 Eric Ubil
Eric Ubil Kashif Rafiq Zahid
Kashif Rafiq Zahid- Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, United States
Immunotherapy is a promising therapeutic tool that promotes the elimination of cancerous cells by a patient’s own immune system. However, in the clinical setting, the number of cancer patients benefitting from immunotherapy is limited. Identification and targeting of other immune subsets, such as tumor-associated macrophages, and alternative immune checkpoints, like Mer, may further limit tumor progression and therapy resistance. In this review, we highlight the key roles of macrophage Mer signaling in immune suppression. We also summarize the role of pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes in tumor onset and progression and how Mer structure and activation can be targeted therapeutically to alter activation state. Preclinical and clinical studies focusing on Mer kinase inhibition have demonstrated the potential of targeting this innate immune checkpoint, leading to improved anti-tumor responses and patient outcomes.
Introduction
Over the last 50 years, the 5-year survival rates of patients with most forms of cancer have improved by more than 15%. However, cancer remains the second leading cause of death in the United States and a leading cause of death worldwide (1). While improvements in diagnosis and treatment have driven increased patient survival, it is clear that further advances are necessary. In recent years, immunotherapies, such as those targeting the adaptive immune response, have improved survival for patients with some forms of cancer (2). Unfortunately, checkpoint blockade has limited efficacy for the treatment of several solid tumors (3, 4) with the success of therapy correlated with overall tumor mutational burden (5). Despite current limits of efficacy, approximately 44% of cancer patients in the United States are eligible for, or have received, immune checkpoint blockade (ICB) therapy. However only 13% of all cancer patients will respond (6). The reasons for the low response rates are still under investigation, but clinical data demonstrates the therapeutic potential of targeting immune checkpoints.
The role of macrophages in cancer progression
Though innate immune cell targeted therapies lag behind adaptive immunotherapies in the clinic (7), there are well established roles for cells like macrophages in tumor progression. Often one of the most populous intra-tumoral immune cell subsets (8), macrophages are known to adopt pro-inflammatory (M1) or pro-wound healing (M2) phenotypes in the tumor environment. Single cell RNA sequencing has shown that the classical either/or polarization paradigm is not necessarily reflective of the complexity of intra-tumoral polarization states of macrophages, particularly in regard to plasticity and local context. However, an increased presence of M2 macrophages in the tumor environment has been associated with more rapid tumor progression, worse patient outcomes, and increased resistance to adaptive immunotherapy (9). This is problematic because in human malignancies, tumor associated macrophages (TAMs) typically adopt M2 phenotypes and promote cancer progression and metastasis (10). Mechanistically, this can be because M2 TAMs facilitate immune suppression [e.g., through secretions like IL-4, IL-13, CSF-1 and TGF-β (11, 12)] or promote angiogenesis (13). M2 macrophages are also known to remodel the extracellular matrix (ECM) to allow increased tumor growth and metastasis (14). In addition, they have been implicated in reducing the efficacy of adaptive immunotherapy by altering properties of the ECM that allow immune cell infiltration (15, 16). In contrast, M1 macrophages are known to directly limit tumor growth through production of reactive oxygen species or by coordinating the anti-tumor immune response via secretion of inflammatory cytokines (e.g., TNF-α and IFN-γ) (17).
Mer is an innate immune checkpoint
Macrophages are being actively targeted therapeutically because of their role in orchestrating anti-tumor immune responses (18, 19). One area of intense research is identification of other immune checkpoints. We and others have described Mer (MerTK) (20, 21) as an innate immune checkpoint in macrophages that can be targeted by cancer cells to limit the anti-tumor immune response. However, there are several Mer structure function relationships that have not been fully elucidated. This review describes structural features of Mer, data supporting its roles as an innate immune checkpoint, and pharmacologic therapeutics targeting Mer in the treatment of cancer.
Structural features of Mer
Mer belongs to the Tyro3/Axl/Mer family of receptor tyrosine kinases (RTKs) and is structurally related to the other receptors. Beginning at the amino terminus, Mer is comprised of 2 immunoglobin-like domains, 2 fibronectin type III domains, and a transmembrane domain followed by cytoplasmic kinase domain (22) (Figure 1). Between the second fibronectin type III domain and the transmembrane domain is a cleavage site (Figure 1). Cleavage at this site yields a soluble domain capable of acting as a decoy receptor through binding to Mer ligands (23). Elements of Mer are also capable of translocating to the nucleus to perform functions that are still unclear (24). Lastly, Mer has an adapter binding site between the kinase domain and the carboxy terminus (Figure 1). While some reports indicate kinase-dependent adapter protein recruitment (25), others demonstrate a potentially kinase-independent role for this protein domain (26). In the following sections, findings related to each Mer domain and their prospective roles in contributing to an innate immune checkpoint are described.
