 Natália S. Vellozo
Natália S. Vellozo- Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brazil
During Trypanosoma cruzi infection, macrophages phagocytose parasites and remove apoptotic cells through efferocytosis. While macrophage 1 (M1) produces proinflammatory cytokines and NO and fights infection, M2 macrophages are permissive host cells that express arginase 1 and play a role in tissue repair. The regulation of M1 and M2 phenotypes might either induce or impair macrophage-mediated immunity towards parasite control or persistence in chronic Chagas disease. Here, we highlight a key role of macrophage activation in early immune responses to T. cruzi that prevent escalating parasitemia, heart parasitism, and mortality during acute infection. We will discuss the mechanisms of macrophage activation and deactivation, such as T cell cytokines and efferocytosis, and how to improve macrophage-mediated immunity to prevent parasite persistence, inflammation, and the development of chagasic cardiomyopathy. Potential vaccines or therapy must enhance early T cell-macrophage crosstalk and parasite control to restrain the pathogenic outcomes of parasite-induced inflammation in the heart.
1 Introduction
The protozoan Trypanosoma cruzi infects humans and animals, establishes chronic infection, and causes Chagas disease by affecting the heart in 30% of patients (1, 2). Although 13% of the Latin American population is at risk of infection (1, 3), there is no available vaccine or effective treatment for chronic infection and established pathology (2, 4, 5). Moreover, difficulties in treating and following human patients for decades before the onset of disease symptoms, as well as the costs of human trials for neglected tropical diseases, hamper drug development, despite advances in preclinical research (3–5). Likewise, translation from drug and vaccine research towards human benefits has been delayed owing to unsolved scientific controversies about the mechanisms of Chagas disease pathogenesis (6).
Complex interactions between the parasite, the host, and the immune system underlie the development of heart pathology in Chagas disease, characterized by inflammation and fibrosis, which lead to heart malfunctioning, heart failure, and death (1, 6). Parasite infection contributes to pathology by destroying infected cells, including myocytes, and by stimulating pathogenic immune responses that kill infected cells and cause inflammation (6–8). The immune system is necessary to control T. cruzi infection, thereby reducing parasite spread and parasite-induced inflammation (9, 10). Nonetheless, immune responses are involved in the pathogenesis of Chagas disease by causing tissue damage and inflammation (immunopathology) (7, 8, 10, 11), whereas immunoregulatory mechanisms control immunity and/or immunopathology. The dissection of the immune response components in T. cruzi infection and their roles in immunopathogenesis is crucial for the development of new vaccines or therapeutic tools without stimulating immunopathology.
Macrophages play multiple and key roles as dedicated phagocytes that clear tissues from parasites and apoptotic cells, act as M1 effectors or M2 permissive host cells, and promote inflammation, tissue repair (12), and fibrosis (13). Here, we focused on the molecular mechanisms of macrophage activation and deactivation, the dual role of M1 and M2 macrophages in antiparasitic immunity, and their modulation by T cell cytokines and apoptotic cells. We consider classically activated macrophages to be M1, which express IL-12 and induced NO synthase (iNOS), produce NO, and exhibit microbicidal activity (14). In contrast, alternatively activated M2 macrophages are susceptible to parasite infection (15), express arginase 1 (Arg1) (16), and play a role in tissue repair (12). Macrophage phenotypes are complex, plastic, and interchangeable in response to diverse environmental conditions. Previously published articles provided deeper information on the full spectrum of macrophage phenotypes beyond the M1 and M2 extremes obtained under defined Th1 and Th2 cytokine conditions (17–20).
2 Defective M1 macrophage-mediated immunity plays a pathogenic role in Chagas disease
During acute infection, both innate and adaptive immunity are required to fight T. cruzi parasites in the blood, heart, and other organs (9, 10), yet parasites resist in tissue reservoirs and establish chronic infection (21). Monocytes are mobilized and recruited to the heart, where macrophages dominate early protective inflammatory responses (22). Parasite infection targets myocytes, fibroblasts, and various cell types, while macrophages continuously collect parasites released by infected cells. Macrophages can be detected in the proximity or even inside myocyte parasite nests, whereas some macrophages interact with lymphocytes or contain intracellular parasites (22, 23). In rat and mouse experimental models, macrophage depletion upon silica treatment increased parasitemia, heart parasitism, tissue damage, and mortality (23, 24), highlighting macrophage protective immunity in T. cruzi infection.
CD4 and CD8 T cells play a protective role by inducing NO-producing M1 macrophages (6, 9, 10). In contrast, Th2 cytokines and Arg1-expressing M2 macrophages increase susceptibility to T. cruzi infection (16). Therefore, the activation of M1 and M2 macrophage phenotypes might critically affect disease outcomes. Next, we will discuss how the mechanisms that govern macrophage recruitment and M1/M2 phenotypes induce either protective immunity or parasite persistence and disease progression.
Macrophage activation towards microbicidal M1 responses relies on macrophage receptors for pathogen-associated molecular patterns (PAMPs) and T cell-derived cytokines, such as IFN-γ and TNF-α, which induce iNOS expression and help to control intracellular infection. In addition, M1 macrophages secrete IL-12 and induce IFN-γ production by both NK and T cells, further enhancing type 1 responses (9, 10). Early seminal studies showed that mice deficient in IFN-γ have increased parasitemia, heart parasitism and mortality (25), even after infection with less virulent T. cruzi strains (26). Importantly, mice with macrophages insensitive to IFN-γ (MIIG) fail to control parasite infection in vitro and show increased parasitemia and mortality (27). Higher mortality, parasitemia, and nervous system inflammation were also observed upon genetic ablation of IL-12 (25). Moreover, IL-12-defective macrophages were more susceptible to T. cruzi infection, expressing a reduced NO response to IFN-γ and increased TGF-β production, similar to M2 macrophages (15).
A series of studies addressed the factors that influence inflammatory responses in the heart during acute T. cruzi infection. Silva et al. showed that mice deficient in the inflammasome components ASC/Caspase-1 and IL-1R have defective recruitment of CD11b+/F4/80+ macrophages to the heart associated with increased heart parasitism and mortality (28). In addition, a direct role of IL-1β in the induction of NO and parasite killing was suggested as part of macrophage protective responses (28). Possibly owing to unrestricted parasite infection, ASC, Caspase-1 or IL-1R knockout (KO) mice developed increased inflammation and tissue damage during late acute infection (28).
