 Guoqiang Zhong
Guoqiang Zhong Chengcheng He
Chengcheng He Shanping Wang
Shanping Wang Chuangzhen Lin
Chuangzhen Lin Mingsong Li
Mingsong Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 August 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1241262
This article is part of the Research Topic Pathogenetic mechanism and therapeutic target for inflammation in autoimmune disease View all 22 articles
Inflammatory bowel disease (IBD), a general term encompassing Crohn’s disease (CD) and ulcerative colitis (UC), and other conditions, is a chronic and relapsing autoimmune disease that can occur in any part of the digestive tract. While the cause of IBD remains unclear, it is acknowledged that the disease has much to do with the dysregulation of intestinal immunity. In the intestinal immune regulatory system, Cholesterol-25-hydroxylase (CH25H) plays an important role in regulating the function of immune cells and lipid metabolism through catalyzing the oxidation of cholesterol into 25-hydroxycholesterol (25-HC). Specifically, CH25H focuses its mechanism of regulating the inflammatory response, signal transduction and cell migration on various types of immune cells by binding to relevant receptors, and the mechanism of regulating lipid metabolism and immune cell function via the transcription factor Sterol Regulator-Binding Protein. Based on this foundation, this article will review the function of CH25H in intestinal immunity, aiming to provide evidence for supporting the discovery of early diagnostic and treatment targets for IBD.
Inflammatory bowel disease (IBD) including Crohn’s disease (CD) and ulcerative colitis (UC) is a chronic and relapsing inflammatory digestive disease. As the incidence of IBD has rapidly increased, an increasing number of experts and scholars are investigating the etiology of the disease. Although IBD is founded to be closely related to external factors such as people’s dietary habits and living environment (1), its internal factors, namely its specific cause and exact mechanism, remain to be explored. It is acknowledged that IBD is primarily associated with environmental factors, genetic predisposition, the dysbiosis of intestinal microbiota, and immune dysfunction. CH25H has been demonstrated to have the ability to regulate intestinal immunity and is associated with IBD (2). Previous studies have found that cholesterol-25-hydroxylase (CH25H) can catalyze the production of 25-hydroxycholesterol (25-HC) from cholesterol (3). Sterol regulatory-element binding proteins (SREBPs) is also conducive to transcription in the intestinal immune regulatory system. SREBPs are a family of transcriptive factors that regulate the expression of genes involved in lipogenic processes. They also get involved in numerous cellular processes and pathologies such as reactive oxygen species generation, endoplasmic reticulum stress, apoptosis, and autophagy (4). 25-HC can inactivate SREBPs to inhibit lipid synthesis or exert anti-inflammatory effects. In addition to its role in cholesterol metabolism, CH25H and its downstream products can bind to relevant receptors and participate in regulating immune system, including the direct regulation of inflammatory programming, the development of immune responses, and the signaling transduction. Previous studies have highlighted the significance of CH25H in immunology, arguing that CH25H plays a protective role against viral and bacterial infections (5). However, further investigation is needed to examine the function of CH25H in intestinal immunity, particularly in relation to IBD. Based on this foundation, this article is intended to provide an overview of the research progress of CH25H in intestinal immunity.
CH25H has some biological characteristics, including CH25H gene, CH25H protein, and 25-HC. Specifically, CH25H gene is a member of interferon-stimulating genes (ISGs), which has crucial functions in inflammation, innate immunity, and adaptive immune responses via interferon (IFN) signaling. CH25H protein belongs to the redox enzyme family and is a 31.6-kDa endoplasmic reticulum (ER)-associated hydroxylase that can catalyze cholesterol to produce 25-HC (6). 25-HC is an endogenous oxysterol produced by the oxidation of CH25H. Its function is to regulate the activity of SREBP, thereby inhibiting the biosynthesis of cholesterol and reducing its accumulation. Furthermore, 25-HC can also regulate the activity of nuclear receptors, which enables the regulation of immune response processes and lipid metabolism (3, 7). Regarding the regulation of CH25H expression, Diczfalusy et al. (8) have found that lipopolysaccharide (LPS) can induce an increase in the level of CH25H in macrophages of both mice and human volunteers. In a similar vein, Park et al. (9) conducted a further investigation, discovering that the expression of CH25H is mainly up-regulated by type I IFN via JAK/STAT1 signaling pathway. Through our observations, we have noted that the rapid induction of CH25H subsequently leads to the production of 25-HC, indicating that CH25H plays a significant role in lipid metabolism, gene expression, and immune activation (Figure 1A). Since previous studies have shown that CH25H plays a vital role in antiviral infections and various immune system-related diseases (3, 7), these regulatory mechanisms mentioned above have the possibility to be involved in the pathogenesis of IBD.
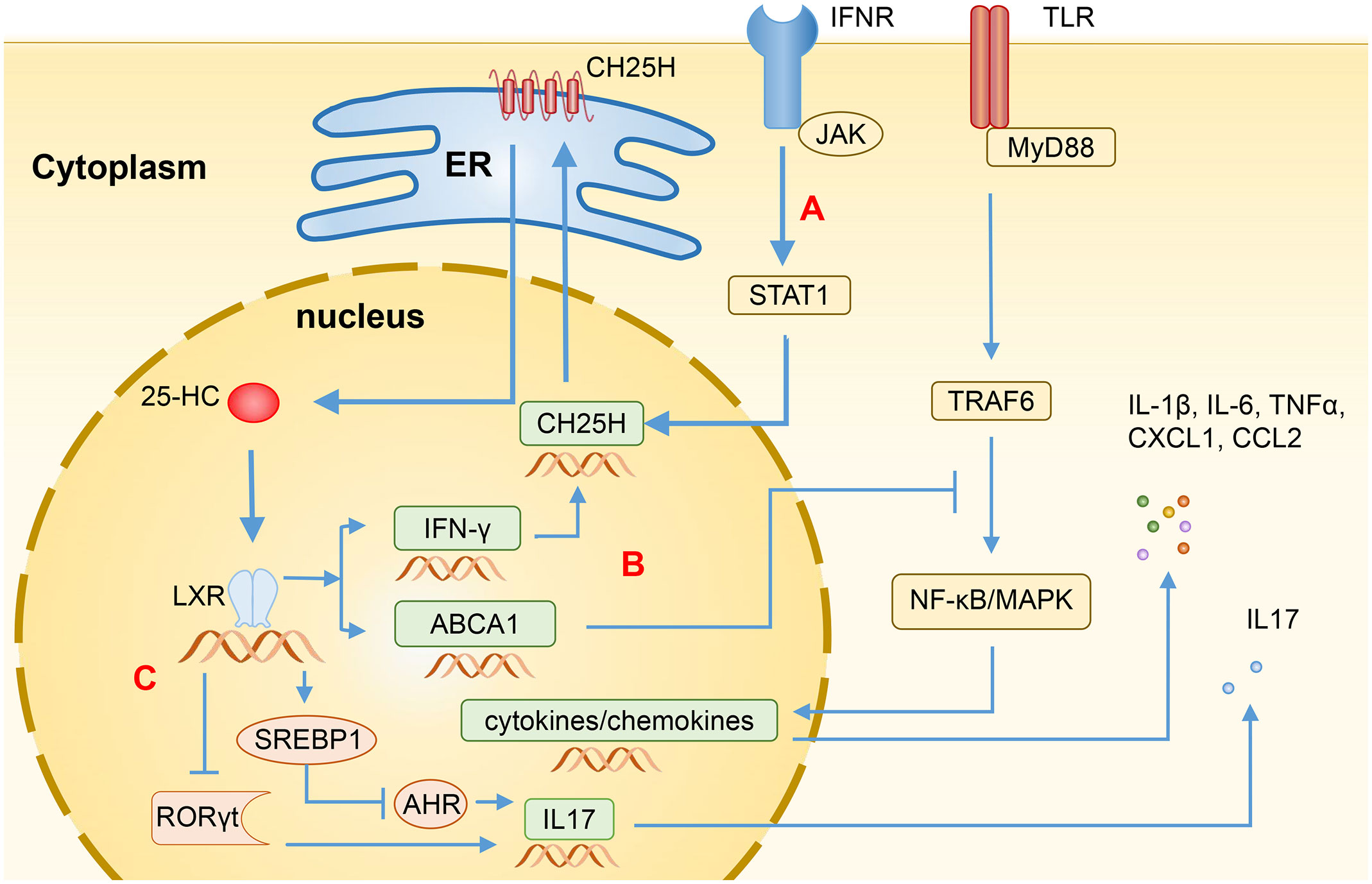
Figure 1 Regulation of immune function by 25-HC through LXR. (A) The transcription levels of CH25H are induced by Type I IFN through the STAT1-dependent signaling pathway, resulting in the production of 25-HC via cholesterol oxidation. (B) Binding LXR, 25-HC can induce the expression of ABCA1, disrupting the recruitment of MyD88 and TRAF6, and subsequently blocking the TLR-induced activation of NF-κB and MAPK, thereby suppressing the production of various inflammatory mediators such as IL-1β, IL-6, IL-17, TNFα, CXCL1, and CCL2. Additionally, 25-HC can also induce the expression of IFN-γ, thereby enhancing the CH25H expression through feedback regulation. (C) In CD4+ T cells, 25-HC activates SREBP1 or inhibits RORγt through LXR, leading to the inhibition of IL-17 expression and Th17 cell differentiation. 25-HC, 25-hydroxycholesterol; CH25H, Cholesterol-25-hydroxylase; LXR, Liver X receptor; IFN, Interferon; ABCA1, ATP-binding cassette transporters A1; MyD88, myeloid differentiation primary response protein 88; TRAF6, TNF Receptor Associated Factor 6; TLR, Toll-like receptor; NF-κB, Nuclear factor-κB, MAPK, Mitogen-activated protein kinase; TNFα, Tumor necrosis factor α; CXCL1, C-X-C motif chemokine ligand 1; CCL2, C-C motif chemokine ligand 2; SREBP1, Sterol regulatory-element binding protein 1; RORγt, Retinoic acid receptor-related orphan receptor γt; AHR, Aryl hydrocarbon receptor.
CH25H is widely expressed in various immune cells in physiological conditions, including macrophages, dendritic cells (DCs), neutrophils, and many others (10), whereas 25-HC is mainly produced in macrophages (11, 12) and regulates the functions and phenotypes of various immune cells, including macrophages, B cells, T cells, DCs, and neutrophils through its receptors (13). Notably, 25-HC has both pro-inflammatory and anti-inflammatory effects, and these differences may depend on its concentration and microenvironment, thereby exerting different functions in regulating gene expression, cellular proliferation, differentiation, and apoptosis (7).
