 Katarzyna Sarad1,2
Katarzyna Sarad1,2 Monika Stefańska3
Monika Stefańska3 Izabela Kraszewska1
Izabela Kraszewska1 Krzysztof Szade4
Krzysztof Szade4 Judith C. Sluimer5,6
Judith C. Sluimer5,6 Przemysław Błyszczuk3,7
Przemysław Błyszczuk3,7 Józef Dulak1
Józef Dulak1 Agnieszka Jaźwa-Kusior1*
Agnieszka Jaźwa-Kusior1*- 1Department of Medical Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków, Poland
- 2Jagiellonian University, Doctoral School of Exact and Natural Sciences, Kraków, Poland
- 3Department of Clinical Immunology, Jagiellonian University Medical College, Kraków, Poland
- 4Laboratory of Stem Cell Biology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków, Poland
- 5Cardiovascular Research Institute Maastricht (CARIM), Department of Pathology, Maastricht University Medical Center (UMC), Maastricht, Netherlands
- 6BHF Centre for Cardiovascular Science, University of Edinburgh, Edinburgh, United Kingdom
- 7Department of Rheumatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcriptional regulator of antioxidant and anti-inflammatory response in all cell types. It also activates the transcription of genes important for macrophage function. Nrf2 activity declines with age and has been closely linked to atherosclerosis, but its specific role in this vascular pathology is not clear. Atherosclerotic plaques contain several macrophage subsets with distinct, yet not completely understood, functions in the lesion development. The aim of this study was to analyze the transcriptome of diverse Nrf2-deficient macrophage subpopulations from murine atherosclerotic aortas. Mice with transcriptionally inactive Nrf2 in Cdh5-expressing cells (Nrf2Cdh5tKO) were used in the experiments. These mice lack transcriptional Nrf2 activity in endothelial cells, but also in a proportion of leukocytes. We confirmed that the bone marrow-derived and tissue-resident macrophages isolated from Nrf2Cdh5tKO mice exhibit a significant decline in Nrf2 activity. Atherosclerosis was induced in Nrf2Cdh5tKO and appropriate control mice via adeno-associated viral vector (AAV)-mediated overexpression of murine proprotein convertase subtilisin/kexin type 9 (Pcsk9) in the liver and high-fat diet feeding. After 21 weeks, live aortic cells were sorted on FACS and single-cell RNA sequencing (scRNA-seq) was performed. Unsupervised clustering singled out 13 distinct aortic cell types. Among macrophages, 9 subclusters were identified. Differential gene expression analysis revealed cell subtype-specific expression patterns. A subset of inflammatory macrophages from atherosclerotic Nrf2Cdh5tKO mice demonstrated downregulation of DNA replication genes (e.g. Mcm7, Lig1, Pola1) concomitant with upregulation of DNA damage sensor Atr gene. Atherosclerotic Nrf2Cdh5tKO Lyve1+ resident macrophages showed strong upregulation of IFN-stimulated genes, as well as changes in the expression of death pathways-associated genes (Slc40a1, Bcl2a1). Furthermore, we observed subtype-specific expression of core ferroptosis genes (e.g. Cp, Hells, Slc40a1) in inflammatory versus tissue resident macrophages. This observation suggested a link between ferroptosis and inflammatory microenvironment appearing at a very early stage of atherogenesis. Our findings indicate that Nrf2 deficiency in aortic macrophages leads to subtype-specific transcriptomic changes associated with inflammation, iron homeostasis, cell injury or death pathways. This may help understanding the role of aging-associated decline of Nrf2 activity and the function of specific macrophage subtypes in atherosclerotic lesion development.
Introduction
Atherosclerosis is an inflammatory disease of the wall of large- and medium-sized arteries. It can begin in childhood with the development of fatty streaks due to an accumulation of lipids in the intimal layer of the artery (1). With time, the fatty streak evolves into established plaque with the thick fibrous cap produced by vascular smooth muscle cells. Then, increasing lipid content and numbers of inflammatory macrophages, enlarged necrotic core and thinner fibrous cap lead to the formation of unstable plaque and sometimes its rupture. That end stage disease is typical for humans, but practically does not occur in mouse models of atherosclerosis (2).
Inflammation and oxidative stress are crucial elements in progression of atherosclerosis. In fact, these two processes are interrelated and form a vicious circle during atherogenesis. Reactive oxygen species induce the expression of inflammatory cytokines, chemokines and soluble mediators of inflammation via activation of various transcription factors (3). In turn, cytokines and chemokines produced by inflammatory cells recruit additional inflammatory cells to the sites of inflammation, what increases oxidative stress and exacerbates this adverse cycle.
Macrophages are the most numerous immune cells in the pathogenesis of atherosclerosis, present through all stage of the disease from lesion initiation to plaque rupture (4). Lipoproteins sequestered by macrophages in the arterial wall undergo various modifications including oxidation and aggregation. These pro-inflammatory particles lead to activation of the overlying endothelial cells and recruitment of monocytes differentiating to macrophages inside the vessel wall (5). However, lesional macrophages accumulate not only through recruitment and differentiation of circulating monocytes, but also via local proliferation, (trans)-differentiation of vascular smooth muscle cells or local progenitors (4). During atherosclerosis, macrophages are exposed to many environmental signals, which modulate their functional phenotypes. For e.g. cholesterol crystals, found not only in advanced plaques, but also at early stages of atherosclerotic lesions, were shown to activate the caspase-1-activating-NLRP3 inflammasome and thus were shown to act as a proinflammatory stimulus in LPS-primed human peripheral blood mononuclear cells in culture and in vivo in mice (6).
Recent research involving the single-cell transcriptomics identified several distinct macrophage clusters within human and mouse atherosclerotic plaques (7–9). These populations may play either beneficial or harmful functions in atherosclerosis, which may depend on the stage of the disease (10, 11). Undeniably, the beneficial role of plaque macrophages, especially at the early stage of the lesion development, results from their ability to scavenge cytotoxic lipoproteins and remove dead cells. But, similarly to other cells in the plaque, macrophages also undergo different forms of cell death. It is currently proposed that targeting the diverse types of macrophage death may affect different stages of atherosclerosis development (12).
Nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor encoded in humans by the NFE2L2 gene, is regarded as a master transcriptional regulator of cellular redox homeostasis. Nrf2 induces the expression of a battery of genes involved in defence against oxidative stress (13). This transcription factor is ubiquitously expressed in vascular cells, where it regulates expression of various atheroprotective enzymes, such as heme oxygenase-1 (14) or peroxiredoxin-1 (15). Moreover, Nrf2 is involved in regulation of iron metabolism. It may affect the labile iron pool via regulation of the expression of ferroportin, an iron transporter (16, 17) and ferritin, an iron storage protein (18). Nrf2 also plays an important role in biosynthesis and degradation of iron-containing heme via regulation of the expression of genes such as ATP binding cassette subfamily B member 6 or ferrochelatase (19), as well as the above-mentioned heme degrading enzyme - heme oxygenase-1 (14). Noteworthy, Nrf2 activity declines with age what may lead to disturbed redox homeostasis, cell senescence and even cell death (18, 20, 21).
The data on the role of Nrf2 in atherosclerosis are contradictory. Nrf2 was shown to confer protection against foam cells formation by regulating the expression of antioxidant proteins and scavenger receptors in bone marrow transplantation model (22). However, a decreased susceptibility to apolipoprotein E (ApoE)-mediated atherosclerotic plaque formation was shown in Nrf2−/−ApoE−/− mice (23, 24). On the other hand, increased atherosclerosis in low-density lipoprotein receptor–deficient (Ldlr–/–) mice was observed following Nrf2−/− bone marrow transplantation (25). Another study demonstrated that the ablation of Nrf2 in the bone marrow-derived cells suppresses atherosclerotic lesion area (26). More recently, global Nrf2 deficiency was shown to promote the signs of plaque instability in Ldlr−/−ApoB100/100 mice via increased inflammation and oxidative stress (27).
The contradictory data on the role of Nrf2 in atherosclerosis may result from different approaches in atherosclerosis induction, the genetic background of mice and/or a combination of systemic and local effects in conventional Nrf2 knockout mice. Thus, it is crucial to investigate the association between Nrf2 signaling and fate of specific vascular cell types in atherosclerosis. In this study we aimed to analyze at the single-cell level the transcriptome of diverse macrophage subpopulations from murine atherosclerotic aortas in the early stages of the disease development. Our data indicate that decreased Nrf2 transcriptional activity affects expression of genes involved in inflammatory pathways, cell proliferation and programmed cell death in a subtype-specific manner.
Materials and methods
Animals
All mouse experiments were carried out in accordance with Directive 2010/63/UE of the European Parliament on the protection of animals used for scientific purposes and approved by the 2nd Institutional Animal Care and Use Committee (IACUC) in Kraków, Poland (approval number 142/2020). The animals were housed in specific pathogen-free (SPF) conditions with water and food available ad libitum. To generate mice with Nrf2 transcriptional knockout in Cdh5-expressing cells (Nrf2Cdh5tKO) and control mice (Nrf2flox/flox), C57BL/6-Nfe2l2tm1.1Sred/SbisJ mice (The Jackson Laboratory, Strain #:025433) were crossed with B6;129-Tg(Cdh5-cre)1Spe/J mice (The Jackson Laboratory, Strain #: 017968). Genotyping of animals was performed by PCR on the DNA isolated from the tail tips.
AAV8-Pcsk9 vector production
Production of the adeno-associated viral vector serotype 8 coding for proprotein convertase subtilisin/kexin type 9 (AAV8-Pcsk9) was performed using CellRoll Roller Bottle system (Integra) in three-plasmid Helper-free system as previously described (25). Briefly, AAV293 cells seeded on collagen-precoated Roller Bottles (Corning) were cultured until 60% confluence and transfected with 130 µg of pHelper (Stratagene), 100 µg of pAAV2/8 (kindly provided by Prof. James Wilson, University of Pennsylvania), and 90 µg of pAAV/D377Y-mPCSK9. The plasmid encoding murine Pcsk9 under the control of liver-dependent promoter (pAAV/D377Y-mPCSK9) was a gift from Jacob Bentzon (Addgene plasmid # 58376; http://n2t.net/addgene:58376; RRID: Addgene_58376). According to Addgene specification, the plasmid possessed an alternate 5’ITR sequence which, in our hands, prevented AAV production. Thus, to replace the alternate 5’ITR with the functional 5’ITR, the pAAV-MCS (Stratagene) and pAAV/D377Y-mPCSK9 were digested with Not1 (New England Biolabs) according to the vendor’s protocol. The fragment encoding functional 5’ITR from pAAV-MCS (backbone) and the fragment with Pcsk9 coding gene from pAAV/D377Y-mPCSK9 (GOI) were excised from 1% agarose gel and purified using Zymoclean Gel DNA Recovery Kit (ZymoResearch). Then the backbone was dephosphorylated and ligated with GOI. The proper modified plasmid sequence was confirmed by restriction analysis and functional tests. AAV293 cell transfection was performed using PEI MAX (2.58 mg/ml, 1 µl per 1 µg of DNA). Then, 72 hours after transfection, the cells were detached and washed with PBS. The cells were suspended in a small volume (2-3 ml) of PBS containing calcium and magnesium and subjected to 3 freeze-thaw cycles by placing them in liquid nitrogen and thawing in a 37°C water bath with vigorous mixing after each cycle. The lysate was incubated with HS nuclease (50 U/ml; MoBiTec) for 1 hour at 37°C. Then lysate was centrifuged (4000 × g, 30 min, 4°C) twice, the supernatant was collected and stored at -20°C. Next, AAV8-Pcsk9 vector was purified by Iodixanol (OptiPrep™, Sigma-Aldrich) gradient ultracentrifugation as previously described (28).
The AAV8-Pcsk9 titer was determined using quantitative PCR (qPCR). Briefly, the DNA was isolated from 5 µl of stock using the phenol-chloroform extraction method. Quantification of genome copies in the sample was performed using TaqMan-quantitative real-time PCR. The reaction was performed using primers recognizing the sequence of promoter (HCR_F: 5’-TGGAGTGCAGTGACACAATC-3’, HCR_R: 5’-AGGCCTGTAATCCCAGTTAC-3’), the custom Taqman probe (HCR_probe: 5’-6-FAM-GTAGCTGGGATTACAAGCATGTGC-BHQ-1-3’) and TaqMan Gene Expression Master Mix (Applied Biosystems). The serial dilutions of linearized modified pAAV/D377Y-mPCSK9 were used to generate a standard curve. The reaction conditions were applied according to the manufacturer’s protocol. The AAV titer was calculated as the number of vector genomes per µl of purified stock vector preparation.
Atherosclerosis model
The experiment started when the mice were 3-month-old. To induce hyperlipidemia and atherosclerosis, male Nrf2Cdh5tKO and Nrf2flox/flox mice received a single tail vein injection of AAV8-Pcsk9 at a dose of 1×1011 vector genomes (vg) per mouse and were subsequently placed on a high-fat diet (35% fat; 60% kcal from fat; ZooLab, DP-1E-60S) for 21 weeks. Normocholesterolemic 3-month-old male Nrf2Cdh5tKO and Nrf2flox/flox control mice received a single tail vein injection of saline and were fed a standard diet. At the end of the experiment, the mice were euthanized by inhalation of an overdose of isoflurane. Next, the blood was collected by cardiac puncture using a syringe containing heparin (10 U/ml) and used for blood cell count analysis (ABC Vet, Horiba ABX) and preparation of plasma. Plasma was obtained by centrifugation (2000 × g, 10 min, 4°C) and stored at -80°C. After blood collection, the mice were perfused with PBS containing 0.5 U/ml heparin and the aortic arch, heart, and liver samples were collected for further analysis. In the western blot analysis of liver samples of both AAV8-Pcsk9-treated atherogenic groups we observed highly efficient depletion of the LDL receptor compared with control animals that did not receive AAV8-Pcsk9 (not shown).
Plasma cholesterol and triglyceride analysis
The concentration of cholesterol of plasma LDL/VLDL lipoproteins was evaluated with HDL and LDL/VLDL Quantitation Kit (Sigma-Aldrich) according to vendor’s protocol. The level of triglycerides was determined with Liquick-Cor TG kit (Cormay, Poland) according vendor’s instruction.
Histological examination
The hearts and brachiocephalic arteries (BCAs) collected from the atherosclerotic Nrf2Cdh5tKO and Nrf2flox/flox mice were embedded in OCT compound. Sequential cross sections (8 µm thick) were fixed in formalin, immersed in 60% isopropyl alcohol, and stained with freshly prepared Oil Red O working solution (0.5% Oil Red O in isopropanol was mixed with distilled water in a ratio 3:2) for 5 min. Then the sections were briefly immersed in 60% isopropyl alcohol solution and subsequently examined under a light microscope.
Isolation and culture of bone marrow-derived macrophages
The healthy 5-month-old Nrf2Cdh5tKO and Nrf2flox/flox mice were euthanized by inhalation of an overdose of isoflurane. The BMDM were obtained according to a protocol described previously (29), with modifications. Briefly, bone marrow cells were collected by flushing the femurs and tibias with sterile physiological saline. The cells were washed with PBS, red blood cells were lysed and the remaining cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS, Biowest), penicillin (100 U/ml) and streptomycin (100 µg/ml; pen-strep, Gibco), and 10 ng/ml of purified human recombinant M-CSF (R&D Systems) for 5 days.