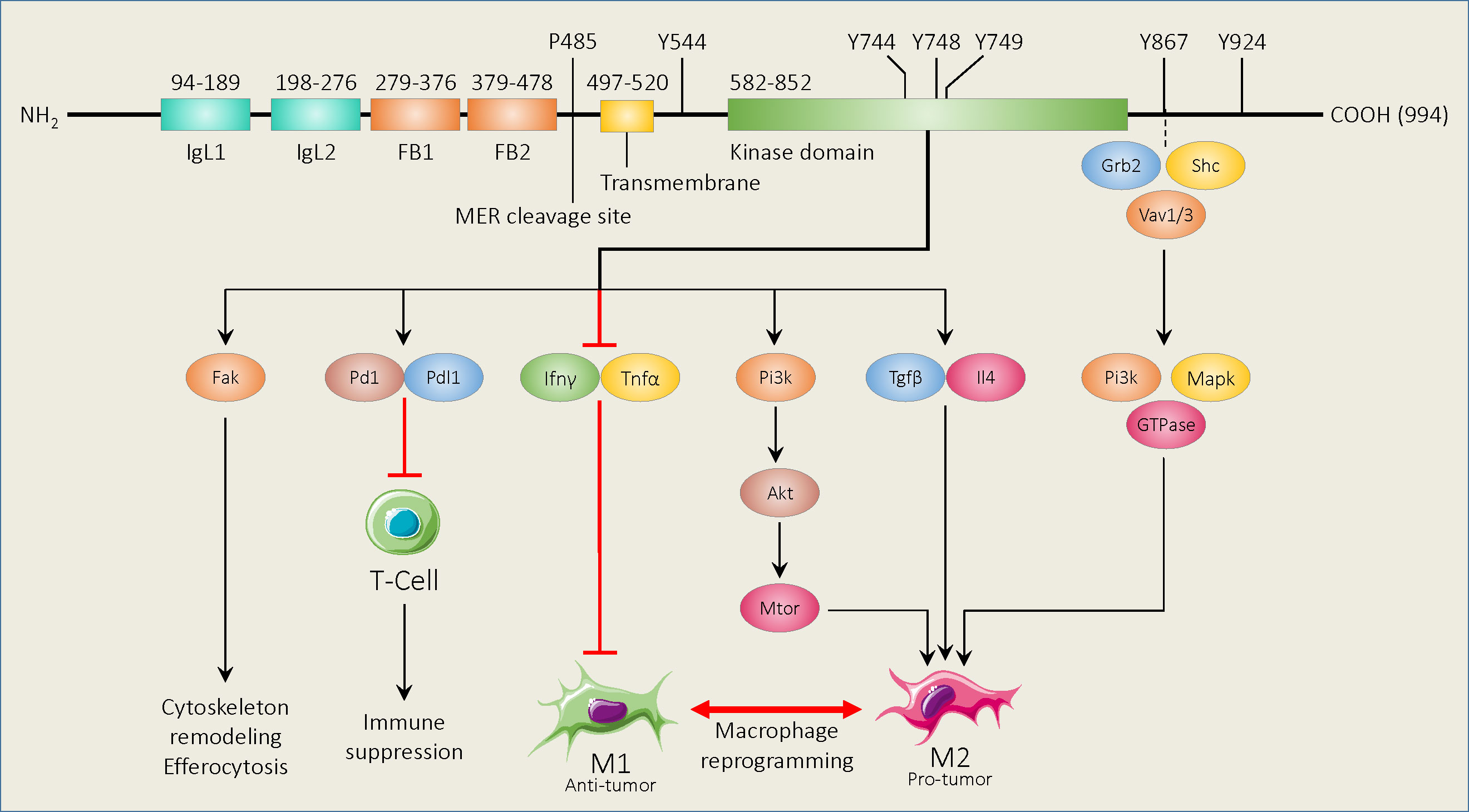
Figure 1 Schematic of murine Mer functional domains and known binding and phosphorylation sites, and downstream signaling pathways. Beginning at the amino terminus, Mer contains 2 immunoglobin-like domains (IgL1,2), 2 fibronectin type III domains (FB1,2), and a transmembrane domain followed by a cytoplasmic kinase region. The Mer cleavage site is found between the second fibronectin type III domain and the transmembrane domain. The Mer adapter binding site is between the kinase domain and the carboxy terminus. Amino acid positions for each functional area are shown above the diagram. Mer activation promotes various cellular processes and immune suppression using the downstream pathways shown. Mer signaling mediated by adapter binding protein binding also promotes a macrophage M2 phenotype.
Activation and effects of the Mer kinase domain
MER binding with known ligands, including Growth arrest specific 6 (Gas6) (27), Protein S (Pros1) (28), Tubby (Tub1) (29), Tubby-like protein 1 (Tulp1) (30), and Galectin 3 (Gal3) (31) can induce autophosphorylation of the tyrosine residues within the activation loop of the kinase domain (Tyr749/53/54, Human) (32). Kinase activation initiates the process of efferocytosis, the engulfment of apoptotic material, through coordinated cytoskeletal rearrangement (33). While ligand binding to apoptotic material promotes efferocytosis, engulfment can still be induced by ligands alone, but efficiency is reduced (34). Studies have also shown that efferocytosis can limit NF-kB signaling in macrophages to reduce inflammatory cytokine expression and limit pro-inflammatory (M1) polarization (35).
Alternatively, efferocytosis has been shown to promote a macrophage pro-wound healing (M2) phenotype, including increased secretion of immune suppressive cytokines (e.g., IL-4/10 and TGF-β).This can downregulate the local and systemic immune responses and allow for tumor immune escape (36). Mer-mediated efferocytosis can also increase immune suppression by causing macrophage upregulation of programmed death-ligand 1 (PD-L1) (37).
Through clearance of apoptotic material, Mer reduces the presence of Damage Associated Molecular Patterns (DAMPs), such as extracellular ATP, endogenous nucleic acids, and transcription factors like HMGB-1 (38), which could potentially activate the M1 response (39, 40). Clearance of tumor cell debris may further deprive macrophages and other antigen presenting cells of tumor antigenic peptides which would otherwise be presented to T cells, either through Major Histocompatibility Complex II (MHCII) to CD4+ T cells, or through cross-presentation to CD8+ T cells (41, 42). That process may limit potential T cell effector functions and diminish the overall anti-tumor response.
As an example of how both features contribute to immune escape, a recent study shows that Mer-mediated efferocytosis leads to tumor progression and immune tolerance in osteosarcoma by increasing M2 polarization and PD-L1 expression (43).
Mer can be cleaved to yield a soluble form of the protein
Macrophage membrane-bound Mer can be cleaved at Pro485-Ser486 in murine macrophages (44) by the metalloproteases ADAM10/17 to yield a soluble version of Mer (sMER) (23, 44). Subsequently, cleaved Mer contributes to defective efferocytosis in leukemia (23). After cleavage, sMer can act as a competitive Mer inhibitor and serve as a decoy for its ligand Gas6 (23). Alternatively, sMer can be localized to multiple intra-cellular compartments, including the cytoplasm, the nucleus, and the proteasome (45). While the role of Mer cleavage is not fully understood in the cancer context, the presence of soluble Mer has the potential to add further nuances to TAM signaling.
Mer can be localized in the nucleus
Mer is generally thought to be maintained at the cell membrane or in endosomes, though some studies have found that Mer can be translocated into nucleus. While not described in macrophages, Borgman et al. showed ligand dependent translocation of MER into the nucleus of human dendritic cells (DCs). Further, the authors found that MER acts as a transcription factor and that the amount of MER in the nucleus is associated with the differentiation state of DCs. Interestingly, the process was also regulated by the transmembrane receptor LRP-1 (46). A similar nuclear translocation phenomenon was observed in cancer cells, though nuclear translocation was determined to be regulated by the phosphorylation of deglycosylated MER, fueling the proliferation and transformation of hepatocellular carcinoma cells (HCC) (47). Similarly, Mer nuclear translocation was observed in leukemic cells (48).
