
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 02 November 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1251452
This article is part of the Research TopicImmunity and Inflammatory Responses in Kidney Disease: from Mechanisms to Novel Therapeutic StrategiesView all 17 articles
 Aspasia Pefanis1,2,3
Aspasia Pefanis1,2,3 Anjan K. Bongoni1
Anjan K. Bongoni1 Jennifer L. McRae1
Jennifer L. McRae1 Evelyn J. Salvaris1
Evelyn J. Salvaris1 Nella Fisicaro1
Nella Fisicaro1 James M. Murphy4,5,6Francesco L. Ierino2,3
James M. Murphy4,5,6Francesco L. Ierino2,3 Peter J. Cowan1,2*
Peter J. Cowan1,2*Necroptosis, a pathway of regulated necrosis, involves recruitment and activation of RIPK1, RIPK3 and MLKL, leading to cell membrane rupture, cell death and release of intracellular contents causing further injury and inflammation. Necroptosis is believed to play an important role in the pathogenesis of kidney ischemia-reperfusion injury (IRI). However, the dynamics of necroptosis in kidney IRI is poorly understood, in part due to difficulties in detecting phosphorylated MLKL (pMLKL), the executioner of the necroptosis pathway. Here, we investigated the temporal and spatial activation of necroptosis in a mouse model of unilateral warm kidney IRI, using a robust method to stain pMLKL. We identified the period 3-12 hrs after reperfusion as a critical phase for the activation of necroptosis in proximal tubular cells. After 12 hrs, the predominant pattern of pMLKL staining shifted from cytoplasmic to membrane, indicating progression to the terminal phase of necroptotic cell death. Mlkl-ko mice exhibited reduced kidney inflammation at 12 hrs and lower serum creatinine and tubular injury at 24 hrs compared to wild-type littermates. Interestingly, we observed increased apoptosis in the injured kidneys of Mlkl-ko mice, suggesting a relationship between necroptosis and apoptosis in kidney IRI. Together, our findings confirm the role of necroptosis and necroinflammation in kidney IRI, and identify the first 3 hrs following reperfusion as a potential window for targeted treatments.
Ischemia-reperfusion injury (IRI) is a pathological process that occurs when the blood supply to an organ is temporarily reduced and then restored (1). Cellular damage occurs during both the ischemic period and subsequent reperfusion of the kidney. During ischemia, there is a switch from aerobic to anaerobic metabolism (2), resulting in acidosis (3). Cellular ion transport mechanisms are compromised, with increased intracellular calcium, sodium and water causing cellular oedema (4–6). During reperfusion aerobic metabolism is restored and pH is normalised, but reactive oxygen species (ROS) are generated, damaging functional cellular components and ultimately inducing cell death (7–9). The kidney, a highly vascular and metabolically active organ, is particularly susceptible to IRI which may occur during episodes of hypotension, vascular surgery, sepsis, cardiac events or during kidney retrieval for transplantation (10). Kidney IRI causes inflammation (11, 12), immune system activation (13–15), microvascular dysfunction (16–19) and fibrosis (20, 21). IRI has both immediate and early clinical consequences (acute kidney failure) (22, 23) and contributes to progressive long term fibrosis and chronic kidney disease (CKD) (24). Kidney IRI causes significant patient morbidity and mortality, contributing to increasing healthcare costs (25–31). One of the hallmarks of kidney IRI is acute tubular necrosis, which occurs predominantly in the proximal tubules (9).
Recent studies have established that necrosis can occur in a regulated manner via several pathways including necroptosis (32), ferroptosis (33), pyroptosis (34, 35), mitochondrial permeability transition (MPT)-driven necrosis (36), and parthanatos (37, 38). In contrast to apoptosis, these forms of regulated necrosis lead to cell membrane rupture and release of damage-associated molecular patterns (DAMPs), resulting in inflammation and immune activation [reviewed in (39)]. Necroptosis is a caspase-independent regulated necrosis pathway triggered by a range of extracellular stimuli, including death receptor ligands (e.g. TNF) or pathogen patterns (e.g. LPS) binding to their respective cell surface receptors, or intracellular stimuli such as viral nucleic acids binding to the intracellular receptor ZBP1 (reviewed in (40, 41). In scenarios where the pro-NF-κB activity of the cIAP E3 ubiquitin ligase family and the pro-apoptotic function of Caspase-8 protease are diminished, the intracellular kinases Receptor Interacting Protein Kinase 1 (RIPK1) and Receptor Interacting Protein Kinase 3 (RIPK3) assemble into a high molecular weight intracellular platform termed the necrosome via their RIP homology interaction motifs (RHIMs) (42, 43). Recruitment of subcomplexes comprising RIPK3 and the terminal effector in the pathway – the Mixed lineage kinase domain-like (MLKL) pseudokinase – from the cytosol to the necrosome prompts RIPK3-mediated phosphorylation of the MLKL pseudokinase domain at T357/S358 in human MLKL and S345 in mouse MLKL (44–46). Phosphorylation is the critical step in MLKL activation, inducing MLKL to undergo a conformational change, disengage from the necrosome, and assemble into pro-necroptotic oligomers (47–50). MLKL oligomers are trafficked from the necrosome to the plasma membrane, where they accumulate in hotspots that perturb the membrane to kill cells once a phospho-MLKL (pMLKL) threshold is exceeded (50). Accordingly, phosphorylation of MLKL has become synonymous with necroptosis pathway and MLKL activation, and is considered a hallmark of necroptosis pathway activation. Many of the inflammatory molecules released during necroptosis promote further necroptotic cell death and inflammation (51), creating an auto-amplification loop termed necroinflammation (52).
Necroptosis is believed to play an important role in the pathogenesis of kidney IRI (reviewed in (39) (53). Studies by Linkermann et al. (54) and Newton et al. (55) showed lower mortality of Ripk3-deficient mice compared to wild-type (WT) mice. In the absence of mouse-specific inhibitors of MLKL, the executioner protein of necroptosis, in vivo data for its role come from studies of necroptosis-deficient Mlkl knockout (Mlkl-ko) mice (55, 56). Müller et al. used a bilateral renal pedicle clamping model of IRI to demonstrate improved kidney function in Mlkl-ko mice compared with wild-type (WT) mice at 48 and 72 hrs post-reperfusion, but not at the earlier timepoints of 6, 12, and 24 hrs (56). Although there was a time-dependent increase in total MLKL protein expression in WT kidneys commencing at 12 hrs post-reperfusion, the authors were unable to detect pMLKL by immunostaining or Western blotting (56). This inability to reliably detect pMLKL in mouse kidney sections has been a major hindrance to understanding the dynamics of necroptosis in kidney IRI. To address this problem, we developed a reproducible method to stain pMLKL in formalin-fixed kidney tissue sections and used it to track the temporal and spatial activation of necroptosis in the kidneys of WT mice subjected to unilateral IRI. Mice were analyzed at different timepoints post-reperfusion (0, 3, 12, 24, 48, 72 hrs, and 4 weeks). The pMLKL staining pattern at each timepoint was correlated with kidney function, tubular injury, and the expression of a range of genes relevant to kidney injury, inflammation, and necroptosis. Finally, we compared Mlkl-ko mice with WT littermate controls at each timepoint to assess the degree and mechanism of protection from IRI afforded by deletion of MLKL.
Mlkl-ko mice on a C57BL/6J background (45) and WT littermate controls were generated by heterozygous matings and screened by PCR of tail tip genomic DNA by the Walter and Eliza Hall Institute for Medical Research (WEHI) (45). Mice were housed in microisolator cages in a pathogen-free facility with a 12-hour light-dark cycle under standard conditions of temperature and humidity and fed commercial mouse chow diet with free access to drinking water. All animal experiments were conducted in compliance with the Australian Code of Practice for the Care and Use of Laboratory Animals for Scientific Purposes (Eighth edition, 2013) and the Prevention of Cruelty to Animals Act 1986, Victoria, Australia. All experiments were carried out with the approval of the Animal Ethics Committee of St. Vincent’s Hospital Melbourne.
