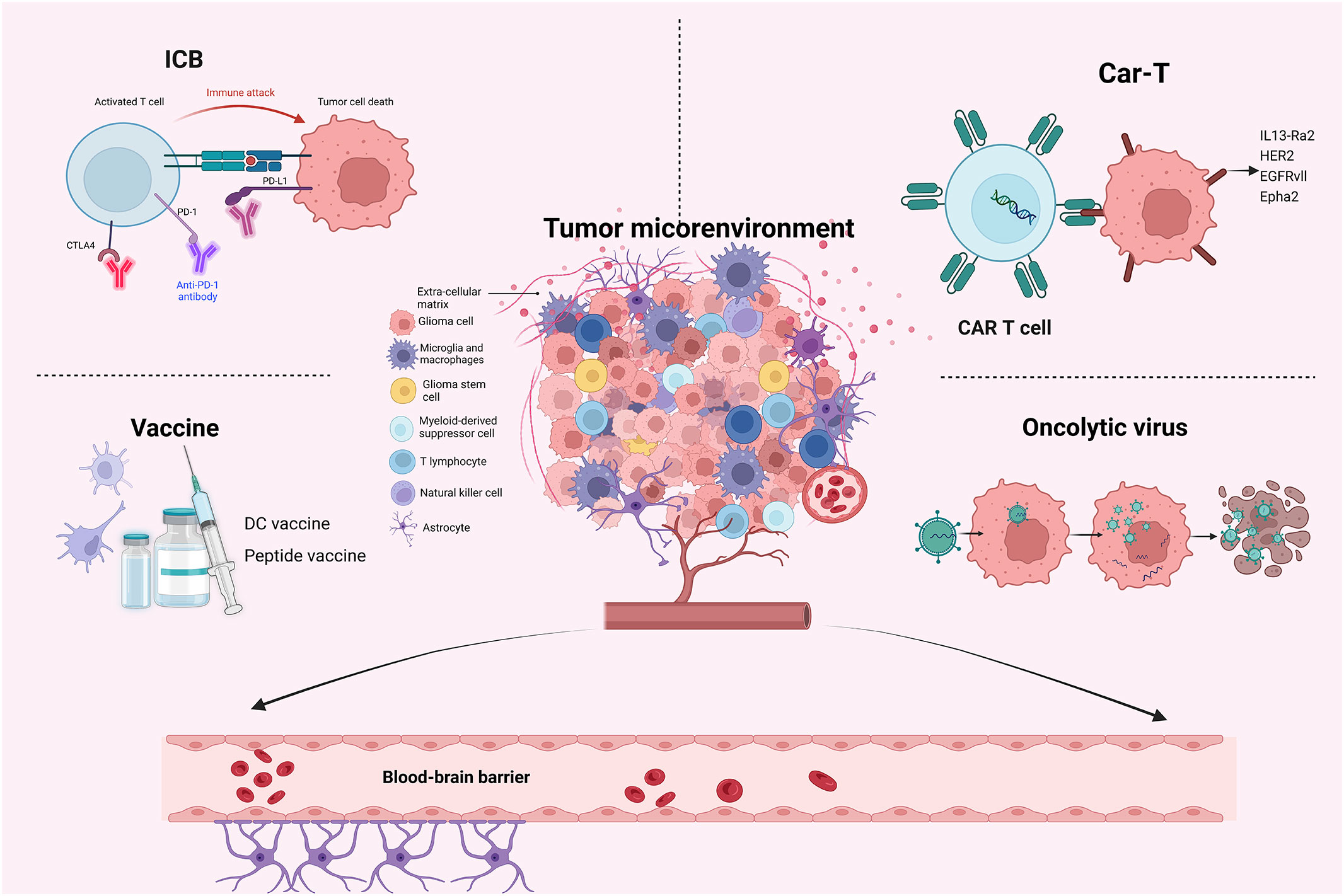
Figure 1 The illustration of four main immunotherapies and TME in gliomas.
