 Stefan Forster1,2
Stefan Forster1,2 Ramin Radpour
Ramin Radpour Adrian F. Ochsenbein
Adrian F. Ochsenbein
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 September 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1243997
This article is part of the Research TopicInteraction of Cell Subtypes in Tumor Microenvironment, and Implications for ImmunotherapyView all 12 articles
Multiple myeloma (MM) is a hematologic malignancy characterized by the proliferation of clonal plasma cells in the bone marrow (BM). It is known that early genetic mutations in post-germinal center B/plasma cells are the cause of myelomagenesis. The acquisition of additional chromosomal abnormalities and distinct mutations further promote the outgrowth of malignant plasma cell populations that are resistant to conventional treatments, finally resulting in relapsed and therapy-refractory terminal stages of MM. In addition, myeloma cells are supported by autocrine signaling pathways and the tumor microenvironment (TME), which consists of diverse cell types such as stromal cells, immune cells, and components of the extracellular matrix. The TME provides essential signals and stimuli that induce proliferation and/or prevent apoptosis. In particular, the molecular pathways by which MM cells interact with the TME are crucial for the development of MM. To generate successful therapies and prevent MM recurrence, a thorough understanding of the molecular mechanisms that drive MM progression and therapy resistance is essential. In this review, we summarize key mechanisms that promote myelomagenesis and drive the clonal expansion in the course of MM progression such as autocrine signaling cascades, as well as direct and indirect interactions between the TME and malignant plasma cells. In addition, we highlight drug-resistance mechanisms and emerging therapies that are currently tested in clinical trials to overcome therapy-refractory MM stages.
Multiple Myeloma (MM) belongs to the heterogeneous group of plasma cell disorders comprising monoclonal gammopathy of undetermined significance (MGUS), smoldering myeloma (SMM), plasma cell myeloma, plasmacytoma, monoclonal immunoglobulin deposition disease and plasma cell neoplasms associated with paraneoplastic syndromes (1). Plasma cell myeloma/MM is the most frequent plasma cell disorder and is characterized by the uncontrolled expansion of malignant plasma cells in the bone marrow (BM) causing distinct clinical symptoms such as bone pain, spontaneous fractures, and renal impairment (1, 2). To date, MM is still considered incurable. However, due to the introduction of novel immunotherapies that directly target plasma cell surface markers such as CD38, the overall and progression-free survival of myeloma patients has substantially improved within the last years (3–5). For the diagnosis of MM, the international myeloma working group (IMWG) requires 10% BM plasma cells or a biopsy-proven bone or extramedullary plasmacytoma with at least one or more CRAB criteria (calcium elevation, renal insufficiency, anemia, bone lesions) and/or myeloma-defining molecular aberrations (6). MGUS and SMM are considered as the precursor lesions of MM (2, 6, 7). Patients with MGUS and SMM have increased BM plasma cell counts and/or monoclonal proteins, but lack MM-defining symptoms (6, 7). Early genetic alterations in the malignant transformation process include hyperdiploidy or translocations involving the immunoglobulin heavy chain (IGH) locus on chromosome 14 that lead to the overexpression or overactivation of defined oncogenes stimulating plasma cell proliferation and/or preventing apoptosis (8–11).
As a consequence of early cytogenetic alterations that occur in postgerminal center B/plasma cells, a small group of abnormal cells, so called founder clones, expand and initiate the process of myelomagenesis. Subsequently, the clonal evolution of malignant plasma cells emerges through a series of additional copy number modifications, epigenetic changes, and the acquisition of additional secondary mutations that enhance the intratumoral heterogeneity finally resulting in the co-existence of multiple clonal subpopulations with various selection and/or fitness advantages (2, 12). Indeed, the spatio-temporal clonal landscape drastically changes in the course of MM progression thus favoring disease-promoting clones during repetitive cycles of myeloma cell engraftment, dissemination and re-engraftment at another BM site, as previously shown for MM xenografts grown in severe combined immunodeficiency (SCID) mice (12). Moreover, so called focal lesions - clonally heterogeneous and spatially distributed tumor clusters - are seen as mutational “hot spots” in MM, consistent with the regional outgrowth of advanced tumor clones (13, 14). In relapsed and therapy-refractory MM end-stages, single myeloma clones might even lose their BM dependency, survive and expand in the circulation, or spread to distant body regions, resulting in plasma cell leukemia or extramedullary myeloma (15, 16). Both, plasma cell leukemia and extramedullary myeloma are defined by rapid disease onset, poor therapy response, and an overall poor prognosis (15).
Multiple molecular mechanisms might influence the outgrowth of MM clonal subsets. Based on specific cytogenetic alterations that occur in clonal plasma cells during MM progression, myeloma cells may develop resistance to standard MM treatments such as proteasome inhibitors and immunomodulatory agents driving the selection of drug-resistant populations that outcompete drug-sensitive populations. Furthermore, the activation of autocrine signaling loops might inhibit tumor cell death or enhance cell proliferation of clonal subsets that co-express the corresponding ligand-receptor pairs. Finally, the cross-talk between tumor cells and the tumor microenvironment (TME) promotes the dissemination of distinct clonal populations that are seen as the main drivers of MM progression (12). In summary, this review outlines the central pathways and mechanisms that drive myelomagenesis and contribute to the clonal evolution and expansion of malignant plasma cells finally leading to MM progression and therapy-refractory end-stages.
Cancer stem cells (CSCs) are a small subpopulation of cancer cells harboring an unlimited capacity of self-renewal. Low proliferation or quiescence make CSCs resistant to radiation and chemotherapies that predominantly target rapidly reproducing cancer cells. As a result, CSCs are often considered crucial for disease recurrence (17–20). CSCs have been identified in a variety of cancer entities including colorectal cancer, lung cancer, pancreatic cancer and acute myeloid leukemia (21–26). However, the presence and (immuno-) phenotypic characterization of MM CSCs or cancer-stem cell like cells have been controversially discussed in the past (27). So far, no particular cellular or molecular markers have been identified to accurately distinguish myeloma CSCs from the remaining tumor mass. Plasma cells, both normal and malignant, are terminally differentiated cells. Thus, myeloma stem cells are thought to be derived from abnormal postgerminal center B cells (27, 28). Rasmussen et al. discovered clonotypic memory B cells in most MM patients (29). In immunodeficient mice, injection of blood-derived CD19+ CD27+ B cells from myeloma patients successfully initiated the disease, whereas CD138+ plasma cells failed to engraft in vivo (30). Myeloma-derived CD19+ CD138- leukemic cells engrafted in NOD/SCID mice, indicating a role of clonotypic late-stage B-cells in disease initiation (31). In line, RPMI8226 and NCI H929 cell line derived CD138- cell fractions had increased ALDH1 enzyme activity and superior clonogenic potential both in vitro and in vivo (32). In colony formation assays – an approach to investigate clonogenicity, stemness and self-renewal – MM cell line and patient derived CD138- fractions showed increased colony formation upon serial replatings compared to CD138+ fractions (33). In contrast, other studies postulated that myeloma stem cells are characterized by a CD38+ CD138+ CD19- CD45- immunophenotype suggesting that stem cell related markers might undergo dynamic changes or differ between MM patients and the experimental model systems (34, 35). Indeed, environmental factors influence the expression of distinct plasma cell surface molecules. Xenografts created by the injection of CD19+ CD138- myeloma cell fractions showed partial CD138 re-expression in the primary engrafted tumor, whereas CD138 expression was almost non-existent in circulating B cells, suggesting that CD138 expression is dependent on environmental cues (30, 31). Nutrient deprivation changes CD138 surface expression, and CD138+ Vk*MYC murine myeloma cells showed better engraftment and tumor development, whereas CD138- cells were characterized by increased motility, intravasation, and dissemination (36). In addition, low oxygen levels decrease CD138 expression and induce a mature B cell-like transcriptional signature with upregulation of PAX5 and BCL6 in myeloma cells, as previously described by Kawano et al. (37). Hypoxia resulted in the upregulation of the stem cell-related transcription factors OCT4 and SOX2 in the MM cell line RPMI8226 (37).
A small side population (SP) of clonogenic myeloma cells has been revealed by reduced Hoechst 33342 staining (30, 38, 39). Multiple independent studies demonstrated that SP cells harbor stem cell like features with decreased drug sensitivity and over-activation of stem cell related pathways including Notch-, Hedgehog-, PI3K/Akt or Wnt-signaling that are also found enriched in most MM patients (40–50). In line with previous findings, Wang et al. reported that SP myeloma cells are more resistant to bortezomib or melphalan chemotherapy and that SP abundance and clonogenicity are regulated by the activated-leukocyte-cell-adhesion-molecule (ALCAM)/EGFR-EGF signaling axis (49, 51). Interestingly, combination therapy with melphalan and an EGFR inhibitor resulted in a reduction of the SP and a significantly lower disease burden in 5TGM1 myeloma bearing mice compared to monotherapies (49). Furthermore, the expression of specific molecules such as CD24, CD34 and ATP binding cassette subfamily G member 2 (ABCG2) have been linked to a MM stem cell-like phenotype (52–54). CD24 expression is increased in MM SP and the stem cell related genes KLF4, OCT4, NANOG and SOX2 were enriched in isolated CD24+ cells. In vivo limiting dilution assays revealed that CD24+ myeloma cells showed a significantly higher tumor initiating capacity in NOD-Rag1null mice compared to CD24- fractions (55).
While the concept of MM founder clones – defined as early mutation harboring clones that initiate myelomagenesis - is widely accepted and supported by numerous research studies - no unified characterization and terminology of MM CSCs – that maintain and propagate the disease - have been reached (27, 56, 57). In this regard, high cancer cell plasticity with dynamic expression profiles of stem-cell related markers – in parts driven by environmental conditions - might be a fundamental challenge in defining and eventually targeting MM CSCs.
The clonal evolution of MM occurs through a series of genetic alterations, including chromosomal abnormalities, somatic mutations, and epigenetic modifications. Early genetic events are divided in two groups. (1) Translocations involving the IGH locus on chromosome 14 such as t(11;14), t(4;14), t(14;16) leading to the overexpression of the oncogenic drivers CCND1, MMSET/FGFR3 or cMAF, respectively. Overexpression of the MMSET/FGFR3 fusion gene is caused by the translocation t(4;14), one of the most important chromosomal abnormalities in MM (58–64). (2) Hyperdiploidy of the odd-numbered chromosomes 3, 5, 7, 9, 11, 15 or 19 might also trigger myelomagenesis (58, 65). Moreover, single-cell RNA-sequencing and single-molecule long-read sequencing revealed early IFITM2 and ANK1 alterations in clonal plasma cells (66). Multiple evolutionary branches emerge from a single abnormal postgerminal center B-/plasma cell during MM development, driven in part by TME-related interactions (67). In this regard, the clonal evolution of abnormal plasma cells is thought being initiated by genetic mutations occurring in postgerminal center B cells in the course of somatic hypermutation and isotype class switching. After homing to the BM, these mutation-harboring B-cells become so called founder clones that start the clonal evolution process (56, 57). Interestingly, the number of founder clones and the patterns of clonal evolution differs in-between patients. Whole exome sequencing of paired biopsies at diagnosis and progression revealed that in most cases two founder clones are present at disease onset (range one to three founder clones) (67, 68). After disease initiation, the clonal evolution most often follows a branching pattern, where multiple heterogeneous clones develop independently and distinct mutations are lost or gained in the course of MM progression. Less frequently, the clonal structure persists between early and late disease stages, referred as a stable evolutionary pattern (69, 70). Notably, clonal heterogeneity can be found throughout all steps of myeloma progression starting from MGUS (71, 72). CyTOF analyses of distinct markers involved in B cell regulation (e.g. MMSET or sXBP1), stemness (e.g. SOX2) and abnormal plasma cell differentiation (e.g. CD56) revealed different clusters among the B cell/plasma cell compartment and heterogeneous expression profiles within these defined clusters, in-between patients and among MGUS, SMM, MM or relapsed MM stages (72). Longitudinal whole-exome sequencing analyses of plasma cells from paired MGUS/SMM and MM patients revealed intraclonal heterogeneity at the early MGUS/SMM stages characterized by the detection of multiple co-existing clonal subsets each harboring a distinct set of driver gene mutations (71). Further, Dutta et al. described a subclonal stability between MGUS/SMM and MM stages – meaning that the subclonal architecture and heterogeneity found at advanced disease stages is often already present at MGUS/SMM (71). After disease onset with MM manifestation, the further clonal evolution of malignant plasma cell subsets is driven by late stage (or secondary) somatic mutations involving KRAS, NRAS, BRAF, TP53, and DIS3 that are among the most frequent mutated genes in MM. These mutations have an impact on key signaling pathways that control cell survival, proliferation, and drug resistance, including MAPK/ERK, PI3K/AKT, and NF-ҡB (67, 73). In addition, copy number gains/amplifications or deletions of distinct chromosomal regions are acquired in the course of myeloma progression and further promote the clonal evolution of malignant plasma cells. In this regard, alterations in RAS or TP53 genes, as well as the 1q21 chromosomal amplification, are important drivers in the clonal selection process and are detected more frequently after MM treatment (67, 73). Patients treated with high-dose melphalan had the highest rate of tumor mutations (73). Clonal evolution analysis of matched samples from time of diagnosis and MM relapse revealed that clonal selection was detected in relapsed stages of all patients that had undergone high-dose melphalan treatment (74).
Aside from drug-induced genomic alterations, the genomic instability is also influenced by external or environmental factors. In this regard, focal lesions (FLs) are seen as clonally heterogeneous and spatially distributed tumor foci that can be found in up to 84% of MM patients at time of diagnosis. The presence of FLs is linked to myeloma dissemination, disease relapse and adverse outcomes (75). For this reason, FLs are considered as mutational “hot spots” in MM, consistent with the regional outgrowth of advanced tumor clones (13, 75, 76). The increased acquisition of mutations within FLs might generate subclones with enhanced capacities for immune evasion, drug resistance and dissemination thus driving disease progression. Hypoxic tension within the tumor microenvironment and (epi-) genetic changes are associated with MM resistance and both factors are highly induced within FLs (77–79). In line with these findings, multi-region sequencing of matched and synchronously taken iliac crest and FLs biopsies from MM patients revealed spatial clonal heterogeneity in MM. Somatic aberrations such as the deletion del(17p) or MYC translocations – alterations that are frequently found in advanced MM stages - were exclusively present at one biopsy site only. In contrast, early genetic alterations such as t(4;14) were uniformly detected at both sites suggesting spatio-temporal evolution from a common subclone (13, 14, 80). Moreover, the longitudinal analyses of FLs and matched samples at later onset of MM relapse showed a high similarity in the clonal composition between both manifestations further supporting the role of FLs as mutational “hotspots” and origins of clonal evolution (14).