The adapter binding site can modulate downstream signaling and cytoskeletal rearrangement
More than 20 years ago, Georgescu et al. (49) developed a fusion protein with a CD8 extracellular domain coupled to the Mer intracellular domain in Ba/F3 cells. Point mutations of known Mer phosphorylation sites (i.e., amino acids 544, 614, 825, 867, and 924) were generated within the fusion protein to determine their respective roles in constitutively activated Mer. The study showed that genetic loss of Mer, or mutations of Y867F or Y924F, reduced survival in IL-3 dependent cells cultured in the absence of IL-3. Further analysis identified position 867 as a binding site for the adapter protein Grb2, which is known to mediate Fak, Rac, and PI3K signaling (50). Georegescu and colleagues also determined that Mer 867 was essential for NF-kB activation (48). A later study by Tibrewal et al. (51) conflicted with Georgescu et al. in that NF-kB was not found to be regulated by Mer 867 in Mer mutant transfected RAW264 macrophages. Instead, Tibrewal et al. showed that Gas6 activated Mer 867 mediates PLCγ and Fak signaling and modulated p130 activation (50). Further studies have shown that other SH2 adapter proteins can interact with the intracellular domain of Mer to facilitate cytoskeletal remodeling. One such study showed that Gas6 can induce the release of constitutively bound Vav1 from Mer, to facilitate cellular reorganization for phagocytosis (52). Shelby and colleagues later utilized a screening strategy to identify Mer downstream adapter proteins. Primarily through the study of retinal pigment epithelium, they confirmed that Mer interacts with Grb2, but also with Vav1/3, PIK3R1 and Src to facilitate cellular remodeling (53). Taken together, these studies indicate that, in addition to the Mer kinase domain, there are additional molecular features important to downstream signaling.
Pharmacologically targeting Mer to improve outcomes
Because of its various roles in tumor immune progression, Mer is actively being targeted to increase cancer patient survival. As described in the following sections, Mer-directed therapeutics are in various stages of development, with some already in clinical trials.
Mer kinase inhibition promotes better outcomes in preclinical models
Several groups have shown that by pharmacologically targeting Mer kinase activity, efferocytosis and other immune suppressive processes may be reduced and lead to better outcomes in preclinical models. Mer-targeted compounds, such as UNC2250 (54), UNC9253 (55), SA4488 (56), and UNC2025 (57), have been investigated to inhibit Mer kinase activity in different murine tumor models like melanoma, prostate, pancreatic, and breast cancer (26). Zhou et al. have also reported that blockade of efferocytosis in TME mediates a switch from apoptosis/efferocytosis to immunogenic cell death of tumor cells (58).
Hsu et al. investigated the effect of the Mer kinase inhibitor XL092 in various human xenograft murine models. They found that XL092 monotherapy alone or in combination with immune checkpoint inhibitors (ICIs) significantly inhibited tumor growth. XL092 treatment led to decreased tumor cell proliferation and angiogenesis and fostered a less immune-permissive TME, including macrophage reprogramming from M2 to M1 polarization and suppression of efferocytosis (59).
BMS794833, a potent Mer inhibitor, was also found to be very effective in suppressing Mer activation and Mer-induced efferocytosis in an animal model (60). Further, in a preclinical model of glioblastoma, the Mer inhibitor UNC2025 results in induction of an inflammatory macrophage phenotype and reduced the fraction of TAMs expressing the anti-inflammatory marker CD206 in the tumor microenvironment (TME) (61). In addition to TAMs, Mer also regulates other immune cells such as T cells, natural killer cells (NKs), and myeloid derived suppressor cells (MDSCs) (62, 63). In most of these immune cells, Mer plays an immunosuppressive function.
Mer expression has been linked to leukemogenesis and therapy resistance in leukemia models (54, 64). These findings led to the use of Mer inhibitors in the treatment of preclinical leukemia models (65). For example, Mer kinase inhibition using MRX2843 prevented efferocytosis in a model of acute myeloid leukemia (AML). Results showed that Mer blockade diminished macrophage immunosuppressive traits by reducing expression of M2 markers, including TIM-3, PD-L1, Arginase-1, and CD163, and promoting macrophage polarization toward an antitumor (M1) phenotype by limiting STAT6 phosphorylation. In addition, Mer inhibition increased the production of cytokines involved in T cell activation, such as IFN-β, IL-1β, and IL-18 via NF-kB activation and prolonged leukemia-free survival (66). Taken together, these findings support the potential of Mer kinase inhibition as a means of increasing the anti-tumor immune response.
Antibody-based approaches to targeting Mer
Multiples groups have targeted the Mer kinase domain using small molecules, whereas other researchers have developed specific Mer-targeted monoclonal antibodies (mAbs) which have been tested in preclinical models (67, 68). Davara et al. investigated the effect of mAbs in a murine breast cancer model to block Mer function and found that mice treated with Mer neutralizing mAb displayed decreased tumor growth and metastasis. Mechanistically, Mer blockade with mAb or Mer knockout led to suppressed efferocytosis, reduced M2 macrophages, and enhanced infiltration of CD8+ T cells into tumors, indicating an anti-Mer mAb can foster host antitumor immunity (69). In breast and colon tumor murine models, antibody mediated blockade of Mer phagocytic engulfment of apoptotic cells also results in dramatic induction of Type 1 IFN response and increased antitumor T cells immunity, as well as enhanced efficacy of ICI therapy (58). Anti-Mer-antibody has significantly inhibited tumor growth either as single agent or along with anti-PD-1 (58). In a murine lung cancer model, combination radiotherapy, anti-PD-1 and anti-Mer, led to suppressed tumor growth, enhanced survival rates, and an overall decrease in metastasis by enhancing the population of CD8+ CD103+ TRM cells at distal tumor sites. Triple therapy also reprogrammed macrophage to the M1 phenotype and stimulated activation of CD8+ T cells and NK cells at metastatic sites (70). Preclinical studies reinforce the idea that Mer-blockade, either by targeting the kinase domain or by using mAbs, can improve outcomes, particularly when combined with other therapies like radiation or checkpoint blockade to shift the immunosuppressive TME into a more immunogenic state, thereby inducing the adaptive immune response for long lasting anti-tumor immunity.
