
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 October 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1250719
This article is part of the Research Topic Molecular and Cellular Control of B Cell responses: Germinal Center and Extrafollicular Responses for Cellular Outputs View all 6 articles
 Shu Horiuchi1†
Shu Horiuchi1† Takuya Koike1†
Takuya Koike1† Hirofumi Takebuchi1
Hirofumi Takebuchi1 Katsuaki Hoshino2,3Izumi Sasaki4Yuri Fukuda-Ohta4
Katsuaki Hoshino2,3Izumi Sasaki4Yuri Fukuda-Ohta4 Tsuneyasu Kaisho3,4
Tsuneyasu Kaisho3,4 Daisuke Kitamura1*
Daisuke Kitamura1*Generation of memory B cells is one of the key features of adaptive immunity as they respond rapidly to re-exposure to the antigen and generate functional antibodies. Although the functions of memory B cells are becoming clearer, the regulation of memory B cell generation and maintenance is still not well understood. Here we found that transcription factor SpiB is expressed in some germinal center (GC) B cells and memory B cells and participates in the maintenance of memory B cells. Overexpression and knockdown analyses revealed that SpiB suppresses plasma cell differentiation by suppressing the expression of Blimp1 while inducing Bach2 in the in-vitro-induced germinal center B (iGB) cell culture system, and that SpiB facilitates in-vivo appearance of memory-like B cells derived from the iGB cells. Further analysis in IgG1+ cell-specific SpiB conditional knockout (cKO) mice showed that function of SpiB is critical for the generation of late memory B cells but not early memory B cells or GC B cells. Gene expression analysis suggested that SpiB-dependent suppression of plasma cell differentiation is independent of the expression of Bach2. We further revealed that SpiB upregulates anti-apoptosis and autophagy genes to control the survival of memory B cells. These findings indicate the function of SpiB in the generation of long-lasting memory B cells to maintain humoral memory.
1. GC and memory B cells express SpiB and SpiB expression is induced by CD40 stimulation in vitro.
2. SpiB suppresses plasma cell differentiation while inducing the expression of Bach2 in vitro.
3. SpiB overexpression enhances and SpiB knockdown diminishes Bmem-like cell generation.
4. SpiB Cγ1-cKO mice generates less IgG1+ late Bmem cells with diminished memory antibody response.
5. SpiB suppresses plasma cell differentiation independent of Bach2.
6. SpiB Cγ1-cKO B cells express lower levels of anti-apoptosis and autophagy genes with less ability to survive.
Upon immunization with T-cell-dependent (TD) antigens, antigen-activated B cells undergo proliferation and class switching of their B cell receptor (BCR) and differentiate into effector B cell populations. Some B cells differentiate to short-lived plasma cells (PCs) that produce low affinity antibodies at the early stage of the immune response. Other B cells differentiate to germinal center (GC) B cells to form GCs. In GCs, B cells undergo somatic hypermutation (SHM) in variable regions of immunoglobulin genes, encoding BCR and antibodies, and selection based on the affinity of the resultant BCR to the antigen. Among the selected B cells, those expressing BCR with relatively higher affinity differentiate into long-lived PCs (LLPCs) whereas those with relatively lower affinity to memory B (Bmem) cells. In addition, Bmem cells are also generated independently of GCs. LLPCs and Bmem cells, of either GC-experienced or not, contribute to long-term humoral immunity (1–3).
Bmem cells are maintained in the body for a long time and activated rapidly when re-exposed to the primary antigen, partly owing to IL-9 autocrine stimulation (4, 5). The typical recall response for class-switched Bmem cells is to produce a large amount of antigen-specific antibodies in a short time. A small proportion of the class-switched Bmem cells and most IgM+ Bmem cells differentiate to GC B cells to undergo further SHM and affinity maturation during the recall response. Although regulatory mechanisms of the Bmem cell recall response through BCR and other receptors are becoming clearer (4), the mechanisms for Bmem cell development, including external signals and transcription factors that induce differentiation to Bmem cells, are largely unknown. Bach2 is a transcriptional suppressor that regulates the expression of Blimp1, a gene encoding a transcription factor known to induce PC development. Bach2 was shown to be required for GC B-cell differentiation toward Bmem cells albeit in a specific genetic background, namely Blimp1-deficiency (6).
To elucidate the mechanisms for Bmem cell development, we established an experimental system in which naive B cells can be committed to either Bmem cells or PCs in vitro. In this system, naïve B cells are propagated on the feeder cells expressing exogenous CD40L and BAFF (termed 40LB) in the presence of IL-4 for the first 4-5 days. This culture allows massive expansion of B cells expressing GC B-cell markers, which we call ‘induced GC B (iGB)-4 cells’, and efficient class switching either to IgG1 or IgE. The following culture with IL-21 allows further proliferation of iGB cells (now called iGB-21) for another 4-5 days, gradually increasing the proportion of CD138+ plasmablasts. Importantly, when transferred into irradiated wild type mice, the iGB-4 cells differentiate into memory-like B cells termed “induced memory B (iMB) cells”, which are phenotypically and functionally equivalent to bona fide Bmem cells and survive more than 2 months in the spleens and lymph nodes of the recipient mice. Notably, IgG1+ and IgM+, but not IgE+, iGB-4 cells become iMB cells in vivo. In contrast, iGB-21 cells fail to develop into iMB cells but instead differentiate into PCs in the bone marrow of the recipients. The iGB cells tertiary cultured for another 2 days without any cytokines (iGB-m) partly regain the ability to form iMB cells in vivo (7). From the data set of microarray analysis comparing iMB cells and naive follicular B cells, we selected the genes expressed in excess in iMB cells, and further selected those expressed at a higher level in IgG1+ iGB-4 cells than in IgG1+ iGB-21 cells, and not in IgE+ iGB-4/iGB-21 cells, aiming to identify the genes that regulate Bmem cell development. Among the genes selected in this approach, we focused on SpiB.
SpiB is an ETS family transcription factor expressed in various types of immune cells (8–10). In B cells, it has been reported to regulate the differentiation of pre-B cells in the bone marrow and follicular B cells (11, 12). SpiB is also required for proper BCR signaling (13) and mature B cell survival, although being compensated by PU.1 (12), and for sustaining GC B cells in the primary, and antibody production in the secondary, TD immune response (14, 15). In an in vitro study, SpiB suppresses PC differentiation in human and mice (16, 17). However, the role of SpiB in B cells after the generation of the GC is unknown.
Here we show our finding that SpiB is critical for Bmem cell maintenance. SpiB mRNA is expressed in GC B cells and Bmem cells, and its expression is upregulated by CD40 stimulation. Overexpression and knockdown of SpiB in iGB cells revealed that SpiB induces the expression of Bach2 and suppresses the induction of Blimp1 expression and the differentiation of CD138+ PCs, independently of Bach2, and that SpiB facilitates iMB cell formation from iGB-4 cells in vivo. A conditional mouse strain that lacked SpiB only in IgG1+ B cells (SpiB cKO) exhibited defective formation of IgG1+ late Bmem cells after primary immunization and attenuated IgG1 recall responses upon the secondary challenge. Additionally, Bmem cells in the SpiB cKO mice showed lower expression of genes required for anti-apoptosis and autophagy, suggesting that SpiB is critical for long-term survival of Bmem cells.
C57BL/6 NCrSlc (B6) mice were purchased from Sankyo Labo Service. All the following mouse strains were backcrossed to B6 or congenic B6 CD45.1+ mice: SpiBf/f (see below), Blimp1f/f, Blimp1-GFP (18), and Bach2-/- (19). SpiBf/f and Blimp1f/f mice were crossed with Cγ1-cre (20) and CD19-cre mice (21), respectively, to generate each conditional knockout strain. As the control for the SpiBf/f Cγ1-cre (here called ‘SpiB cKO’) mice, SpiB+/+ Cγ1-cre (here called ‘SpiB WT’) mice were used. Sex-matched 7-12 week-old mice were immunized i.p. with 100 μg of 4-hydroxy-3-nitrophenyl acetyl (NP)32-chicken gamma globulin (CGG) in alum or NP-conjugated sheep red blood cell (SRBC) where indicated. Mice were maintained in the Tokyo University of Science mouse facility under specific pathogen-free conditions. Mouse procedures were performed under protocols approved by the Animal Care and Use Committee of Tokyo University of Science (Approval No S13009).
To generate the Spib-floxed (SpiBf/f) mice, a targeting vector was designed to delete exon 6 encoding the Ets domain of Spi-B. To construct the targeting vector, C57BL/6N mouse genomic DNA fragments were amplified by PCR using KOD -plus- DNA polymerase (Toyobo, Osaka, Japan). An 8.0 kbp long arm containing exon 6, which has an insertion of a loxP site in the intron downstream of the exon 6, was connected to an MC1 promoter-driven neomycin resistance gene (neo) cassette flanked by two FRT sites and a loxP site. Then, a 1.9 kbp short arm containing exons 3-5 was ligated upstream of the neo cassette. A herpes simplex virus thymidine kinase gene (HSV-TK) was attached at the downstream end of the long arm. The targeting vector was linearized with SalI and electroporated into the Bruce4 ES cells. Clones with resistance to G418 and gancyclovir were screened for homologous recombination by PCR and confirmed by Southern blot analysis. ES cell clones carrying the targeted allele were used to generate Spib mutant mice. To delete the neo cassette flanked by two FRT sites, the Spib-targeted mice were intercrossed with C57BL/6-Tg(CAG-flpe)36Ito/ItoRbrc (RBRC01834) (RIKEN BioResource Research Center, Japan) (Supplementary Figure 1A).