Isolation of Kupffer cells
The Kupffer cells were isolated from the liver of the same animals, which were used for BMDM isolation. Kupffer cells isolation was performed as previously described (28).
RT-qPCR
The cells were lysed in Fenozol (A&A Biotechnology). Total RNA was isolated using Total RNA Mini kit (A&A Biotechnology) and then reverse transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instruction. The qPCR reaction was performed with primers targeting Nrf2 exon 3, Nrf2 exon 5 sequence, Nqo1 gene, Gclm gene, Eef2 reference gene (Nrf2_ex3_F: 5’-CAGAGACATTCCCATTTGTAG-3’, Nrf2_ex3_R: 5’-ATTCGGGAATGGAAAATAGC-3’, Nrf2_ex5_F: 5’-CATTCCCGAATTACAGTGTC-3’, Nrf2_ex5_R: 5’-GGAGATCGATGAGTAAAAATGG-3’, Nqo1_F: 5’-GGTTTGGAGTCCCTGCCATT-3’ Nqo1_R: 5’-GTGGATCCCTTGCAGAGAGT-3’, Gclm_F: 5’-TGGAGTTCCCAAATCAGCCC-3’, Gclm_R: 5’-CAACTCCAAGGACGGAGCAT-3’, Eef2_F: 5’-GACATCACCAAGGGTGTGCAG-3’, Eef2_R: 5’-TCAGCACACTGGCATAGAGGC-3’) and AceQ qPCR SYBR Green Master Mix (Vazyme) in StepOne Plus Real-Time PCR system (Applied Biosystems). All the procedures were performed according to the manufacturer’s instructions. The level of Nrf2 transcriptional activity (exon 5 deletion) was calculated as a relative expression of Nrf2 exon 5 in comparison to Nrf2 exon 3 expression.
Preparation of single cells suspension from mouse aortas
The aortic arches with main branches including the perivascular tissue were isolated from atherosclerotic Nrf2Cdh5tKO and Nrf2flox/flox mice (n=3 per group) and control normocholesterolemic (no AAV transduction and standard diet) Nrf2Cdh5tKO and Nrf2flox/flox mice (n=4 per group). The aortas of each group of mice were pooled, cut into small pieces, and digested in 2.5 ml enzyme mix from Multi Tissue Dissociation Kit 2 (Miltenyi Biotec) prepared as described in the protocol provided by the vendor. The samples were placed at 37°C for 30 min with shaking (250 RPM). The suspension was gently pipetted every 15 min. Then the cells were filtered through a 70 μm strainer, centrifuged at 500 × g for 5 min, and resuspended in red blood cells lysis buffer (BioLegend). After 5 min, the cells were centrifuged at 500 × g for 5 min, washed with PBS, and resuspended in 300 µl of PBS containing 2% FBS and 200 ng/ml DAPI. To exclude dead cells and collect only the viable ones, the DAPI-negative cells were sorted with MoFlo XPD (Beckman Coulter) cell sorter.
Library preparation/data analysis/subclustering/DEG analysis
A total of ~12.000 DAPI-negative cells per sample were loaded on a chromium single cell controller to generate single cell GEMs and then cDNA libraries were prepared using Chromium™ Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 (10x Genomics, PN-1000121) following manufacturers protocol. Following QC analysis, cDNA libraries were sequenced on Illumina NovaSeq instrument (paired end, single indexing, 28-8-0-91 cycles) at desired sequencing depth of 50 000 reads/cell. Following sequencing output fastq files were mapped to the mm10 mouse reference genome using 10x Genomics Cell Ranger (cellranger-6.1.2) software. Subsequent downstream analysis was performed in R and R studio softwares (packages: Seurat, Patchwork, Dplyr, Harmony and ggplot2). Any possible cell doublets (nFeature>6000) as well as cells expressing less than 200 genes and more than 15% of mitochondrial genes were excluded from analysis. Cells were clustered using Seurat package (v4.3.0). Results were displayed by UMAP and Heatmaps. Main cell populations were annotated based on the expression of 30 marker genes with highest expression within the cluster. Differentially expressed genes between comparable groups were found using FindMarkers function with the parameters test.use=wilcox, min.pct=0.2, tresh.use=0.25, the genes were filtered based on the p value <0.05.
Kegg pathways analysis
The differentially expressed genes (DEGs) were uploaded to the KEGG Pathways (DAVID; version DAVID 2021, Dec. 2021) (30) to identify biological pathways enriched in specific mononuclear phagocyte subtypes of 1) atherosclerotic Nrf2flox/flox mice in comparison to normocholesterolemic Nrf2flox/flox mice and 2) atherosclerotic Nrf2Cdh5tKO mice in comparison to atherosclerotic Nrf2flox/flox mice. The threshold for the statistical significance was p<0.05. The plots were drawn using the “ggplot2” packages in the R software (version 4.2.1).
Statistical analysis
Results are presented as mean ± standard deviation (SD) unless stated otherwise. The Student’s t-test was used to compare the means of two groups and a value of p<0.05 was considered statistically significant. Statistical analyses were performed with GraphPad Prism (GraphPadSoftware Inc., San Diego, CA).
Results
Expression of Nrf2 is significantly decreased in macrophages from Nrf2Cdh5tKO mice
Previous studies investigating the effect of macrophage-specific loss of Nrf2 on atherosclerosis were based on bone marrow transplantation of Nrf2-deficient bone marrow to lethally irradiated Ldlr−/− (25, 31) or ApoE−/− mice (26). Unfortunately, these results did not provide a clear answer on the role of macrophage-specific Nrf2 in atherosclerosis. Here we used animals with transcriptionally inactive Nrf2 in cadherin 5 (Cdh5)-expressing cells (Nrf2Cdh5tKO) and the control Nrf2flox/flox mice. In Nrf2Cdh5tKO mice, Cre recombinase excises exon 5 of Nrf2 gene, which is responsible for DNA binding and activation of transcription, making Nrf2 transcriptionally inactive. As a major component of the endothelial cell adherens junction, Cdh5 (VE-cadherin) plays an important role in vascular permeability and angiogenesis (32). Thus, Cdh5-Cre is frequently used for endothelial gene deletion. However, Cdh5-dependent constitutive Cre recombinase expression is present also in a proportion of hematopoietic cells (33–35). To verify whether Nrf2 transcriptional activity is affected in macrophages in our mouse model, we isolated macrophages of two different origins: bone marrow-derived macrophages (BMDM) and Kupffer cells from the liver. For both cell types, we confirmed a decreased level of Nrf2 exon 5 (Figures 1A, B) in comparison to Nrf2 exon 3 (the one not affected by Cre recombinase). In support of the decreased Nrf2 activity, we observed lower expression of the two selected Nrf2 target genes: NAD(P)H quinone dehydrogenase 1 (Nqo1) and glutamate-cysteine ligase modifier subunit (Gclm) in BMDM (Figures 1C, E, respectively), as well as in Kupffer cells (Figures 1D, F, respectively). In addition, the complete blood count analysis showed no difference in white blood cells (WBC, Figure 1G) and monocytes (Figure 1H) between analyzed healthy mice. It indicates that in steady state conditions decreased transcriptional activity of Nrf2 in hematopoietic and endothelial lineages does not affect formation of WBC and monocytes.
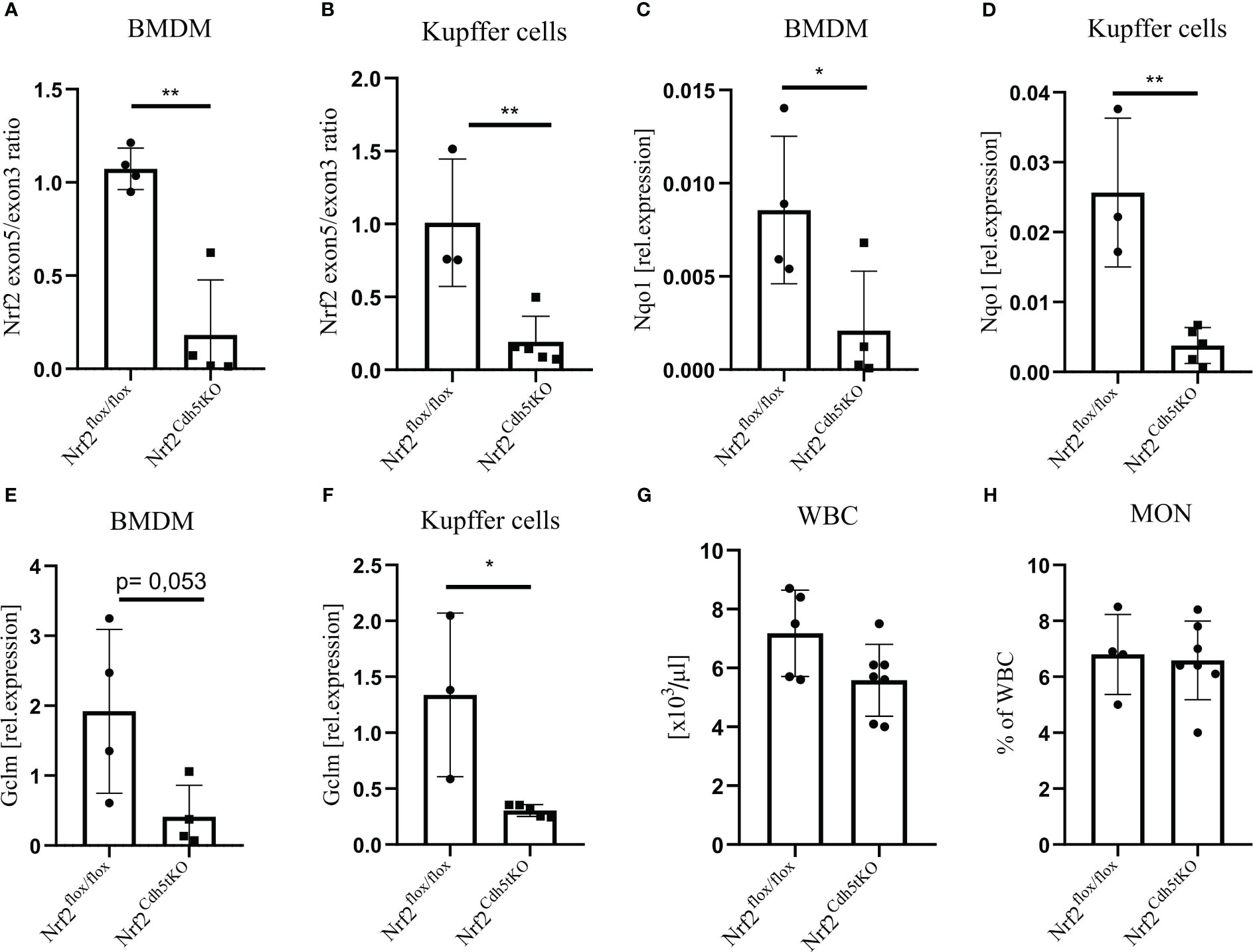
Figure 1 Evaluation of Nrf2 expression in macrophages from Nrf2Cdh5tKO mice. (A) The qPCR confirming excision of exon 5 in Nrf2Cdh5tKO BMDM presented as a ratio of exon 5 to exon 3. (B) The qPCR confirming the excision of exon 5 Nrf2Cdh5tKO Kupffer cells presented as a ratio of exon 5 to exon 3. (C) The qPCR analysis of Nqo1 in BMDM. (D) The qPCR analysis of Nqo1 in Kupffer cells. (E) The qPCR analysis of Gclm in BMDM. (F) The qPCR analysis of Gclm in Kupffer cells. (G) The number of WBC in control Nrf2flox/flox and Nrf2Cdh5tKO mice. (H) The percentage of blood monocytes in control Nrf2flox/flox and Nrf2Cdh5tKO mice. Results are presented as mean ± SD (*p<0.05, ** p<0.01).
Pcsk9 overexpression and high fat feeding changes the blood lipids level and induces early stage plaque development in the aortic arch branches of Nrf2flox/flox and Nrf2Cdh5tKO mice
Our atherosclerosis model was based on intravenous delivery of the AAV8 vector encoding Pcsk9, a protein controlling the expression of LDL receptor in the liver what, in combination with high-fat feeding, induces hypercholesterolemia in mice (36). Two groups of mice were subjected to this treatment: Nrf2Cdh5tKO and Nrf2flox/flox. The mouse plasma samples were collected 21 weeks after introducing proatherogenic factors. The analysis revealed increased level of plasma cholesterol carried by LDL/VLDL lipoproteins (Figure 2A) and increased level of triglycerides (Figure 2B) in atherosclerotic mice when compared to control littermates, with no significant differences between both genotypes. The Oil Red O (ORO) staining of aortic roots cross-sections confirmed the presence of plaques and atherosclerotic phenotype in both groups of mice (Figure 2C). The ORO staining of brachiocephalic artery cross-sections showed small lipid deposits within the aortic wall corresponding to an early stage of plaque development in the aortic arch branches of both genotypes (Figure 2D). No lipid deposits were detected within the aortic walls of normocholesterolemic Nrf2flox/flox and Nrf2Cdh5tKO mice (data not shown). After 21 weeks of atherosclerosis induction, the complete blood count analysis showed comparable numbers of WBC (Figure 2E) and monocytes (Figure 2F) in Nrf2flox/flox and Nrf2Cdh5tKO mice.