The innate and adaptive immune responses in intestinal immune system are typically tightly regulated. Imbalances in the immune system will result in the development and progression of IBD (14). The immunological dysregulation present in IBD is characterized by epithelial damage, the expansion of inflammation facilitated by intestinal microbiota, and the infiltration of various types of cells into lamina propria, including T cells, B cells, macrophages, DCs, and neutrophils. These activated cells within the lamina propria produce proinflammatory cytokines such as tumor necrosis factor (TNF), Interleukin-1β (IL-1β), IFN-γ, and the IL-6 family of cytokines, leading to a high level of inflammation in the local tissue (15). CH25H can potentially contribute to the pathogenesis of IBD as it is widely expressed in various immune cells (10, 13). Intestinal dysbiosis and IBD often have a mutually causal relationship. The dysbiosis observed in individuals with IBD is associated with the increased production of LPS, and damage to the gut epithelial barrier allows LPS produced by the microbiota to enter the bloodstream, resulting in metabolic endotoxemia (16). Upon LPS stimulation, the TLR4/TRIF dependent pathway upregulates the expression of CH25H (8). The CH25H metabolite can act on multiple membrane receptors and nuclear receptors, exerting regulatory functions. Among these receptors, the liver X receptor (LXR), G protein-coupled receptor 183 (GPR183), and retinoic acid receptor-related orphan receptor (ROR) are pivotal in immune regulation.
LXRs, as the nuclear receptor of ligand-activated transcription factors, is capable of participating in various metabolic processes, including lipid, cholesterol, and carbohydrate metabolism. There are two LXR isoforms: LXRα and LXRβ (7, 17). LXRα may primarily play a role in the innate immune response via macrophages and dendritic cells, whereas LXRβ is likely responsible for the immune response in colonic epithelial cells and the adaptive immune response through CD3/CD4 lymphocytes (17). The activation of LXRs in a mouse colitis model can suppress the expression of inflammatory factors such as IL-1β, IL-6, IL-17A, TNFα, and chemokines such as CXCL1/KC, CXCL2/MIP2, CXCL8/IL-8, CCL2/MCP1 and CXCL10 in the colon tissue (17). In addition, LXRs can aid in the polarization of M2 macrophages and enhance the phagocytosis of macrophages and DCs, thereby increasing the clearance of senescent neutrophils and maintaining the neutrophil homeostasis (18, 19).
Being 25-HC an LXR ligand, it can induce the expression of cholesterol efflux transporters, such as ATP-binding cassette transporters A1 (ABCA1) and ATP-binding cassette transporters G1 (ABCG1). 25-HC can also upregulate the expression of Apolipoprotein E (ApoE) and Cytochrome P450 family 7 subfamily A member 1 (CYP7A1), which are important for cholesterol clearance (20). Additionally, 25-HC can induce the expression of IFN-γ in an LXR-dependent mechanism. The activated IFN-γ can subsequently enhance the expression of CH25H, forming a positive feedback loop (21). Ito et al. (22) have also found that the activation of LXRs can induce the expression of ABCA1, which could impede the recruitment of myeloid differentiation primary response protein 88 (MyD88) and TNF Receptor Associated Factor 6 (TRAF6), and subsequently block the Toll-like receptor (TLR)-induced activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways. Consequently, this leads to the inhibition of the production of several inflammatory mediators, such as IL-1β, IL-6, IL-17, TNFα, CXCL1, and CCL2 (Figure 1B).
LXRs are associated with the innate immunity against pathogens. Research has shown that LXRs can protect macrophages from pathogen-induced cell death, thereby reducing pathogen infection (23, 24). There are two types of macrophages involving in host defense and tissue homoeostasis: M1 and M2 macrophages. M2 macrophages possess an anti-inflammatory property, contributing to maintaining tissue homeostasis. M2 macrophages can be polarized due to the activation of LXR by 25-HC. This polarization is generated due to the presence of intracellular amino-acid sufficiency signal and the extrinsic IL-4 signal (19). On the other hand, under the stimulation of pathogenic microorganisms, IFN-γ or TLR ligands can activate macrophages to become M1 macrophages, which play a role in initiating and sustaining inflammation (19). However, both IFN-γ or TLR ligands can also lead to the production of 25-HC, which can promote the polarization of M2 macrophages. It has been reported that the activation of TLR4 by LPS can increase the level of 25-HC, thereby regulating the LXR-dependent lipid metabolism and immune response, and inhibiting the expression of inflammatory mediators in macrophages such as COX-2, IL-6, IL-1β, and G-CSF (25).
LXRs also affect the function of neutrophils. Smoak et al. (26) have shown that the activation of LXRs can inhibit neutrophil motility, thus impairing pulmonary antibacterial host defense, and worsening the survival of mice. Korf et al. (27) found that mice lacking LXRs showed a deficiency in the early neutrophilic airway response to Mycobacterium tuberculosis infection, an impaired Th1/Th17 function, and a higher susceptibility to disseminated systemic infection. Reboldi et al. (28) discovered that the recruitment of neutrophils to the peritoneum relies on IL-1β, whereas 25-HC functions in suppressing IL-1β-mediated peritoneal inflammation by acting the downstream of type I IFN. The findings indicate that the signaling pathway of LXRs, which is activated by 25-HC, plays multiple roles in the regulation of innate immunity.
Anti-aging gene Sirtuin 1 (SIRT1) is associated with autoimmune diseases and irreversible programmed cell death in various cells and tissues (29). SIRT1 is regulated by environmental factors, diet, stress, lifestyle factors and bacterial LPS (30, 31). SIRT1 has to do with LXRs, in the sense that SIRT1 can deacetylate LXRs and promote their ubiquitination, thereby regulating ABCA1 and SREBP1c, which are involved in cellular cholesterol homeostasis (30). Studies have discovered that metformin can alleviate hepatic inflammation by activating CH25H in a SIRT1-dependent manner in that CH25H can promote cholesterol catabolism and the subsequent increased levels of 25-HC can inhibit inflammation driven by IL-1β. Moreover, metformin can suppress the polarization of M1 macrophages in a SIRT1-dependent manner (32). These findings suggest that SIRT1 gets involved in the regulation of cholesterol homeostasis through the LXR/ABCA1 pathway relevant to the regulation of CH25H and its downstream products.
The helper T cell plays an important role in the pathogenesis of IBD. The activation of LXRs leads to an increase in the expression of SREBP1, which then translocate into the nucleus and disrupts aryl hydrocarbon receptor (AHR), an essential positive regulator of Th17 cell differentiation. As a result, this negatively regulates the Th17 cell differentiation and inhibits the IL-17 promoter to reduce the IL-17 transcription (20, 33) (Figure 1C). 25-HC can also inhibit the differentiation of Th17 cells by suppressing the production of IL-1, which works synergistically with transforming the growth of factor-β (TF-β) to induce Th17 cells (28, 34). Jakobsson et al. (17) observed that LXR-deficient mice are more susceptible to suffer dextran sodium sulfate (DSS)- and 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis and showed a slower recovery and decreased survival, while LXRs agonists can inhibit the expression of inflammatory cytokines, i.e. TNFα, and the recruitment of CD11b+ immune cell populations, and reduce the infiltration of pro-inflammatory Th17 cells in the colon epithelium. The activation of LXRs can also induce the differentiation of regulatory T cells, thereby reducing the severity of colitis (35). It has been found that the LXR level in the colon of IBD patients is significantly repressed, indicating the involvement of LXR in the development of IBD (17). 25-HC acts as a natural agonist of LXR, showing potential therapeutic effects for IBD.
GPR183, also known as Epstein-Barr virus-induced G-protein-coupled receptor 2 (EBI2), is activated by its ligand 7α,25-dihydroxycholesterol (7α,25-HC). CH25H and Cytochrome P450 family 7 subfamily B member 1 (CYP7B1) are critical enzymes required for the generation of 7α,25-HC, which binds to GPR183 to exert its immune regulatory function by inducing the localization of immune cells in the spleen and lymph nodes. GPR183 is expressed in B cells, T cells, DCs, macrophages, natural killer (NK) cells, neutrophils, and plays an important role in immune system (10, 36).
The concentration of 7α,25-HC is a key factor inducing the migration of GPR183+ cells. Immune cells such as B cells, T cells, DCs, etc., migrate to lymphoid tissues and lymphoid organs according to the concentration gradient of 7α,25-HC, thus enhancing the efficacy of adaptive immune responses (37). Dietary cholesterol absorption and microbiome recognition can result in the production of 7α,25-HC in duodenal intestinal epithelial cells. The elevated levels of 7α,25-HC can influence the migration and positioning of intestinal plasma cells by binding to GPR183. Additionally, 7α,25-HC can reduce the expression of CD98 in these cells and impair their ability to secrete IgA (38). Innate lymphoid cells (ILCs) also express GPR183 and migrate to the solitary intestinal lymphoid tissue in colonic and small intestinal tissues according to the concentration gradient of 7α,25-HC. Among the different types of ILCs, ILC3s are dominant and express higher levels of GPR183. They are characterized by the expression of RORγt and can produce IL-22, playing an protective role in intestinal barrier integrity and immunity (37). Liu et al. (10) found that under the LPS stimulation, CH25H and CYP7B1 were highly up-regulated in spleen DCs, macrophages, and especially NK cells and neutrophils, indicating that these cells are likely to be the source of 7α,25-HC production. Additionally, since these cells also express GPR183, they may also be regulated by 7α,25-HC. Chen et al. (39) have recently discovered that lymphatic stromal cells are the primary source of 7α,25-HC within lymphoid organs, thus facilitating the migration of B cells and DCs, and stimulating the effective adaptive immunity and antibody production. Similarly, fibroblast stromal cells within the colonic lymphoid tissue can produce 7α,25-HC, which induces the migration of ILC3 and helps maintain colonic immune homeostasis (40) (Figure 2).