MER is a biomarker of therapy resistance
Likely due to its various roles in suppressing the anti-tumor response, MER has been associated with resistance to anti-cancer therapies. For instance, MER overexpression has been detected in patients receiving chemotherapy or targeted therapy as first-line treatment. In colorectal tumors, MER is a predictive biomarker against MEK1/2 inhibitors resistance (71). For melanoma patients, MER upregulation is associated with resistance to MEK and BRAF inhibitors (72). Osimertinib has been proposed as a front-line targeted therapy for NSCLC patients with either T790M mutations or activating mutations because of superior efficacy and enhanced overall survival compared with earlier generation EGFR tyrosine kinase inhibitors (TKIs), such as erlotinib or gefitinib (73). Unfortunately, osimertinib is facing clinical challenges similar to first generation inhibitors, as only 3% of NSCLC patients showed complete response with osimertinib treatment while most of the patients exhibited residual tumor (74). A recent study has identified MER as a driver of osimertinib resistance and residual tumor growth in EGFRMT NSCLC patients. These findings showed that MER and its ligand GAS6 are upregulated in EGFRMT tumors following osimertinib treatment in both the NSCLC patients samples and xenograft models (75).
Utilizing MER kinase inhibition to treat cancer patients
Supported by preclinical findings, both MER specific and multi-kinase targeted therapies that also target MER are currently in clinical trials for the treatment of cancer. Clinical trials using MER selective inhibitors such as INCB081776 (NCT03522142) (76), ONO-7475 (NCT03176277) (77), and MRX-2843 (NCT03510104) (78) are being conducted for the treatment of different cancers. The selective inhibitor ONO-7475, which targets MER, AXL, and FLT3, is in Phase I clinical trials for advanced solid tumors and acute leukemia after suppressing progression of both forms of cancer in preclinical models (79). INCB081776 is a potent inhibitor of AXL and MER which is being tested along with nivolumab in advanced solid tumors (NCT03522142) (80).
While targeting of MER using small molecular inhibitors has shown clinical promise, issues with drug toxicity and off-target effects have limited their clinical application. Cabozantinib is a multi-kinase inhibitor that has also shows potent activity against AXL and MET (81). Cabozantinib treatment was found to increase hemoglobin levels in ovarian and prostate cancer patients (82, 83). Some patient deaths were also associated with cabozantinib treatment (84).
He et al. conducted the Phase I clinical trial (NCT01285037) of multi-kinase inhibitor merestinib (LY2801653), which also targets MER, to investigate the therapeutic effect and safety of this novel agent in advanced tumor patients. In this study, the authors tested the combination therapy of merestinib along with other therapeutic agents, including cisplatin, gemcitabine, and cetuximab. Results showed that 32% of tumor patients responded to this therapeutic agent and a 120 mg daily dose of merestinib demonstrated a tolerable safety profile and significant antitumor activity (85). CT053PTSA is a multi-kinase inhibitor being tested in a Phase I clinical trial (NCT04577703) in advanced solid tumor patients. Research findings of the trial suggested that CT053PTSA is well tolerated and safe for patients. This Phase I clinical trial has some limitations in that it was performed at a single center and that all the participants were Chinese, which might affect the generalizability of research findings to a broader population (86). Sitravantinib is a multi-kinase novel inhibitor which potentiates ICI therapy by modulating innate and adaptive immune cell changes in TME, thereby significantly improving efficacy of ICI therapy (87). Sitravantinib works partly by reprogramming M2 macrophages to the M1 phenotype (88). A Phase II clinical trial (NCT02954991) of sitravatinib with the immune checkpoint inhibitor nivolumab was conducted in nonsquamous NSCLC patients who experienced disease progression following ICI therapy or chemotherapy (89). Combination therapy of sitravatinib with nivolumab showed a tolerable safety profile and significant antitumor activity and also improved the survival of ICI therapy experienced NSCLC patients. This study has some limitation because it is based on single arm-design, which precluded tumor patients’ randomization to standard or care.
MER inhibitors possess complex and pleotropic action in human tumors and their therapeutic potential depends on local immune microenvironment, mutation burden, tumor type, and drug resistance. For example, a Phase 1/1b clinical trial of sitravatinib monotherapy demonstrated modest clinical outcomes in advanced NSCLC patients (NCT02219711) (90). Alternatively, the Phase 1 SNOW window-of-opportunity clinical trial of combining sitravatinib with nivolumab for treatment of oral cavity squamous cell carcinoma tumor patients demonstrated a tolerable safety profile and meaningful clinical and pathological outcomes (NCT03575598) (91). Importantly, combining sitravatinib with nivolumab resulted in a decreased immunosuppressive TME and led to macrophage reprogramming toward an M1 phenotype in tumor patients who responded to combination therapy (92).
A summary of ongoing clinical trials is presented in Table 1.