Aside from genetic alterations, the clonal evolution process is regulated by epigenetic changes such as DNA methylation and histone modifications. DNA hypermethylation can result in the downregulation/loss of important tumor suppressor genes such as CDH1 or SHP1 or alter the expression of genes that influence the sensitivity towards anti-myeloma drugs (81–84). The translocation t(4;14) leads to Nuclear Receptor Binding SET Domain Protein 2 (NSD2) overexpression with subsequent NSD2-driven epigenetic downregulation of specific genes that desensitize affected cells for bortezomib therapy (85, 86). Epigenetic alterations can not only affect myeloma cells but also occur in the cellular components of the TME, especially in bone marrow stromal cells (BMSCs) (87, 88). Direct co-culture of BMSCs from healthy individuals together with the myeloma cell line MM.1S has been reported to induce DNA methylation changes in BMSCs. These epigenetic modifications in BMSCs were predominantly located in osteogenic gene loci, regulating the differentiation process of BMSCs to osteoblasts. Thus, myeloma cells might inhibit the formation of new bone-forming osteoblasts that have been proposed to keep myeloma cells in quiescent states or induce their apoptosis (16, 89, 90). In this regard, separated transwell-based cultures of BMSCs and myeloma cells resulted in similar epigenetic alterations in BMSCs as direct co-culture experiments suggesting that soluble factors induce epigenetic changes in surrounding stromal cells thus creating a tumor-supportive microenvironment stimulating myeloma cell growth and survival (90). As a consequence, targeting of enzymes that are involved in DNA methylation (i.e. DNA methyltransferases or histone methyltransferase G9a) led to significantly lower tumor burden and reduction of bone lysis in immunodeficient mice injected with MM cell lines (90). The genomic instability and multistep molecular pathogenesis involved in MM clonal evolution and expansion are illustrated in Figure 1.
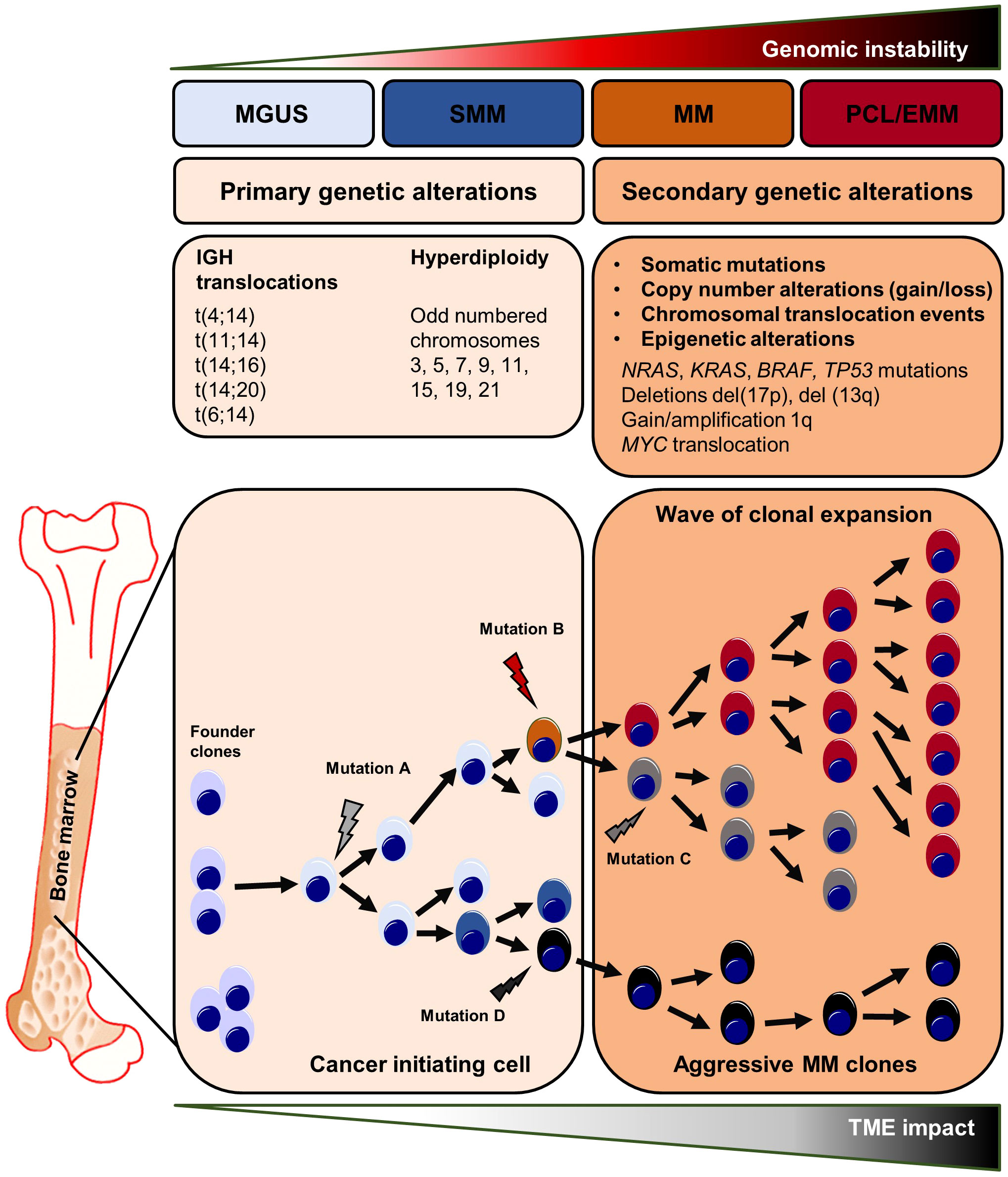
Figure 1 Genetic abnormalities in myeloma initiation and clonal evolution. Primary and secondary genetic processes involved in the development and progression of MGUS and SMM to MM and ultimately to plasma cell leukemia and extramedullary myeloma. Chromosomal translocations are seen at the early steps of the malignant transformation process. The development of MGUS to MM is frequently related with c-MYC overexpression, RAS mutations, and chromosome 13 deletion. The final step, PCL/EMM manifestation, is accompanied by NF-ҡB pathway activating mutations, chromosomal translocation involving the c-MYC gene, 1q gain, loss of 1p and deletion of 17p where the TP53 gene is situated, BRAF mutations, and epigenetic alterations. EMM, extramedullary myeloma; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; PCL, plasma cell leukemia; SMM, smoldering multiple myeloma.
The growth and survival of malignant plasma cells is highly dependent on the abundance of defined cytokines and growth-promoting factors that are primarily provided by the BM microenvironment. However, myeloma cells can also produce some of these soluble factors by themselves thus stimulating their proliferation or preventing spontaneous and stress-induced apoptosis. In this regard, autocrine signaling may emerge from the release of cytokines and the simultaneous expression of the cytokine-specific receptor on the same cell. Furthermore, the co-expression of growth-stimulatory ligand-receptor pairs may promote the activation of autonomous signaling (91). Clonal subsets that co-express a growth-promoting ligand with its corresponding receptor might harbor a crucial selection advantage. Exemplary, amplification of the chromosomal region 1q21 - frequently detected in advanced disease stages - is often accompanied by the expression of the IL-6 receptor (whose encoding gene is located on the 1q21 region) (92). IL-6 receptor (IL-6R) expression is reportedly associated with a poor prognosis in MM (93). Moreover, myeloma cell lines that express the IL-6R respond positively to induction of IL-6R signaling, with increased proliferation and reduced apoptosis (94, 95). The expression levels of microRNAs such as miR-197-3p and miR-451 influence bortezomib resistance by inhibiting the IL-6/IL-6R signaling pathway (94, 96). In addition, the availability of IL-6 is crucial for the survival and treatment sensitivity of malignant plasma cells (97). Multiple studies have shown that malignant plasma cells can generate and secrete IL-6 hence promoting their own survival (98–100). IL-6 enhances pro-survival signaling and decreases bortezomib sensitivity because it activates STAT3, which acts as a transcription factor for the anti-apoptotic proteins Bcl-2 and Bcl-xL (97, 101). Taken together, IL-6R expressing subclones might undergo a positive selection process based on their enhanced responsiveness to IL-6 that is provided by the TME or secreted by themselves subsequently preventing spontaneous or stress-induced cell apoptosis. In line, Ryu et al. reported frequent IL6/IL-6R co-expression in extramedullary MM that no longer require IL-6 provided by the BM niche (102). Approaches to target the IL-6/IL-6R signaling pathway have been tested in a cohort of transplant-eligible newly diagnosed MM patients using the anti-IL6 antibody siltuximab in combination with lenalidomide, bortezomib and dexamethasone (Table 1). Complete remission or very good partial remission were achieved in 2/11 (18%) and 2/11 (18%) of patients, respectively (103).
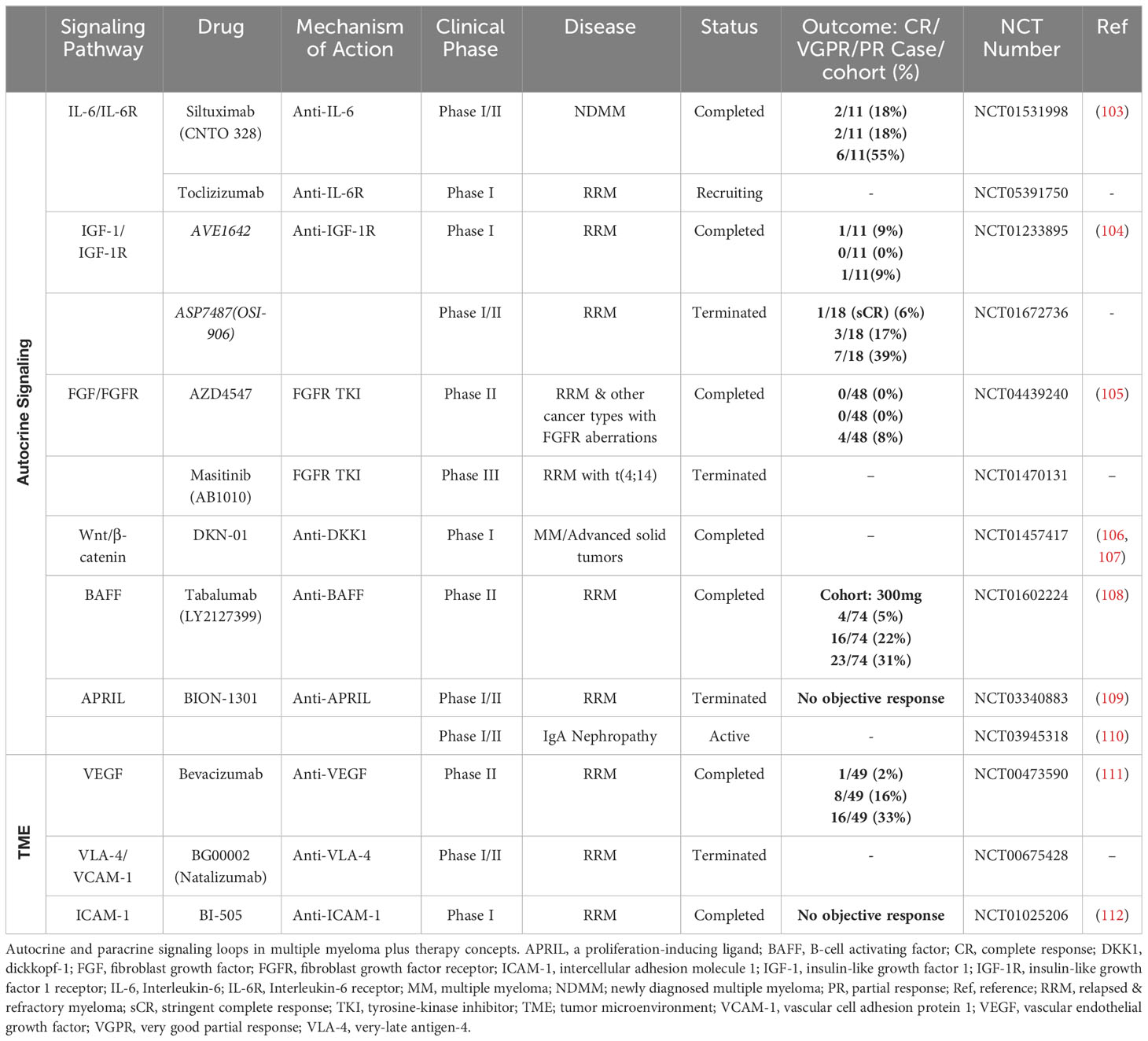
Table 1 Overview clinical studies that target MM autocrine and TME-mediated signaling pathways.
