
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 September 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1240275
This article is part of the Research TopicThe Therapeutic Inhibition of Macrophage CheckpointsView all 8 articles
 Tobias Zeller1
Tobias Zeller1 Ira A. Münnich1
Ira A. Münnich1 Roland Windisch1Patricia Hilger1Denis M. Schewe2Andreas Humpe1
Roland Windisch1Patricia Hilger1Denis M. Schewe2Andreas Humpe1 Christian Kellner1*
Christian Kellner1*Immune checkpoint blockade is a compelling approach in tumor immunotherapy. Blocking inhibitory pathways in T cells has demonstrated clinical efficacy in different types of cancer and may hold potential to also stimulate innate immune responses. A novel emerging potential target for immune checkpoint therapy is leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1). LILRB1 belongs to the superfamily of leukocyte immunoglobulin-like receptors and exerts inhibitory functions. The receptor is expressed by a variety of immune cells including macrophages as well as certain cytotoxic lymphocytes and contributes to the regulation of different immune responses by interaction with classical as well as non-classical human leukocyte antigen (HLA) class I molecules. LILRB1 has gained increasing attention as it has been demonstrated to function as a phagocytosis checkpoint on macrophages by recognizing HLA class I, which represents a ‘Don’t Eat Me!’ signal that impairs phagocytic uptake of cancer cells, similar to CD47. The specific blockade of the HLA class I:LILRB1 axis may provide an option to promote phagocytosis by macrophages and also to enhance cytotoxic functions of T cells and natural killer (NK) cells. Currently, LILRB1 specific antibodies are in different stages of pre-clinical and clinical development. In this review, we introduce LILRB1 and highlight the features that make this immune checkpoint a promising target for cancer immunotherapy.
Immunotherapies are established as valid therapeutic options in the treatment of cancer (1). Various approaches have been evaluated pre-clinically and individual concepts have been translated into clinical application. Many novel approaches aim to unleash T cell responses in patients. Beside bispecific T cell engagers and chimeric antigen receptor (CAR) T cells, immune checkpoint inhibitors revealed therapeutic efficacy and their approval for clinical use opened up new avenues (2–4).
Immune checkpoint inhibitors are a class of therapeutic antibodies that block inhibitory interactions between receptors on immune cells and their ligands expressed on cancer cells (4). Immune checkpoint blockade in T cells using antibodies that target cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) or its ligand PD-L1, and more recently lymphocyte-activation gene 3 (LAG3) has proven therapeutic efficacy in different cancer types (4, 5). Yet, while in certain tumor entities considerable improvements were achieved, overall therapeutic response rates are still unsatisfactory (4). Beside T cells, also innate immune cells such as natural killer (NK) cells and macrophages exert a pivotal role in the recognition and elimination of malignant cells in the tumor microenvironment (6, 7). Thus, approaches that modulate innate immune cells are promising. Like T cells, innate immune cells are regulated by an interplay of activating and inhibitory receptors, which may serve as target antigens for therapeutic intervention. While currently the contribution of innate immune cells in CTLA-4 or PD-1 immune checkpoint therapies are under investigation (8), various antibodies or antibody-based fusion proteins targeting emerging immune checkpoints such as CD47, T cell immunoreceptor with Ig and ITIM domains (TIGIT) or natural killer group 2 member A (NKG2A) are developed and characterized for their effector functions (9–14).
In particular, approaches that orchestrate both innate and adaptive immune responses are of interest. Here, leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1), also named immunoglobulin-like transcript (ILT) 2, monocyte/macrophage Ig-like receptor (MIR) 7 and CD85j, may represent an attractive target (9, 15, 16). This inhibitory receptor for human leukocyte antigen (HLA) class I is expressed by a variety of immune cells including certain cytotoxic lymphocytes and macrophages (17, 18). Thus, the engagement of this receptor was shown to impair cytotoxicity of LILRB1-expressing NK cells and CD8-positive T cells. More recently, HLA class I expression was demonstrated to protect cancer cells from phagocytosis by macrophages via interaction with LILRB1, rendering LILRB1 also a phagocytosis checkpoint (9, 19).
The therapeutic potential of antibodies targeting phagocytosis checkpoints has lately been highlighted by promising pre-clinical and clinical results obtained with antibodies targeting the ‘Don`t Eat Me!’ signal CD47 or its myeloid receptor signal regulatory protein (SIRP) α (9, 20, 21). Antibody blockade of either of them was shown to notably enhance antibody-dependent cellular phagocytosis (ADCP) by macrophages in vitro and in murine tumor xenograft models (20, 22–26). Clinically, encouraging results were achieved with the CD47 antibody magrolimab (hu5F9 G4) in combination with the CD20 antibody rituximab in a clinical phase Ib study in lymphoma patients and in combination with azacitidine and venetoclax in a phase I/II trial in acute myeloid leukemia (AML) patients, providing a rationale for further investigation of phagocytosis checkpoint inhibitors and their clinical development (27, 28).
Since interference with LILRB1 signaling may offer the opportunity to modulate various immune cell populations and to promote both innate and adaptive anti-tumoral immune responses, the LILRB1 immune checkpoint receives growing interest. Currently, the receptor is evaluated as a target pre-clinically and first clinical trials with individual LILRB1 targeted agents have been initiated to evaluate the potential of LILRB1 blockade in cancer patients. Here, the immune checkpoint LILRB1 is introduced putting a focus on its role in the regulation of monocytes, macrophages and cytotoxic lymphocytes, and perspectives for LILRB1 targeting in the treatment of cancer are outlined.
LILRB1 is a member of the family of leukocyte inhibitory receptor (LIR) genes comprising eleven protein encoding members and two pseudo genes (18, 29–31). The genes are clustered within the leukocyte receptor complex (LRC) in proximity to genes of several related receptors such as killer cell immunoglobulin (Ig)-like receptors (KIR) and leukocyte-associated Ig-like receptors on human chromosome 19q13.4. LIR are grouped into two subfamilies (29, 32, 33). Subfamily A consists of five cell surface receptors (LILRA1, LILRA2, LILRA4-6) that exert activating functions by association with the immunoreceptor tyrosine-based activation motif (ITAM) containing fragment crystallizable (Fc) region receptor (FcR) γ chain and the soluble member LILRA3. The subfamily B comprises five inhibitory receptors (LILRB1-5), which are type I transmembrane proteins characterized by two to four extracellular Ig-like domains for ligand binding and two to four intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIM) for signal transduction (18, 32, 34). LILRB are specific to primates and humans, but orthologs exist in other species such as paired immunoglobulin-like receptor B (PirB) 18 and gp49B1 in mice (31, 34). LILRB1 together with LILRA2 (ILT1) were the first members of the LIR receptor family identified in 1997 by Samaridis and Colonna (35). In the same year, LILRB1 was shown to be a receptor for cellular HLA class I and for the human cytomegalovirus (CMV) UL18 gene product, a viral homolog of HLA class I (17, 36, 37).
LILRB1 was identified as a 110-120 kDa, 651 amino acid glycoprotein, which is composed of four Ig-like domains in its extracellular part and a cytoplasmic tail of 167 amino acids containing four ITIM or ITIM-like sequences (Figure 1) (36). LILRB1 exists in 13 different isoforms due to alternative splicing. Like other LIR, LILRB1 is expressed also as a soluble isoform, which is capable of ligand binding and may interfere with interactions between ligand and cell surface LILRB1 (39). As related KIR, LILRB family members are polymorphic, although to a lesser extent, with LILRB1 showing a considerable allelic diversity (33, 40, 41).