The powerful immunosuppressive effect in the TME of gliomas is caused by several mechanisms, including the abnormal function of cells such as the existence of immunosuppressive cells (M2 GAMs, Tregs, and MDSCs) and immunosuppressive cytokines (TGF-β, IL-10), low number of TILs, and the high expression of inhibitory immune checkpoint molecules such as PD-1, TIM-3, and LAG-3 (38, 109–112). The most abundant cells in GBM are GAMs, which are divided into the M1 phenotype (proinflammatory) and M2 phenotype (immunosuppressive) (107). These GAMs contribute to tumor heterogeneity and progression. On the one hand, M2 GAMs can produce a lot of IL-10 and TGF-β and low levels of IL-12, which can play an immunosuppressive role in the TME (109). GAMs can also promote the proliferation of GSCs, which is closely related to drug resistance (113).
Compared with other tumors, research on TILs in gliomas is relatively less (110). After T cells infiltrate the TME, dysfunction occurs through different mechanisms, including aging, tolerance, and incompetence (111). Additionally, glioma cells and certain immune cells emit a variety of immunosuppressive substances into the TME, including TGF-β and IL-10. Then these factors attract and stimulate immunosuppressive cells such as TAMs and Treg cells and inhibit the activation of APCs and effector immune cells (114, 115).
Glioma stem cells (GSCs), namely cancer stem cells in gliomas, also play a vital role in the TME. GSCs have the ability of self-renewal and multi-differentiation, which are regarded as the primary cause of tumor occurrence, development, drug resistance and recurrence, and heterogeneity (116–118). GSCs can regulate cell metabolism in the TME of gliomas to increase resistance to challenging conditions in addition to reprogramming associated cells to promote tissue remodeling (116–118). The intimate interaction between GSCs and glioma TME is a critical factor to the resistance of immunotherapy, and targeting both GSCs and TME can enhance the immunotherapeutic effect.
The BBB is also found closely connected with the immune response in gliomas. BBB is a semi-permeable physiological boundary formed by parenchymal capillary endothelial cells, capillary astrocytic endfeet, and capillary basal membrane pericytes (119, 120). The presence of BBB benefits the central nervous system’s special immune privilege, which is characterized by the lack of traditional lymphatic structures, a dearth of APC, low levels of MHC molecule expression, and the constitutive expression of immunosuppressive cytokines like IL10 and TGF-β (121–123). Moreover, BBB can also largely limit the delivery of most therapeutic drugs in treating gliomas (124). In addition to being a barrier to both immunity and drug delivery, several studies on tumor brain metastasis proved that the properties of BBB can be fundamentally regulated by some components of the TME (125).
4 Conclusion and prospect
This review analyzed the present landscape of immunotherapies for gliomas, focusing on four main therapeutic approaches: ICBs, CAR-T cell therapy, vaccine therapy, and OV therapy. Among these, the efficacy of the dendritic cell vaccine DCVax-L and OV G47Δ has been validated in phase II and III clinical trials, respectively. Notably, G47Δ received conditional and time-limited approval from the Japanese Ministry of Health, Labor, and Welfare (MHLW) in 2021 for the treatment of malignant glioma. This review also briefly introduces gene therapy and bispecific antibody therapy, both closely aligned with immunotherapy, as well as combined therapies incorporating immunotherapy. The critical role of the tumor microenvironment (TME) in the effectiveness of immunotherapies for gliomas has been emphasized in this review.
Immunotherapy, given its present standing and rapid evolution, is heralded as a promising avenue for glioma treatment. However, significant challenges must be overcome to realize its full potential. These include:
Understanding Molecular Biology: Investigations into the molecular biology of GBM, TME, and the BBB are vital for designing innovative immunotherapeutic drugs with potential antitumor effects.
Exploring TME-Immunotherapy Relationships: Continued and profound exploration of the relationship between TME and immunotherapy is necessary for the success of immunotherapies in gliomas.
Combination Therapies: Active and rational exploration of combined therapies is likely to become the focal point in this field, offering synergistic advantages.
Personalized Treatment: Tailoring treatments to individual patients with distinct molecular characteristics may be essential to optimize therapeutic outcomes.