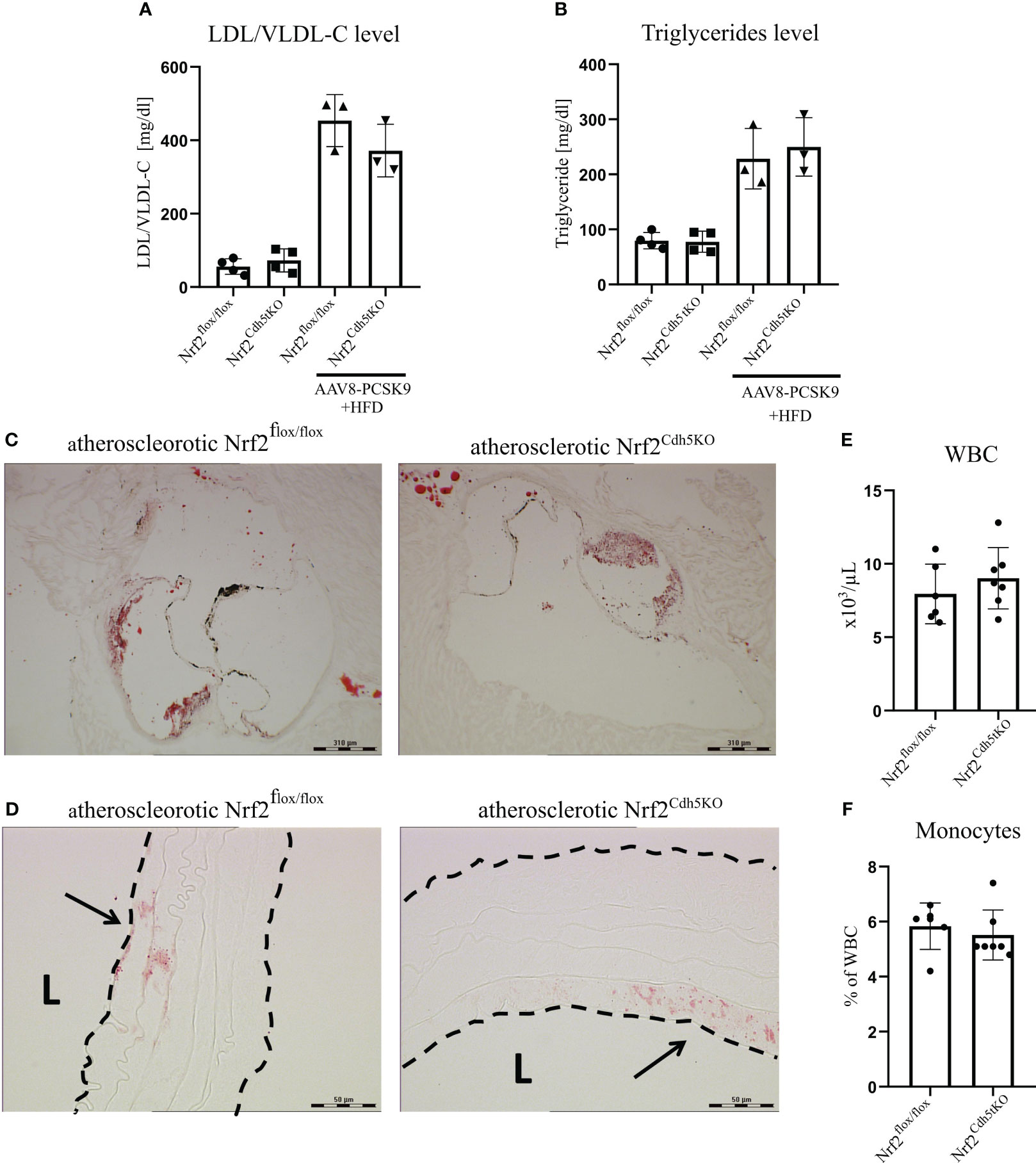
Figure 2 Characterization of atherosclerotic phenotype of Nrf2flox/flox and Nrf2Cdh5tKO mice. (A) The level of plasma cholesterol carried by LDL/VLDL lipoproteins in control healthy and atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO mice. (B) The level of plasma triglycerides in control healthy and atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO mice. (C) The staining of lipid deposits within aortic root of Nrf2flox/flox and Nrf2Cdh5tKO mice 21 weeks after AAV8-Pcsk9 injection and HFD. (D) The staining of lipid deposits in the brachiocephalic artery of Nrf2flox/flox and Nrf2Cdh5tKO mice 21 weeks after AAV8-Pcsk9 injection and HFD. Lumen of the blood vessel is marked with an ‘L’. The arrows indicate lipid deposits. (E) The number of WBC in the blood of Nrf2flox/flox and Nrf2Cdh5tKO mice 21 weeks after AAV8-Pcsk9 injection and HFD. (F) The percentage of monocytes in WBC population in the blood of atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO mice 21 weeks after AAV8-Pcsk9 injection and HFD. Results are presented as mean ± SD.
Single-cell RNA sequencing identifies several types of mononuclear phagocytes in the aortas of Nrf2flox/flox and Nrf2Cdh5tKO mice
Previous studies using single-cell RNA sequencing (scRNA-seq) highlighted the complexity of macrophage biology and their heterogeneity in human and murine aortas (7, 9). In our study we applied this technology to analyze the transcriptomic changes in different macrophage subpopulations deficient in Nrf2 transcriptional activity in the context of their possible contribution to atherosclerosis development. We collected control and atherosclerotic aortic arches with branches from Nrf2flox/flox and Nrf2Cdh5tKO mice. Then, the pooled fragments of aortas of each group were subjected to scRNA-seq (Figure 3A). The unsupervised clustering of 14 125 cells identified, among others, two clusters of macrophages. They were annotated as proinflammatory and tissue-resident based on the expression of Pf4, Mrc1, C1qa, C1qb, C1qc genes, and Lyz1, Rentla, Lyz2, Cd74 genes, respectively. Within the merged (proinflammatory and tissue-resident) macrophage population of 1821 cells, 92.3% were Cd45(Ptprc)-positive, 92.5% were Cd68-positive, 82.3% were F4/80(Adgre1)-positive, and 73.1% were Cd11b(Itgam)-positive (Figure 3B). The variation in the content of these immune cell markers suggested that the population is heterogenous and, besides macrophages, consists of other cell types. The subclustering analysis of this heterogenous population, here called mononuclear phagocytes, revealed 9 subpopulations (Figure 3C) with a high abundance of lymphatic vessel endothelial receptor-1 (Lyve1+) resident macrophages, proinflammatory macrophages, cavity macrophages and monocytes (Figure 3D). The other subtypes - the population of stem, endothelial and mesenchymal (SEM) cells, hematopoietic progenitor cells (HPC)/fibrocytes, aortic intima resident (AIR) macrophages, conventional type 1 dendritic cells (cDC1) and conventional type 2 dendritic cells (cDC2) were less abundant (Figure 3D). Lyve1+ resident macrophages were characterized according to previous reports (8, 9, 37) by the expression of Lyve1, Folr2, Hmox1, Maf, and Cd209f, whereas cavity macrophages (8) by Lyz1, Fn1, Ear2, Rentla, and Lpl (Figure 3E). The expression of Cxcl1, Nlrp3, Sdc4, Cxcl16, and Ccl3 genes characterized both proinflammatory and AIR macrophages (8), but the presence of typical AIR macrophage markers Cd72, Ccrl2, Ctss, Acp5, Mpeg1 (8) distinguished this population from the proinflammatory subset (Figure 3E). Monocyte cluster (8) showed high expression of Plac8, Chil3, Msrb1, Clec4e, and Slpi (Figure 3E); HPC/fibrocytes (38) were enriched in Dcn, Sparc, Gsn, Col3a1, Mgp (Figure 3E), while SEM cluster (39) highly expressed Lum, Runx1, Apoe, Acta2, Ctsb (Figure 3E). For cDC1 (8) and cDC2 (8) we found overlapping expression of classical marker genes with higher expression of Naaa, Cst3, Ifi205, Cd24a, H2afz for cDC1 (Figure 3E) and Napsa, H2-ab1, H2-eb1, Traf1, Wnt11 for cDC2 (Figure 3E).
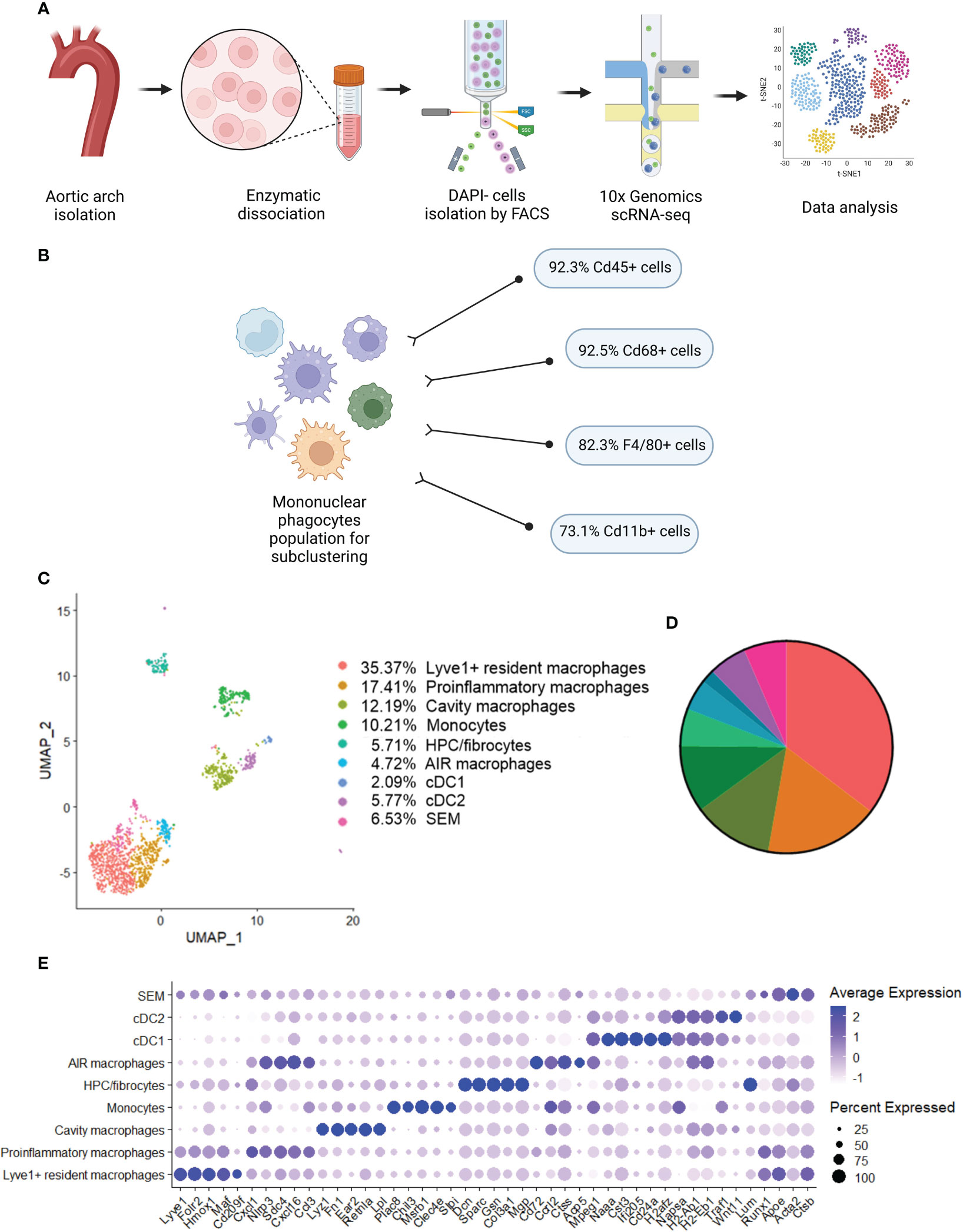
Figure 3 Identification of distinct mononuclear phagocytes in the analyzed fragments of aortas of Nrf2flox/flox and Nrf2Cdh5tKO mice. (A) Schematic depiction of the scRNA-seq procedure. (B) The percentage of cells expressing classical macrophage markers: Cd45, Cd68, F4/80, Cd11b within the whole macrophage population before subclustering. (C) UMAP representation of integrated scRNA-seq gene expression data from normocholesterolemic Nrf2flox/flox (n=4) and Nrf2Cdh5tKO (n=4), as well as atherosclerotic Nrf2flox/flox (n=3) and Nrf2Cdh5tKO (n=3) murine aortic arches and branches with identification of the major cell types. (D) Proportions of distinct cell types singled out among whole macrophage population. (E) Dot plot of average gene expression of the indicated marker transcripts for each cell cluster.
The analysis of classical macrophage markers - Cd45, Cd68, F4/80, Cd11b (Figures 4A–D, respectively) revealed their lowest expression in HPC/fibrocytes and SEM cells. Moreover, the level of Cd68, F4/80, and Cd11b (Figures 4B–D, respectively) was moderately or strongly decreased in cDC1 and cDC2 cell clusters. Analysis of the abundance of various populations of mononuclear phagocytes in the aortas of healthy and atherosclerotic mice of both genotypes revealed a higher percentage of monocytes (9.18% and 5.25% respectively for normocholesterolemic Nrf2flox/flox and Nrf2Cdh5tKO vs. 27.06% and 10.17% respectively for atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO) and cDC2 cells (2.05% and 2.62% respectively for normocholesterolemic Nrf2flox/flox and Nrf2Cdh5tKO vs. 11.76% and 6.10% respectively for atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO) in atherogenic conditions (Figure 4E). Interestingly, in atherosclerotic Nrf2Cdh5tKO mice a lower proportion of monocytes was associated with a higher proportion of proinflammatory macrophages (17.44% for atherosclerotic Nrf2Cdh5tKO vs. 7.65% for atherosclerotic Nrf2flox/flox).
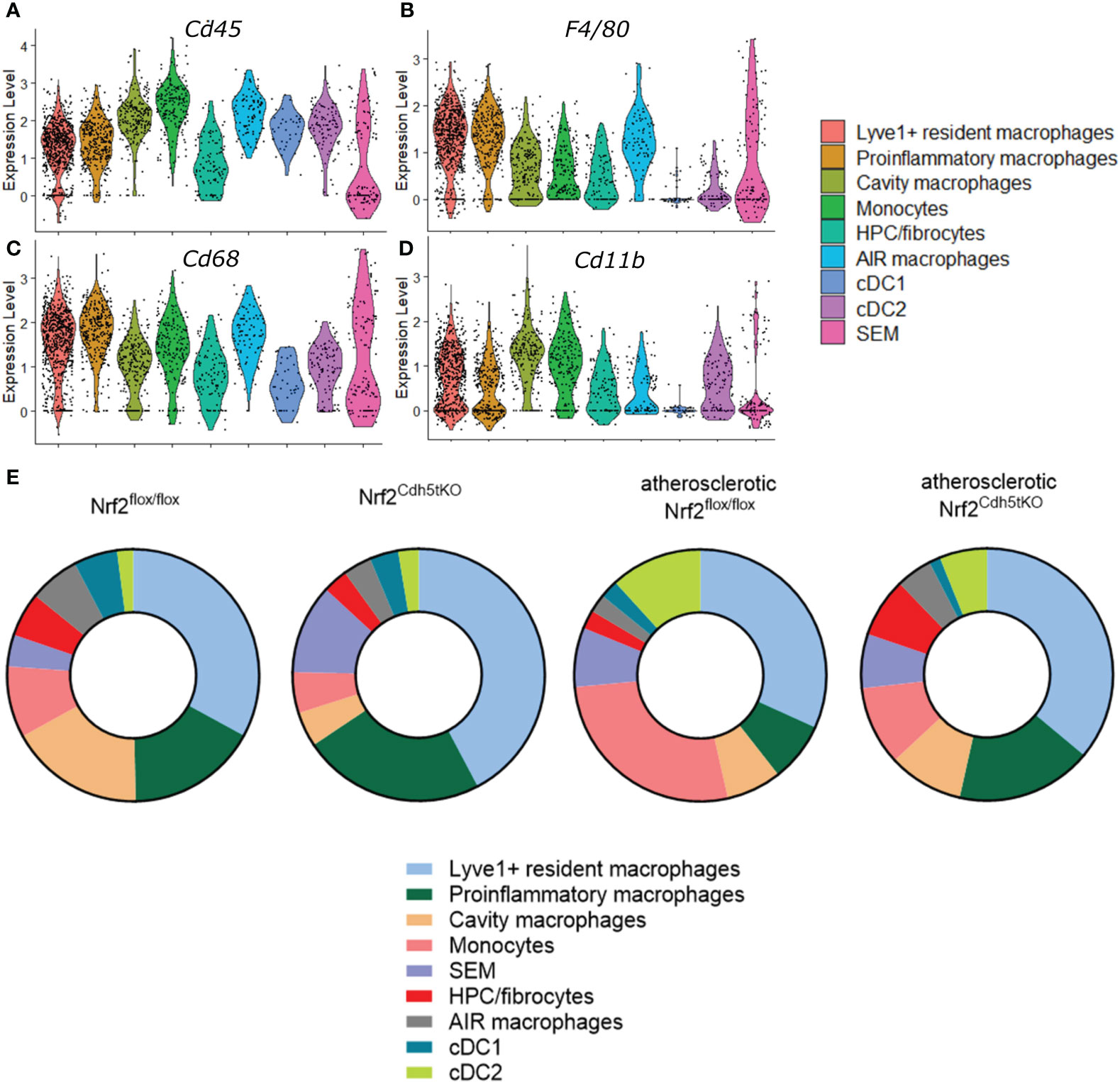
Figure 4 Characteristics of the identified mononuclear phagocyte clusters. The expression of typical macrophage genes (A) Cd45, (B) F4/80, (C) Cd68, (D) Cd11b within particular mononuclear phagocyte population shown in the violin plot. (E) Proportions of identified cell subsets in the analyzed fragments of aortas of control normocholesterolemic and atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO mice.