Genotyping of Spib mutant mice was performed by PCR using the primers listed below. PCR using primers #1 and #2 generates 301bp and 475 bp bands in the wild-type and floxed alleles, respectively. PCR using primers #1 and #3 generates a 454 bp band in the deleted allele. Although PCR using primers #1 and #3 should also generate a band with larger than 2700 bp in both wild-type and floxed alleles, it was too large to be detected (Supplementary Figures 1B, C).
Primer #1: 5’-AACCAGGCCCAACTCCATTGTGAAAAC-3’.
Primer #2: 5’-GCCAGCTTCTGATACGTCATGCGCTTG-3’.
Primer #3: 5’-AGTCCTGTCACACCATTGGTTGCAGTG-3’.
Single-cell suspensions from spleens were depleted of RBCs by ammonium chloride lysis, incubated with mAb to FcγRII/III (2.4G2; BD Pharmingen) to block FcγRs and were stained for 30 min on ice in PBS containing 0.5% BSA, 2 mM EDTA, 0.05% sodium azide with various combinations of antibodies and reagents. Intracellular SpiB staining was performed with FoxP3 Staining Buffer Set according to the manufacturer’s protocol (eBioscience) with anti-SpiB sheep polyclonal antibody (R&D systems, AF7204) followed by Alexa 647-labeled donkey anti-sheep IgG (Invitrogen, A-21448). All samples were analyzed by either FACS Callibur or FACS Canto II (BD Biosciences). The data were analyzed by FlowJo (Tree Star).
Splenic naïve B cells were purified as described previously (7). B cells were cultured in RPMI-1640 medium (Wako) supplemented with 10% FCS, 10 mM HEPES, 1 mM sodium pyruvate, 5.5 × 10−5 M 2-ME, 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO). For iGB cell culture, naive B cells were cultured on 40LB feeder cells with IL-4 (10 ng/ml) or IL-21 (10 ng/ml) where indicated. To purify iGB cells or to change the culture condition, 40LB feeder cells were removed from the total cells of iGB cultures by anti-H-2Kd (BioLegend) Ab, Streptavidin Particle Plus DM and the IMag system (BD Biosciences).
Early Bmem and GC B cells were purified from spleen of the mice immunized with NP-SRBC, 7 or 14 days previously, as follows: splenocytes were stained with anti-CD19, anti-CD38, anti-GL7, and NP-BSA-biotin, followed by fluorescence-labeled streptavidin. Bmem cells (CD19+ NP+ CD38+ GL7–) and GC B cells (CD19+ NP+ CD38– GL7+) were sorted by FACS AriaII. For iGB cells, cultured cells depleted of 40LB feeder cells as described above, were stained with anti-IgG and anti-IgE and sorted by FACS Aria II.
Retroviruses were produced in Plat-E cells transfected with pMXs-based vectors with Fugene HD (Promega) or PEI “Max” (Mw 40,000; Polysciences). The virus-containing supernatant was collected 2 days after transfection and added to iGB cells cultured for 2 days previously. Cells were spin-infected at 2,000 rpm, room temperature for 90 minutes with 10 μg/ml DOTAP Liposomal Transfection Reagent (Roche) and IL-4 (10 ng/ml). One day later, cells were collected and cultured on new 40LB feeder layers with IL-4 for further culture.
iMB cells were generated as previously described (7). Retrovirally transduced iGB cells from CD45.1+ B6 mice were collected by removing 40LB feeder cells with anti-H-2Kd Ab (BioLegend), Streptavidin Particle Plus DM, and the IMag system (BD Biosciences). The iGB cells were adoptively transferred to γ-irradiated (6.5Gy) B6 mice (CD45.2+). Splenocytes were harvested from the recipient mice after 2 weeks or later for further analysis.
The procedures for RNA extraction and reverse transcription to cDNA have been described previously (7). Quantitative real-time PCR was performed with a 7500 fast Real-time PCR system (Applied Biosystems). Gene expression levels were determined by the relative standard curve method and normalized to that of either Gapdh or 18S rRNA.
Total and high affinity NP-specific IgG1 was detected by the method described previously (7). NP13.6-BSA or NP2.7-BSA for total or high affinity antibodies, respectively, was coated on 96 well flat bottom plate. Bound Abs were detected by HRP-conjugated goat anti-mouse IgG1 Ab (Southern Biotechnology) and TMB substrate (Sigma).
To understand the regulation of Bmem cells by transcription factors, we analyzed the gene-expression profiles of induced memory-like B (iMB) cells and compared the result with publicly available gene expression data (ImmGen). We noticed that mRNA for the transcription factor SpiB was expressed selectively high in Bmem cells among B cells, while hardly expressed in plasmablasts and PCs. In contrast, Bcl6 was expressed substantially lower in Bmem cells than in GC B cells (Supplementary Figure 2A). To confirm these expression patterns, we analyzed the mRNA levels of SpiB, Bach2 and Bcl6 in early Bmem and GC B cells from mice immunized with NP-CGG, and naïve follicular B cells from unimmunized mice. In slight contrast to the ImmGen data, SpiB expression in the early Bmem cells was lower than that in GC B cells, but appeared to be higher than that in follicular B cells. These expression patterns are similar to those of Bach2 and Bcl6 (Figure 1A; Supplementary Figures 2B, C).
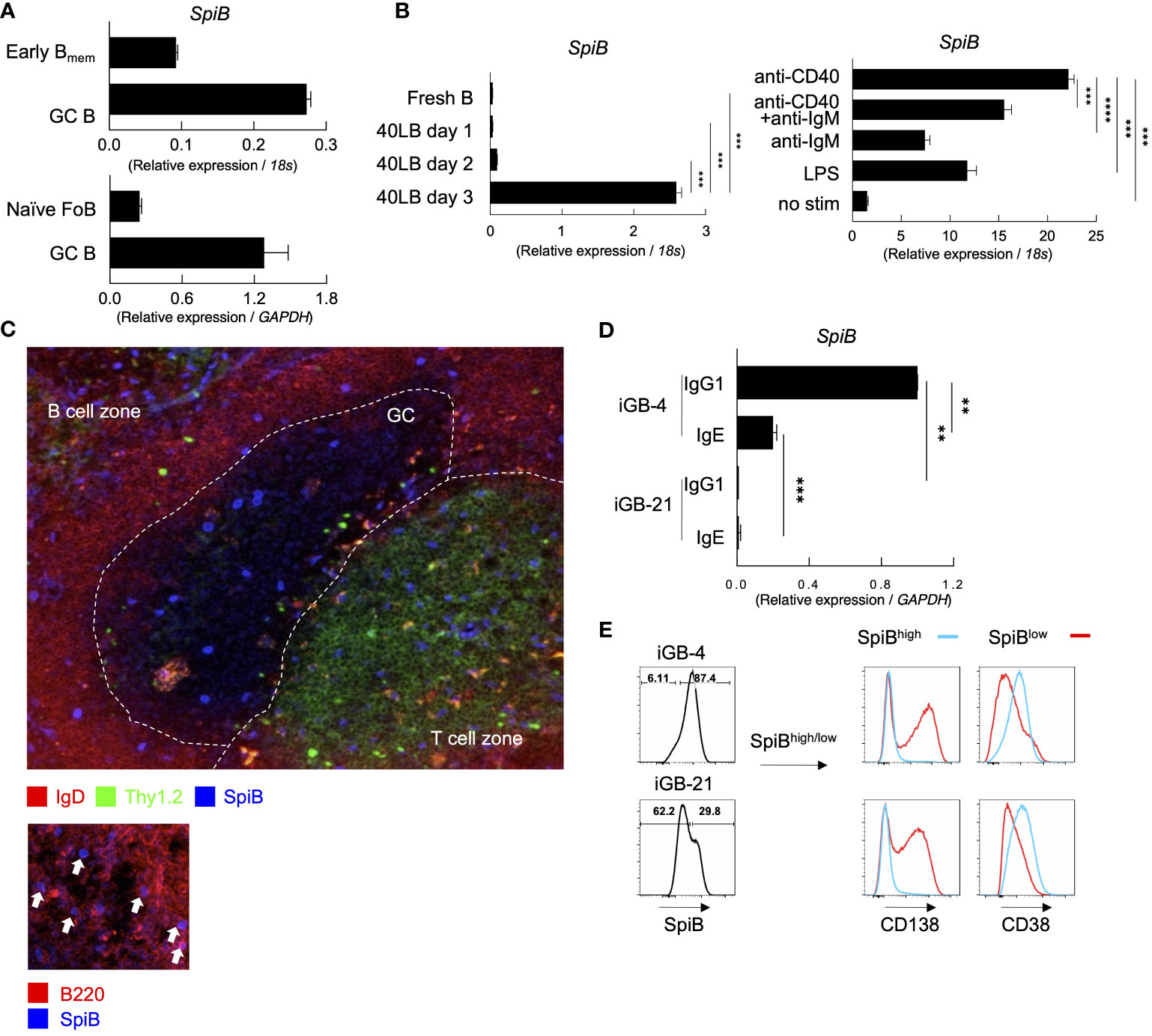