Figure 1 Structure of LILRB1 and immune inhibitory functions. (A) LILRB1 contains four extracellular immunoglobulin (Ig)-like domains (D1-D4). The binding site for HLA class I/β2-microglobulin (β2m) molecules localizes to the apical D1-D2 region. The intracellular portion comprises four immunoreceptor tyrosine-based inhibitory motifs (ITIM). Critical phosphorylation tyrosine residues involved in the recruitment of SHP-1 and CSK within ITIM domains are indicated. (B) Illustration of the LILRB1 D1-D2 domains (blue) bound to HLA-A2/β2m (depicted in yellow and green, respectively) according to the previously published crystal structure of the complex (38). The LILRB1 D1 domain mainly interacts with the HLA class I α3 domain and the D1-D2 interdomain hinge region with the conserved β2m subunit. The contact sites in LILRB1, HLA-A2 and β2m are highlighted in red, pink and purple, respectively. The ribbon drawing was generated using the pdb file 1P7Q as a template and Discovery Studio Visualizer software (Dassault Systèmes Biovia Corp). (C) LILRB1 is expressed by various immune cell populations including subpopulations of T cells and natural killer (NK) cells, B cells, macrophages and dendritic cells (DC), and contributes to the regulation of diverse immune cell functions.
LILRB1 functions as an inhibitory receptor by suppressing the activity of intracellular kinases. Upon LILRB1 ligation, tyrosines within the ITIM are phosphorylated and the phosphatase SRC-homology 2 domain-containing phosphatase (SHP) 1 is recruited (17, 36, 42, 43). The tyrosine residues Y562, Y614 and Y644 were shown to be involved in SHP-1 recruitment with Y614 being the main docking site, while tyrosine phosphorylation of the receptor involved Y533 (44, 45). In addition, recruitment of SHP-2 has been reported (46). The activated phosphatases dephosphorylate ITAM and suppress key kinases involved in the activation of immune cells including spleen tyrosine kinase (SYK), SRC, zeta-chain associated protein kinase 70-kDa, phosphatidylinositol-4-phosphate 3-kinase and others (43). LILRB1 also interacts with the C-terminal SRC kinase (CSK), an important negative regulator of SRC kinases, which preferentially binds to the ITIM containing Y562 (45). In certain situations, LILRB1 was demonstrated to exert stimulatory function (34, 47). The underlying mechanistic events have not been unraveled yet, but as an explanation, the C-terminal ITIM was suggested to function as an immunotyrosine-based switch (ITSM) domain, which converts inhibitory into activating signaling.
In contrast to KIR, which are restricted to the recognition of distinct HLA class I allele variants, LILRB1 is a receptor for a wide spectrum of HLA class I molecules. Thus, LILRB1 not only interacts broadly with allelic variants of classical HLA-A, HLA-B and HLA-C antigens but also binds the non-classical HLA-E, HLA-F and HLA-G molecules (38, 48–52). Yet, affinities vary, and equilibrium binding constants are between 2 and 100 µM (51–54). HLA-F and HLA-G, the latter of which plays a crucial role in tumor immune escape owing to its immune suppressive function, were suggested to bind with a particular high affinity. Importantly, LILRB1 does not recognize β2-microglobulin (β2m)-free forms of HLA molecules.
The binding site for HLA class I localizes to the apical LILRB1 D1-D2 region, which contains two Ig-like domains (Figure 1A) (38, 52, 55). Crystal structure analysis showed that the D3-D4 domains are not involved (56, 57). Binding of HLA molecules may occur both in cis and in trans, due to a highly flexible interdomain hinge region (55, 57, 58). The observed broad specificity for HLA class I molecules results from the distinct binding sites within the HLA complex. As revealed by crystallographic studies, LILRB1 binds the comparably low polymorphic α3 domain of HLA via the tip of its D1 domain and the conserved β2-microglobulin (β2m) subunit mainly via the D1-D2 interdomain hinge region (Figure 1B) (38, 52, 55). Yet, LILRB1 polymorphisms affect HLA class I binding and allelic variants differing in their D1-D2 region exert different affinities (40). In addition, allelic variation in the α3 domains of HLA class I molecules influences binding affinity (48). Of note, also other members of the LILR family (i.e. LILRB2, LILRB5, LILRA1-3) bind HLA class I molecules, but differ in terms of β2m dependency (59). For example, LILRB2 binds in a β2m-independent mode and also recognizes open conformers lacking β2m due to a few differences in its amino acid sequence resulting in a predominant recognition of the α3 domain (48, 60).
As other members of the LILRB subfamily LILRB1 has multiple ligands (18, 34). Thus, LILRB1 also binds the two calcium-binding cellular proteins S100A8 and S100A9 (61) and interacts with pathogen-derived ligands. These include the above-mentioned UL18 (36, 62), dengue virus and bacterial antigens (29, 63) as well as certain Plasmodium falciparum repetitive interspersed families of polypeptides (RIFINs) (64).
LILRB1 is the most widely expressed member of the LILRB subfamily and is found in various immune cell populations including monocytes, macrophages, dendritic cells (DC), granulocytes, mast cell progenitors, osteoclast precursors as well as B cells and subpopulations of T cells and NK cells (17, 58, 65–68). LILRB1 is involved in the regulation of diverse processes including immune cell differentiation and proliferation, cytokine release, antigen presentation, phagocytosis, cytotoxicity as well as antibody production (Figure 1C) (17, 18, 69–72). Like other members of the LIR family, LILRB1 plays a role in different diseases including pathogen infection, certain autoimmune diseases and cancer (18, 29, 34, 59, 73–75). Particularly its expression by monocytes/macrophages and cytotoxic lymphocytes, which represent relevant effector cell populations in current immunotherapeutic approaches, renders this receptor an attractive target for cancer immunotherapy.
Monocytes and macrophages express all members of the LILRB subfamily (32) with LILRB1 being uniformly expressed in monocytes as well as in ex vivo differentiated macrophages (17, 19, 76). LILRB1 cross-linking reduced Ca2+ mobilization upon HLA-DR activation (17), and importantly, interfered with activation through stimulation of FcγR (42). Thus, co-ligation of LILRB1 and the activating FcγRI inhibited protein tyrosine kinases and led to reduced tyrosine phosphorylation of the FcRγ-chain and SYK, resulting in decreased intracellular Ca2+ mobilization. Therefore, LILRB1 activation was suggested to impair FcγR-mediated effector functions of therapeutic antibodies such as ADCP. Indeed, LILRB1 plays an important role in the regulation of phagocytosis by macrophages (19). The function of LILRB1 as a phagocytosis checkpoint was identified in an attempt to unravel inhibitory pathways that impair the pro-phagocytic activity of the CD47 antibody magrolimab using a panel of solid tumor cell lines (19). LILRB1 gene knockout experiments showed that LILRB1 represents the major receptor for HLA molecules in the regulation of phagocytosis by macrophages.
LILRB1 may also be involved in the regulation of macrophage polarization. Macrophages exist in a range of activation states between the two extremes of classically activated, pro-inflammatory M1 and alternatively activated M2 macrophages (77, 78). A regulatory role was suggested for LILRB1, since knockout of the LILRB1 gene in macrophages in vitro resulted in a higher proportion of macrophages displaying an M1 immunophenotype, possibly by abrogation of interactions of LILRB1 with HLA molecules in cis (19). This function has already been demonstrated for LILRB2. LILRB2 antagonism has been reported to drive tumor-infiltrating myeloid cell polarization toward an inflammatory, M1-like phenotype and to enhance pro-inflammatory and adaptive immune responses (79).