The association between the presence of the chemokine receptors CCR2/CCR5 and the chemokines CCL2/CCL3/CCL5/CXCL9 and macrophages in the hearts of T. cruzi-infected mice indicates that chemokines and their receptors play a role in macrophage recruitment or activation to fight infection (29–33). In agreement with this idea, mice deficient in CCR2, CCR5, CXCL9, CCL3, and CCL2 developed increased parasitemia and/or heart parasitism (29–33), whereas higher mortality was also observed in infected CCR5 and CCL2 KO mice (30, 32). Moreover, the transfer of CCR5+ splenocytes to CCR5 KO mice rescued macrophage recruitment to the heart and early protective inflammatory responses (30). In contrast, the transfer of CCR5-/- splenocytes failed to generate macrophages in the heart. By addressing the role of CCL2 in experimental Chagas disease, Paiva et al. showed that CCL2 is expressed on the heart inflammatory foci (32). Accordingly, CCL2 KO mice have reduced inflammatory foci and macrophage activation in the heart, despite increased systemic cytokine responses secondary to uncontrolled parasitemia and tissue parasitism (32).
By studying the relevant mechanisms for parasite killing in T. cruzi infection, Sharma et al. showed that macrophages defective in phospholipase 2 β (PLA2β) have reduced NO production and increased parasite replication, whereas parasite nests are abundant in the hearts of PLA2β KO mice (34). Recently, Silva et al. addressed the role of phosphatidylinositol 3-kinase-γ (PI3Kγ), which is important for macrophage-mediated immunity, as highlighted by reduced NO production and increased parasite infection in PI3Kγ-defective or inhibitor-treated macrophages (35). Infected PI3Kγ KO mice exhibited increased weight loss, parasitism, heart inflammation and malfunction, tissue damage, and mortality (35). Defective downstream PI3Kγ signalling in macrophage conditional AKT1 KO mice also increased parasitism and mortality (35). Moreover, macrophages are the major players in protective immune responses mediated by PI3Kγ (35). Interestingly, infected PI3Kγ KO mice benefited from treatment with anti-inflammatory or antiparasitic drugs (35). These results suggest that both parasites and inflammatory responses contribute to disease secondary to PI3Kγ deficiency.
Next, we will discuss the downregulation/inhibition of macrophage activation in experimental BALB/c models of T. cruzi infection. By using CD73 KO mice and/or pharmacological CD73 inhibition, Ponce et al. showed that the CD73 ectonucleotidase deactivates macrophages during infection (36). CD73 genetic deficiency or inhibitor restored M1 responses in the heart and reduced heart parasitism, inflammation, tissue damage, and arrythmia (36). Calderon et al. addressed the role of SLAMF1, a factor that downregulates NADPH oxidase in T. cruzi infection (37). They show that macrophages from SLAMF1 KO mice show better control of parasite replication and that SLAMF1 KO mice have reduced arginase expression in their hearts and reduced parasitism, tissue damage, and mortality (37).
Altogether, these studies suggest a major protective role of M1-mediated immunity to T. cruzi during acute infection that reduces infection, mortality, and pathology (Table 1). Conversely, macrophage failure to fight parasites might be implicated in parasite persistence throughout chronic infection and more severe infection outcomes (38), with continuous or intermittent release of infected cell contents and antigens further insufflating inflammation and heart pathology (39).
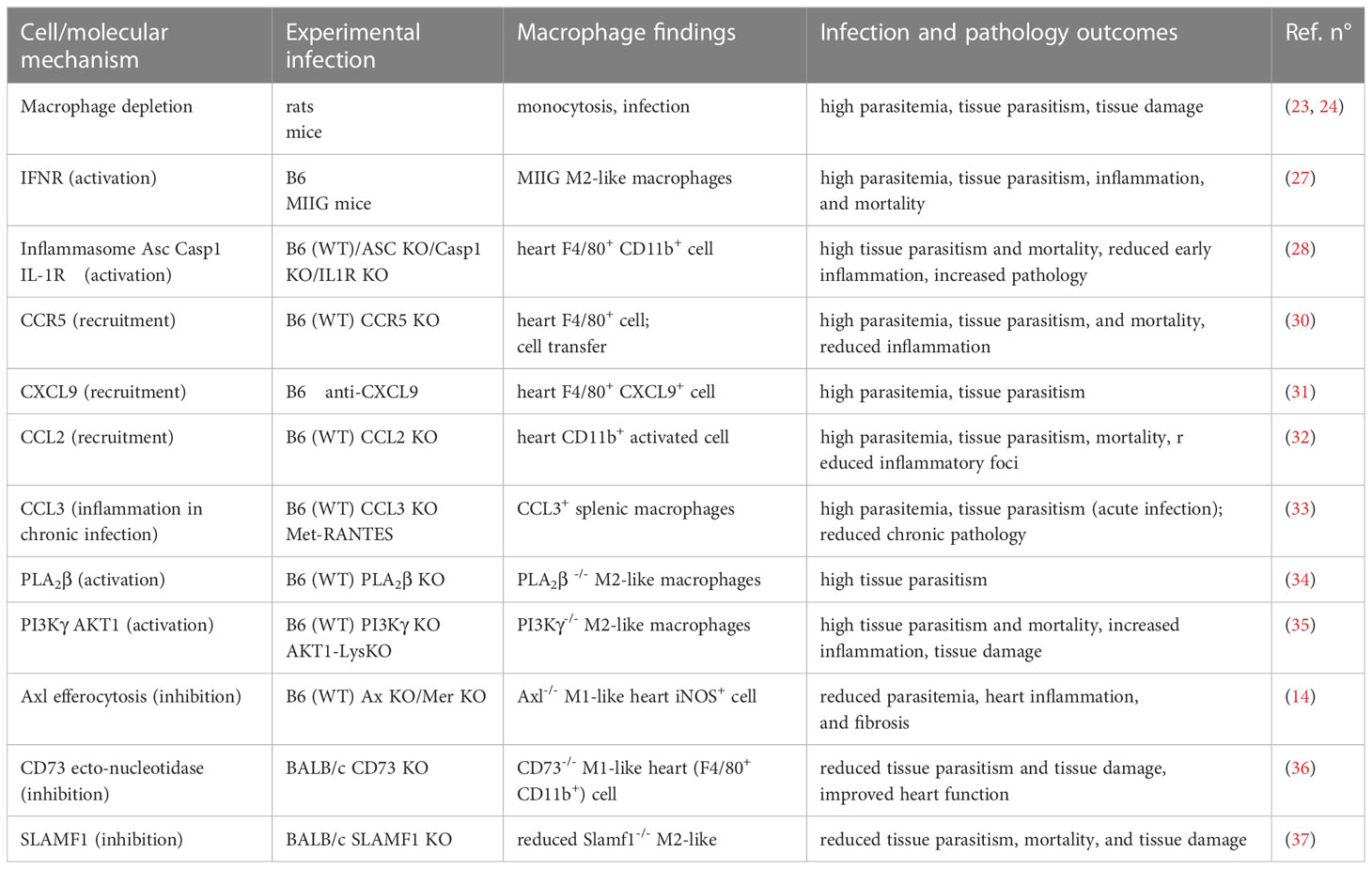
Table 1 Macrophage 1 provides immunity whereas Macrophage 2 promotes infection and pathology.