10–12-week-old male mice were anaesthetised by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (15 mg/kg), followed by right nephrectomy prior to left renal pedicle clamping for 18 min using a microvascular clamp (Roboz, Rockville, MD). After removal of the clamp, the kidney was assessed for even reperfusion prior to abdominal wound closure. Mice received 200 µL warm normal saline (37°C) i.p. post-operatively, and core body temperature was maintained at 35.5 - 36.5°C throughout. Sham mice had a right nephrectomy but the left renal pedicle was not clamped. Separate cohorts of mice were euthanised by anesthetic overdose (ketamine/xylazine) and exsanguination immediately following injury (baseline), at 3, 12, 24, 48, 72 hours or 4 weeks after reperfusion. Blood and kidney samples were obtained to assess kidney function, kidney injury, inflammation, and necroptosis. Group sizes varied depending on availability of homozygous Mlkl-ko and WT littermates during experimental procedures. Specific group sizes are specified in all figure legends.
Serum creatinine was measured using a kinetic colorimetric assay on a COBAS Integra 400 Plus analyser (Roche, Castle Hill, NSW, Australia).
Kidney tissue blocks were fixed overnight in 10% formalin and embedded in paraffin wax. 3 µm sections were cut and de-waxed prior to Periodic Acid Schiff (PAS) staining (57). Each section was divided into 12 regions. A representative area of each region was viewed under 400X magnification, with a focus on the corticomedullary junction. A score was derived by calculating the number of damaged proximal tubules as a percentage of total proximal tubules manually counted in each area (58). Markers of a damaged tubule included tubular atrophy, tubular dilatation, tubular cast formation, vacuolization, and tubular cell degeneration with loss of brush border or thickening of tubular basement membranes. An average of the 12 scores obtained per section was calculated. Scoring was performed under blinded conditions by personnel trained by an experienced veterinarian pathologist.
3 µm paraffin sections were de-waxed prior to antigen retrieval in a citrate buffer (pH 6.0) using a pressure cooker at 125°C for 90 s (pMLKL staining) or 180 s (cC3 staining), and left to cool to room temperature. Slides were then washed on a shaker prior to incubation in 10 μL 3% H2O2 v/v in double-distilled water for 5 min and blocked with 50 μL 10% swine serum for 1 hr at room temperature. Sections were then incubated with rabbit monoclonal anti-phospho-MLKL (pSer345) (ab196436, 1:100, Abcam, Melbourne, Australia) or rabbit monoclonal anti-human/mouse cleaved Caspase-3 (cC3) (Asp175) (IC835G, 1:200, RD Systems, Noble Park, Australia) in 2% swine serum at 4°C overnight. After incubation with DAKO anti-rabbit IgG HRP secondary antibody (Envision+ System, K4003, 1:1, Dako, Santa Clara, USA) for 1 hr, sections were developed using DAB and counterstained with hematoxylin. pMLKL- or cC3-stained sections were scanned using an Aperio ScanScope (Leica Biosystems) to generate a digitized image of the whole section. 12 representative areas of the cortex were analysed (4 upper pole, 4 mid pole and 4 lower pole) at X400 magnification. A score was manually calculated by counting the number of tubules with pMLKL or cC3 staining as a percentage of the total number of tubules in the section.
Harvested kidneys were stored in RNA Later® at 4°C for 24-48 h, followed by storage at -80°C until processing. Total RNA was extracted using the ReliaPrep ™ RNA Tissue Miniprep system (Promega Australia, Alexandria, NSW, Australia) according to manufacturer’s instructions. RNA concentration and quality were measured using a Fluorostar Omega multimode microplate reader (BMG Labtech, Mornington, Australia). First-strand complementary DNA (cDNA) was generated in a reaction volume of 22 µL containing 1 µg oligo (dT), 1 µg random hexamers (Invitrogen, Carlsbad, CA), 12 µg of RNA and sterile Milli-Q H2O. The reaction was incubated for 10 min at 70°C. Following this, a 28 µL mix comprising 2.5 µL 10 mM dNTPs, 1 µL SuperScript III recombinant reverse transcriptase, 1 µL RNaseOUT recombinant ribonuclease inhibitors, 2.5 µL 0.1 M DTT, 10 µL 5 x first strand buffer (Invitrogen, Carlsbad, USA) and 11 µL Milli-Q H2O was added to the first reaction. Reverse transcription was performed at 42°C for 1 hr and 70°C for 10 min. The cDNA was stored at -20°C. Quantitative real-time PCR was performed using the TaqMan Universal PCR Master Mix system and TaqMan Gene Expression Assays for Kidney injury molecule 1 (Kim1) (Mm00506686_m1), Neutrophil gelatinase-associated lipocalin (Ngal) (Mm01324470_m1), Tumour necrosis factor alpha (Tnfa) (Mm00443258_m1), Interleukin 1 beta (Il1b) (Mm00434228_m1), Interleukin 33 (Il33) (Mm00505403_m1), Interleukin 6 (Il6) (Mm00446190_m1), Macrophage inflammatory protein-2 (Mip2) (Mm00436450_m1), Receptor interacting protein kinase 1 (Ripk1) (Mm00436354_m1), Receptor interacting protein kinase 3 (Ripk3) (Mm00444947_m1), Mixed lineage kinase domain-like protein (Mlkl) (Mm01244222_m1), Toll-like receptor 2 (Tlr2) (Mm00442346_m1), Toll-like receptor 4 (Tlt4) (Mm00445273_m1), and Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (Mm99999915_g1) (Applied Biosystems, Carlsbad, CA), using a 7400Fast Real-Time PCR System (Applied Biosystems).
Descriptive statistics were used to summarise the kinetic changes at different time points after reperfusion. Results were expressed as mean ± SEM unless otherwise indicated. A Kruskal-Wallis test and Dunn multiple comparisons test were used when comparing > 2 groups. Comparisons between 2 groups were performed using a Mann-Whitney U-test. Statistical analyses were performed using GraphPad Prism version 9.3 (GraphPad, San Diego, CA) with p<0.05 considered significant.
C57BL/6J WT mice were subjected to right nephrectomy and clamping of the left renal pedicle for 18 min, and cohorts were analyzed at 7 timepoints post-reperfusion (0, 3, 12, 24, 48, 72 hrs, and 4 weeks). Kidney injury was assessed by measuring kidney function (serum creatinine), tubular injury (semi-quantitative morphological analysis), and mRNA expression of the genes for the kidney injury markers KIM-1 and NGAL. Serum creatinine (Figure 1A) and tubular injury (Figure 1B; Supplementary Figure 2) were elevated at 3 hrs, although these increases did not reach statistical significance and there was no change in expression of Kim1 (Figure 1C) or Ngal (Figure 1D) at this early timepoint. By 12 hrs, creatinine levels and tubular injury were significantly increased, and expression of both Kim1 and Ngal was massively upregulated. All parameters remained elevated at 24, 48 and 72 hrs before returning to baseline by 4 weeks.
