
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 17 October 2023
Sec. Alloimmunity and Transplantation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1248027
This article is part of the Research TopicEffects of cell interactions at the endothelium in leukocyte differentiation and polarization during inflammationView all 13 articles
 Shaoxun Wang1,2,3†
Shaoxun Wang1,2,3† Guiyu Song1,2,4†
Guiyu Song1,2,4† Mahsa Nouri Barkestani1,2†
Mahsa Nouri Barkestani1,2† Zuzana Tobiasova5Qianxun Wang1,2
Zuzana Tobiasova5Qianxun Wang1,2 Quan Jiang1,2Roberto Lopez6Yasmin Adelekan-Kamara7
Quan Jiang1,2Roberto Lopez6Yasmin Adelekan-Kamara7 Matthew Fan5
Matthew Fan5 Jordan S. Pober5George Tellides3
Jordan S. Pober5George Tellides3 Dan Jane-wit1,2*
Dan Jane-wit1,2*Introduction: Ischemia reperfusion injury (IRI) confers worsened outcomes and is an increasing clinical problem in solid organ transplantation. Previously, we identified a “PtchHi” T-cell subset that selectively received costimulatory signals from endothelial cell-derived Hedgehog (Hh) morphogens to mediate IRI-induced vascular inflammation.
Methods: Here, we used multi-omics approaches and developed a humanized mouse model to resolve functional and migratory heterogeneity within the PtchHi population.
Results: Hh-mediated costimulation induced oligoclonal and polyclonal expansion of clones within the PtchHi population, and we visualized three distinct subsets within inflamed, IRI-treated human skin xenografts exhibiting polyfunctional cytokine responses. One of these PtchHi subsets displayed features resembling recently described T peripheral helper cells, including elaboration of IFN-y and IL-21, expression of ICOS and PD-1, and upregulation of positioning molecules conferring recruitment and retention within peripheral but not lymphoid tissues. PtchHi T cells selectively homed to IRI-treated human skin xenografts to cause accelerated allograft loss, and Hh signaling was sufficient for this process to occur.
Discussion: Our studies define functional heterogeneity among a PtchHi T-cell population implicated in IRI.
Ischemia-reperfusion injury (IRI) is a condition where donor tissues subjected to protracted ex vivo hypoxia develop complement-mediated damage upon surgical revascularization. Tissues subjected to IRI show poor peri-operative function and have worsened short- and long-term survival (1–5). In an effort to increase organ availability, recent program changes to organ allocation have newly allowed tissues from suboptimal and geographically distant donors to become eligible for transplantation (6–9). Post-hoc analyses showed that these newly eligible tissues were susceptible to developing IRI, and the cumulative prevalence of IRI has consequently risen by ~25% (10–13). IRI is now estimated to affect between 40% and 50% of transplant recipients and is an increasing problem in the field of transplantation.
To address this emerging issue, we have investigated how IRI affected the capacity of donor endothelial cells (ECs) to stimulate allogeneic T cells (14–16). During IRI, developmental pathways, including Hedgehog (Hh) signaling become re-activated within injured tissues, ostensibly to promote wound repair (17, 18). Hh signaling supports prenatal organogenesis, and postnatal reactivation of Hh signaling specifies programs for cell proliferation and migration to regulate tissue regeneration (19). Mural cells including ECs, pericytes, smooth muscle cells, and fibroblasts, respond to Hh-mediated growth and positioning signals to promote angiogenesis and wound closure (20–22).
CD4+ T cells, while lacking primary cilium, organelles mediating Hh signaling, highly express Hh pathway components, including Ptch, Smo, and Gli effectors, and are capable of both receipt and transmission of Hh inputs, possibly via signals derived from immune synapses (23). Hh signaling encodes programs for positive costimulation (24, 25) to regulate type 1 (14), Th2 (26), Treg (27), and Th17 (28) effector polarization.
Due to increasing rates of IRI, recent studies have explored the effects of exogenous Hh ligands as therapies for mitigating IRI-induced tissue injury. These reports showed that Hh ligands, while enhancing peri-operative tissue function (29–31), paradoxically exacerbated post-operative inflammation and led to accelerated graft loss following IRI (32–35). To explain these paradoxical findings, we hypothesized that Hh programs specifying proliferation and migration and intended for wound repair are pathologically subsumed by allogeneic T cells as a costimulatory signal. Following the acquisition of Hh costimulation, respondent Tmem went on to acquire Hh-related growth and migratory phenotypes to mediate IRI-associated tissue injury.
We previously explored this notion using a humanized mouse model incorporating IRI-treated human coronary artery xenografts (14, 15). We found that soluble and surface-bound Hh ligands, including Sonic, Desert, and Indian Hedgehog, were upregulated by complement membrane attack complexes (MACs) during the reperfusion phase of IRI, and the same Hh ligands delivered costimulatory but not mitogenic signals to memory CD4+CD45RO+ T cells (Tmem), endowing these cells with heightened effector and migratory responses (14).
Hh signaling upregulates the expression of its own component signaling molecules (19), and exploiting this, we found that the observed functional alterations induced by Hh selectively occurred on Tmem highly expressing Ptch1, the surface receptor for Hh ligands. We found that surface levels of Ptch1, a marker for Hh costimulatory strength, were directly proportional to the ability of these cells to mediate chronic inflammation of IRI-treated human artery xenografts (14).
Frequencies of PtchHi T cells were increased in patients with delayed graft function (DGF), a clinical manifestation of IRI in renal transplantation (14), and the PtchHi population contained a subset displaying a PD-1hiICOShi CCR2+CCR5+CXCR5− surface phenotype (14). This subset exhibited features characteristic of recently described T peripheral helper (TPH) cells (35), including expression of ICOS, elaboration of IFN-γ and IL-21, and vigorous homing to inflamed tissues (15). To understand the pathogenic effects of PtchHi Tmem, we sought to identify populations within the PtchHi subset showing similar effector and homing phenotypes relevant to IRI. To do this, we applied multi-omics approaches and tested the relevance of PtchHi subsets using a humanized mouse model. This model incorporated human IRI-treated allogeneic skin and autologous lymphoid tissues to allow interrogation of the effector and migratory properties of subsets contained within PtchHi Tmem.
All protocols were approved by the Yale Institutional Review Board (Protocol No. 0601000969). PBMCs were isolated from leukopacks obtained from the Red Cross (Philadelphia, PA) using density centrifugation as described previously and cryopreserved in liquid nitrogen. CD4+CD45RO+ T cells were isolated from thawed cryovials using magnetic bead separation kits (Miltenyi, Charlestown, MA, USA) with HLA-DR Ab (clone L243, Novus No. NB100-77855) and CD45RA Ab-negative depletion (10 μL per cryovial, eBiosciences, 14-0458-82, San Diego, CA, USA). Where indicated, flat-bottom 96-well microtiter plates were coated with anti-CD3 Ab (1 μg/mL, eBiosciences, No. 16003785) in sterile PBS at 4°C overnight, and the following day, isolated CD4+CD45RO+ T cells (Tmem) were pretreated with soluble anti-CD28 Ab (1 μg/mL, eBiosciences, No. 16-0281-82) or SAG (15 μM) for 1 h prior to addition to anti-CD3 Ab-coated plates. Tmem were stimulated for 48 h prior to use in downstream applications including TCR deep sequencing, single-cell proteomics, mass cytometry, and passive transfer into humanized mice.
For TCR deep sequencing, Tmem were isolated as above and seeded in 96-well flat-bottomed microtiter plates at 50,000 cells/well. PtchHi and PtchLo Tmem were treated with anti-CD3 Ab and soluble anti-CD28 Ab or SAG were stimulated for 48 h as above, and relevant PtchHi and PtchLo populations were FACS sorted, homogenized via QiaShredder spin columns (Qiagen, #79656, Hilden, Germany) for 2 min at room temperature, and total DNA was isolated using spin columns (Qiagen). Purified total DNA at 200 ng–1 µg was suspended in nuclease-free H2O (30 μL) and submitted for TCR deep sequencing (Adaptive Biotechnologies, Seattle, WA, USA). The CDR3 region of the TCR-α and TCR-β chains were separately amplified in multi-plexed PCR reactions using a panel of proprietary primers.
The analysis results, quality control, and summary statistics of CDR3 sequences (templates) were performed using the online ImmunoSEQ Analyzer portal provided by Adaptive Biotechnologies (http://www.immunoseq.com). All samples were quantified for the absolute total rearrangements, which is the sum of unique detectable CDR3 sequences (or rearrangements) in the TCR-α and TCR-β chains.
T-cell surface staining used the following primary antibodies at 1:100 for 1 h at 4°C in PBS containing 3% BSA and 0.01% NaN3: CD4+ (Biolegend, San Diego, CA, USA), HLA-DR FITC (Biolegend, No. 307604), PD-1 PE (eBiosciences, No. 12-2799-42), CCR2 APC (Biolegend, No. 257208), CXCR5 Pacific Blue (Biolegend, No. 356918), and Ptch1 (Cell Signal, clone C53A3, No. 2468T, Cell Signaling Technology, Danvers, MA, USA). For intracellular staining, T cells were fixed and permeabilized using a FOXP3 Staining Kit according to the manufacturer’s specifications (R&D Systems, Cell Signaling Technology, Danvers, MA, USA). Prior to fixation and permeabilization, cells were stained using the following antibodies at 1:100 for 1 h at 4°C: Gli1 (Cell Signal, clone C68H3, No. 3538T), NLRP3 (Cell Signal, clone DRD8T, No. 15101), and cleaved caspase-1 (Cell Signal, Cat. No. 4199). For cellular staining, antibodies included IFN-γ PE (Biolegend), IL-4 PE/Cy7 (Biolegend, No. 500824), and cleaved caspase-1 (Cell Signaling, clone D57A2, No. 4199). T cells were stimulated for 2 h with PMA/ionomycin prior to the addition of GolgiStop (Becton Dickinson, Franklin Lakes, NJ, USA) for 4 h prior to harvest and FACS analysis as previously described (14, 15). T cells were stained with CFSE (Molecular Probes, Fanklin Lakes, NJ, USA) as previously described (36). T cells were gated and analyzed using an LSR II flow cytometer (Becton Dickinson) and sorted using a FACSAria cell sorter (Becton Dickinson).
Transplanted skin and lymph nodes were embedded into an optimal cutting temperature compound (OCT) prior to sectioning. Human CD31 and Sonic Hedgehog were detected using RNA probes Hs-PECAM1-O2 (No. 858151, Advanced Cell Diagnostics, CA, Westborough, MA, USA) and Hs-SHH-C2 (No. 600951-C2 per manufacturer’s specification prior to target amplification by RNAscope Fluorescent Multiplex Reagent Kit (No. 320851, Advanced Cell Diagnostics, CA). Imaging was conducted by laser scanning confocal microscopy using a Leica TCS SP8 Gated STED 3X super-resolution microscope (Leica Microsystems Inc., IL, Wetzlar, Germany). Images were taken at ×400 magnification. Z-stacks were taken at 1 μm apart. A total of 15 Z images were taken, and the Z-projection images were shown. Merged images were deconvoluted into red, green, and, in the case of triple colocalization studies, blue color channels. Pixels ≤1,500 were selected for analysis in the case of Sonic Hedgehog (Shh) and CD31 and sequentially analyzed for colocalization, with colocalized pixels being defined as < 50 pixel diameters.
Polystyrene beads containing fluorescent dyes were coated with a capture antibody specific to a given protein analyte. Color-coded beads were then analyzed using a bead analyzer (Bio-Plex 200) containing a dual-laser system where the fluorescent dye within each bead is activated and a second laser excites the fluorescent conjugate (streptavidin-PE) that has been bound to the beads during the assay. The amount of conjugate detected by the analyzer is in direct proportion to the amount of the target analyte, which can be quantified using a standard curve (Eve Technologies, Calgary, Canada). Due to wide ranges in quantities among elaborated cytokines, heatmap colors for each row were computed relative to the overall average for that one particular cytokine.