DC differentiated ex vivo from monocytes (moDC) or CD34-positive progenitor cells strongly express LILRB1 (17, 69, 70, 80). During differentiation, LILRB1 was shown to be upregulated and expression was preserved upon DC maturation (70). LILRB1 is also expressed by most human dendritic subsets from the peripheral blood (65). Thus, LILRB1 was found on type 2 conventional DC, plasmacytoid DC and 6-sulfo-LacNAc-positive DC. In these DC subtypes, LILRB1 is upregulated upon toll-like receptor (TLR) stimulation with lipopolysaccharide (LPS) or imiquimod. In contrast, type 1 conventional DC lack LILRB1 expression, even after exposure to TLR ligands. LILRB1 has been suggested to modulate the differentiation of DC and to regulate their functions (70). Thus, DC differentiated from monocytes in the presence of a LILRB1 ligand remained CD14 expression and exerted low HLA-DR levels. The cells did not respond to LPS-induced maturation, did not secrete characteristic cytokines such as interleukin (IL)-10, IL-12 or TGFβ, and exerted a weak T cell stimulating activity (70, 81). Moreover, LILRB1 engagement was shown to inhibit activation by stimulation of the osteoclast-associated receptor (OSCAR), an activating FcR γ- chain associated myeloid receptor involved in antigen presentation (69). Thus, co-ligation of LILRB1 and OSCAR resulted in reduced Ca2+ mobilization, impaired cytokine secretion, and a diminished potency to stimulate the proliferation of antigen-specific T cells in vitro (69).
In contrast to monocytes, only a subpopulation of NK cells displays LILRB1 and at lower levels (17). Cell type specific divergent expression was attributed to different promotors in monocytes and NK cells (82). In healthy conditions, LILRB1 expression varies considerably between individuals. The receptor was found on the cell surface of 23% to 77% of NK cells (17), and the observed variation between individuals was linked to polymorphisms in the LILRB1 gene (83). In general, LILRB1 expression differs in NK cell subpopulations and is higher in CD56dim NK cells, which express FcγRIIIA and are predominant in the peripheral blood, than in CD56bright NK cells, which lack FcγRIIIA expression (84). In particular, LILRB1 is expressed by terminally differentiated NK cells expressing CD57 and numerous KIR and by virus-induced adaptive NK cells (85–88). Recent findings show that at least under certain conditions LILRB1 contributes to NK cell education - a process that counterbalances the responsiveness of individual NK cells with their sensitivity for inhibition by cognate HLA class I molecules (89, 90). Importantly, LILRB1 engagement by HLA-G impairs the formation of the NK cell activating synapse by inhibiting the polarization of lytic granules and the microtubule organizing centre and impaired filamentous actin accumulation (91). Upon ligation, LILRB1 inhibits Ca2+ mobilization, the release of inflammatory cytokines such as interferon (IFN) γ and impairs both natural cytotoxicity and ADCC (17, 91–96).
LILRB1 is also displayed by certain T cells, yet a considerable variation in the frequency of T cells expressing cell surface LILRB1 exists between individuals (97). In healthy donors, LILRB1 is found on the cell surface of about 20% of αβ T cells in younger individuals, but this can increase up to over 50% with age or upon chronic infections (98, 99). LILRB1 surface expression is found predominantly in CD8-positive T cells, in contrast to CD4-positive T cells, which rarely display LILRB1 (98, 100). LILRB1-positive T cells revealed a reduced potential to proliferate but were capable of producing IFN-γ upon T cell receptor (TCR) stimulation (101). Further expression analysis revealed that in CD8-positive T cells LILRB1 was preferentially displayed by the terminally differentiated population of effector memory T cells re-expressing CD45RA (TEMRA) and marks a T cell population with strong effector functions and a high content of intracytoplasmic perforin (99, 101–103). In addition, cell surface LILRB1 was found also on a subset of effector memory T cells (TEM). Interestingly, LILRB1 and PD-1 are expressed on distinct T cell populations and induced expression of PD-1 by TCR stimulation was found preferentially in LILRB1-negative T cells in vitro (102).
In experiments with superantigen pulsed antigen presenting cells (APC) LILRB1 was found to colocalize with the T cell receptor (TCR) at the immunological synapse (104). Engagement of LILRB1 interfered with TCR/CD3 signaling and impaired actin cytoskeleton reorganization, cytotoxicity, cytokine production, and attenuated proliferation (17, 93, 94, 104–106). Moreover, LILRB1 was shown to compete with CD8 for binding to HLA class I molecules (53). Thus, in addition to impairing T cell responses by ITIM signaling, LILRB1 may interfere with T cell activation by blocking the binding of CD8. T cells can also acquire LILRB1 from monocytes by trogocytosis (107). The acquired LILRB1 molecules had signaling properties and were shown to regulate T cell functions. LILRB1 is also expressed by subsets of γδ T cells and contributes to their regulation (17, 108). For example, gene knock-down of β2m or antibody blockade of LILRB1 enhanced lysis of lymphoma cells by γδ T cell clones (108).
LILRB1 expression in B cells increases throughout maturation in the bone marrow and is maintained at the plasma cell stage (16, 82). In peripheral blood, mature B cell populations including naïve and memory B cells homogenously express LILRB1, whereas only a fraction of transitional B cells were stained positive for LILRB1 (82). LILRB1 has been suggested to participate in the regulation of B cell functions. Thus, antibody-mediated crosslinking of LILRB1 interfered with B cell receptor activation resulting in reduced Ca2+ mobilization (17). Subsequent findings indicated that LILRB1 activation dampened IgG production, inhibited isotype switching and impaired cytokine production (71). In addition, HLA-G aggregated on nanoparticles interfered with B cell responses by binding to LILRB1 (72). In these experiments, inhibition of T cell-dependent as well as T cell-independent B cell responses was observed and aggregated HLA-G reduced B cell proliferation, antibody secretion and cytokine release.
Increasing evidence indicates that LILRB1 like other members of the LILRB family plays a role in cancer development, treatment and tumor immunotherapies (43). In the tumor microenvironment, LILRB1 is expressed by different immune cells and was suggested to support tumor growth indirectly by contributing to the repression of multiple anti-tumoral functions. LILRB1-mediated immune suppression relies on the expression of HLA class I molecules and their expression levels. Interestingly, also malignant cells from certain tumor entities, in particular lymphomas and leukemias, express LILRB1.
Alterations in HLA expression that emerge during the evolution of immune escape variants are frequently found in tumors (109, 110). Down-modulation of classical HLA molecules enables tumor cells to evade from attack by T cells, which typically recognize peptides presented on HLA molecules. In contrast, HLA class I molecules function as markers of self and inhibit NK cell functions (11). As a consequence, reduced or loss of HLA class I expression renders tumor cells susceptible to NK cell cytotoxicity. Typically, HLA class I recognition involves KIR and NKG2A receptors, but also LILRB1 plays a role. Thus, masking of LILRB1 enhanced NK cell cytotoxicity against HLA class I-expressing leukemia cells, in particular when KIR or NKG2A were blocked simultaneously (111). LILRB1 was suggested to interfere with activation of NK cells upon engagement of natural killer group 2 member D (NKG2D), an important stimulatory NK cell receptor that regulates natural cytotoxicity, and to inhibit lysis of leukemic cells with ectopic expression of the NKG2D ligand MHC class I chain-related protein A (MIC A) (92).