In conclusion, while the rapid advancement of immunotherapy provides hope, the complexities of gliomas necessitate an equally nuanced approach to treatment. We ardently hope that immunotherapy, with continued research and development, will become a formidable tool in the fight against GBM in the foreseeable future.
Author contributions
FY: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Writing – original draft, Writing – review & editing. YX: Funding acquisition, Writing – review & editing, Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft. HG: Data curation, Investigation, Methodology, Writing – original draft. RG: Supervision, Writing – review & editing, Conceptualization, Supervision. LY: Supervision, Writing – review & editing, Conceptualization, Supervision. ZL: Supervision, Writing – review & editing, Conceptualization, Supervision. HW: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
This work was supported by Natural Science Foundation of China grants (81500116) to HW, Jilin Science and Technique development grants (20200201472JC) to HW, Jilin Science and Technique development grants (YDZJ202301ZYTS092) to RG, National College Students’ innovation and entrepreneurship training program (202210183300) to YX, and College Students’ innovation and entrepreneurship training program of Jilin University (X202210183572) to FY.
Acknowledgments
We greatly appreciate Professor Jifan Hu from Stanford University for modifying the language of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol (2020) 22(12 Suppl 2):iv1–iv96. doi: 10.1093/neuonc/noaa200
2. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
3. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015-2019. Neuro Oncol (2022) 24(Suppl 5):v1–v95. doi: 10.1093/neuonc/noac202
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
5. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
6. Redman JM, Gibney GT, Atkins MB. Advances in immunotherapy for melanoma. BMC Med (2016) 14:20. doi: 10.1186/s12916-016-0571-0
7. Reck M, Remon J, Hellmann MD. First-line immunotherapy for non-small-cell lung cancer. J Clin Oncol (2022) 40(6):586–97. doi: 10.1200/JCO.21.01497
8. Ferrall L, Lin KY, Roden RBS, Hung CF, Wu TC. Cervical cancer immunotherapy: facts and hopes. Clin Cancer Res (2021) 27(18):4953–73. doi: 10.1158/1078-0432.CCR-20-2833
9. Mahmoud AB, Ajina R, Aref S, Darwish M, Alsayb M, Taher M, et al. Advances in immunotherapy for glioblastoma multiforme. Front Immunol (2022) 13:944452. doi: 10.3389/fimmu.2022.944452
10. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: A phase 3 prospective externally controlled cohort trial. JAMA Oncol (2023) 9(1):112–21. doi: 10.1001/jamaoncol.2022.5370
11. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med (2018) 16(1):142. doi: 10.1186/s12967-018-1507-6
12. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent gli oblastoma: a phase 2 trial. Nat Med (2022) 28(8):1630–9. doi: 10.1038/s41591-022-01897-x
13. Dagher OK, Schwab RD, Brookens SK, Posey AD Jr. Advances in cancer immunotherapies. Cell (2023) 186(8):1814–.e1. doi: 10.1016/j.cell.2023.02.039
14. Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol (2020) 11:683. doi: 10.3389/fimmu.2020.00683
15. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol (2018) 15(7):422–42. doi: 10.1038/s41571-018-0003-5
16. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
17. Rong L, Li N, Zhang Z. Emerging therapies for glioblastoma: current state and future directions. J Exp Clin Cancer Res (2022) 41(1):142. doi: 10.1186/s13046-022-02349-7
18. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discovery (2022) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
19. Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res (2013) 19(19):5300–9. doi: 10.1158/1078-0432.CCR-13-0143
20. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
21. Berger KN, Pu JJ. PD-1 pathway and its clinical application: A 20year journey after discovery of the complete human PD-1 gene. Gene (2018) 638:20–5. doi: 10.1016/j.gene.2017.09.050
22. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271
23. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med (2019) 25(3):477–86. doi: 10.1038/s41591-018-0337-7
24. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol (2020) 6(7):1003–10. doi: 10.1001/jamaoncol.2020.1024
25. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
26. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol (2022) 24(11):1935–49. doi: 10.1093/neuonc/noac116
27. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol (2023) 25(1):123–34. doi: 10.1093/neuonc/noac099
28. Duerinck J, Schwarze JK, Awada G, Tijtgat J, Vaeyens F, Bertels C, et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial. J Immunother Cancer (2021) 9(6):e002296. doi: 10.1136/jitc-2020-002296
29. Brown NF, Ng SM, Brooks C, Coutts T, Holmes J, Roberts C, et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer (2020) 20(1):198. doi: 10.1186/s12885-020-6624-y
30. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol (2018) 20(5):674–86. doi: 10.1093/neuonc/nox208
31. Zen K, Guo Y, Bian Z, Lv Z, Zhu D, Ohnishi H, et al. Inflammation-induced proteolytic processing of the SIRPalpha cytoplasmic ITIM in neutrophils propagates a proinflammatory state. Nat Commun (2013) 4:2436. doi: 10.1038/ncomms3436
32. Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS One (2016) 11(4):e0153550. doi: 10.1371/journal.pone.0153550
33. Gholamin S, Youssef OA, Rafat M, Esparza R, Kahn S, Shahin M, et al. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate Immun (2020) 26(2):130–7. doi: 10.1177/1753425919876690
34. Azambuja JH, Schuh RS, Michels LR, Iser IC, Beckenkamp LR, Roliano GG, et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol Immunother (2020) 69(9):1801–12. doi: 10.1007/s00262-020-02569-w
35. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med (2020) 26(1):39–46. doi: 10.1038/s41591-019-0694-x
36. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology (2018) 7(8):e1466769. doi: 10.1080/2162402X.2018.1466769
37. Puigdelloses M, Garcia-Moure M, Labiano S, Laspidea V, Gonzalez-Huarriz M, Zalacain M, et al. CD137 and PD-L1 targeting with immunovirotherapy induces a potent and durable antitumor immune response in glioblastoma models. J Immunother Cancer (2021) 9(7):e002644. doi: 10.1136/jitc-2021-002644
38. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.CCR-17-1846
39. Woroniecka KI, Rhodin KE, Dechant C, Cui X, Chongsathidkiet P, Wilkinson D, et al. 4-1BB agonism averts TIL exhaustion and licenses PD-1 blockade in glioblastoma and other intracranial cancers. Clin Cancer Res (2020) 26(6):1349–58. doi: 10.1158/1078-0432.CCR-19-1068
40. Morton DL, Barth A. Vaccine therapy for Malignant melanoma. CA Cancer J Clin (1996) 46(4):225–44. doi: 10.3322/canjclin.46.4.225
41. Cunto-Amesty G, Monzavi-Karbassi B, Luo P, Jousheghany F, Kieber-Emmons T. Strategies in cancer vaccines development. Int J Parasitol (2003) 33(5-6):597–613. doi: 10.1016/S0020-7519(03)00054-7
42. Zhao B, Wu J, Li H, Wang Y, Wang Y, Xing H, et al. Recent advances and future challenges of tumor vaccination therapy for recurrent glioblastoma. Cell Commun Signal (2023) 21(1):74. doi: 10.1186/s12964-023-01098-0
43. Swartz AM, Batich KA, Fecci PE, Sampson JH. Peptide vaccines for the treatment of glioblastoma. J Neurooncol (2015) 123(3):433–40. doi: 10.1007/s11060-014-1676-y