Several other autonomous and autocrine signaling pathways play a key role in the expansion of clonal plasma cells (Figure 2A). Expression of the insulin-like growth factor 1 receptor (IGF1R) was described in the majority of patients with extramedullary disease manifestation and positive expression profiles were associated with the occurrence of the high-risk cytogenetic alterations t(4;14), t(14;16) and were linked to reduced overall survival suggesting a potential selection advantage of IGF1R positive clones in the course of MM progression (113). Indeed, IGF1R and IGF1 are frequently co-expressed in myeloma cells and Chiron et al. discovered that blocking IGF1R signaling inhibits the self-renewal of myeloma cells in vitro. In addition, inhibition of self-renewal and colony formation in certain myeloma cell lines after treatment with a c-KIT receptor antagonist indicates the presence of a SCF/c-KIT autocrine signaling loop (114). Myeloma expansion is further regulated by the Wnt/β-catenin signaling pathway that is often over-activated in MM, partially driven by the autocrine release of Wnt ligands and/or by the (over-) expression of distinct molecules such as Syndecan-1 (CD138) or Leucine Rich Repeat Containing G Protein-Coupled Receptor 4 (LGR4) on cancer cells (44–46, 115). Wnt-ligands can directly bind to heparan sulfate side chains of Syndecan-1 thus mediating abnormal Wnt-signaling activity in MM. Indeed, CRISPR/Cas9-mediated deletion of Syndecan-1 heparan sulfate side chains inhibits Wnt-signaling activity and reduces the growth of MM cell lines suggesting autonomous Wnt-signaling stimulation via the autocrine release of Wnts (45). Malignant plasma cell clones may prevent both spontaneous and stress/drug-induced apoptosis by autocrine secretion of various pro-survival factors such as TNF-α, MIP-1, sonic hedgehog or GAS6 (116–119). Autocrine TNF-α/MCP-1/TNF-R2 signaling further enhances trans-endothelial migration of MM cell lines and primary myeloma samples (120, 121). B-cell activating factor (BAFF) and a proliferation inducing ligand (APRIL) are essential survival factors for myeloma cells (122). While these cytokines are primarily produced by cells of the BM microenvironment, previous research has revealed that malignant plasma cells may express and release BAFF and APRIL in an autocrine manner (116, 122). The translocation event t(4;14) (p16.3;q32) results in the overexpression of the fibroblast-growth factor receptor 3 (FGFR3) and can be detected in about 15% of all myeloma patients (61, 123). Different studies demonstrated that myeloma cells produce FGF and that autocrine FGF/FGFR signaling prevents oxidative stress-induced apoptosis (124, 125). In line, t(4;14) is seen as a high-risk cytogenetic alteration that occurs in advanced MM stages and FGFR expression might promote the positive selection of t(4;14) carrying subclones. Therapeutically, tyrosine kinase inhibitor-mediated FGF/FGFR signaling disruption abrogated growth and dissemination of MM cell lines in vivo (125). In addition, treatment approaches targeting FGF/FGFR signaling have been tested in phase 3 clinical trials using the tyrosine kinase inhibitor masitinib. Other monoclonal antibodies or small molecules targeting autocrine signaling pathways or MM-promoting components of the TME are currently tested in phase 1 and 2 clinical trials.
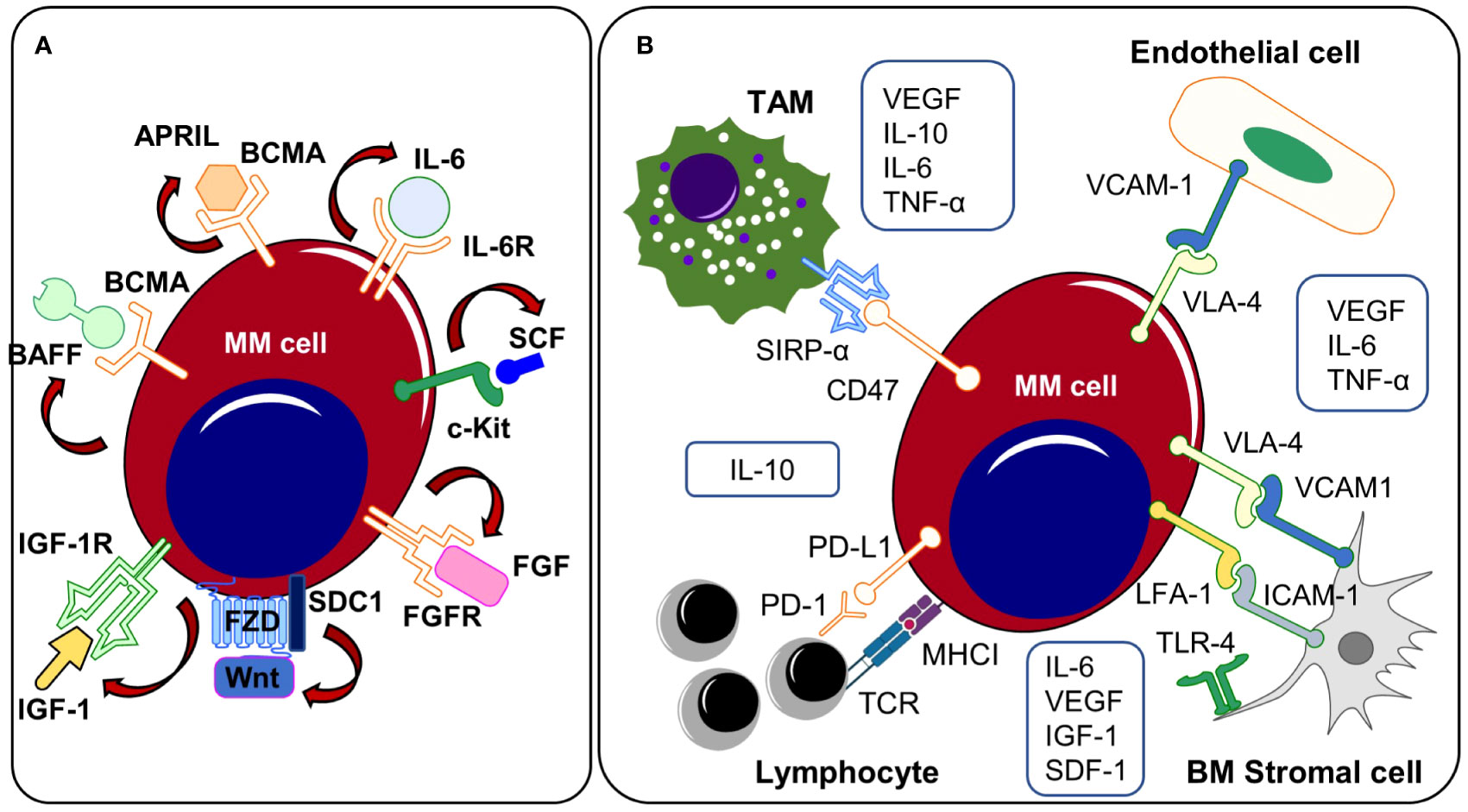
Figure 2 (A) Autocrine signaling and (B) TME related mechanisms. Both autocrine signaling cascades and interactions with the TME promote the outgrowth of clonal subsets that (co-)express growth promoting ligands/receptors on the cell surface. APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; BCMA, B cell maturation antigen; BM, bone marrow; c-Kit, receptor tyrosine kinase; FGF, fibroblast growth factor, FGFR, fibroblast growth factor receptor; ICAM-1, intercellular adhesion molecule 1; IGF-1, insulin-like growth factor 1; IGF1R, insulin-like growth factor 1 receptor, IL-6, Interleukin-6; IL-6R, Interleukin-6 receptor; IL-10, Interleukin-10; LFA-1, lymphocyte function-associated antigen 1; MM, multiple myeloma; PD-1, programmed Cell Death 1; PD-L1, programmed Cell Death Ligand 1; SCF, stem cell factor; SDC1, Syndecan 1, SIRPα, signal regulatory protein α; SDF-1, stromal cell-derived factor 1; TAM, tumor-associated macrophage; TLR-4, toll-like receptor 4; TME, tumor microenvironment; TNF-α, tumor necrosis factor alpha. VCAM-1, vascular cell adhesion protein 1; VEGF, vascular endothelial growth factor; VLA-4, very-late antigen-4.
In summary, genomic instability with the acquisition of specific secondary mutations might lead to the expression of distinct ligand-receptor pairs in myeloma subclones inducing cell proliferation or protecting from spontaneous and/or drug-induced apoptosis thus favoring the expansion of the latter.
Malignant plasma cells are surrounded by a variety of cell types and extracellular matrix in the BM. These elements work together to form a cytokine-rich and growth-promoting micromilieu or “niche” known as the tumor microenvironment. The TME is composed of the cellular fractions (e.g., fibroblasts, endothelial cells, osteoblasts, osteoclasts, hematopoietic cells, immune cells and neuronal cells). In addition, the extracellular matrix and its liquid milieu (cytokines, chemokines, and growth factors) are also essential components (16, 126–128). The TME promotes the survival and expansion of myeloma cells via different mechanisms. Furthermore, it has a crucial impact on the genomic instability in MM (129). In general, the BM environment is characterized by low oxygen levels (130). Due to the rapid expansion of malignant plasma cells within the BM during MM progression and the generation of abnormal neo-vessels, the oxidative stress within the TME increases. The latter may lead to the accumulation of reactive oxygen species (ROS), which can cause DNA damage. During the anti-tumor response, activated immune cells of the TME additionally release ROS, further exacerbating genomic instability in cancer cells and generating possible selection advantages (131, 132). In addition, hypoxia has been shown to induce myeloma cell dissemination and impair treatment sensitivity (77, 78, 133, 134). Hypoxia-inducible factors (HIFs) can alter the transcription of key regulators involved in cancer stemness, cell proliferation, drug resistance and/or cell survival (37, 77, 135). Moreover, increased HIF1A gene expression was detected in end-stage extramedullary MM cases (102). Taken together, areas with low oxygen levels might be “hotspots” for the generation of new clonal subpopulations that are prone to disseminate to distant BM sites and lead to MM progression.
While subcutaneously injected MM cell lines do not enter the blood circulation or disseminate in immunodeficient mice, myeloma cells that are grown in subcutaneously implanted bone chips maintain their capacities to disseminate and re-engraft at distant BM sites, indicating that the interactions with specific compartments of the BM TME play a fundamental role in MM spread and progression, as previously shown by Shen et al. (12). Here, they found that the clonal selection of myeloma cells occurs primarily in distant BM sites by using an implanted bone chip xenograft model combined with a fluorescence-based tracking system of clonal subsets enabling the simultaneous assessment of myeloma cell dissemination and clonal heterogeneity. While the primary tumor site showed a high degree of clonal heterogeneity with the co-occurrence of multiple clonal subsets, distant BM metastases displayed a markedly reduced heterogeneity with predominance of a single clonal population. Compared to myeloma cells from the primary site, disseminated clusters were characterized by an enrichment of genes that are linked to MM progression, suggesting that only a small number of plasma cell clones harbor the capacities to leave the primary tumor and disseminate to distant BM sites (12). In this regard, various direct and indirect interactions with the TME might promote the outgrowth and dissemination of single plasma cell clones (Figure 2B). The growth promoting cytokines IL-6 and APRIL are provided by different sources of the TME including bone marrow stromal cells (BMSCs), perivascular cells, eosinophils, mast cells and megakaryocytes (128, 136–140). Apart from IL-6, BMSCs additionally produce VEGF, IGF1 or SDF-1, soluble factors that have been shown to directly or indirectly affect myeloma growth, migration and invasion (141–144). Induction of the Toll-like receptor-4 (TLR-4) that has been found overexpressed on MM-BMSCs leads to the release of IL-6 by BMSCs, which promotes cell growth and survival in MM cells. Targeting these pathways has been demonstrated to offer therapeutic potential for the management of multiple myeloma. In this regard, selective blockade of TLR-4 reduced myeloma progression in murine myeloma models (145). Interleukin-10 (IL-10) is an anti-inflammatory cytokine that is produced by macrophages and lymphocytes (146, 147). In newly diagnosed MM patients, elevated IL-10 serum levels negatively correlated with both, therapy response and overall survival suggesting an apoptosis preventing function of IL-10 (147). M2 polarized tumor-associated macrophages (TAMs) promote myeloma progression via the release of multiple soluble factors and cytokines that directly influence cell growth (148). Clodronate-based depletion of M2 TAMs in xenograft-bearing nude mice resulted in significantly decreased tumor growth and reduced microvessel density. VEGFA serum levels were significantly lower in M2 depleted mice (148). Moreover, release of IL-6 and TNF-α by TAMs increases the vascular leakiness of newly formed tumor vessels in Vk*MYC myeloma mice thus facilitating the entrance of single plasma cell clones into the blood circulation (149). TNF-α has been shown to not only affect the vessel wall permeability but also directly enhances trans-endothelial myeloma cell migration (121).
Aside from cytokine or growth-factor driven mechanisms, direct cell-cell and cell-matrix interactions mediated through surface molecules and adhesion receptors play a critical role in MM expansion and dissemination. In advanced MM, platelets are highly activated and have been reported to enhance myeloma proliferation and engraftment through an IL-1β dependent mechanism (150). Myeloma cell adhesion to BMSCs or extracellular matrix components by expression of adhesion molecules such as Syndecan-1, intercellular adhesion molecule 1 (ICAM-1) or vascular cell adhesion protein 1 (VCAM-1) prevent apoptosis, resulting in cell-adhesion driven drug resistance (151–156). Myeloma cells express high levels of very late antigen-4 (VLA-4)/integrin α4β1 that binds to its receptor ICAM-1 expressed on endothelial cells. The VLA-4/ICAM-1 interaction enables myeloma cell adhesion to the vessel wall followed by trans-endothelial migration and engraftment (157, 158). Therapy concepts that prevent the vicious circle of myeloma cell dissemination and re-engraftment are currently tested in pre-clinical studies. Injection of VLA-4 deficient 5TGM1 murine myeloma cells into syngeneic recipients leads to higher extra-and reduced intramedullary disease burden with improved survival (158). Targeting VLA-4 with nanoparticles overcomes cell adhesion mediated drug resistance in myeloma cell lines and enhances chemotherapy response in 5TGM1 myeloma bearing mice (159).