Importantly, the use of experimental models to follow disease development shows that the role played by protective versus pathogenic immune responses is timing dependent in acute versus chronic infection. During acute infection, CCL3-chemokine KO mice express increased parasitemia and heart parasitism, indicating that early CCL3-mediated recruitment of immune cells to the heart protects against parasite infection (33). In contrast, chronically infected mice deficient in CCL3 or treated with a chemokine receptor antagonist show reduced cardiac inflammation and tissue damage and restored heart function (33).Therefore, whereas early CCL3 expression in macrophages correlates with protective immune responses, continuous CCL3-mediated inflammation throughout chronic infection is deleterious to the host in Chagas disease (33). These results are consistent with clinical studies of chronic Chagas disease that show that heart expression of the proinflammatory cytokines IFN-γ and TNF-α, as well as chemokines, correlates with severe chagasic cardiomyopathy (1, 40).
3 Apoptosis underlies defective T cell help to macrophages in T. cruzi infection
Immunoregulatory mechanisms that defeat T cell-mediated immunity, such as the death of cytokine-producing T cells, might affect their ability to help macrophages infected with T. cruzi (41). By searching for defects in the immune responses underlying parasite persistence, we found that splenic T cells from infected mice proliferate less than T cells from healthy mice in response to T cell receptor (TCR) agonists (42). Moreover, during acute infection, T cells undergo activation-induced cell death, which correlates with reduced proliferative responses upon TCR engagement (43, 44). Other groups also reported increased T and B cell apoptosis in lymphoid organs during T. cruzi infection (45–51). Furthermore, apoptosis and defective proliferative responses occur in T cells from patients with chronic cardiac Chagas disease and heart failure (52, 53). Importantly, apoptotic cells were found in the hearts both in experimental models and in human patients (54–56).
The molecular mechanisms involved in programmed cell death have been investigated as potential targets to restore immunity during parasitic diseases (57, 58). T cells from T. cruzi-infected mice express increased levels of proapoptotic molecules, such as Fas (CD95) and Fas ligand (FasL, CD95L), as well as caspase-8 activity and activated caspase-3 (59–63). The extrinsic apoptotic pathway ensues during T. cruzi infection through FasL binding to the death receptor Fas in CD4 and CD8 T cells (59, 62). The antagonist anti-FasL mAb (62), the caspase-8 inhibitor zIETD, and the pan caspase inhibitor zVAD (60, 61) prevent activation-induced death in T cells from infected mice. T cell proliferation increases in the presence of anti-FasL and in T cells from infected FasL-deficient gld mutant mice (59, 62). These findings indicate that Fas-mediated apoptosis might counteract T cell expansion during infection. Moreover, activation-induced cell death and FasL-Fas expression underlie defective proliferation in patient T cells (52, 53).
Terminally differentiated effector cells undergo apoptosis to abbreviate the breadth of potentially pathogenic immune responses. Nonetheless, early apoptosis of effector T cells might curtail their ability to kill infected cells or help infected macrophages. During acute infection, antigen-specific effector CD8 T cells from infected mice express Fas and a proapoptotic phenotype (63). Likewise, CD4 T cells undergo Fas-mediated apoptosis and express a reduced ability to help infected macrophages (41, 59). The use of anti-FasL or CD4 T cells from infected gld mice allowed macrophages to control intracellular infection (41, 59). Pharmacological approaches used injections of anti-FasL and zVAD in T. cruzi-infected mice to evaluate their effects on immune responses during parasite infection (61, 62, 64). Treatment with zVAD during acute infection reduced parasitemia and apoptosis in splenocytes (61). Similarly, injection of anti-FasL reduced peak parasitemia and apoptosis in splenic CD8 T cells (62). Moreover, in both cases, infected mice had increased cytokine responses, and their macrophages expressed an improved ability to control parasite infection (61, 62, 64).
To directly address whether CD8 T cells cooperate with macrophages to fight T. cruzi parasites, splenic or peritoneal CD8 T cells and macrophages from infected mice were cocultured to evaluate IFN-γ production, T cell apoptosis and macrophage responses (64). Upon T cell activation, the failure of macrophages to produce NO and restrict parasite infection correlated with increased CD8 T cell apoptosis and development of the M2 phenotype (64). Treatment in vitro or in vivo with anti-FasL reduced T cell apoptosis, improved M1 responses, and restored macrophage-mediated immunity to T. cruzi infection (64). Altogether, these results suggest that the induction of T cell apoptosis during infection contributes to defective T cell and macrophage immune responses, allowing a permissive environment for parasite persistence towards the development of chronic infection.
Although these studies are useful as a proof of principle that apoptosis negatively regulates protective immune responses mediated by T cells, pharmacological and genetic ablation of apoptosis pathways during infection opens a “Pandora box” of undesirable effects such as the onset of autoimmunity in gld/lpr models (65–67) or increased inflammation in the hearts of T. cruzi-infected mice treated with anti-FasL (58). Genetic inhibition of the FasL-Fas or caspase-8 pathways also dysregulated Th2 cytokine responses and increased parasite infection (48, 59, 60). Moreover, these studies revealed that caspase-8 is also required for CD8 T cell expansion during T. cruzi infection (60). Finally, Bim-deleted mice are more susceptible to T. cruzi infection, most likely owing to defective macrophage and T cell responses (68). Therefore, the translation of apoptosis inhibition into treatment for chronic diseases is unlikely so far. Nonetheless, vaccine approaches might be useful to prevent the development of proapoptotic T cells, thereby improving antiparasitic immune responses (63).
4 Efferocytosis suppresses macrophage-mediated immunity
Apoptotic cells express ‘eat me signals’ in the outer membrane, such as phosphatidylserine, allowing their detection and clearance by phagocytes, a process named efferocytosis. The phagocytosis and dismounting of apoptotic cells prevent their accumulation in tissues and the release of proinflammatory cell content through secondary necrosis (69, 70). Several receptors might be involved in the detection of phosphatidylserine and phagocytosis (69, 70). In addition, apoptotic cells actively signal through macrophage receptors and induce anti-inflammatory responses (71, 72). How these receptors cooperate with each other and engage signaling pathways to convey proper responses to apoptotic cells is a complex scenario under investigation (72–74).