The transcriptome of distinct aortic mononuclear phagocytes is differently affected by decreased Nrf2 transcriptional activity under atherogenic conditions
Next, we analyzed macrophage subtype-specific gene expression using scRNA-seq technology in the aortic arches and branches from control normocholesterolemic and atherosclerotic mice of two genotypes - Nrf2flox/flox and Nrf2Cdh5tKO. We focused mostly on three highly abundant clusters that play an important role in plaque development: Lyve1+ resident macrophages, proinflammatory macrophages and monocytes (Figure 3C).
Lyve1+ resident macrophages
Lyve1+ resident macrophages are considered to originate from embryonic erythro-myeloid progenitors (40) but, under certain circumstances, circulating monocytes can differentiate into self-maintaining tissue-resident macrophages that resemble their embryonic counterparts. In our analysis, this macrophage subset is characterized by high expression of Lyve1, Folr2, Hmox1, Maf, and Cd209f (Figures 3E, 5A), and was the most numerous subpopulation in all samples subjected to scRNA-seq (Figure 4E). First, we analyzed how atherosclerosis itself affects gene expression in this macrophage subset. In Lyve1+ resident macrophages of atherosclerotic Nrf2flox/flox mice, we identified 16 downregulated and 38 upregulated genes when compared to normocholesterolemic Nrf2flox/flox controls (Table 1). The KEGG pathway analysis (Figure 5B) showed that many differentially expressed genes (DEGs) were significantly enriched in cytokine-cytokine receptor interaction (Ccl9, Bmp2, Gdf15, Il1b, Ccl4, Cxcl13, Ccr2), C-type lectin receptor signaling pathway (Cd209d, Clec4b1, Cd209b, Il1b, Lsp1), chemokine signaling pathway (Lyn, Ccl9, Ccl4, Cxcl13, Ccr2), NF-kappa B signaling pathway (Lyn, Il1b, Ccl4). The majority of the identified DEGs were associated with a pro-inflammatory cell phenotype.
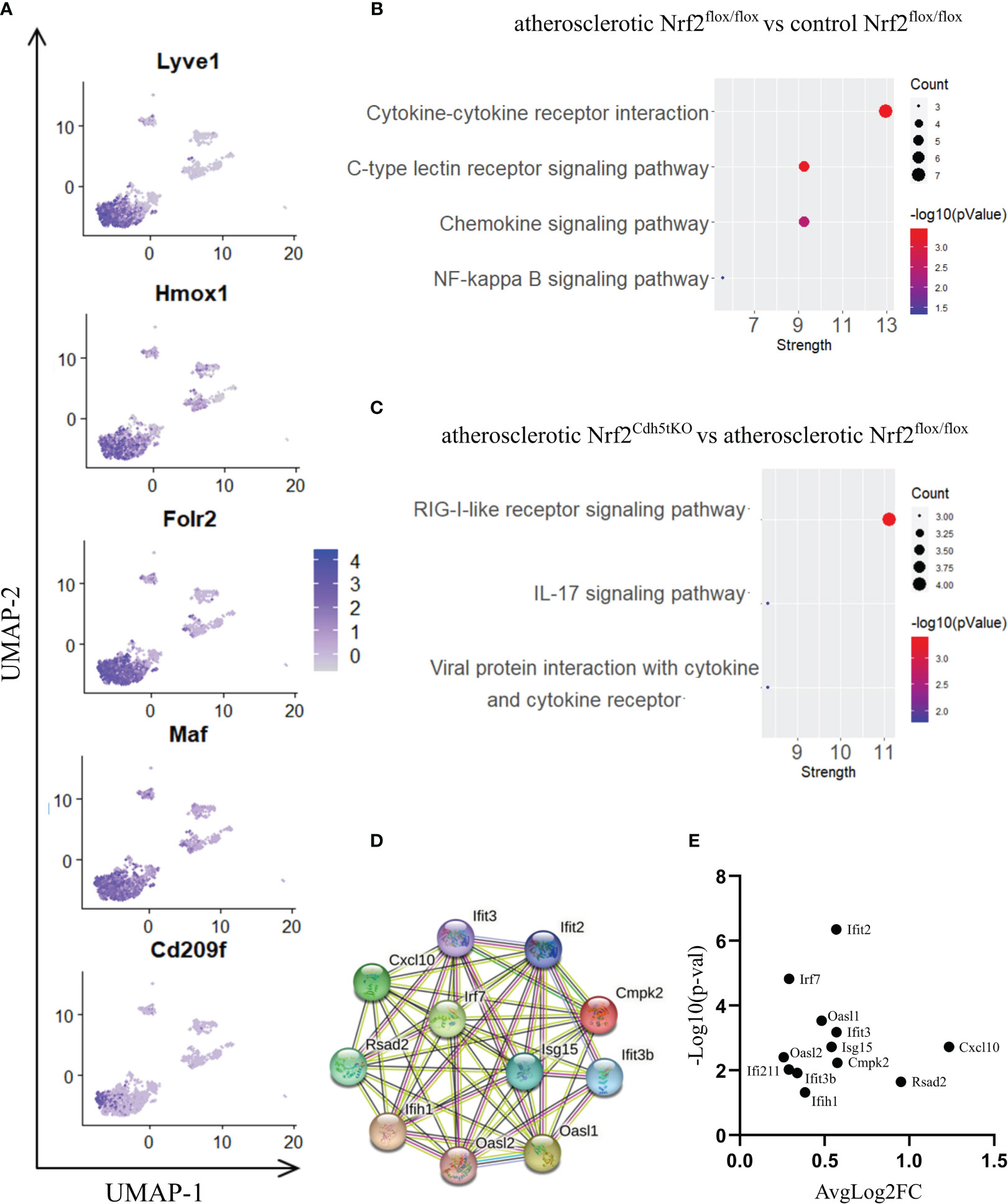
Figure 5 The analysis of transcriptomic changes in Lyve1+ resident macrophages. (A) Expression of marker genes identified for Lyve1+ resident macrophages projected onto the UMAP plot. (B) KEGG pathway enrichment analysis of upregulated and downregulated genes for atherosclerotic Nrf2flox/flox vs. control Nrf2flox/flox and (C) atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox Lyve1+ resident macrophages. The x-axis represents the strength (Log10(observed/expected)) of enriched pathways, whereas the color denotes –log10(pValue) and dot size corresponds to gene count in each pathway. (D) STRING interaction network - graphical representation of Interferon-simulated genes (ISG) cluster. (E) Volcano plot of the top differentially expressed ISG in atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox Lyve1+ resident macrophages.
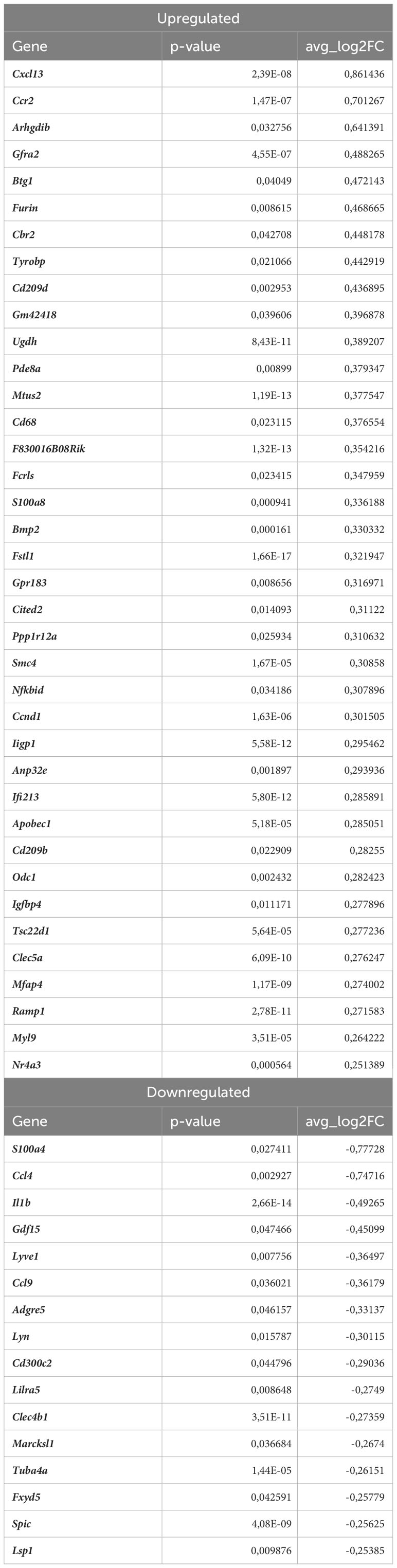
Table 1 DEGs in Lyve1+ resident macrophages (atherosclerotic Nrf2flox/flox vs. normocholesterolemic Nrf2flox/flox).
Next, we investigated the effect of Nrf2 deficiency on the transcriptome of Lyve1+ resident macrophages under atherogenic conditions. The scRNAseq analysis revealed that in this macrophage subset, 17 genes were downregulated and 20 were upregulated in atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox mice (Table 2). The DEGs were subjected to KEGG functional enrichment analysis (Figure 5C). Among the significantly enriched KEGG pathways, we found retinoic acid-inducible gene I (RIG-I)-like receptor signaling pathway (Ifih1, Cxcl10, Irf7, Isg15), IL-17 signaling pathway (Cxcl10, Ccl7, S100a8), viral protein interaction with cytokine and cytokine receptor (Cxcl10, Ccl7, Ccr2).
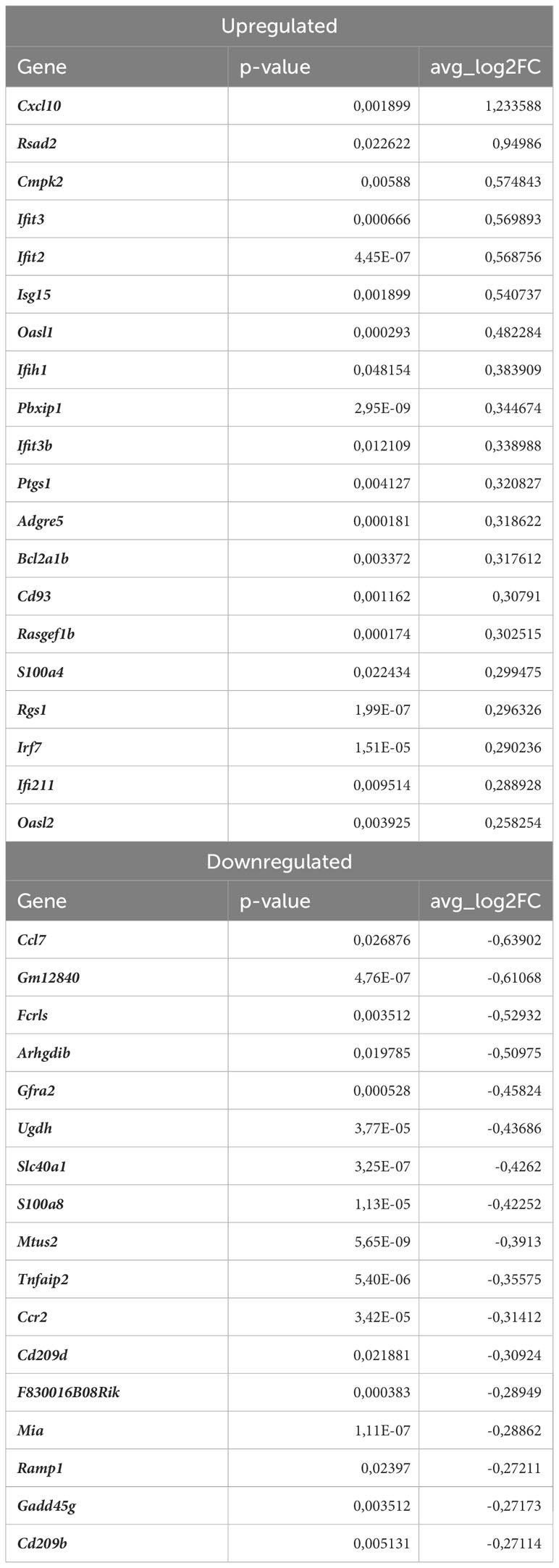
Table 2 DEGs in atherosclerotic Nrf2Cdh5tKO Lyve1+ resident macrophages (atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox).
RIG-I not only participates in antiviral signaling pathways, but may also affect non-viral diseases such as atherosclerosis (41, 42). Once activated, RIG-I initiates a signaling cascade that leads to the activation of transcription factors, including interferon regulatory factors (IRFs) and nuclear factor kappa B (NF-κB), which induce the expression of interferon-related genes and pro-inflammatory cytokines (43). In fact, our results confirmed that observation revealing upregulation of interferon-stimulated genes (ISGs, Figures 5D, E), Rsad2, Cxcl10, Irf7, Ifit2, Ifit3, Ifit3b, Ifih1, Isg15, Oasl1, Oasl2, Cmpk2, in Nrf2Cdh5tKO Lyve1+ resident macrophages highlighting that over 50% of upregulated genes were associated with downstream IFN-response. Among them, the key transcription factor interferon regulatory factor 7 (Irf7) was upregulated. Interestingly, comparison of normocholesterolemic Nrf2Cdh5tKO vs. normocholesterolemic Nrf2flox/flox mice revealed increased, among others, expression of Rsad2 and Oasl1 (Table 3) suggesting that Nrf2-defficient Lyve1+ resident macrophages can be predisposed to develop an interferon-stimulated response under favorable proatherogenic conditions. In terms of atherosclerosis, we also found that the top upregulated in the atherosclerotic Nrf2Cdh5tKO mice Cxcl10 gene, was involved in all the most enriched KEGG pathways. Its altered expression, together with other chemokine and chemokine receptors such as Ccl7, Ccr2, Cd97, Rgs1 and S100a8, may play a role in leukocyte chemotaxis and migration.