Intriguingly, HLA class I molecules not only inhibit NK cell-mediated lysis of tumor cells but also function as ‘Don`t Eat Me!’ signals that prevent malignant cells from phagocytic uptake by macrophages through LILRB1 engagement (19). In the tumor microenvironment, tumor-associated macrophages (TAM) contribute to cancer progression by supporting cancer cell survival and proliferation, promotion of angiogenesis, and suppression of immune responses (77, 78, 112). Yet, macrophages are also able to eliminate malignant cells by phagocytosis and to promote anti-tumoral functions of lymphocytes. The role of MHC class I in the regulation of anti-tumoral responses by macrophages was demonstrated in xenograft studies using genetically modified human tumor cells expressing a mouse human chimeric β2m protein to confer interaction with murine macrophages (19). These experiments revealed that MHC class I protected tumor cells from macrophage attack in vivo. Genetic deletion of HLA class I cell surface molecules augmented ADCP by anti-epithelial cell adhesion molecule (EpCAM) or anti-epidermal growth factor receptor (EGFR) antibodies, suggesting that the HLA class I:LILRB1 axis may compromise the therapeutic efficacy of tumor targeting antibodies. Thus, loss of HLA class I may sensitize tumor cells to phagocytosis.
Tumors may also impair immune cells by neo-expression of non-classical HLA molecules such as HLA-G. HLA-G expression was described in various tumors including gastric, colorectal, lung, breast, hepatocellular and pancreatic cancer as well as chronic lymphocytic leukemia and was associated with tumor progression and a worse outcome (113–116). Both cell membrane-bound HLA-G and its soluble form exert pleiotropic immune suppressive functions on various immune cells including T cells, NK cells and APC, which involve LILRB1 signaling (114). For instance, in NK cells, interaction of HLA-G and LILRB1 was shown to counteract signaling and induction of cytotoxicity by interaction of the activating NK cell receptor NKG2D with its ligand MIC A on tumor cells (117). Interestingly, HLA-G upregulates the expression of LILRB1 in NK cells, T cells and APC in vitro (118), which may further potentiate immune suppression.
The analysis of cells in the tumor microenvironment of different types of solid tumors confirmed that LILRB1 was mainly expressed in the tumor stroma with TAM representing the major LILRB1 expressing immune cell population (113). Of note, LILRB1 was found to be displayed by TAM from different types of solid tumors including colon carcinoma, head and neck cancer, non–small cell lung cancer, renal cell carcinoma and by lymphoma associated macrophages (19, 76, 113). The analysis of the tumor microenvironment in gastric cancer revealed that LILRB1-positive TAM had an M2-like phenotype. Their numbers correlated with the levels of immune suppressing cytokines and were associated with T cell exhaustion and increased expression of inhibitory immune checkpoints such as PD-1 and CTLA-4 (119). Beside TAM, LILRB1 was expressed by a significant number of NK cells (113). In certain tumors, the fraction of LILRB1 expressing NK cells was increased in the tumor microenvironment. For example, in prostate cancer, a higher percentage of tumor infiltrating NK cells expressed LILRB1 in comparison to NK cells from control tissues (120). Tumor-induced upregulation of LILRB1 was supported further by subsequent findings revealing that certain tumor cells, i.e. prostate cancer cells and glioblastoma cells, are capable of inducing the expression of LILRB1 on NK cells in vitro (120, 121). In addition, cell-to-cell contact with M2 polarized macrophages augmented LILRB1 expression in NK cells, which led to reduced cytotoxicity and cytokine production (122). In contrast, LILRB1 was more rarely expressed in tumor infiltrating T cells and rather restricted to CD8-positive T cells with a TEMRA phenotype. Of note, in the peripheral blood of cancer patients an increased frequency of LILRB1 expressing NK and T cells was observed compared to healthy controls (92, 96, 113, 120, 123, 124).
The expression of LILRB1 in the tumor microenvironment may have prognostic features. In gastric cancer for example, elevated LILRB1 expression was associated with advanced tumor stages, increased risk of recurrence and inferior survival (119). In prostate cancer, enhanced levels of LILRB1 mRNA were associated with a shorter biochemical recurrence-free survival according to prostate specific antigen levels in the blood (125). Moreover, in ovarian cancer a high content of LILRB1-positive immune cells was correlated with shorter survival, and worse adjuvant chemotherapy responses (126).
LILRB1 is also expressed by malignant cells of different tumor types (43). Thus, LILRB1 expression was reported in AML (127), certain T cell lymphomas (128–130), B-lineage lymphomas and leukemias (131–133), gastric cancer (134), ovarian cancer (126), and renal cell carcinoma (135). LILRB1 expression by tumor cells may have different consequences. For example, LILRB1 expression protected cutaneous T-cell lymphoma cells from CD3/TCR activation induced cell death (128), while in malignant B cells LILRB1 engagement dampened tumor cell proliferation and induced cell cycle blockade (132). Intriguingly, the expression of LILRB1 may increase the susceptibility of cancer cells to an attack by immune cells. In multiple myeloma, loss of LILRB1 has been suggested to contribute to immune escape by reducing their susceptibility to NK cell mediated lysis (133). The LILRB1 ligand S100A9 expressed by NK cells was suggested to play a role and effects of LILRB1 expression on cytotoxicity were diminished by S100A9 blockade. Moreover, LILRB1 expressed by lymphoma cells sensitized these cells to lysis by certain γδ T cells, presumably by co-stimulation of the effector cells through ligation of HLA class I on γδ T cells (131).
The important inhibitory function of the HLA class I:LILRB1 axis on phagocytosis and its role in the regulation of NK cells and T cells may offer the opportunity for therapeutic intervention using immune checkpoint inhibitors (9). LILRB1 antagonism may diminish inhibitory signaling and shift the balance towards activating signaling in immune cells – thereby promoting phagocytosis and/or cellular cytotoxicity. In combination with other therapeutic monoclonal or bispecific antibodies targeting tumor cells such inhibitors may enhance the efficacy of antibody therapy by enhancing key functions such as ADCP or ADCC (Figure 2).