Figure 1 SpiB is expressed in GC B and Bmem cells and induced by CD40 stimulation in vitro. (A) SpiB mRNA expression in B cell subsets from mouse spleen collected 14 days post NP-CGG immunization. B cells were sorted as CD19+ Igκ- and NP+ CD38+ GL7- (early Bmem cells), NP+ CD38-GL7+ (GC B cells), or CD19+ CD21lo CD23hi AA4.1- [naive follicular B (FoB) cells]. Comparison of early Bmem and GC B cells (top) and naïve FoB and GC B cells (bottom) are shown. (B) SpiB mRNA expression in B cells cultured on 40LB feeder cells for 0-3 days (left) and B cells stimulated with the indicated antibodies or LPS (right). (C) Immunofluorescence microscopy of frozen sections of spleens from mice immunized with NP-SRBC 7 days previously. The sections were stained for SpiB (blue), IgD (red), and Thy1.2 (green) (top), or for SpiB (blue) and B220 (red) (bottom). (D) SpiB mRNA expression in sorted IgG1+ and IgE+ from iGB cells cultured with IL-4 (iGB-4) for 4 days and IL-21 (iGB-21) for the following 4 days. (E) FCM analysis of the expression of CD138 and CD38 in SpiBhigh (blue) and SpiBlow (red) iGB-4 and iGB-21 cells. SpiB staining profile and gating of iGB-4 and iGB-21 cells are shown on the left and the expression of CD138 and CD38 on the gated fractions are on the right. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Our previous finding proposed that the quantity of CD40 stimulation on B cells during the primary response is a key factor in regulating Bmem cells (22). Additionally, SpiB is reported to be induced by OBF-1 which is upregulated by CD40 signaling (23, 24). Thus, we assessed whether CD40 stimulation induces the expression of SpiB in B cells. By culturing naïve B cells on feeder cells expressing CD40L and BAFF (40LB) without any cytokines, the expression of SpiB was induced after 3 days (Figure 1B). We then tested other stimuli known to activate naïve B cells, namely, anti-CD40 or/and anti-IgM antibodies, or LPS, and observed that anti-CD40 induced the highest levels of SpiB expression among these stimuli, and no synergistic effect was seen between anti-CD40 and anti-IgM antibodies (Figure 1B). These results indicate that CD40 stimulation is sufficient to induce SpiB expression in B cells. Immunofluorescence microscopy analysis of spleens from mice immunized with NP-SRBC 7 days earlier showed that a small number of discernible SpiB+ cells were scattered in GC, B cell follicles, and T cell zones in the white pulp, while SpiB expression was undetectable in the vast majority of GC and follicular B cells (Figure 1C, top). At a closer look, nuclear SpiB+ cells in the B cell follicle were also B220+ (Figure 1C, bottom), either IgD+ or IgD–, and those in the GC and T cell zones were discernible as IgD– (Figure 1, top). Together with our mRNA expression data and previous reports showing that SpiB mRNA expression is limited to B cells and plasmacytoid dendritic cells (9; ImmGen), these results suggest that a significant level of SpiB is expressed in selected GC B cells, class-switched or unswitched Bmem cells in the B cell follicle, and plasmacytoid dendritic cells in the T cell zone.
It has been shown that SpiB suppresses the expression of Blimp1 in human cells (17) and potentially in mice (16, 25). To further infer the role of SpiB in the late B-cell development, we analyzed the expression of SpiB in B cells cultured in the iGB system (7). The expression of SpiB mRNA was readily detected in IgG1+ B cells cultured on 40LB feeder cells with IL-4 (iGB-4 cells), and in IgE+ iGB-4 cells albeit at a reduced level, but declined to an almost undetectable level following culture with IL-21 (iGB-21 cells) (Figure 1D). SpiB protein expression was also decreased in the iGB-21 cells as compared to iGB-4 cells (Figure 1E). Co-staining of SpiB protein with memory/naive B cell marker (CD38) and PC marker (CD138) revealed that SpiBhigh cells expressed higher levels of CD38 and little CD138, whereas SpiBlow cells expressed lower CD38 and were mostly CD138+ (Figure 1E). Among the iGB-4 cells, IgG1+ cells were largely SpiB+ CD38high CD138–, whereas IgE+ cells contained a substantial population of SpiB– CD38low CD138+ cells, which markedly increased in iGB-21 cells, either of IgG1+ or IgE+ (Supplementary Figures 2D, E). This result corresponded to the notion that IgE+ B cells rapidly differentiate into PCs (26, 27) and IL-21 facilitates PC differentiation (7). These results suggest that SpiB may have a function to suppress PC differentiation while inducing the phenotype of Bmem cells.
To better understand the function of SpiB in B cell differentiation, we performed retroviral overexpression or knockdown of SpiB in iGB cells (Figure 2A). Overexpression of SpiB resulted in the decrease, while knockdown of SpiB resulted in the increase, of the proportion of CD138+ cells in both IgE– (mostly IgG1+) and IgE+ fractions of iGB-4 and iGB-21 cells (Figure 2B; Supplementary Figure 3A). PC differentiation is known to be induced by a transcription factor Blimp1 which is coded by the gene Prdm1 (28). To further elucidate the mechanism of SpiB function, we analyzed the expression of Prdm1 gene by using iGB cells derived from Blimp1-reporter (Blimp1-GFP) mice. Overexpression of SpiB resulted in the decrease, whereas knockdown of SpiB resulted in the increase, of the proportion of Blimp1 GFP+ cells within IgE+, IgM+ and IgE– IgM– (mostly IgG1+) B cells in iGB-4 and iGB-21 cells (Figure 2C). These results suggest the function of SpiB to suppress Blimp1 expression at a transcription level and thus PC differentiation.
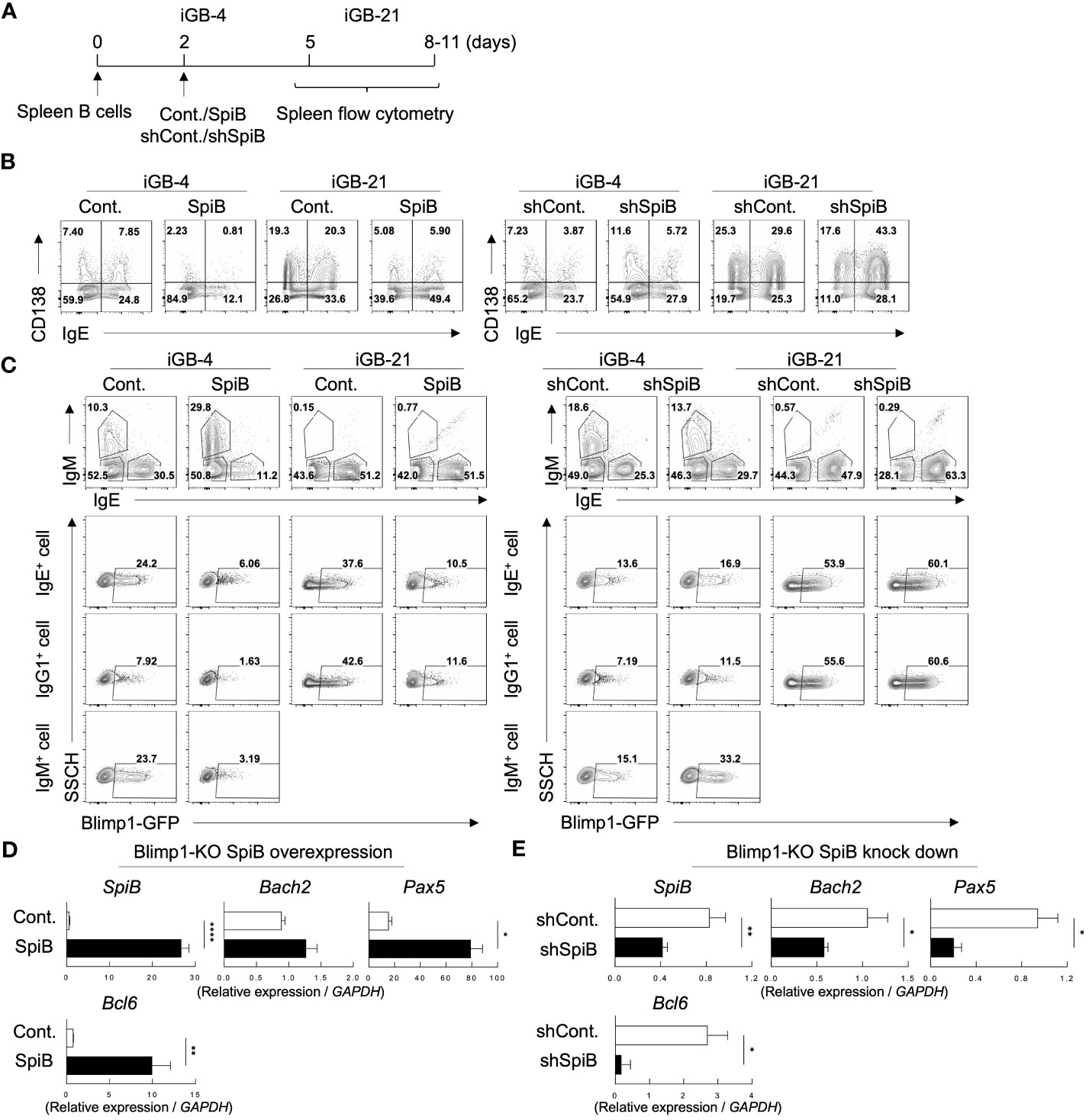
Figure 2 SpiB suppresses PC differentiation and Blimp1 expression while upregulates Bach2 expression in vitro. (A) Experimental protocol for retroviral overexpression and knockdown of SpiB in iGB cells. Spleen B cells were cultured on 40LB feeder cells with IL-4 for 5 days (IGB-4), to which the overexpression or knockdown retroviral vectors were introduced on day 2, then followed by the culture with IL-21 for 3- 5 days (iGB-21). (B) FCM analysis of CD138 and IgE expression on SpiB-overexpressing (left) or SpiB-knockdown (right) IGB-4 and iGB-21 cells. (C) FCM analysis of Blimp1-GFP expression in SpiB-overexpressing (left) or SpiB-knockdown (right) IGB-4 and iGB-21 cells. IgE+, IgM+ and IgE- IgM- (mostly IgG1+) populations were gated on iGB-4 and iGB-21 cells (top panels) and the expression of GFP in each population (bottom) were analyzed by FCM (bottom). (D, E) qRT-PCR analysis of mRNA expression of B cell-related genes in SpiB-overexpressng (D) and SpiB-knockdown (E) Blimp1-KO iGB-4 cells. *p < 0.05, **p < 0.01, ****p < 0.0001.