Cancer cells are targeted by immune cells and thus have developed strategies to evade the host’s immune response. Both, direct mechanisms - such as upregulation of immune checkpoints on cancer cells – and indirect mechanisms (e.g. an overall immunosuppressive tumor microenvironment) are involved in cancer cell immune evasion. As direct mechanisms, upregulation of the immune checkpoints PD-L1 (CD274) and CD276 have been reported in MM patients (160–162). In addition, PD-1, an activation and exhaustion marker, is present on CD8+ T cells invading myeloma foci (163). In advanced MM stages, activating mutations in MYC oncogenes are frequently detected (164). In genetically engineered MM mouse models, early transgenic activation of the MYC oncogene was associated with enhanced numbers of MM infiltrating PD-1+ TIGIT+ LAG3+ CD8+ T cells, whereas late MYC activation was linked to significantly decreased numbers of activated/exhausted CD8+ T cells (165). Accordingly, anti-PD-1 therapies significantly reduced MM burden in mice with early transgenic MYC activation but had no effect on disease burden in mice with late oncogenic MYC activation. Moreover, pharmacological inhibition of MYC resulted in a downregulation of PD-L1 expression in malignant plasma cell clones upon early transgenic MYC activation, indicating that early myeloma subclones with alterations in the MYC oncogene may express PD-L1 thus preventing their elimination by PD-1+ CD8+ cytotoxic T cells (165). Further, HIF1α has been found to induce PD-L1 in cancer cells and might thus provide an advantage to myeloma subclones that are exposed to hypoxic tension in the BM or at extramedullary sites (102). In addition, the expression of IL-32γ by myeloma cells has been reported to induce the PD-L1 expression on tumor-associated macrophages, which suppresses CD8+ effector T cells. Increased IL-32γ expression levels were predominantly observed in relapsed MM stages, indicating that IL-32γ may be upregulated in clonal populations during the progression of MM and provide a selective advantage based on its immunosuppressive effect on infiltrating immune cells (166). Moreover, myeloma cell clones may directly prevent phagocytosis by TAMs via upregulation of the “don’t eat me” molecule CD47, which binds to the inhibitory receptor SIRPα expressed on macrophages (167). In preclinical studies, CD47 blockade resulted in increased phagocytosis and reduced growth of CD47 expressing myeloma cell lines both in vitro and in vivo (167). Clinical trials are currently conducted to test the efficacy of the anti-CD47 monoclonal antibody magrolimab in combination with other anti-myeloma drugs in relapsed and therapy refractory MM patients (168). In addition, natural killer (NK) cells were found to be deficient in perforin, CCLA5, and granzyme B and myeloma cells have been shown to directly induce inhibitory molecules in NK cells (e.g. by the expression of leukocyte immunoglobulin-like family proteins) (66, 102).
Aside from mechanisms that are mediated by myeloma cells to directly inhibit effector cell functions, an immunosuppressive tumor microenvironment – in parts induced by cancer cells themselves - may further promote MM immune evasion. Malignant plasma cells can promote the expression of CD84 on cells of the TME by releasing macrophage migration inhibitory factor (MIF). CD84 upregulation induces the differentiation and expansion of myeloid-derived suppressor cells (MDSCs), which inhibit T cell activity (169). IL-10 and TGFβ, primarily released by cells of the TME, have been shown to inhibit T cell mediated immunity (170–172). However, malignant plasma cells also secrete TGFβ by themselves thus creating an immunosuppressive environment facilitating immune evasion and expansion (173). Autocrine and/or paracrine IL-6 secretion induces IL-10 production, further suppressing T cell function, hence promoting myeloma clonal expansion (174–176). IL-10 may also affect TAM polarization towards an immunosuppressive M2 phenotype (176). The production of IL-10 and TGFβ by regulatory T cells (Tregs) has been shown to mediate their immunosuppressive activity on T effector cells in MM (177–179). MDSC numbers in MM patients’ BM are significantly higher and they have been shown to induce Treg differentiation and promote myeloma clonal selection and progression (128, 180–182). Furthermore, Leone et al. reported that dendritic cells accumulate in the BM of myeloma patients, where they promote cancer cell immune evasion by downregulating proteasome subunits (183). Therefore, understanding how immunosurveillance influences clonal selection in MM is crucial. By unraveling the dynamic interactions between the TME, immune system and malignant plasma cells, we can gain insights into the mechanisms underlying the immunosurveillance- related clonal evolution in MM.
Therapy resistance is a fundamental challenge in the management of MM. Although the majority of MM patients responds well to first-line therapies, almost all patients eventually experience MM recurrence and therapy-refractory disease states (184). Based on the acquisition of cytogenetic alterations or distinct mutations, a small proportion of malignant plasma cell clones might become resistant to standard MM therapies such as proteasome inhibitors (PIs), immunomodulatory imide drugs (IMiDs), glucocorticoids, and monoclonal antibodies and subsequently outcompete drug-sensitive populations (Figure 3). Misund et al. performed whole exome and RNA sequencing analyses for purified CD138+ cells from paired MM patients before and after therapy with different anti-myeloma drugs to compare genomic and transcriptomic changes as well as clonal evolution under treatment pressure. Changes in the clonal composition occurred in 82% of patients under therapy and alterations were primarily detected among RAS, 1q21 and TP53 (73). Ryu et al. detected overexpression of proteasome components in clonal subpopulations from relapsed and therapy refractory MM patients that had received bortezomib-containing combination therapies indicating selection and outgrowth of PI-resistant subclones under therapy (102). Corre et al. investigated the clonal heterogeneity among patients that had undergone a homogeneous treatment regimen of VTD (bortezomib, thalidomide and dexamethasone) followed by melphalan. Here, the frequencies of KRAS, NRAS and TP53 mutations were increased in relapsed MM patients compared to initial manifestations suggesting an induction of mutations by the treatment itself or increased resistance of mutation harboring subclones against VTD (185). Deletion of the chromosomal region 17p (del17p) is considered a high-risk cytogenetic alteration and is associated with mono-allelic loss of TP53 and shorter overall survival rates (186, 187). Co-occurrence of del17p with a TP53 mutation on the second allele – detected in around 6-8% of MM patients at the time of diagnosis - results in a bi-allelic inactivation and complete loss of p53 that is reportedly accompanied with worse outcomes compared to mono-allelic inactivations (187, 188). In line, AMO-1 MM cell line derived clones with bi-allelic TP53 inactivation out-competed AMO-1 clones with mono-allelic TP53 aberrations in comparative in vitro studies indicating a selection advantage of TP53 double-hit myeloma cell clones (187, 189). Walker et al. characterized newly diagnosed MM patients with bi-allelic TP53 aberrations as a distinct high-risk subgroup with rapid disease progression and extremely aggressive behavior despite the use of combination treatments of PIs, IMiDs, glucocorticoids and cyclophosphamide (190). These findings highlight the need to adapt MM risk-stratification criteria and the therapy of choice in accordance to the constellation of both cytogenetic and somatic alterations. However, so far, no specific treatment options are available for high-risk MM patients with TP53 aberrations (191). For this reason, clinical trials are urgently needed to investigate whether high-risk patients with TP53 mutations may profit from early intensified therapy regimens involving novel immunotherapies such as bispecific T-cell engagers or chimeric antigen receptor T cells. In addition, further preclinical studies need to be conducted to test the potential of drugs inducing p53-dependent synthetic lethality in MM. In this regard, recent research has shown that myeloma cells with p53 deficiency are more vulnerable to Chk1 inhibition compared to p53 proficient cancer cells (192).
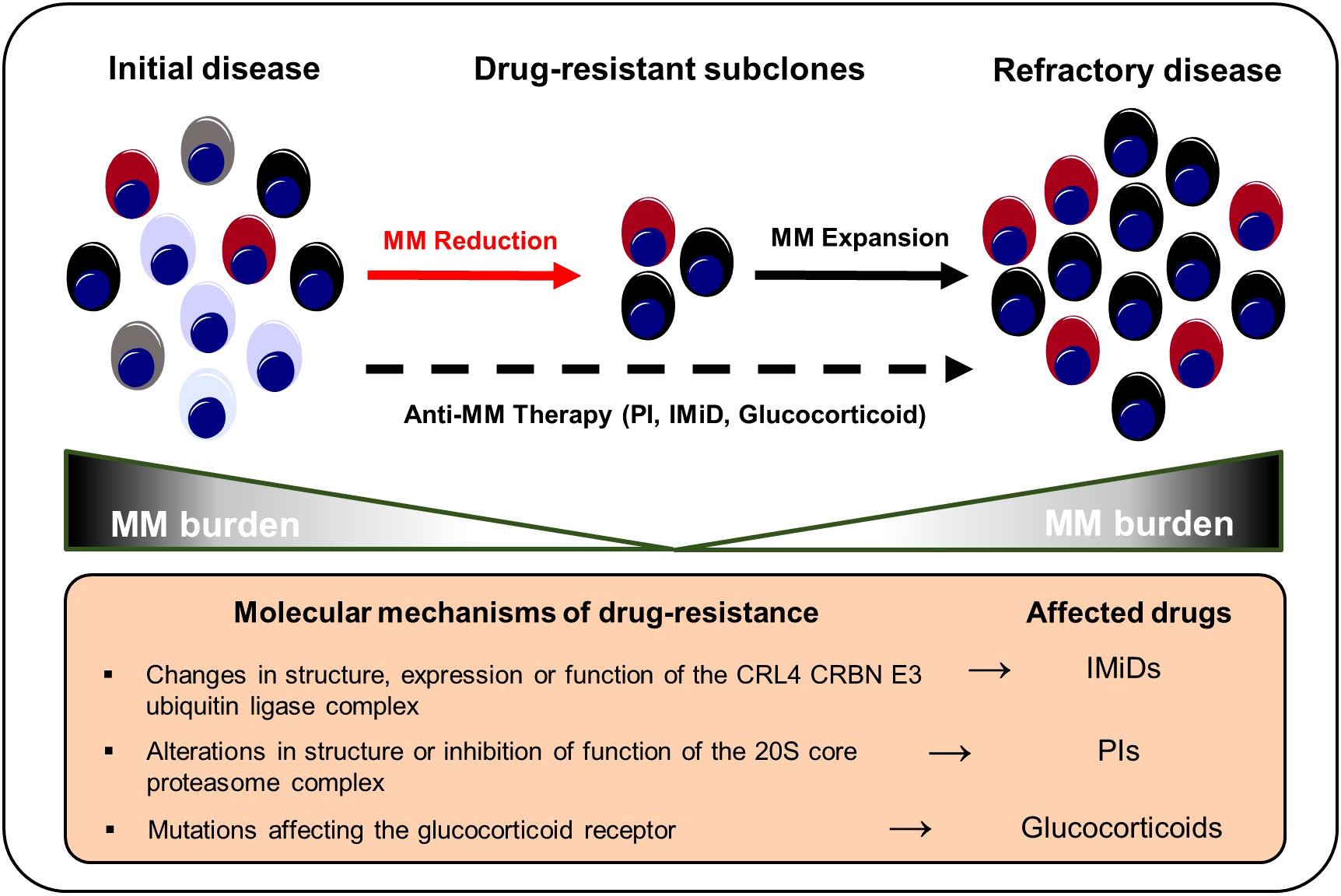
Figure 3 Selection and expansion of drug-resistant subclones under anti-MM therapy. Genetic instability increases the repertoire of drug-resistant subclones in the course of MM progression. Anti-myeloma therapies result in a selection of therapy-refractory and more aggressive clonal subsets that ultimately result in MM recurrence. IMiD, immunomodulatory imide drug; MM, multiple myeloma; PI, proteasome inhibitor.
Various tumor-related mechanisms that promote anti-myeloma drug resistance have been described. Thalidomide has been shown to inhibit tumor angiogenesis and dampen inflammation thus counteracting the release of cytokines such as TNF-α that are essential for myeloma cell growth (193). In addition, IMiDs also directly affect myeloma cell survival by binding to Cereblon (CRBN) – a component of the CRL4CRBN E3 ubiquitin ligase complex (CUL4–ROC1–DDB1–CRBN). Subsequent ubiquitination and degradation of the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3), which control myeloma survival and proliferation genes (e.g., MYC or IRF4), result in growth limitation and death (194–198). Changes in the structure, expression or function of CRL4CRBN E3 may generate IMiD resistant clonal plasma cell populations. Overall, IMiD resistance affects 10-20% of relapsed myeloma patients (196, 198–200). CRBN and CUL4 mutations prevent IMiDs from binding to CRL4CRBN, reducing IMiD efficiency (201). Lenalidomide-refractory MM patients had a higher incidence of COP9 signalosome gene loss, whose products are essential for CUL4-ROC1-DDB1-CRBN E3 ubiquitin ligase maintenance and activity. Approximately, 16% of IMiD patients lost the COP9 signalosome gene region on chromosome 2q37 while none of the patients in the control arm developed a 2q37 chromosomal aberration (202). MM patients receiving IMiD-based therapy had a greater prevalence of IKZF1 mutations (196). Ng et al. showed that overexpression of CDK6 in MM cell lines increases lenalidomide and pomalidomide resistance. Inhibition of CDK6 kinase functions, on the other hand, increases IMiD sensitivity. In addition, relapsed MM bone marrow samples from lenalidomide-pretreated individuals showed CDK6 upregulation. Therapeutically, the combination of pomalidomide with palbociclip (a CDK4/6 inhibitor) increased the overall survival of MM.1S tumor-bearing mice (198).
Glucocorticoids are a fundamental pillar in most MM therapy regimens (203). Glucocorticoids have been shown to trigger apoptosis in myeloma cells by suppressing anti-apoptotic genes such Bcl-xL and nuclear factor kappa B (NF-ҡB) (203, 204). However, malignant plasma cell clones may develop resistance to glucocorticoids like dexamethasone and prednisone. Dexamethasone and prednisone’s pro-apoptotic and anti-proliferative effects are blocked by genetic anomalies and/or mutations occurring in the glucocorticoid receptor NR3C1 in relapsed multiple myeloma patients. Genetic alterations in NR3C1 undermine the NF-ҡB mediated pro-apoptotic and anti-proliferative actions of dexamethasone and prednisone thus favoring NR3C1-mutation harboring clones (201, 204). Overexpression of MDR1 and Survivin (BIRC5), as well as downregulation of the apoptosis activator BIM (BCL2L11) were found in established dexamethasone-resistant MM cell lines. MDR1 inhibition or Survivin knockdown re-sensitized myeloma cells for dexamethasone (205).
PIs target the proteasome 20S subunit beta 5 (PSMB5). Point mutations affecting PSMB5 or other components of the proteasome complex play a central role in MM drug resistance (206). Aside from that, other resistance mechanisms to PIs (e.g., bortezomib and carfilzomib) have been described. PI resistance is frequently associated with chromosome 1q21 gain or amplification occurring in advanced MM stages. Various genes that are located at the 1q21 chromosomal region reportedly influence bortezomib sensitivity such as PSMD4 (207–209). Overexpression of interferon-stimulated 20 kD exonuclease-like 2 (ISG20L2) on chromosome 1q is associated with poor response owing to its high affinity in binding bortezomib (BTZ) and inhibiting the proteasome complex (210). In addition, carfilzomib sensitivity in MM cell lines is reduced by increased 1q21 S100 family overexpression localized within the 1q21 region (211). Overexpression of SRC-3 causes BTZ resistance via interacting with NSD2. SI-2 disrupted NSD2-induced SRC-3 stability to overcome BTZ resistance in t(4;14)-positive MM cell lines (212). Hypoxia has been reported to induce SENP1, which increases SRC-3 stability and thereby enhances PI resistance (134).