44. Li L, Zhou J, Dong X, Liao Q, Zhou D, Zhou Y. Dendritic cell vaccines for glioblastoma fail to complete clinical translation: Bottlenecks and potential countermeasures. Int Immunopharmacol (2022) 109:108929. doi: 10.1016/j.intimp.2022.108929
45. Sayegh ET, Oh T, Fakurnejad S, Bloch O, Parsa AT. Vaccine therapies for patients with glioblastoma. J Neurooncol (2014) 119(3):531–46. doi: 10.1007/s11060-014-1502-6
46. Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discovery (2007) 6(5):404–14. doi: 10.1038/nrd2224
47. Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer (2014) 14(2):92–107. doi: 10.1038/nrc3655
48. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
49. Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, et al. Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer (2014) 134(10):2437–47. doi: 10.1002/ijc.28576
50. Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A (2007) 104(31):12867–72. doi: 10.1073/pnas.0705158104
51. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
52. Lee-Chang C, Lesniak MS. Next-generation antigen-presenting cell immune therapeutics for gliomas. J Clin Invest (2023) 133(3):e163449. doi: 10.1172/JCI163449
53. Bregy A, Wong TM, Shah AH, Goldberg JM, Komotar RJ. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat Rev (2013) 39(8):891–907. doi: 10.1016/j.ctrv.2013.05.007
54. Wylie B, Macri C, Mintern JD, Waithman J. Dendritic cells and cancer: from biology to therapeutic intervention. Cancers (Basel) (2019) 11(4):521. doi: 10.3390/cancers11040521
55. Frederico SC, Hancock JC, Brettschneider EES, Ratnam NM, Gilbert MR, Terabe M. Making a cold tumor hot: the role of vaccines in the treatment of glioblastoma. Front Oncol (2021) 11:672508. doi: 10.3389/fonc.2021.672508
56. Zhang M, Choi J, Lim M. Advances in immunotherapies for gliomas. Curr Neurol Neurosci Rep (2022) 22(1):1–10. doi: 10.1007/s11910-022-01176-9
57. Chen D, Yang J. Development of novel antigen receptors for CAR T-cell therapy directed toward solid Malignancies. Transl Res (2017) 187:11–21. doi: 10.1016/j.trsl.2017.05.006
58. Li L, Zhu X, Qian Y, Yuan X, Ding Y, Hu D, et al. Chimeric antigen receptor T-cell therapy in glioblastoma: current and future. Front Immunol (2020) 11:594271. doi: 10.3389/fimmu.2020.594271
59. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
60. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
61. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
62. Bagley SJ, Desai AS, Linette GP, June CH, O'Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol (2018) 20(11):1429–38. doi: 10.1093/neuonc/noy032
63. Liu P, Wang Y, Wang Y, Kong Z, Chen W, Li J, et al. Effects of oncolytic viruses and viral vectors on immunity in glioblastoma. Gene Ther (2022) 29(3-4):115–26. doi: 10.1038/s41434-020-00207-9
64. Qi Z, Long X, Liu J, Cheng P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front Cell Neurosci (2022) 16:819363. doi: 10.3389/fncel.2022.819363
65. Martikainen M, Essand M. Virus-based immunotherapy of glioblastoma. Cancers (Basel) (2019) 11(2):186. doi: 10.3390/cancers11020186
66. Aurelian L. Oncolytic viruses as immunotherapy: progress and remaining challenges. Onco Targets Ther (2016) 9:2627–37. doi: 10.2147/OTT.S63049
67. Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent Malignant gliomas, in the adjuvant setting. Mol Ther (2004) 10(5):958–66. doi: 10.1016/j.ymthe.2004.07.021
68. Zhu Z, Gorman MJ, McKenzie LD, Chai JN, Hubert CG, Prager BC, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med (2017) 214(10):2843–57. doi: 10.1084/jem.20171093
69. Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst (2007) 99(18):1410–4. doi: 10.1093/jnci/djm102
70. Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res (2011) 17(11):3686–96. doi: 10.1158/1078-0432.CCR-10-3142
71. Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J (2012) 18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9
72. Angelova AL, Barf M, Geletneky K, Unterberg A, Rommelaere J. Immunotherapeutic potential of oncolytic H-1 parvovirus: hints of glioblastoma microenvironment conversion towards immunogenicity. Viruses (2017) 9(12):382. doi: 10.3390/v9120382
73. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med (2018) 10(422):eaam7577. doi: 10.1126/scitranslmed.aam7577
74. Fan X, Lu H, Cui Y, Hou X, Huang C, Liu G. Overexpression of p53 delivered using recombinant NDV induces apoptosis in glioma cells by regulating the apoptotic signaling pathway. Exp Ther Med (2018) 15(5):4522–30. doi: 10.3892/etm.2018.5935
75. Aref S, Bailey K, Fielding A. Measles to the rescue: A review of oncolytic measles virus. Viruses (2016) 8(10):294. doi: 10.3390/v8100294
76. Vasileva N, Ageenko A, Dmitrieva M, Nushtaeva A, Mishinov S, Kochneva G, et al. Double recombinant vaccinia virus: A candidate drug against human glioblastoma. Life (Basel) (2021) 11(10):1084. doi: 10.3390/life11101084
77. Brown MC, Gromeier M. Cytotoxic and immunogenic mechanisms of recombinant oncolytic poliovirus. Curr Opin Virol (2015) 13:81–5. doi: 10.1016/j.coviro.2015.05.007
78. Ulasov IV, Zhu ZB, Tyler MA, Han Y, Rivera AA, Khramtsov A, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther (2007) 18(7):589–602. doi: 10.1089/hum.2007.002
79. Su KY, Balasubramaniam V. Zika virus as oncolytic therapy for brain cancer: myth or reality? Front Microbiol (2019) 10:2715. doi: 10.3389/fmicb.2019.02715
80. Nguyen HM, Saha D. The current state of oncolytic herpes simplex virus for glioblastoma treatment. Oncolytic Virother (2021) 10:1–27. doi: 10.2147/OV.S268426
81. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A (2001) 98(11):6396–401. doi: 10.1073/pnas.101136398
82. Natsume A, Yoshida J. Gene therapy for high-grade glioma: current approaches and future directions. Cell Adh Migr (2008) 2(3):186–91. doi: 10.4161/cam.2.3.6278
83. Varela ML, Comba A, Faisal SM, Argento A, Franson A, Barissi MN, et al. Gene therapy for high grade glioma: the clinical experience. Expert Opin Biol Ther (2022) 23(2):145-161. doi: 10.1080/14712598.2022.2157718
84. Caffery B, Lee JS, Alexander-Bryant AA. Vectors for glioblastoma gene therapy: viral & Non-viral delivery strategies. Nanomaterials (Basel) (2019) 9(1):105. doi: 10.3390/nano9010105
85. Lowenstein PR, Castro MG. Evolutionary basis of a new gene- and immune-therapeutic approach for the treatment of Malignant brain tumors: from mice to clinical trials for glioma patients. Clin Immunol (2018) 189:43–51. doi: 10.1016/j.clim.2017.07.006
86. AAV-mediated gene delivery of LIGHT prolongs survival in glioblastoma. Cancer Discovery (2023) 13(7):1511. doi: 10.1158/2159-8290.CD-RW2023-078
87. Kushiya H, Hiraoka K, Suzuki T, Inoko K, Inagaki A, Niwa H, et al. Retroviral replicating vector toca 511 (Vocimagene amiretrorepvec) for prodrug activator gene therapy of lung cancer. Cancers (Basel) (2022) 14(23):5820. doi: 10.3390/cancers14235820
88. Birocchi F, Cusimano M, Rossari F, Beretta S, Rancoita PMV, Ranghetti A, et al. Targeted inducible delivery of immunoactivating cytokines reprograms glioblastoma microenvironment and inhibits growth in mouse models. Sci Transl Med (2022) 14(653):eabl4106. doi: 10.1126/scitranslmed.abl4106
89. Tian T, Liang R, Erel-Akbaba G, Saad L, Obeid PJ, Gao J, et al. Immune checkpoint inhibition in GBM primed with radiation by engineered extracellular vesicles. ACS Nano (2022) 16(2):1940–53. doi: 10.1021/acsnano.1c05505