We further analyzed the mRNA expression levels of B-cell-related genes in the iGB-4 cells over-expressing SpiB and confirmed that the expression of Prdm1 was suppressed by SpiB (Supplementary Figure 3B). Of note, SpiB overexpression resulted in upregulation of the expression of Bach2, Pax5, and Bcl6, which encode transcription factors known to reciprocally suppress the expression of Blimp1 (6, 29–31) (Supplementary Figure 3B). Blimp1 is known to suppress several B cell-related genes to positively regulate PC differentiation (32–34). To clarify whether the SpiB-mediated upregulation of Bach2, Pax5, and Bcl6 is independent of Blimp1, we used B cells from B-cell-specific Blimp1-deficient mice (Blimp1f/f CD19-cre; here called Blimp1-KO mice) for iGB cell culture. Overexpression of SpiB in the Blimp1-KO iGB-4 cells resulted in an increase of the expression of Bach2, Pax5, and Bcl6, whereas knockdown of SpiB resulted in the opposite outcome (Figures 2D, E). These results suggest that SpiB functions to suppress the expression of Blimp1 and independently induce Bach2, Pax5, and Bcl6 in B cells.
The above results indicating that SpiB suppresses the expression of Blimp1 while inducing the expression of Bach2, suggesting that SpiB functions to generate or maintain Bmem cells. To address this possibility, we applied our system in which iGB-4 cells differentiate to memory-like B (iMB) cells in the recipient mice (7). We retrovirally overexpressed or knocked down SpiB in iGB-4 cells and transferred these cells to irradiated mice respectively to generate iMB cells (Figure 3A). SpiB overexpression resulted in an increase in the frequency of the iMB cells (Figure 3B). On the contrary, SpiB knockdown resulted in the reduced frequency of the iMB cells (Figure 3C). These data indicated that SpiB promoted the generation or maintenance of iMB cells in a dose-dependent manner. These results strongly suggest that SpiB is a factor that regulates the generation or maintenance of Bmem cells.
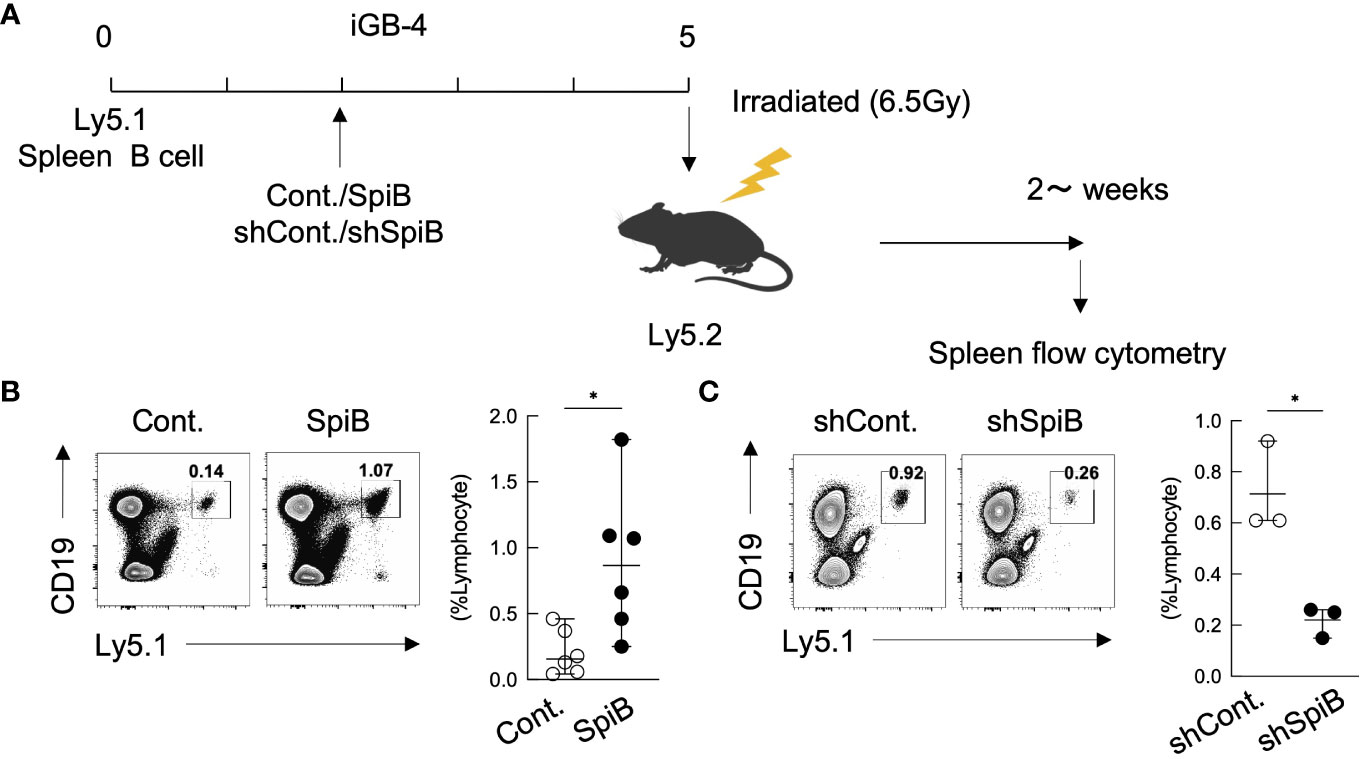
Figure 3 SpiB is critical for Bmem cell generation from iGB cells in vivo. (A) Experimental protocol for the generation of iMB cells from gene-transduced iGB-4 cells. iGB-4 cells derived from Ly5.1 congenic C57BL/6 (B6) mice were transduced with retroviral vectors expressing SpiB or shSpiB, or empty vectors as a control (cont.). These iGB-4 cells on day 5 were adaptively transferred to irradiated Ly5.2 congenic B6 mice. Spleen cells were analyzed 4 weeks later (for SpiB overexpression) or 2 weeks later (for SpiB knockdown) by FCM. (B, C) Frequencies of CD19+ Ly5.1+ iMB cells in the spleens of the recipient mice. A representative FCM data (left) and the frequencies among lymphocytes (right) [(B): n = 6; (C): n = 3]. *p < 0.05.
To confirm this, we next analyzed the generation of Bmem cells from SpiB-deficient B cells. Since SpiB null-knockout mice were previously reported to have defective GCs (15), we analyzed SpiB-floxed (SpiBf/f) mice (see Materials and Methods 2.2) crossed with Cγ1-cre mice expressing cre recombinase only in B cells class-switched to IgG1 (20) (here called ‘SpiB cKO’ mice). SpiB+/+ Cγ1-cre (called ‘SpiB WT’) mice were used as controls. The deletion of SpiB was confirmed in IgG1+ B cells in the immunized mice and in iGB cells (Supplementary Figures 4A–E). We first examined the iMB cell generation from SpiB cKO iGB-4 cells in vivo. As expected from the result of SpiB-knockdown experiments (Figure 3C), very few IgG1+ iMB cells were generated from SpiB cKO iGB-4 cells (Figure 4A).