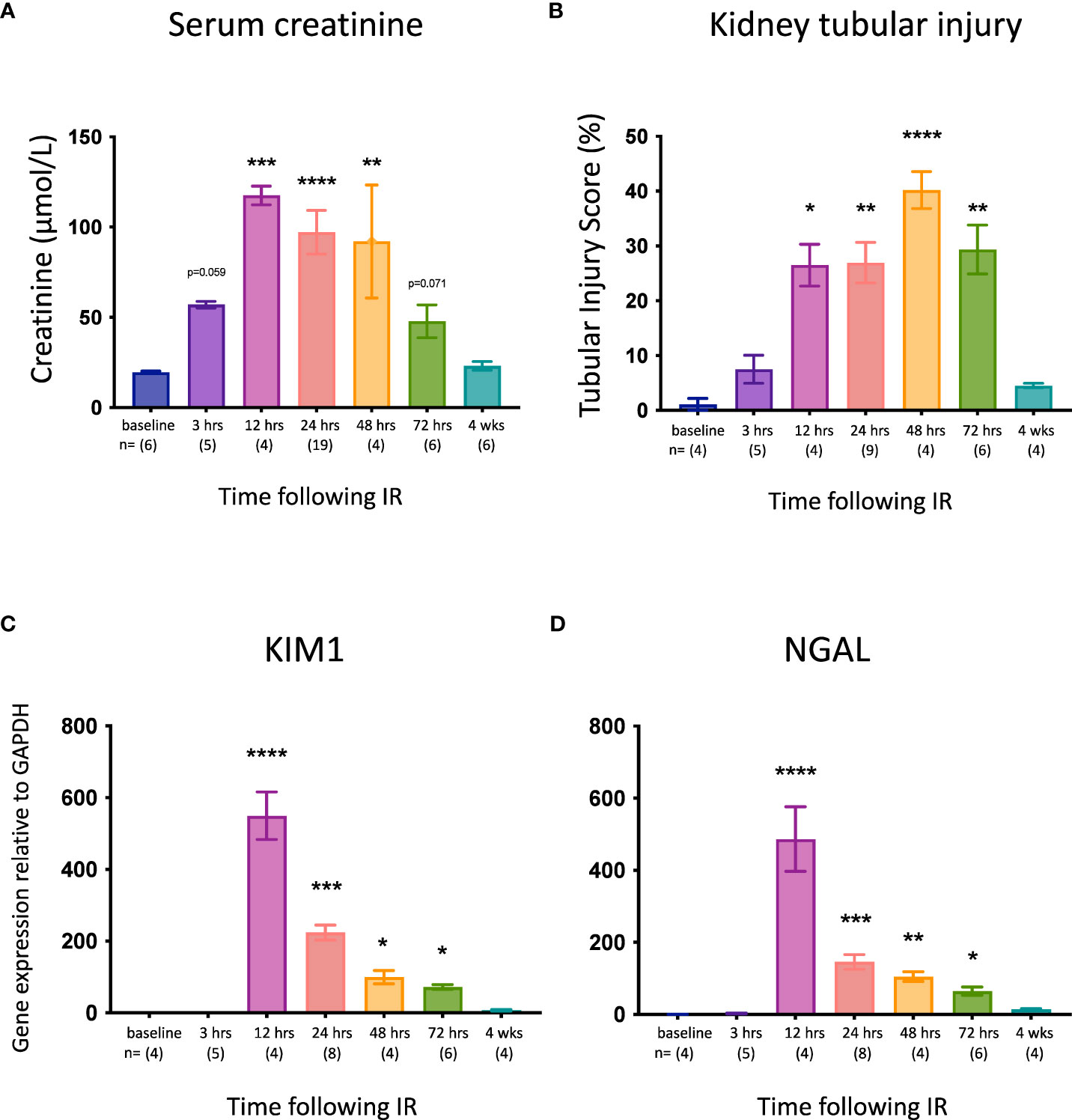
Figure 1 Time course of kidney injury after IR in WT mice. (A): serum creatinine; (B): tubular injury score; (C): mRNA expression of Kim1; (D): mRNA expression of Ngal. Kruskal-Wallis test with Dunn multiple comparisons test with * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to baseline control. Data presented as mean ± SEM.
We then examined the expression of several pro-inflammatory genes relevant to necroptosis and/or kidney IRI. TNFα is the best characterized activator of necroptosis (53) and has been described as a necroptosis-associated alarmin (59). Tnfa expression was significantly increased at 12, 48 and 72 hrs, and remained high at 4 weeks (Figure 2A). Interleukin-6 (IL-6) is an acute inflammatory cytokine that is upregulated in necrotic cells but not in apoptotic cells (60). Il6 expression was elevated earlier than Tnfa (3 hrs) and returned to baseline levels by 72 hrs (Figure 2B). IL-1β and IL-33 are released by necroptotic cells (61, 62). Il1b expression was upregulated at 12, 24 and 48 hrs (Figure 2C), whereas Il33 was increased later, from 24 hrs (Figure 2D). Signalling via Toll-like receptor 4 (TLR4) can activate necroptosis, whereas TLR2 has not been shown to play a role in necroptotic cell death (53). Both Tlr4 (Figure 2E) and Tlr2 (Figure 2F) were upregulated relatively late (48 and 72 hrs, respectively), with Tlr2 expression remaining high at 4 weeks. Macrophage inflammatory protein-2 (MIP-2) is a neutrophil chemoattractant/activator that is upregulated in kidney IRI (63). Mip2 expression was significantly increased at all timepoints except 48 hrs, peaking at 12 hrs (Figure 2G).
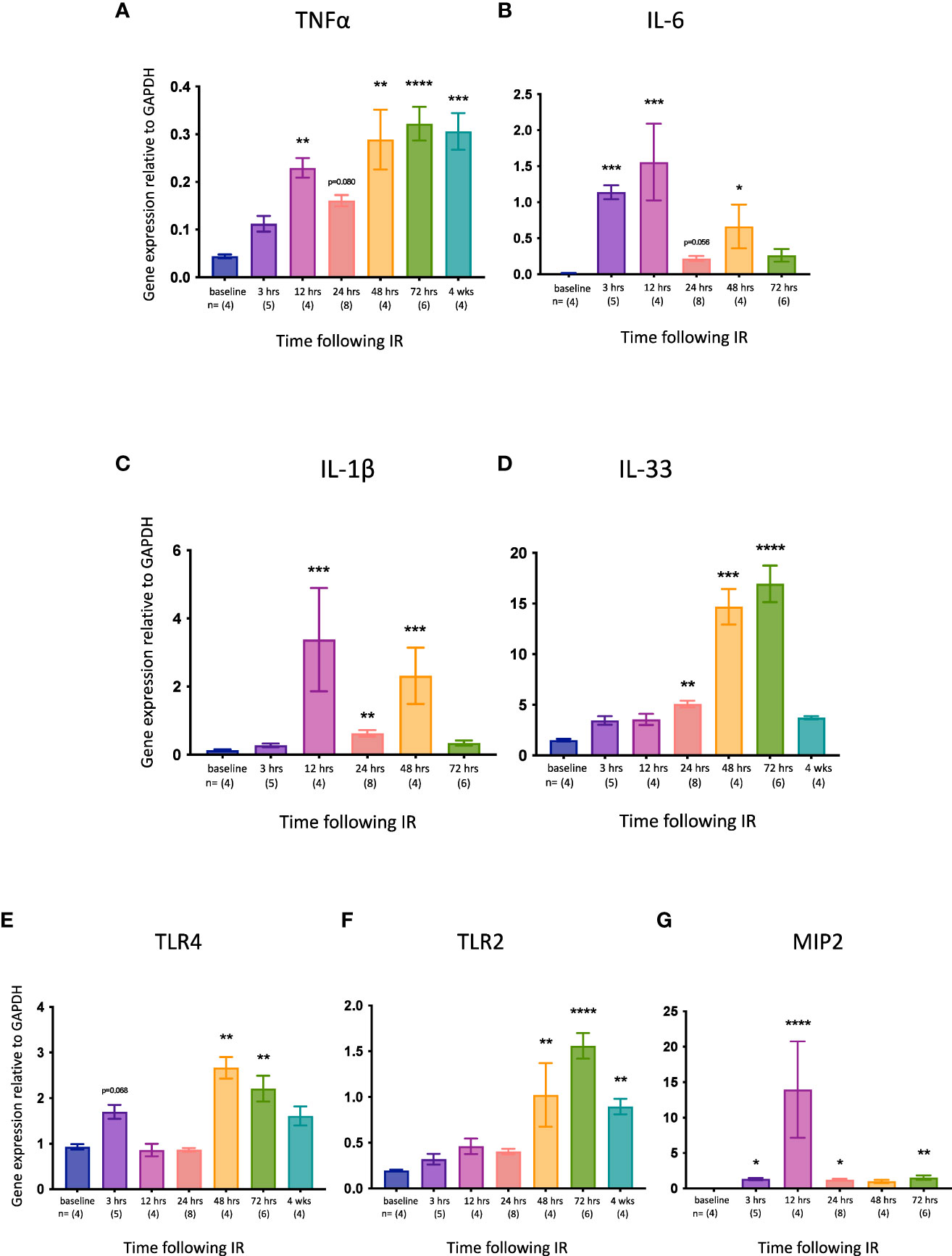
Figure 2 Time course of pro-inflammatory gene expression in the left kidney after IR in WT mice. (A): Tnfa; (B): Il6; (C): Il1b; (D): Il33; (E): Tlr4; (F): Tlr2. (G): Mip2. Kruskal-Wallis test with Dunn multiple comparisons test with * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to baseline control. Data presented as mean ± SEM.