Naïve Tmem were stimulated with anti-CD3 (1 μg/mL) in the presence of SAG (1 μM) for 48 h, and cells highly expressing Ptch1 were FACS sorted as “PtchHi” Tmem. As a comparator, a second set of Tmem stimulated with anti-CD3 in the presence of soluble anti-CD28 (1 μg/mL) for 48 h, and cells showing low expression of Ptch1 from these samples were similarly FACS sorted as “PtchLo” Tmem. PtchHi and PtchLo Tmem were subsequently subjected to 32-plex single-cell proteomics (Isoplexis, Branford, CT, USA). Viable T cells were washed and resuspended in complete RPMI media at a density of 1 × 106 cells/mL. Approximately 100 µL of T-cell suspension were seeded into the 96-well flat-bottom plate and stimulated with PMA/ionomycin for 4 h at 37°C. Approximately 30 µL of the cell suspension was loaded into an IsoCode chip containing ~12,000 microchambers prepatterned with a complete copy of a 32-plex antibody array. After 16-h-on-chip incubation at 37°C, protein signals from ~1,000 live single cells were captured and analyzed by fluorescence ELISA-based assay. Cytokine effector responses, including measures of polyfunctionality, were analyzed by IsoSpeak software. Advanced data visualization algorithms, including polyfunctional heatmap, polyfunctional activation topology-principal component analysis (PAT-PCA), and t-SNE were performed using the IsoSpeak software suite.
Fresh tonsillar and skin tissues were obtained as de-identified samples from the Yale Department of Pathology under IRB-approved protocols approved by the Yale Human Investigations Committee (No. 25173, No. 2000031850). Human tonsillar tissues were obtained from children aged 5–15 years old. Human skin tissues were obtained from adults aged 20–40 years old undergoing mammoplasty. All animal protocols were approved by the Yale Institutional Animal Care and Use Committee (Protocol No.: 2021-20175) and were performed in accordance with institutional guidelines. A portion of fresh, human tonsillar tissue was implanted subcutaneously in the ear of SCID/beige immunodeficient hosts (H-2d) lacking T cells and B cells and containing dysfunctional NK cells, and the remainder of the tonsillar tissue from the same host was cryopreserved. Upon notification of availability, fresh human skin tissues were obtained and subdermal fat tissues were removed. To induce IRI, full-thickness skin segments were then placed at 0% FiO2 and 5% CO2 for 37°C for 8 h in DMEM in organ culture prior to implantation onto the dorsal flanks of SCID/bg hosts containing subcutaneous human LN implants.
For skin transplantation, full-thickness human skin was prepared using a dermatome, grafted via sutures on the dorsal flank of murine hosts, and secured with a bandage for 7 days. For passive transfer, the cryopreserved portions of human LN tissues were placed into single-cell suspension, stimulated with anti-CD3 (1 μg/mL) in the presence of anti-CD28 (1 μg/mL) or SAG (15 μM) for 48 h prior to FACS sorting of relevant PtchHi and PtchLo populations, labeling with intravital dyes (IVIS 680, PerkinElmer; IVIS 770, PerkinElmer, Waltham, MA, USA), and passive transfer of these cells at 1 × 106 cells per host via jugular vein injection. Passively transferred cells were autologous to implanted human LN and allogeneic to IRI-treated skin grafts. Where indicated, following passive transfer, hosts receive once-daily injections of SAG (5 mg) i.p. for 3 days. Sample sizes for all studies were n = 4–5 hosts per group to enable the calculation of standard deviations. Skin xenografts from the same human donor were randomized among treatment groups. The percentage of necrosis area of the skin graft was calculated over time, and ≥80% skin necrosis was operationally used to define rejection (37).
The relevant metal was conjugated to each antibody according to the manufacturer’s specifications (Fluidigm Maxpar, South San Francisco, CA, USA). Antibody–metal pairs are shown in Table 1. Following isolation of CD4+CD45RO+ Tmem via magnetic bead separation, Tmem were resuspended with T-cell media containing 1:10,000 benzonase (25 U/mL), washed, blocked with Fc-receptor blocking solution (Miltenyi), and Ab cocktail was added at a volume of 50 μL to each sample for a total staining volume of 100 μL for 30 mintes at room temperature. Samples were then washed and incubated with 1:500 dilution of 25 cisplatin for a final concentration of 50 μM for 1 min at room temperature. Samples were then washed, fixed, and permeabilized (eBioscience) at 4°C overnight. The next day, Ir-intercalator (Fluidigm) was added at 1:1,000 dilution in 1 mL fixation/permeabilization butter for a final concentration of 125 nM at 4°C overnight. Cells were then washed and resuspended in MilliQ water prior to analysis using the CyTOF Helios instrument (Fluidigm). Metal-conjugated antibodies used in the study are listed in Table 1.
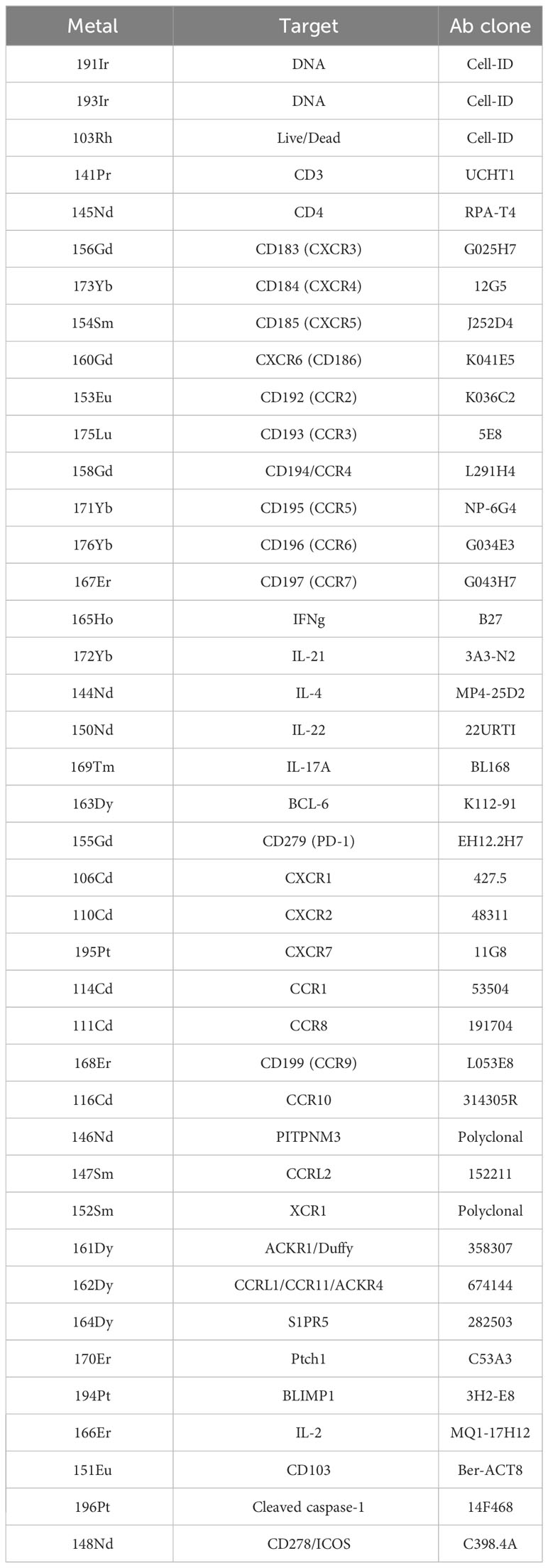
Table 1 CyTOF antibodies.
TCR repertoire richness was determined by applying a clonality score, adapted from the Gini–Simpson index, calculated as the square root of the Simpson diversity metric based on productive CDR3 rearrangements. Simpson’s clonality was calculated as the square root of the sum of all observed rearrangements over the square fractional abundances of each rearrangement and was used due to its robustness across differences in sampling depths. Simpson’s clonality values range between 0 and 1, with values approaching 1 representing a monoclonal population. Clone distribution slopes were calculated as the slope of the best-fit line on a log–log plot comparing the range of clonal frequencies to the number of clones at each frequency as previously described (38). An increased clone distribution slope represents increased populational diversity as more rare clones become expanded. We assessed the differences in total rearrangements and clonality among different groups using the nonparametric Wilcoxon signed-rank tests. Comparisons between groups in mass cytometric analyses were performed using a two-sample Student’s t-test using Origin computer software. Standard deviations are reported throughout the text.
All data and methods are available from the authors upon reasonable request. The following transcriptomic datasets were retrieved from the Gene Expression Omnibus: GSE147089 (n = 224) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147089], GSE112943 (n = 21) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112943], and GSE97779 (n = 23) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97779]. TCR deep sequencing data are available at https://clients.adaptivebiotech.com/pub/wang-2023-fi. All data are available from the authors upon reasonable request.
Human ECs, unlike rodent ECs, express costimulatory molecules including ICOS-L and LFA-3 to enable direct allorecognition, a process where ECs act as antigen-presenting cells (APCs) to directly prime CD4+CD45RO+ memory T cells (Tmem). This process may modulate both the growth and migratory responses of cognate Tmem.
We previously found that human ECs produce Hh ligands in vitro and in vivo (14) following IRI and that these Hh ligands acted as costimulatory signals to a defined T-cell subset highly expressing Ptch1, a population we termed PtchHi Tmem (14). While producing Hh ligands as costimulators, ECs lack CD80/86, costimulatory ligands for CD28 expressed on Tmem. In contrast, professional APCs like dendritic cells express CD80/86 and traffic to draining lymph nodes to present EC-derived alloantigens to cognate Tmem (39). To model the effects of these distinct routes of costimulation in vitro, we exposed αCD3 Ab-treated Tmem to smoothened agonist (SAG) to mimic the effects of Hh ligands, all three of which were induced by MACs in IRI-treated ECs (14); and as controls we exposed αCD3 Ab-treated Tmem to αCD28 Ab to model APC : Tmem interactions in draining LNs.
To gain insight into the effects of Hh costimulation on PtchHi T cells, we initially examined the PtchHi repertoire via deep sequencing of the TCR-α and TCR-β chains. Because we postulate that Hh costimulation occurs within tissues, we focused on Tmem from peripheral blood, which likely forms interchangeable pools with populations recruited to tissues. In contrast to certain autoimmune conditions where one or a few HLA-restricted driver clones dictate the populational response, alloimmune responses against EC-derived HLA molecules are highly polyclonal. To model these effects, we polyclonally stimulated Tmem from three separate donors with plate-bound αCD3 in the presence of either SAG or αCD28 Ab. Following activation, we sorted the respective populations shown in Figure 1A, which we termed PtchHi and PtchLo Tmem, respectively, and performed unbiased sequencing of the TCR-α and TCR-β chains.