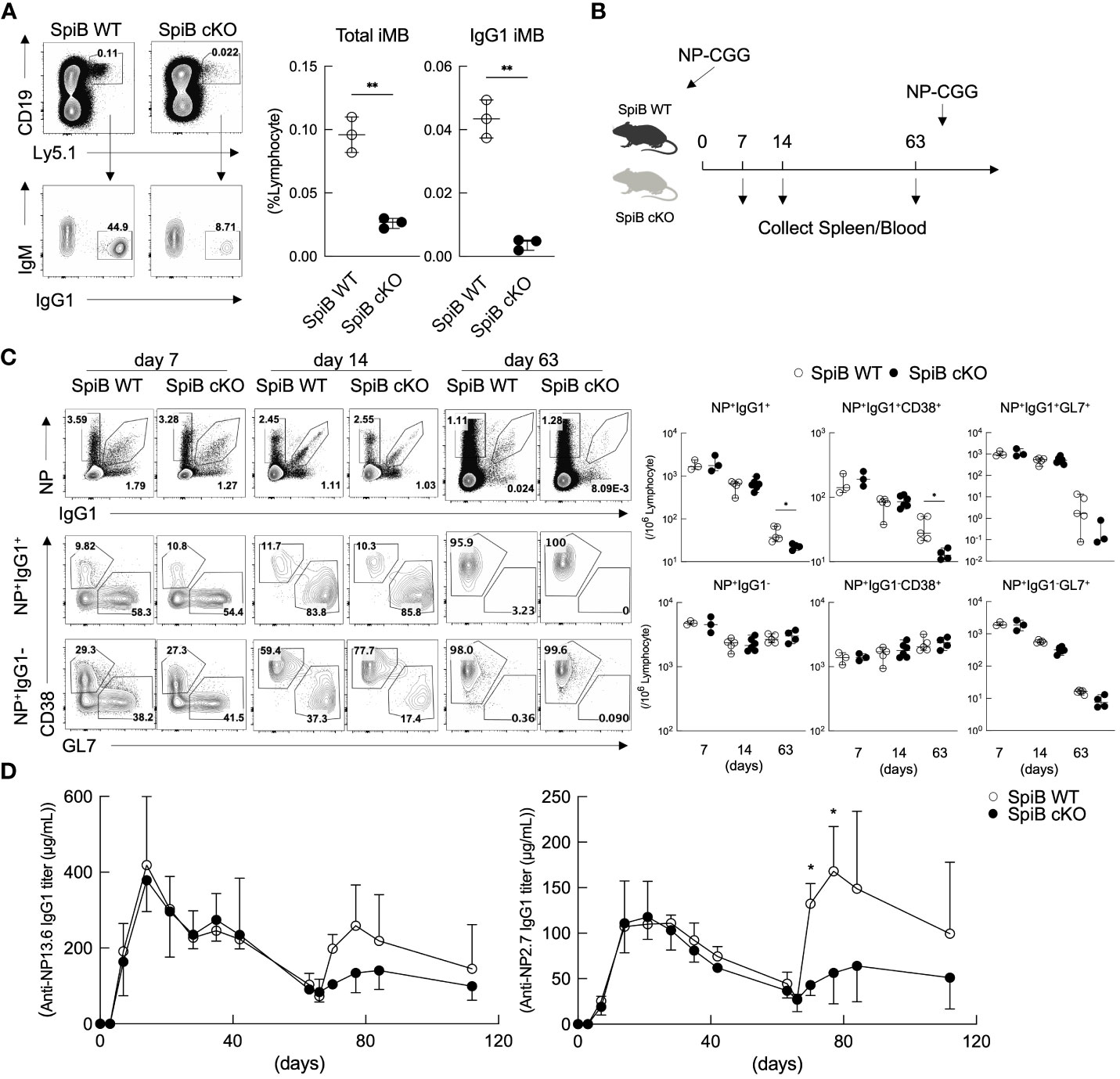
Figure 4 Long-lived Bmem cells and memory antibody response are defective in SpiB cKO mice. (A) Frequencies of CD19+ Ly5.1+ (total iMB) cells and CD19+ Ly5.1+ IgG1+ iMB cells in the spleens of the recipient mice transferred with iGB-4 cells derived from SpiB WT or SpiB cKO mouse B cells 4 weeks earlier, analyzed by FCM. A representative FCM data (left) and the frequencies among lymphocytes (right) (N =3). (B) A protocol of a mice immunization experiment. SpiB WT and SpiB cKO mice were intraperitoneally immunized with NP-CGG in alum and boosted with NP-GCC without alum to induce memory response at 66 days after the primary immunization. Spleen cells and blood were collected before immunization and at the indicated timepoints (spleen) and as indicated in (D) (blood). (C) Frequencies of NP+ IgG1+ or NP+ IgG1-, total B, Bmem and GC B cells in the spleens at the indicated time points after NP-CGG immunization in SpiB WT or SpiB cKO mice. Spleen cells were gated on B220+ Igκlow, NP+ IgG1+ or NP+ IgG1- cells before gating on Bmem (GL7- CD38+) or GC B (GL7+ CD38-) cells. A representative FCM data (left) and the frequencies among lymphocytes of three to six mice (right) are shown. (D) Concentrations of serum antibodies of total anti-NP-IgG1 (left) and high-affinity anti-NP-IgG1 (right) from SpiB WT or SpiB cKO mice primarily immunized with NP-CGG in alum and secondarily with NP-CGG alone on day 66. (n = 4) *p < 0.05, **p < 0.01.
Next, we analyzed T cell-dependent (TD) immune response in the SpiB cKO mice by immunizing the mice with a typical TD antigen, NP-CGG, precipitated in alum. On day 7, 14, and 63 after the primary immunization, we analyzed GC B cells (GL7+ CD38–) and Bmem cells (GL7– CD38+) within NP-specific IgG1+ and IgG1– B cells by FCM (Figure 4B). The frequency and the absolute number of GC B and Bmem cells did not significantly differ between SpiB WT and SpiB cKO mice in both NP+ IgG1+ and NP+ IgG1– B cell populations at 7- and 14-days post-immunization (Figure 4C). The concentration of blood anti-NP-IgG1 antibody was also similar between the two strains during the primary immune response (Figure 4D). On day 63 post-immunization, when GC B cells almost disappeared, the number of Bmem cells in the NP+ IgG1+ population, but not in the NP+ IgG1– population, was significantly reduced in SpiB cKO mice as compared to SpiB WT mice. We further analyzed the memory antibody response in these mice to determine the effect of the reduction of IgG1+ Bmem cells. After re-immunization of the mice with NP-CGG without adjuvants, the generation of serum anti-NP IgG1 antibodies was attenuated in SpiB cKO mice during the secondary immune response, which was highlighted when we detected only high-affinity antibodies that are mostly produced from Bmem cells (Figure 4D). These results suggest that SpiB functions to maintain, rather than to induce, the antigen-specific Bmem cells.
It is known that Bach2 functions as a suppressor for Blimp1 and regulates Bmem cell differentiation (6, 35). Our in-vitro analysis demonstrated that SpiB functions to suppress PC differentiation by suppressing Blimp1 expression while inducing the expression of Bach2 (Figures 2C–E). To elucidate the mechanism of SpiB function in B cells, we focused on the relationship of SpiB and Bach2. We first analyzed expression of SpiB and CD138 in the iGB-4 cells derived from splenic B cells of normal B6 (WT), Blimp1-KO, Bach2-KO, and Blimp1/Bach2-double KO mice by FCM. As expected, CD138+ cells were induced in Bach2 KO iGB cells. However, the induction of CD138+ cells was strongly suppressed in Blimp1/Bach2 double KO iGB-4 cells irrespective of their BCR isotype, indicating that Bach2 suppresses PC differentiation through Blimp1 suppression (Figure 5A; Supplementary Figure 5A).