By using electron and light microscopy and immunofluorescence, we detected apoptotic lymphocytes inside macrophages from the spleen (75) and peritoneum (64) during T. cruzi infection. We found an apoptotic CD8 T cell inside a peritoneal macrophage and macrophages containing both parasites and apoptotic bodies during T. cruzi infection (64). To investigate how efferocytosis directly affects macrophage ability to fight T. cruzi parasites, we added apoptotic T cells to peritoneal macrophages from infected mice and evaluated endogenous infection by assessing parasites released from macrophages (76). Treatment with apoptotic but not necrotic cells exacerbated T. cruzi infection within macrophages and increased parasitemia upon injection in infected mice (76). Moreover, the receptor αvβ3 mediates apoptotic cell uptake and macrophage responses, such as the production of TGF-β and PGE2 and ornithine decarboxylase activity (76). These findings indicated that efferocytosis diverts L-arginine metabolism towards polyamine synthesis, which favors parasite survival and replication (76).
To address the role of efferocytosis during T. cruzi infection, we employed two mouse strains individually defective in Axl and Mer, two out of three TAM receptors involved in efferocytosis (14). Double Mer-/-Axl-/- and single Mer-defective strains have been previously used in Leishmania infection to show that infected neutrophils transfer Leishmania parasites to macrophages or DCs through efferocytosis and reduce macrophage and T cell responses (77, 78).
By employing bone marrow-derived macrophages treated with a TAM receptor inhibitor or Mer- and Axl-defective macrophages, we investigated macrophage responses to T cells from T. cruzi-infected mice, which bear both effector activity and proapoptotic cells (14). Efferocytosis of apoptotic T cells was blocked by a TAM receptor inhibitor, whereas Mer or Axl deficiency partially inhibited efferocytosis (14). Remarkably, TAM inhibition and Axl but not Mer deficiency improved M1 responses to T cells from T. cruzi-infected mice (14). These results indicate that Axl downregulates M1 macrophages, despite predominant Mer expression and the major role of Mer in efferocytosis. More importantly, Axl suppressed the expression of iNOS, NO production, and the ability of macrophages to fight parasite infection (14).
Zagorska et al. (79) previously reported that Mer and Axl play distinct roles in macrophage function. At homeostasis, constitutive Mer expression is important for the clearance of continuously generated apoptotic cells and to prevent inflammatory responses upon secondary necrosis. During immune responses, macrophage activation induces Axl expression to counteract increased inflammatory responses (79). To address the role of Axl in the removal of apoptotic cells during T. cruzi infection, we treated peritoneal macrophages from infected WT and Axl-/- mice with fluorescent apoptotic T cells. Detection of apoptotic cells undergoing efferocytosis was reduced in Axl-defective macrophages from infected mice (14). In addition, the overaccumulation of splenic apoptotic T cells in infected Axl-/- mice is further evidence of defective Axl-mediated efferocytosis (14).
During T. cruzi infection, Axl-/- mice expressed reduced peak parasitemia coupled with increased M1 responses in the spleen, peritoneum, and heart tissues (14). Furthermore, hearts collected from infected Axl-/- but not Mer-/- mice had reduced inflammation and fibrosis characteristics of heart pathology in Chagas disease (14). Overall, these findings indicate that Axl disrupts M1-mediated immunity to T. cruzi, fostering inflammatory responses and fibrosis in the heart (Figure 1).
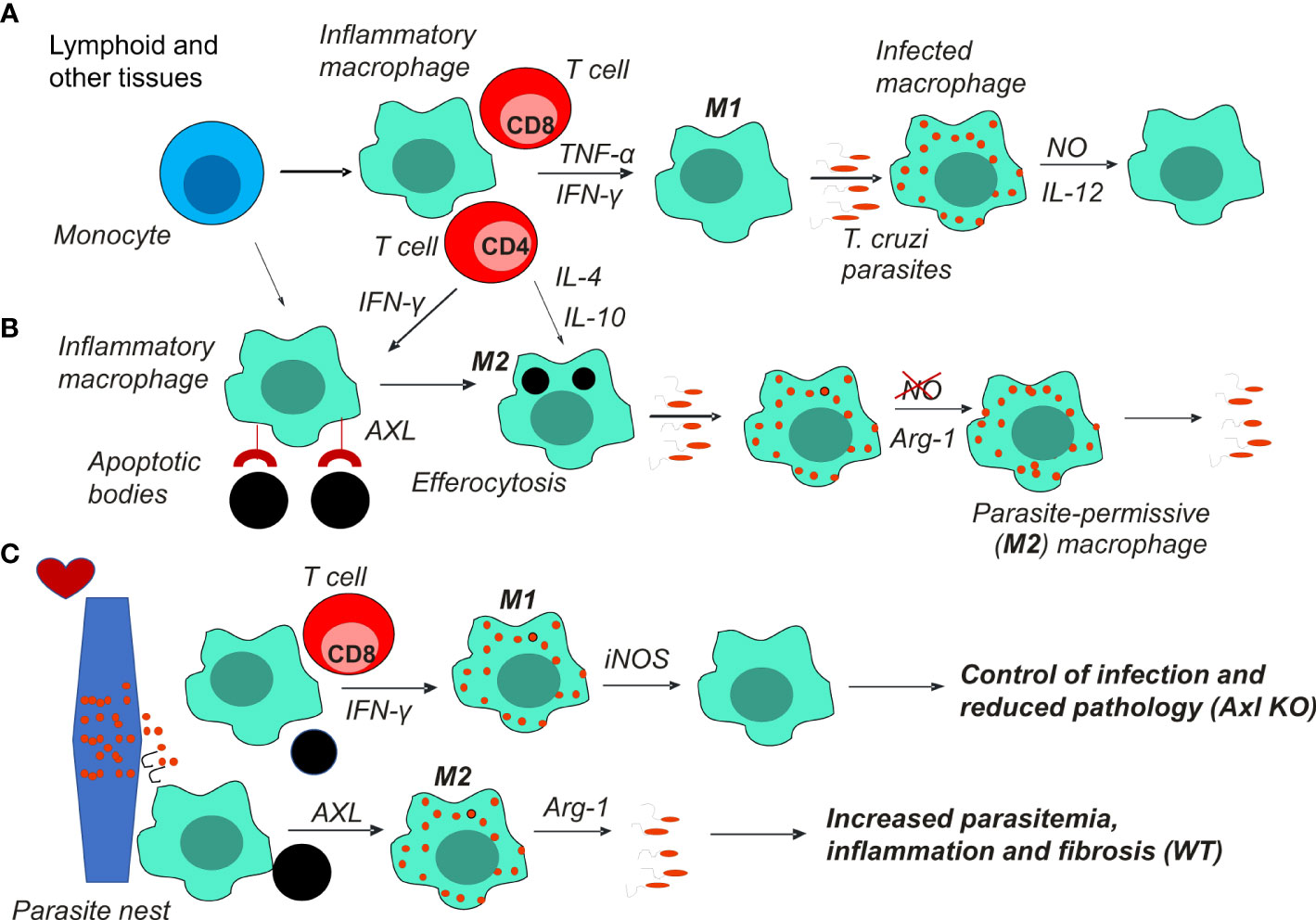
Figure 1 Macrophages play a key role in T. cruzi infection and Chagas disease pathology. (A) Monocytes recruited to lymphoid and other tissues differentiate into M1 macrophages under stimulation by the type 1 cytokines IFN-γ and TNF-α produced by T cells. M1 macrophages produce NO to control parasite infection and proinflammatory cytokines, such as IL-12 and TNF-α. (B) Alternatively, Th2 cytokines or the uptake of apoptotic cells (efferocytosis) induce parasite permissive M2 host cells that express Arg1 and fail to produce NO. The balance between M1 and M2 macrophages determines parasite control or escape and the development of chronic infection. (C) Pseudocysts of parasites within myocytes can rupture and release parasites in the heart. Foci of inflammatory macrophages clear heart tissues from parasites and apoptotic cells. M1 macrophages that control parasites prevent further inflammation and fibrosis, thereby reducing heart pathology. Otherwise, suppressed macrophages promote infection, inflammation, and fibrosis in the heart. Axl receptor-mediated efferocytosis might underlie macrophage suppression by apoptotic cells.