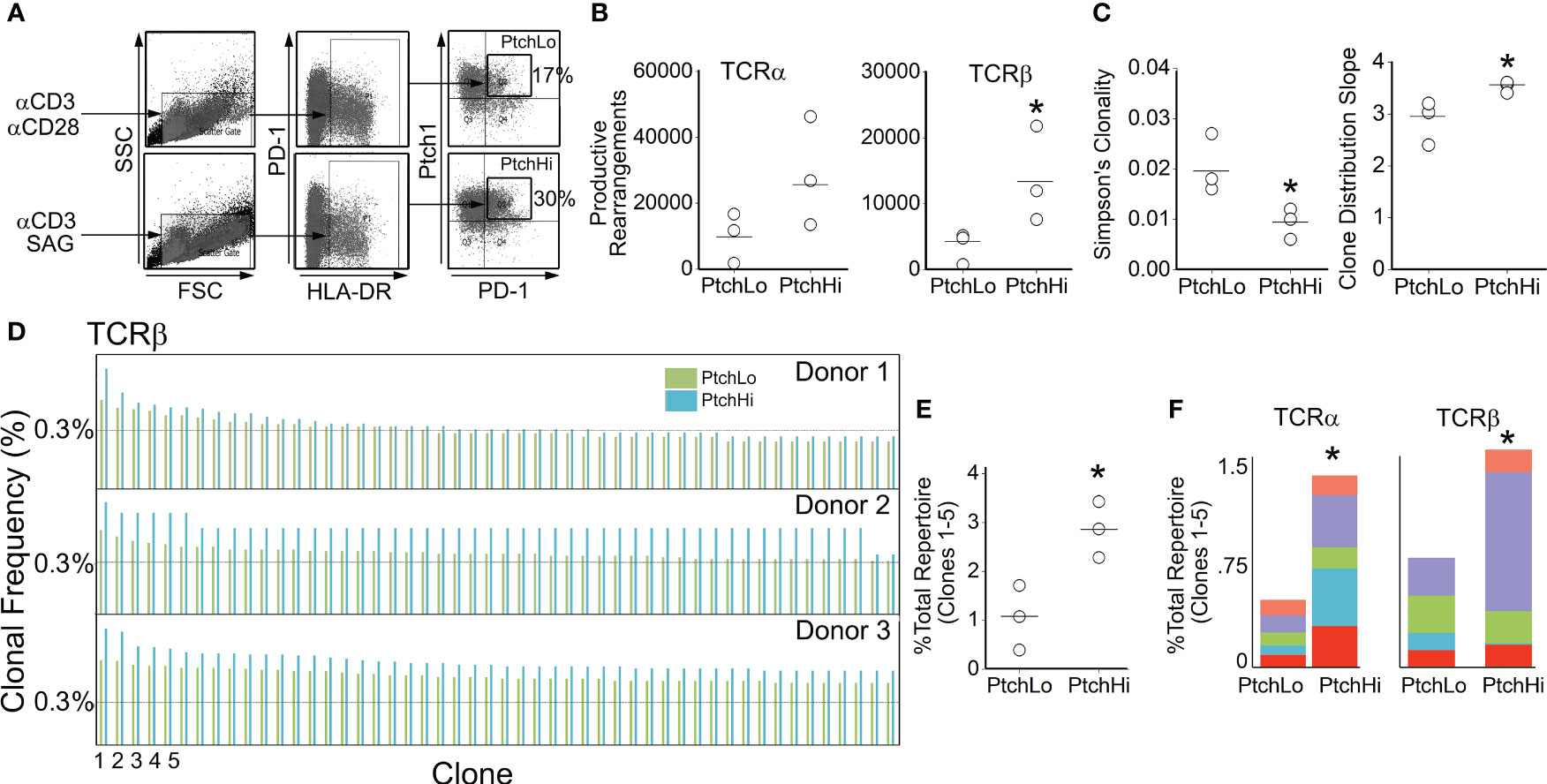
Figure 1 Hh costimulation elicits oligoclonal and polyclonal expansion of Tmem. Human CD4+CD45RO+ T cells (Tmem) were stimulated with plate-bound αCD3 Ab (1 μg/mL) in the presence of soluble αCD28 (1 μg/mL) or SAG (15 μM) for 48 h to generate PtchLo and PtchHi Tmem, respectively (A). CDR3 analysis of TCR-α and TCR-β chains showed increased clonal diversity in PtchHi Tmem compared to PtchLo Tmem (B, C). PtchHi Tmem showed oligoclonal and polyclonal expansion (D–F). (A–F) n = 3 donors tested. *p < 0.05. For (B, C, E), Student’s t-test was used for statistical comparisons.
PtchHi Tmem highly coexpressed PD-1 and showed increased expression of both surface Ptch1 and PD-1 (Supplementary Figure S1A) in alignment with our prior findings (14). We call the T-cell population expanded by SAG, “PtchHi” Tmem, and we call its comparator subset generated in the presence of αCD28 and showing lower Ptch1 expression, “PtchLo” Tmem. General survey statistics are shown in Table 2. Among three separate donors, we detected a total of 19,462 productive rearrangements involving the TCR-α chain and 52,707 productive rearrangements involving the TCR-β chain. Hh costimulation significantly increased the number of productive TCR-β rearrangements (Figure 1B). The most frequently utilized J isoforms in both the TCR-α and TCR-β chains were not significantly altered by Hh costimulation (Supplementary Figure S1B); however, usage of certain low-frequency J isoforms was significantly increased by Hh (Supplementary Figure S1C). CDR3 lengths showed Gaussian distributions in all groups examined, and CDR3 lengths were unchanged in PtchHi vs. PtchLo Tmem (Supplementary Figure S1D). The clonal repertoire of PtchLo vs. PtchHi showed comparatively higher repertoire similarity according to Simpson’s clonality metric within a given donor than between donors (Supplementary Figure S1E).
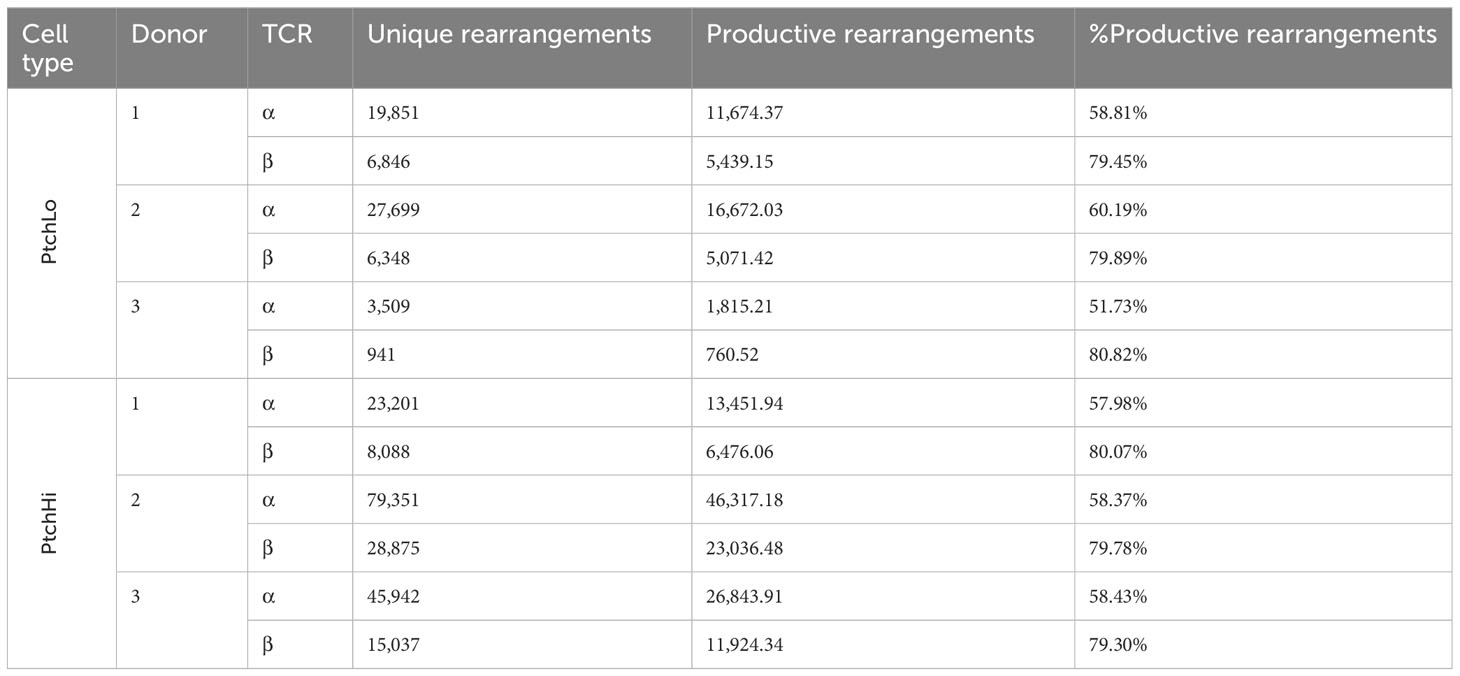
Table 2 General TCR survey statistics.
As above, Tmem subjected to Hh costimulation showed an increased number of productive rearrangements (Figure 1B), and clonal analyses using two separate metrics previously applied to alloresponses (38) revealed that PtchHi Tmem showed increased repertoire diversity involving both TCR-α and TCR-β (Figure 1C). In frequency analyses, we observed that Hh costimulation had induced a disproportionate, oligoclonal expansion of two to five of the highest-frequency clones within each donor while polyclonally expanding 347–365 lower-frequency clones. To illustrate this, the top 50 rearrangements for each donor for TCR-β are shown in Figure 1D. The top 5 highest frequency clones, when analyzed in combination (Figure 1E) or separately (Figure 1F), for TCR-α and TCR-β were significantly expanded by Hh costimulation. The highest expanded clones in each donor whose sequences are shown in Supplementary Figure S1E showed low inter-donor CDR3 sequence similarity as measured by Morista indices (Supplementary Figure S1F). This feature of low inter-donor similarity was recapitulated at a repertoire level (Supplementary Figure S1G). The polyclonally expanded repertoire among all three donors did, however, contain shared clones in PtchHi populations, and these clones were universally of low frequency (Supplementary Figure S1H). Hh costimulation diversifies the TCR repertoire of PtchHi Tmem by inducing both oligoclonal and polyclonal expansion of respondent clones.
Hh costimulation diversified the TCR repertoire, and we tested how this may have affected the cytokine effector response. To test this, we performed multiplexed laser bead assay in conjunction with single-cell proteomics via bead-based capture of barcoded PtchHi vs. PtchLo Tmem. These dual approaches were employed to concurrently obtain information regarding quantities of inflammatory mediators as well as frequencies of cells producing such mediators. PtchHi and PtchLo Tmem were generated under nonpolarizing conditions by plate-bound αCD3 with concurrent exposure to SAG or αCD28. Following this, culture supernatants were analyzed via a multiplexed laser bead assay, and cells were subjected to single-cell proteomics.
We found that Hh costimulation significantly potentiated the elaboration of cytokine mediators involved in IRI (Figure 2A). Due to the wide range in quantities of elaborated cytokines, heatmap colors for each row were computed relative to the overall average for that one particular cytokine, and raw values are shown in Table 3. Frequencies of cytokine-elaborating clones were similarly increased for many inflammatory mediators (Figure 2B). We tested correlations among cytokine levels and corresponding frequencies of cytokine-producing cells. Among the cytokines analyzed, cytokines implicated in IRI-mediated tissue pathology, including IFN-γ, IL-17, and IL-21, were significantly elevated in both multiplexed laser bead and single-cell proteomic assays. In UMAP plots, PtchHi and PtchLo populations showed remarkably distinct effector profiles (Figure 2C). We segregated analyses of effector function based on cytokines vs. chemokines to ask which of these responses drove the divergent effector responses. In this analysis, we found that the frequencies of polyfunctional clones elaborating ≥2 cytokines were significantly enhanced by Hh costimulation (Figure 2D). Among polyfunctional clones elaborating cytokines defining Th2, Th9, and Th17 subsets, we observed a marked increase in clones elaborating IFN-γ, a hallmark of the type 1 response that predominates in IRI (Figure 2E). The heightened type 1 response induced by Hh costimulation aligned with our prior observations (14, 15) and may have occurred due to the potentiated release of cytokines like IL-9 and IL-21, whose increased levels promote T-Bet. Combinatorial frequencies of effector clones among PtchLo and PtchHi populations are shown in Figure 2F. Many polyfunctional subsets were expanded by Hh costimulation, and in particular, we noted two populations that were present in PtchHi but not PtchLo Tmem and that were highly polyfunctional, elaborating ≥4 cytokines each (arrows). In these analyses, cytokine-producing cells below the pre-defined threshold for detection for the assay are listed in Table 4. In a prior report (14), we previously tested the effects of dual Hh costimulation plus CD28 costimulation, which might occur in instances where tissue-infiltrating Tmem become activated via indirect allorecognition involving professional APCs positioned near IRI-treated ECs. In these prior studies, dual costimulation with αCD28 Ab plus SAG phenocopied Hh costimulation alone, and respondent HLA-DR+ Tmem highly elaborated IFN-γ (14).