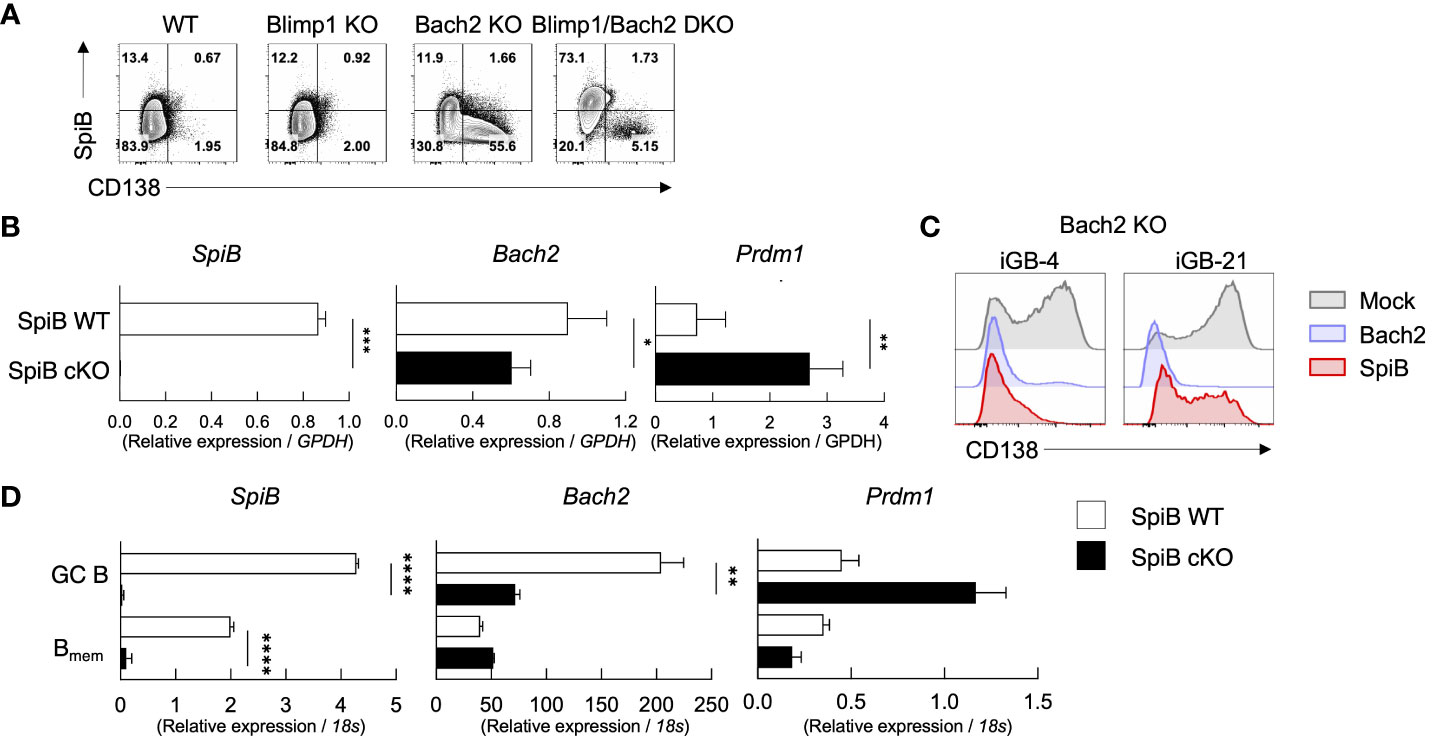
Figure 5 SpiB suppresses PC differentiation independently of Bach2. (A) Expression of intracellular SpiB and cell-surface CD138 in SpiB WT, Blimp1-KO, Bach2-KO and Blimp1/Bach2 DKO iGB-4 cells analyzed by FCM. (B) mRNA expression of SpiB, Bach2, and Prdm1 in SpiB WT and SpiB cKO iGB-4 cells analyzed by qRT-PCR. (C) FCM analysis of CD138 expression on Bach2-KO iGB-4 (left) and iGB-21 (right) cells transduced with mock-, Bach2-, SpiB-expressing retroviral vectors. (D) qRT-PCR analysis for mRNA expression of SpiB, Bach2, and Prdm1 in IgG1+ GC B and Bmem cells from spleens of SpiB WT and SpiB cKO mice immunized with NP-CGG in alum 14 days earlier. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We next analyzed the expression of Bach2 and Prdm1 along with other B cell-related genes, such as Bcl6, Pax5 and IRF4, in SpiB cKO and SpiB WT iGB-4 cells. SpiB-deficiency resulted in decrease of the expression of Bach2 while increase of Prdm1 and IRF4 (Figure 5B, Supplementary Figure 5B). To verify the functional meaning of the SpiB-regulated expression of Bach2 and Blimp1, we retrovirally transduced SpiB and Bach2 into Bach2 KO iGB cells. The frequency of CD138+ cells was strongly suppressed by SpiB overexpression in Bach-2 KO iGB-4 cells as observed by Bach2 reconstitution, and to a lesser extent in iGB-21 cells, indicating that SpiB can suppress PC differentiation independently of Bach2 (Figure 5C). To further analyze the relation between Bach2 and SpiB in vivo, we sorted IgG1+ GC and early Bmem cells from SpiB WT and SpiB cKO mice 14 days after immunization with NP-CGG in alum, and analyzed the expression of Bach2, Prdm1 and other B cell-related genes. In GC B cells, the expression of Bach2 was decreased while that of Prdm1 was increased by the SpiB-deficiency, with other B cell-related genes largely unaffected, resembling the result with iGB-4 cells. In the early Bmem cells, however, the expression of Bach2 and Prdm1, as well as of other genes, was not apparently affected by the SpiB-deficiency (Figure 5D; Supplementary Figure 5C). These results indicate that SpiB upregulates Bach2 expression and suppresses Blimp1 expression and PC differentiation independently of Bach2 in GC B cells, which may render the longevity to the descendant Bmem cells.
Since the phenotype of SpiB cKO mice in the TD immune response was apparent only in the late Bmem cells and their recall response, we suspected the function of SpiB to be more related to the maintenance of Bmem cells rather than their generation. Previous studies have shown that autophagy is one of the mechanisms to inhibit apoptosis and to support the survival of memory T and B cells (36–39). Therefore, we analyzed the expression levels of anti-apoptotic genes and autophagy genes in GC B and Bmem cells sorted from SpiB cKO and SpiB WT mice 14 days after immunization with NP-CGG. As a result, SpiB cKO GC B cells express lower levels of anti-apoptotic genes (Bcl-XL, Bcl2, Mcl1) and some autophagy genes (Atg5, Atg6, Atg7) as compared to SpiB WT GC B cells and the difference was more profound in Bmem cells, except for Bcl2 (Figures 6A, B).
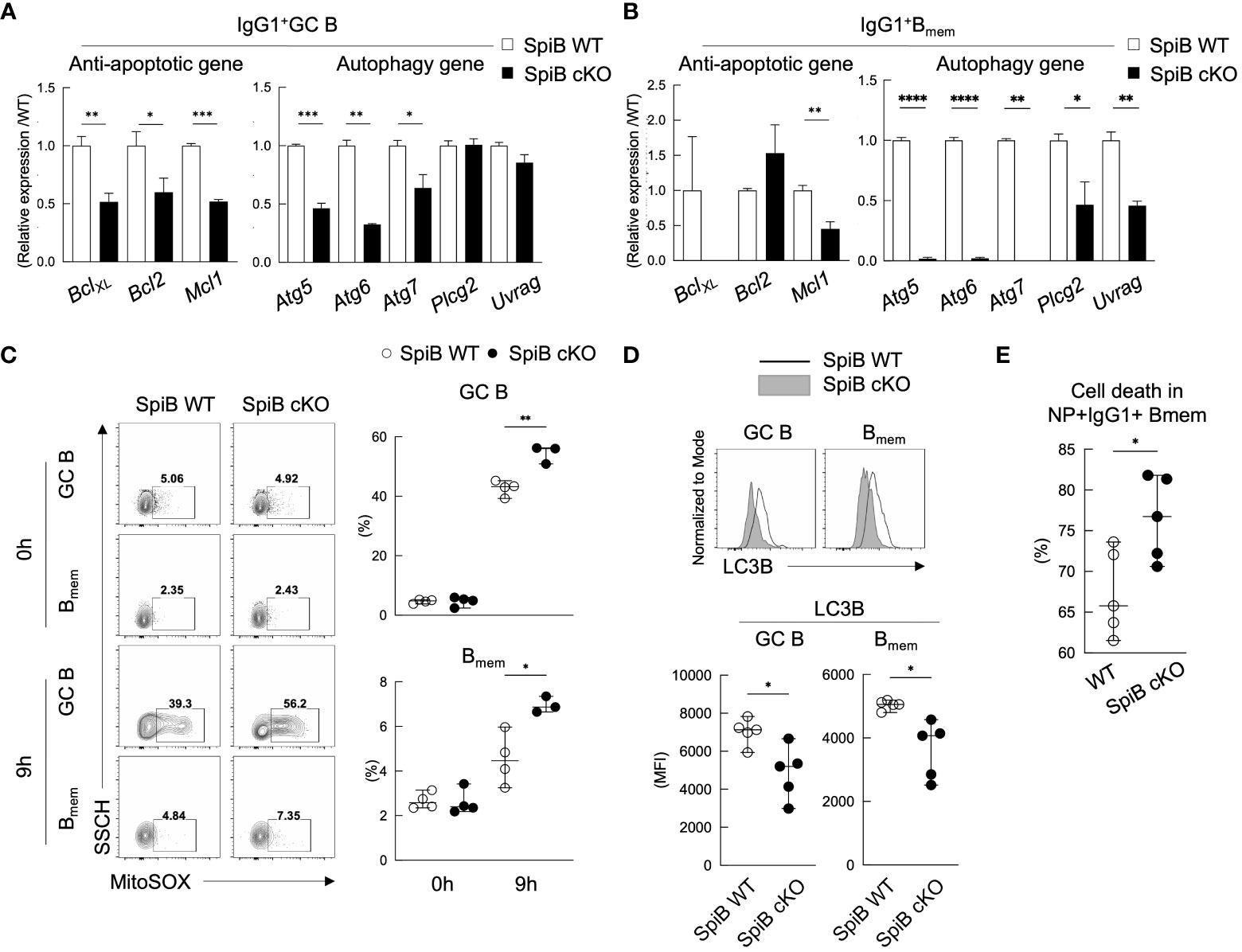
Figure 6 SpiB-deficient GC B and Bmem cells express lower levels of anti-apoptotic and autophagy genes and are more prone to die. (A, B) qRT-PCR analysis for mRNA expression of indicated anti-apoptotic genes (left) and autophagy genes (right) in IgG1+ GC B cells (A) and IgG1+ Bmem cells (B) from spleens of SpiB WT and SpiB cKO mice immunized with NP-CGG in alum 14 days earlier. (C) FCM analysis for MitoSOX staining of the same cells as in (A, B) Cells were incubated for 0 or 9 hours in non-FCS medium before staining. Representative FCM data (left) and the frequencies of MitoSOX-positive cells (n = 3 or 4) (right). (D) FCM analysis for the expression of LC3B in the same cells as in (A, B) Representative FCM data (top) and mean fluorescence intensity (MFI) of the LC3B stained cells (bottom) are shown (n = 5). (E) Frequencies of dead cells in NP+ IgG1+ CD38+ Bmem cells from spleens of SpiB WT and SpiB cKO mice immunized with NP-CGG in alum 14 days earlier. Percentages of the dead cells detected microscopically following Trypan Blue staining after 9 hours’ culture in non-FCS medium as compared to those before the culture (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Autophagy is known to be necessary for the clearance of dysfunctional mitochondria that produce mitochondrial reactive oxygen species (mROS) (40) which is one of the factors to induce apoptosis (41). Given the almost undetectable levels of the autophagy genes, Atg5, Atg6, Atg7, in SpiB cKO Bmem cells, we assessed mROS production in GC B and Bmem cells sorted from immunized SpiB WT and SpiB cKO mice by staining them with MitoSox and analyzing by FCM. MitosSox positive cell population was small and similar between SpiB WT and SpiB cKO GC B and Bmem cells before culture (0 h), but the frequency of MitoSox-positive cells increased after 9 hours’ culture more abundantly in SpiB cKO GC B and Bmem cells than SpiB WT ones (Figure 6C). We next addressed autophagy activity in SpiB cKO GC B and Bmem cells by evaluating the expression of a known autophagy marker LC3B (42–44). When compared with those from SpiB WT mice, GC B and Bmem cells from SpiB cKO mice showed reduced expression of LC3B, indicating lower activity of autophagy in the absence of SpiB in these cells (Figure 6D). Finally, we found that IgG1+ Bmem cells in the immunized SpiB cKO mice contained more dead cells than those in the immunized SpiB WT mice after 9-hour incubation in the plain medium (Figure 6E). Taken together, these results indicate that the lack of SpiB in GC B and Bmem cells results in reduction of gene expression and activity for autophagy, leading to the accumulation of mROS and increased susceptibility to apoptosis.