Immunotherapies and targeted treatments are being developed to overcome therapy resistance and improve the prognosis for patients with relapsed or refractory MM. CD38 has multiple functions in MM cells and immune cells such as promoting tumor cell proliferation, cell adhesion, and survival (213). In MM patients, monoclonal antibodies (mAbs) targeting CD38 (e.g., daratumumab), bi-specific mAbs or chimeric antigen receptor T (CAR-T) cells against the B cell maturation antigen (BCMA) or G protein–coupled receptor, class C, group 5, member D (GPRC5D) have been proven beneficial, particularly in advanced disease stages (Table 2) (3, 4, 224). While normal non-lymphoid tissues lack SLAMF7 expression, normal and malignant plasma cells uniformly express high levels of SLAMF7 making it a promising target for immunotherapies (225). SLAMF7 belongs to the group of the signaling lymphocytic activation molecule (SLAM) transmembrane receptors and has been functionally linked to promoting myeloma cell expansion (226, 227). Elotuzumab – an antibody-dependent cellular cytotoxicity (ADCC) anti-SLAMF7 mAb – has been approved for MM treatment (228). In addition, the efficacy of SLAMF7-CAR-T cells has been tested in preclinical studies resulting in the initiation of currently running Phase I/II clinical trials (226, 229).
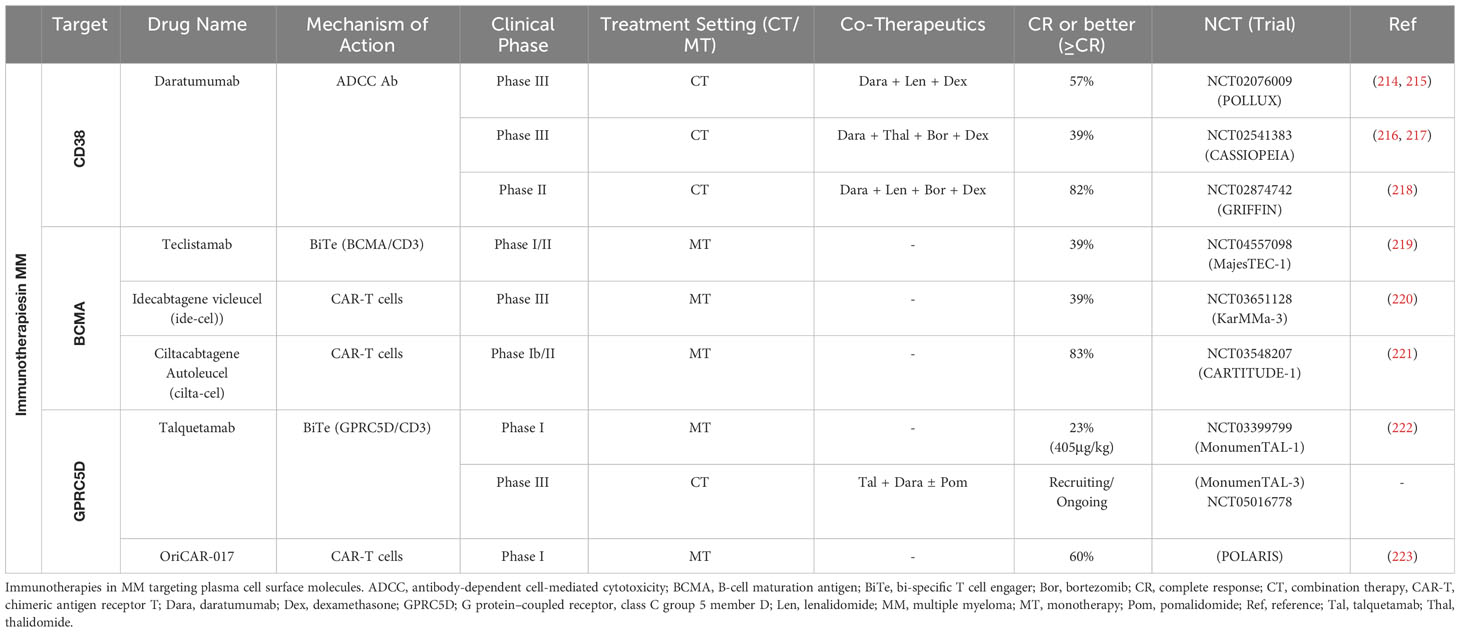
Table 2 Immunotherapies in MM.
However, most MM patients eventually progress after immunotherapies. In this regard, protein structural changes, immune checkpoint inhibitor overexpression, and/or an immunosuppressive environment may all exert an influence on the patients’ response to immunotherapy treatments (5). Exemplary, BCMA downregulation was identified in approximately 70% of MM patients undergoing BCMA CAR T cell therapy treatment suggesting that specific mutations can result in the loss of BCMA expression on malignant plasma cell clones that are then selected under therapy (5, 230, 231). Indeed, homozygous deletion of TNFRSF17 - encoding BCMA - results in a complete loss of BCMA expression and has been described in MM patients after BCMA CAR T cell therapy (232). GPRC5D downregulation or loss was found in six patients with progressive disease after initial response to GPRC5D -targeted CAR T cells suggesting a positive selection of GPRC5D non-expressing subclones (4). CD38 expression on malignant plasma cells reportedly decreases upon anti-CD38 daratumumab treatment (233). Moreover, the efficacy of the ADCC - antibody daratumumab is highly dependent on patients’ NK cell functionality. Verkleij et al. recently showed that non-responding MM patients have higher frequencies of TIM-3+ HLA-DR+ activated/exhausted NK cells and that NK cells are rapidly depleted upon daratumumab treatment initiation (233).
Overall, different tumor-and non-tumor related mechanisms or their respective interactions, promote drug resistance in clonal plasma cells. Those clones with survival advantages such as point mutations that affect drug binding sites or complete downregulation/loss of surface targets eventually outcompete their drug-sensitive counterparts and become the dominant clonal population.
In conclusion, MM is characterized by an extensive intratumor heterogeneity starting in the earliest phases of the disease. The (epi-) genetic aberrations are the primary driver of the considerable intratumor heterogeneity and clonal evolution seen in MM. However, the TME crucially contributes to the clonal evolution of specific clones that depend on soluble factors such as chemokines or growth factors or cell-cell interactions provided by BM resident cells. The selection process is further supported by IMiDs, proteasome inhibitors and glucocorticoids. Although, the TME in MM is composed of various immune cells, the immune response is often dysfunctional or suppressed. In contrast, immunotherapy with mAbs, bispecific Abs or CAR-T cells select for resistant clones often characterized as antigen-loss variants. The high intratumor heterogeneity that evolves during disease progression and treatment is responsible for the fact that although many very efficacious treatments have been developed recently, MM still remains an incurable disease.
SF, RR & AFO wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vogelsberg A, Schürch CM, Fend F. Multiples Myelom aus Sicht der Pathologie. Radiologe (2022) 62(1):12–9. doi: 10.1007/s00117-021-00926-z
2. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos M-V, et al. Multiple myeloma. Nat Rev Dis Prim (2017) 3:17046. doi: 10.1038/nrdp.2017.46
3. Zhao W-H, Liu J, Wang B-Y, Chen Y-X, Cao X-M, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol (2018) 11(1):141.
4. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med (2022) 387(13):1196–206. doi: 10.1056/NEJMoa2209900
5. Atilla PA, Atilla E. Resistance against anti-CD19 and anti-BCMA CAR T cells: Recent advances and coping strategies. Transl Oncol (2022) 22:101459. doi: 10.1016/j.tranon.2022.101459
6. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M-V, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol (2014) 15(12):e538–48. doi: 10.1016/S1470-2045(14)70442-5
7. Ho M, Patel A, Goh CY, Moscvin M, Zhang L, Bianchi G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia (2020) 34(12):3111–25. doi: 10.1038/s41375-020-01051-x
8. Manier S, Salem K, Glavey SV, Roccaro AM, Ghobrial IM. Genomic Aberrations in Multiple Myeloma BT - Plasma Cell Dyscrasias. Roccaro AM, Ghobrial IM, editors. Cham: Springer International Publishing (2016) p. 23–34.
9. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol (2017) 14(2):100–13. doi: 10.1038/nrclinonc.2016.122
10. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood (2018) 132(6):587–97. doi: 10.1182/blood-2018-03-840132
11. Diamantidis MD, Papadaki S, Hatjiharissi E. Exploring the current molecular landscape and management of multiple myeloma patients with the t(11;14) translocation. Front Oncol (2022) 12:934008. doi: 10.3389/fonc.2022.934008
12. Shen YJ, Mishima Y, Shi J, Sklavenitis-Pistofidis R, Redd RA, Moschetta M, et al. Progression signature underlies clonal evolution and dissemination of multiple myeloma. Blood (2021) 137(17):2360–72. doi: 10.1182/blood.2020005885
13. Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun (2017) 8(1):268. doi: 10.1038/s41467-017-00296-y
14. Rasche L, Schinke C, Maura F, Bauer MA, Ashby C, Deshpande S, et al. The spatio-temporal evolution of multiple myeloma from baseline to relapse-refractory states. Nat Commun (2022) 13(1):4517. doi: 10.1038/s41467-022-32145-y
15. Varettoni M, Corso A, Pica G, Mangiacavalli S, Pascutto C, Lazzarino M. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol Off J Eur Soc Med Oncol (2010) 21(2):325–30. doi: 10.1093/annonc/mdp329
16. Forster S, Radpour R. Molecular impact of the tumor microenvironment on multiple myeloma dissemination and extramedullary disease. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.941437
17. Kim CH, Broxmeyer HE. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood (1998) 91(1):100–10. doi: 10.1182/blood.V91.1.100
18. Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood (2009) 114(6):1150. doi: 10.1182/blood-2009-01-202606
19. Holthof LC, Mutis T. Challenges for immunotherapy in multiple myeloma: bone marrow microenvironment-mediated immune suppression and immune resistance. Cancers (2020) 17;12(4):988. doi: 10.3390/cancers12040988
20. Marquardt S, Solanki M, Spitschak A, Vera J, Pützer BM. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin Cancer Biol (2018) 53:90–109. doi: 10.1016/j.semcancer.2018.06.006
21. Abdou Hassan W, Muqresh MA, Omer M. The potential role of CD44 and CD133 in colorectal stem cell cancer. Cureus (2022) 14(10):e30509–9.
22. MacDonagh L, Gray SG, Breen E, Cuffe S, Finn SP, O’Byrne KJ, et al. Lung cancer stem cells: The root of resistance. Cancer Lett (2016) 372(2):147–56. doi: 10.1016/j.canlet.2016.01.012
23. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res (2007) 67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030
24. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
25. Radpour R. Tracing and targeting cancer stem cells: New venture for personalized molecular cancer therapy. World J Stem Cells (2017) 9(10):169–78. doi: 10.4252/wjsc.v9.i10.169
26. Forster S, Radpour R. Molecular immunotherapy: promising approach to treat metastatic colorectal cancer by targeting resistant cancer cells or cancer stem cells. Front Oncol (2020) 10:569017. doi: 10.3389/fonc.2020.569017
27. Johnsen HE, Bøgsted M, Schmitz A, Bødker JS, El-Galaly TC, Johansen P, et al. The myeloma stem cell concept, revisited: from phenomenology to operational terms. Haematologica (2016) 101(12 SE-Review Articles):1451–9. doi: 10.3324/haematol.2015.138826
28. Kellner J, Liu B, Kang Y, Li Z. Fact or fiction - identifying the elusive multiple myeloma stem cell. J Hematol Oncol (2013) 6(1):91. doi: 10.1186/1756-8722-6-91
29. Rasmussen T, Lodahl M, Hancke S, Johnsen HE. In multiple myeloma clonotypic CD38 – /CD19 + /CD27 + Memory B cells recirculate through bone marrow, peripheral blood and lymph nodes. Leuk Lymphoma (2004) 45(7):1413–7. doi: 10.1080/10428190410001655157
30. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res (2008) 68(1):190–7. doi: 10.1158/0008-5472.CAN-07-3096
31. Pilarski LM, Seeberger K, Coupland RW, Eshpeter A, Keats JJ, Taylor BJ, et al. Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice. Exp Hematol (2002) 30(3):221–8. doi: 10.1016/S0301-472X(01)00788-3
32. Reghunathan R, Bi C, Liu SC, Loong KT, Chung T-H, Huang G, et al. Clonogenic multiple myeloma cells have shared stemness signature associated with patient survival. Oncotarget (2013) 4(8):1230–40. doi: 10.18632/oncotarget.1145
33. Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood (2004) 103(6):2332–6. doi: 10.1182/blood-2003-09-3064
34. Kim D, Park CY, Medeiros BC, Weissman IL. CD19–CD45low/–CD38high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia (2012) 26(12):2530–7. doi: 10.1038/leu.2012.140
35. Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood (1999) 94(10):3576–82. doi: 10.1182/blood.V94.10.3576.422k01_3576_3582
36. Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, Fooksman DR. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia (2020) 34(1):245–56. doi: 10.1038/s41375-019-0519-4
37. Kawano Y, Kikukawa Y, Fujiwara S, Wada N, Okuno Y, Mitsuya H, et al. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int J Oncol (2013) 43(6):1809–16. doi: 10.3892/ijo.2013.2134
38. Goodell MA. Stem cell identification and sorting using the Hoechst 33342 Side Population (SP). Curr Protoc Cytom (2005) 34(1):9.18.1–9.18.11.