Although elevated expression of the necroptotic pathway genes Ripk1, Ripk3 and Mlkl does not necessarily equate with increased necroptosis in vivo (53), it may indicate an increased propensity for this form of cell death. We therefore measured IRI-associated changes in the expression of these genes in the kidney at different times post-reperfusion. All 3 genes were significantly upregulated at most timepoints, with the highest expression observed at 48 hrs (Figure 3).
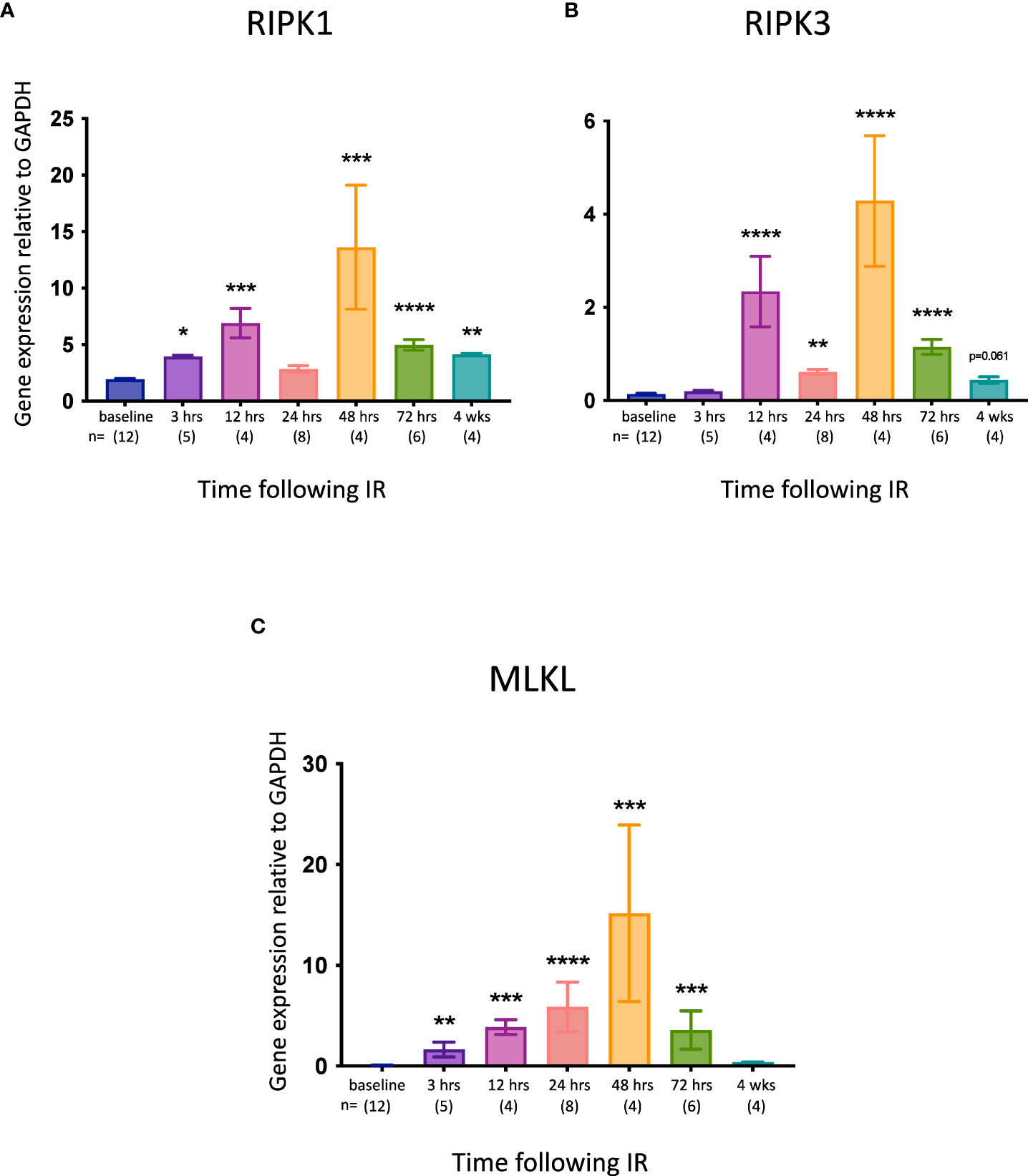
Figure 3 Time course of necroptotic pathway gene expression in the left kidney after IR in WT mice. (A): Ripk1; (B): Ripk3; (C): Mlkl. Kruskal-Wallis test with Dunn multiple comparisons test with * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to baseline control. Data presented as mean ± SEM.
Sections were cut from formalin-fixed left kidney tissue of WT mice from each cohort and stained for pMLKL using a rabbit monoclonal antibody [ab196436, EPR9515 (2)] This antibody has previously been used to detect pMLKL in methanol-fixed mouse dermal fibroblast cell lines (64) and in formalin-fixed mouse cecal tissue (65). Reproducible staining was achieved after optimization of the antigen retrieval step based initially on a method described by He et al., 2021 (65). Mlkl-ko mice subjected to IRI were used as a control to validate the antibody; Mlkl-ko kidneys showing substantial morphological injury were negative for pMLKL staining at each timepoint (Supplementary Figure 3). pMLKL was not detected in WT/IRI kidneys at 0 or 3 hrs post-reperfusion (Figures 4A, B). At 12 hrs, numerous tubules containing cells displaying dense punctate cytoplasmic staining were observed (Figure 4C). Interestingly, the tubules at this timepoint were either completely positive or completely negative. At 24 hrs, the pattern of staining had changed from cytoplasmic to predominantly membrane/intraluminal i.e., on the apical surface of tubular epithelial cells (Figure 4D). At 48 hrs, strong staining of proteinaceous casts within the tubules was observed, along with patchy intraluminal staining (Figure 4E). At 72 hrs (Figure 4F) and 4 weeks (Figure 4G), staining was weaker and mainly restricted to casts.