Having been faced with the global pandemic of SARS-CoV-2, fundamental regulation of Bmem cell survival after vaccination is gathering more attention than ever. How Bmem cells behave after the re-exposure to the antigen has recently becoming clearer. For example, Bmem cell subsets characterized by BCR isotypes as well as the expression of CD80/PD-L2 were shown to behave differently in the recall response in both human and mice (45, 46). However, mechanisms of Bmem cell generation and maintenance are largely unknown. While it was reported that Bach2 is required for the generation of Bmem cells, Bach2 was expressed in majority of GC B cells as well as naive B cells (6). Thus, the specific function of Bach2 in the Bmem cell development remains obscure.
To tackle this difficult problem, we used our original in-vitro-induced GC B (iGB) cell culture and induced memory B (iMB) cell system for the functional analysis of transcription factors by retroviral overexpression and knockdown. In this study, we focused on transcription factors whose expression levels were higher in the Bmem-like iMB cells than naive B cells, and identified SpiB that acts as a key factor for the maintenance of Bmem cells. Overexpression of SpiB suppressed differentiation of CD138+ PCs by inhibiting the expression of Blimp1, while knockdown of SpiB facilitated the PC differentiation, in the iGB cells in vitro. We further assessed in-vivo development of iMB cells from these manipulated iGB cells. Overexpression of SpiB in iGB cells increased the frequency of iMB cells, while knockdown of SpiB decreased it. These results indicated the role of SpiB in Bmem cell generation and/or maintenance well before generation and analysis of SpiB KO mice, demonstrating the iGB-iMB system as a powerful and easier tool to investigate the molecular mechanisms for GC B cell differentiation in vivo. In accord with this result, the number of long-lived Bmem cells was significantly reduced in SpiB cKO mice, consistent with the supposed role of SpiB in Bmem cell maintenance.
The mechanism of the maintenance of Bmem still remains unclear. It has been supposed that anti-apoptotic protein Bcl2 promotes survival of Bmem cells mainly from the results showing increased number of Bmem cells in Bcl2-transgenic mice. However, it has been reported that transgenic Bcl2 inhibits apoptosis and promotes proliferation of GC B cells, resulting in an increased number of Bmem cells (47, 48). Therefore, the contribution of Bcl2 to Bmem cell survival will remain unclear until the Bmem-cell-specific Bcl2-knockout mice become available. Our data demonstrated that the expression of Bcl2 gene was not reduced in the SpiB-deficient Bmem cells being affected in their long-liveness, instead the expression of BclxL gene was almost abrogated. Although BclxL was reported to be dispensable for development of GC B and Bmem cells (49), BclxL may play a redundant role in collaboration with other factors that are regulated by SpiB.
It has been reported that autophagy protein Atg7 regulates Bmem cell survival (39, 50, 51) and that the Atg7-dependent autophagy is independent of Bcl2-mediated anti-apoptosis pathway (39, 51). We found that the expression of autophagy genes, Atg5, Atg6, and Atg7 was markedly reduced in the SpiB-deficient Bmem cells, which exhibited a phenotype with diminished autophagy activity, increased mitochondrial ROS, and augmented cell death. Thus, attenuated mitophagy resulting in the over-production of ROS, together with the abolished BclxL expression, may render Bmem cells prone to die earlier in SpiB-KO mice.
Direct binding and regulation by SpiB of its possible target genes in GC B cells remain to be determined, for example, by ChIP-Seq analysis. Regarding this, it was reported that PU.1 and SpiB share some binding targets in B cell lymphoma, suggesting that they may work together as co-factors to regulate target genes in GC B cells (52, 53). In mice lacking both PU.1 and SpiB in B cells, the number of follicular B cells was decreased and the GC response was abrogated, while these defects were ameliorated in mice lacking PU.1 alone in B cells, and not apparent in mice lacking SpiB alone (53). This result suggests that PU.1 is mainly responsible for the generation of follicular and GC B cells, but SpiB compensates this function to some extent when PU.1 is absent. Thus, SpiB does not seem to be responsible for these functions but for the maintenance of Bmem cells under normal conditions.
In conclusion, we found that SpiB plays a critical role in the maintenance of Bmem cells. Further studies are needed to fully understand the mechanisms underlying SpiB-mediated Bmem cell maintenance and the potential therapeutic implications of this finding. Investigating of the detailed function of SpiB and its relationship with genes involved in metabolism, will provide a better understanding of the regulation of Bmem cells, ultimately leading to the development of vaccines that can generate longer-lasting humoral memory.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Animal Care and Use Committee of Tokyo University of Science. The study was conducted in accordance with the local legislation and institutional requirements.
DK supervised and funded the project. SH designed and performed the experiments. TKo and HT contributed to conducting the experiments. KH, IS, YF-O and TKa were involved in methodology, investigation, and writing of the original draft, on SpiB cKO mice. SH and DK wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas from MEXT, Japan (to DK, JP25118730).