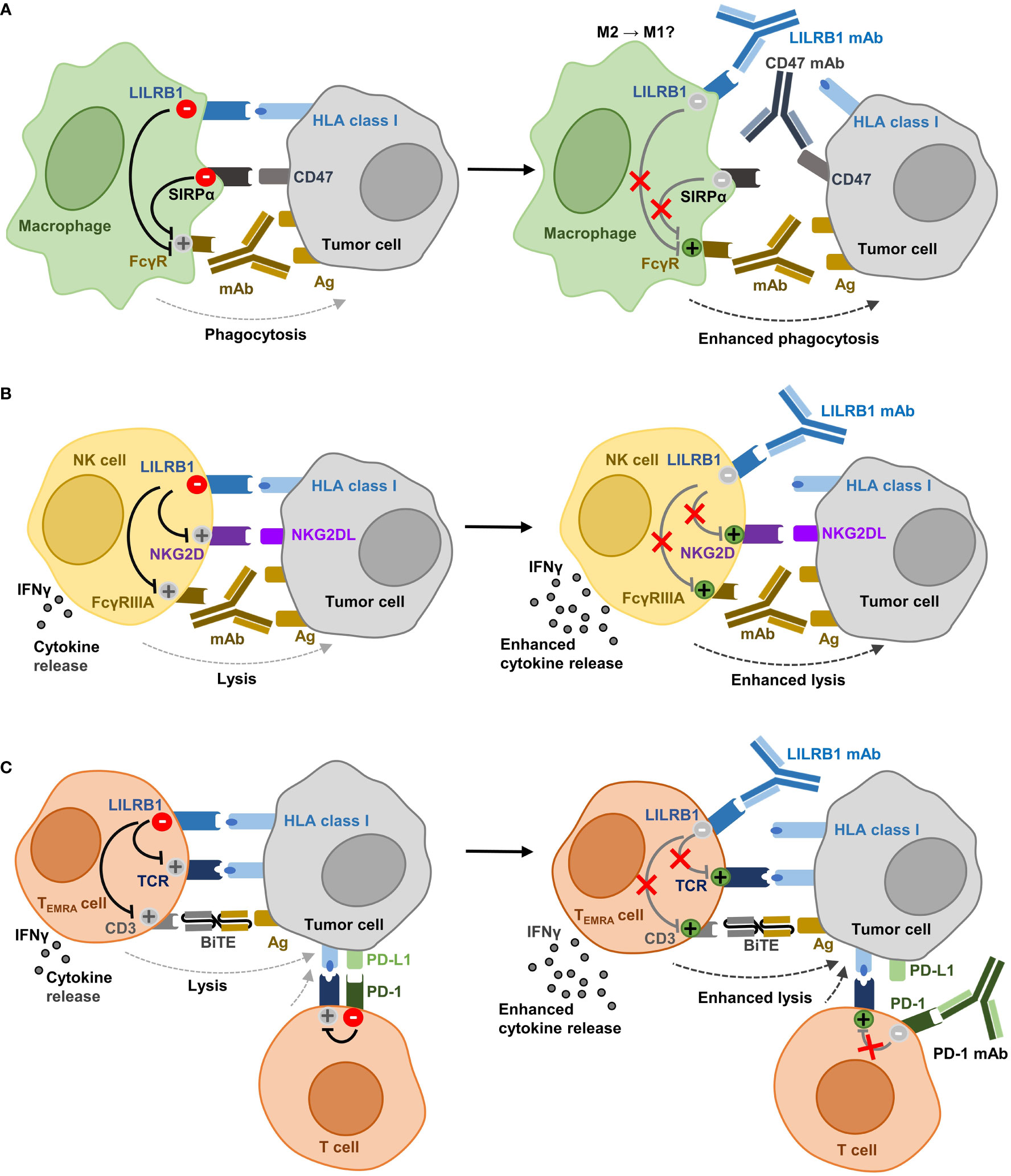
Figure 2 Potential effector functions of anti-LILRB1 antibodies in cancer immunotherapy. (A) Recognizing HLA class I molecules, LILRB1 functions as an immune checkpoint in macrophages and inhibits together with the CD47 receptor SIRPα phagocytosis of tumor cells. Antagonistic LILRB1 antibodies enhance phagocytosis and promote ADCP by therapeutic antibodies that target an antigen expressed by the tumor and which provide an ‘Eat Me!’ signal by ligation of activating FcγR. LILRB1 blockade also cooperates with CD47 antibodies in enhancing ADCP. Since a role for LILRB1 has been implicated in macrophage polarization, the blockade of the HLA class I:LILRB1 axis may support re-programming of TAM from an M2-like phenotype towards M1. (B) In NK cells, LILRB1 engagement impairs both cytokine release and cytotoxicity. LILRB1 ligation hampers lysis of cancer cells induced by NK cell lysis receptors such as NKG2D or by activation of FcγRIIIA through therapeutic antibodies targeting a tumor-expressed antigen. LILRB1 antibody blockade enhances cytokine release, natural cytotoxicity and ADCC. (C) In CD8-positive T cells, mainly TEMRA cells, LILRB1 inhibits cytokine release and cytotoxic functions triggered by activation of the T cell receptor. LILRB1 ligation also impairs tumor cell lysis induced by therapeutic BiTE molecules specific for a tumor cell expressed antigen and activating CD3. Antagonistic LILRB1 antibodies promote cytokine release and T cell cytotoxicity and may be employed to enhance the efficacy BiTE molecules. The expression of PD-1 and LILRB1 on different T cell subsets renders LILRB1 blockade attractive for combination with anti-PD-1 antibodies to abrogate inhibitory signaling in both T cell populations (Ag, antigen; NKG2D, natural killer group 2 member D; NKG2DL, NKG2D ligand; FcγRIIIA, Fcγ receptor IIIA; TCR, T cell receptor; BiTE, bispecific T cell engager; PD-1, programmed cell death 1; PD-L1, PD-1 ligand 1).
Clinically applied immune checkpoint inhibitors are often IgG4 antibodies, which exert lower affinity to FcγR than IgG1, the most commonly used isotype for therapeutic antibodies. Fc engineered, Fc-silent versions of IgG antibodies, in which both FcγR and complement binding is abolished by introduction of distinct amino acid substitutions, may represent an alternative (136, 137). Such engineered antibodies may prevent undesired binding to various FcγR expressed by different immune cell populations and abrogate Fc-dependent elimination of LILRB1 expressing effector cell populations by ADCC, ADCP or complement dependent cytotoxicity. The pronounced homology between LILRB1 and LILRB2, which is more restricted to the myeloid lineage in its expression, allowed the generation of antibodies that bind both receptors (138). Such dual antagonist strategies may be beneficial in respect of the HLA specificity and inhibitory functions of both receptors in myeloid cells. In addition to monoclonal antibodies, receptor-ligand interactions can also be abrogated using recombinant fusion proteins between a cognate ligand and the human Fc domain (139, 140). Corresponding HLA-β2m-Fc fusion proteins may also offer the opportunity to block other HLA class I receptors, including LILRB2, concomitantly (141).
In initial experiments, Barkal and colleagues employed the murine hybridoma antibody clone GHI/75 to disrupt the HLA class I:LILRB1 axis (19). Similar to HLA class I-specific fragments antigen binding (Fab), this anti-LILRB1 antibody enhanced the phagocytosis of HLA class I-expressing solid tumor cell lines in vitro, but required combination with the CD47 directed IgG4 antibody magrolimab to become effective (19). Notably, CD47 IgG4 antibodies may exert dual function by blocking CD47 at the tumor cell site and engaging activating FcγR at the effector cell side to provide an ‘Eat Me!’ signal (9, 19, 142). Regarding the suggested interplay between the two axes CD47:SIRPα and HLA class I:LILRB1 in counteracting effective ADCP of tumor cells as demonstrated for anti-EpCAM or anti-EGFR antibodies (19), additional antibody combinations were studied. For example, a chimeric, Fc-silent variant of antibody GHI/75 with abrogated FcγR binding was generated, to prevent potential occurring effects by binding of this antibody to FcγR (76). In combination with a CD20 antibody (i.e. rituximab or obinutuzumab) for FcγR engagement and an Fc-silent CD47 antibody to inhibit SIRPα signaling, LILRB1 antibody blockade further enhanced the phagocytic uptake of lymphoma cells by macrophages in vitro compared to the combination of CD20 antibodies and CD47 blockade only. This was observed in studies with non-polarized macrophages as well as macrophages that were polarized towards M1 or M2 phenotypes ex vivo, and lymphoma-associated macrophages isolated from the bone marrow of lymphoma patients. The blockade of LILRB1 alone however was not sufficient to induce phagocytosis in this study. In addition, the LILRB1 antibody was not effective when combined only with either a CD20 or a CD47 antibody. These results suggested that both FcγR engagement by the tumor targeting antibody and blockade of the CD47:SIRPα axis were required (76). In contrast, characterization of the humanized IgG4 anti-LILRB1 antibody BND-22 revealed that this antibody was effective also as single agent in inducing phagocytosis of tumor cells by human macrophages both in vitro and in a xenograft in vivo model in which both human macrophages and cancer cells were transferred (113). In addition, the antibody enhanced ADCP by the anti-EGFR antibody cetuximab in vitro, thereby providing further evidence that LILRB1 blockade may enhance the therapeutic efficacy of tumor targeting antibodies by enhancing their ADCP function.