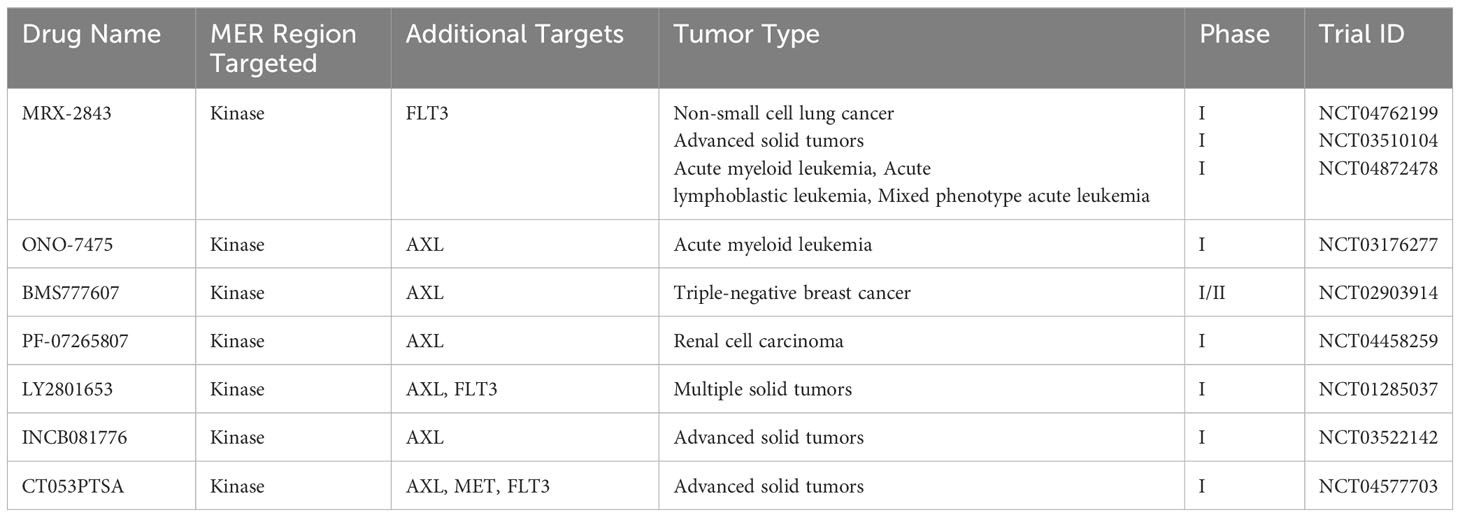
Table 1 Clinical trials investigating MER inhibitors for the treatment of various malignancies.
Potential challenges and alternative benefits of MER kinase inhibitors
MER inhibition has the possibility of on-target off-tumor effects because of the receptor’s role in tissue repair, platelet aggregation, and innate immune regulation. A previous study reported that Mer inhibition by using small molecule inhibitor UNC-569 causes ultra-structural changes in the mouse retina (93). Moreover, MER mutations are associated with human retinitis pigmentosa in various family cohorts (94), suggesting that kinase inhibition may have an effect on vision as well as cancer.
As described previously, MER kinase inhibitors can also have effects on other targets including FLT3, AXL, and MET (Table 1). In some instances, these secondary targets can lead to increased drug efficacy as they affect other clinically relevant features of cancerous cells. For instance, FLT3 is often mutated in hematologic malignancies like acute myeloid leukemia (95) and is a drug target in its own right (96). Similarly, AXL has been shown to confer selective advantage to cancerous cells in tumor progression (97) and simultaneously targeting AXL and MER could show increased benefit for patients. Because of structural similarities between MER, AXL, and TYRO3, it can be challenging to develop selective MER inhibitors (98). While relatively less is known about the role of TYRO3 in cancer, targeting MER and AXL may be advantageous.
Conclusion
With multiple known ligands, structural similarity to Tyro3 and Axl, and several functional domains that can modify downstream functions, Mer signaling is rich in complexity. However, preclinical and clinical studies focusing primarily on MER kinase inhibition have demonstrated the potential to target MER as an innate immune checkpoint, thereby improving the anti-tumor response and patient outcomes. While it is difficult to fully disaggregate the effects of Mer inhibition in immune cells from those taking place concurrently in tumor cells, reviewed elsewhere (99), Mer remains a promising therapeutic target for anti-cancer therapy alone, or in combination with other immunotherapies.
Author contributions
KZ and EU jointly wrote and edited the manuscript. KZ prepared the figure and table. All authors contributed to the article and approved the submitted version.
Funding
Preparation of this review was supported by the National Cancer Institute of the National Institutes of Health under award number CA262241.
Acknowledgments
We gratefully acknowledge Dr. Umar Raza for assistance in preparing the figure and for editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang A, Miao K, Sun H, Deng C-X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int J Biol Sci (2022) 18(7):3019. doi: 10.7150/ijbs.72534
2. Xu MM, Pu Y, Zhang Y, Fu Y-X. The role of adaptive immunity in the efficacy of targeted cancer therapies. Trends Immunol (2016) 37(2):141–53. doi: 10.1016/j.it.2015.12.007
3. Sharma P, Pachynski RK, Narayan V, Fléchon A, Gravis G, Galsky MD, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell (2020) 38(4):489–99.e3. doi: 10.1016/j.ccell.2020.08.007
4. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
5. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: is more always better? Tumor mutational burden as an immunotherapy biomarker. Clin Cancer Res (2021) 27(5):1236–41. doi: 10.1158/1078-0432.CCR-20-3054
6. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA network Open (2019) 2(5):e192535–e. doi: 10.1001/jamanetworkopen.2019.2535
7. Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting innate immunity in cancer therapy. Vaccines (2021) 9(2):138. doi: 10.3390/vaccines9020138
8. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
9. Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Zong Z, et al. PD-L1-mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front Immunol (2020) 11:588552. doi: 10.3389/fimmu.2020.588552
10. Chen Y, Zhang S, Wang Q, Zhang X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J Hematol Oncol (2017) 10:1–13. doi: 10.1186/s13045-017-0408-0
11. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
12. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
13. Fu L-Q, Du W-L, Cai M-H, Yao J-Y, Zhao Y-Y, Mou X-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
14. Kim H, Chung H, Kim J, Choi DH, Shin Y, Kang YG, et al. Macrophages-triggered sequential remodeling of endothelium-interstitial matrix to form pre-metastatic niche in microfluidic tumor microenvironment. Advanced Science (2019) 6(11):1900195. doi: 10.1002/advs.201900195
15. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci Trans Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604
16. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
17. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
18. Xiao N, Zhu X, Li K, Chen Y, Liu X, Xu B, et al. Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol (2021) 10(1):1–14. doi: 10.1186/s40164-021-00230-5
19. Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer science (2010) 101(8):1913–9. doi: 10.1111/j.1349-7006.2010.01614.x
20. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti–PD-1 therapy in melanomaTAM RTK inhibition reverses MDSC suppression and augments PD-1. Cancer Immunol Res (2019) 7(10):1672–86. doi: 10.1158/2326-6066.CIR-19-0008