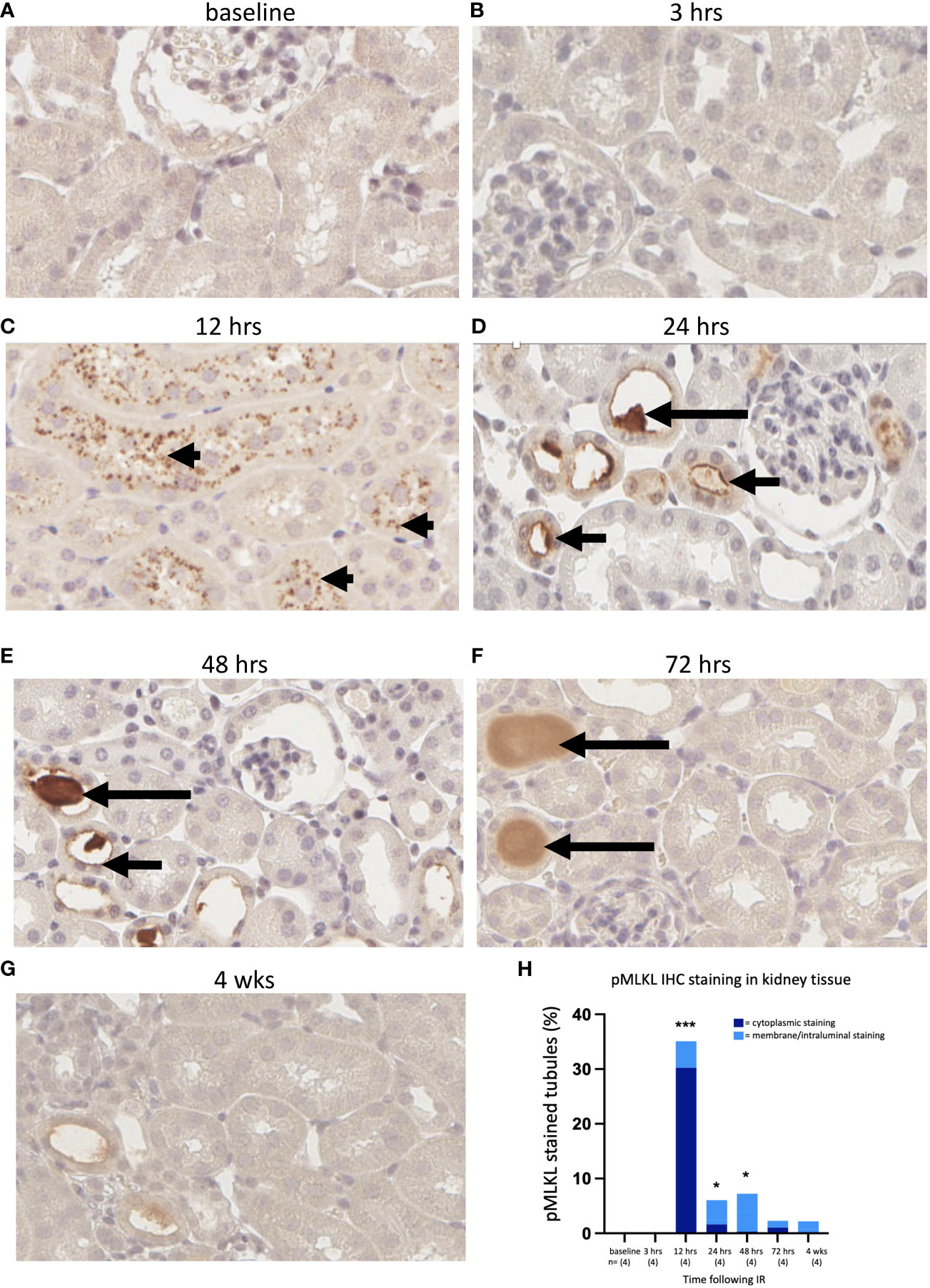
Figure 4 Time course of pMLKL staining in the left kidney after IR in WT mice. Representative sections from 0 hrs (A), 3 hrs (B), 12 hrs (C), 24 hrs (D), 48 hrs (E), 72 hrs (F), and 4 weeks (G) post-reperfusion. (H): percentage of proximal tubules staining positive for pMLKL, stratified according to cellular location (dark blue = cytoplasmic; light blue = membrane/intraluminal). Examples of cytoplasmic, membrane and proteinaceous cast staining are indicated by arrowheads and short and long arrows, respectively. Kruskal-Wallis test with Dunn multiple comparisons test with * p<0.05, *** p<0.001 compared to baseline control. Image magnification x 400.
We performed a semi-quantitative analysis by counting the number of pMLKL-positive tubules as a percentage of the total number of tubules. The degree of positivity was highest at 12 hrs and remained significantly elevated at 24 and 48 hrs (Figure 4H). The data were further stratified into cytoplasmic and intraluminal staining, likely representing the activation and execution phases of necroptosis, respectively. On this basis, maximum activation of necroptosis was observed at 12 hrs, while the terminal stage of necroptotic cell death was relatively constant at 12, 24 and 48 hrs, and returned to close to baseline by 72 hrs (Figure 4H).
Compared to WT mice, Mlkl-ko mice showed reduced kidney injury at 24 hrs post-reperfusion, evidenced by significant decreases in serum creatinine (Figure 5A) and tubular injury score (Figure 5B). Kidney function and injury were not significantly different at the other timepoints. Expression of Kim1 (Figure 5C) and Ngal (Figure 5D) were reduced at the earlier timepoint of 12 hrs, although only the former was statistically significant. Expression of the pro-inflammatory cytokine genes Tnfa, Il1b and Mip2 was also significantly lower at 12 hrs in Mlkl-ko mice compared to WT mice, whereas expression of Il6, Il33, Tlr2 and Tlr4, and of the necroptosis pathway genes Ripk1 and Ripk3, was generally similar in Mlkl-ko and WT mice at all timepoints (Supplementary Figure 1). As expected, Mlkl expression and pMLKL staining were undetectable in kidneys from Mlkl-ko mice (data not shown).