5 Concluding remarks
The development of antiparasitic therapy to treat T. cruzi infection has progressed recently (4). Likewise, the new vaccine generations tested for COVID-19 will help vaccine development for Chagas disease and other neglected diseases. Since early macrophage responses to T cell cytokines and apoptotic cells control macrophage M1/M2 responses and disease outcomes, the use of appropriate vaccine adjuvants to target macrophage activation and dampen regulatory circuits might upregulate early protective responses. In agreement with this, experimental vaccines induced protective cytokine responses by T cells and macrophages and reduced parasitemia, tissue parasitism, heart pathology, and mortality (80, 81). Improved T cell and macrophage responses induced upon vaccination in endemic areas might help to prevent the formation of larger parasite reservoirs in tissues and intermittent cycles of infection that underly inflammatory responses (38, 39) and the pathogenesis of Chagas disease.
Author contributions
NV cowrote the manuscript; TM-S discussed and reviewed the manuscript; ML analysed the literature and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Brazilian National Research Council (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq) and the Rio de Janeiro State Science Foundation (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, FAPERJ). ML is a research fellow at CNPq, Brazil. We also received fellowships from CNPq (TM-S), FAPERJ (NV and TM-S) and the American Association of Immunologists (NV and ML).
Acknowledgments
We thank George Alexandre DosReis (in memoriam) for his contributions to this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nunes MCP, Beaton A, Acquatella H, Bern C, Bolger AF, Echeverria LE, et al. Chagas cardiomyopathy: an update of current clinical knowledge and management: A scientific statement from the american heart association. Circulation (2018) 138(12):e169–209. doi: 10.1161/CIR.0000000000000599
2. Bustamante JM, Padilla AM, White B, Auckland LD, Busselman RE, Collins S, et al. Prophylactic low-dose, bi-weekly benznidazole treatment fails to prevent Trypanosoma cruzi infection in dogs under intense transmission pressure. PloS Negl Trop Dis (2022) 16(10):e0010688. doi: 10.1371/journal.pntd.0010688
3. Nunes MCP, Buss LF, Silva JLP, Martins LNA, Oliveira CDL, Cardoso CS, et al. Incidence and predictors of progression to chagas cardiomyopathy: long-term follow-up of trypanosoma cruzi-seropositive individuals. Circulation (2021) 144(19):1553–66. doi: 10.1161/CIRCULATIONAHA.121.055112
4. Padilla AM, Wang W, Akama T, Carter DS, Easom E, Freund Y, et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat Microbiol (2022) 7(10):1536–46. doi: 10.1038/s41564-022-01211-y
5. Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr., Rosas F, et al. Randomized trial of benznidazole for chronic chagas’ Cardiomyopathy. N Engl J Med (2015) 373(14):1295–306. doi: 10.1056/NEJMoa1507574
6. De Alba-Alvarado MC, Torres-Gutierrez E, Reynoso-Ducoing OA, Zenteno-Galindo E, Cabrera-Bravo M, Guevara-Gomez Y, et al. Immunopathological mechanisms underlying cardiac damage in chagas disease. Pathogens (2023) 12(2):335. doi: 10.3390/pathogens12020335
7. Silverio JC, de-Oliveira-Pinto LM, da Silva AA, de Oliveira GM, Lannes-Vieira J. Perforin-expressing cytotoxic cells contribute to chronic cardiomyopathy in Trypanosoma cruzi infection. Int J Exp Pathol (2010) 91(1):72–86. doi: 10.1111/j.1365-2613.2009.00670.x
8. Silverio JC, Pereira IR, Cipitelli Mda C, Vinagre NF, Rodrigues MM, Gazzinelli RT, et al. CD8+ T-cells expressing interferon gamma or perforin play antagonistic roles in heart injury in experimental Trypanosoma cruzi-elicited cardiomyopathy. PloS Pathog (2012) 8(4):e1002645. doi: 10.1371/journal.ppat.1002645
9. Macaluso G, Grippi F, Di Bella S, Blanda V, Gucciardi F, Torina A, et al. A Review on the Immunological Response against Trypanosoma cruzi. Pathogens (2023) 12(2):282. doi: 10.3390/pathogens12020282
10. Pellegrini A, Guinazu N, Giordanengo L, Cano RC, Gea S. The role of Toll-like receptors and adaptive immunity in the development of protective or pathological immune response triggered by the Trypanosoma cruzi protozoan. Future Microbiol (2011) 6(12):1521–33. doi: 10.2217/fmb.11.122
11. Tarleton RL, Koller BH, Latour A, Postan M. Susceptibility of beta 2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature (1992) 356(6367):338–40. doi: 10.1038/356338a0
12. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science (2017) 356(6342):1072–6. doi: 10.1126/science.aai8132
13. Choudhuri S, Garg NJ. Trypanosoma cruzi Induces the PARP1/AP-1 Pathway for Upregulation of Metalloproteinases and Transforming Growth Factor beta in Macrophages: Role in Cardiac Fibroblast Differentiation and Fibrosis in Chagas Disease. mBio (2020) 11(6):e01853-20. doi: 10.1128/mBio.01853-20
14. Rigoni TS, Vellozo NS, Guimaraes-Pinto K, Cabral-Piccin M, Fabiano-Coelho L, Matos-Silva TC, et al. Axl receptor induces efferocytosis, dampens M1 macrophage responses and promotes heart pathology in Trypanosoma cruzi infection. Commun Biol (2022) 5(1):1421. doi: 10.1038/s42003-022-04401-w