39. Loh YS, Mo S, Brown RD, Yamagishi T, Yang S, Joshua DE, et al. Presence of Hoechst low side populations in multiple myeloma. Leuk Lymphoma (2008) 49(9):1813–6. doi: 10.1080/10428190802272676
40. Du J, Liu S, He J, Liu X, Qu Y, Yan W, et al. MicroRNA-451 regulates stemness of side population cells via PI3K/Akt/mTOR signaling pathway in multiple myeloma. Oncotarget (2015) 6(17):14993–5007. doi: 10.18632/oncotarget.3802
41. Yang Y, Shi J, Gu Z, Salama ME, Das S, Wendlandt E, et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res (2015) 75(3):594–604. doi: 10.1158/0008-5472.CAN-14-2362
42. Jundt F, Pröbsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood (2004) 103(9):3511–5. doi: 10.1182/blood-2003-07-2254
43. Fan J-L, Zhang J, Dong L-W, Fu W-J, Du J, Shi H-G, et al. URI regulates tumorigenicity and chemotherapeutic resistance of multiple myeloma by modulating IL-6 transcription. Cell Death Dis (2014) 5(3):e1126–6. doi: 10.1038/cddis.2014.93
44. Derksen PWB, Tjin E, Meijer HP, Klok MD, Mac Gillavry HD, van Oers MHJ, et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc Natl Acad Sci (2004) 101(16):6122–7. doi: 10.1073/pnas.0305855101
45. Ren Z, van Andel H, de Lau W, Hartholt RB, Maurice MM, Clevers H, et al. Syndecan-1 promotes Wnt/β-catenin signaling in multiple myeloma by presenting Wnts and R-spondins. Blood (2018) 131(9):982–94. doi: 10.1182/blood-2017-07-797050
46. van Andel H, Kocemba KA, Spaargaren M, Pals ST. Aberrant Wnt signaling in multiple myeloma: molecular mechanisms and targeting options. Leukemia (2019) 33(5):1063–75. doi: 10.1038/s41375-019-0404-1
47. Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood (2012) 120(25):5002–13. doi: 10.1182/blood-2011-07-368142
48. Sabol HM, Delgado-Calle J. The multifunctional role of Notch signaling in multiple myeloma. J Cancer Metastasis Treat (2021) 7:20. doi: 10.20517/2394-4722.2021.35
49. Wang F, Dan Z, Luo H, Huang J, Cui Y, Ding H, et al. ALCAM regulates multiple myeloma chemoresistant side population. Cell Death Dis (2022) 13(2):136. doi: 10.1038/s41419-022-04556-8
50. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci (2007) 104(10):4048–53. doi: 10.1073/pnas.0611682104
51. Luo H, Zhang D, Wang F, Wang Q, Wu Y, Gou M, et al. ALCAM-EGFR interaction regulates myelomagenesis. Blood Adv (2021) 5(23):5269–82. doi: 10.1182/bloodadvances.2021004695
52. Gilad N, Zukerman H, Pick M, Gatt ME. The role of CD24 in multiple myeloma tumorigenicity and effects of the microenvironment on its expression. Oncotarget (2019) 10(52):5480–91. doi: 10.18632/oncotarget.27190
53. Wang L, Lin N, Li Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol Rep (2019) 41(3):1678–90. doi: 10.3892/or.2019.6968
54. Serizawa K, Tanaka H, Ueda T, Fukui A, Kakutani H, Taniguchi T, et al. CD34+ myeloma cells with self-renewal activities are therapy-resistant and persist as MRD in cell cycle quiescence. Int J Hematol (2022) 115(3):336–49. doi: 10.1007/s12185-021-03261-0
55. Gao M, Bai H, Jethava Y, Wu Y, Zhu Y, Yang Y, et al. Identification and characterization of tumor-initiating cells in multiple myeloma. J Natl Cancer Inst (2020) 112(5):507–15. doi: 10.1093/jnci/djz159
56. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood (2015) 125(20):3049–58. doi: 10.1182/blood-2014-11-568881
57. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer (2012) 12(5):335–48. doi: 10.1038/nrc3257
58. Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood (2012) 120(5):1060–6. doi: 10.1182/blood-2012-01-405977
59. Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood (1996) 88(2):674–81. doi: 10.1182/blood.V88.2.674.bloodjournal882674
60. Chesi M, Nardini E, Lim RSC, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3and a novel gene, MMSET, resulting in igH/MMSET hybrid transcripts. Blood (1998) 92(9):3025–34. doi: 10.1182/blood.V92.9.3025
61. Chesi M, Nardini E, Brents LA, Schröck E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet (1997) 16(3):260–4.
62. Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent Dysregulation of the c-maf Proto-Oncogene at 16q23 by Translocation to an Ig Locus in Multiple Myeloma. Blood (1998) 91(12):4457–63. doi: 10.1182/blood.V91.12.4457.412k48_4457_4463
63. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer (2002) 2(3):175–87. doi: 10.1038/nrc746
64. Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene (2001) 20(40):5611–22. doi: 10.1038/sj.onc.1204641
65. Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C, Hématologique on behalf of the GF de C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood (2001) 98(7):2229–38. doi: 10.1182/blood.V98.7.2229
66. Liang Y, He H, Wang W, Wang H, Mo S, Fu R, et al. Malignant clonal evolution drives multiple myeloma cellular ecological diversity and microenvironment reprogramming. Mol Cancer (2022) 21(1):182. doi: 10.1186/s12943-022-01648-z
67. Kaur G, Jena L, Gupta R, Farswan A, Gupta A, Sriram K. Correlation of changes in subclonal architecture with progression in the MMRF CoMMpass study. Transl Oncol (2022) 23:101472. doi: 10.1016/j.tranon.2022.101472
68. Farswan A, Jena L, Kaur G, Gupta A, Gupta R, Rani L, et al. Branching clonal evolution patterns predominate mutational landscape in multiple myeloma. Am J Cancer Res (2021) 11(11):5659–79.
69. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun (2014) 5:2997. doi: 10.1038/ncomms3997
70. Salomon-Perzyński A, Barankiewicz J, Machnicki M, Misiewicz-Krzemińska I, Pawlak M, Radomska S, et al. Tracking clonal evolution of multiple myeloma using targeted next-generation DNA sequencing. Biomedicines (2022) 10(7):1674. doi: 10.3390/biomedicines10071674
71. Dutta AK, Fink JL, Grady JP, Morgan GJ, Mullighan CG, To LB, et al. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia (2019) 33(2):457–68. doi: 10.1038/s41375-018-0206-x
72. Jakubikova J, Cholujova D, Beke G, Hideshima T, Klucar L, Leiba M, et al. Heterogeneity of B cell lymphopoiesis in patients with preMalignant and active myeloma. JCI Insight (2023) 8(3):e159924. doi: 10.1172/jci.insight.159924
73. Misund K, Hofste op Bruinink D, Coward E, Hoogenboezem RM, Rustad EH, Sanders MA, et al. Clonal evolution after treatment pressure in multiple myeloma: heterogenous genomic aberrations and transcriptomic convergence. Leukemia (2022) 36(7):1887–97. doi: 10.1038/s41375-022-01597-y
74. Samur MK, Roncador M, Aktas Samur A, Fulciniti M, Bazarbachi AH, Szalat R, et al. High-dose melphalan treatment significantly increases mutational burden at relapse in multiple myeloma. Blood (2023) 141(14):1724–36. doi: 10.1182/blood.2022017094
75. Rasche L, Angtuaco EJ, Alpe TL, Gershner GH, McDonald JE, Samant RS, et al. The presence of large focal lesions is a strong independent prognostic factor in multiple myeloma. Blood (2018) 132(1):59–66. doi: 10.1182/blood-2018-04-842880
76. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res (2012) 72(19):4875–82. doi: 10.1158/0008-5472.CAN-12-2217
77. Vandyke K, Zeissig MN, Hewett DR, Martin SK, Mrozik KM, Cheong CM, et al. HIF-2α Promotes dissemination of plasma cells in multiple myeloma by regulating CXCL12/CXCR4 and CCR1. Cancer Res (2017) 77(20):5452–63. doi: 10.1158/0008-5472.CAN-17-0115
78. Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood (2012) 119(24):5782–94. doi: 10.1182/blood-2011-09-380410
79. Kitajima S, Lee KL, Fujioka M, Sun W, You J, Chia GS, et al. Hypoxia-inducible factor-2 alpha up-regulates CD70 under hypoxia and enhances anchorage-independent growth and aggressiveness in cancer cells. Oncotarget (2018) 9(27):19123–35. doi: 10.18632/oncotarget.24919
80. Schürch CM, Rasche L, Frauenfeld L, Weinhold N, Fend F. A review on tumor heterogeneity and evolution in multiple myeloma: pathological, radiological, molecular genetics, and clinical integration. Virchows Arch (2020) 476(3):337–51. doi: 10.1007/s00428-019-02725-3
81. Braggio E, Maiolino A, Gouveia ME, Magalhães R, Souto Filho JT, Garnica M, et al. Methylation status of nine tumor suppressor genes in multiple myeloma. Int J Hematol (2010) 91(1):87–96. doi: 10.1007/s12185-009-0459-2
82. Chim C-S, Fung T-K, Cheung W-C, Liang R, Kwong Y-L. SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood (2004) 103(12):4630–5. doi: 10.1182/blood-2003-06-2007
83. Rastgoo N, Abdi J, Hou J, Chang H. Role of epigenetics-microRNA axis in drug resistance of multiple myeloma. J Hematol Oncol (2017) 10(1):121. doi: 10.1186/s13045-017-0492-1
84. Furukawa Y, Kikuchi J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int J Hematol (2016) 104(3):281–92. doi: 10.1007/s12185-016-2048-5
85. Wang J, Zhu X, Dang L, Jiang H, Xie Y, Li X, et al. Epigenomic reprogramming via HRP2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J Clin Invest (2022) 132(4)::e149526. doi: 10.1172/JCI149526
86. Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun (2019) 10(1):1911. doi: 10.1038/s41467-019-09555-6
87. Allegra A, Casciaro M, Barone P, Musolino C, Gangemi S. Epigenetic crosstalk between Malignant plasma cells and the tumour microenvironment in multiple myeloma. Cancers (2022) 14(11):2597. doi: 10.3390/cancers14112597
88. Schütt J, Nägler T, Schenk T, Brioli A. Investigating the interplay between myeloma cells and bone marrow stromal cells in the development of drug resistance: dissecting the role of epigenetic modifications. Cancers (Basel) (2021) 13(16):4069.
89. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun (2015) 6:8983. doi: 10.1038/ncomms9983
90. Garcia-Gomez A, Li T, de la Calle-Fabregat C, Rodríguez-Ubreva J, Ciudad L, Català-Moll F, et al. Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. Nat Commun (2021) 12(1):421. doi: 10.1038/s41467-020-20715-x
91. Segers VFM, De Keulenaer GW. Autocrine signaling in cardiac remodeling: A rich source of therapeutic targets. J Am Heart Assoc (2021) 10(3):e019169. doi: 10.1161/JAHA.120.019169
92. Teoh PJ, Chung T-H, Chng PYZ, Toh SHM, Chng WJ. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica (2020) 105(5):1391–404. doi: 10.3324/haematol.2019.221176
93. Kim SY, Min HJ, Park HK, Oh B, Kim TY, She CJ, et al. Increased copy number of the interleukin-6 receptor gene is associated with adverse survival in multiple myeloma patients treated with autologous stem cell transplantation. Biol Blood Marrow Transplant (2011) 17(6):810–20. doi: 10.1016/j.bbmt.2011.01.002
94. Zhong L, Xu Z, Jin X, He Y, Zhang J, Jiang T, et al. miR−451a suppression of IL−6R can inhibit proliferation and increase apoptosis through the JAK2/STAT3 pathway in multiple myeloma. Oncol Lett (2020) 20(6):339.
95. Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly Malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood (2001) 97(2):483–9. doi: 10.1182/blood.V97.2.483
96. Liu Y, Cheng P, Zhao W, Zhu L, Sui J, Dai Y, et al. MiR-197-3p reduces bortezomib resistance in multiple myeloma by inhibiting IL-6 expression in a MEAF6-dependent manner. Leuk Res (2022) 114:106785. doi: 10.1016/j.leukres.2022.106785
97. Chong PSY, Zhou J, Lim JSL, Hee YT, Chooi J-Y, Chung T-H, et al. IL6 promotes a STAT3-PRL3 feedforward loop via SHP2 repression in multiple myeloma. Cancer Res (2019) 79(18):4679–88. doi: 10.1158/0008-5472.CAN-19-0343
98. Kawano M, HIrano T, Matsuda T, Taga T, Horii Y, Iwato K, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature (1988) 332(6159):83–5. doi: 10.1038/332083a0
99. Anderson KC, Jones RM, Morimoto C, Leavitt P, Barut BA. Response patterns of purified myeloma cells to hematopoietic growth factors. Blood (1989) 73(7):1915–24. doi: 10.1182/blood.V73.7.1915.1915
100. Matthes T, Manfroi B, Huard B. Revisiting IL-6 antagonism in multiple myeloma. Crit Rev Oncol Hematol (2016) 105:1–4. doi: 10.1016/j.critrevonc.2016.07.006
101. Brocke-Heidrich K, Kretzschmar AK, Pfeifer G, Henze C, Löffler D, Koczan D, et al. Interleukin-6–dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family–independent survival pathway closely associated with Stat3 activation. Blood (2004) 103(1):242–51. doi: 10.1182/blood-2003-04-1048
102. Ryu D, Kim SJ, Hong Y, Jo A, Kim N, Kim H-J, et al. Alterations in the transcriptional programs of myeloma cells and the microenvironment during extramedullary progression affect proliferation and immune evasion. Clin Cancer Res (2020) 26(4):935–44. doi: 10.1158/1078-0432.CCR-19-0694
103. Shah JJ, Feng L, Thomas SK, Berkova Z, Weber DM, Wang M, et al. Siltuximab (CNTO 328) with lenalidomide, bortezomib and dexamethasone in newly-diagnosed, previously untreated multiple myeloma: an open-label phase I trial. Blood Cancer J (2016) 6(2):e396–6. doi: 10.1038/bcj.2016.4
104. Moreau P, Cavallo F, Leleu X, Hulin C, Amiot M, Descamps G, et al. Phase I study of the anti insulin-like growth factor 1 receptor (IGF-1R) monoclonal antibody, AVE1642, as single agent and in combination with bortezomib in patients with relapsed multiple myeloma. Leukemia (2011) 25(5):872–4. doi: 10.1038/leu.2011.4
105. Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EP, et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH trial (EAY131) subprotocol W. J Clin Oncol (2020) 38(21):2407–17. doi: 10.1200/JCO.19.02630
106. Klempner SJ, Bendell JC, Villaflor VM, Tenner LL, Stein SM, Rottman JB, et al. Safety, efficacy, and biomarker results from a phase Ib study of the anti-DKK1 antibody DKN-01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther (2021) 20(11):2240–9. doi: 10.1158/1535-7163.MCT-21-0273
107. Edenfield WJ, Richards DA, Vukelja SJ, Weiss GJ, Sirard CA, Landau SB, et al. A phase 1 study evaluating the safety and efficacy of DKN-01, an investigational monoclonal antibody (Mab) in patients (pts) with advanced non-small cell lung cancer. J Clin Oncol (2014) 32(15_suppl):8068. doi: 10.1200/jco.2014.32.15_suppl.8068
108. Raje NS, Moreau P, Terpos E, Benboubker L, Grząśko N, Holstein SA, et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br J Haematol (2017) 176(5):783–95. doi: 10.1111/bjh.14483
109. Bensinger W, Raptis A, Berenson JR, Spira AI, Nooka AK, Chaudhry M, et al. Safety and tolerability of BION-1301 in adults with relapsed or refractory multiple myeloma. J Clin Oncol (2019) 37(15_suppl):8012. doi: 10.1200/JCO.2019.37.15_suppl.8012
110. Barratt J, Hour B, Kooienga L, Roy S, Schwartz B, Siddiqui A, et al. POS-109 interim results of phase 1 and 2 trials to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics, and clinical activity of BION-1301 in patients with IgA nephropathy. Kidney Int Rep (2022) 7(2):S46. doi: 10.1016/j.ekir.2022.01.121
111. White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer (2013) 119(2):339–47. doi: 10.1002/cncr.27745
112. Hansson M, Gimsing P, Badros A, Niskanen TM, Nahi H, Offner F, et al. A phase I dose-escalation study of antibody BI-505 in relapsed/refractory multiple myeloma. Clin Cancer Res (2015) 21(12):2730–6. doi: 10.1158/1078-0432.CCR-14-3090
113. Bataille R, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P. CD221 (IGF-1R) is aberrantly expressed in multiple myeloma, in relation to disease severity. Haematologica (2005) 90(5 SE-Letters to the Editor):706–7.