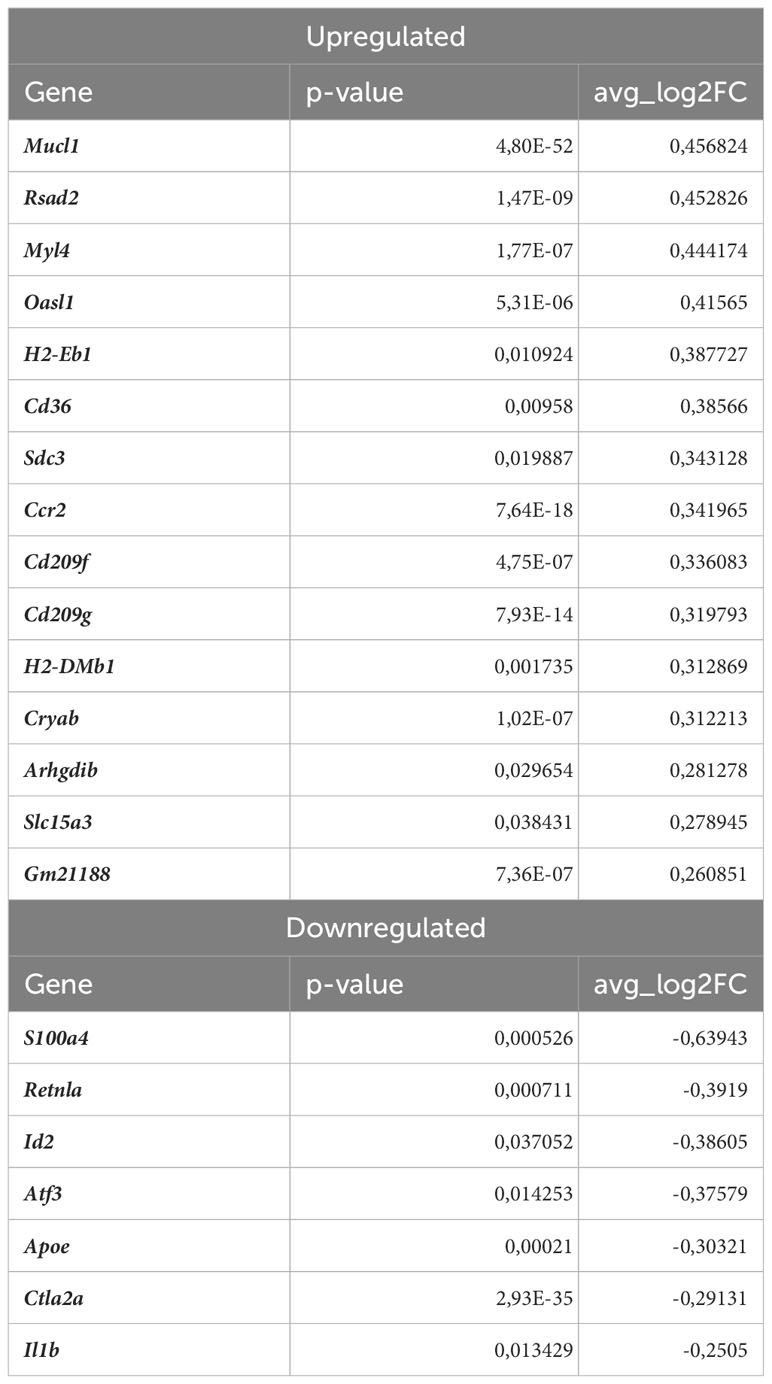
Table 3 DEGs in normocholesterolemic Nrf2Cdh5tKO Lyve1+ resident macrophages (normocholesterolemic Nrf2Cdh5tKO vs. normocholesterolemic Nrf2flox/flox).
Among DEGs in atherosclerotic Nrf2Cdh5tKO Lyve1+ resident aortic macrophages (Table 2), we also identified genes involved in different aspects of cell death regulation. Slc40a1, whose expression may be indirectly regulated by Nrf2 (18), encodes ferroportin and has been implicated in the regulation of iron homeostasis and ferroptotic cell death (44). In addition, we detected the upregulation of Pbxip1 gene which has been shown to promote apoptosis by inhibiting anti-apoptotic Bcl-2 family members, while on the other hand, the anti-apoptotic Bcl2a1b gene was upregulated. We also observed changes in Gadd45g, involved in promoting cell death in response to DNA damage and other stress signals (45); Ifi211 that may inhibit cell growth via p53/TP53 and RB1-dependent and independent pathways (46); Tnfaip2 that may regulate cell death in the context- and cell-dependent manner (47, 48).
Proinflammatory macrophages
Proinflammatory macrophages are the subtype of immune cells mostly derived from monocytes infiltrating the tissues in response to different environmental stimuli. In our scRNAseq analysis, proinflammatory macrophages were the second most abundant cell population within aortic wall, characterized by Cxcl1, Nlrp3, Sdc4, Cxcl16, Ccl3 marker genes (Figures 3E, 6A). The atherogenic conditions led to the downregulation of 29 and upregulation of 38 genes (Table 4) in proinflammatory macrophages of Nrf2flox/flox mice when compared to the normocholesterolemic Nrf2flox/flox controls. Among the KEGG enriched pathways (Figure 6B), we identified cytokine-cytokine receptor interaction (Cxcl10, Cxcl9, Cd4, Cxcl12, Ccl6, Il1b, Il21r, Il2rg, Tnfsf13b, Ccr2), chemokine signaling pathway (Lyn, Cxcl10, Hck, Itk, Cxcl9, Cxcl12, Ccl6, Ccr2), viral protein interaction with cytokine and cytokine receptor (Cxcl10, Cxcl9, Cxcl12, Ccl6, Il2rg, Ccr2), NF-kappa B signaling pathway (Lyn, Cxcl12, Lck, Il1b, Tnfsf13b). Here, similarly to Lyve1+ resident macrophages, the proatherogenic conditions altered mainly the expression of genes involved in immune responses and proinflammatory cell phenotype.
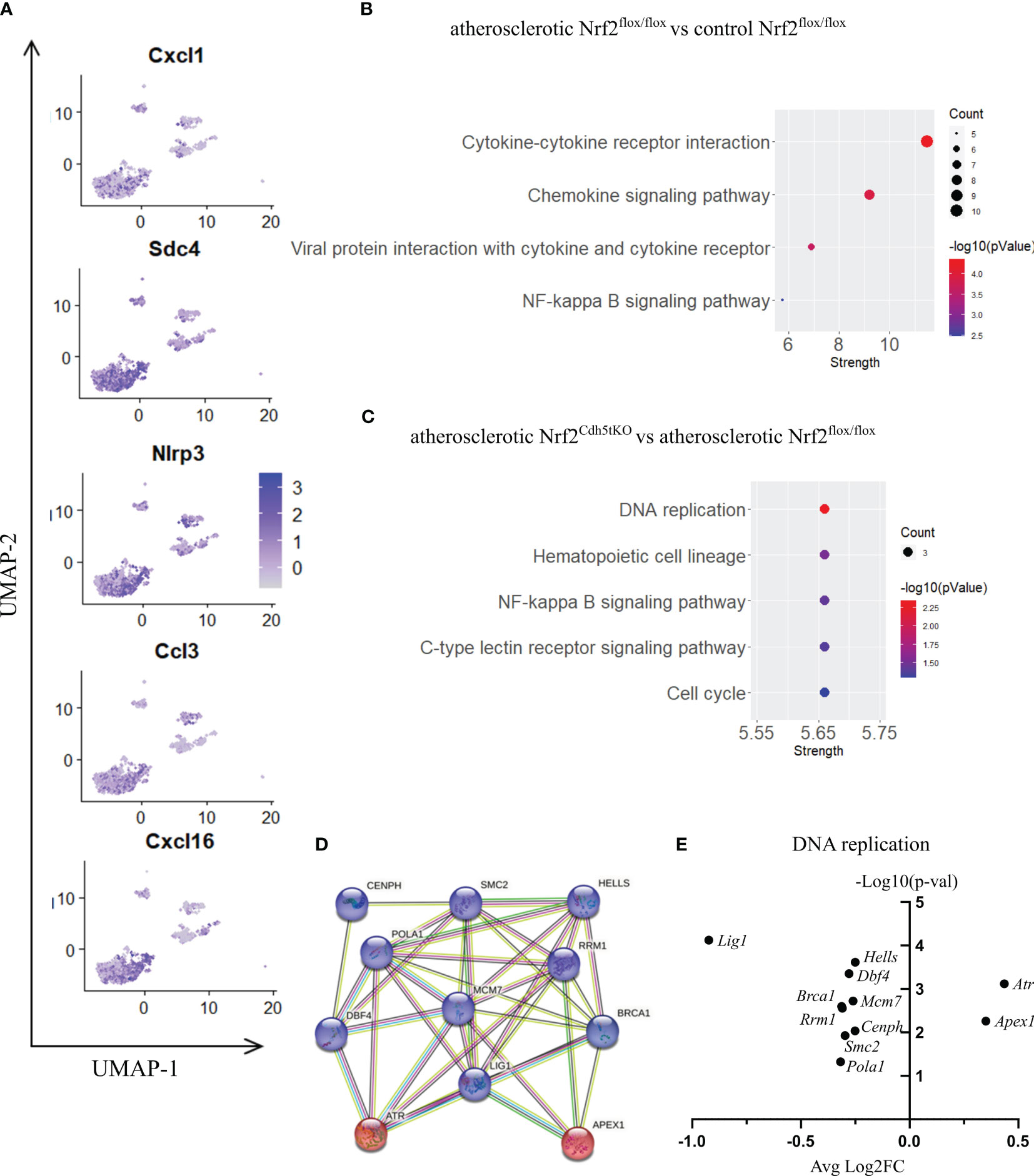
Figure 6 The analysis of transcriptomic changes in proinflammatory macrophages. (A) Expression of marker genes identified for proinflammatory macrophages projected onto the UMAP plot. (B) KEGG pathway enrichment analysis of upregulated and downregulated genes for atherosclerotic Nrf2flox/flox vs. control Nrf2flox/flox and (C) atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox proinflammatory macrophages. The x-axis represents the strength (Log10(observed/expected)) of enriched pathways, whereas the color denotes –log10(pValue) and dot size corresponds to gene count in each pathway. (D) STRING interaction network - graphical representation of DNA replication and repair genes cluster. The upregulated genes are presented in red color, the downregulated in blue. (E) Volcano plot of the top differentially expressed DNA replication and repair genes in atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox proinflammatory macrophages.
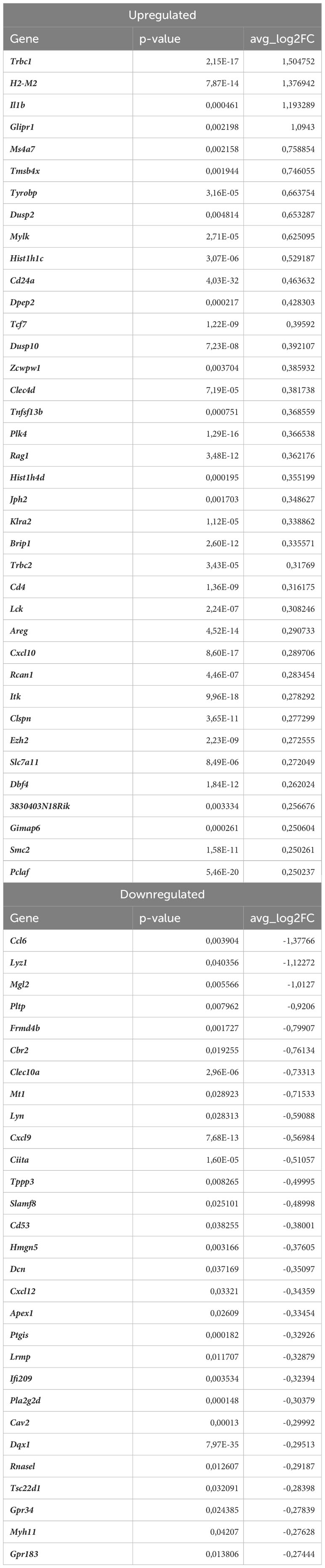
Table 4 DEGs in proinflammatory macrophages (atherosclerotic Nrf2flox/flox vs. normocholesterolemic Nrf2flox/flox).
Nrf2 deficiency additionally modified the transcriptome of these cells. The scRNAseq analysis revealed that 38 genes were downregulated and 15 genes were upregulated in proinflammatory macrophages of atherosclerotic Nrf2Cdh5tKO in comparison to atherosclerotic Nrf2flox/flox (Table 5). The KEGG pathway analysis (Figure 6C) revealed that the majority of DEGs were associated with DNA replication (Pola1, Lig1, Mcm7), hematopoietic cell lineage (Cd14, Il7r, Cd34), NF-kappa B signaling pathway (Lyn, Lck, Cd14), C-type lectin receptor signaling pathway (Clec4d, Cd209f, Clec4e) and cell cycle (Dbf4, Mcm7, Atr). Interestingly, comparison of normocholesterolemic Nrf2Cdh5tKO vs. normocholesterolemic Nrf2flox/flox mice revealed that the top upregulated gene in proinflammatory macrophages was Il1b (Table 6). This, together with other DEGs involved in cytokine-cytokine receptor interaction, antigen processing and presentation, cell adhesion molecules, or toll-like receptor pathway, may suggest a more inflammatory phenotype of this Nrf2-deficient macrophage subtype already in steady state conditions.
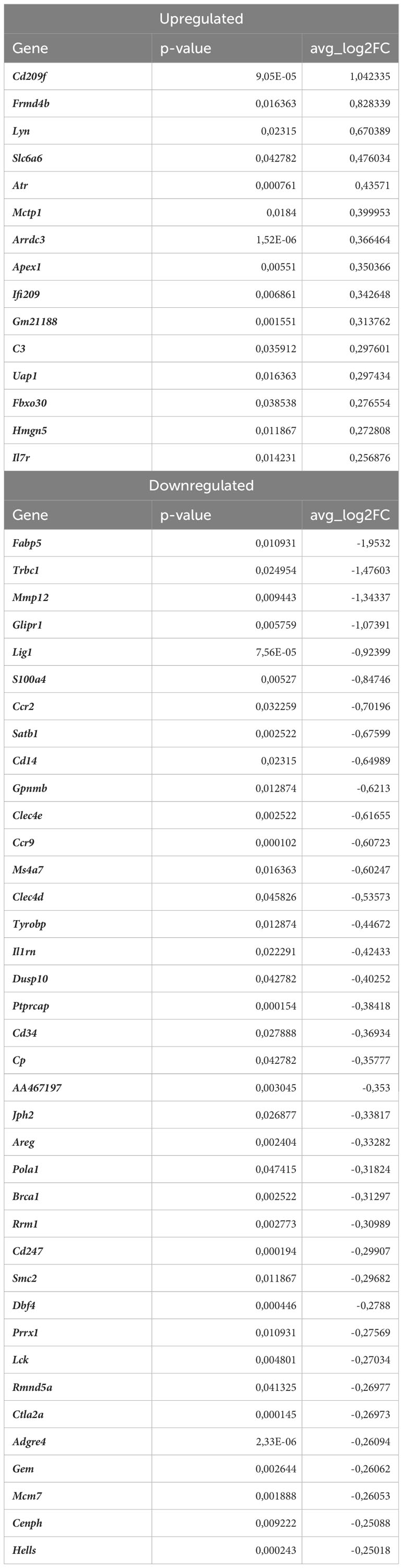
Table 5 DEGs in atherosclerotic Nrf2Cdh5tKO proinflammatory macrophages (atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox).