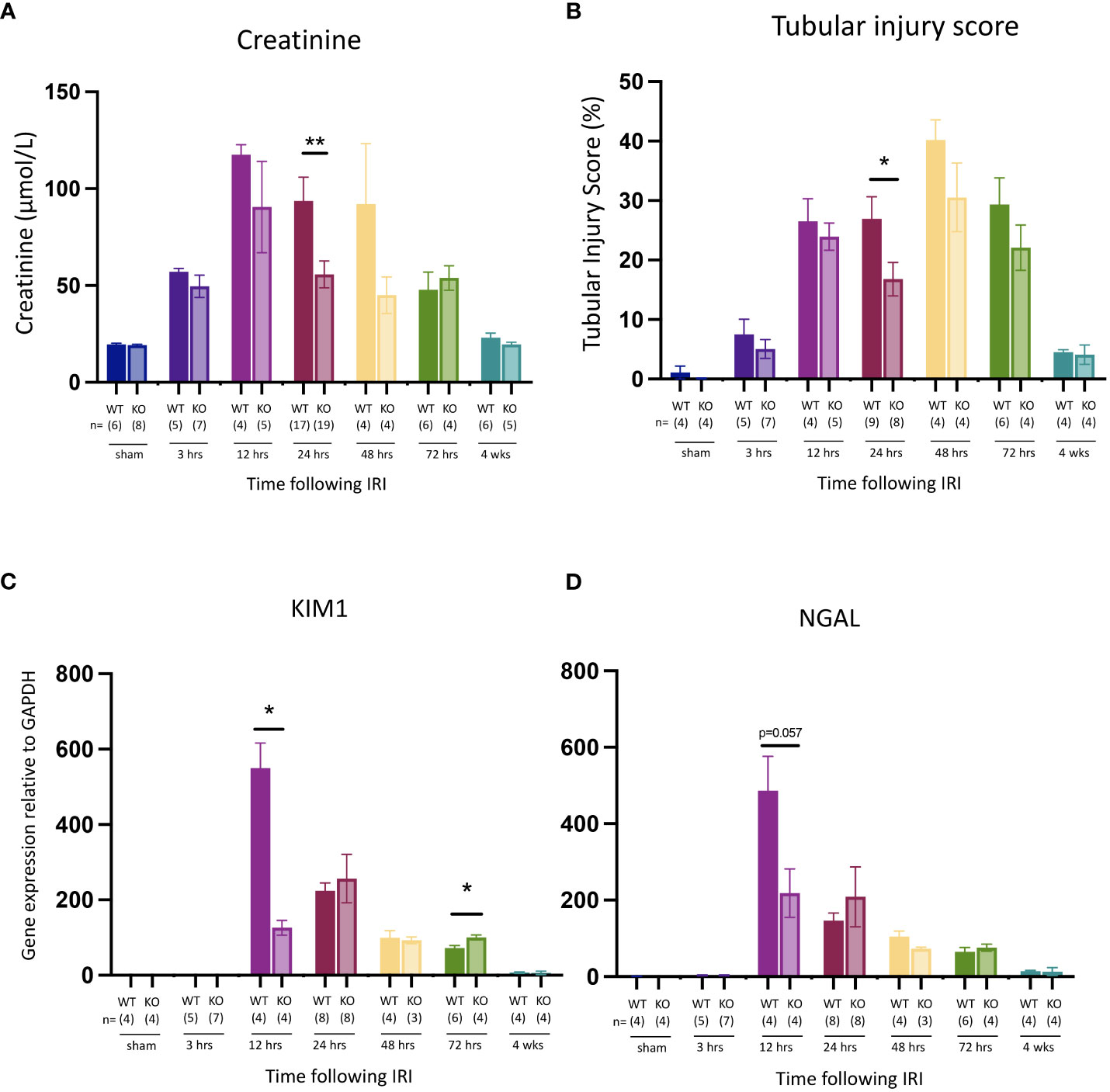
Figure 5 Comparison of kidney injury time course after IR in Mlkl-ko and WT mice. (A): serum creatinine; (B): tubular injury score; (C): mRNA expression of Kim1; (D): mRNA expression of Ngal. Mann Whitney U test comparing with Mlkl-ko and WT mice at each timepoint; * p<0.05, ** p<0.01. Data presented as mean ± SEM.
To investigate the mode of IRI-associated cell death in a necroptosis-deficient setting, kidney sections from the 24 hr timepoint were stained for cleaved Caspase-3 (cC3) as a marker of apoptosis. While WT kidneys showed a significant but modest increase in apoptotic tubules compared to sham kidneys, Mlkl-ko kidneys showed a major increase (Figure 6), suggesting a greater role for apoptosis in this model when necroptosis is inactivated. The pattern of cC3 staining in Mlkl-ko tubules at 24 hrs was like that of pMLKL staining in WT tubules at 12 hrs (Figure 4C) i.e., tubules were either completely positive or negative.

Figure 6 Comparison of cleaved Caspase-3 staining in WT and Mlkl-ko mice at 24 hrs post-reperfusion. (A): WT; (B): Mlkl-ko; (C): sham; (D): semi-quantitative analysis. Mann Whitney U test with * p<0.05 compared to sham procedure and # p<0.05 compared to WT mice. Data presented as mean ± SEM. Image magnification x 200.
Although several studies employing mouse models have implicated necroptosis in the pathogenesis of kidney IRI (54, 55, 66, 67), this is the first study to definitively demonstrate activation of the terminal stage of the necroptotic pathway (i.e., phosphorylation of MLKL) in this setting and to explore the dynamics of the process. We performed a time course analysis in a mouse model of unilateral warm IRI to detail the kinetics of kidney injury, inflammation and necroptosis. Our data suggest that the critical phase lies within 3-12 hrs following reperfusion. During this period, there was major activation of necroptosis as evident by strong pMLKL staining within proximal tubular cells (PTCs) at 12 hrs. Some of the PTCs exhibited pMLKL staining at the cell surface, suggesting progression to the pre-lytic/lytic phase of necroptosis. At the same time, there was upregulated expression of the necroptosis pathway genes Ripk1, Ripk3 and Mlkl and the pro-inflammatory cytokine genes Tnfa, Il6, Il1b and Mip2. This, together with activation of the NLRP3 inflammasome by pMLKL (68), would be expected to create favourable conditions for further necroptosis and inflammation via DAMP release, driving the cycle of necroinflammation.
Of interest was the pattern of pMLKL staining. Epithelial cell injury associated with kidney IR is most apparent in the proximal tubules (1), as PTCs have low anaerobic glycolytic capacity and are located in areas with low partial pressure of oxygen such as the outer medulla and inner cortex (9). Consistent with this, we observed that tubular injury was focused in the proximal tubules in the cortico-medullary region. Necroptosis, as indicated by pMLKL staining, was also restricted to PTCs. This is in contrast to the findings of a recent single-cell analysis suggesting that necroptosis in kidney IRI occurs mainly in non-PTC cell types, with secondary effects on PTCs (69). Furthermore, injured tubules were clustered together, and necroptosis appeared to be activated in all cells within affected tubules. This suggests either a location-dependent effect, or communication between adjacent cells undergoing necroptosis resulting in the spread of necroptosis from cell to cell within affected tubules. pMLKL staining from 24 hrs appeared polarised, with staining of the apical but not basal surface of PTCs. The precise mechanism by which pMLKL clustering at the apical surface may promote cellular communication within the lumen of affected tubules remains to be further explored, with roles for pMLKL in promoting plasma membrane remodelling at intercellular junctions (50) and generation of extracellular vesicles (70, 71) previously proposed.
In the absence of necroptosis (i.e., in Mlkl-ko mice), early inflammation and kidney injury were reduced, providing further evidence that necroptosis and the associated inflammation play a role in kidney damage following IR. The absence of pMLKL staining at 3 hrs in WT mice indicated that the initial injury did not involve necroptosis. It is therefore unsurprising that there was no difference in serum creatinine [which is a delayed marker of kidney injury (72)] between WT and Mlkl-ko mice at 3 hrs. The inability of Mlkl-ko mice to activate necroptosis in the critical 3-12 hr phase is likely to account for the subsequent reduction in kidney inflammation at 12 hrs, and lower serum creatinine and tubular injury scores at 24 hrs. Interestingly, Mlkl-ko mice were protected from kidney IRI at 24 hrs but not at 48 or 72 hrs. This contrasts with an earlier study where protection was noted at 48 and 72 hrs but not at 24 hrs (56). Model differences, including the type (unilateral versus bilateral) and degree of ischemia (18 min versus 35 min), anesthesia (ketamine/xylazine versus the more IRI-protective isoflurane), and WT controls (littermates versus non-littermates), may account for this discrepancy.
While we have further defined the role of necroptosis in kidney IRI, the stimulus that triggers necroptosis in this setting remains unclear. Engagement of death receptors such as TNF receptor 1, activation of TLRs, interferon signalling, and intracellular stimuli in response to viruses, are all able to initiate necroptosis (73). A recent in vitro study using a genetically manipulated myeloid cell system confirmed TNF as an alarmin molecule released following necroptotic cell death to drive further inflammation (59). We suggest that TNF released by inflammatory cells triggers necroptosis during the 3-12 hours post-reperfusion, while TNF and DAMP release following necroptotic cell death stimulate subsequent waves of necroptosis. We showed upregulation of Tnfa gene expression in WT mice, which was reduced in Mlkl-ko mice, leading us to hypothesise that TNF activates necroptosis and drives ongoing necroinflammation in kidney IRI.