114. Chiron D, Maïga S, Surget S, Descamps G, Gomez-Bougie P, Traore S, et al. Autocrine insulin-like growth factor 1 and stem cell factor but not interleukin 6 support self-renewal of human myeloma cells. Blood Cancer J (2013) 3(6):e120–0. doi: 10.1038/bcj.2013.18
115. van Andel H, Ren Z, Koopmans I, Joosten SPJ, Kocemba KA, de Lau W, et al. Aberrantly expressed LGR4 empowers Wnt signaling in multiple myeloma by hijacking osteoblast-derived R-spondins. Proc Natl Acad Sci (2017) 114(2):376–81. doi: 10.1073/pnas.1618650114
116. Tsubaki M, Komai M, Itoh T, Imano M, Sakamoto K, Shimaoka H, et al. Inhibition of the tumour necrosis factor-alpha autocrine loop enhances the sensitivity of multiple myeloma cells to anticancer drugs. Eur J Cancer (2013) 49(17):3708–17. doi: 10.1016/j.ejca.2013.07.010
117. Lentzsch S, Gries M, Janz M, Bargou R, Dörken B, Mapara MY. Macrophage inflammatory protein 1-alpha (MIP-1α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood (2003) 101(9):3568–73. doi: 10.1182/blood-2002-08-2383
118. Liu Z, Xu J, He J, Zheng Y, Li H, Lu Y, et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood (2014) 124(13):2061–71. doi: 10.1182/blood-2014-03-557298
119. Furukawa M, Ohkawara H, Ogawa K, Ikeda K, Ueda K, Shichishima-Nakamura A, et al. Autocrine and paracrine interactions between multiple myeloma cells and bone marrow stromal cells by growth arrest-specific gene 6 cross-talk with interleukin-6*. J Biol Chem (2017) 292(10):4280–92. doi: 10.1074/jbc.M116.733030
120. Sati HIA, Greaves M, Apperley JF, Russell RGG, Croucher PI. Expression of interleukin-1β and tumour necrosis factor-α in plasma cells from patients with multiple myeloma. Br J Haematol (1999) 104(2):350–7. doi: 10.1046/j.1365-2141.1999.01193.x
121. Jöhrer K, Janke K, Krugmann J, Fiegl M, Greil R. Transendothelial migration of myeloma cells is increased by tumor necrosis factor (TNF)-α via TNF receptor 2 and autocrine up-regulation of MCP-1. Clin Cancer Res (2004) 10(6):1901–10. doi: 10.1158/1078-0432.CCR-1053-03
122. Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood (2004) 103(8):3148–57. doi: 10.1182/blood-2003-06-1984
123. Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J (2012) 2(9):e89. doi: 10.1038/bcj.2012.37
124. Bisping G, Leo R, Wenning D, Dankbar B, Padró T, Kropff M, et al. Paracrine interactions of basic fibroblast growth factor and interleukin-6 in multiple myeloma. Blood (2003) 101(7):2775–83. doi: 10.1182/blood-2002-09-2907
125. Ronca R, Ghedini GC, Maccarinelli F, Sacco A, Locatelli SL, Foglio E, et al. FGF Trapping Inhibits Multiple Myeloma Growth through c-Myc Degradation–Induced Mitochondrial Oxidative Stress. Cancer Res (2020) 80(11):2340–54. doi: 10.1158/0008-5472.CAN-19-2714
126. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
127. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
128. García-Ortiz A, Rodríguez-García Y, Encinas J, Maroto-Martín E, Castellano E, Teixidó J, et al. The role of tumor microenvironment in multiple myeloma development and progression. Cancers (Basel) (2021) 13(2):217. doi: 10.3390/cancers13020217
129. Sonugür FG, Akbulut H. The role of tumor microenvironment in genomic instability of Malignant tumors. Front Genet (2019) 10.
130. Eliasson P, Jönsson J-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J Cell Physiol (2010) 222(1):17–22. doi: 10.1002/jcp.21908
131. Chen G, Wu K, Li H, Xia D, He T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.961637
132. Degtyareva NP, Heyburn L, Sterling J, Resnick MA, Gordenin DA, Doetsch PW. Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase zeta-dependent only for adenines and guanines. Nucleic Acids Res (2013) 41(19):8995–9005. doi: 10.1093/nar/gkt671
133. Muz B, Azab F, de la Puente P, Landesman Y, Azab AK. Selinexor overcomes hypoxia-induced drug resistance in multiple myeloma. Transl Oncol (2017) 10(4):632–40. doi: 10.1016/j.tranon.2017.04.010
134. Guo J, Lv Y, Wang S, Peng Z, Xie Y, Wang Y, et al. Hypoxia induces chemoresistance to proteasome inhibitors through orchestrating deSUMOylation and ubiquitination of SRC-3 in multiple myeloma. Oncogene (2022) 41(45):4971–9. doi: 10.1038/s41388-022-02494-5
135. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckland NZ) (2015) 3:83–92. doi: 10.2147/HP.S93413
136. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell (2019) 177(7):1915–1932.e16. doi: 10.1016/j.cell.2019.04.040
137. Kawano MM, Mihara K, Huang N, Tsujimoto T, Kuramoto A. Differentiation of early plasma cells on bone marrow stromal cells requires interleukin-6 for escaping from apoptosis. Blood (1995) 85(2):487–94. doi: 10.1182/blood.V85.2.487.487
138. Chu VT, Fröhlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol (2011) 12(2):151–9. doi: 10.1038/ni.1981
139. Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood (2010) 116(11):1867–75. doi: 10.1182/blood-2009-12-259457
140. Vyzoukaki R, Tsirakis G, Pappa CA, Devetzoglou M, Tzardi M, Alexandrakis MG. The impact of mast cell density on the progression of bone disease in multiple myeloma patients. Int Arch Allergy Immunol (2015) 168(4):263–8. doi: 10.1159/000443275
141. Chen W-C, Hu G, Hazlehurst LA. Contribution of the bone marrow stromal cells in mediating drug resistance in hematopoietic tumors. Curr Opin Pharmacol (2020) 54:36–43. doi: 10.1016/j.coph.2020.08.006
142. Rø TB, Holien T, Fagerli U-M, Hov H, Misund K, Waage A, et al. HGF and IGF-1 synergize with SDF-1α in promoting migration of myeloma cells by cooperative activation of p21-activated kinase. Exp Hematol (2013) 41(7):646–55.
143. Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)–dependent migration and homing in multiple myeloma. Blood (2006) 109(7):2708–17.
144. Qiang Y-W, Yao L, Tosato G, Rudikoff S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood (2004) 103(1):301–8. doi: 10.1182/blood-2003-06-2066
145. Lemaitre L, Hamaidia M, Descamps J-G, Do Souto Ferreira L, Joubert M-V, Gadelorge M, et al. Toll-like receptor 4 selective inhibition in medullar microenvironment alters multiple myeloma cell growth. Blood Adv (2022) 6(2):672–8. doi: 10.1182/bloodadvances.2020003704
146. Musolino C, Allegra A, Innao V, Allegra AG, Pioggia G, Gangemi S. Inflammatory and anti-inflammatory equilibrium, proliferative and antiproliferative balance: the role of cytokines in multiple myeloma. Giovarelli M, editor. Mediators Inflamm (2017) 2017:1852517. doi: 10.1155/2017/1852517
147. Yue X, Huang L, Yang Y, Zhao Y, He D, Han X, et al. High levels of serum IL-10 indicate disease progression, extramedullary involvement, and poor prognosis in multiple myeloma. J Zhejiang Univ Sci B (2022) 23(11):968–74. doi: 10.1631/jzus.B2200277
148. Sun M, Xiao Q, Wang X, Yang C, Chen C, Tian X, et al. Tumor-associated macrophages modulate angiogenesis and tumor growth in a xenograft mouse model of multiple myeloma. Leuk Res (2021) 110:106709. doi: 10.1016/j.leukres.2021.106709
149. Akhmetzyanova I, Aaron T, Galbo P, Tikhonova A, Dolgalev I, Tanaka M, et al. Tissue-resident macrophages promote early dissemination of multiple myeloma via IL-6 and TNFα. Blood Adv (2021) 5(18):3592–608. doi: 10.1182/bloodadvances.2021005327
150. Takagi S, Tsukamoto S, Park J, Johnson KE, Kawano Y, Moschetta M, et al. Platelets enhance multiple myeloma progression via IL-1β Upregulation. Clin Cancer Res (2018) 24(10):2430–9. doi: 10.1158/1078-0432.CCR-17-2003
151. Katz B-Z. Adhesion molecules—The lifelines of multiple myeloma cells. Semin Cancer Biol (2010) 20(3):186–95. doi: 10.1016/j.semcancer.2010.04.003
152. Okada T, Hawley RG, Kodaka M, Okuno H. Significance of VLA-4–VCAM-1 interaction and CD44 for transendothelial invasion in a bone marrow metastatic myeloma model. Clin Exp Metastasis (1999) 17(7):623–9. doi: 10.1023/A:1006715504719
153. Di Marzo L, Desantis V, Solimando AG, Ruggieri S, Annese T, Nico B, et al. Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget (2016) 7(37)::60698–711. doi: 10.18632/oncotarget.10849
154. Boyd AW, Wawryk SO, Burns GF, Fecondo JV. Intercellular adhesion molecule 1 (ICAM-1) has a central role in cell-cell contact-mediated immune mechanisms. Proc Natl Acad Sci (1988) 85(9):3095–9. doi: 10.1073/pnas.85.9.3095
155. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer (2007) 7(8):585–98. doi: 10.1038/nrc2189
156. Sherbenou DW, Su Y, Behrens CR, Aftab BT, Perez de Acha O, Murnane M, et al. Potent activity of an anti-ICAM1 antibody–drug conjugate against multiple myeloma. Clin Cancer Res (2020) 26(22):6028–38. doi: 10.1158/1078-0432.CCR-20-0400
157. Sanz-Rodríguez F, Ruiz-Velasco N, Pascual-Salcedo D, Teixidó J. Characterization of VLA-4-dependent myeloma cell adhesion to fibronectin and VCAM-1. Br J Haematol (1999) 107(4):825–34. doi: 10.1046/j.1365-2141.1999.01762.x
158. Hathi D, Chanswangphuwana C, Cho N, Fontana F, Maji D, Ritchey J, et al. Ablation of VLA4 in multiple myeloma cells redirects tumor spread and prolongs survival. Sci Rep (2022) 12(1):30. doi: 10.1038/s41598-021-03748-0
159. Fontana F, Scott MJ, Allen JS, Yang X, Cui G, Pan D, et al. VLA4-targeted nanoparticles hijack cell adhesion-mediated drug resistance to target refractory myeloma cells and prolong survival. Clin Cancer Res (2021) 27(7):1974–86. doi: 10.1158/1078-0432.CCR-20-2839
160. Lin L, Cao L, Liu Y, Wang K, Zhang X, Qin X, et al. B7-H3 promotes multiple myeloma cell survival and proliferation by ROS-dependent activation of Src/STAT3 and c-Cbl-mediated degradation of SOCS3. Leukemia (2019) 33(6):1475–86. doi: 10.1038/s41375-018-0331-6
161. Yousef S, Marvin J, Steinbach M, Langemo A, Kovacsovics T, Binder M, et al. Immunomodulatory molecule PD-L1 is expressed on Malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J (2015) 5(3):e285–5. doi: 10.1038/bcj.2015.7
162. Costa F, Vescovini R, Marchica V, Storti P, Notarfranchi L, Dalla Palma B, et al. PD-L1/PD-1 pattern of expression within the bone marrow immune microenvironment in smoldering myeloma and active multiple myeloma patients. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.613007
163. Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol (2016) 9(1):116. doi: 10.1186/s13045-016-0345-3
164. Pawlyn C, Morgan GJ. Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer (2017) 17(9):543–56. doi: 10.1038/nrc.2017.63
165. Larrayoz M, Garcia-Barchino MJ, Celay J, Etxebeste A, Jimenez M, Perez C, et al. Preclinical models for prediction of immunotherapy outcomes and immune evasion mechanisms in genetically heterogeneous multiple myeloma. Nat Med (2023) 29(3):632–45. doi: 10.1038/s41591-022-02178-3
166. Liu Y, Yan H, Gu H, Zhang E, He J, Cao W, et al. Myeloma-derived IL-32γ induced PD-L1 expression in macrophages facilitates immune escape via the PFKFB3-JAK1 axis. Oncoimmunology (2022) 11(1):2057837.
167. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia (2012) 26(12):2538–45. doi: 10.1038/leu.2012.141
168. Paul B, Liedtke M, Khouri J, Rifkin R, Gandhi MD, Kin A, et al. A phase II multi-arm study of magrolimab combinations in patients with relapsed/refractory multiple myeloma. Futur Oncol (2023) 19(1):7–17. doi: 10.2217/fon-2022-0975
169. Lewinsky H, Gunes EG, David K, Radomir L, Kramer MP, Pellegrino B, et al. CD84 is a regulator of the immunosuppressive microenvironment in multiple myeloma. JCI Insight (2021) 6(4):e141683. doi: 10.1172/jci.insight.141683
170. Alexandrakis MG, Goulidaki N, Pappa CA, Boula A, Psarakis F, Neonakis I, et al. Interleukin-10 induces both plasma cell proliferation and angiogenesis in multiple myeloma. Pathol Oncol Res (2015) 21(4):929–34. doi: 10.1007/s12253-015-9921-z
171. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8+ T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity (2018) 48(2):299–312.e5. doi: 10.1016/j.immuni.2018.01.006
172. Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic Malignancies. Blood (2006) 107(12):4589–96. doi: 10.1182/blood-2005-10-4169
173. Rana PS, Soler DC, Kort J, Driscoll JJ. Targeting TGF-β signaling in the multiple myeloma microenvironment: Steering CARs and T cells in the right direction. Front Cell Dev Biol (2022) 10. doi: 10.3389/fcell.2022.1059715
174. Gadó K, Domján G, Hegyesi H, Falus A. Role of interleukin-6 in the pathogenesis of multiple myeloma. Cell Biol Int (2000) 24(4):195–209. doi: 10.1006/cbir.2000.0497
175. Kim J, Denu RA, Dollar BA, Escalante LE, Kuether JP, Callander NS, et al. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol (2012) 158(3):336–46. doi: 10.1111/j.1365-2141.2012.09154.x
176. Sun J, Park C, Guenthner N, Gurley S, Zhang L, Lubben B, et al. Tumor-associated macrophages in multiple myeloma: advances in biology and therapy. J Immunother Cancer (2022) 10(4):e003975. doi: 10.1136/jitc-2021-003975
177. Ho M, Xiao A, Yi D, Zanwar S, Bianchi G. Treating multiple myeloma in the context of the bone marrow microenvironment. Curr Oncol (2022) 29:8975–9005. doi: 10.3390/curroncol29110705
178. Feyler S, Scott GB, Parrish C, Jarmin S, Evans P, Short M, et al. Tumour cell generation of inducible regulatory T-cells in multiple myeloma is contact-dependent and antigen-presenting cell-independent. PloS One (2012) 7(5):e35981. doi: 10.1371/journal.pone.0035981
179. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
180. Giallongo C, Tibullo D, Parrinello NL, La CP, Di RM, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget (2016) 7(52):85764–75. doi: 10.18632/oncotarget.7969
181. Wang Z, Zhang L, Wang H, Xiong S, Li Y, Tao Q, et al. Tumor-induced CD14+HLA-DR–/low myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol Immunother (2015) 64(3):389–99. doi: 10.1007/s00262-014-1646-4
182. Kuwahara-Ota S, Shimura Y, Steinebach C, Isa R, Yamaguchi J, Nishiyama D, et al. Lenalidomide and poMalidomide potently interfere with induction of myeloid-derived suppressor cells in multiple myeloma. Br J Haematol (2020) 191(5):784–95. doi: 10.1111/bjh.16881
183. Leone P, Berardi S, Frassanito MA, Ria R, De Re V, Cicco S, et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood (2015) 126(12):1443–51. doi: 10.1182/blood-2015-01-623975
184. Robak P, Drozdz I, Szemraj J, Robak T. Drug resistance in multiple myeloma. Cancer Treat Rev (2018) 70:199–208. doi: 10.1016/j.ctrv.2018.09.001
185. Corre J, Cleynen A, Robiou du Pont S, Buisson L, Bolli N, Attal M, et al. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia (2018) 32(12):2636–47. doi: 10.1038/s41375-018-0153-6
186. Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J Clin Oncol (2015) 33(26):2863–9. doi: 10.1200/JCO.2015.61.2267
187. Flynt E, Bisht K, Sridharan V, Ortiz M, Towfic F, Thakurta A. Prognosis, biology, and targeting of TP53 dysregulation in multiple myeloma. Cells (2020) 9(2):287. doi: 10.3390/cells9020287
188. Thanendrarajan S, Tian E, Qu P, Mathur P, Schinke C, van Rhee F, et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica (2017) 102(9 SE-Online Only Articles):e364–7. doi: 10.3324/haematol.2017.168872
189. Munawar U, Rasche L, Müller N, Vogt C, Da-Via M, Haertle L, et al. Hierarchy of mono- and biallelic TP53 alterations in multiple myeloma cell fitness. Blood (2019) 134(10):836–40. doi: 10.1182/blood.2019000080
190. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia (2019) 33(1):159–70. doi: 10.1038/s41375-018-0196-8
191. Marcon C, Simeon V, Deias P, Facchin G, Corso A, Derudas D, et al. Experts’ consensus on the definition and management of high risk multiple myeloma. Front Oncol (2023) 12.
192. Teoh PJ, An O, Chung T-H, Vaiyapuri T, Raju A, Hoppe MM, et al. p53-NEIL1 co-abnorMalities induce genomic instability and promote synthetic lethality with Chk1 inhibition in multiple myeloma having concomitant 17p13(del) and 1q21(gain). Oncogene (2022) 41(14):2106–21. doi: 10.1038/s41388-022-02227-8
193. Kumar S, Rajkumar SV. Thalidomide and lenalidomide in the treatment of multiple myeloma. Eur J Cancer (2006) 42(11):1612–22. doi: 10.1016/j.ejca.2006.04.004
194. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science (2014) 343(6168):301–5. doi: 10.1126/science.1244851
195. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science (2014) 343(6168):305–9. doi: 10.1126/science.1244917
196. Barrio S, Munawar U, Zhu YX, Giesen N, Shi CX, Da Viá M, et al. IKZF1/3 and CRL4CRBN E3 ubiquitin ligase mutations and resistance to immunomodulatory drugs in multiple myeloma. Haematologica (2020) 105:e237–41. doi: 10.3324/haematol.2019.217943
197. Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature (2008) 454(7201):226–31. doi: 10.1038/nature07064
198. Ng YLD, Ramberger E, Bohl SR, Dolnik A, Steinebach C, Conrad T, et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat Commun (2022) 13(1):1009. doi: 10.1038/s41467-022-28515-1
199. Kortüm KM, Mai EK, Hanafiah NH, Shi C-X, Zhu Y-X, Bruins L, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood (2016) 128(9):1226–33. doi: 10.1182/blood-2016-02-698092
200. Liu J, Song T, Zhou W, Xing L, Wang S, Ho M, et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia (2019) 33(1):171–80. doi: 10.1038/s41375-018-0205-y
201. Vo JN, Wu Y-M, Mishler J, Hall S, Mannan R, Wang L, et al. The genetic heterogeneity and drug resistance mechanisms of relapsed refractory multiple myeloma. Nat Commun (2022) 13(1):3750. doi: 10.1038/s41467-022-31430-0
202. Gooding S, Ansari-Pour N, Kazeroun MH, Karagoz K, Polonskaia A, Angulo Salazar M, et al. Loss of COP9-signalosome genes at 2q37 is associated with IMiD agent resistance in multiple myeloma. Blood (2022) 140(16):1816–21. doi: 10.1182/blood.2022015909
203. Burwick N, Sharma S. Glucocorticoids in multiple myeloma: past, present, and future. Ann Hematol (2019) 98(1):19–28. doi: 10.1007/s00277-018-3465-8
204. Chauhan D, Auclair D, Robinson EK, Hideshima T, Li G, Podar K, et al. Identification of genes regulated by Dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene (2002) 21(9):1346–58. doi: 10.1038/sj.onc.1205205
205. Tsubaki M, Satou T, Itoh T, Imano M, Komai M, Nishinobo M, et al. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk Res (2012) 36(10):1315–22. doi: 10.1016/j.leukres.2012.07.003
206. Allmeroth K, Horn M, Kroef V, Miethe S, Müller R-U, Denzel MS. Bortezomib resistance mutations in PSMB5 determine response to second-generation proteasome inhibitors in multiple myeloma. Leukemia (2021) 35(3):887–92. doi: 10.1038/s41375-020-0989-4
207. An G, Xu Y, Shi L, Shizhen Z, Deng S, Xie Z, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica (2014) 99(2):353–9. doi: 10.3324/haematol.2013.088211
208. Shaughnessy JD Jr, Qu P, Usmani S, Heuck CJ, Zhang Q, Zhou Y, et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3. Blood (2011) 118(13):3512–24. doi: 10.1182/blood-2010-12-328252
209. Hanamura I. Gain/Amplification of chromosome arm 1q21 in multiple myeloma. Cancers (Basel) (2021) 13(2):256. doi: 10.3390/cancers13020256
210. Yang Y, Gao Y, Huang J, Yang Z, Luo H, Wang F, et al. ISG20L2 suppresses bortezomib anti-myeloma activity by attenuating bortezomib binding to PSMB5. JCI Insight (2022) 7(19):e157081. doi: 10.1172/jci.insight.157081
211. Liu M, Wang Y, Miettinen JJ, Kumari R, Majumder MM, Tierney C, et al. S100 calcium binding protein family members associate with poor patient outcome and response to proteasome inhibition in multiple myeloma. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.723016
212. Liu J, Xie Y, Guo J, Li X, Wang J, Jiang H, et al. Targeting NSD2-mediated SRC-3 liquid–liquid phase separation sensitizes bortezomib treatment in multiple myeloma. Nat Commun (2021) 12(1):1022. doi: 10.1038/s41467-021-21386-y
213. van de Donk NWCJ, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02134
214. Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Overall survival with daratumumab, lenalidomide, and dexamethasone in previously treated multiple myeloma (POLLUX): A randomized, open-label, phase III trial. J Clin Oncol (2023) 41(8):1590–9. doi: 10.1200/JCO.22.00940
215. Kaufman JL, Usmani SZ, San-Miguel J, Bahlis N, White DJ, Benboubker L, et al. Four-year follow-up of the phase 3 pollux study of daratumumab plus lenalidomide and dexamethasone (D-Rd) versus Lenalidomide and Dexamethasone (Rd) alone in Relapsed or Refractory Multiple Myeloma (RRMM). Blood (2019) 134(Supplement_1):1866. doi: 10.1182/blood-2019-123483
216. Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet (2019) 394(10192):29–38. doi: 10.1016/S0140-6736(19)31240-1
217. Moreau P, Hulin C, Perrot A, Arnulf B, Belhadj K, Benboubker L, et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): an open-label, randomised, phase 3 trial. Lancet Oncol (2021) 22(10):1378–90. doi: 10.1016/S1470-2045(21)00428-9
218. Laubach JP, Kaufman JL, Sborov DW, Reeves B, Rodriguez C, Chari A, et al. Daratumumab (DARA) plus lenalidomide, bortezomib, and dexamethasone (RVd) in patients (Pts) with transplant-eligible newly diagnosed multiple myeloma (NDMM): updated analysis of griffin after 24 months of maintenance. Blood (2021) 138:79. doi: 10.1182/blood-2021-149024
219. Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med (2022) 387(6):495–505. doi: 10.1056/NEJMoa2203478
220. Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med (2023) 388(11):1002–14. doi: 10.1056/NEJMoa2213614
221. Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene autoleucel, an anti–B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol (2022) 41(6):1265–74.
222. Chari A, Minnema MC, Berdeja JG, Oriol A, van de Donk NWCJ, Rodríguez-Otero P, et al. Talquetamab, a T-cell–redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med (2022) 387(24):2232–44. doi: 10.1056/NEJMoa2204591
223. Zhang M, Wei G, Zhou L, Zhou J, Chen S, Zhang W, et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol (2023) 10(2):e107–16. doi: 10.1016/S2352-3026(22)00372-6
224. D’Agostino M, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia (2020) 34(1):21–34. doi: 10.1038/s41375-019-0669-4
225. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res (2008) 14(9):2775–84. doi: 10.1158/1078-0432.CCR-07-4246
226. Gogishvili T, Danhof S, Prommersberger S, Rydzek J, Schreder M, Brede C, et al. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood (2017) 130(26):2838–47. doi: 10.1182/blood-2017-04-778423
227. Kikuchi J, Hori M, Iha H, Toyama-Sorimachi N, Hagiwara S, Kuroda Y, et al. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia (2020) 34(1):180–95. doi: 10.1038/s41375-019-0525-6
228. Poh A. Multiple myeloma gets three new drugs. Cancer Discovery (2016) 6(1):4. doi: 10.1158/2159-8290.CD-NB2015-169
229. Danhof S, Rasche L, Mottok A, Steinmüller T, Zhou X, Schreder M, et al. Elotuzumab for the treatment of extramedullary myeloma: a retrospective analysis of clinical efficacy and SLAMF7 expression patterns. Ann Hematol (2021) 100(6):1537–46. doi: 10.1007/s00277-021-04447-6
230. Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma. J Clin Invest (2019) 129(6):2210–21. doi: 10.1172/JCI126397
231. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood (2016) 128(13):1688–700. doi: 10.1182/blood-2016-04-711903
232. Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med (2021) 27(4):616–9. doi: 10.1038/s41591-021-01245-5
Keywords: multiple myeloma, malignant plasma cell, clonal evolution, tumor microenvironment, TME, immunotherapy, drug resistance
Citation: Forster S, Radpour R and Ochsenbein AF (2023) Molecular and immunological mechanisms of clonal evolution in multiple myeloma. Front. Immunol. 14:1243997. doi: 10.3389/fimmu.2023.1243997
Received: 21 June 2023; Accepted: 21 August 2023;
Published: 06 September 2023.
Edited by:
Takatsugu Ishimoto, Kumamoto University, JapanReviewed by:
Kentaro Minagawa, The Pennsylvania State University, United StatesCopyright © 2023 Forster, Radpour and Ochsenbein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramin Radpour, cmFtaW4ucmFkcG91ckB1bmliZS5jaA==; Adrian F. Ochsenbein, YWRyaWFuLm9jaHNlbmJlaW5AaW5zZWwuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.