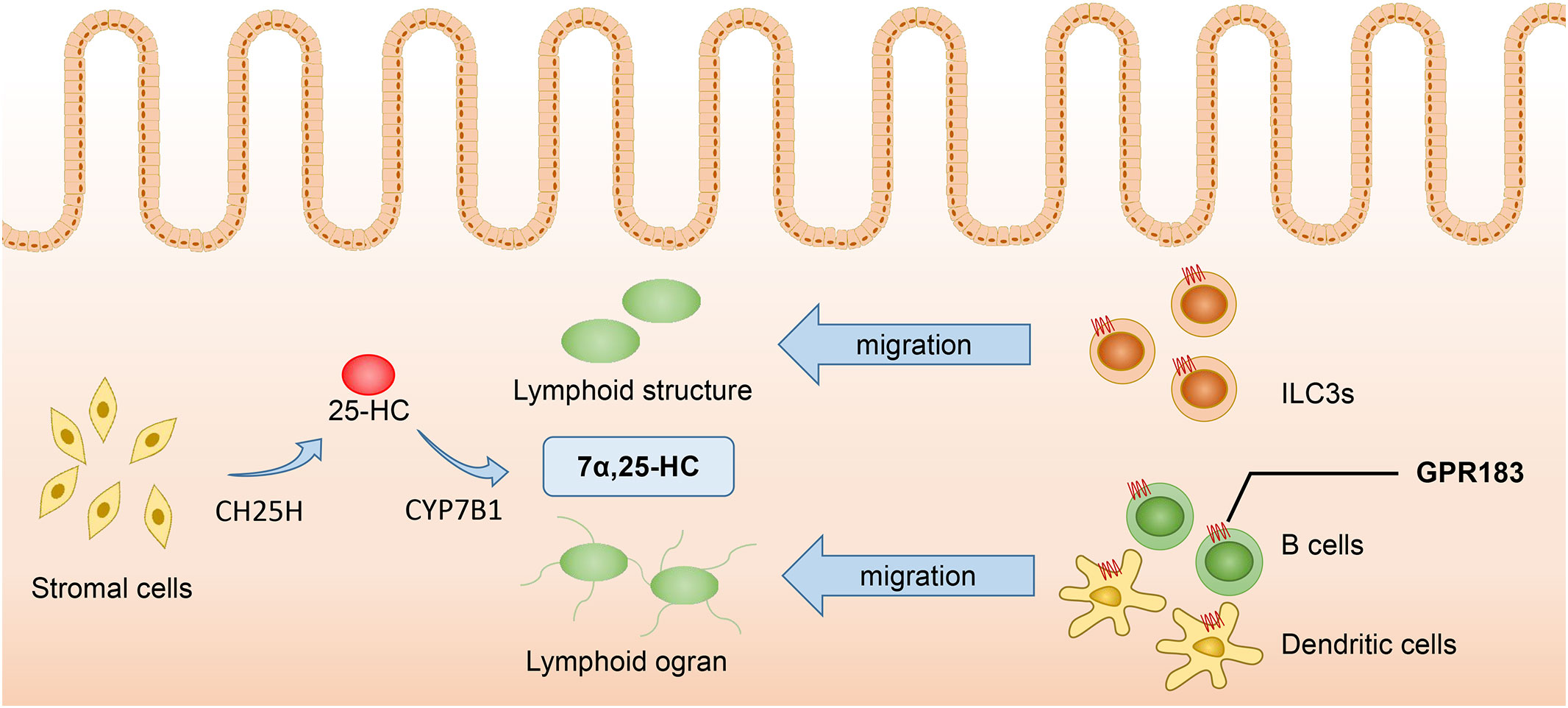
Figure 2 7α,25-HC inducing the migration of GPR183+ cells. Stromal cells serve as the primary source of 7α,25-HC, which is synthesized from cholesterol through hydroxylation by CH25H and CYP7B1 enzymes. Immune cells such as ILC3s, B cells, and DCs, migrate to colonic lymphoid tissues and lymphoid organs in response to the concentration gradient of 7α,25-HC. Proper localization of immune cells enhances the effectiveness of adaptive immune responses. 7α,25-HC, 7α,25-dihydroxycholesterol; GPR183, G protein-coupled receptor 183; CH25H, Cholesterol-25-hydroxylase; CYP7B1, Cytochrome P450 family 7 subfamily B member 1; ILC3s, Type 3 innate lymphoid cells.
Despite its importance in regulating intestinal immune homeostasis, the overexpression of GPR183 is considered a pathological mechanism in colitis. Emgard et al. (40) found that GPR183 facilitates the formation of cryptopatches and isolated lymphoid follicles in the colon by ILC3s, both in steady-state conditions and during inflammation. The increase levels of GPR183 ligand 7α,25-HC produced by CH25H and CYP7B1 can exacerbate the inflammatory response through the GPR183-dependent activation of ILC migration, the recruitment of myeloid cells, and tissue remodeling, and knocking out of GPR183 was able to alleviate the severity of CD40-Ab-induced colitis. Wyss et al. (41) further investigated the function of GPR183 and found that GPR183 also plays a role in promoting lymphoid tissue formation in IL-10-/- colitis. However, in the DSS colitis model, GPR183 knockout resulted in a reduction of colonic lymphoid structures but did not alleviate the severity of acute or chronic inflammation induced by DSS. Therefore, it is speculated that the destruction of the epithelial barrier and innate immune response are the main factors involved in the pathogenesis of the DSS colitis model, while GPR183 plays a minor role. On the other hand, in ILCs-related colitis models such as CD40 colitis, IL-10 colitis, T-bet (-/-) Rag2 (-/-) ulcerative colitis (TRUC), GPR183 is one of the pathogenic mechanisms.
The 7α,25-HC-GPR183 pathway is critical for the formation of colonic lymphoid structures, but it is dispensable in small intestine, as other factors can compensate for its absence (40). Although GPR183 primarily influences ILCs-related colitis models, it is considered a potential therapeutic target for the treatment of IBD.
RORs belong to the nuclear receptor family of transcription factors binding oxysterols, which includes three different isoforms: RORα, RORβ, and RORγ. RORγ also has variants, including RORγ1 and RORγ2 (also known as RORγt) (42). RORγt participates in the differentiation process of IL-17-producing cells such as Th17 cells and γδT cells, and plays an important role in various pathological conditions. Oxysterols such as 20α-HC, 22R-HC, 25-HC, and 27-HC can act as the agonists of RORγ nuclear receptors, especially the RORγt isoforms, thereby affecting the function of lymphocytes (43). Soroosh et al. (44) found that 7β,27-HC and 7α,27-HC can promote the differentiation of Th17 cells through RORγt, confirming that oxysterols can directly regulate lymphocyte differentiation through RORγt. Therefore, 25-HC, a ROR receptor agonist, also appears to be involved in the differentiation of IL17-producing lymphocytes (20, 45). In contrast, Cui et al. (33) found that the activation of LXR can lead to a decrease in the level of RORγt expressions, inhibiting the differentiation of Th17 cells (Figure 1C). Indicating the dual role of 25-HC in immune system.
The generation and function of ILC3s depend on the expression of RORγt and are involved in the pathogenesis of IBD. The activated DCs produce IL-6, IL-23 and TNF-α, which promote the movement of ILC3s into or out of cryptopatches, starting an inflammatory immune cascade and causing intestinal inflammation (46, 47). 25-HC can inhibit the production of pro-inflammatory cytokines in macrophages and DCs, as well as the expression of RORγt in ILC3, thereby potentially impacting the occurrence and development of IBD.
SREBPs regulate genes that are involved in lipogenic processes, such as fatty acid and cholesterol synthesis. There are three isoforms of SREBPs, of which SREBP1a and SREBP1c are mainly involved in genes that relate to fatty acid synthesis, while SREBP2 regulates cholesterol synthesis (20). The overexpression of CH25H can suppress cholesterol biosynthesis through SREBP. In the absence of cholesterol, SREBPs bind to the SREBP cleavage-activating protein (SCAP) in the endoplasmic reticulum to form SREBP/SCAP complexes. These complexes are then transported to the Golgi apparatus and cleaved into mature transcription factor forms by site 1 protease (S1P) and site 2 protease (S2P), which promotes the expression of the rate-limiting enzyme, HMG-CoA reductase, and increases cholesterol biosynthesis. In the presence of excessive cholesterol, 25-HC binds to insulin-induced gene 2 (INSIG2), which is an endoplasmic reticulum anchor protein. This binding promotes the formation of SREBP/INSIG2/SCAP complexes that prevent SREBP from transporting to the Golgi apparatus, reducing intracellular cholesterol production (Figure 3A) (3, 20).
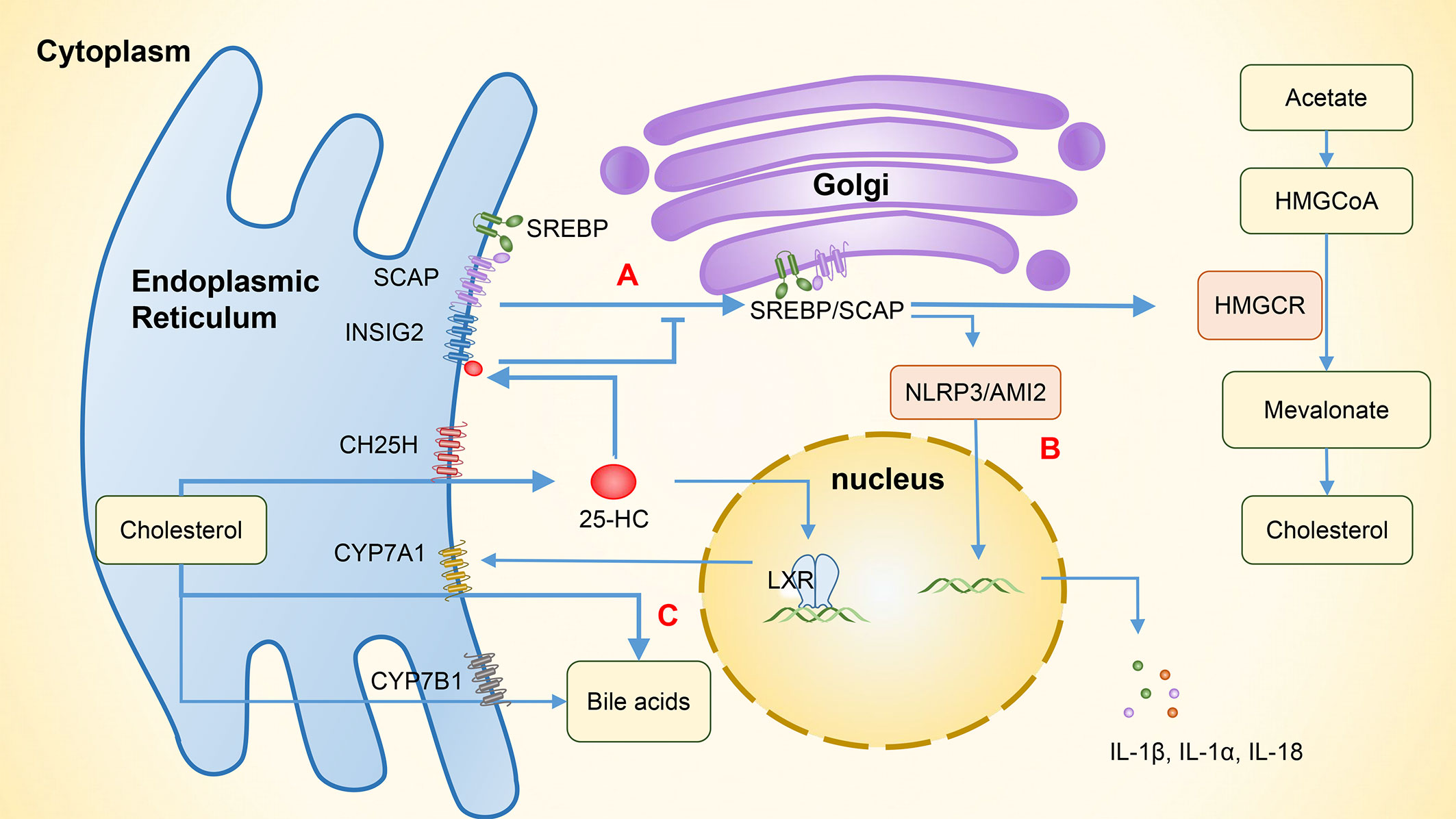
Figure 3 The involvement of 25-HC in the regulation of lipid metabolism and related immunity. (A) The binding of 25-HC with INSIG2 protein forms the SREBP/INSIG2/SCAP complex, which inhibits the transportation of SREBP to the Golgi apparatus. As a result, the expression of HMG-CoA reductase is suppressed and intracellular cholesterol production is reduced. (B) By inhibiting SREBP, the cholesterol synthesis is reduced, thereby inhibiting the activation of NLRP3 and AIM2 inflammasomes and subsequently suppressing the expression of IL-1β, IL-1α, and IL-18. (C) 25-HC increases the expression of CYP7A1 by activating LXR and enhances the synthesis of bile acids through the classical pathway. In addition to its role in hydroxylating 25-HC to 7α,25-HC, CYP7B1 also functions as an enzyme involved in the alternative bile acid pathway. 25-HC, 25-hydroxycholesterol; 7α,25-HC, 7α,25-dihydroxycholesterol; INSIG2, Insulin-induced gene 2, SREBP, Sterol regulatory-element binding protein; SCAP, SREBP cleavage-activating protein; HMG-CoA, 3-hydroxy-3-methyl glutaryl coenzyme A reductase; HMGCR, HMG-CoA reductase; NLRP3, NOD-like receptor protein 3; AIM2, Absent in melanoma 2; LXR, Liver X receptor; CYP7A1, Cytochrome P450 family 7 subfamily A member 1; CYP7B1, Cytochrome P450 family 7 subfamily B member 1.