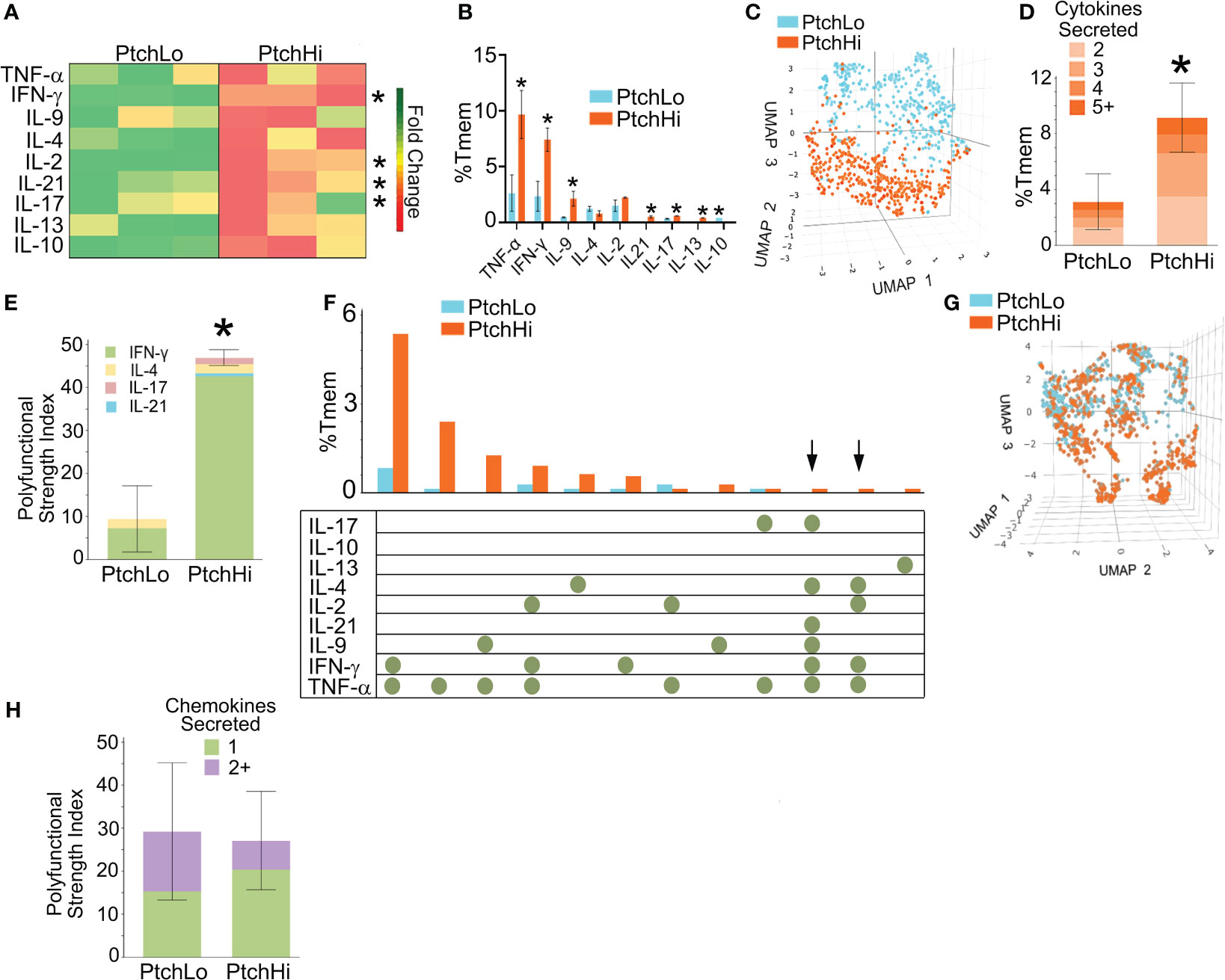
Figure 2 Hh costimulation elicits polyfunctional cytokine responses in PtchHi Tmem. Culture supernatants from PtchLo and PtchHi Tmem were analyzed using a multiplexed laser bead assay (A). Single-cell proteomics showed quantitative (B) and qualitative (C) differences in the frequencies of the effector PtchLo and PtchHi Tmem. Compared to PtchLo Tmem, PtchHi Tmem showed polyfunctional cytokine responses (D) that were strongly driven by IFN-γ (E). Subset frequencies based on cytokine function among PtchLo and PtchHi Tmem (F). Chemokine responses in PtchHi vs. PtchLo Tmem did not show qualitative differences (G) or changes in polyfunctionality (H). In (A), heatmap colors for each row were computed relative to the overall average for that particular cytokine. n = 3 separate donors tested. *p < 0.05, *** p < 0.001. For (D, E, G, H), Student’s t-test was used for statistical comparisons.
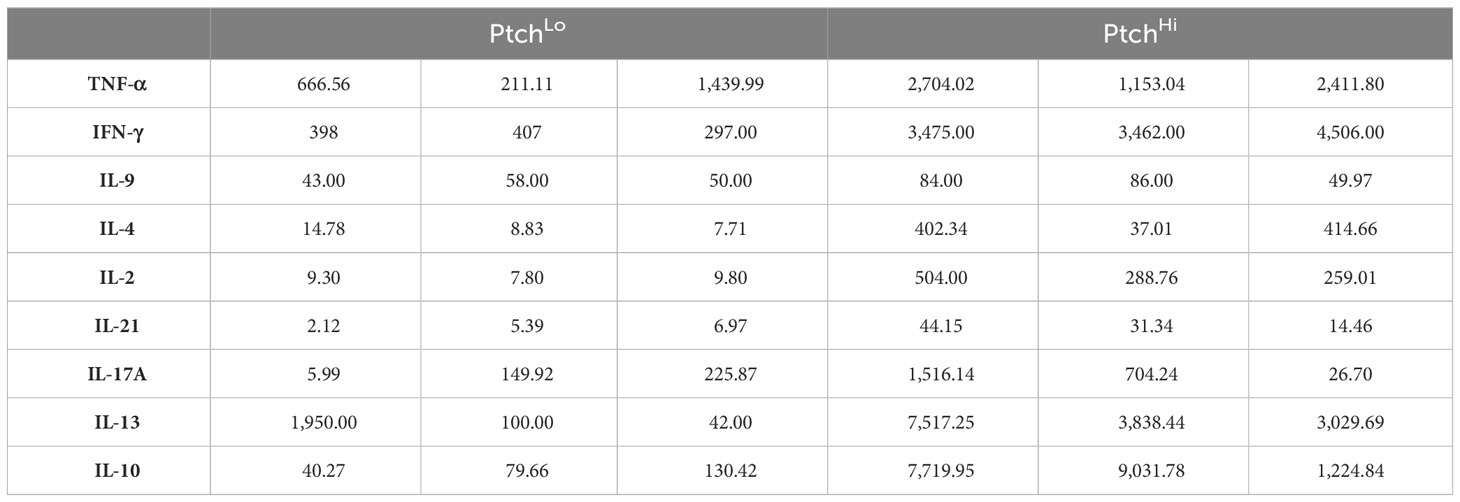
Table 3 Cytokine concentration (pg/mL).
Table 4 Undetected mediators.
We performed the same analysis based on chemokine levels and frequencies of chemokine-expressing clones in PtchHi Tmem. While a majority of PtchHi clones elaborated type 1-dependent chemokines, including MIP-1α, PtchHi Tmem did not show qualitatively distinct chemokine responses compared to PtchLo following Hh costimulation (Figure 2G). Moreover, in contrast to cytokine effectors, Hh costimulation did not generate polyfunctional chemokine responses (Figure 2H). These data indicated that the differential effector response among PtchLo vs. PtchHi Tmem in Figure 2C was principally driven by a diversified cytokine effector profile in PtchHi Tmem. Hh costimulation diversifies the cytokine response of PtchHi Tmem, generating subsets displaying heightened and polyfunctional responses, particularly those involving IFN-γ.
We have previously utilized a human artery xenograft model to examine EC-mediated direct allorecognition, a response unique to human ECs. Using this model, we found that human artery xenografts subjected to IRI showed enhanced Tmem recruitment to an expanded neointima in vivo (14, 15). To define subset(s) recruited to IRI-treated tissues, we examined spatial positioning molecules that were expressed on polyfunctional PtchHi Tmem. To do this, we performed multi-parameter analyses of surface chemokine receptors, integrins, and lipid receptors using mass cytometry.
In initial studies performed in vitro to allow optimization, PtchHi Tmem was stained for a total of 38 molecules via mass cytometry (Table 1). Following the identification of viable, CD3+CD4+Ptch1+PD-1+ singlets, we gated on HLA-DR+ subsets that produced IFN-γ, IL-17, and IL-21, cytokines appearing in the multiplexed laser bead and single-cell proteomic assays above and that have been implicated in IRI. This gating strategy revealed three distinct subsets, clusters 1–3 (blue, Figure 3A). In addition to the cytokines above, clusters 2 and 3 additionally elaborated IL-2, IL-4, and IL-22 and showed cleaved caspase-1 whose activity was previously detected in PtchHi Tmem to allow these cells to elaborate IL-1β (Figure 3B) (14). Relative to Tmem lacking cytokine elaboration (orange), PtchHi cells in clusters 2 and 3 (blue) highly coexpressed costimulatory molecules, including PD-1 and ICOS (Figure 3C). Tmem within cluster 2 (blue) broadly expressed canonical chemokine receptors conferring homing to the gut (CCR9), skin (CCR6), and inflamed tissues (CXCR3, CCR11, Figure 3D); atypical chemokine receptors (ACKR, XCR4, Figure 3E); and positioning molecules allowing homing to lymphoid organs, including CXCR5 and CCR7 (Figure 3F).