Regarding the tumor promoting and immune suppressive functions of TAM, reprogramming TAM to a pro-inflammatory state may inhibit tumor growth. In fact, LILRB2 antagonistic antibodies were shown to exert this immunomodulatory function (79). Yet, whether LILRB1 blockade allows promoting polarization towards an inflammatory M1-like phenotype and whether this may offer the opportunity to also reprogram TAM, has not been addressed experimentally to our knowledge and will require further investigation (Figure 2A).
NK cells play an important role in immunosurveillance against cancer due to their natural cytotoxic functions and represent an important effector cell population for therapeutic antibodies as mediators of ADCC. LILRB1 blockade may allow both promoting natural cytotoxicity and enhancing ADCC (Figure 2B). For example, antibody masking of LILRB1 was demonstrated to promote the activation of NK cells from chronic lymphocytic leukemia (CLL) patients, to enhance NK cell proliferation when combined with the immunomodulatory drug lenalidomide and to promote lysis of CLL cells (96). Similarly, enhanced NK cell cytotoxicity against glioblastoma cells was observed in vitro when LILRB1 was blocked (121). A humanized, Fc-engineered anti-LILRB1 IgG antibody, B1-176, with reduced FcγR binding was shown to disrupt LILRB1 signaling in co-culture experiments with LILRB1 reporter cells and various tumor cell lines (92). The antibody enhanced natural cytotoxicity by the NK cell line NKL against HLA class I-positive leukemia cells and particularly supported lysis induced by NKG2D engagement. In addition, antibody B1-176 promoted anti-tumoral activities of NK cells in ALL or multiple myeloma xenograft experiments in vivo. Regarding ADCC, LILRB1 was shown to hamper cetuximab-mediated ADCC by NK cells. Thus, antagonistic LILRB1 antibodies restored the cytotoxic activity of NK cells from triple-negative breast cancer patients and enhanced ADCC (123).
T cells are an attractive effector cell population in cancer immunotherapy, in which T cell checkpoint inhibitors or bispecific T cell engager (BiTE) molecules have shown promising results (3, 4). The ability of anti-LILRB1 antibodies to enhance T cell activation was demonstrated in mixed lymphocyte reactions with allogeneic T cells and DC, in which the antibody BND-22 enhanced T cell activation as verified by IFN-γ release (113). Recent findings suggest that LILRB1 engagement may impair the efficacy of both BiTE and PD-1 immune checkpoint inhibitors (Figure 2C). LILRB1-expressing TEMRA cells represent an important T cell population for CD3 bispecific antibodies with a high cytotoxicity (102). The anti-LILRB1 antibody GHI/75 has shown potential to enhance T cell cytotoxic activity mediated by an [anti-MART-1 × CD3] BiTE molecule by disrupting the interaction of LILRB1 with HLA-G expressed by malignant melanoma cells (102). Regarding PD-1 immune checkpoint blockade, LILRB1 upregulation has been discussed to be a potential mechanism of resistance. Thus, increased expression of LILRB1 was reported in nearly half of malignant melanoma patients after treatment with the PD-1 directed antibody nivolumab (113). Interestingly, combining anti-PD-1 and anti-LILRB1 antibodies synergistically enhanced the secretion of TNF-α by autologous peripheral blood mononuclear cells co-incubated with colon cancer cells in vitro and improved therapeutic effects were observed for the antibody combination in vivo (113). Moreover, since PD-1 and LILRB1 are described to be expressed by different T cell subsets, co-blockade of both receptors may provide an opportunity to abrogate inhibitory signaling pathways in both subsets (102).
The promising pre-clinical results achieved by targeting LILRB1 have paved the way for first clinical trials with individual LILRB1 antagonists in cancer patients (Table 1). The anti-LILRB1 antibody BND-22/SAR444881 (Biond Biologics/Sanofi) is a humanized monoclonal antibody of the IgG4 isotype (113). The antibody was shown to bind LILRB1 selectively and not to cross react with other members of the LILR family. By binding the LILRB1 D1-D2 domains, the antibody prevented engagement of LILRB1 by both classical HLA class I and HLA-G molecules. In vitro, BND-22/SAR444881 enhanced NK cell-mediated tumor cell lysis, T cell activation and phagocytosis. BND-22 improved the therapeutic efficacy of anti-EGFR or anti-PD-1 antibodies in murine tumor models. The antibody is currently evaluated in an ongoing phase 1/2 clinical trial (clinical trials.gov identifier: NCT04717375) in patients with advanced solid tumors. BND-22/SAR444881 is applied alone or in combination with either the anti-EGFR antibody cetuximab or the anti-PD-1 antibody pembrolizumab.
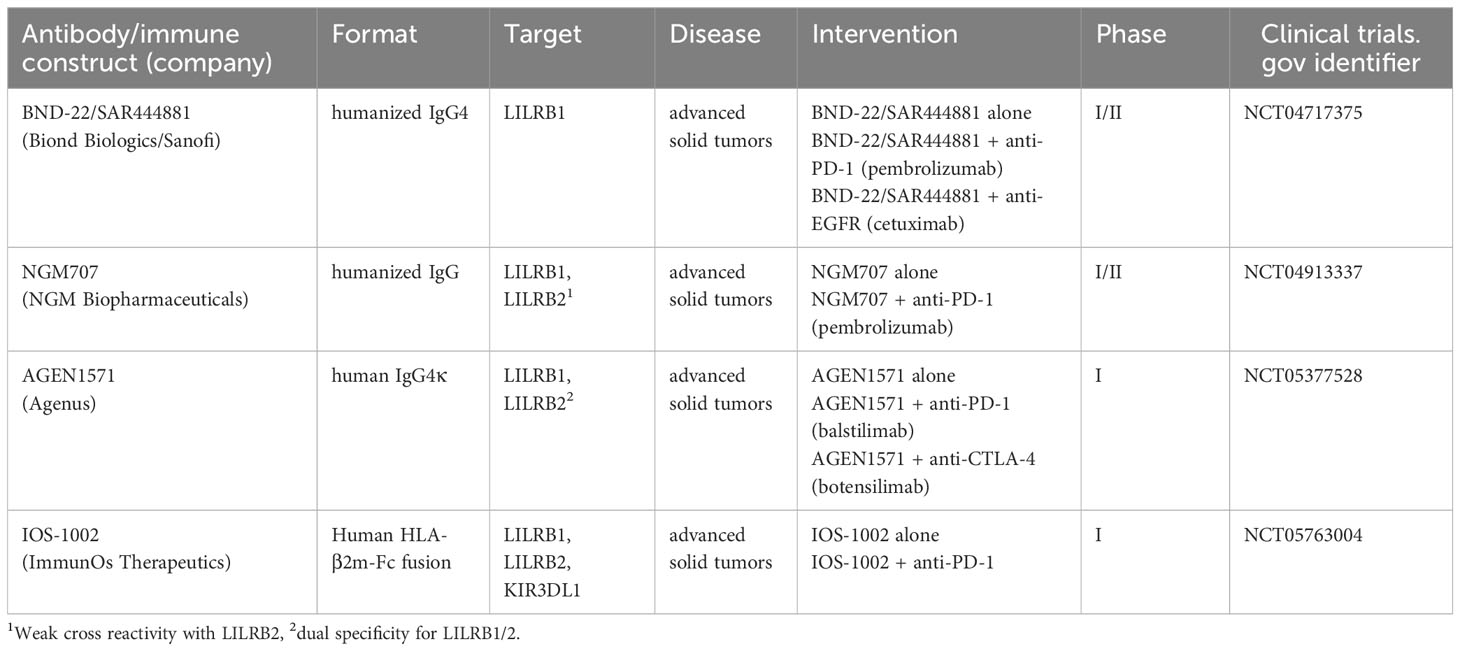
Table 1 Current clinical studies with LILRB1 targeting antibodies or fusion proteins.