15. Bastos KR, Alvarez JM, Marinho CR, Rizzo LV, Lima MR. Macrophages from IL-12p40-deficient mice have a bias toward the M2 activation profile. J Leukoc Biol (2002) 71(2):271–8. doi: 10.1189/jlb.71.2.271
16. Abad Dar M, Holscher C. Arginase-1 is responsible for IL-13-mediated susceptibility to trypanosoma cruzi infection. Front Immunol (2018) 9:2790. doi: 10.3389/fimmu.2018.02790
17. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
18. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
19. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
20. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
21. Lewis MD, Kelly JM. Putting infection dynamics at the heart of chagas disease. Trends Parasitol (2016) 32(11):899–911. doi: 10.1016/j.pt.2016.08.009
22. Melo RC. Acute heart inflammation: ultrastructural and functional aspects of macrophages elicited by Trypanosoma cruzi infection. J Cell Mol Med (2009) 13(2):279–94. doi: 10.1111/j.1582-4934.2008.00388.x
23. Melo RC, MaChado CR. Trypanosoma cruzi: peripheral blood monocytes and heart macrophages in the resistance to acute experimental infection in rats. Exp Parasitol (2001) 97(1):15–23. doi: 10.1006/expr.2000.4576
24. Kierszenbaum F, Knecht E, Budzko DB, Pizzimenti MC. Phagocytosis: a defense mechanism against infection with Trypanosoma cruzi. J Immunol (1974) 112(5):1839–44. doi: 10.4049/jimmunol.112.5.1839
25. Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and interferon-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol (2001) 159(5):1723–33. doi: 10.1016/s0002-9440(10)63019-2
26. Marinho CR, Nunez-Apaza LN, Martins-Santos R, Bastos KR, Bombeiro AL, Bucci DZ, et al. IFN-gamma, but not nitric oxide or specific IgG, is essential for the in vivo control of low-virulence Sylvio X10/4 Trypanosoma cruzi parasites. Scand J Immunol (2007) 66(2-3):297–308. doi: 10.1111/j.1365-3083.2007.01958.x
27. Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, et al. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol (2010) 184(2):877–85. doi: 10.4049/jimmunol.0902346
28. Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FR, et al. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1beta response and host resistance to Trypanosoma cruzi infection. J Immunol (2013) 191(6):3373–83. doi: 10.4049/jimmunol.1203293
29. Hardison JL, Kuziel WA, Manning JE, Lane TE. Chemokine CC receptor 2 is important for acute control of cardiac parasitism but does not contribute to cardiac inflammation after infection with Trypanosoma cruzi. J Infect Dis (2006) 193(11):1584–8. doi: 10.1086/503812
30. Hardison JL, Wrightsman RA, Carpenter PM, Kuziel WA, Lane TE, Manning JE. The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi. Infect Immun (2006) 74(1):135–43. doi: 10.1128/IAI.74.1.135-143.2006
31. Hardison JL, Wrightsman RA, Carpenter PM, Lane TE, Manning JE. The chemokines CXCL9 and CXCL10 promote a protective immune response but do not contribute to cardiac inflammation following infection with Trypanosoma cruzi. Infect Immun (2006) 74(1):125–34. doi: 10.1128/IAI.74.1.125-134.2006
32. Paiva CN, Figueiredo RT, Kroll-Palhares K, Silva AA, Silverio JC, Gibaldi D, et al. CCL2/MCP-1 controls parasite burden, cell infiltration, and mononuclear activation during acute Trypanosoma cruzi infection. J Leukoc Biol (2009) 86(5):1239–46. doi: 10.1189/jlb.0309187
33. Gibaldi D, Vilar-Pereira G, Pereira IR, Silva AA, Barrios LC, Ramos IP, et al. CCL3/macrophage inflammatory protein-1alpha is dually involved in parasite persistence and induction of a TNF- and IFNgamma-enriched inflammatory milieu in trypanosoma cruzi-induced chronic cardiomyopathy. Front Immunol (2020) 11:306. doi: 10.3389/fimmu.2020.00306
34. Sharma J, Blase JR, Hoft DF, Marentette JO, Turk J, McHowat J. Mice with genetic deletion of group VIA phospholipase A2beta exhibit impaired macrophage function and increased parasite load in trypanosoma cruzi-induced myocarditis. Infect Immun (2016) 84(4):1137–42. doi: 10.1128/IAI.01564-15
35. Silva MC, Davoli-Ferreira M, Medina TS, Sesti-Costa R, Silva GK, Lopes CD, et al. Canonical PI3Kgamma signaling in myeloid cells restricts Trypanosoma cruzi infection and dampens chagasic myocarditis. Nat Commun (2018) 9(1):1513. doi: 10.1038/s41467-018-03986-3
36. Ponce NE, Sanmarco LM, Eberhardt N, Garcia MC, Rivarola HW, Cano RC, et al. CD73 inhibition shifts cardiac macrophage polarization toward a microbicidal phenotype and ameliorates the outcome of experimental chagas cardiomyopathy. J Immunol (2016) 197(3):814–23. doi: 10.4049/jimmunol.1600371
37. Calderon J, Maganto-Garcia E, Punzon C, Carrion J, Terhorst C, Fresno M. The receptor Slamf1 on the surface of myeloid lineage cells controls susceptibility to infection by Trypanosoma cruzi. PloS Pathog (2012) 8(7):e1002799. doi: 10.1371/journal.ppat.1002799
38. Marinho CR, D’Imperio Lima MR, Grisotto MG, Alvarez JM. Influence of acute-phase parasite load on pathology, parasitism, and activation of the immune system at the late chronic phase of Chagas’ disease. Infect Immun (1999) 67(1):308–18. doi: 10.1128/IAI.67.1.308-318.1999