The concept that one regulated cell death pathway can compensate for another in acute kidney injury has previously been proposed for necroptosis and ferroptosis, although treatment of Mlkl-ko mice with a ferroptosis inhibitor did not provide further protection from kidney IRI (56). In our model, apoptosis appeared to become the dominant cell death pathway in the absence of necroptosis, suggesting a relationship between necroptosis and apoptosis in kidney IRI. However, a limitation of our study was that it focused on the role of necroptosis without investigating the contribution of other regulated necrosis pathways, notably ferroptosis.
Our study is the first to describe the dynamics of necroptosis and necroinflammation in a mouse model of kidney IRI. Our results suggest that treatments targeting the key necroptosis component, MLKL, may reduce kidney injury following IR, although it should be noted that no specific inhibitors of mouse MLKL are currently available. In addition, our results suggest that such treatments may be effective even if given 3 hrs after reperfusion. This is advantageous in a clinical setting, as in many instances IRI is unpredictable and cannot be treated prophylactically. Finally, combination therapy with inhibitors of MLKL and apoptosis may provide additive protection, with the ultimate aim of preventing acute and chronic kidney disease following kidney IR.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by St Vincent’s Hospital Melbourne Animal Ethics Committee. The study was conducted in accordance with the local legislation and institutional requirements.
AP, JM, FI and PC designed the experimental studies. AP, AB, JM, NF, ES conducted the experimental studies. AB, JM and AP performed the IRI surgery. AP, AB, ES and JM analysed the results. AP and PC drafted the manuscript. All authors contributed to the article and approved the submitted version.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded in part by grant number 88279 from the St Vincent’s Hospital Melbourne Research Endowment Fund. AP was supported by a postgraduate scholarship and JMM by an Investigator Grant (1172929) from the National Health & Medical Research Council (NHMRC) of Australia. JMM acknowledges infrastructure support from the NHMRC (IRIISS 9000719) and the Victorian State Government Operational Infrastructure Support Scheme.
We thank Dr Andre Samson, Dr Ueli Nächbur, and Dr Anne Hempel for helpful discussion on necroptosis and provision of experimental animals; Dr Fenella Munz for advice on veterinary matters and tubular injury scoring; and staff at the Bioresources Centre, St Vincent’s Hospital, Melbourne, for animal care.
Authors JMM and PC have received research funding from Anaxis Pharma Pty Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1251452/full#supplementary-material
Supplementary Figure 1 | Comparison of pro-inflammatory and necroptotic gene expression after IR in Mlkl-ko and WT mice. (A): Tnfa; (B): Il6; (C): Il1b; (D): Il33; (E): Tlr4; (F): Tlr2; (G): Mip2; (H): Ripk1; (I): Ripk3. Mann Whitney U test comparing with Mlkl-ko and WT mice at each timepoint; * p<0.05. Data presented as mean ± SEM.
Supplementary Figure 2 | Representative PAS stained kidney tissue at baseline, 3 hrs, 12 hrs, 24 hrs, 48 hrs, 72 hrs and 4 weeks following IRI. Image magnification x 40.
Supplementary Figure 3 | Representative pMLKL stained kidney tissue of Mlkl-ko mice taken at baseline, 3 hrs, 12 hrs, 24 hrs, 48 hrs, 72 hrs and 4 weeks following IRI. Image magnification x 40.
1. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest (2011) 121(11):4210–21. doi: 10.1172/JCI45161
2. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol (2012) 298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
3. Kosieradzki M, Rowinski W. Ischemia/reperfusion injury in kidney transplantation: mechanisms and prevention. Transplant Proc (2008) 40(10):3279–88. doi: 10.1016/j.transproceed.2008.10.004
4. Murphy E, Steenbergen C. Ion transport and energetics during cell death and protection. Physiol (Bethesda) (2008) 23:115–23. doi: 10.1152/physiol.00044.2007
5. Croall DE, Ersfeld K. The calpains: modular designs and functional diversity. Genome Biol (2007) 8(6):218. doi: 10.1186/gb-2007-8-6-218
6. Kako K, Kato M, Matsuoka T, Mustapha A. Depression of membrane-bound Na+-K+-ATPase activity induced by free radicals and by ischemia of kidney. Americal Physiol Soc (1988) 1988:330–7. doi: 10.1152/ajpcell.1988.254.2.C330
7. Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet (2004) 364(9447):1814–27. doi: 10.1016/S0140-6736(04)17406-0
8. Castaneda MP, Swiatecka-Urban A, Mitsnefes MM, Feuerstein D, Kaskel FJ, Tellis V, et al. Activation of mitochondrial apoptotic pathways in human renal allografts after ischemiareperfusion injury. Transplantation (2003) 76(1):50–4. doi: 10.1097/01.TP.0000069835.95442.9F
9. Scholz H, Boivin FJ, Schmidt-Ott KM, Bachmann S, Eckardt KU, Scholl UI, et al. Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat Rev Nephrol (2021) 17(5):335–49. doi: 10.1038/s41581-021-00394-7
10. Brady HR, Singer GG. Acute renal failure. Lancet (1995) 346(8989):1533–40. doi: 10.1016/S0140-6736(95)92057-9
11. Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev (2007) 220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x
12. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81(1):1–5. doi: 10.1189/jlb.0306164
13. Li L, Okusa MD. Blocking the immune response in ischemic acute kidney injury: the role of adenosine 2A agonists. Nat Clin Pract Nephrol (2006) 2(8):432–44. doi: 10.1038/ncpneph0238
14. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev (2008) 223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x
15. Chadha R, Heidt S, Jones ND, Wood KJ. Th17: contributors to allograft rejection and a barrier to the induction of transplantation tolerance? Transplantation (2011) 91(9):939–45. doi: 10.1097/TP.0b013e3182126eeb
16. Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int (2004) 66(2):496–9. doi: 10.1111/j.1523-1755.2004.761_5.x
17. Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influence long-term function. Am J Physiol Renal Physiol (2001) 281(5):887–99. doi: 10.1152/ajprenal.00050.2001
18. Legrand M, Mik EG, Johannes T, Payen D, Ince C. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol Med (2008) 14(7-8):502–16. doi: 10.2119/2008-00006.Legrand
19. Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol (2011) 300(3):F721–33. doi: 10.1152/ajprenal.00546.2010
20. Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int (2000) 58(6):2351–66. doi: 10.1046/j.1523-1755.2000.00419.x
21. Gueler F, Gwinner W, Schwarz A, Haller H. Long-term effects of acute ischemia and reperfusion injury. Kidney Int (2004) 66(2):523–7. doi: 10.1111/j.1523-1755.2004.761_11.x
22. LaFrance JP, Miller DR. Defining acute kidney injury in database studies: the effects of varying the baseline kidney function assessment period and considering CKD status. Am J Kidney Dis (2010) 56(4):651–60. doi: 10.1053/j.ajkd.2010.05.011
23. Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med (2015) 41(8):1411–23. doi: 10.1007/s00134-015-3934-7
24. See EJ, Jayasinghe K, Glassford N, Bailey M, Johnson DW, Polkinghorne KR, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int (2019) 95(1):160–72. doi: 10.1016/j.kint.2018.08.036
25. Yang F, Zhang L, Wu H, Zou H, Du Y. Clinical analysis of cause, treatment and prognosis in acute kidney injury patients. PloS One (2014) 9(2):e85214. doi: 10.1371/journal.pone.0085214