25-HC can inhibit the activation of NOD-like receptor protein 3 (NLRP3) inflammasomes and the expression of cytokines IL-1β, IL-1α, and IL-18 by suppressing SREBP translocation (28). Dang et al. (48) have shown that the cholesterol overload in macrophages can trigger the release of mitochondrial DNA and activate the absent in melanoma 2 (AIM2) inflammasomes, leading to an increase in IL-1β production. However, 25-HC can maintain the integrity of mitochondria by inhibiting SREBP2-mediated cholesterol synthesis, which in turn prevents the activation of AIM2 inflammasomes and the IL-1β-induced inflammation (Figure 3B). LPS activates macrophage TLR4 to induce the synthesis and secretion of 25-HC. This can inhibit the activity of the transcription factor SREBP2 in intestinal germinal center B cells, suppressing their differentiation into IgA plasma cells and reducing the production of IgA, and consequently impairing antigen-specific IgA responses during intestinal infections (12, 49). SREBPs also play a significant role in Th17 cell differentiation. Cholesterol synthesis induced by SREBPs increases intracellular sterol that serves as RORγ receptor agonists. By inhibiting SREBPs and reducing cholesterol metabolism, 25-HC can indirectly inhibit Th17 cell differentiation (45, 50). In addition, 25-HC can activate SREBP1 through LXRs, which can bind to the IL-17 promoter and interact with the positive regulator of Th17 differentiation, leading to the inhibition of Th17 cell differentiation and transcriptional antagonism of IL-17 (20, 33). As 25-HC has a weak but specific RORγ receptor activity, it can influence Th17 cell differentiation through various pathways (45).
When ER cholesterol is more than 5 moles% of total ER lipids, INSIG binds to SCAP/SREBP complex, preventing the complex from transporting to the Golgi and reducing the cholesterol synthesis and uptake. However, ER only contains about 1% of the total cellular cholesterol and no more than 5% of total membrane lipids. Similarly, 25-HC accounts for a small portion of the total cellular sterol; it can still regulate cellular lipid metabolism quickly and accurately (51). Das et al. (51) proposed that cholesterol in plasma membrane can be divided into three pools: accessible pool, sphingomyelin (SM)-sequestered pool, and essential pool. Of these, the accessible pool is labile, serving as a primary site for the excessive cholesterol absorption. Subsequently, this excess cholesterol is transported to the pool in endoplasmic reticulum, thereby regulating cholesterol metabolism. Abrams et al. (52) found that 25-HC produced by IFN-γ-stimulated macrophages can increase the activity of acyl-CoA: cholesterol acyltransferase (ACAT), which can convert cholesterol in the ER to cholesteryl esters and induce the formation of cholesterol-rich lipid droplets. This decreases the level of the free cholesterol in the ER, triggering the internalization of accessible cholesterol from the plasma membrane and reducing the cholesterol synthesis and uptake. In addition, 25-HC can result in a long-term suppression of accessible cholesterol through inhibiting SREBP2. The cell surface accessible cholesterol is crucial for bacteria to penetrate adjacent cells, and the antibacterial activity of 25-HC is based on the decrease in accessible cholesterol on the plasma membrane (52). Ormsby et al. (53) conducted further research and found that by reducing accessible cholesterol on the plasma membrane through ACAT, 25-HC can limit pore formation and cytolysis caused by pore-forming toxins, thus protecting tissues from pathogenic bacteria.
CH25H exerts its antivirus function through inhibiting cholesterol biosynthesis, interacting with viral components, and modulating inflammation and immunity (3). Gastrointestinal symptoms are also observed in patients with SARS-CoV-2 when the virus infects intestinal epithelial cells. This is because differentiated enterocytes express high levels of the SARS-CoV-2 receptor, angiotensin-converting enzyme 2 (ACE2) (54). Studies have shown that 25-HC engages in anti-SARS-CoV-2 activities. Wang et al. (55) proposed that 25-HC inhibits the fusion of the viral envelope with the plasma membrane by activating ACAT, which can reduce the accessible cholesterol in the plasma membrane. In a similar vein, the research conducted by Zang et al. (56) demonstrated that 25-HC is capable of reducing the fusion of viral membrane by inhibiting Niemann-Pick C1 (NPC1), which is responsible for the reduction of accessible cholesterol in the membrane of the endosomal/lysosomal compartment. However, SARS-CoV-2 infection can also increase the level of 7α,25-HC in lungs, attracting monocytes/macrophages through a GPR183-dependent mechanism and subsequently leading to inflammation. Inhibiting GPR183 can reduce SARS-CoV-2 loads, macrophage infiltration, and inflammatory cytokine expression (57).
Bile acids are synthesized in the liver from cholesterol. Evidence shows that the altered composition of primary bile acids and secondary bile acids may contribute to the development of IBD through differential effects on epithelial and immune cells (58). Metabolomics studies have found that primary bile acids (PBAs) are increased while secondary bile acids (SBAs) are decreased in patients with IBD (58, 59). Dong et al. (60) have shown that 25-HC can increase bile acid synthesis through the activation of LXRs, which is regulated by the classic pathway of CYP7A1, and secondary bile acids also increase due to the involvement of gut bacteria. In addition, CH25H can regulate innate immune responses via the SREBP regulation and alleviate the liver inflammation caused by a high-fat diet, indicating that CH25H has a regulatory role in bile acid metabolism and anti-inflammation (Figure 3C). In addition to hydroxylating 25-HC to 7α,25-HC, CYP7B1 is also an enzyme involved in the alternative bile acid pathway (61). In humans, the major SBAs are lithocholic acid (LCA) and deoxycholic acid (DCA). Studies (62, 63) have found that gut bacteria and corresponding enzymes can convert the LCA into 3-oxoLCA and isoLCA, which have different effects on the differentiation of immune cells. Specifically, 3-oxoLCA can inhibit the differentiation of Th17 cells via RORγt, while isoLCA promotes the differentiation of Treg cells by inducing the production of mitochondrial reactive oxygen species (mitoROS) and subsequently increasing the FoxP3 expression. These effects help reduce the inflammation in intestine, and both bile acid metabolites were found to be significantly reduced in patients with IBD. However, previous studies have shown that although CH25H can regulate the cholesterol metabolism in a tissue-specific fashion, it contributes little to the overall bile acid synthesis (64). Further research is needed to examine whether CH25H can help regulate the inflammatory responses in IBD patients by affecting the bile acid pathway.
The research above indicates that 25-HC can inhibit the production of inflammatory factors through LXR or SREBP. In addition, 25-HC can also interact with myeloid differentiation protein 2 (MD2) to prevent LPS from binding to TLR4, thereby inhibiting the transcription of NF-κB and AP-1 and leading to the inhibition of inflammatory responses (65).
Regarding the pro-inflammatory effect of 25-HC, researchers (66, 67) have found that 25-HC can induce the secretion of pro-inflammatory cytokines and chemokines in macrophage cells, such as IL-1β and IL-6. When cocultured with smooth muscle cells and monocytic cells, 25-HC can induce monocytes to produce IL-1, which in turn synergistically increases the levels of IL-6 and MCP-1 expressions produced by smooth muscle cells (67, 68). Bai et al. (68) discovered that 25-HC can also enhance the IL-8 promoter activity caused by IL-1β in Caco-2 cells. Further research (69) has shown that 25-HC can bind to the lipid raft domains of the plasma membrane and promote Ca2+ influx, leading to the activation of calcium-dependent kinase PYK2, and consequently activating the MAPK ERK1/2 pathway and AP-1 transcription, which promoted the transcription of IL-8. Another study concluded that 25-HC promotes NLRP3 inflammasome assembly and its activation through potassium efflux, mitoROS and LXR-mediated pathways (70). Moreover, 25-HC can activate the FAK signaling pathway by binding to α5β1/αvβ3 integrins directly, leading to an increase in the production of pro-inflammatory cytokines, such as TNF and IL-6 (71).
Scholars (72, 73) have found that 25-HC can enhance the immune response mediated by TLR3 in airway epithelial cells, stimulate the release of IL-8 and IL-6 via the NF-κB pathway, and participate in neutrophilic airway inflammation. Friedrich et al. (74) have shown that for patients with IBD who did not respond to anti-TNF therapy or corticosteroids, their inflamed tissues were characterized by neutrophil infiltrates, fibroblast activation, and loss of epithelial cells. In addition, the high amount of IL-1β was expressed in colon ulcers, but not in NR3C1, ITGA4, or TNF. Fibroblast IL-1R signaling drives the recruitment of neutrophils, which then induces inflammation, indicating that targeting the neutrophil-attractant program in fibroblasts by blocking IL-1R could be used as an alternative treatment for patients with refractory IBD. CH25H can inhibit the secretion of IL-1β by macrophages and may have a potential role in regulating the migration and function of neutrophils, which are involved in the pathogenesis of IBD. However, there is a lack of relevant research on the mechanism by which CH25H regulates neutrophils.