39. Higuchi Mde L, De Brito T, Martins Reis M, Barbosa A, Bellotti G, Pereira-Barreto AC, et al. Correlation between Trypanosoma cruzi parasitism and myocardial inflammatory infiltrate in human chronic chagasic myocarditis: Light microscopy and immunohistochemical findings. Cardiovasc Pathol (1993) 2(2):101–6. doi: 10.1016/1054-8807(93)90021-S
40. Koh CC, Neves EGA, de Souza-Silva TG, Carvalho AC, Pinto CHR, Galdino AS, et al. Cytokine networks as targets for preventing and controlling chagas heart disease. Pathogens (2023) 12(2):171. doi: 10.3390/pathogens12020171
41. Nunes MP, Andrade RM, Lopes MF, DosReis GA. Activation-induced T cell death exacerbates Trypanosoma cruzi replication in macrophages cocultured with CD4+ T lymphocytes from infected hosts. J Immunol (1998) 160(3):1313–9. doi: 10.4049/jimmunol.160.3.1313
42. Lopes MF, dos Reis GA. Trypanosoma cruzi-induced immunosuppression: blockade of costimulatory T-cell responses in infected hosts due to defective T-cell receptor-CD3 functioning. Infect Immun (1994) 62(4):1484–8. doi: 10.1128/iai.62.4.1484-1488.1994
43. Lopes MF, da Veiga VF, Santos AR, Fonseca ME, DosReis GA. Activation-induced CD4+ T cell death by apoptosis in experimental Chagas’ disease. J Immunol (1995) 154(2):744–52. doi: 10.4049/jimmunol.154.2.744
44. Lopes MF, DosReis GA. Trypanosoma cruzi-induced immunosuppression: selective triggering of CD4+ T-cell death by the T-cell receptor-CD3 pathway and not by the CD69 or Ly-6 activation pathway. Infect Immun (1996) 64(5):1559–64. doi: 10.1128/iai.64.5.1559-1564.1996
45. Zuniga E, Motran C, Montes CL, Diaz FL, Bocco JL, Gruppi A. Trypanosoma cruzi-induced immunosuppression: B cells undergo spontaneous apoptosis and lipopolysaccharide (LPS) arrests their proliferation during acute infection. Clin Exp Immunol (2000) 119(3):507–15. doi: 10.1046/j.1365-2249.2000.01150.x
46. Zuniga E, Motran CC, Montes CL, Yagita H, Gruppi A. Trypanosoma cruzi infection selectively renders parasite-specific IgG+ B lymphocytes susceptible to Fas/Fas ligand-mediated fratricide. J Immunol (2002) 168(8):3965–73. doi: 10.4049/jimmunol.168.8.3965
47. Martins GA, Cardoso MA, Aliberti JC, Silva JS. Nitric oxide-induced apoptotic cell death in the acute phase of Trypanosoma cruzi infection in mice. Immunol Lett (1998) 63(2):113–20. doi: 10.1016/s0165-2478(98)00066-2
48. Martins GA, Petkova SB, MacHado FS, Kitsis RN, Weiss LM, Wittner M, et al. Fas-FasL interaction modulates nitric oxide production in Trypanosoma cruzi-infected mice. Immunology (2001) 103(1):122–9. doi: 10.1046/j.1365-2567.2001.01216.x
49. Martins GA, Vieira LQ, Cunha FQ, Silva JS. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infection: a possible role in immune response control. Infect Immun (1999) 67(8):3864–71. doi: 10.1128/IAI.67.8.3864-3871.1999
50. Mucci J, Hidalgo A, Mocetti E, Argibay PF, Leguizamon MS, Campetella O. Thymocyte depletion in Trypanosoma cruzi infection is mediated by trans-sialidase-induced apoptosis on nurse cells complex. Proc Natl Acad Sci U.S.A. (2002) 99(6):3896–901. doi: 10.1073/pnas.052496399
51. de Meis J, Mendes-da-Cruz DA, Farias-de-Oliveira DA, Correa-de-Santana E, Pinto-Mariz F, Cotta-de-Almeida V, et al. Atrophy of mesenteric lymph nodes in experimental Chagas’ disease: differential role of Fas/Fas-L and TNFRI/TNF pathways. Microbes Infect (2006) 8(1):221–31. doi: 10.1016/j.micinf.2005.06.027
52. Rodrigues V Jr., Agrelli GS, Leon SC, Silva Teixeira DN, Tostes S Jr., Rocha-Rodrigues DB. Fas/Fas-L expression, apoptosis and low proliferative response are associated with heart failure in patients with chronic Chagas’ disease. Microbes Infect (2008) 10(1):29–37. doi: 10.1016/j.micinf.2007.09.015
53. Chaves AT, de Assis Silva Gomes Estanislau J, Fiuza JA, Carvalho AT, Ferreira KS, Fares RC, et al. Immunoregulatory mechanisms in Chagas disease: modulation of apoptosis in T-cell mediated immune responses. BMC Infect Dis (2016) 16:191. doi: 10.1186/s12879-016-1523-1
54. Tostes S Jr., Bertulucci Rocha-Rodrigues D, de Araujo Pereira G, Rodrigues V. Myocardiocyte apoptosis in heart failure in chronic Chagas’ disease. Int J Cardiol (2005) 99(2):233–7. doi: 10.1016/j.ijcard.2004.01.026
55. Zhang J, Andrade ZA, Yu ZX, Andrade SG, Takeda K, Sadirgursky M, et al. Apoptosis in a canine model of acute Chagasic myocarditis. J Mol Cell Cardiol (1999) 31(3):581–96. doi: 10.1006/jmcc.1998.0893
56. de Souza EM, Araujo-Jorge TC, Bailly C, Lansiaux A, Batista MM, Oliveira GM, et al. Host and parasite apoptosis following Trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res (2003) 314(2):223–35. doi: 10.1007/s00441-003-0782-5
57. Lopes MF, Guillermo LV, Silva EM. Decoding caspase signaling in host immunity to the protozoan Trypanosoma cruzi. Trends Immunol (2007) 28(8):366–72. doi: 10.1016/j.it.2007.06.004
58. Guillermo LV, Pereira WF, De Meis J, Ribeiro-Gomes FL, Silva EM, Kroll-Palhares K, et al. Targeting caspases in intracellular protozoan infections. Immunopharmacol Immunotoxicol. (2009) 31(2):159–73. doi: 10.1080/08923970802332164
59. Lopes MF, Nunes MP, Henriques-Pons A, Giese N, Morse HC 3rd, Davidson WF, et al. Increased susceptibility of Fas ligand-deficient gld mice to Trypanosoma cruzi infection due to a Th2-biased host immune response. Eur J Immunol (1999) 29(1):81–9. doi: 10.1002/(SICI)1521-4141(199901)29:01<81::AID-IMMU81>3.0.CO;2-Y