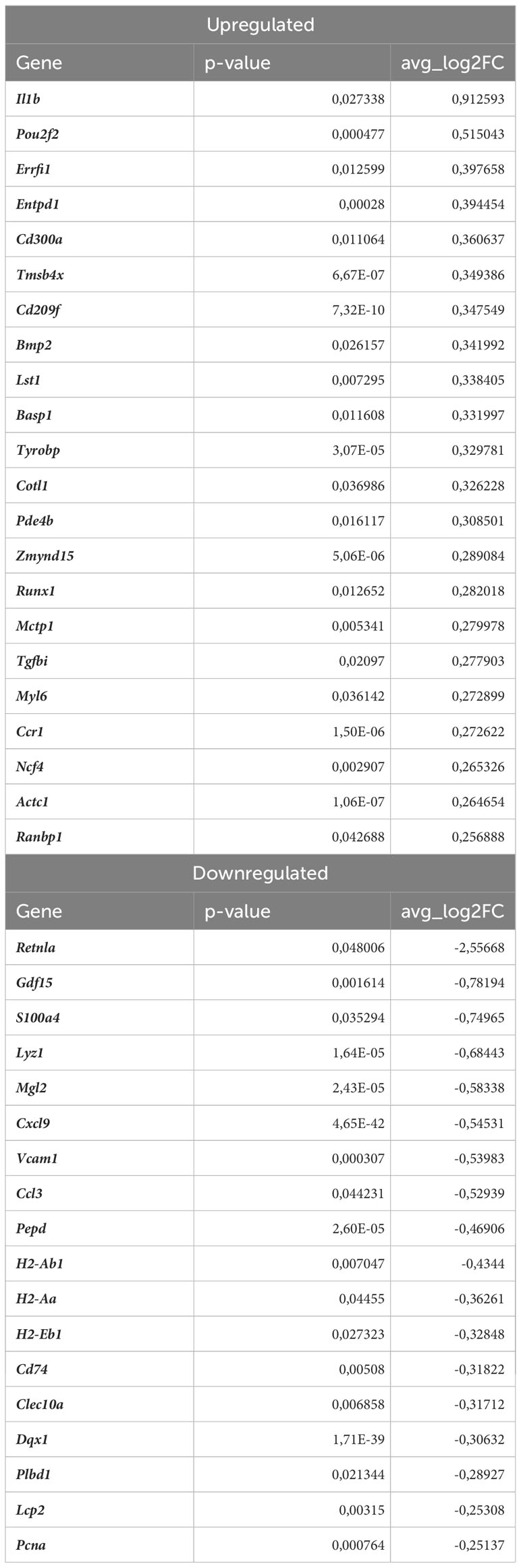
Table 6 DEGs in normocholesterolemic Nrf2Cdh5tKO proinflammatory macrophages (normocholesterolemic Nrf2Cdh5tKO vs. normocholesterolemic Nrf2flox/flox).
Moreover, in atherogenic conditions, several DEGs in proinflammatory macrophages of Nrf2Cdh5tKO vs. Nrf2flox/flox mice (Table 5) were identified as hub genes associated with DNA replication and maintenance (downregulation of Brca1, Smc2, Cenph, Lig1, Mcm7, Rrm1, Dbf4, Pola1, Hells and upregulation of Atr, Apex1, Figures 6D, E). As the Atr and Apex1 genes are the main sensors of oxidative stress-induced DNA damage, we checked for any other genes associated with increased oxidative stress in Nrf2Cdh5tKO proinflammatory macrophages. Among these, we observed upregulation of Lyn, which is involved in response to DNA damage/redox imbalance (49); Fbxo3, which is a part of SCF complex and may be involved in DNA damage response (50); Hmgn5, which may regulate histone modification, DNA replication, repair, and gene transcription through binding to chromatin regulators (51). Among the downregulated DEGs, we found Dbf4 gene which decreases S-phase checkpoint signaling and maintains DNA replication and cell cycle progression induced by DNA damage (52), DNA damage-induced Areg (53), Satb1 that is involved in DNA repair (54), and Gem which regulates chromatin remodeling and DNA replication (51).
Some DEGs were associated with the C-type lectin receptor (CLR) signaling pathway. CLRs are widely expressed by myeloid cells. The expression of macrophage-specific members of CLR family were decreased in Nrf2Cdh5tKO proinflammatory macrophages. Among them, we identified Clec4e (macrophage inducible C-type lectin; Mincle) and Clec4d (macrophage C-type lectin; MCL). The first one is activated by dead cells/necrosis and both of them are able to sense damage-associated molecular patterns (55).
Within the Src family kinases, Lck was downregulated, the level of Ptprcap, that stabilizes the association of CD45 with Lck (56), was decreased, similarly to Tyrobp (Dap12) that may be phosphorylated by Src kinases. On the other hand, the other Src family proteins, Cd14 and Lyn were upregulated.
Monocytes
Monocytes possess the ability to infiltrate the tissues and mature into macrophages. In our scRNAseq analysis they were characterized by high expression of Plac8, Chil3, Msrb1, Clec4e, Slpi (Figures 3E, 7A) and were one of the most abundant mononuclear phagocyte subset. The monocytes of Nrf2flox/flox mice were strongly affected by the atherogenic conditions what was reflected by the number of DEGs – 146 genes (57 down- and 89 upregulated vs. normocholesterolemic Nrf2flox/flox mice; Table 7). The KEGG pathway analysis (Figure 7B) revealed that many DEGs were related to inflammatory responses: viral protein interaction with cytokine and cytokine receptor (Ccr1, Il10, Il6, Ccl7, Ccl5, Ccl2, Il2rg, Cxcl3, Pf4), cytokine-cytokine receptor interaction (Ccr1, Il10, Il1r2, Inhba, Cxcl3, Il2rg, Cxcl16, Il6, Ccl7, Ccl5, Ccl2, Il7r, Pf4), chemokine signaling pathway (Ccr1, Grk3, Gngt2, Ccl7, Ccl5, Ccl2, Cxcl3, Cxcl16, Pf4), IL-17 signaling pathway (Il6, Ccl7, Ccl2, Cxcl3, S100a9, S100a8), complement and coagulation cascades (C1qb, C4b, C1qa, Serpinb2, C3ar1, C1qc), genes associated with TNF signaling pathway (Il6, Ccl5, Ccl2, Cxcl3, Creb5).
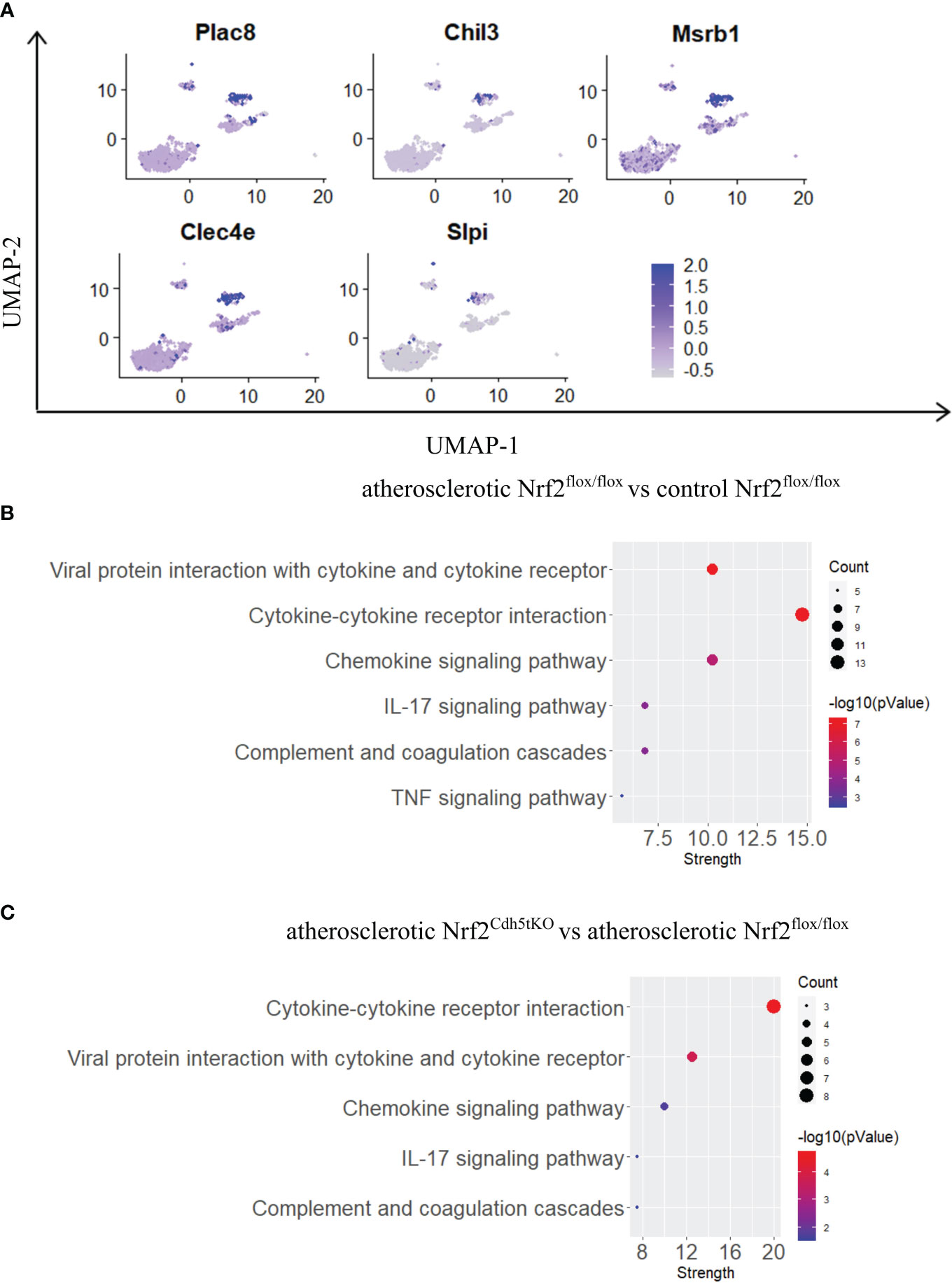
Figure 7 The analysis of transcriptomic changes in monocytes. (A) Expression of marker genes identified for monocytes projected onto the UMAP plot. (B) KEGG pathway enrichment analysis of upregulated and downregulated genes for atherosclerotic Nrf2flox/flox vs. control Nrf2flox/flox and (C) atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox monocytes. The x-axis represents the strength (Log10(observed/expected)) of enriched pathways, whereas the color denotes –log10(pValue) and dot size corresponds to gene count in each pathway.

Table 7 DEGs in monocytes (atherosclerotic Nrf2flox/flox vs. normocholesterolemic Nrf2flox/flox).
In monocytes of atherosclerotic Nrf2Cdh5tKO mice, we found 19 downregulated and 24 upregulated genes when compared to atherosclerotic Nrf2flox/flox (Table 8). The KEGG pathway analysis (Figure 7C) uncovered 5 enriched pathways. Among these, we found cytokine-cytokine receptor interaction (Ccr1, Il6, Il1rn, Ccl7, Gdf15, Il1r2, Ccl3, Cxcl13), viral protein interaction with cytokine and cytokine receptor (Ccr1, Il6, Ccl7, Ccl3, Cxcl13), chemokine signaling pathway (Ccr1, Ccl7, Ccl3, Cxcl13), IL-17 signaling pathway (Il6, Ccl7, S100a9), complement and coagulation cascades (C4b, Serpinb2, Vsig4). The last pathway was the only one to be enriched when normocholesterolemic Nrf2Cdh5tKO and Nrf2flox/flox mice were compared (Table 9).
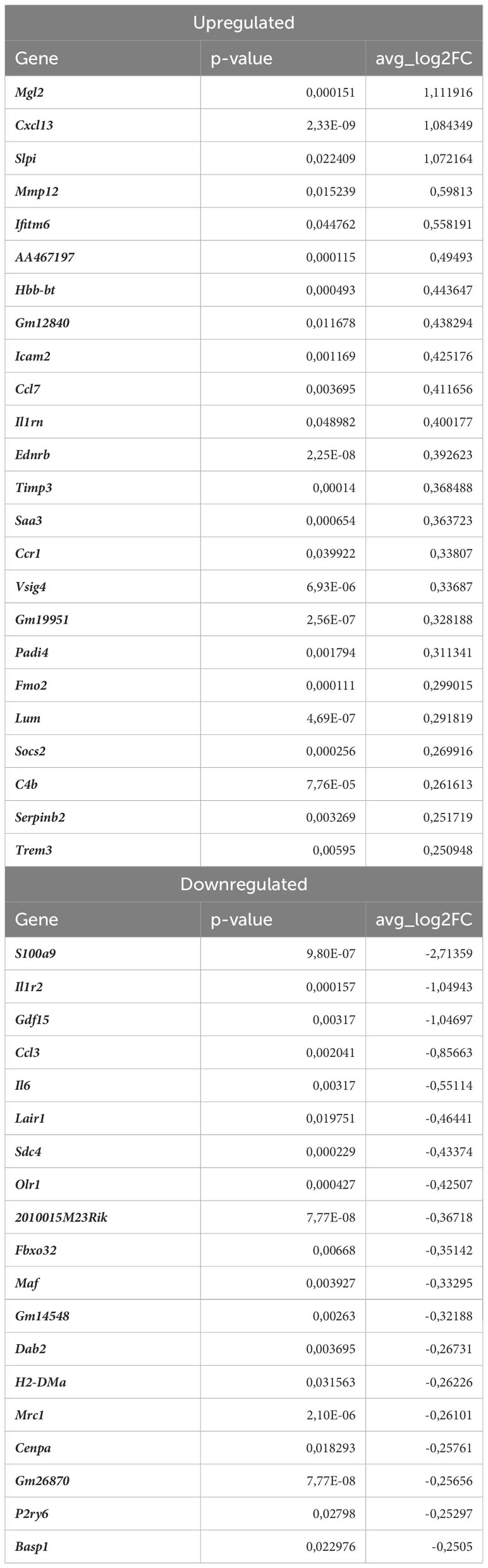
Table 8 DEGs in atherosclerotic Nrf2Cdh5tKO monocytes (atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox).