CH25H can enhance the progression and metastasis of colorectal cancer (CRC). A reduced level of CH25H was observed in the cancer stroma, particularly in the intratumoral endothelial cells (ECs), among CRC patients (75). It is worth noting that intercellular biomolecule transfer (ICBT), facilitated by tumor-derived extracellular vesicles (TEVs), is also a crucial factor that influences the progression and prognosis of CRC. CH25H is capable of inhibiting of the fusion lipid membrane, thereby hindering the uptake of TEVs. As a result, CH25H restricts the ICBT-induced angiopoietin-2 (ANGPT2)-dependent activation of ECs, and inhibits intratumoral angiogenesis. Furthermore, it has also been observed that the administration of reserpine as a treatment can enhance the expression of CH25H in TEV-treated cells. This subsequently leads to a reduction in the ICBT between malignant and benign cells, ultimately resulting in the suppression of angiogenesis (75). Additionally, a study has revealed that LINC01915, a type of long non-coding RNA, inhibited the uptake of TEVs by normal fibroblasts (NFs), cancer-associated fibroblasts (CAFs) activation, and tumor angiogenesis through the miR-92a-3p/KLF4/CH25H axis, thereby impeding tumor growth. These findings indicate that CH25H has an inhibitory effect on CRC (76).
It is accepted that 25-HC as a soluble factor can function both as an autocrine and paracrine agent. Specifically, Doms et al. (77) employed transient transfection to conduct an exogenous expression of CH25H in HeLa cells using a plasmid. They observed that the exogenous expression of CH25H reduced the proportion of cells infected with reovirus, particularly in the cells expressing CH25H. Moreover, they found that reovirus infection decreased in cells that do not express CH25H, indicating that 25-HC may be secreted from cells to limit infection in CH25H non-expressing cells. On the other hand, Canfrán-Duque et al. (78) have found that in atherosclerosis, activated macrophages release 25-HC which acts on SMC through paracrine action. This interaction blunts SMC migration by altering platelet-derived growth factor (PDGF) signaling and ultimately promotes plaque instability. Thus, the potential role of extracellular 25-HC in intestinal immunity needs to be further explored.
CH25H may have either pro-inflammatory or anti-inflammatory effects, depending on the experimental protocols used, such as the concentration of 25-HC or treatment time. For instance, the high concentrations of 25-HC were used in the study that discovered its pro-inflammatory effect, while relatively low concentrations were used in the study that found its anti-inflammatory effect (28, 70).
Although the pathogenesis of IBD is still unclear, it is mainly associated with genetic susceptibility, intestinal microbiota, environmental factors, and immunological dysregulation characterized by abnormal infiltration of T cells, B cells, macrophages, DCs, and neutrophils, which produce high levels of proinflammatory cytokines such as TNF, IL-1β, IFN-γ, and cytokines of the IL-23/Th17 pathway (15). As an oxysterol produced through oxidation by CH25H, 25-HC has various effects on immune cell regulation, but its specific role in IBD has not been fully ascertained.
Macrophages are among the most abundant types of leukocytes, found in the intestines of all mammalian species. Macrophages play a crucial role in maintaining local homeostasis and the balance of commensal microbiota. Moreover, they are significant factors in the development of IBD (79, 80). Resident macrophages in the lamina propria of the intestine have the ability to capture and degrade bacteria in a non-inflammatory manner. This helps prevent commensal bacteria from crossing the intestinal epithelial barrier. Additionally, macrophages can maintain intestinal homeostasis through the production of anti-inflammatory cytokines like IL-10 and TGF-β, debris scavenging, angiogenesis, and wound repair (79–81). Intestinal macrophages are also capable of inducing T cells to become anergic or differentiate into Tregs to promote immune tolerance, and mediate Th1, Th2, and Th17 to help participate in the adaptive immune response (80).
In colitis models, monocytes and immature macrophages can migrate to intestinal mucosa through the CCL2 or MCP-1 mediated recruitment and produce large quantities of inflammatory mediators such as IL-1, IL-6, and TNFα, as well as inflammatory chemokines like CCL2 and CCL3 to coordinate the recruitment of other innate and adaptive immune cells, such as neutrophils, Th1, and Th17 cells (82). In IL-10-/- mice with spontaneous chronic colitis, macrophages differentiate into pro-inflammatory subsets that produce large amounts of IL-12 and IL-23 in response to bacterial stimulation, inducing Th1 cell polarization (81). Similar findings have been observed in patients with IBD. The inflamed mucosa of patients with IBD shows an increase in the number of macrophages, and several phenotypic and functional characteristics of these macrophages differ from those under physiological conditions. For example, these macrophages display the expression of T cell costimulatory molecules like CD40, CD80, and CD86 (83), as well as pathogen-associated molecular pattern (PAMP) receptors like TLR2, TLR4, CD89, TREM1, and CD14 (84, 85). The abnormal expression of CD14 in macrophages enhances their pro-inflammatory activities induced by LPS, ultimately leading to the secretion of the significant amounts of IL-23 and TNF-α, which further promotes the release of IFN-γ by lamina propria mononuclear cells. IFN-γ then drives macrophage differentiation toward an IL-23-hyperproducing phenotype and forms a positive feedback, thereby playing a pivotal role in the pathogenesis of IBD (84, 86).
ILCs are an important component of intestinal organs and contribute to antibacterial defense, immune regulation, maintenance of barrier function, and intestinal homeostasis (87). There are three subtypes of ILCs: type 1 ILCs (including NK cells and ILC1), type 2 ILCs (ILC2), and type 3 ILCs (including ILC3 and lymphoid tissue-inducing cells (LTis)) (47). Unlike intestinal B, T, and NK cells, the intestinal population of ILCs does not continuously replenish from circulation (88). They are maintained by self-renewal in physiological conditions and continuously produce cytokines and other soluble factors that can have a direct impact on the function of epithelial cells at steady state, such as ILC1 producing IFNγ, ILC2 producing IL-5 and IL-13, and ILC3 producing IL-22 and IL-17 (47, 87). Type 1 ILCs are predominantly located in upper gastrointestinal tract, type 2 ILCs are distributed over the intestine in relatively small proportions, and type 3 ILCs are mainly found in ileum and colon (89). Any imbalance in ILC subtypes can result in the disruption of intestinal homeostasis and lead to intestinal inflammation.
In patients with CD, the number of IL-17/IL-22-producing type 3 ILCs is reduced in inflamed intestinal tissues, while there is an accumulation of IFNγ-producing type 1 ILCs. This could be ascribed to a decrease in the expression of RORγt which is a marker for type 3 ILCs, followed by the expression of T-bet, NK1.1, and NKp46, resulting in the acquisition of type 1 ILC phenotype (46). Type 1 ILCs produce a significant amount of IFN-γ, which induces the migration of neutrophils and activates lymphocytes, macrophages, endothelial cells, and affects the tight junction function, resulting in the damage to the epithelial barrier, thereby exacerbating the induction and progression of inflammation (46). RORγt type 3 ILCs also involve in the pathogenesis of IBD. When stimulated by TNF-α, IL-23, and IL-6, RORγt type 3 ILCs enter and exit crypts, which may initiate inflammatory immune cascades that lead to intestinal inflammation (46, 47). The increase of IFN-γ–producing ILC1s and IL-17–producing ILC3s, and the decrease of IL-22–producing ILC3s, are associated with the level of inflammation in patients with IBD.
CD4+ Th cells play a crucial role in adaptive immune response. Naive CD4+ T cells differentiate into various types of Th cells in response to different cytokines. Under the stimulation of IL-12 or IL-27, they differentiate into Th1 cells which primarily secrete IFN-γ. In contrast, under the stimulation of IL-4, they differentiate into Th2 cells which produce a range of interleukins, including IL-4, IL-5, IL-13, and IL-25. When exposed to both IL-4 and TGF-β, they differentiate into Th9 cells, which secrete IL-9, IL-10, and IL-21. Conversely, with the stimulation from IL-1, IL-6, IL-23, and TGF-β, they differentiate into Th17 cells that secrete IL-17A, IL-17F, IL-21, and IL-22. Finally, with the induction of IL-2 and TGF-β, they develop into Treg cells that maintain immune tolerance and regulate the homeostasis, activation, and function of lymphocytes (90, 91). Studies have shown that Th1 cells are primarily involved in the development of CD, while Th2 cells are associated with UC (92, 93). Additionally, the pathogenesis of UC involves the participation of Th9 cells (94), and both Th17 and Treg cells contribute to the pathogenesis of UC and CD (95). In IBD, the chronic inflammatory environment and the local hypoxic condition result in the upregulation of hypoxia-inducible-factor-1-alpha (HIF-1α). This upregulation can impair Th17 regulatory responses to AHR ligation by increasing ABC transporter levels, thus promoting the pro-inflammatory phenotype in T cells (96).
DCs are regarded as the most potent professional antigen-presenting cells within human body. They possess the ability to internalize, process, and present antigens to T cells while also orchestrating innate and adaptive immune responses. The increased expression of chemokines and adhesion molecules observed in the intestinal mucosa of patients with IBD results in an accumulation of DCs in inflammatory sites (97). In patients with CD, the failure to control IL-12 secretion by the activated DCs will lead to undesirable Th1 inflammatory responses (98). In mouse colitis, DCs within the colonic lamina propria express higher levels of costimulatory molecules (CD40, CD80, and CD86) and generate increased amounts of IL-12p40 and IL-23p19, which ultimately combine to form IL-23, thereby promoting Th17 differentiation (99). Additionally, the intestinal tissues of patients with CD exhibit a decreased abundance of CD11c DCs, resulting in an enhanced ability to generate Th1/Th2/Th17 responses (100).
Neutrophils are the first line of defense in the innate immunity of the intestinal mucosa. They are the most abundant immune cells and can be quickly recruited to sites of infection or inflammation (100). Kuhl et al. (101) discovered that blocking neutrophil adhesion and migration or neutrophil depletion could exacerbate TNBS/DNBS-induced colitis in mice. This indicates that neutrophils are an important factor in mediating wound healing in IBD. Further research (102, 103) has revealed that neutrophils can maintain mucosal barrier function and immune response in the gut by depleting local oxygen and stabilizing the transcription factor HIF, or by altering nucleotide signaling to promote mucosal inflammatory resolution and epithelial restitution.