21. Akalu YT, Rothlin CV, Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev (2017) 276(1):165–77. doi: 10.1111/imr.12522
22. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res (2008) 100:35–83. doi: 10.1016/S0065-230X(08)00002-X
23. Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood (2007) 109(3):1026–33. doi: 10.1182/blood-2006-05-021634
24. Tanim K, DeRyckere D, Graham DK. Phosphorylation of MERTK is required for nuclear localization in non-small cell lung cancer (NSCLC). Cancer Res (2023) 83(7_Supplement):4991–. doi: 10.1158/1538-7445.AM2023-4991
25. Ling L, Kung H-J. Mitogenic signals and transforming potential of Nyk, a newly identified neural cell adhesion molecule-related receptor tyrosine kinase. Mol Cell Biol (1995) 15(12):6582–92. doi: 10.1128/MCB.15.12.6582
26. Ubil E, Caskey L, Holtzhausen A, Hunter D, Story C, Earp HS. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J Clin Invest (2018) 128(6):2356–69. doi: 10.1172/JCI97354
27. Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene (1997) 14(17):2033–9. doi: 10.1038/sj.onc.1201039
28. Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem (1996) 271(47):30022–7. doi: 10.1074/jbc.271.47.30022
29. Caberoy NB, Alvarado G, Li W. Tubby regulates microglial phagocytosis through MerTK. J neuroimmunol (2012) 252(1-2):40–8. doi: 10.1016/j.jneuroim.2012.07.009
30. Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J (2010) 29(23):3898–910. doi: 10.1038/emboj.2010.265
31. Caberoy NB, Alvarado G, Bigcas JL, Li W. Galectin-3 is a new MerTK-specific eat-me signal. J Cell Physiol (2012) 227(2):401–7. doi: 10.1002/jcp.22955
32. Ling L, Templeton D, Kung H-J. Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J Biol Chem (1996) 271(31):18355–62. doi: 10.1074/jbc.271.31.18355
33. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411(6834):207–11. doi: 10.1038/35075603
34. Tsou W-I, Nguyen K-QN, Calarese DA, Garforth SJ, Antes AL, Smirnov SV, et al. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J Biol Chem (2014) 289(37):25750–63. doi: 10.1074/jbc.M114.569020
35. Ge Y, Huang M, Yao Y-m. Efferocytosis and its role in inflammatory disorders. Front Cell Dev Biol (2022) 10. doi: 10.3389/fcell.2022.839248
36. Cheng L, Weng B, Jia C, Zhang L, Hu B, Deng L, et al. The expression and significance of efferocytosis and immune checkpoint related molecules in pancancer samples and the correlation of their expression with anticancer drug sensitivity. Front Pharmacol (2022) 13:977025. doi: 10.3389/fphar.2022.977025
37. Nguyen K-QN, Tsou W-I, Calarese DA, Kimani SG, Singh S, Hsieh S, et al. Overexpression of MERTK receptor tyrosine kinase in epithelial cancer cells drives efferocytosis in a gain-of-function capacity. J Biol Chem (2014) 289(37):25737–49. doi: 10.1074/jbc.M114.570838
38. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis (2017) 22(10):1189–204. doi: 10.1007/s10495-017-1413-z
39. Werfel TA, Cook RS. Efferocytosis in the tumor microenvironment. Semin Immunopathol. (2018) 40(6):545–554. doi: 10.1007/s00281-018-0698-5
40. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest (2013) 123(8):3231–42. doi: 10.1172/JCI67655
41. Blachère NE, Darnell RB, Albert ML. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PloS Biol (2005) 3(6):e185. doi: 10.1371/journal.pbio.0030185
42. Kim R, Emi M, Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther (2005) 4(9):924–33. doi: 10.4161/cbt.4.9.2101
43. Lin J, Xu A, Jin J, Zhang M, Lou J, Qian C, et al. MerTK-mediated efferocytosis promotes immune tolerance and tumor progression in osteosarcoma through enhancing M2 polarization and PD-L1 expression. Oncoimmunology (2022) 11(1):2024941. doi: 10.1080/2162402X.2021.2024941
44. Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem (2011) 286(38):33335–44. doi: 10.1074/jbc.M111.263020
45. Merilahti JA, Elenius K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene (2019) 38(2):151–63. doi: 10.1038/s41388-018-0465-z
46. Borgman KJ, Flórez-Grau G, Ricci MA, Manzo C, Lakadamyali M, Cambi A, et al. Membrane receptor MerTK is a newly identified transcriptional regulator that associates to chromatin as nanoclusters during human DC differentiation. bioRxiv (2020) 2020:044974. doi: 10.1101/2020.04.16.044974
47. Liu Y, Lan L, Li Y, Lu J, He L, Deng Y, et al. N-glycosylation stabilizes MerTK and promotes hepatocellular carcinoma tumor growth. Redox Biol (2022) 54:102366. doi: 10.1016/j.redox.2022.102366
48. Migdall-Wilson J, Bates C, Schlegel J, Brandão L, Linger RM, DeRyckere D, et al. Prolonged exposure to a Mer ligand in leukemia: Gas6 favors expression of a partial Mer glycoform and reveals a novel role for Mer in the nucleus. PloS One (2012) 7(2):e31635. doi: 10.1371/journal.pone.0031635
49. Georgescu M-M, Kirsch KH, Shishido T, Zong C, Hanafusa H. Biological effects of c-Mer receptor tyrosine kinase in hematopoietic cells depend on the Grb2 binding site in the receptor and activation of NF-κB. Mol Cell Biol (1999) 19(2):1171–81. doi: 10.1128/MCB.19.2.1171
50. Belov AA, Mohammadi M. Grb2, a double-edged sword of receptor tyrosine kinase signaling. Sci Signaling (2012) 5(249):pe49–pe. doi: 10.1126/scisignal.2003576
51. Tibrewal N, Wu Y, D'mello V, Akakura R, George TC, Varnum B, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-κB transcriptional activation. J Biol Chem (2008) 283(6):3618–27. doi: 10.1074/jbc.M706906200