60. Silva EM, Guillermo LV, Ribeiro-Gomes FL, De Meis J, Pereira RM, Wu Z, et al. Caspase-8 activity prevents type 2 cytokine responses and is required for protective T cell-mediated immunity against Trypanosoma cruzi infection. J Immunol (2005) 174(10):6314–21. doi: 10.4049/jimmunol.174.10.6314
61. Silva EM, Guillermo LV, Ribeiro-Gomes FL, De Meis J, Nunes MP, Senra JF, et al. Caspase inhibition reduces lymphocyte apoptosis and improves host immune responses to Trypanosoma cruzi infection. Eur J Immunol (2007) 37(3):738–46. doi: 10.1002/eji.200636790
62. Guillermo LV, Silva EM, Ribeiro-Gomes FL, De Meis J, Pereira WF, Yagita H, et al. The Fas death pathway controls coordinated expansions of type 1 CD8 and type 2 CD4 T cells in Trypanosoma cruzi infection. J Leukoc Biol (2007) 81(4):942–51. doi: 10.1189/jlb.1006643
63. Vasconcelos JR, Bruna-Romero O, Araujo AF, Dominguez MR, Ersching J, de Alencar BC, et al. Pathogen-induced proapoptotic phenotype and high CD95 (Fas) expression accompany a suboptimal CD8+ T-cell response: reversal by adenoviral vaccine. PloS Pathog (2012) 8(5):e1002699. doi: 10.1371/journal.ppat.1002699
64. Cabral-Piccin MP, Guillermo LV, Vellozo NS, Filardy AA, Pereira-Marques ST, Rigoni TS, et al. Apoptotic CD8 T-lymphocytes disable macrophage-mediated immunity to Trypanosoma cruzi infection. Cell Death Dis (2016) 7(5):e2232. doi: 10.1038/cddis.2016.135
65. Davidson WF, Holmes KL, Roths JB, Morse HC 3rd. Immunologic abnorMalities of mice bearing the gld mutation suggest a common pathway for murine nonMalignant lymphoproliferative disorders with autoimmunity. Proc Natl Acad Sci U.S.A. (1985) 82(4):1219–23. doi: 10.1073/pnas.82.4.1219
66. Boyer MH, Hoff R, Kipnis TL, Murphy ED, Roths JB. Trypanosoma cruzi: susceptibility in mice carrying mutant gene lpr (lymphoproliferation). Parasite Immunol (1983) 5(2):135–42. doi: 10.1111/j.1365-3024.1983.tb00731.x
67. Cohen PL, Eisenberg RA. The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunol Today (1992) 13(11):427–8. doi: 10.1016/0167-5699(92)90066-G
68. Hernandez-Torres M, Silva do Nascimento R, Reboucas MC, Cassado A, Matteucci KC, D’Imperio-Lima MR, et al. Absence of Bim sensitizes mice to experimental Trypanosoma cruzi infection. Cell Death Dis (2021) 12(7):692. doi: 10.1038/s41419-021-03964-6
69. Henson PM. Cell removal: efferocytosis. Annu Rev Cell Dev Biol (2017) 33:127–44. doi: 10.1146/annurev-cellbio-111315-125315
70. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
71. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15(11):1266–72. doi: 10.1038/nm.2048
72. Roszer T. Transcriptional control of apoptotic cell clearance by macrophage nuclear receptors. Apoptosis (2017) 22(2):284–94. doi: 10.1007/s10495-016-1310-x
73. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front Immunol (2020) 11:553. doi: 10.3389/fimmu.2020.00553
74. Trahtemberg U, Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol (2017) 8:1356. doi: 10.3389/fimmu.2017.01356
75. DosReis GA, Fonseca ME, Lopes MF. Programmed T-cell death in experimental chagas disease. Parasitol Today (1995) 11(10):391–4. doi: 10.1016/0169-4758(95)80011-5
76. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403(6766):199–203. doi: 10.1038/35003208
77. Chaves MM, Lee SH, Kamenyeva O, Ghosh K, Peters NC, Sacks D. The role of dermis resident macrophages and their interaction with neutrophils in the early establishment of Leishmania major infection transmitted by sand fly bite. PloS Pathog (2020) 16(11):e1008674. doi: 10.1371/journal.ppat.1008674
78. Ribeiro-Gomes FL, ROmano A, Lee S, Roffe E, Peters NC, Debrabant A, et al. Apoptotic cell clearance of Leishmania major-infected neutrophils by dendritic cells inhibits CD8(+) T-cell priming in vitro by Mer tyrosine kinase-dependent signaling. Cell Death Dis (2015) 6(12):e2018. doi: 10.1038/cddis.2015.351
79. Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol (2014) 15(10):920–8. doi: 10.1038/ni.2986
80. Rodrigues da Cunha GM, Azevedo MA, Nogueira DS, Climaco MC, Valencia Ayala E, Jimenez Chunga JA, et al. alpha-Gal immunization positively impacts Trypanosoma cruzi colonization of heart tissue in a mouse model. PloS Negl Trop Dis (2021) 15(7):e0009613. doi: 10.1371/journal.pntd.0009613
81. Quijano-Hernandez IA, Castro-Barcena A, Vazquez-Chagoyan JC, Bolio-Gonzalez ME, Ortega-Lopez J, Dumonteil E. Preventive and therapeutic DNA vaccination partially protect dogs against an infectious challenge with Trypanosoma cruzi. Vaccine (2013) 31(18):2246–52. doi: 10.1016/j.vaccine.2013.03.005
Keywords: apoptosis, Chagas disease, efferocytosis, fibrosis, heart pathology, inflammation, M1 macrophages, T lymphocytes
Citation: Vellozo NS, Matos-Silva TC and Lopes MF (2023) Immunopathogenesis in Trypanosoma cruzi infection: a role for suppressed macrophages and apoptotic cells. Front. Immunol. 14:1244071. doi: 10.3389/fimmu.2023.1244071
Received: 21 June 2023; Accepted: 04 August 2023;
Published: 17 August 2023.
Edited by:
Roberta Olmo Pinheiro, Oswaldo Cruz Foundation (FIOCRUZ), BrazilReviewed by:
Patrícia Santos, Federal University of Pernambuco, BrazilCopyright © 2023 Vellozo, Matos-Silva and Lopes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela F. Lopes, marcelal@biof.ufrj.br