Neutrophils participate in the development of IBD. Neutrophils cause the damage to the epithelial barrier and inflammation by producing high levels of ROS. They also release proteases, proinflammatory cytokines, and mediators such as IL-8, TNF-α, and leukotriene B4, which further damages the epithelial barrier and recruit monocytes and additional neutrophils to the inflamed tissue (100, 104). Neutrophil extracellular traps (NETs) are released by neutrophils during the infection and inflammation as a protective response to inhibit foreign pathogens. However, they can also cause damage to the intestinal barrier and activate proinflammatory functions of neutrophils through the phosphorylation of Akt, ERK1/2, and p38 (104, 105). The level of NETs increases in the inflamed intestinal mucosa, blood, and stool of IBD patients, especially during the active stage of the disease (104). NETs can activate macrophages to release cytokines such as IL-1β, TNF-α, IL-6, induce the activation of platelets and intestinal epithelial cells, promoting colitis and thrombosis. This ultimately causes the damage to intestinal epithelial and vascular endothelial cells (106). Studies have also shown that butyrate, a microbial metabolite in the intestines, can improve intestinal inflammation by inhibiting neutrophil migration and NETs formation, as well as reducing pro-inflammatory mediator production (107).
Macrophages are the main source of 25-HC (9, 11). Through interactions with membrane receptors, nuclear receptors, and the regulation of lipid metabolism and other pathways, 25-HC can influence the differentiation and function of immune cells, including macrophages, T cells, B cells, DCs, and neutrophils. Additionally, 7α,25-HC can induce the migration of immune cells, such as ILCs, DCs, and B cells, as well as regulating gut immune homeostasis and inflammation. Therefore, CH25H is believed to be involved in the pathogenesis of IBD.
Except for being generated in cells and tissues through enzymatic or nonenzymatic reactions, oxysterols are also found in various foodstuffs, particularly in cholesterol-rich foods. The most commonly represented oxysterols in cholesterol-rich foods are 7-oxygenated sterols and 5,6-oxygenated sterols, while 25-HC is present in smaller amounts, and both dietary and endogenous oxysterols have potentially proapoptotic, pro-oxidant and cytotoxic effects, leading to the loss of epithelial colonic cells and the impairment of the intestinal barrier function (108–110).
Chalubinski et al. (111) noted that although 25-HC can cause slight damage to the integrity of Caco2 cell monolayers, it does not have a significant effect on cell viability or apoptosis. This suggests that 25-HC is not the primary factor disrupting epithelial barrier function. Guillemot et al. (112) discovered that in both the DSS or TNBS-induced mouse colitis model and patients with IBD, there is a disturbance in the enzymatic metabolism of oxysterols. CH25H levels increased in mouse colon tissue, plasma, liver, and human colon tissue, as did the 7α,25-HC. This indicates that the increase in CH25H may serve as a form of internal homeostatic regulation to alleviate inflammatory reaction. One of the main characteristics of IBD is the destruction of the intestinal barrier. The damaged intestinal barrier allows unrestricted entry of microbiota into both lamina propria and bloodstream (113). A study conducted by Sheng et al. (114) revealed that intestinal tight junction protein expression is reduced in CH25H-/- mice, which leads to the disruption of the intestinal epithelial barrier function and increases susceptibility to and severity of DSS-induced colitis, while supplementing exogenous 25-HC can alleviate colitis in mice and improve the integrity of the intestinal barrier. This suggests that CH25H may have a protective role in colitis and is associated with intestinal epithelial regeneration and tissue reconstruction. IBD is usually accompanied by dysbiosis, and persistent dysbiosis can worsen inflammation. Conversely, chronic inflammation contributes to dysbiosis by altering the oxidative and metabolic environment of the intestine (115). The administration of medications, such as anti-TNFα antibody therapy, for the treatment of IBD has been found to impact the composition of intestinal microbiota (116). While CH25H has been shown to improve the intestinal inflammation and the integrity of the intestinal barrier, its impact on the composition of intestinal microbiota remains unknown.
The SIRT1-CH25H pathway is one of the mechanisms that contribute to the metformin-induced alleviation of hepatic inflammation (32), and the nuclear receptor SIRT1 has been shown to play a role in the pathogenesis of IBD. Caruso et al. (117) demonstrated that SIRT1 is downregulated in the inflamed tissue of patients with IBD and colitis models by TNF-α and IL-21. T cells and macrophages deficient in SIRT1 are hyperactivated and produce significant amounts of inflammatory cytokines. Conversely, the activation of SIRT1 can reduce the acetylation of NF-κBp65 and subsequently decrease NF-kB activation, thereby decreasing the levels of inflammatory cytokines such as IFN-γ, IL-17A, and IL-21. However, SIRT1 also has deleterious effects on IBD. The Inhibition of SIRT1 may reduce the severity of colitis by promoting the production of Foxp3+T-regulatory cells, as well as paneth and goblet cells, which play crucial roles in maintaining gastrointestinal homeostasis (118). Therefore, SIRT1 imbalances in the immune system can result in the development and progression of IBD, and it is speculated that SIRT1 decreases the production of inflammatory cytokines by macrophage and Th1/Th17 cells through the activation of CH25H, and enhance the LXR/ABCA1 pathway to exert anti-inflammatory effects. Since SIRT1 serves as the upstream regulator of CH25H, the role of SIRT1 activators and inhibitors may influence the effects of CH25H on IBD relevant to gastrointestinal immune homeostasis. SIRT1 is relevant to the impact of CH25H on IBD through immune cells, the detection of both SIRT1 and CH25H can assist in the early diagnosis and treatment of the disease.
However, studies have shown that the overexpression of CH25H in ILC-related colitis models can stimulate the pro-inflammatory ILC3 activity and aggravate inflammatory reactions (40, 41). Additionally, 25-HC, the product of CH25H can contribute to the development of intestinal fibrosis, resulting in complications such as intestinal stenosis (119). In other conditions, such as obesity and diabetes, CH25H is upregulated in adipose tissue while associating with insulin resistance and adipose tissue inflammation. This is because 25-HC can induce inflammatory gene expressions in macrophages and preadipocytes from patients with diabetes (120). These results suggest that the role of CH25H may be different or even opposite in different disease models, possibly related to the pathogenesis and microenvironment of the disease.
CH25H and its downstream products have been found to have various functions, such as immunomodulation and lipid metabolism. The downstream product of CH25H exerts its function by binding to multiple membrane receptors and nuclear receptors, such as LXR, GPR183 and ROR. This helps regulate the immune cell function in maintaining gastrointestinal homeostasis. Additionally, 25-HC can regulate the activity of SREBP, thereby inhibiting the biosynthesis of fatty acids and cholesterol and playing a role in lipid metabolism regulation. 25-HC can also regulate immune function via SREBP.
The immunological dysregulation caused by IBD is characterized by epithelial damage, the expansion of inflammation and the infiltration of various types of immune cells, including macrophages, ILCs, T cells, DCs and neutrophils. 25-HC can influence the differentiation and function of these immune cells, while 7α,25-HC can induce their migration. These effects of CH25H metabolite may be different or even opposite in different conditions. The dual role of CH25H may depend on the pathogenesis and microenvironment of IBD. This highlights the importance of maintaining CH25H balance in the immune system for the treatment of IBD.
The pathogenesis of IBD remains unclear, and the high expression of CH25H in patients with IBD makes it a potential target for future diagnosis and treatment. While the regulation of CH25H and its downstream products have been extensively investigated, its role and function in the immune system and related diseases are not fully understood. Although it has been confirmed that various immune cells play a role in the development of IBD, research on discussing the relationship between CH25H and IBD is still insufficient. Further investigation on the impact of CH25H on IBD through immune cells is essential for the early diagnosis and treatment of IBD.
GZ wrote the first draft of the manuscript. CH, SW, CL contributed to writing-review and editing. ML supervised the study. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant number 82204480 and 82070565) and Guangdong Basic and Applied Basic Research Foundation (grant number 2021A1515110714).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jairath V, Feagan BG. Global burden of inflammatory bowel disease. Lancet Gastroenterol Hepatol (2020) 5(1):2–3. doi: 10.1016/s2468-1253(19)30358-9
2. Liu Z, Liu R, Gao H, Jung S, Gao X, Sun R, et al. Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat Genet (2023) 55(5):796–806. doi: 10.1038/s41588-023-01384-0
3. Zhao J, Chen J, Li M, Chen M, Sun C. Multifaceted functions of CH25H and 25HC to modulate the lipid metabolism, immune responses, and broadly antiviral activities. Viruses (2020) 12(7):727. doi: 10.3390/v12070727
4. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol (2017) 13(12):710–30. doi: 10.1038/nrendo.2017.91
5. Griffiths WJ, Wang Y. Cholesterol metabolism: from lipidomics to immunology. J Lipid Res (2022) 63(2):100165. doi: 10.1016/j.jlr.2021.100165
6. Ke W, Fang L, Jing H, Tao R, Wang T, Li Y, et al. Cholesterol 25-hydroxylase inhibits porcine reproductive and respiratory syndrome virus replication through enzyme activity-dependent and -independent mechanisms. J Virol (2017) 91(19):e00827-17. doi: 10.1128/JVI.00827-17