26. See EJ, Toussaint ND, Bailey M, Johnson DW, Polkinghorne KR, Robbins R, et al. Risk factors for major adverse kidney events in the first year after acute kidney injury. Clin Kidney J (2021) 14(2):556–63. doi: 10.1093/ckj/sfz169
27. Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int (2012) 81(5):442–8. doi: 10.1038/ki.2011.379
28. Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol (2012) 23(6):967–9. doi: 10.1681/ASN.2012030222
29. Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl (2022) 12(1):7–11. doi: 10.1016/j.kisu.2021.11.003
30. Mortality GBD, Causes of Death C. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 385(9963):117–71. doi: 10.1016/S0140-6736(14)61682-2
31. Rhee CM, Kovesdy CP. Epidemiology: Spotlight on CKD deaths-increasing mortality worldwide. Nat Rev Nephrol (2015) 11(4):199–200. doi: 10.1038/nrneph.2015.25
32. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1(2):112–9. doi: 10.1038/nchembio711
33. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
34. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
35. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
36. Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol (2009) 297(3):F749–59. doi: 10.1152/ajprenal.00239.2009
37. Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA (2006) 103(48):18308–13. doi: 10.1073/pnas.0606526103
38. David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (2009) 14:1116–28. doi: 10.2741/3297
39. Pefanis A, Ierino FL, Murphy JM, Cowan PJ. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int (2019) 96(2):291–301. doi: 10.1016/j.kint.2019.02.009
40. Horne CR, Samson AL, Murphy JM. The web of death: the expanding complexity of necroptotic signaling. Trends Cell Biol (2023) 33(2):162–74. doi: 10.1016/j.tcb.2022.05.008
41. Samson AL, Garnish SE, Hildebrand JM, Murphy JM. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci Signal (2021) 14(668):eabc6178. doi: 10.1126/scisignal.abc6178
42. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell (2012) 150(2):339–50. doi: 10.1016/j.cell.2012.06.019
43. Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, et al. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell (2018) 173(5):1244–53.e10. doi: 10.1016/j.cell.2018.03.032
44. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
45. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity (2013) 39(3):443–53. doi: 10.1016/j.immuni.2013.06.018
46. Meng Y, Davies KA, Fitzgibbon C, Young SN, Garnish SE, Horne CR, et al. Human RIPK3 maintains MLKL in an inactive conformation prior to cell death by necroptosis. Nat Commun (2021) 12(1):6783. doi: 10.1038/s41467-021-27032-x
47. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun (2018) 9(1):2422. doi: 10.1038/s41467-018-04714-7
48. Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE, et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc Natl Acad Sci USA (2020) 117(15):8468–75. doi: 10.1073/pnas.1919960117
49. Garnish SE, Meng Y, Koide A, Sandow JJ, Denbaum E, Jacobsen AV, et al. Conformational interconversion of MLKL and disengagement from RIPK3 precede cell death by necroptosis. Nat Commun (2021) 12(1):2211. doi: 10.1038/s41467-021-22400-z
50. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun (2020) 11(1):3151. doi: 10.1038/s41467-020-16887-1
51. Nakano H, Murai S, Moriwaki K. Regulation of the release of damage-associated molecular patterns from necroptotic cells. Biochem J (2022) 479(5):677–85. doi: 10.1042/BCJ20210604
52. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol (2014) 14(11):759–67. doi: 10.1038/nri3743
53. Kolbrink B, von Samson-Himmelstjerna FA, Murphy JM, Krautwald S. Role of necroptosis in kidney health and disease. Nat Rev Nephrol (2023) 19(5):300–14. doi: 10.1038/s41581-022-00658-w
54. Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA (2013) 110(29):12024–9. doi: 10.1073/pnas.1305538110
55. Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ (2016) 23(9):1565–76. doi: 10.1038/cdd.2016.46
56. Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci (2017) 74(19):3631–45. doi: 10.1007/s00018-017-2547-4
57. Crikis S, Lu B, Murray-Segal LM, Selan C, Robson SC, D'Apice AJ, et al. Transgenic overexpression of CD39 protects against renal ischemia-reperfusion and transplant vascular injury. Am J Transplant (2010) 10(12):2586–95. doi: 10.1111/j.1600-6143.2010.03257.x
58. McRae JL, Vikstrom IB, Bongoni AK, Salvaris EJ, Fisicaro N, Ng M, et al. Blockade of the G-CSF receptor is protective in a mouse model of renal ischemia-reperfusion injury. J Immunol (2020) 205(5):1433–40. doi: 10.4049/jimmunol.2000390
59. Pinci F, Gaidt MM, Jung C, Nagl D, Kuut G, Hornung V. Tumor necrosis factor is a necroptosis-associated alarmin. Front Immunol (2022) 13:1074440. doi: 10.3389/fimmu.2022.1074440
60. Vanden Berghe T, Kalai M, Denecker G, Meeus A, Saelens X, Vandenabeele P. Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal (2006) 18(3):328–35. doi: 10.1016/j.cellsig.2005.05.003
61. Li L, Shan S, Kang K, Zhang C, Kou R, Song F. The cross-talk of NLRP3 inflammasome activation and necroptotic hepatocyte death in acetaminophen-induced mice acute liver injury. Hum Exp Toxicol (2021) 40(4):673–84. doi: 10.1177/0960327120961158
62. Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M, et al. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J (2019) 286(3):507–22. doi: 10.1111/febs.14738
63. Lemay S, Rabb H, Postler G, Singh AK. Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation (2000) 69(5):959–63. doi: 10.1097/00007890-200003150-00049
64. Samson AL, Fitzgibbon C, Patel KM, Hildebrand JM, Whitehead LW, Rimes JS, et al. A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells. Cell Death Differ (2021) 28(7):2126–44. doi: 10.1038/s41418-021-00742-x
65. He P, Ai T, Yang ZH, Wu J, Han J. Detection of necroptosis by phospho-MLKL immunohistochemical labeling. STAR Protoc (2021) 2(1):100251. doi: 10.1016/j.xpro.2020.100251
66. Liu W, Chen B, Wang Y, Meng C, Huang H, Huang XR, et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc Natl Acad Sci USA (2018) 115(7):E1475–E84. doi: 10.1073/pnas.1716959115
67. Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol (2014) 35:2–13. doi: 10.1016/j.semcdb.2014.08.010
68. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA (2017) 114(6):E961–E9. doi: 10.1073/pnas.1613305114
69. Balzer MS, Doke T, Yang YW, Aldridge DL, Hu H, Mai H, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun (2022) 13(1):4018. doi: 10.1038/s41467-022-31772-9
70. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity (2017) 47(1):51–65.e7. doi: 10.1016/j.immuni.2017.06.001
71. Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell (2017) 169(2):286–300.e16. doi: 10.1016/j.cell.2017.03.020
72. Moran SM, Myers BD. Course of acute renal failure studied by a model of creatinine kinetics. Kidney Int (1985) 27(6):928–37. doi: 10.1038/ki.1985.101
Keywords: necroptosis, ischemia-reperfusion, necroinflammation, mlkl, acute kidney injury, chronic kidney disease
Citation: Pefanis A, Bongoni AK, McRae JL, Salvaris EJ, Fisicaro N, Murphy JM, Ierino FL and Cowan PJ (2023) Dynamics of necroptosis in kidney ischemia-reperfusion injury. Front. Immunol. 14:1251452. doi: 10.3389/fimmu.2023.1251452
Received: 01 July 2023; Accepted: 19 October 2023;
Published: 02 November 2023.
Edited by:
Xu-jie Zhou, Peking University, ChinaReviewed by:
Kenta Moriwaki, Toho University, JapanCopyright © 2023 Pefanis, Bongoni, McRae, Salvaris, Fisicaro, Murphy, Ierino and Cowan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Cowan, UGV0ZXIuQ293YW5Ac3ZoYS5vcmcuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.