AGEN1571 (Agenus Inc.) is a fully human IgG4κ monoclonal antibody (143). AGEN1571 demonstrated weak cross reactivity to LILRB2, but did not bind other LILR. The antibody was shown to promote M1 polarization and enhanced cytokine release and activation of NK and CD8-positive T cells alone and in combination with a PD-1 blocking antibody in vitro. AGEN1571 is currently tested in a phase I clinical trial (NCT05377528) in patients with advanced solid tumors. AGEN1571 is evaluated as a single agent or in combination with either the PD-1 inhibitor balstilimab and/or the anti-CTLA-4 antibody botensilimab.
NGM707 (NGM Biopharmaceuticals, Inc.) is a humanized IgG antibody with dual specificity for both LILRB1 and LILRB2 (138). The antibody exerts dual antagonistic functions by blocking the interaction of both receptors with classical HLA class I as well as HLA-G molecules. NGM707 promoted phagocytosis of cancer cells by macrophages and increased cytotoxicity by NK and CD8-positive T cells. In combination with pembrolizumab, NGM707 enhanced activation of T cells by macrophages in mixed lymphocyte reactions. NGM707 is currently tested as monotherapy or in combination with pembrolizumab in advanced or metastatic solid tumor malignancies in a phase 1/2 trial (NCT04913337). First results have been reported for the NGM707 monotherapy dose escalation, in which the antibody was well tolerated up to a dose level of 1800 mg, and a maximum tolerated dose was not reached. NGM707 monotherapy demonstrated early signs of efficacy with the best overall responses of stable disease in six patients and non-complete response/non-progressive disease in one patient of 20 response-evaluable patients. Interestingly, signs of myeloid reprogramming in form of reduced CD163 expression were observed post-treatment (144).
IOS-1002 (iosH2, ImmunOs Therapeutics AG) is an optimized fusion protein consisting of an peptide-free engineered variant of HLA-B*57 (amino acid substitutions: A46E/V97R) fused to human β2m and the human IgG4 Fc domain (141). The amino acid substitutions were introduced into the HLA-B moiety to improve stability and production. In vitro, IOS-1002 was demonstrated to bind LILRB1 with nanomolar affinities and to react also with LILRB2 as well as KIR3DL1. The fusion protein blocked the interaction between HLA-G and both LILRB1 and LILRB2, thereby impairing ITIM signaling by reducing phosphorylation of SHP1 and SHP2 adapter molecules. IOS-1002 shifted macrophage polarization towards an inflammatory M1 phenotype, increased tumor cell phagocytosis in vitro as single agent and enhanced cytotoxicity by T and NK cells against tumor cells. Therapeutic efficacy was demonstrated in vivo in syngeneic murine models of colon cancer and in non-small cell lung cancer patient-derived xenografts in human immune system mice. A phase 1a/1b study to evaluate IOS-1002 in adult patients with advanced solid tumors has received approval (NCT05763004) recently. IOS-1002 will be investigated as monotherapy or in combination with a PD-1 antibody.
Pre-clinical investigations have identified the HLA class I:LILRB1 axis as an attractive target for immune checkpoint inhibition, as its blockade may allow to promote both adaptive and innate immune responses. Promising results were obtained with combinations of LILRB1-directed antibodies and tumor targeting antibodies, bispecific CD3 antibodies or other immune checkpoint inhibitors blocking CD47 or PD-1. However, the results were obtained with individual anti-LILRB1 antibodies, combination partners or in models of distinct tumor entities, and further studies are required to draw general conclusions. Interestingly, in different studies individual LILRB1 antibodies differed in their activity to induce phagocytosis of tumor cells as single agent, which may be due to characteristics of target cells or of the antibody such as the isotype or the epitope specificity. Yet, direct comparisons between antibodies are missing, and the ideal format for LILRB1 blocking agents needs to be determined.
Moreover, it is not clear if or to what extent the expression of other inhibitory receptors may impact the efficacy of LILRB1 blockade. Thus, other ‘Don´t Eat Me!’ signals beside HLA class I and CD47, such as PD-L1, CD24, adipocyte plasma membrane-associated protein (APMAP) or signaling lymphocyte activation molecule (SLAM) (9, 145–147), may hamper phagocytosis and diminish the effects by LILRB1 blockade. In addition, the presence of antigens with pro-phagocytotic function such as the ‘Eat Me!’ signal molecule calreticulin may play a role (148). Moreover, knowledge on the interplay between LILRB1 with other inhibitory receptors recognizing HLA or other cellular ligands in NK cells and T cells is still patchy and requires further investigation.
The encouraging preclinical results laid the foundation to evaluate individual LILRB1 antibodies clinically. Yet, current clinical studies focus on solid tumors. LILRB1 blockade may be also effective against leukemias and lymphomas. These may more likely express LILRB1, which needs to be taken into consideration. Potential pitfalls in the clinical application of LILRB1 directed antibodies may arise from antigen sink due to LILRB1 expression by tumor cells or immune cells that may not contribute to eradication or play only minor roles. Also, side effects of LILRB1 blockade may derive from its broad expression pattern including various types of leukocytes and osteoclast precursors. For example, genetic deletion of the murine LILRB ortholog PirB was shown to accelerate osteoclastogenesis in mice, though the development of osteoporosis was not observed in this study (58).
Beside LILRB1 also other LIR represent potential target antigens in immunotherapy. For instance, LILRB2, LILRB3 and LILRB4 were described as myeloid immune checkpoints and anti-LILRB2 and anti-LIRB4 antibodies have entered clinical trials recently (18, 149–155). It will be interesting to see, whether the pre-clinical success of targeting LILRB1 or other LIR can be translated into clinical application.
TZ and CK conceived the article structure and prepared the initial draft of the manuscript. TZ prepared the table and designed figures. All authors contributed to writing, editing and proofreading the manuscript.
This work was funded by the Verein zur Förderung von Wissenschaft und Forschung an der Medizinischen Fakultät der Ludwig-Maximilians-Universität München (to CK).