90. Janjua TI, Rewatkar P, Ahmed-Cox A, Saeed I, Mansfeld FM, Kulshreshtha R, et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv Drug Delivery Rev (2021) 171:108–38. doi: 10.1016/j.addr.2021.01.012
91. Giotta Lucifero A, Luzzi S. Against the resilience of high-grade gliomas: gene therapies (Part II). Brain Sci (2021) 11(8):976. doi: 10.3390/brainsci11080976
92. Karjoo Z, Chen X, Hatefi A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv Drug Delivery Rev (2016) 99(Pt A):113–28. doi: 10.1016/j.addr.2015.05.009
93. Vahabi M, Dehni B, Antomás I, Giovannetti E, Peters GJ. Targeting miRNA and using miRNA as potential therapeutic options to bypass resistance in pancreatic ductal adenocarcinoma. Cancer Metastasis Rev (2023). doi: 10.1007/s10555-023-10127-w
94. van de Donk N, Zweegman S. T-cell-engaging bispecific antibodies in cancer. Lancet (2023) 402(10396):142–58. doi: 10.1016/S0140-6736(23)00521-4
95. Tapia-Galisteo A, Álvarez-Vallina L, Sanz L. Bi- and trispecific immune cell engagers for immunotherapy of hematological Malignancies. J Hematol Oncol (2023) 16(1):83. doi: 10.1186/s13045-023-01482-w
96. Farhangnia P, Ghomi SM, Akbarpour M, Delbandi AA. Bispecific antibodies targeting CTLA-4: game-changer troopers in cancer immunotherapy. Front Immunol (2023) 14:1155778. doi: 10.3389/fimmu.2023.1155778
97. Fan R, Chen C, Mu M, Chuan D, Liu H, Hou H, et al. Engineering MMP-2 activated nanoparticles carrying B7-H3 bispecific antibodies for ferroptosis-enhanced glioblastoma immunotherapy. ACS Nano (2023) 17(10):9126–39. doi: 10.1021/acsnano.2c12217
98. Huang L, He H, Wang K, Ma X, Chen X, Chen W, et al. EGFRvIII-targeted immunotoxin combined with temozolomide and bispecific antibody for the eradication of established glioblastoma. BioMed Pharmacother (2022) 155:113659. doi: 10.1016/j.biopha.2022.113659
99. Iurlaro R, Waldhauer I, Planas-Rigol E, Bonfill-Teixidor E, Arias A, Nicolini V, et al. A novel EGFRvIII T-cell bispecific antibody for the treatment of glioblastoma. Mol Cancer Ther (2022) 21(10):1499–509. doi: 10.1158/1535-7163.MCT-22-0201
100. Sternjak A, Lee F, Thomas O, Balazs M, Wahl J, Lorenczewski G, et al. Preclinical assessment of AMG 596, a bispecific T-cell engager (BiTE) immunotherapy targeting the tumor-specific antigen EGFRvIII. Mol Cancer Ther (2021) 20(5):925–33. doi: 10.1158/1535-7163.MCT-20-0508
101. Yin Y, Rodriguez JL, Li N, Thokala R, Nasrallah MP, Hu L, et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol Ther (2022) 30(7):2537–53. doi: 10.1016/j.ymthe.2022.05.011
102. Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, et al. Bispecific antibodies: from research to clinical application. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.626616
103. Rosenthal M, Balana C, Van Linde ME, Sayehli C, Fiedler WM, Wermke M, et al. Novel anti-EGFRvIII bispecific T cell engager (BiTE) antibody construct in glioblastoma (GBM): Trial in progress of AMG 596 in patients with recurrent or newly diagnosed disease. J Clin Oncol (2019) 37(15_suppl):TPS2071–TPS. doi: 10.1200/JCO.2019.37.15_suppl.TPS2071
104. Rosenthal MA, Balana C, van Linde ME, Sayehli C, Fiedler WM, Wermke M, et al. ATIM-49 (LTBK-01). AMG 596, A NOVEL ANTI-EGFRVIII BISPECIFIC T CELL ENGAGER (BITE®) MOLECULE FOR THE TREATMENT OF GLIOBLASTOMA (GBM): PLANNED INTERIM ANALYSIS IN RECURRENT GBM (RGBM). Neuro-Oncology (2019) 21(Supplement_6):vi283–vi. doi: 10.1093/neuonc/noz219.1195
105. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev (2019) 290(1):60–84. doi: 10.1111/imr.12773
106. Wang X, Lu J, Guo G, Yu J. Immunotherapy for recurrent glioblastoma: practical insights and challenging prospects. Cell Death Disease (2021) 12(4):299. doi: 10.1038/s41419-021-03568-0