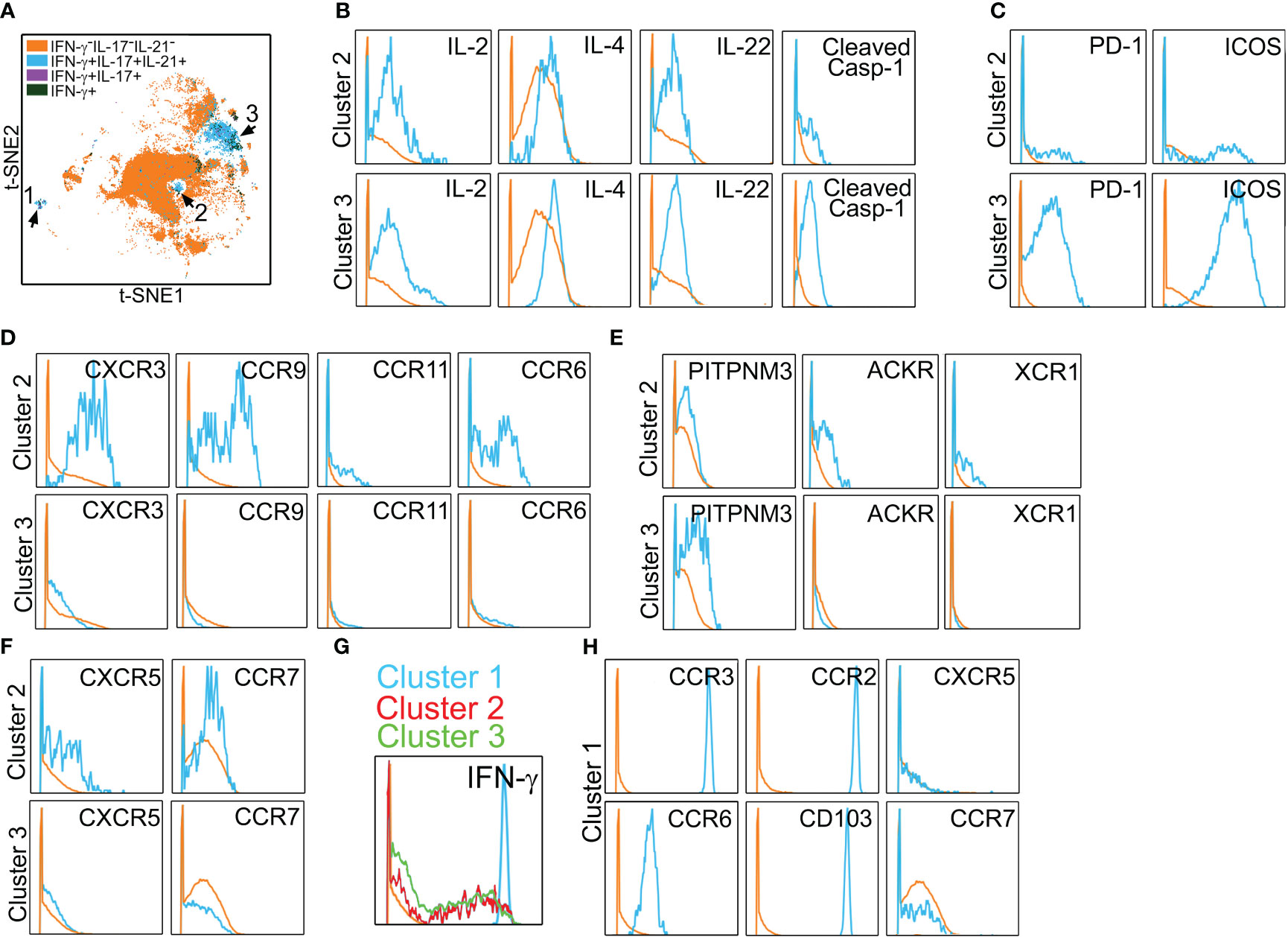
Figure 3 Polyfunctional PtchHi subsets coexpress molecules for peripheral tissue homing. Mass cytometry of PtchHi Tmem was performed, and a concatenated t-SNE plot of PtchHi Tmem from three separate donors uncovered three effector subsets (A). Effector subsets within clusters 2 and 3 show polyfunctional cytokine responses (B). Cluster 3 highly coexpresses costimulatory molecules (C), while cluster 2 expresses a broad array of classical (D) and atypical (E) chemokine receptors for peripheral tissue homing as well as chemokine receptors for homing to lymphoid tissues (F). Relative to clusters 2 and 3, cluster 1 shows strong IFN-γ responses (G) and uniquely expresses molecules for positioning and retention within peripheral but not lymphoid tissues (H).
In t-SNE plots (Figure 3A), cluster 1 was more spatially separated from clusters 2 and 3, and Tmem in cluster 1 showed very strong IFN-γ responses (Figure 3G). Cluster 1 (blue) Tmem uniquely and strongly coexpressed molecules for homing to inflamed (CCR2, CCR3, CCR6, CD103) but not lymphoid tissues (CXCR5, CCR7, Figure 3H) relative to Tmem that did not elaborate cytokines (orange). These data showed that Hh costimulation generates polyfunctional T-cell subsets expressing costimulatory molecules and a diversified pattern of spatial positioning molecules.
We subsequently developed a humanized mouse model to test the effector and migratory properties of the PtchHi Tmem subsets visualized above. In this model, fresh, human tonsillar LNs were implanted as subcutaneous xenografts in proximity to the ear of immunodeficient SCID/beige mice lacking T and B cells, and a portion of the same LN tissues were placed into single-cell suspension and cryopreserved (Figure 4A). Subsequently, human skin that was allogeneic to the implanted tonsillar LN was placed in organ culture and subjected to anoxia, a condition we found was necessary to mimic IRI because skin tissues were of sufficient thickness to be well oxygenated by diffusion. Following this treatment, human skin tissues were placed as full-thickness xenografts on the dorsal flank of SCID/bg hosts. Following implantation of IRI-treated skin, the cryopreserved, tonsillar LN cells above were thawed and used to generate PtchHi or PtchLo cells in vitro via polyclonal stimulation with plate-bound αCD3 Ab in the presence of Hh- or CD28-mediated costimulation. PtchHi and PtchLo cells generated in this fashion were differentially labeled with fluorescent dyes, IVIS770 and IVIS 680, respectively, prior to being passively transferred either separately or cotransferred at a 1:1 ratio as indicated into SCID/bg hosts via jugular vein injection. The differentially labeled PtchHi and/or PtchLo cells were autologous to the implanted LNs and allogeneic to the IRI-treated skin tissues. At various times following the transfer, differentially labeled Tmem contained in peripheral skin tissues and/or LN tissues were imaged in vivo using bioluminescence and recovered for ex vivo analysis.
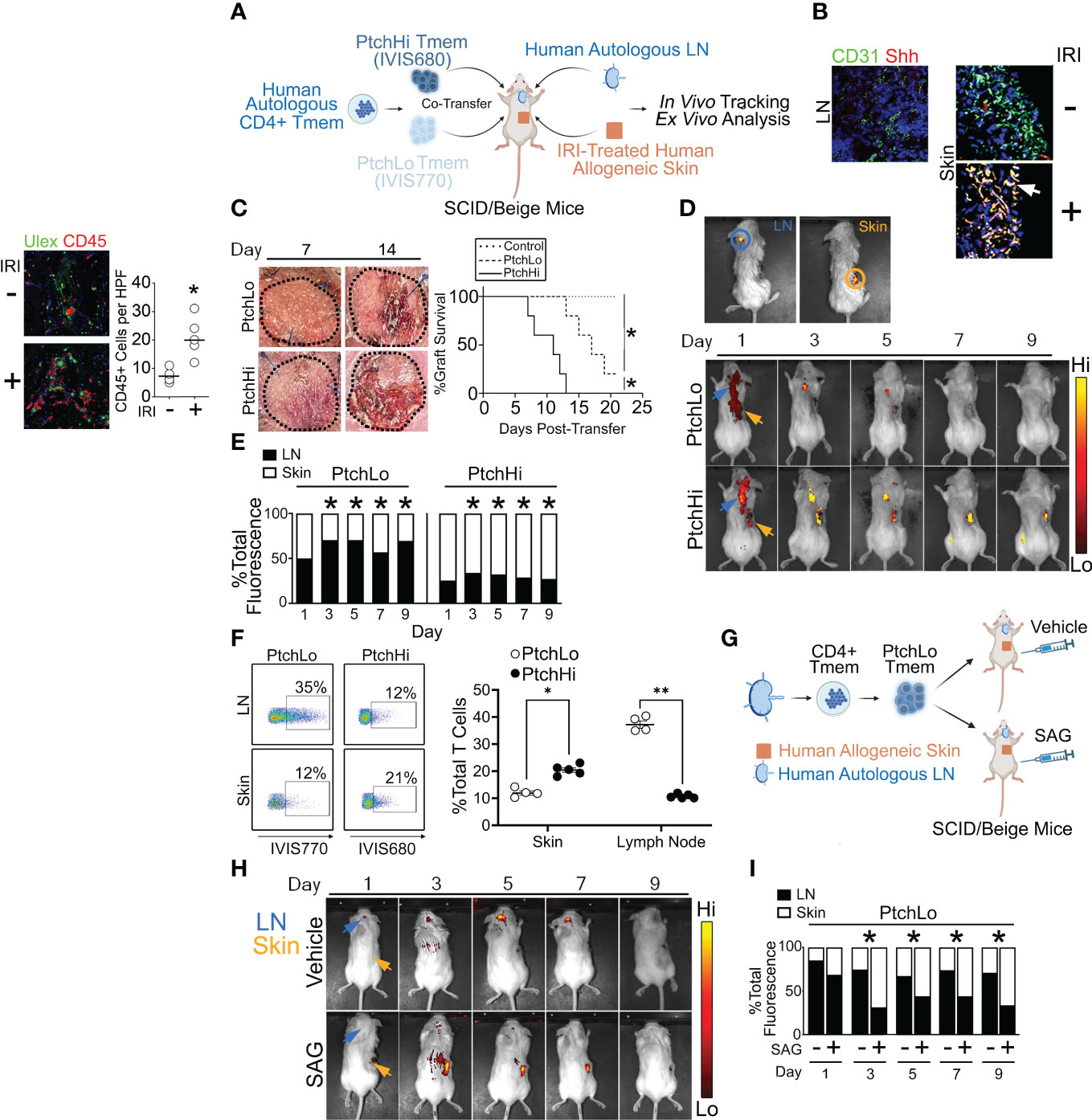
Figure 4 A humanized mouse model examining effector and migratory responses of PtchHi Tmem. Human LN and IRI-treated human skin were implanted into immunodeficient SCID/beige mice that passively received PtchHi and/or PtchLo Tmem that was autologous to LN tissues and allogeneic to skin tissues (A). At the time of passive transfer, Shh transcripts colocalized with CD31+ vessels in IRI-treated human skin but not in LN (B). Hosts receiving PtchHi Tmem only showed significantly accelerated rejection of allogeneic skin xenografts compared to hosts receiving PtchLo Tmem only or hosts receiving no Tmem (C). PtchLo and PtchHi Tmem were differentially labeled with fluorescent membrane dyes, mixed at 1:1 ratios for a total of 2 × 106 cells, and passively cotransferred into SCID/beige mice bearing human LN and IRI-treated human skin. PtchLo and PtchHi Tmem migration was tracked in vivo over time (D). Fractions of bioluminescence in ROIs encompassing LN and skin tissue were calculated over time and statistically compared with respective bioluminescence values at Day 1 post-transfer (E). Percentages of dye-labeled PtchLo and PtchHi Tmem were calculated in LN and skin as a percentage of CD3+CD4+ T cells at Day 5 post-transfer (F). Hosts bearing LN and IRI-treated skin and passively receiving PtchLo Tmem were subsequently injected i.p. with vehicle or SAG (5 mg) for 3 days while undergoing IVIS imaging (G, H). Percentages of dye-labeled cells were calculated at each time point post-transfer (n = 3 per timepoint per group (I). *p < 0.05, **p < 0.01. For (C), Wilcoxon’s rank sum test was used for statistical comparisons. For (E, F), Student’s t-test was used for statistical comparisons.
Female SCID/beige hosts were used as recipients for human tissues due to their higher post-surgery survival compared to males. We used human skin tissues due to their heightened immunogenicity relative to kidney or liver xenografts, thereby permitting discrimination of the effector and migratory properties among PtchHi and PtchLo cells. Additionally, the large surface area of these tissues enabled the recovery of infiltrating cells, a feature that was lacking in human artery xenografts, which we employed previously (14, 15). During the course of optimization, we found that human LN implanted subcapsularly within mouse kidneys showed high rates of engraftment but failed to induce significant T-cell recruitment, precluding their further use. Various anatomical sites for LN implantation were also tested in pilot studies, including the subcapsular subcapsular liver and subcutaneous sites including the dorsal flank and ear. Tissues subjacent to the ear in mice are highly vascularized, and ear tissues are widely examined as readouts for hapten-induced hypersensitivity. Among the tested sites, subcutaneous skin implantation proximal to the ear allowed the highest rates of engraftment.