7. de Freitas FA, Levy D, Reichert CO, Cunha-Neto E, Kalil J, Bydlowski SP. Effects of oxysterols on immune cells and related diseases. Cells (2022) 11(8):1251. doi: 10.3390/cells11081251
8. Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, et al. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res (2009) 50(11):2258–64. doi: 10.1194/jlr.M900107-JLR200
9. Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol (2010) 88(6):1081–7. doi: 10.1189/jlb.0610318
10. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature (2011) 475(7357):519–23. doi: 10.1038/nature10226
11. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity (2013) 38(1):106–18. doi: 10.1016/j.immuni.2012.11.004
12. Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A (2009) 106(39):16764–9. doi: 10.1073/pnas.0909142106
13. Traversari C, Russo V. Control of the immune system by oxysterols and cancer development. Curr Opin Pharmacol (2012) 12(6):729–35. doi: 10.1016/j.coph.2012.07.003
14. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med (2020) 383(27):2652–64. doi: 10.1056/NEJMra2002697
15. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res (2019) 2019:7247238. doi: 10.1155/2019/7247238
16. Candelli M, Franza L, Pignataro G, Ojetti V, Covino M, Piccioni A, et al. Interaction between lipopolysaccharide and gut microbiota in inflammatory bowel diseases. Int J Mol Sci (2021) 22(12):6242. doi: 10.3390/ijms22126242
17. Jakobsson T, Vedin LL, Hassan T, Venteclef N, Greco D, D'Amato M, et al. The oxysterol receptor LXRbeta protects against DSS- and TNBS-induced colitis in mice. Mucosal Immunol (2014) 7(6):1416–28. doi: 10.1038/mi.2014.31
18. Hong C, Kidani Y, A-Gonzalez N, Phung T, Ito A, Rong X, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest (2012) 122(1):337–47. doi: 10.1172/JCI58393
19. Kimura T, Nada S, Takegahara N, Okuno T, Nojima S, Kang S, et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat Commun (2016) 7:13130. doi: 10.1038/ncomms13130
20. Choi C, Finlay DK. Diverse immunoregulatory roles of oxysterols-the oxidized cholesterol metabolites. Metabolites (2020) 10(10):384. doi: 10.3390/metabo10100384
21. Liu Y, Wei Z, Ma X, Yang X, Chen Y, Sun L, et al. 25-Hydroxycholesterol activates the expression of cholesterol 25-hydroxylase in an LXR-dependent mechanism. J Lipid Res (2018) 59(3):439–51. doi: 10.1194/jlr.M080440
22. Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife (2015) 4:e08009. doi: 10.7554/eLife.08009
23. Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell (2004) 119(2):299–309. doi: 10.1016/j.cell.2004.09.032
24. Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci U S A (2004) 101(51):17813–8. doi: 10.1073/pnas.0407749101
25. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med (2003) 9(2):213–9. doi: 10.1038/nm820
26. Smoak K, Madenspacher J, Jeyaseelan S, Williams B, Dixon D, Poch KR, et al. Effects of liver X receptor agonist treatment on pulmonary inflammation and host defense. J Immunol (2008) 180(5):3305–12. doi: 10.4049/jimmunol.180.5.3305
27. Korf H, Vander Beken S, ROmano M, Steffensen KR, Stijlemans B, Gustafsson JA, et al. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest (2009) 119(6):1626–37. doi: 10.1172/JCI35288
28. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (2014) 345(6197):679–84. doi: 10.1126/science.1254790
29. Martins IJ. Appetite control and biotherapy in the management of autoimmune induced global chronic diseases. Clin Immunol Res (2018) 2(1):1–4. doi: 10.33425/2639-8494.1014
30. Martins IJ. Anti-aging genes improve appetite regulation and reverse cell senescence and apoptosis in global populations. Adv Aging Res (2016) 05(01):9–26. doi: 10.4236/aar.2016.51002
31. Martins IJ. Nutrition therapy regulates caffeine metabolism with relevance to NAFLD and induction of type 3 diabetes. Diabetes Metab Disord (2017) 4(1):1–9. doi: 10.24966/dmd-201x/100019
32. Guo WR, Liu J, Cheng LD, Liu ZY, Zheng XB, Liang H, et al. Metformin alleviates steatohepatitis in diet-induced obese mice in a SIRT1-dependent way. Front Pharmacol (2021) 12:704112. doi: 10.3389/fphar.2021.704112
33. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest (2011) 121(2):658–70. doi: 10.1172/JCI42974
34. Mufazalov IA, Schelmbauer C, Regen T, Kuschmann J, Wanke F, Gabriel LA, et al. IL-1 signaling is critical for expansion but not generation of autoreactive GM-CSF+ Th17 cells. EMBO J (2017) 36(1):102–15. doi: 10.15252/embj.201694615
35. Herold M, Breuer J, Hucke S, Knolle P, Schwab N, Wiendl H, et al. Liver X receptor activation promotes differentiation of regulatory T cells. PLoS One (2017) 12(9):e0184985. doi: 10.1371/journal.pone.0184985
36. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475(7357):524–7. doi: 10.1038/nature10280
37. Misselwitz B, Wyss A, Raselli T, Cerovic V, Sailer AW, Krupka N, et al. The oxysterol receptor GPR183 in inflammatory bowel diseases. Br J Pharmacol (2021) 178(16):3140–56. doi: 10.1111/bph.15311
38. Ceglia S, Berthelette A, Howley K, Li Y, Mortzfeld B, Bhattarai SK, et al. An epithelial cell-derived metabolite tunes immunoglobulin A secretion by gut-resident plasma cells. Nat Immunol (2023) 24(3):531–44. doi: 10.1038/s41590-022-01413-w
39. Chen H, Huang W, Li X. Structures of oxysterol sensor EBI2/GPR183, a key regulator of the immune response. Structure (2022) 30(7):1016–24.e5. doi: 10.1016/j.str.2022.04.006
40. Emgard J, Kammoun H, Garcia-Cassani B, Chesne J, Parigi SM, Jacob JM, et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity (2018) 48(1):120–32.e8. doi: 10.1016/j.immuni.2017.11.020
41. Wyss A, Raselli T, Perkins N, Ruiz F, Schmelczer G, Klinke G, et al. The EBI2-oxysterol axis promotes the development of intestinal lymphoid structures and colitis. Mucosal Immunol (2019) 12(3):733–45. doi: 10.1038/s41385-019-0140-x
42. Duc D, Vigne S, Pot C. Oxysterols in autoimmunity. Int J Mol Sci (2019) 20(18):4522. doi: 10.3390/ijms20184522
43. Jin L, Martynowski D, Zheng S, Wada T, Xie W, Li Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol Endocrinol (2010) 24(5):923–9. doi: 10.1210/me.2009-0507
44. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, et al. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci U S A (2014) 111(33):12163–8. doi: 10.1073/pnas.1322807111
45. Santori FR, Huang P, van de Pavert SA, Douglass EF Jr., Leaver DJ, Haubrich BA, et al. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell Metab (2015) 21(2):286–98. doi: 10.1016/j.cmet.2015.01.004
46. Wu Y, Shen J. Innate lymphoid cells in crohn's disease. Front Immunol (2020) 11:554880. doi: 10.3389/fimmu.2020.554880
47. Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. Int J Mol Sci (2021) 22(14):7618. doi: 10.3390/ijms22147618
48. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell (2017) 171(5):1057–71.e11. doi: 10.1016/j.cell.2017.09.029
49. Trindade BC, Ceglia S, Berthelette A, Raso F, Howley K, Muppidi JR, et al. The cholesterol metabolite 25-hydroxycholesterol restrains the transcriptional regulator SREBP2 and limits intestinal IgA plasma cell differentiation. Immunity (2021) 54(10):2273–87.e6. doi: 10.1016/j.immuni.2021.09.004
50. Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol (2015) 11(2):141–7. doi: 10.1038/nchembio.1714
51. Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife (2014) 3:e02882. doi: 10.7554/eLife.02882
52. Abrams ME, Johnson KA, Perelman SS, Zhang LS, Endapally S, Mar KB, et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat Microbiol (2020) 5(7):929–42. doi: 10.1038/s41564-020-0701-5
53. Ormsby TJR, Owens SE, Clement L, Mills TJ, Cronin JG, Bromfield JJ, et al. Oxysterols protect epithelial cells against pore-forming toxins. Front Immunol (2022) 13:815775. doi: 10.3389/fimmu.2022.815775
54. Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. SARS-CoV-2 productively infects human gut enterocytes. Science (2020) 369(6499):50–4. doi: 10.1126/science.abc1669
55. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, et al. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J (2020) 39(21):e106057. doi: 10.15252/embj.2020106057
56. Zang R, Case JB, Yutuc E, Ma X, Shen S, Gomez Castro MF, et al. Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc Natl Acad Sci (2020) 117(50):32105–13. doi: 10.1073/pnas.2012197117
57. Foo CX, Bartlett S, Chew KY, Ngo MD, Bielefeldt-Ohmann H, Arachchige BJ, et al. GPR183 antagonism reduces macrophage infiltration in influenza and SARS-CoV-2 infection. Eur Respir J (2023) 61(3):2201306. doi: 10.1183/13993003.01306-2022
58. Thomas JP, Modos D, Rushbrook SM, Powell N, Korcsmaros T. The emerging role of bile acids in the pathogenesis of inflammatory bowel disease. Front Immunol (2022) 13:829525. doi: 10.3389/fimmu.2022.829525
59. Di Vincenzo F, Puca P, Lopetuso LR, Petito V, Masi L, Bartocci B, et al. Bile acid-related regulation of mucosal inflammation and intestinal motility: from pathogenesis to therapeutic application in IBD and microscopic colitis. Nutrients (2022) 14(13):2664. doi: 10.3390/nu14132664
60. Dong Z, He F, Yan X, Xing Y, Lei Y, Gao J, et al. Hepatic reduction in cholesterol 25-hydroxylase aggravates diet-induced steatosis. Cell Mol Gastroenterol Hepatol (2022) 13(4):1161–79. doi: 10.1016/j.jcmgh.2021.12.018
61. Chiang JYL, Ferrell JM. Bile acid metabolism in liver pathobiology. Gene Expr (2018) 18(2):71–87. doi: 10.3727/105221618X15156018385515
62. Paik D, Yao L, Zhang Y, Bae S, D'Agostino GD, Zhang M, et al. Human gut bacteria produce TauEta17-modulating bile acid metabolites. Nature (2022) 603(7903):907–12. doi: 10.1038/s41586-022-04480-z
63. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
64. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
65. Ouyang W, Zhou H, Liu C, Wang S, Han Y, Xia J, et al. 25-Hydroxycholesterol protects against acute lung injury via targeting MD-2. J Cell Mol Med (2018) 22(11):5494–503. doi: 10.1111/jcmm.13820
66. Rosklint T, Ohlsson BG, Wiklund O, Noren K, Hulten LM. Oxysterols induce interleukin-1beta production in human macrophages. Eur J Clin Invest (2002) 32(1):35–42. doi: 10.1046/j.1365-2362.2002.00931.x
67. Fu H, Spieler F, Grossmann J, Riemann D, Larisch M, Hiebl B, et al. Interleukin-1 potently contributes to 25-hydroxycholesterol-induced synergistic cytokine production in smooth muscle cell-monocyte interactions. Atherosclerosis (2014) 237(2):443–52. doi: 10.1016/j.atherosclerosis.2014.10.002