52. Mahajan NP, Earp HS. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: a mechanism for localizing guanine nucleotide-exchange factor action. J Biol Chem (2003) 278(43):42596–603. doi: 10.1074/jbc.M305817200
53. Shelby SJ, Colwill K, Dhe-Paganon S, Pawson T, Thompson DA. MERTK interactions with SH2-domain proteins in the retinal pigment epithelium. PLoS One (2013) 8(2):e53964. doi: 10.1371/journal.pone.0053964
54. DeRyckere D, Lee-Sherick AB, Huey MG, Hill AA, Tyner JW, Jacobsen KM, et al. UNC2025, a MERTK small-molecule inhibitor, is therapeutically effective alone and in combination with methotrexate in leukemia modelsTherapeutic effects of UNC2025 in leukemia. Clin Cancer Res (2017) 23(6):1481–92. doi: 10.1158/1078-0432.CCR-16-1330
55. Zheng H, Zhao J, Li B, Zhang W, Stashko MA, Minson KA, et al. UNC5293, a potent, orally available and highly MERTK-selective inhibitor. Eur J Medicinal Chem (2021) 220:113534. doi: 10.1016/j.ejmech.2021.113534
56. Pyo K-H, Cho H-N, Xin C-F, Cho JS, Kang HN, Yun J, et al. Efficacy of MERTK inhibitor in combination with pembrolizumab in lung cancer. Cancer Res (2019) 79(13_Supplement):4081–. doi: 10.1158/1538-7445.AM2019-4081
57. Hoque M, Wong SW, Recasens A, Abbassi R, Nguyen N, Zhang D, et al. MerTK activity is not necessary for the proliferation of glioblastoma stem cells. Biochem Pharmacol (2021) 186:114437. doi: 10.1016/j.bcp.2021.114437
58. Zhou Y, Fei M, Zhang G, Liang W-C, Lin W, Wu Y, et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity (2020) 52(2):357–73.e9. doi: 10.1016/j.immuni.2020.01.014
59. Hsu J, Chong C, Serrill J, Goon L, Balayan J, Johnson EN, et al. Preclinical characterization of XL092, a novel receptor tyrosine kinase inhibitor of MET, VEGFR2, AXL, and MER. Mol Cancer Ther (2023) 22(2):179–91. doi: 10.1158/1535-7163.MCT-22-0262
60. Bae S-H, Kim J-H, Park TH, Lee K, Lee BI, Jang H. BMS794833 inhibits macrophage efferocytosis by directly binding to MERTK and inhibiting its activity. Exp Mol Med (2022) 54(9):1450–60. doi: 10.1038/s12276-022-00840-x
61. Wu J, Frady LN, Bash RE, Cohen SM, Schorzman AN, Su Y-T, et al. MerTK as a therapeutic target in glioblastoma. Neuro-oncology (2018) 20(1):92–102. doi: 10.1093/neuonc/nox111
62. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
63. Chirino LM, Kumar S, Okumura M, Sterner DE, Mattern M, Butt TR, et al. TAM receptors attenuate murine NK-cell responses via E3 ubiquitin ligase Cbl-b. Eur J Immunol (2020) 50(1):48–55. doi: 10.1002/eji.201948204
64. Lee-Sherick A, Eisenman K, Sather S, McGranahan A, Armistead P, McGary C, et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene (2013) 32(46):5359–68. doi: 10.1038/onc.2013.40
65. Minson KA, Huey MG, Hill AA, Perez I, Wang X, Frye S, et al. Bone marrow stromal cell mediated resistance to mertk inhibition in acute leukemia. Blood (2016) 128(22):2819. doi: 10.1182/blood.V128.22.2819.2819
66. Cruz JC, Allison KC, Page LS, Jenkins AJ, Wang X, Earp HS, et al. Inhibiting efferocytosis reverses macrophage-mediated immunosuppression in the leukemia microenvironment. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1146721
67. Alvarado D, Vitale L, Murphy M, O'Neill T, Proffitt A, Lillquist J, et al. Monoclonal antibodies targeting the TAM family of receptor tyrosine kinases. Cancer Res (2019) 79(13_Supplement):1555–. doi: 10.1158/1538-7445.AM2019-1555
68. Takeda S, Andreu-Agullo C, Sridhar S, Halberg N, Lorenz IC, Tavazoie S, et al. Abstract LB-277: Characterization of the anti-cancer and immunologic activity of RGX-019, a novel pre-clinical stage humanized monoclonal antibody targeting the MERTK receptor. Cancer Res (2019) 79(13_Supplement):LB–277-LB-. doi: 10.1158/1538-7445.AM2019-LB-277
69. Davra V, Kumar S, Geng K, Calianese D, Mehta D, Gadiyar V, et al. Axl and Mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host antitumor immunity. Cancer Res (2021) 81(3):698–712. doi: 10.1158/0008-5472.CAN-20-2066
70. Caetano MS, Younes AI, Barsoumian HB, Quigley M, Menon H, Gao C, et al. Triple therapy with merTK and PD1 inhibition plus radiotherapy promotes abscopal antitumor immune responsesRT with merTK and PD1 blockade promotes abscopal responses. Clin Cancer Res (2019) 25(24):7576–84. doi: 10.1158/1078-0432.CCR-19-0795
71. Frejno M, Zenezini Chiozzi R, Wilhelm M, Koch H, Zheng R, Klaeger S, et al. Pharmacoproteomic characterisation of human colon and rectal cancer. Mol Syst Biol (2017) 13(11):951. doi: 10.15252/msb.20177701
72. Xue G, Kohler R, Tang F, Hynx D, Wang Y, Orso F, et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget (2017) 8(41):69204. doi: 10.18632/oncotarget.18213
73. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. New Engl J Med (2020) 382(1):41–50. doi: 10.1056/NEJMoa1913662
74. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. New Engl J Med (2018) 378(2):113–25. doi: 10.1056/NEJMoa1713137
75. Yan D, Huelse JM, Kireev D, Tan Z, Chen L, Goyal S, et al. MERTK activation drives osimertinib resistance in EGFR-mutant non–small cell lung cancer. J Clin Invest (2022) 132(15):e150517. doi: 10.1172/JCI150517
76. Rios-Doria J, Favata M, Lasky K, Feldman P, Lo Y, Yang G, et al. A potent and selective dual inhibitor of AXL and MERTK possesses both immunomodulatory and tumor-targeted activity. Front Oncol (2020) 10:598477. doi: 10.3389/fonc.2020.598477