Compatible with our prior findings (14), Hh costimulation strongly modulated both T-cell effector function (Figure 2) and spatial positioning molecules (Figure 3). Due to these strong effects, we tested anatomical sites for Hh ligand production in vivo. We performed multiplexed in situ hybridization and found that LNs surgically implanted in the ear showed low levels of Sonic Hedgehog (Shh) as did freshly obtained human skin prior to IRI. In contrast, skin tissues subjected to IRI were diffusely stained with Shh in both the epidermis and dermis and heavily colocalized with transcripts for CD31, a marker of ECs (Figure 4B). These data aligned with our prior results showing Shh production by ECs in human renal tissues from DGF patients in IRI-treated human artery xenografts (14). ECs within IRI-treated human skin are a source of Hh ligands in our humanized mouse model.
In our initial studies, we examined the effects of IRI on the survival of allogeneic skin grafts. PtchLo and PtchHi Tmem were cotransferred at 1:1 ratios into mice bearing allogeneic skin grafts subjected either to anoxia to simulate IRI or to normoxia as controls prior to surgical implantation (n = 5 per group). As expected, we found that skin grafts subjected to IRI showed accelerated rejection defined as ≥80% scab formation compared to normoxia-treated skin grafts (Supplementary Figure S2A). Upon analysis at post-transfer day 14, IRI-treated grafts showed increased immune cell infiltrates (Supplementary Figure S2B) and significantly increased epidermal thickening (Supplementary Figure S2C), a skin pathology induced by IFN-γ. In alignment with our prior model using human artery xenografts (14), IRI significantly increased Tmem tissue infiltration.
We next assessed the effects of PtchLo and PtchHi Tmem on the survival of allogeneic skin grafts. Hosts bearing autologous LN and IRI-treated allogeneic skin tissues passively received PtchLo Tmem, PtchHi Tmem, or no Tmem, and skin rejection was assessed over time (n = 5 per group, Figure 4C). Compatible with prior results using IRI-treated human artery xenografts (14) and in accord with the data in Figures 2, 3, showing heightened and polyfunctional cytokine responses, hosts receiving PtchHi Tmem showed significantly increased perivascular (Ulex) CD45+ Tmem infiltration at day 7 (Supplementary Figure S2D) and developed significantly accelerated allograft rejection compared to PtchLo Tmem (Figure 4C). In contrast to the above, hosts receiving no Tmem did not show rejection during the monitoring period.
We next examined the respective migratory behavior of PtchHi vs. PtchLo Tmem. PtchHi and PtchLo Tmem were isolated by FACS sorting; differentially labeled using IVIS770 and IVIS680 dyes, respectively; and passively cotransferred at 1:1 ratios into SCID/beige hosts bearing autologous LN and IRI-treated allogeneic skin. Following passive transfer, we were able to use ex vivo bioluminescence to spatially track PtchHi and PtchLo cells in the same host. Regions of interest (ROIs) were drawn around sites of autologous human LN (blue circle) and allogeneic human skin (orange circle), and bioluminescent intensities were calculated within these ROIs.
At post-transfer day 1, we observed bioluminescence signals for both PtchLo and PtchHi Tmem in regions outside implanted human tissues. This signal may have reflected cells in the hematogenous and/or lymphatic circulation (Figures 4D, E). By day 3, bioluminescence signals for both PtchHi and PtchLo Tmem became more restricted to implanted tissues, enabling analyses of bioluminescence ratios between tissue LN and skin tissues. At this time, PtchLo Tmem showed increased LN:skin ratios of bioluminescence values, while PtchHi showed significantly reduced LN:skin bioluminescence, and this remarkably divergent pattern was maintained up to ~10–12 days post-transfer, after which time fluorescent labels became too weak to permit reliable ex vivo imaging. We obtained similar, divergent recruitment of PtchHi vs. PtchLo Tmem has similar kinetics when using human splenic tissues in lieu of human LNs (Supplementary Figure S2E), indicating that cellular transit between LN and skin tissues likely reflected hematogenous and not lymphatic trafficking.
Dye-labeled Tmem were recovered from LN and IRI-treated skin tissues at day 5 and analyzed by flow cytometry. Supporting bioluminescence findings, in IRI-treated skin tissues, PtchHi Tmem were detected at approximately twofold increased frequencies compared to PtchLo Tmem (Figure 4F). In contrast, autologous LN tissues showed an approximately fourfold comparative enrichment of PtchLo Tmem compared to PtchHi Tmem. These differential trafficking phenotypes were reproducibly observed across multiple murine hosts. These data showed that PtchHi and PtchLo Tmem show divergent homing to peripheral vs. lymphoid tissues in vivo.
To test whether the differential migratory capacity of PtchLo vs. PtchHi Tmem was mediated by Hh costimulation, we passively transferred PtchLo Tmem receiving αCD28 Ab costimulation into hosts bearing IRI-treated human skin and autologous LN who then received daily treatments of the vehicle or SAG (Figure 4G). Compared to vehicle-treated hosts where PtchLo Tmem primarily homed to autologous LN, SAG-treated hosts showed increased migration of PtchLo Tmem to skin tissues, phenocopying the migratory responses of PtchHi Tmem (Figure 4H). At each time point, ratios of dye-labeled Tmem were calculated in LN and skin tissues. We found that Hh costimulation significantly increased the ratios of PtchLo Tmem homing to skin vs. LN starting at Day 3, and these responses were durable up to day 9 post-transfer (Figure 4I, n = 3 per timepoint per group). In PtchLo Tmem receiving antecedent costimulation with αCD28 Ab, Hh costimulation was sufficient for inducing migratory homing to IRI-treated peripheral tissues.
We used mass cytometry to examine PtchHi Tmem recovered from IRI-treated skin tissues. Five days post-transfer, PtchHi Tmem elaborating IFN-γ, IL-21, and IL-17 were gated and analyzed for spatial positioning molecules. This gating strategy, as before, revealed three distinct effector populations, which may have been derived from clusters 1–3 visualized prior to transfer (Figure 3). We call these PtchHi Tmem clusters, clusters A–C (Figure 5A). Among these clusters, clusters A and C showed highly polyfunctional responses, elaborating IL-2, IL-4, IL-2, IL-22, and cluster C highly expressed cleaved caspase-1 (Figure 5B). Cluster A highly coexpressed PD-1 and ICOS (Figure 5C), as well as chemokine receptors for peripheral tissue homing (Figure 5D; Supplementary Figure S3A).
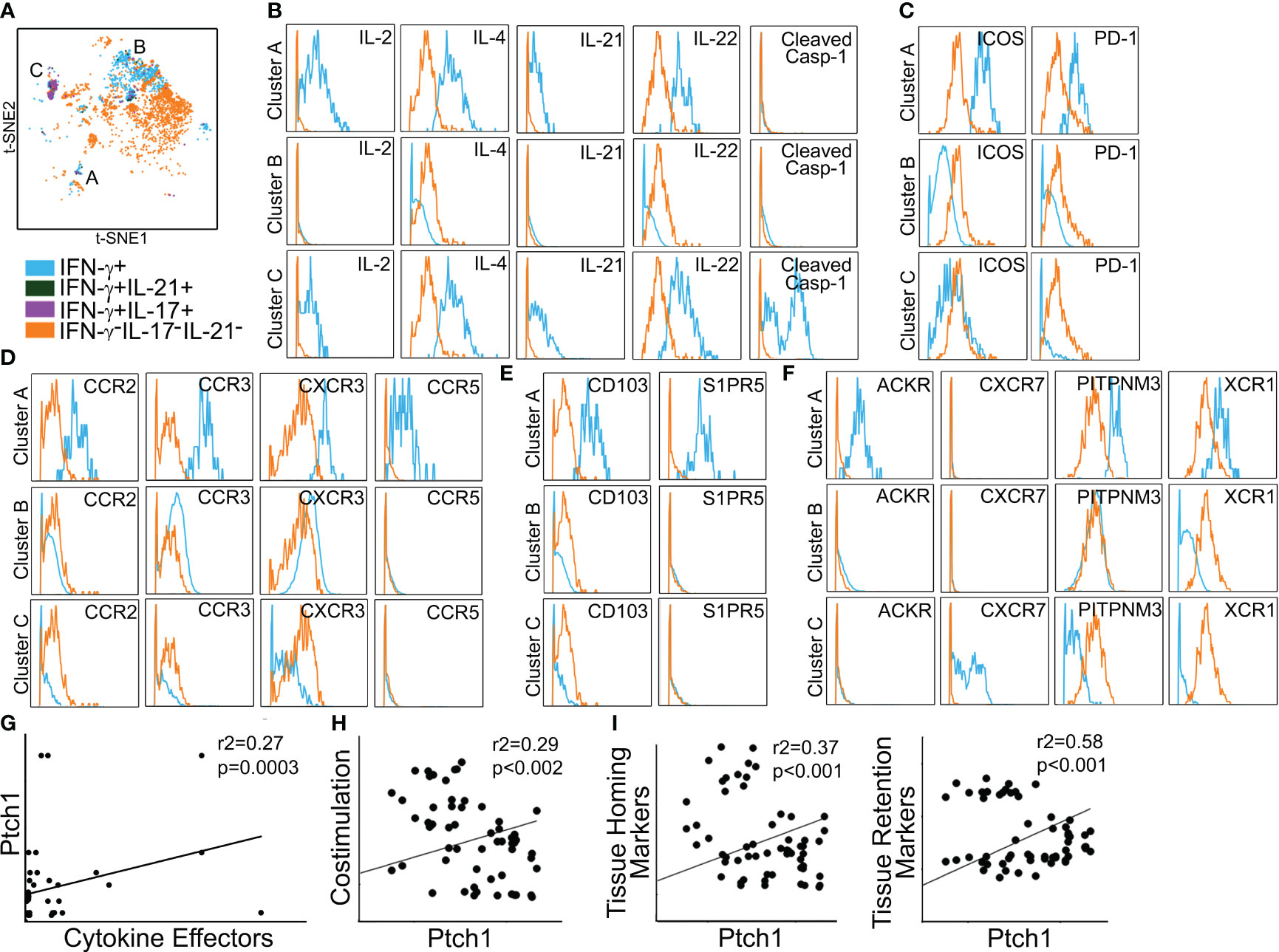
Figure 5 Polyfunctional PtchHi subsets preferentially home to peripheral tissues in vivo. PtchHi Tmem recovered from IRI-treated human skin 5 days post-transfer were analyzed ex vivo via mass cytometry. Concatenated t-SNE plots from n = 3 separate donors identified three effector subsets (A). Cluster A coexpresses effector cytokines (B), costimulatory markers (C), and chemokine receptors (D) compatible with T peripheral helper cells. Cluster A expresses markers for peripheral tissue retention (E) and atypical chemokine receptors (F). Correlation analyses were performed on three public RNA seq datasets involving (n = 268 patients) as specified in the Materials and methods section. Transcript abundance of Ptch1 showed significant correlations with an index of effector cytokines (G, IFN-G, IL-17, IL-21); costimulatory molecules (H, ICOS, PDCD1); as well as tissue homing (I, left, CCR2, CCR4) and tissue retention (I, right, CD103, S1PR5) molecules.