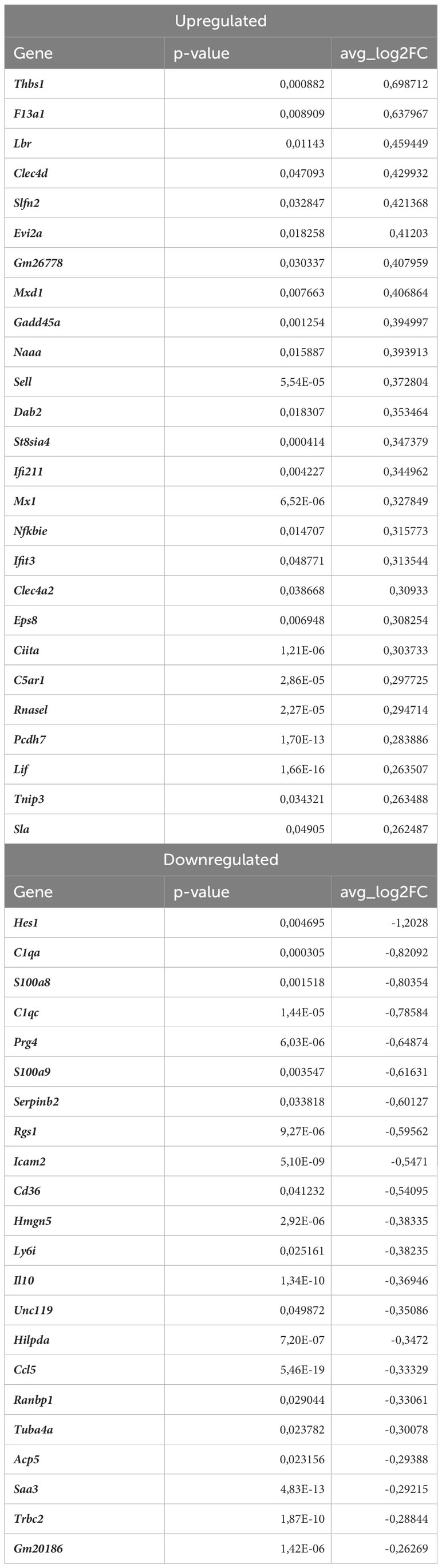
Table 9 DEGs in normocholesterolemic Nrf2Cdh5tKO monocytes (normocholesterolemic Nrf2Cdh5tKO vs. normocholesterolemic Nrf2flox/flox).
The transcriptome of monocytes from atherosclerotic Nrf2Cdh5tKO mice (Table 8) was strongly changed in terms of the expression of genes encoding chemokines, cytokines, and their receptors. Besides the genes associated with cytokine-cytokine receptor interaction (downregulated Il6, Gdf15, Il1r2, Ccl3 and upregulated Ccr1, Il1rn, Ccl7, Cxcl13), in atherosclerotic Nrf2Cdh5tKO monocytes we also identified upregulation of Ednrb, which is a receptor for endothelin and angiotensin (57). Interestingly, in monocyte DEGs between atherosclerotic Nrf2Cdh5tKO and Nrf2flox/flox mice, we also found several protease inhibitors, such as Timp3, Serpinb2, and Slpi, that may change the activity of peptidases and contribute to vascular remodeling.
Discussion
Macrophages drive atherosclerosis at all stages of plaque development. It was previously shown that local tissue environment shapes the transcriptome and identity of macrophages and various macrophage types may differentially respond to the same stimuli (58). One of the stimuli acting in atherosclerotic plaques is oxidative stress, which has been implicated in the disease progression from the early fatty streak lesions to advanced atherosclerotic plaques (22). The accumulation of reactive oxygen species within the cells can affect different signaling pathways regulating cell cycle, migration and survival leading to cellular dysfunction and even cell death (59).
In this study, we analyzed the transcriptomic changes caused by atherogenic conditions in different populations of murine aortic mononuclear phagocytes deficient in Nrf2 transcriptional activity. In physiological conditions this redox-activated transcription factor provides protection via activation of expression of a number of genes mediating antioxidant response (22). However, Nrf2 effects in atherosclerosis seem to be more complex and currently not completely understood. Here, we analyzed the data obtained from mice with transcriptionally inactive Nrf2 in Cdh5-expressing cells and their progeny. The prevailing scientific consensus is that hematopoiesis, the process of blood cell formation, primarily occurs through two distinct mechanisms during development: yolk-sac hematopoiesis and hemogenic endothelium-derived hematopoiesis. The exact contribution of these sources to adult hematopoiesis is still a subject of active research and ongoing debate (40, 60). Nevertheless, according to the literature data even 50-96% of CD45+ adult bone marrow–derived cells may be affected by Cdh5-dependent Cre recombinase active during embryonic development (34, 35). Therefore, the Cre-driven mutation is usually present not only in endothelial cells, but also in a number of leukocytes. In our mouse model, macrophages of two different origins – BMDM and Kupffer cells - were characterized by significantly decreased level of Nrf2 exon 5 responsible for the transcriptional Nrf2 activity.
Atherosclerosis was induced via AAV8-mediated Pcsk9 overexpression and a high-fat diet (36). This approach successfully elevated the cholesterol level in the LDL/VLDL plasma lipoproteins, as well as the level of triglycerides in the mouse plasma in the range previously described for similar proatherogenic approaches (61–63). In the fragments of the aorta (aortic arch and branches) used for scRNAseq procedure, we observed lipids accumulation within the first layers of intima and media corresponding to fatty streaks, which represent an early stage of atherosclerotic plaque formation. In such early stage lesions, macrophage infiltration, initiation of inflammation and chemoattraction of other cell types is usually observed.
Nowadays, mainly with the use of scRNA-seq technology, several macrophage subtypes were identified in mouse and human atherosclerotic plaques, each with distinct functions and properties (7–9, 37, 38). It is well-recognized that monocyte-derived inflammatory macrophages are not the only players in atherosclerotic plaque development. Thus, we performed subclustering analysis to visualize the macrophage heterogeneity and macrophage subtype-specific effects of decreased Nrf2 transcriptional activity. That strategy uncovered 9 subpopulations of mononuclear phagocytes, in majority comprising macrophages of different origin, associated with different layers of aortic wall or peritoneal cavity. Each subpopulation was characterized by the typical markers described in previous reports (8, 9, 37, 38).
We selected three most abundant subtypes, namely Lyve1+ resident macrophages, proinflammatory macrophages and monocytes, for in-depth transcriptomic analysis in proatherogenic conditions. Tissue-resident macrophages maintain tissue homeostasis, remove pathogens and abnormal (apoptotic or senescent) cells, participate in tissue repair and regeneration (60). In the aortic wall, the tissue-resident macrophages are present mainly in adventitia in steady-state conditions, whereas in inflammation they can migrate towards the media and intima layer (64). The proinflammatory macrophages release several proinflammatory cytokines and ingest modified lipids what results in foam cells formation (64). Monocytes are blood-circulating cells that play an important role in atherosclerosis as protagonists of plaque development. When the lining of the artery is damaged, monocytes are intensively recruited to the site of injury.
The comparison between atherosclerotic Nrf2flox/flox and control normocholesterolemic Nrf2flox/flox mice revealed that each analyzed mononuclear phagocytes subset contained DEGs encoding cytokines, chemokines and their receptors involved in immune responses. This confirms that inflammation is an important component in the pathogenesis of atherosclerosis already in the early stage lesions (4, 5). In our analysis, the highest number of DEGs induced by atherogenic conditions was identified in the monocyte subset. Interestingly, among monocyte DEGs between atherosclerotic Nrf2flox/flox and control normocholesterolemic Nrf2flox/flox mice we found upregulation of NOD-like receptor pyrin domain-containing protein 3 (Nlrp3) and interleukin 1β (Il1b), two components of Nlrp3 inflammasome activation, which normally leads to Il-1β release. Inhibition of this cytokine production is currently of particular interest for the secondary prevention of atherosclerotic events (65).
Next, we investigated the effect of decreased Nrf2 transcriptional activity on the transcriptomic changes in monocytes and selected macrophage subsets in atherosclerotic mice. Previous in vitro studies indicated that Nrf2 activity may promote the anti-inflammatory phenotype of macrophages (66, 67), but the effect of Nrf2 on tissue-resident macrophages is still poorly investigated. It was shown that in alveolar macrophages Nrf2 increases phagocytic ability (68), macrophage-driven efferocytosis and apoptotic neutrophil clearance (69), and may protect from ferroptosis in sepsis-induced acute lung injury (20). Other studies performed on dermal resident macrophages showed the involvement of Nrf2 in IL-23–IL-17A–TRPV1 axis and pain perception (70) or wound repair (71). In our study, the RIG-I like pathway was the top altered pathway in atherosclerotic Nrf2Cdh5tKO Lyve1+ resident macrophages. RIG-I may activate IRFs and NF-κB, and then induce the expression of antiviral genes, such as type I interferons and pro-inflammatory cytokines (43). The latest research shows that RIG-I-like pathway is not only triggered by viral nucleic acids, but also by damage-associated molecular patterns (72). It was suggested that Nrf2 can interact with RIG-I-like pathway and modulate its function. Specifically, Nrf2 has been shown to regulate the expression of genes involved in the antiviral response, including interferon-stimulated genes (ISGs) that are downstream effectors of RIG-I-like signaling (41). Interestingly, in Lyve1+ resident macrophages of atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox, we also observed upregulation of Bcl2a1b, an IFN-dependent master switch for the function of Cd11b+ cells exerting anti-apoptotic and pro-survival effects (42). In addition, in Lyve1+ resident macrophages from atherosclerotic Nrf2Cdh5tKO mice we observed downregulation of ferroportin gene Slc40a1. This is in accordance with previous reports describing involvement of Nrf2 in regulation of this gene expression, as well as in macrophage resistance towards ferroptosis (16, 17). In this context, decreased Nrf2 activity might promote ferroptotic death of Lyve1+ resident macrophages at early stages of plaque development and in this way contribute to disease progression. This, however, would require further investigation.
In terms of bone marrow-derived macrophages, the role of Nrf2 is better recognized. Previous in vivo research demonstrated strong evidence that Nrf2 works as a negative upstream regulator of macrophage proinflammatory phenotype, while the majority of in vitro data indicates that Nrf2 deficiency promotes macrophage polarization towards proinflammatory M1 phenotype (25, 26, 31, 66). Macrophages are generally considered as non-dividing cells with limited capacity for self-renewal. However, our current data demonstrate that in Nrf2Cdh5tKO proinflammatory macrophages the pathway associated with DNA replication was significantly enriched. The precise role of DNA replication in macrophage proliferation and activation (especially in atherosclerosis) is still not fully understood. There are indications that macrophages may undergo DNA replication and proliferation upon certain, usually considered as harmful or danger, stimuli (73, 74). The upregulation of Atr and Apex1 in Nrf2Cdh5tKO proinflammatory macrophages may suggest increased oxidative stress and DNA damage that often accompany Nrf2 deficiency (74). Some data indicate that high level of DNA synthesis aiming at DNA repair in macrophages is a response to oxidative DNA damage and is highly associated with apoptotic cell death (11). In chronic inflammation, DNA synthesis may raise generation of polyploid macrophages (73). In addition, we observed that Nrf2 deficiency in proinflammatory macrophages was associated with upregulation of genes from the CLR family, including Mincle and MCL, involved in lipids recognition (including cholesterol) and their endocytosis (75). Our transcriptomic analysis of proinflammatory macrophages from atherosclerotic Nrf2Cdh5tKO mice also revealed decreased expression of genes connected with autophagy (Mcm7, S100a4 and Gpnmb; Table 5) and genes described as ferroptosis-related (Hells and Cp; Table 5). Observations of the last years indicate that the process of autophagy and ferroptosis are intertwined (76). Autophagy promotes intracellular lipid hydrolysis and cholesterol efflux thereby inhibiting development of macrophage-derived foam cells. Inhibition of autophagy in macrophages leads to foam cells formation, macrophage death, pro-inflammatory factor release and contributes to atherogenesis (77). Autophagy plays a key role in inhibiting ferroptosis of macrophages via maintaining cellular iron homeostasis and cellular reactive oxygen species generation, and alleviates atherosclerosis (78, 79). Overall, our data indicate that Nrf2 deficiency in atherosclerotic proinflammatory macrophages affects the expression of genes involved in DNA replication and repair mechanisms, as well as in the recognition of lipids and cell death pathways, what may impact these cells activation, phagocytic ability, and survival.
In atherosclerotic conditions, Nrf2-deficiency caused transcriptomic changes also in the monocyte subset. Some proinflammatory genes were downregulated and anti-inflammatory were upregulated, whereas on the other hand, the expression of some chemoattractants was increased when compared to monocytes from the aortas of atherosclerotic Nrf2flox/flox mice. The observed changes suggest modulation of the immune phenotype of monocytes, but not necessarily towards the proinflammatory one. In addition, in atherosclerotic Nrf2Cdh5tKO vs. atherosclerotic Nrf2flox/flox monocytes, increased level of Mmp12 and protease inhibitors may suggest their involvement in ECM remodeling and monocyte infiltration.
In conclusion, this exploratory study identified several subtype-specific differences in monocytes and two selected, the most numerous in the aortic wall, types of macrophages from atherosclerotic Nrf2Cdh5tKO mice. The introduced proatherogenic factors caused significant changes particularly in the expression of genes encoding inflammatory cytokines and chemokines. Comparison of atherosclerotic Nrf2flox/flox and Nrf2Cdh5tKO mice enabled identification of Nrf2-dependent macrophage subtype-specific transcriptomic changes associated with inflammation, iron homeostasis, DNA repair, cell injury and death pathways. Our data demonstrate a possible link between ferroptosis and inflammatory microenvironment appearing at a very early stage of atherogenesis. A limitation of this study can be our mouse model based on Cdh5-dependent Cre activity. First, because Nrf2-deficient endothelial cells may affect the microenvironment and their interaction with other cell types (e.g. monocytes) may differ. Second, this mouse model is characterized by a significant downregulation, but not a complete knockout of Nrf2 in mononuclear phagocytes. Although, we think that such model better reflects the observed in humans aging-associated decline of Nrf2 activity, in the future it may be worth to reproduce some of the findings in mice with the lysozyme 2 (LysM) Cre-driven recombination.
Data availability statement
The data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE245820.
Ethics statement
The animal study was approved by the 2nd Institutional Animal Care and Use Committee (IACUC) in Kraków, Poland. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
KSa and AJ-K designed the study. KSa, MS, IK, and KSz performed the experiments. MS and KSa performed the bioinformatic analyses and data visualization. KSa and AJ-K were involved in data interpretation and wrote the manuscript. JCS, PB, and JD provided the intellectual contribution and revised the manuscript. AJ-K secured funding and coordinated the project. All authors provided constructive feedback, helped shape the manuscript and approved the submitted version.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant ERA-CVD/MEND-AGE/6/2019 from the National Centre for Research and Development and project No. 2021/43/B/NZ5/03336 financed from the funds of the National Science Center, Poland.