77. Okura N, Nishioka N, Yamada T, Taniguchi H, Tanimura K, Katayama Y, et al. ONO-7475, a novel AXL inhibitor, suppresses the adaptive resistance to initial EGFR-TKI treatment in EGFR-mutated non–small cell lung cancerONO-7475 suppresses resistance to initial EGFR-TKI. Clin Cancer Res (2020) 26(9):2244–56. doi: 10.1158/1078-0432.CCR-19-2321
78. Minson KA, Smith CC, DeRyckere D, Libbrecht C, Lee-Sherick AB, Huey MG, et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight (2016) 1(3):e85630. doi: 10.1172/jci.insight.85630
79. Ruvolo PP, Ma H, Ruvolo VR, Zhang X, Mu H, Schober W, et al. Anexelekto/MER tyrosine kinase inhibitor ONO-7475 arrests growth and kills FMS-like tyrosine kinase 3-internal tandem duplication mutant acute myeloid leukemia cells by diverse mechanisms. haematologica (2017) 102(12):2048. doi: 10.3324/haematol.2017.168856
80. Favata M, Lasky K, Lo Y, Feldman P, Li J, Chen Y, et al. Characterization of INCB081776, a potent and selective dual AXL/MER kinase inhibitor. Cancer Res (2018) 78(13_Supplement):3759–. doi: 10.1158/1538-7445.AM2018-3759
81. Hellerstedt BA, Vogelzang NJ, Kluger HM, Yasenchak CA, Aftab DT, Ramies DA, et al. Results of a phase ii placebo-controlled randomized discontinuation trial of cabozantinib in patients with non–small-cell lung carcinoma. Clin Lung Cancer (2019) 20(2):74–81.e1. doi: 10.1016/j.cllc.2018.10.006
82. Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol (2013) 31(4):412. doi: 10.1200/JCO.2012.45.0494
83. Vergote IB, Smith DC, Berger R, Kurzrock R, Vogelzang NJ, Sella A, et al. A phase 2 randomised discontinuation trial of cabozantinib in patients with ovarian carcinoma. Eur J Cancer (2017) 83:229–36. doi: 10.1016/j.ejca.2017.06.018
84. Schöffski P, Gordon M, Smith DC, Kurzrock R, Daud A, Vogelzang NJ, et al. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur J Cancer (2017) 86:296–304. doi: 10.1016/j.ejca.2017.09.011
85. He AR, Cohen RB, Denlinger CS, Sama A, Birnbaum A, Hwang J, et al. First-in-human phase I study of merestinib, an oral multikinase inhibitor, in patients with advanced cancer. oncologist (2019) 24(9):e930–e42. doi: 10.1634/theoncologist.2018-0411
86. Ma Y-X, Liu F-R, Zhang Y, Chen Q, Chen Z-Q, Liu Q-W, et al. Preclinical characterization and phase I clinical trial of CT053PTSA targets MET, AXL, and VEGFR2 in patients with advanced solid tumors. Front Immunol (2022) 13:1024755. doi: 10.3389/fimmu.2022.1024755
87. Yoshizawa T, Tanaka K, Yasuhiro T, Fujikawa R, Ri S, Kawabata K. Abstract LB-218: Development of Axl/Mer inhibitor, ONO-9330547: preclinical evidence supporting the combination with immunotherapeutics. Cancer Res (2016) 76(14_Supplement):LB–218-LB-. doi: 10.1158/1538-7445.AM2016-LB-218
88. Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight (2018) 3(21):e124184. doi: 10.1172/jci.insight.124184
89. He K, Berz D, Gadgeel SM, Iams WT, Bruno DS, Blakely CM, et al. MRTX-500 phase 2 trial: sitravatinib with nivolumab in patients with nonsquamous NSCLC progressing on or after checkpoint inhibitor therapy or chemotherapy. J Thorac Oncol (2023) 18(7):907–921. doi: 10.1016/j.jtho.2023.02.016
90. Bauer T, Cho BC, Heist R, Bazhenova L, Werner T, Goel S, et al. First-in-human phase 1/1b study to evaluate sitravatinib in patients with advanced solid tumors. Investigational New Drugs (2022) 40(5):990–1000. doi: 10.1007/s10637-022-01274-y
91. Oliva M, Chepeha D, Araujo DV, Diaz-Mejia JJ, Olson P, Prawira A, et al. Antitumor immune effects of preoperative sitravatinib and nivolumab in oral cavity cancer: SNOW window-of-opportunity study. J Immunother Cancer (2021) 9(10):e003476. doi: 10.1136/jitc-2021-003476
92. Bernal MO, Chepeha D, Prawira A, Vines D, Spreafico A, Bratman S, et al. Abstract CT124: Sitravatinib and nivolumab in oral cavity cancer window of opportunity study (SNOW). Cancer Res (2019) 79(13_Supplement):CT124–CT. doi: 10.1158/1538-7445.AM2019-CT124
93. Sayama A, Okado K, Yamaguchi M, Samata N, Imaoka M, Kai K, et al. The impact of the timing of dosing on the severity of UNC569-induced ultrastructural changes in the mouse retina. Toxicologic Pathology (2020) 48(5):669–76. doi: 10.1177/0192623320931415
94. Bhatia S, Kaur N, Singh IR, Vanita V. A novel mutation in MERTK for rod-cone dystrophy in a North Indian family. Can J Ophthalmol (2019) 54(1):40–50. doi: 10.1016/j.jcjo.2018.02.008
95. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Science (2020) 111(2):312–22. doi: 10.1111/cas.14274
96. Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev (2022) 52:100905. doi: 10.1016/j.blre.2021.100905
97. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol cancer (2019) 18:1–22. doi: 10.1186/s12943-019-1090-3
98. Mikolajczyk A, Mitula F, Popiel D, Kaminska B, Wieczorek M, Pieczykolan J. Two-front war on cancer—Targeting TAM receptors in solid tumour therapy. Cancers (2022) 14(10):2488. doi: 10.3390/cancers14102488
Keywords: MerTK, macrophage, cancer, immune, MerTK inhibitors, clinical trials
Citation: Ubil E and Zahid KR (2023) Structure and functions of Mer, an innate immune checkpoint. Front. Immunol. 14:1244170. doi: 10.3389/fimmu.2023.1244170
Received: 22 June 2023; Accepted: 09 October 2023;
Published: 23 October 2023.
Edited by:
Alexandre P. A. Theocharides, University Hospital Zürich, SwitzerlandReviewed by:
Rodwell Mabaera, Dartmouth Hitchcock Medical Center, United StatesDerek Brandon Oien, AstraZeneca, United States
Copyright © 2023 Ubil and Zahid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Ubil, ZXJpY3ViaWxAdWFiLmVkdQ==