Prior to passive transfer, we detected a subset of PtchHi Tmem (cluster 1) that concurrently expressed molecules for both peripheral (CCR2, CCR3, CCR6, CD103) and lymphoid (CXCR5) homing. In contrast to this cluster visualized prior to passive transfer, Tmem in cluster A recovered from IRI-treated skin showed downregulated expression of CXCR5 (Supplementary Figure S3B). The effector, costimulatory, and chemokine receptor profile of cluster A was compatible with recently described T peripheral helper cells (TPH cells) (35). In support of this notion, among the three effector clusters, cluster A showed the lowest bcl6:BLIMP1 ratios (Supplementary Figure S3C). In addition to the markers above, cluster A was uniquely labeled by molecules allowing tissue retention (CD103, S1PR5, Figure 5E) and atypical chemokine receptors (XCR1, PITPNM3, ACKR, Figure 5F). Thus, IRI is a complement-mediated condition, and to test the relevance of our findings, we used public RNA seq datasets and performed correlations of effector cytokines, costimulatory molecules, and spatial positioning molecules with Ptch1, the salient marker for PtchHi Tmem. We analyzed datasets from three separate conditions involving complement-mediated tissue injury, including antibody-mediated rejection, a complication of DGF; rheumatoid arthritis; and systemic lupus erythematosus. Pearson’s r2 showed low–moderate and moderate–high correlations of Ptch1 transcript abundance with various genes indicative of observed features of PtchHi Tmem (Figures 5G–I). We observed significant correlations involving transcript abundances of Ptch1 with cytokine effectors (Figure 5G, IFN-G, IL-17, IL-21), costimulatory molecules (Figure 5H, ICOS, PDCD1), and molecules conferring homing (Figure 5I, left, CCR2, CCR4) and retention (Figure 5I, right, CD103, S1PR5) within peripherally inflamed tissues.
In parallel to the above, we analyzed spatial positioning molecules on PtchLo Tmem, which preferentially homed to autologous LN tissues. Following costimulation with αCD28 Ab PtchLo Tmem labeled with IVIS770 dye, these were passively transferred into hosts bearing autologous LN and allogeneic, IRI-treated skin tissues. PtchLo Tmem were recovered from LN tissues 5 days post-transfer and analyzed via mass cytometry (Figure 6). We found that, in contrast to PtchHi Tmem, PtchLo Tmem localized to autologous LN tissues showed only one salient, cytokine-producing cluster, which we termed cluster D (Figure 6A). Cluster D Tmem showed polyfunctional cytokine responses including IL-2, IL-4, IL-21, IL-22, and cleaved casp-1, whose activity generates IL-1β (Figure 6B). Cluster D Tmem also showed increased expression of costimulatory molecules (Figure 6C). In contrast to cluster A Tmem, Tmem in cluster D showed a distinct pattern of expression of spatial positioning molecules, showing relatively lower expression of positioning molecules conferring peripheral tissue homing including decreased typical chemokine receptors (CXCR3, CCR2, and CCR5, Figure 6D), atypical chemokine receptors (ACKR, PITNP3, XCR1, Figure 6E), and tissue retention molecules (CD103, S1PR5, Figure 6F) when compared to non-cytokine producing PtchLo Tmem. PtchLo Tmem lacking cytokine expression showed increased expression of CCR7 and CXCR5, allowing lymphoid homing (Figure 6D). Together, these data indicate that Tmem shows markedly divergent cytokine effector responses and homing patterns in vivo based on the presence or absence of Hh costimulation.
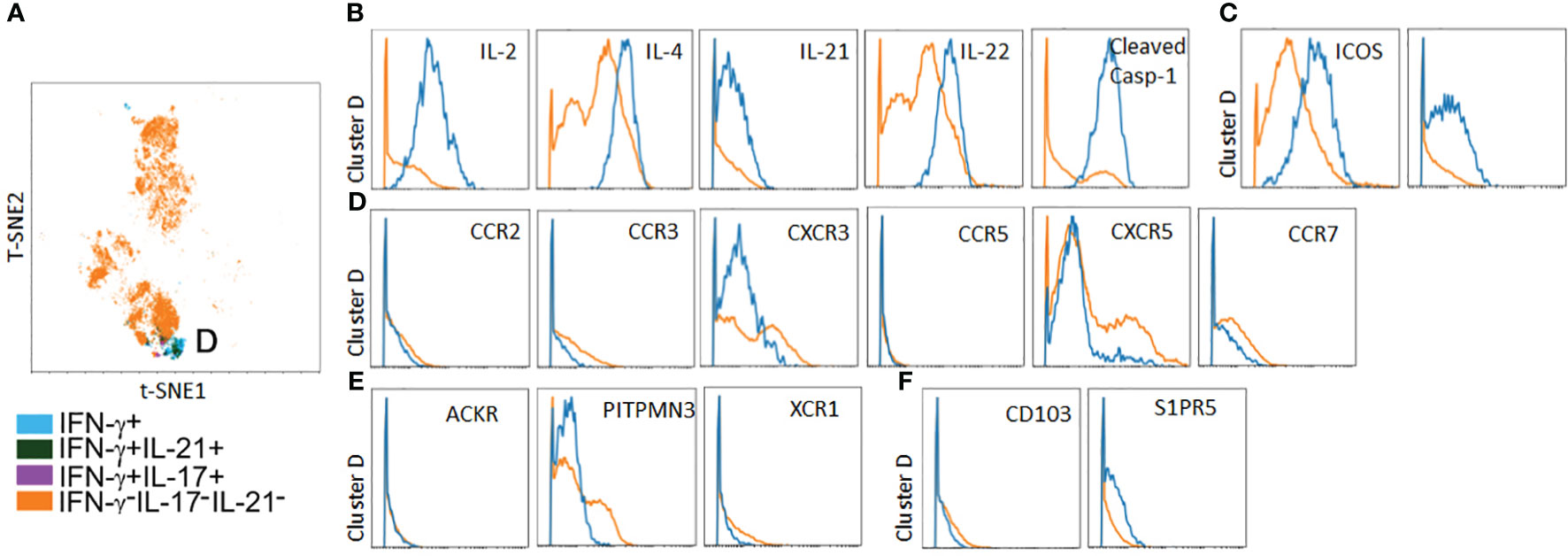
Figure 6 A polyfunctional PtchLo subset preferentially home to lymphoid tissues in vivo. PtchLo Tmem recovered from autologous LN tissues 5 days post-transfer were analyzed ex vivo via mass cytometry. Concatenated t-SNE plots from n = 3 separate donors identified one salient effector subset, cluster D (A). Cluster D Tmem coexpresses effector cytokines (B) and costimulatory markers (C) but shows low expression of chemokine receptors (D) and atypical chemokine receptors (E), tissue retention molecules (F) as well as upregulated expression of CXCR5 (D).
In this study, we use multi-omic approaches in conjunction with a novel, humanized mouse model to resolve functional heterogeneity among a pathogenic PtchHi T-cell population implicated in IRI. Human ECs release Hh morphogens following IRI, and these same ligands deliver potent costimulatory signals to heighten the effector and migratory responses of PtchHi Tmem. In this study, we found that Hh costimulation induced oligoclonal and polyclonal expansion of PtchHi Tmem, and these Hh-responsive clones showed highly polyfunctional cytokine but not chemokine profiles typified by markedly increased IFN-γ. Relative to PtchLo Tmem receiving costimulation by CD28, PtchHi Tmem differentially expressed molecules favoring homing to peripheral tissues and accordingly showed enhanced infiltration into IRI-treated human skin xenografts. Informed by multiplexed laser bead assays, single-cell proteomics, and mass cytometry, we visualized three clusters of PtchHi Tmem that recapitulated effector features observed in vitro, including enhanced polyfunctionality and expression of positioning molecules, allowing homing to IRI-treated peripheral tissues. Our studies resolve heterogeneity within PtchHi Tmem, an alloimmune T-cell subset mediating IRI-associated tissue injury.
We used human LNs as prototypical lymphoid tissues due to the availability of tissue samples and their high rates of surgical engraftment. A limitation on the use of these tissues is that, while becoming vascularized, implanted LNs do not form lymphatic drainage channels from peripheral skin tissues and thus do not reflect “draining” LNs. We found that human spleen implants elicited qualitatively similar homing with preferential recruitment of PtchLo Tmem, and this occurred under similar kinetics. These observations suggested that LN tissues functionally recapitulate hematogenous trafficking of immune cells, similar to spleen tissues. Another limitation of our model is the inability to distinguish immune effects between major, i.e., MHC I/II, and minor alloantigens. In our system, skin and LN tissues are obtained from different donors to induce full MHC mismatch(es), the strength of which likely far outweighs immune responses induced by minor alloantigens like H-Y or other polymorphic alleles such as MICA.
Analyses of PtchHi Tmem within inflamed skin tissues uncovered a subset, cluster 1, which demonstrated features of TPH cells (Figure 5). This population, variably termed T follicular helper-like cells, expresses costimulatory molecules including ICOS, IL-21, and homing markers including CCR2/CCR5, but not CXCR5. Based on these phenotypes, TPH cells are postulated to support Ab formation within peripheral tissues (35). We previously showed that IRI-treated ECs expanded TPH cells in a human artery xenograft model (14) and that these cells could support the production of isotype-switched IgG binding to donor tissues. Our current multi-omics approaches confirmed that Hh costimulation expands TPH cells, thus placing TPH cells as a subset contained within PtchHi Tmem. TPH cells are expanded in numerous inflammatory conditions and may be similarly expanded via tissue-derived Hh costimulation, and this premise is yet to be explored.
Via differential approaches including mAb production (40) and limiting dilution (41), other groups have demonstrated that B cells contained within human donor tissues may produce Abs directed against antigens on donor tissues. As our prior humanized model lacked human lymphoid tissues, our current humanized model may enable focused interrogation as to how the PtchHi population containing TPH cells might contribute to alloAb responses.
The immune mechanism(s) underlying IRI of skin tissues are highly relevant to clinical settings involving extensive trauma and burns where skin allografts are frequently employed. While vascular composite allografts (VCAs) involving facial skin tissues are newly utilized and currently constitute a minority of transplanted tissues (42), our studies may show relevance to the rejection of these tissues (43), the immune mechanisms of which are becoming increasingly investigated. PtchHi Tmem was newly described by our group (14) and as such, there are no antecedent reports in the literature describing its roles in VCAs. However, tissue-infiltrating subsets including PtchHi Tmem (14), TPH cells (35), and a recently described granzyme K+ T-cell population (44) show surface phenotypes compatible with enhanced peripheral tissue homing, and we surmise that one or more of these Tmem subsets may principally mediate VCA rejection.
While toleragenic strategies have been proposed, including CAR T and autologous Treg therapies (37, 43, 45), targeted therapies toward Tmem showing enhanced peripheral tissue homing, inclusive of PtchHi Tmem, have not been explored in the setting of VCAs. Blockade of EC-mediated anti-VCAM1 interactions via biologics, including natalizumab and vedolizumab, has proven effective in ameliorating T-cell homing to inflamed tissues in autoimmune disease and may show promise in blocking immune complications attendant to VCAs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE147089, GSE112943, GSE97779 (GEO).
The studies involving humans were approved by Yale Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by Yale Institutional Animal Care & Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
SW, QW, and DJ-W performed all in vitro experiments, in vivo experiments, and data analysis. GS, QJ, MN, and MF drafted the manuscript. JP and GT proofread the manuscript. ZT contributed to the methodology. DJ-W designed all of the experiments. All authors contributed to the article and approved the submitted version.