Acknowledgments
We kindly acknowledge Dr. Anna Grochot-Przęczek (Department of Medical Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University) for providing C57BL/6-Nfe2l2tm1.1Sred/SbisJ and B6;129-Tg(Cdh5-cre)1Spe/J mice for breeding. Staff members in the Animal Facility of the Faculty of Biochemistry, Biophysics and Biotechnology are kindly acknowledged for mice breeding and technical assistance with intravenous AAV8-Pcsk9 delivery. Dr. Aleksandra Kopacz (Department of Medical Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University) is kindly acknowledged for any advice rendered in conducting experiments. Dr. Krzysztof Guzik (Department of Immunology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University) is kindly acknowledged for comments and suggestions regarding the manuscript. Schematic Figure 3A and 3B were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Strong JP, Malcom GT, Newman WP, Oalmann MC. Early lesions of atherosclerosis in childhood and youth: natural history and risk factors. J Am Coll Nutr (1992) 11(sup1):51S–4S. doi: 10.1080/07315724.1992.10737984
2. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
3. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med (2010) 49(11):1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
4. Cochain C, Zernecke A. Macrophages in vascular inflammation and atherosclerosis. Pflüg Arch - Eur J Physiol (2017) 469(3–4):485–99. doi: 10.1007/s00424-017-1941-y
5. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
6. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464(7293):1357–61. doi: 10.1038/nature08938
7. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E ad D, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med (2019) 25(10):1576–88. doi: 10.1038/s41591-019-0590-4
8. Zernecke A, Erhard F, Weinberger T, Schulz C, Ley K, Saliba AE, et al. Integrated single-cell analysis-based classification of vascular mononuclear phagocytes in mouse and human atherosclerosis. Cardiovasc Res (2022), 119(8):1676–89. doi: 10.1093/cvr/cvac161
9. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res (2018) 122(12):1661–74. doi: 10.1161/CIRCRESAHA.117.312509
10. Schrijvers D, Demeyer G, Herman A, Martinet W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res (2007) 73(3):470–80. doi: 10.1016/j.cardiores.2006.09.005
11. Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res (2000) 45(3):736–46. doi: 10.1016/S0008-6363(99)00235-7
12. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol (2019) 10:306. doi: 10.3389/fphar.2019.00306
13. Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernández-Ruiz J, Cuadrado A. Pharmacological targeting of the transcription factor nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signal (2011) 14(12):2347–60. doi: 10.1089/ars.2010.3731
14. Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, et al. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res (2007) 100(12):1703–11. doi: 10.1161/CIRCRESAHA.107.151720
15. Kisucka J, Chauhan AK, Patten IS, Yesilaltay A, Neumann C, Van Etten RA, et al. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ Res (2008) 103(6):598–605. doi: 10.1161/CIRCRESAHA.108.174870
16. Namgaladze D, Fuhrmann D, Brüne B. Interplay of Nrf2 and BACH1 in inducing ferroportin expression and enhancing resistance of human macrophages towards ferroptosis. Cell Death Discovery (2022) 8:327. doi: 10.1038/s41420-022-01117-y
17. Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica (2010) 95(8):1261–8. doi: 10.3324/haematol.2009.020123
18. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol (2020) 27(4):436–47. doi: 10.1016/j.chembiol.2020.03.011
19. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal (2018) 29(17):1756–73. doi: 10.1089/ars.2017.7176
20. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discovery (2022) 8(1):1–11. doi: 10.1038/s41420-021-00807-3
21. Kopacz A, Kloska D, Targosz-Korecka M, Zapotoczny B, Cysewski D, Personnic N, et al. Keap1 governs ageing-induced protein aggregation in endothelial cells. Redox Biol (2020) 34:101572. doi: 10.1016/j.redox.2020.101572
22. Ooi B, Goh B, Yap W. Oxidative stress in cardiovascular diseases: involvement of nrf2 antioxidant redox signaling in macrophage foam cells formation. Int J Mol Sci (2017) 18(11):2336. doi: 10.3390/ijms18112336
23. Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, et al. NF-E2–related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler Thromb Vasc Biol (2011) 31(1):58–66. doi: 10.1161/ATVBAHA.110.210906
24. Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, et al. Disruption of nrf2, a key inducer of antioxidant defenses, attenuates apoE-mediated atherosclerosis in mice. PloS One (2008) 3(11):e3791. doi: 10.1371/journal.pone.0003791
25. Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Mäkinen PI, et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res (2013) 98(1):107–15. doi: 10.1093/cvr/cvt008
26. Harada N, Ito K, Hosoya T, Mimura J, Maruyama A, Noguchi N, et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic Biol Med (2012) 53(12):2256–62. doi: 10.1016/j.freeradbiomed.2012.10.001
27. Ruotsalainen AK, Lappalainen JP, Heiskanen E, Merentie M, Sihvola V, Näpänkangas J, et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc Res (2019) 115(1):243–54. doi: 10.1093/cvr/cvy143
28. Tomczyk M, Kraszewska I, Mąka R, Waligórska A, Dulak J, Jaźwa-Kusior A. Characterization of hepatic macrophages and evaluation of inflammatory response in heme oxygenase-1 deficient mice exposed to scAAV9 vectors. PloS One (2020) 15(10):e0240691. doi: 10.1371/journal.pone.0240691
29. Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. Cold Spring Harb Protoc (2008) 2008(12):pdb.prot5080. doi: 10.1101/pdb.prot5080
30. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc (2009) 4(1):44–57. doi: 10.1038/nprot.2008.211
31. Collins AR, Gupte AA, Ji R, Ramirez MR, Minze LJ, Liu JZ, et al. Myeloid deletion of nuclear factor erythroid 2–related factor 2 increases atherosclerosis and liver injury. Arterioscler Thromb Vasc Biol (2012) 32(12):2839–46. doi: 10.1161/ATVBAHA.112.300345
32. Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci (2013) 126(Pt 12):2545–9. doi: 10.1242/jcs.124529
33. He Q, Li X, Singh K, Luo Z, Meija-Cordova M, Jamalpour M, et al. The Cdh5-CreERT2 transgene causes conditional Shb gene deletion in hematopoietic cells with consequences for immune cell responses to tumors. Sci Rep (2019) 9(1):7548. doi: 10.1038/s41598-019-44039-z
34. Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature (2009) 457(7231):887–91. doi: 10.1038/nature07619
35. Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, et al. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn Off Publ Am Assoc Anat (2006) 235(3):759–67. doi: 10.1002/dvdy.20643
36. Bjørklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res (2014) 114(11):1684–9. doi: 10.1161/CIRCRESAHA.114.302937
37. Liu X, Chen W, Zhu G, Yang H, Li W, Luo M, et al. Single-cell RNA sequencing identifies an Il1rn+/Trem1+ macrophage subpopulation as a cellular target for mitigating the progression of thoracic aortic aneurysm and dissection. Cell Discovery (2022) 8(1):1–21. doi: 10.1038/s41421-021-00362-2
38. Harasymowicz NS, Rashidi N, Savadipour A, Wu CL, Tang R, Bramley J, et al. Single-cell RNA sequencing reveals the induction of novel myeloid and myeloid-associated cell populations in visceral fat with long-term obesity. FASEB J Off Publ Fed Am Soc Exp Biol (2021) 35(3):e21417. doi: 10.1096/fj.202001970R
39. McQueen LW, Ladak SS, Abbasciano R, George SJ, Suleiman MS, Angelini GD, et al. Next-generation and single-cell sequencing approaches to study atherosclerosis and vascular inflammation pathophysiology: A systematic review. Front Cardiovasc Med (2022) 9:849675. doi: 10.3389/fcvm.2022.849675
40. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol (2017) 17(7):451–60. doi: 10.1038/nri.2017.42
41. Calcagno DM, Ng RP, Toomu A, Zhang C, Huang K, Aguirre AD, et al. The myeloid type I interferon response to myocardial infarction begins in bone marrow and is regulated by Nrf2-activated macrophages. Sci Immunol (2020) 5(51):eaaz1974. doi: 10.1126/sciimmunol.aaz1974
42. Medina-Echeverz J, Haile LA, Zhao F, Gamrekelashvili J, Ma C, Métais JY, et al. IFN-γ regulates survival and function of tumor-induced CD11b+Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur J Immunol (2014) 44(8):2457–67. doi: 10.1002/eji.201444497
43. Ireton RC, Gale M. RIG-I like receptors in antiviral immunity and therapeutic applications. Viruses (2011) 3(6):906–19. doi: 10.3390/v3060906
44. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol (2020) 8:590226. doi: 10.3389/fcell.2020.590226
45. Liebermann DA, Hoffman B. Gadd45 in stress signaling. J Mol Signal (2008) 3:15. doi: 10.1186/1750-2187-3-15
46. Nakaya Y, Lilue J, Stavrou S, Moran EA, Ross SR. AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio (2017) 8(4):e00944-17. doi: 10.1128/mBio.00944-17
47. Xie Y, Wang B. Downregulation of TNFAIP2 suppresses proliferation and metastasis in esophageal squamous cell carcinoma through activation of the Wnt/β-catenin signaling pathway. Oncol Rep (2017) 37(5):2920–8. doi: 10.3892/or.2017.5557
48. Jia L, Shi Y, Wen Y, Li W, Feng J, Chen C. The roles of TNFAIP2 in cancers and infectious diseases. J Cell Mol Med (2018) 22(11):5188–95. doi: 10.1111/jcmm.13822
49. Yoshida K, Weichselbaum R, Kharbanda S, Kufe D. Role for Lyn tyrosine kinase as a regulator of stress-activated protein kinase activity in response to DNA damage. Mol Cell Biol (2000) 20(15):5370–80. doi: 10.1128/MCB.20.15.5370-5380.2000
50. Zhang Z, Bao Z, Gao P, Yao J, Wang P, Chai D. Diverse roles of F-boxProtein3 in regulation of various cellular functions. Front Cell Dev Biol (2022) 9:802204. doi: 10.3389/fcell.2021.802204
51. Ohno Y, Suzuki-Takedachi K, Yasunaga S, Kurogi T, Santo M, Masuhiro Y, et al. Manipulation of cell cycle and chromatin configuration by means of cell-penetrating geminin. PloS One (2016) 11(5):e0155558. doi: 10.1371/journal.pone.0155558
52. Tsuji T, Lau E, Chiang GG, Jiang W. The role of Dbf4/Drf1-dependent kinase Cdc7 (Ddk) in DNA damage checkpoint control. Mol Cell (2008) 32(6):862–9. doi: 10.1016/j.molcel.2008.12.005
53. Taira N, Yamaguchi T, Kimura J, Lu ZG, Fukuda S, Higashiyama S, et al. Induction of amphiregulin by p53 promotes apoptosis via control of microRNA biogenesis in response to DNA damage. Proc Natl Acad Sci U.S.A. (2014) 111(2):717–22. doi: 10.1073/pnas.1313675111
54. Vickridge E, Faraco CCF, Tehrani PS, Ramdzan ZM, Djerir B, Rahimian H, et al. The DNA repair function of BCL11A suppresses senescence and promotes continued proliferation of triple-negative breast cancer cells. NAR Cancer (2022) 4(4):zcac028. doi: 10.1093/narcan/zcac028
55. Richardson MB, Williams SJ. MCL and mincle: C-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front Immunol (2014) 5:288. doi: 10.3389/fimmu.2014.00288
56. Hermiston ML, Zikherman J, Zhu JW. CD45, CD148, and lyp/pep: critical phosphatases regulating src family kinase signaling networks in immune cells. Immunol Rev (2009) 228(1):288–311. doi: 10.1111/j.1600-065X.2008.00752.x
57. Guyonnet L, Czopek A, Farrah TE, Baudrie V, Bonnin P, Chipont A, et al. Deletion of the myeloid endothelin-B receptor confers long-term protection from angiotensin II-mediated kidney, eye and vessel injury. Kidney Int (2020) 98(5):1193–209. doi: 10.1016/j.kint.2020.05.042
58. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
59. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, et al. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun (2012) 425(2):121–6. doi: 10.1016/j.bbrc.2012.07.011
60. Wu Y, Hirschi KK. Tissue-resident macrophage development and function. Front Cell Dev Biol (2021) 8:617879. doi: 10.3389/fcell.2020.617879
61. Kumar S, Kang DW, Rezvan A, Jo H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest (2017) 97(8):935–45. doi: 10.1038/labinvest.2017.47
62. Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, et al. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol (2015) 35(1):50–9. doi: 10.1161/ATVBAHA.114.303617
63. Keeter WC, Carter NM, Nadler JL, Galkina EV. The AAV-PCSK9 murine model of atherosclerosis and metabolic dysfunction. Eur Heart J Open (2022) 2(3):oeac028. doi: 10.1093/ehjopen/oeac028
64. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res (2020) 127(3):402–26. doi: 10.1161/CIRCRESAHA.120.316903
65. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
66. Wang L, He C. Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front Immunol (2022) 13:967193. doi: 10.3389/fimmu.2022.967193
67. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7(1):11624. doi: 10.1038/ncomms11624
68. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med (2011) 3(78):78ra32. doi: 10.1126/scitranslmed.3002042
69. Reddy NM, Tamatam CM, Aparna A, Reddy SP. Nrf2 is required for optimal alveolar-macrophage-mediated apoptotic neutrophil clearance after oxidant injury. Antioxidants (2022) 11(2):212. doi: 10.3390/antiox11020212
70. Tanaka T, Okuda H, Isonishi A, Terada Y, Kitabatake M, Shinjo T, et al. Dermal macrophages set pain sensitivity by modulating the amount of tissue NGF through an SNX25–Nrf2 pathway. Nat Immunol (2023), 24(3): 439–51. doi: 10.1038/s41590-022-01418-5
71. Eichenfield DZ, Troutman TD, Link VM, Lam MT, Cho H, Gosselin D, et al. Tissue damage drives co-localization of NF-κB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. eLife (2016) 5:e13024. doi: 10.7554/eLife.13024
72. Wang PT, Li N, Wang XY, Chen JL, Geng CH, Liu ZQ, et al. RIG-I, a novel DAMPs sensor for myoglobin activates NF-κB/caspase-3 signaling in CS-AKI model. Mil Med Res (2021) 8:37. doi: 10.1186/s40779-021-00333-4
73. Horn V, Triantafyllopoulou A. DNA damage signaling and polyploid macrophages in chronic inflammation. Curr Opin Immunol (2018) 50:55–63. doi: 10.1016/j.coi.2017.11.002
74. Zhao H, Song L, Ma N, Liu C, Dun Y, Zhou Z, et al. The dynamic changes of Nrf2 mediated oxidative stress, DNA damage and base excision repair in testis of rats during aging. Exp Gerontol (2021) 152:111460. doi: 10.1016/j.exger.2021.111460
75. Kostarnoy AV, Gancheva PG, Kireev II, Soloviev AI, Lepenies B, Kulibin AY, et al. A mechanism of self-lipid endocytosis mediated by the receptor Mincle. Proc Natl Acad Sci U.S.A. (2022) 119(30):e2120489119. doi: 10.1073/pnas.2120489119
76. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res (2016) 26(9):1021–32. doi: 10.1038/cr.2016.95
77. Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun (2017) 8(1):15750. doi: 10.1038/ncomms15750
78. Yu W, Liu W, Xie D, Wang Q, Xu C, Zhao H, et al. High level of uric acid promotes atherosclerosis by targeting NRF2-mediated autophagy dysfunction and ferroptosis. Oxid Med Cell Longev (2022) 2022:1–21. doi: 10.1155/2022/9304383
Keywords: atherosclerosis, scRNA-seq, Nrf2, macrophages, heterogeneity, monocytes
Citation: Sarad K, Stefańska M, Kraszewska I, Szade K, Sluimer JC, Błyszczuk P, Dulak J and Jaźwa-Kusior A (2023) Single-cell transcriptomics reveals subtype-specific molecular profiles in Nrf2-deficient macrophages from murine atherosclerotic aortas. Front. Immunol. 14:1249379. doi: 10.3389/fimmu.2023.1249379
Received: 28 June 2023; Accepted: 17 October 2023;
Published: 27 October 2023.
Edited by:
David Masson, Université de Bourgogne, FranceReviewed by:
Kobina Essandoh, University of Michigan, United StatesGiulia Chinetti, INSERM U1065 Centre Meíditerraneíen de Meídecine Moleículaire, France
Copyright © 2023 Sarad, Stefańska, Kraszewska, Szade, Sluimer, Błyszczuk, Dulak and Jaźwa-Kusior. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Jaźwa-Kusior, YWduaWVzemthLmphendhQHVqLmVkdS5wbA==