68. Bai B, Yamamoto K, Sato H, Sugiura H, Tanaka T. Combined effect of 25-hydroxycholesterol and IL-1beta on IL-8 production in human colon carcinoma cell line (Caco-2). Inflammation (2005) 29(4-6):141–6. doi: 10.1007/s10753-006-9009-8
69. Lemaire-Ewing S, Berthier A, Royer MC, Logette E, Corcos L, Bouchot A, et al. 7beta-Hydroxycholesterol and 25-hydroxycholesterol-induced interleukin-8 secretion involves a calcium-dependent activation of c-fos via the ERK1/2 signaling pathway in THP-1 cells: oxysterols-induced IL-8 secretion is calcium-dependent. Cell Biol Toxicol (2009) 25(2):127–39. doi: 10.1007/s10565-008-9063-0
70. Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y, et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun (2016) 7:13129. doi: 10.1038/ncomms13129
71. Pokharel SM, Shil NK, Gc JB, Colburn ZT, Tsai SY, Segovia JA, et al. Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nat Commun (2019) 10(1):1482. doi: 10.1038/s41467-019-09453-x
72. Sugiura H, Koarai A, Ichikawa T, Minakata Y, Matsunaga K, HIrano T, et al. Increased 25-hydroxycholesterol concentrations in the lungs of patients with chronic obstructive pulmonary disease. Respirology (2012) 17(3):533–40. doi: 10.1111/j.1440-1843.2012.02136.x
73. Koarai A, Yanagisawa S, Sugiura H, Ichikawa T, Kikuchi T, Furukawa K, et al. 25-Hydroxycholesterol enhances cytokine release and Toll-like receptor 3 response in airway epithelial cells. Respir Res (2012) 13:63. doi: 10.1186/1465-9921-13-63
74. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med (2021) 27(11):1970–81. doi: 10.1038/s41591-021-01520-5
75. Lu Z, Ortiz A, Verginadis II, Peck AR, Zahedi F, Cho C, et al. Regulation of intercellular biomolecule transfer–driven tumor angiogenesis and responses to anticancer therapies. J Clin Invest (2021) 131(10):e144225. doi: 10.1172/jci144225
76. Zhou M, Wang S, Liu D, Zhou J. LINC01915 Facilitates the Conversion of Normal Fibroblasts into Cancer-Associated Fibroblasts Induced by Colorectal Cancer-Derived Extracellular Vesicles through the miR-92a-3p/KLF4/CH25H Axis. ACS Biomaterials Sci Eng (2021) 7(11):5255–68. doi: 10.1021/acsbiomaterials.1c00611
77. Doms A, Sanabria T, Hansen JN, Altan-Bonnet N, Holm GH, López S. 25-hydroxycholesterol production by the cholesterol-25-hydroxylase interferon-stimulated gene restricts mamMalian reovirus infection. J Virol (2018) 92(18):e01047–18. doi: 10.1128/jvi.01047-18
78. Canfrán-Duque A, Rotllan N, Zhang X, Andrés-Blasco I, Thompson BM, Sun J, et al. Macrophage-derived 25-hydroxycholesterol promotes vascular inflammation, atherogenesis, and lesion remodeling. Circulation (2023) 147(5):388–408. doi: 10.1161/circulationaha.122.059062
79. Mowat AM, Bain CC. Mucosal macrophages in intestinal homeostasis and inflammation. J Innate Immun (2011) 3(6):550–64. doi: 10.1159/000329099
80. Danese S. Immune and nonimmune components orchestrate the pathogenesis of inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol (2011) 300(5):G716–22. doi: 10.1152/ajpgi.00472.2010
81. Kamada N, Hisamatsu T, Okamoto S, Sato T, Matsuoka K, Arai K, et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol (2005) 175(10):6900–8. doi: 10.4049/jimmunol.175.10.6900
82. Meroni E, Stakenborg N, Viola MF, Boeckxstaens GE. Intestinal macrophages and their interaction with the enteric nervous system in health and inflammatory bowel disease. Acta Physiol (Oxf) (2019) 225(3):e13163. doi: 10.1111/apha.13163
83. Rugtveit J, Bakka A, Brandtzaeg P. Differential distribution of B7.1 (CD80) and B7.2 (CD86) costimulatory molecules on mucosal macrophage subsets in human inflammatory bowel disease (IBD). Clin Exp Immunol (1997) 110(1):104–13. doi: 10.1046/j.1365-2249.1997.5071404.x
84. Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol (2001) 167(5):2651–6. doi: 10.4049/jimmunol.167.5.2651
85. Hausmann M, Kiessling S, Mestermann S, Webb G, Spottl T, Andus T, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology (2002) 122(7):1987–2000. doi: 10.1053/gast.2002.33662
86. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118(6):2269–80. doi: 10.1172/JCI34610
87. Diefenbach A, Gnafakis S, Shomrat O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity (2020) 52(3):452–63. doi: 10.1016/j.immuni.2020.02.016
88. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi: 10.1126/science.aac9593
89. Forkel M, van Tol S, Hoog C, Michaelsson J, Almer S, Mjosberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn's disease and ulcerative colitis. J Crohns Colitis (2019) 13(1):67–78. doi: 10.1093/ecco-jcc/jjy119
90. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238(1):247–62. doi: 10.1111/j.1600-065X.2010.00951.x
91. Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev (2013) 252(1):104–15. doi: 10.1111/imr.12028
92. Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology (2011) 140(6):1756–67. doi: 10.1053/j.gastro.2011.02.016
93. Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut (2009) 58(8):1152–67. doi: 10.1136/gut.2008.163667
94. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol (2014) 15(7):676–86. doi: 10.1038/ni.2920
95. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi: 10.1038/nature06005
96. Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrügge L, Csizmadia E, et al. HIF-1α-induced xenobiotic transporters promote Th17 responses in Crohn's disease. J Autoimmun (2018) 94:122–33. doi: 10.1016/j.jaut.2018.07.022
97. Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. J Clin Invest (2009) 119(9):2441–50. doi: 10.1172/JCI39134
98. Rimoldi M, Chieppa M, Salucci V, Avogadri F, Sonzogni A, Sampietro GM, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol (2005) 6(5):507–14. doi: 10.1038/ni1192
99. Krajina T, Leithauser F, Moller P, Trobonjaca Z, Reimann J. Colonic lamina propria dendritic cells in mice with CD4+ T cell-induced colitis. Eur J Immunol (2003) 33(4):1073–83. doi: 10.1002/eji.200323518
100. Saez A, Herrero-Fernandez B, Gomez-Bris R, Sanchez-Martinez H, Gonzalez-Granado JM. Pathophysiology of inflammatory bowel disease: innate immune system. Int J Mol Sci (2023) 24(2):1526. doi: 10.3390/ijms24021526
101. Kuhl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology (2007) 133(6):1882–92. doi: 10.1053/j.gastro.2007.08.073
102. Colgan SP. Neutrophils and inflammatory resolution in the mucosa. Semin Immunol (2015) 27(3):177–83. doi: 10.1016/j.smim.2015.03.007
103. Yin J, Ren Y, Yang K, Wang W, Wang T, Xiao W, et al. The role of hypoxia-inducible factor 1-alpha in inflammatory bowel disease. Cell Biol Int (2022) 46(1):46–51. doi: 10.1002/cbin.11712
104. Drury B, Hardisty G, Gray RD, Ho GT. Neutrophil extracellular traps in inflammatory bowel disease: pathogenic mechanisms and clinical translation. Cell Mol Gastroenterol Hepatol (2021) 12(1):321–33. doi: 10.1016/j.jcmgh.2021.03.002
105. Domer D, Walther T, Moller S, Behnen M, Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol (2021) 12:636954. doi: 10.3389/fimmu.2021.636954
106. Li T, Wang C, Liu Y, Li B, Zhang W, Wang L, et al. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohns Colitis (2020) 14(2):240–53. doi: 10.1093/ecco-jcc/jjz132
107. Li G, Lin J, Zhang C, Gao H, Lu H, Gao X, et al. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes (2021) 13(1):1968257. doi: 10.1080/19490976.2021.1968257
108. Baynes JW. Dietary ALEs are a risk to human health–NOT! Mol Nutr Food Res (2007) 51(9):1102–6. doi: 10.1002/mnfr.200600287
109. Vejux A, Malvitte L, Lizard G. Side effects of oxysterols: cytotoxicity, oxidation, inflammation, and phospholipidosis. Braz J Med Biol Res (2008) 41(7):545–56. doi: 10.1590/s0100-879x2008000700001
110. Biasi F, Mascia C, Astegiano M, Chiarpotto E, Nano M, Vizio B, et al. Pro-oxidant and proapoptotic effects of cholesterol oxidation products on human colonic epithelial cells: a potential mechanism of inflammatory bowel disease progression. Free Radic Biol Med (2009) 47(12):1731–41. doi: 10.1016/j.freeradbiomed.2009.09.020
111. Chalubinski M, Zemanek K, Skowron W, Wojdan K, Gorzelak P, Broncel M. The effect of 7-ketocholesterol and 25-hydroxycholesterol on the integrity of the human aortic endothelial and intestinal epithelial barriers. Inflammation Res (2013) 62(12):1015–23. doi: 10.1007/s00011-013-0660-x
112. Guillemot-Legris O, Mutemberezi V, Buisseret B, Paquot A, Palmieri V, Bottemanne P, et al. Colitis alters oxysterol metabolism and is affected by 4beta-hydroxycholesterol administration. J Crohns Colitis (2019) 13(2):218–29. doi: 10.1093/ecco-jcc/jjy157
113. Yu S, Sun Y, Shao X, Zhou Y, Yu Y, Kuai X, et al. Leaky gut in IBD: intestinal barrier-gut microbiota interaction. J Microbiol Biotechnol (2022) 32(7):825–34. doi: 10.4014/jmb.2203.03022
114. Sheng N, Ma Z, Zhou Y, Xu J, Gao Y, Fu XY. Cholesterol 25-hydroxylase protects against experimental colitis in mice by modulating epithelial gut barrier function. Sci Rep (2020) 10(1):14246. doi: 10.1038/s41598-020-71198-1
115. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol (2017) 14(10):573–84. doi: 10.1038/nrgastro.2017.88
116. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol (2018) 11(1):1–10. doi: 10.1007/s12328-017-0813-5
117. Caruso R, Marafini I, Franze E, Stolfi C, Zorzi F, Monteleone I, et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol (2014) 7(6):1467–79. doi: 10.1038/mi.2014.35
118. Devi K, Singh N, Jaggi AS. Dual role of sirtuin 1 in inflammatory bowel disease. Immunopharmacol Immunotoxicol (2020) 42(5):385–91. doi: 10.1080/08923973.2020.1790595
119. Raselli T, Wyss A, Gonzalez Alvarado MN, Weder B, Mamie C, Spalinger MR, et al. The oxysterol synthesising enzyme CH25H contributes to the development of intestinal fibrosis. J Crohns Colitis (2019) 13(9):1186–200. doi: 10.1093/ecco-jcc/jjz039
Keywords: cholesterol-25-hydroxylase, 25-hydroxycholesterol, inflammatory bowel disease, intestinal immunity, mechanism
Citation: Zhong G, He C, Wang S, Lin C and Li M (2023) Research progress on the mechanism of cholesterol-25-hydroxylase in intestinal immunity. Front. Immunol. 14:1241262. doi: 10.3389/fimmu.2023.1241262
Received: 16 June 2023; Accepted: 21 August 2023;
Published: 31 August 2023.
Edited by:
Hai-Feng Pan, Anhui Medical University, ChinaReviewed by:
Marta Vuerich, Novartis Institutes for BioMedical Research, SwitzerlandCopyright © 2023 Zhong, He, Wang, Lin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingsong Li, bGltczY2MTIxNkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.