DJ-W was supported by grants from the NIH (R01HL141137) and the Veterans Hospital Administration (I01 BX005117).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1248027/full#supplementary-material
1. Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transpl (2011) 11(11):2279–96. doi: 10.1111/j.1600-6143.2011.03754.x
2. Taber DJ, DuBay D, McGillicuddy JW, Nadig S, Bratton C. F., Chavin K. D., et al. Impact of the new kidney allocation system on perioperative outcomes and costs in kidney transplantation. J Am Coll Surg (2017) 224(4):585–92. doi: 10.1016/j.jamcollsurg.2016.12.009
3. Rohan VS, Pilch N, McGillicuddy J, White J, Lin A, Dubay D, et al. Early assessment of national kidney allocation policy change. J Am Coll Surg (2022) 234(4):565–70. doi: 10.1097/XCS.0000000000000096
4. Montgomery RA, Loupy A, Segev DL. Antibody-mediated rejection: new approaches in prevention and management. Am J Transpl (2018) 18(3):3–17. doi: 10.1111/ajt.1458436
5. Kim DW, Tsapepas D, King KL, Husain SA, Corvino FA, Dillon A, et al. Financial impact of delayed graft function in kidney transplantation. Clin Transpl (2020) 34(10):e14022. doi: 10.1111/ctr.14022
6. Aubert O, Kamar N, Vernerey D, Viglietti D, Martinez F, Duong-Van-Huyen JP, et al. Long term outcomes of transplantation using kidneys from expanded criteria donors: prospective, population based cohort study. BMJ (2015) 351:h3557. doi: 10.1136/bmj.h3557
7. Yeung MY, Coates PT, Li PK. Kidney organ allocation system: how to be fair. Semin Nephrol (2022) 42(4):151274. doi: 10.1016/j.semnephrol.2022.09.002
8. Cron DC, Husain SA, Adler JT. The new distance-based kidney allocation system: implications for patients, transplant centers, and organ procurement organizations. Curr Transplant Rep (2022) 9(4):302–7. doi: 10.1007/s40472-022-00384-z
9. Friedewald JJ, Schantz K, Mehrotra S. Kidney organ allocation: reducing discards. Curr Opin Organ Transpl (2023) 28(2):145–8. doi: 10.1097/MOT.0000000000001049
10. Han J, Moayedi Y, Henricksen EJ, Waddell K, Valverde-Twiggs J, Kim D, et al. Primary graft dysfunction is associated with development of early cardiac allograft vasculopathy, but not other immune-mediated complications, after heart transplantation. Transplantation (2023) 20:e004551. doi: 10.1097/TP.0000000000004551
11. Fuentes-Valenzuela E, Tejedor-Tejada J, García-Pajares F, Rubiales BM, Nájera-Muñoz R, Maroto-Martín C, et al. Postreperfusion liver biopsy as predictor of early graft dysfunction and survival after orthotopic liver transplantation. J Clin Exp Hepatol (2022) 12(4):1133–41. doi: 10.1016/j.jceh.2021.12.015
12. Zhao H, Alam A, Soo AP, George AJT, Ma D. Ischemia-reperfusion injury reduces long term renal graft survival: mechanism and beyond. EBioMedicine (2018) 28:31–42. doi: 10.1016/j.ebiom.2018.01.025
13. Ito T, Naini BV, Markovic D, Aziz A, Younan S, Lu M, et al. Ischemia-reperfusion injury and its relationship with early allograft dysfunction in liver transplant patients. Am J Transpl (2021) 21(2):614–25. doi: 10.1111/ajt.16219
14. Jiang B, Wang S, Song G, Jiang Q, Fan M, Fang C, et al. Hedgehog-induced ZFYVE21 promotes chronic vascular inflammation by activating NLRP3 inflammasomes in T cells. Sci Signal (2023) 16(777):eabo3406. doi: 10.1126/scisignal.abo3406
15. Liu L, Fang C, Fu W, Jiang B, Li G, Qin L, et al. Endothelial cell-derived interleukin-18 released during ischemia reperfusion injury selectively expands T peripheral helper cells to promote alloantibody production. Circulation (2020) 141(6):464–78. doi: 10.1161/CIRCULATIONAHA.119.042501
16. Qin L, Li G, Kirkiles-Smith N, Clark P, Fang C, Wang Y, et al. Complement C5 inhibition reduces T cell-mediated allograft vasculopathy caused by both alloantibody and ischemia reperfusion injury in humanized mice. Am J Transpl (2016) 16(10):2865–76. doi: 10.1111/ajt.13834
17. Mascharak S, desJardins-Park HE, Davitt MF, Guardino NJ, Gurtner GC, Wan DC, et al. Modulating cellular responses to mechanical forces to promote wound regeneration. Adv Wound Care (New Rochelle) (2022) 11(9):479–95. doi: 10.1089/wound.2021.0040
18. Bielefeld KA, Amini-Nik S, Alman BA. Cutaneous wound healing: recruiting developmental pathways for regeneration. Cell Mol Life Sci (2013) 70(12):2059–81. doi: 10.1007/s00018-012-1152-9
19. Ramsbottom SA, Pownall ME. Regulation of hedgehog signalling inside and outside the cell. J Dev Biol (2016) 4(3):23. doi: 10.3390/jdb4030023
20. Le H, Kleinerman R, Lerman OZ, Brown D, Galiano R, Gurtner GC, et al. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen (2008) 16(6):768–73. doi: 10.1111/j.1524-475X.2008.00430.x
21. Luo JD, Hu TP, Wang L, Chen MS, Liu SM, Chen AF. Sonic hedgehog improves delayed wound healing via enhancing cutaneous nitric oxide function in diabetes. Am J Physiol Endocrinol Metab (2009) 297(2):E525–31. doi: 10.1152/ajpendo.00308.2009
22. Shen X, Peng Y, Li H. The injury-related activation of hedgehog signaling pathway modulates the repair-associated inflammation in liver fibrosis. Front Immunol (2017) 8:1450. doi: 10.3389/fimmu.2017.01450
23. Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol (2007) 7(9):726–35. doi: 10.1038/nri2151
24. Chan VS, Chau SY, Tian L, Chen Y, Kwong SK, Quackenbush J, et al. Sonic hedgehog promotes CD4+ T lymphocyte proliferation and modulates the expression of a subset of CD28-targeted genes. Int Immunol (2006) 18(12):1627–36. doi: 10.1093/intimm/dxl096
25. Lowrey JA, Stewart GA, Lindey S, Hoyne GF, Dallman MJ, Howie SE, et al. Sonic hedgehog promotes cell cycle progression in activated peripheral CD4(+) T lymphocytes. J Immunol (2002) 169(4):1869–75. doi: 10.4049/jimmunol.169.4.1869
26. Yánez DC, Lau CI, Chawda MM, Ross S, Furmanski AL, Crompton T. Hedgehog signaling promotes TH2 differentiation in naive human CD4 T cells. J Allergy Clin Immunol (2019) 144(5):1419–23. doi: 10.1016/j.jaci.2019.07.011
27. Papaioannou E, Yánez DC, Ross S, Lau CI, Solanki A, Chawda MM, et al. Sonic Hedgehog signaling limits atopic dermatitis via Gli2-driven immune regulation. J Clin Invest (2019) 129(8):3153–70. doi: 10.1172/JCI125170
28. Hanna J, Beke F, O’Brien LM, Kapeni C, Chen HC, Carbonaro V, et al. Cell-autonomous Hedgehog signaling controls Th17 polarization and pathogenicity. Nat Commun (2022) 13(1):4075. doi: 10.1038/s41467-022-31722-5
29. Ghaleh B, Thireau J, Cazorla O, Soleti R, Scheuermann V, Bizé A, et al. Cardioprotective effect of sonic hedgehog ligand in pig models of ischemia reperfusion. Theranostics (2020) 10(9):4006–16. doi: 10.7150/thno.40461
30. Zeng Q, Fu Q, Wang X, Zhao Y, Liu H, Li Z, et al. Protective effects of sonic hedgehog against ischemia/reperfusion injury in mouse skeletal muscle via AKT/mTOR/p70S6K signaling. Cell Physiol Biochem (2017) 43(5):1813–28. doi: 10.1159/000484068
31. Paulis L, Fauconnier J, Cazorla O, Thireau J, Soleti R, Vidal B, et al. Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci Rep (2015) 5:7983. doi: 10.1038/srep07983
32. Pratap A, Panakanti R, Yang N, Lakshmi R, Modanlou KA, Eason JD, et al. Cyclopamine attenuates acute warm ischemia reperfusion injury in cholestatic rat liver: hope for marginal livers. Mol Pharm (2011) 8(3):958–68. doi: 10.1021/mp200115v
33. Chen Y, Li X, Tian L, Lui VC, Dallman MJ, Lamb JR, et al. Inhibition of sonic hedgehog signaling reduces chronic rejection and prolongs allograft survival in a rat orthotopic small bowel transplantation model. Transplantation (2007) 83(10):1351–7. doi: 10.1097/01.tp.0000262568.73590.81
34. Bijlsma MF, Spek CA. The Hedgehog morphogen in myocardial ischemia-reperfusion injury. Exp Biol Med (Maywood) (2010) 235(4):447–54. doi: 10.1258/ebm.2009.009303
35. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
36. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-κB signaling in endothelial cells. Circulation (2013) 128(23):2504–16. doi: 10.1161/CIRCULATIONAHA.113.002972
37. Pilat N, Wiletel M, Weijler AM, Steiner R, Mahr B, Warren J, et al. Treg-mediated prolonged survival of skin allografts without immunosuppression. Proc Natl Acad Sci U S A (2019) 116(27):13508–16. doi: 10.1073/pnas.1903165116
38. DeWolf S, Grinshpun B, Savage T, Lau SP, Obradovic, A, Shonts B, et al. Quantifying size and diversity of the human T cell alloresponse. JCI Insight (2018) 3(15):e121256. doi: 10.1172/jci.insight.121256
39. Song G, Wang S, Barkestani MN, Mullan C, Fan M, Jiang B, et al. Membrane attack complexes, endothelial cell activation, and direct allorecognition. Front Immunol. (2022) 13:1020889. doi: 10.3389/fimmu.2022.1020889
40. Asano Y, Daccache J, Jain D, Ko K, Kinloch A, Veselits M, et al. Innate-like self-reactive B cells infiltrate human renal allografts during transplant rejection. Nat Commun (2021) 12(1):4372. doi: 10.1038/s41467-021-24615-6
41. Chatterjee D, Moore C, Gao B, Clerkin KJ, See SB, Shaked D, et al. Prevalence of polyreactive innate clones among graft–infiltrating B cells in human cardiac allograft vasculopathy. J Heart Lung Transpl (2018) 37(3):385–93. doi: 10.1016/j.healun.2017.09.011
42. Milek D, Reed LT, Echternacht SR, Shanmugarajah K, Cetrulo CL Jr, Lellouch AG, et al. A systematic review of the reported complications related to facial and upper extremity vascularized composite allotransplantation. J Surg Res (2023) 281:164–75. doi: 10.1016/j.jss.2022.08.023
43. Win TS, Crisler WJ, Dyring-Andersen B, Lopdrup R, Teague JE, Zhan Q, et al. Immunoregulatory and lipid presentation pathways are upregulated in human face transplant rejection. J Clin Invest (2021) 131(8):e135166. doi: 10.1172/JCI135166
44. Jonsson AH, Zhang F, Dunlap G, Gomez-Rivas E, Watts GFM, Faust HJ, et al. Granzyme K+ CD8 T cells form a core population in inflamed human tissue. Sci Transl Med (2022) 14(649):eabo0686. doi: 10.1126/scitranslmed.abo0686
Keywords: ischemia-reperfusion (I/R) injury, alloimmune, T cell, hedgehog, humanized model mouse
Citation: Wang S, Song G, Barkestani MN, Tobiasova Z, Wang Q, Jiang Q, Lopez R, Adelekan-Kamara Y, Fan M, Pober JS, Tellides G and Jane-wit D (2023) Hedgehog costimulation during ischemia-reperfusion injury potentiates cytokine and homing responses of CD4+ T cells. Front. Immunol. 14:1248027. doi: 10.3389/fimmu.2023.1248027
Received: 26 June 2023; Accepted: 15 September 2023;
Published: 17 October 2023.
Edited by:
Jean Kwun, Duke University, United StatesReviewed by:
Paul Schroder, University of Wisconsin Health, United StatesCopyright © 2023 Wang, Song, Barkestani, Tobiasova, Wang, Jiang, Lopez, Adelekan-Kamara, Fan, Pober, Tellides and Jane-wit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Jane-wit, ZGFuLmphbmUtd2l0QHlhbGUuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.