We thank Dr. Alexander Tarakhovsky (Rockefeller University) for Blimp1f/f mice, Dr. Mitinori Saitou (Kyoto University) for Blimp1-GFP mice, Drs. Akihiko Muto and Kazuhiko Igarashi (Tohoku University) for Bach2-/- mice, and Dr. Klaus Rajewsky (Max-Delbrück Center for Molecular Medicine) for Cγ1-cre and CD19-cre mice. We thank Evan Rossignol for proofreading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1250719/full#supplementary-material
1. Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol (2021) 21(4):209–20. doi: 10.1038/s41577-020-00446-2
2. Young C, Brink R. The unique biology of germinal center B cells. Immunity (2021) 54(8):1652–64. doi: 10.1016/j.immuni.2021.07.015
3. Inoue T, Shinnakasu R, Kurosaki T. Generation of high quality memory B cells. Front Immunol (2021) 12:825813. doi: 10.3389/fimmu.2021.825813
4. Kitamura D. Mechanisms for the regulation of memory B-cell recall responses in mice. Int Immunol (2021) 33(12):791–6. doi: 10.1093/intimm/dxab042
5. Takatsuka S, Yamada H, Haniuda K, Saruwatari H, Ichihashi M, Renauld JC, et al. IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nat Immunol (2018) 19(9):1025–34. doi: 10.1038/s41590-018-0177-0
6. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol (2016) 17(7):861–9. doi: 10.1038/ni.3460
7. Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun (2011) 2:465. doi: 10.1038/ncomms1475
8. Su GH, Ip HS, Cobb BS, Lu MM, Chen HM, Simon MC. The Ets protein Spi-B is expressed exclusively in B cells and T cells during development. J Exp Med (1996) 184(1):203–14. doi: 10.1084/jem.184.1.203
9. Schotte R, Rissoan MC, Bendriss-Vermare N, Bridon JM, Duhen T, Weijer K, et al. The transcription factor Spi-B is expressed in plasmacytoid DC precursors and inhibits T-, B-, and NK-cell development. Blood (2003) 101(3):1015–23. doi: 10.1182/blood-2002-02-0438
10. Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I, et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol (2012) 13(8):729–36. doi: 10.1038/ni.2352
11. DeKoter RP, Geadah M, Khoosal S, Xu LS, Thillainadesan G, Torchia J, et al. Regulation of follicular B cell differentiation by the related E26 transformation-specific transcription factors PU.1, Spi-B, and Spi-C. J Immunol (2010) 185(12):7374–84. doi: 10.4049/jimmunol.1001413
12. Batista CR, Li SK, Xu LS, Solomon LA, DeKoter RP. PU.1 regulates ig light chain transcription and rearrangement in pre-B cells during B cell development. J Immunol (2017) 198(4):1565–74. doi: 10.4049/jimmunol.1601709
13. Garrett-Sinha LA, Su GH, Rao S, Kabak S, Hao Z, Clark MR, et al. PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity (1999) 10(4):399–408. doi: 10.1016/s1074-7613(00)80040-0
14. Hu CJ, Rao S, Ramirez-Bergeron DL, Garrett-Sinha LA, Gerondakis S, Clark MR, et al. PU.1/Spi-B regulation of c-rel is essential for mature B cell survival. Immunity (2001) 15(4):545–55. doi: 10.1016/s1074-7613(01)00219-9
15. Su GH, Chen HM, Muthusamy N, Garrett-Sinha LA, Baunoch D, Tenen DG, et al. Defective B cell receptor-mediated responses in mice lacking the Ets protein, Spi-B. EMBO J (1997) 16(23):7118–29. doi: 10.1093/emboj/16.23.7118
16. Laramee AS, Raczkowski H, Shao P, Batista C, Shukla D, Xu L, et al. Opposing roles for the related ETS-family transcription factors Spi-B and Spi-C in regulating B cell differentiation and function. Front Immunol (2020) 11:841. doi: 10.3389/fimmu.2020.00841
17. Schmidlin H, Diehl SA, Nagasawa M, Scheeren FA, Schotte R, Uittenbogaart CH, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood (2008) 112(5):1804–12. doi: 10.1182/blood-2008-01-136440
18. Ohinata Y, Payer B, O'Carroll D, Ancelin K, Ono Y, Sano M, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature (2005) 436(7048):207–13. doi: 10.1038/nature03813
19. Muto A, Tashiro S, Nakajima O, Hoshino H, Takahashi S, Sakoda E, et al. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature (2004) 429(6991):566–71. doi: 10.1038/nature02596
20. Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci U S A. (2006) 103(19):7396–401. doi: 10.1073/pnas.0602353103
21. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature (1995) 376(6538):352–5. doi: 10.1038/376352a0
22. Koike T, Harada K, Horiuchi S, Kitamura D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. Elife (2019) 8. doi: 10.7554/eLife.44245
23. Bartholdy B, Du Roure C, Bordon A, Emslie D, Corcoran LM, Matthias P. The Ets factor Spi-B is a direct critical target of the coactivator OBF-1. Proc Natl Acad Sci U S A. (2006) 103(31):11665–70. doi: 10.1073/pnas.0509430103
24. Song S, Cao C, Choukrallah MA, Tang F, Christofori G, Kohler H, et al. OBF1 and Oct factors control the germinal center transcriptional program. Blood (2021) 137(21):2920–34. doi: 10.1182/blood.2020010175
25. Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity (2002) 17(1):51–62. doi: 10.1016/S1074-7613(02)00335-7
26. Yang Z, Sullivan BM, Allen CD. Fluorescent in vivo detection reveals that IgE(+) B cells are restrained by an intrinsic cell fate predisposition. Immunity (2012) 36(5):857–72. doi: 10.1016/j.immuni.2012.02.009
27. Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D. Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol (2016) 17(9):1109–17. doi: 10.1038/ni.3508
28. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity (2003) 19(4):607–20. doi: 10.1016/S1074-7613(03)00267-X
29. Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity (2006) 24(3):269–81. doi: 10.1016/j.immuni.2006.01.012
30. Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol (2016) 17(3):331–43. doi: 10.1038/ni.3349
31. Vasanwala FH, Kusam S, Toney LM, Dent AL. Repression of AP-1 function: a mechanism for the regulation of Blimp-1 expression and B lymphocyte differentiation by the B cell lymphoma-6 protooncogene. J Immunol (2002) 169(4):1922–9. doi: 10.4049/jimmunol.169.4.1922
32. Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol (2002) 22(13):4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002
33. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamica control of IRF4. Immunity (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
34. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol (2011) 23(5):341–9. doi: 10.1016/j.smim.2011.08.010
35. Ochiai K, Katoh Y, Ikura T, Hoshikawa Y, Noda T, Karasuyama H, et al. Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. J Biol Chem (2006) 281(50):38226–34. doi: 10.1074/jbc.M607592200
36. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi: 10.1038/ni.3025
37. Murera D, Arbogast F, Arnold J, Bouis D, Muller S, Gros F. CD4 T cell autophagy is integral to memory maintenance. Sci Rep (2018) 8(1):5951. doi: 10.1038/s41598-018-23993-0
38. Watanabe R, Fujii H, Shirai T, Saito S, Ishii T, Harigae H. Autophagy plays a protective role as an anti-oxidant system in human T cells and represents a novel strategy for induction of T-cell apoptosis. Eur J Immunol (2014) 44(8):2508–20. doi: 10.1002/eji.201344248
39. Kodali S, Li M, Budai MM, Chen M, Wang J. Protection of quiescence and longevity of IgG memory B cells by mitochondrial autophagy. J Immunol (2022) 208(5):1085–98. doi: 10.4049/jimmunol.2100969
40. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (2011) 333(6046):1109–12. doi: 10.1126/science.1201940
41. Gottlieb E, Vander Heiden MG, Thompson CB. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol (2000) 20(15):5680–9. doi: 10.1128/MCB.20.15.5680-5689.2000
42. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy (2005) 1(2):84–91. doi: 10.4161/auto.1.2.1697
43. Watanabe K, Ichinose S, Hayashizaki K, Tsubata T. Induction of autophagy by B cell antigen receptor stimulation and its inhibition by costimulation. Biochem Biophys Res Commun (2008) 374(2):274–81. doi: 10.1016/j.bbrc.2008.07.013
44. He C, Wang S, Zhou C, He M, Wang J, Ladds M, et al. CD36 and LC3B initiated autophagy in B cells regulates the humoral immune response. Autophagy (2021) 17(11):3577–91. doi: 10.1080/15548627.2021.1885183
45. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol (2014) 15(7):631–7. doi: 10.1038/ni.2914
46. Weisel NM, Joachim SM, Smita S, Callahan D, Elsner RA, Conter LJ, et al. Surface phenotypes of naive and memory B cells in mouse and human tissues. Nat Immunol (2022) 23(1):135–45. doi: 10.1038/s41590-021-01078-x
47. Smith KG, Light A, O'Reilly LA, Ang SM, Strasser A, Tarlinton D. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J Exp Med (2000) 191(3):475–84. doi: 10.1084/jem.191.3.475
48. Egle A, Harris AW, Bath ML, O'Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood (2004) 103(6):2276–83. doi: 10.1182/blood-2003-07-2469
49. Vikstrom I, Carotta S, Luthje K, Peperzak V, Jost PJ, Glaser S, et al. Mcl-1 is essential for germinal center formation and B cell memory. Science (2010) 330(6007):1095–9. doi: 10.1126/science.1191793
50. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med (2014) 20(5):503–10. doi: 10.1038/nm.3521
51. Chen M, Kodali S, Jang A, Kuai L, Wang J. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol (2015) 194(6):2607–15. doi: 10.4049/jimmunol.1403001
52. Solomon LA, Li SK, Piskorz J, Xu LS, DeKoter RP. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genomics (2015) 16(1):76. doi: 10.1186/s12864-015-1303-0
Keywords: memory B cell, germinal center B cell, SpiB, apoptosis, autophagy, humoral immune response, SpiB conditional knockout mice
Citation: Horiuchi S, Koike T, Takebuchi H, Hoshino K, Sasaki I, Fukuda-Ohta Y, Kaisho T and Kitamura D (2023) SpiB regulates the expression of B-cell-related genes and increases the longevity of memory B cells. Front. Immunol. 14:1250719. doi: 10.3389/fimmu.2023.1250719
Received: 30 June 2023; Accepted: 18 September 2023;
Published: 27 October 2023.
Edited by:
Rinako Nakagawa, The Francis Crick Institute, United KingdomReviewed by:
Mauro Gaya, INSERM U1104 Centre d’immunologie de Marseille-Luminy (CIML), FranceCopyright © 2023 Horiuchi, Koike, Takebuchi, Hoshino, Sasaki, Fukuda-Ohta, Kaisho and Kitamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisuke Kitamura, a2l0YW11cmFAcnMudHVzLmFjLmpw
†Present addresses: Shu Horiuchi, CSL, Waltham, MA, United States
Takuya Koike, Center for Infectious Disease Education and Research, Osaka University, Osaka, Japan
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.