107. Bikfalvi A, da Costa CA, Avril T, Barnier JV, Bauchet L, Brisson L, et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends Cancer (2023) 9(1):9–27. doi: 10.1016/j.trecan.2022.09.005
108. De Vleeschouwer S, Bergers G. Glioblastoma: to target the tumor cell or the microenvironment? In: De Vleeschouwer S, editor. Glioblastoma. Brisbane (AU (2017). doi: 10.15586/codon.glioblastoma.2017.ch16
109. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest (2017) 97(5):498–518. doi: 10.1038/labinvest.2017.19
110. Maddison K, Graves MC, Bowden NA, Fay M, Vilain RE, Faulkner S, et al. Low tumour-infiltrating lymphocyte density in primary and recurrent glioblastoma. Oncotarget (2021) 12(21):2177–87. doi: 10.18632/oncotarget.28069
111. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res (2018) 24(16):3792–802. doi: 10.1158/1078-0432.CCR-18-0047
112. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol (2006) 8(3):261–79. doi: 10.1215/15228517-2006-008
113. Zhang C, Cheng W, Ren X, Wang Z, Liu X, Li G, et al. Tumor purity as an underlying key factor in glioma. Clin Cancer Res (2017) 23(20):6279–91. doi: 10.1158/1078-0432.CCR-16-2598
114. Da Ros M, De Gregorio V, Iorio AL, Giunti L, Guidi M, de Martino M, et al. Glioblastoma chemoresistance: the double play by microenvironment and blood-brain barrier. Int J Mol Sci (2018) 19(10):2879. doi: 10.3390/ijms19102879
115. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol (2015) 17 Suppl 7(Suppl 7):vii9–vii14. doi: 10.1093/neuonc/nov151
116. Biserova K, Jakovlevs A, Uljanovs R, Strumfa I. Cancer stem cells: significance in origin, pathogenesis and treatment of glioblastoma. Cells (2021) 10(3):621. doi: 10.3390/cells10030621
117. Gimple RC, Bhargava S, Dixit D, Rich JN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev (2019) 33(11-12):591–609. doi: 10.1101/gad.324301.119
118. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev (2015) 29(12):1203–17. doi: 10.1101/gad.261982.115
119. Brown LS, Foster CG, Courtney JM, King NE, Howells DW, Sutherland BA. Pericytes and neurovascular function in the healthy and diseased brain. Front Cell Neurosci (2019) 13:282. doi: 10.3389/fncel.2019.00282
120. Serlin Y, Shelef I, Knyazer B, Friedman A. Anatomy and physiology of the blood-brain barrier. Semin Cell Dev Biol (2015) 38:2–6. doi: 10.1016/j.semcdb.2015.01.002
121. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol (2015) 36(10):569–77. doi: 10.1016/j.it.2015.08.006
122. Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res (2014) 20(14):3651–9. doi: 10.1158/1078-0432.CCR-13-2057
123. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
124. Griffith JI, Rathi S, Zhang W, Zhang W, Drewes LR, Sarkaria JN, et al. Addressing BBB heterogeneity: A new paradigm for drug delivery to brain tumors. Pharmaceutics (2020) 12(12):1205. doi: 10.3390/pharmaceutics12121205
Keywords: immunotherapy, glioma, ICB, car-t, oncolytic viruses, vaccine, tumor microenvironment
Citation: Yasinjan F, Xing Y, Geng H, Guo R, Yang L, Liu Z and Wang H (2023) Immunotherapy: a promising approach for glioma treatment. Front. Immunol. 14:1255611. doi: 10.3389/fimmu.2023.1255611
Received: 09 July 2023; Accepted: 24 August 2023;
Published: 07 September 2023.
Edited by:
Mohd Wajid Ali Khan, University of Hail, Saudi ArabiaReviewed by:
Farhad Dastmalchi, University of Florida, United StatesFriedrich Erhart, Medical University of Vienna, Austria
Nasser Khaled Yaghi, Barrow Neurological Institute (BNI), United States
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United States
Copyright © 2023 Yasinjan, Xing, Geng, Guo, Yang, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Wang, Z29vaG9uZ3ppQGpsdS5lZHUuY24=