The Deutsche José Carreras Leukämie-Stiftung and the Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie are gratefully acknowledged for the José Carreras-DGHO scholarship for doctoral research to TZ (05 PSD/2021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget (2014) 5(24):12472–508. doi: 10.18632/oncotarget.2998
2. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
3. Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol (2020) 17(7):418–34. doi: 10.1038/s41571-020-0347-5
4. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, et al. The next decade of immune checkpoint therapy. Cancer Discovery (2021) 11(4):838–57. doi: 10.1158/2159-8290.CD-20-1680
5. Ascierto PA, Lipson EJ, Dummer R, Larkin J, Long GV, Sanborn RE, et al. Nivolumab and relatlimab in patients with advanced melanoma that had progressed on anti-programmed death-1/programmed death ligand 1 therapy: Results from the phase I/IIa RELATIVITY-020 trial. J Clin Oncol (2023), JCO2202072 41(15):2724–35. doi: 10.1200/JCO.22.02072
6. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574(7776):45–56. doi: 10.1038/s41586-019-1593-5
7. Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer (2020) 20(8):437–54. doi: 10.1038/s41568-020-0272-z
8. Liu X, Hogg GD, DeNardo DG. Rethinking immune checkpoint blockade: 'Beyond the T cell'. J Immunother Cancer (2021) 9(1):e001460. doi: 10.1136/jitc-2020-001460
9. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer (2019) 19(10):568–86. doi: 10.1038/s41568-019-0183-z
10. Lentz RW, Colton MD, Mitra SS, Messersmith WA. Innate immune checkpoint inhibitors: The next breakthrough in medical oncology? Mol Cancer Ther (2021) 20(6):961–74. doi: 10.1158/1535-7163.MCT-21-0041
11. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16(1):7–19. doi: 10.1038/nrc.2015.5
12. van den Berg TK, Valerius T. Myeloid immune-checkpoint inhibition enters the clinical stage. Nat Rev Clin Oncol (2019) 16(5):275–6. doi: 10.1038/s41571-018-0155-3
13. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell (2018) 175(7):1731–43. doi: 10.1016/j.cell.2018.10.014
14. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
15. Zhao J, Zhong S, Niu X, Jiang J, Zhang R, Li Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scand J Immunol (2019) 90(5):e12804. doi: 10.1111/sji.12804
16. Banham AH, Colonna M, Cella M, Micklem KJ, Pulford K, Willis AC, et al. Identification of the CD85 antigen as ILT2, an inhibitory MHC class I receptor of the immunoglobulin superfamily. J Leukoc Biol (1999) 65(6):841–5. doi: 10.1002/jlb.65.6.841
17. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med (1997) 186(11):1809–18. doi: 10.1084/jem.186.11.1809
18. De Louche CD, Roghanian A. Human inhibitory leukocyte Ig-like receptors: from immunotolerance to immunotherapy. JCI Insight (2022) 7(2):e151553. doi: 10.1172/jci.insight.151553
19. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol (2018) 19(1):76–84. doi: 10.1038/s41590-017-0004-z
20. Bouwstra R, van Meerten T, Bremer E. CD47-SIRPalpha blocking-based immunotherapy: Current and prospective therapeutic strategies. Clin Transl Med (2022) 12(8):e943. doi: 10.1002/ctm2.943
21. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPalpha immune checkpoint. Immunity (2020) 52(5):742–52. doi: 10.1016/j.immuni.2020.04.011
22. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
23. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142(5):699–713. doi: 10.1016/j.cell.2010.07.044
24. Muller K, Vogiatzi F, Winterberg D, Rosner T, Lenk L, Bastian L, et al. Combining daratumumab with CD47 blockade prolongs survival in preclinical models of pediatric T-ALL. Blood (2022) 140(1):45–57. doi: 10.1182/blood.2021014485
25. Liu J, Xavy S, Mihardja S, Chen S, Sompalli K, Feng D, et al. Targeting macrophage checkpoint inhibitor SIRPalpha for anticancer therapy. JCI Insight (2020) 5(12):e134728. doi: 10.1172/jci.insight.134728
26. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U.S.A. (2012) 109(17):6662–7. doi: 10.1073/pnas.1121623109
27. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by hu5F9-G4 and rituximab in non-hodgkin's lymphoma. N Engl J Med (2018) 379(18):1711–21. doi: 10.1056/NEJMoa1807315
28. Daver N, Senapati J, Maiti A, Loghavi S, Kadia TM, DiNardo CD, et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in patients (pts) with newly diagnosed (ND) older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood 140(Supplement (2022) 1):141–4. doi: 10.1182/blood-2022-170188
29. Abdallah F, Coindre S, Gardet M, Meurisse F, Naji A, Suganuma N, et al. Leukocyte immunoglobulin-like receptors in regulating the immune response in infectious diseases: A window of opportunity to pathogen persistence and a sound target in therapeutics. Front Immunol (2021) 12:717998. doi: 10.3389/fimmu.2021.717998
30. Liu WR, Kim J, Nwankwo C, Ashworth LK, Arm JP. Genomic organization of the human leukocyte immunoglobulin-like receptors within the leukocyte receptor complex on chromosome 19q13.4. Immunogenetics (2000) 51(8-9):659–69. doi: 10.1007/s002510000183
31. Storm L, Bruijnesteijn J, de Groot NG, Bontrop RE. The genomic organization of the LILR region remained largely conserved throughout primate evolution: Implications for health and disease. Front Immunol (2021) 12:716289. doi: 10.3389/fimmu.2021.716289
32. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother (2017) 66(8):1079–87. doi: 10.1007/s00262-017-2023-x
33. Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens (2004) 64(3):215–25. doi: 10.1111/j.0001-2815.2004.00290.x
34. Deng M, Chen H, Liu X, Huang R, He Y, Yoo B, et al. Leukocyte immunoglobulin-like receptor subfamily B: therapeutic targets in cancer. Antib Ther (2021) 4(1):16–33. doi: 10.1093/abt/tbab002
35. Samaridis J, Colonna M. Cloning of novel immunoglobulin superfamily receptors expressed on human myeloid and lymphoid cells: structural evidence for new stimulatory and inhibitory pathways. Eur J Immunol (1997) 27(3):660–5. doi: 10.1002/eji.1830270313
36. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity (1997) 7(2):273–82. doi: 10.1016/S1074-7613(00)80529-4
37. Reyburn HT, Mandelboim O, Vales-Gomez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature (1997) 386(6624):514–7. doi: 10.1038/386514a0
38. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol (2003) 4(9):913–9. doi: 10.1038/ni961
39. Jones DC, Roghanian A, Brown DP, Chang C, Allen RL, Trowsdale J, et al. Alternative mRNA splicing creates transcripts encoding soluble proteins from most LILR genes. Eur J Immunol (2009) 39(11):3195–206. doi: 10.1002/eji.200839080
40. Liu F, Cocker ATH, Pugh JL, Djaoud Z, Parham P, Guethlein LA. Natural LILRB1 D1-D2 variants show frequency differences in populations and bind to HLA class I with various avidities. Immunogenetics (2022) 74(6):513–25. doi: 10.1007/s00251-022-01264-7
41. Oliveira MLG, Castelli EC, Veiga-Castelli LC, Pereira ALE, Marcorin L, Carratto TMT, et al. Genetic diversity of the LILRB1 and LILRB2 coding regions in an admixed Brazilian population sample. HLA (2022) 100(4):325–48. doi: 10.1111/tan.14725
42. Fanger NA, Cosman D, Peterson L, Braddy SC, Maliszewski CR, Borges L. The MHC class I binding proteins LIR-1 and LIR-2 inhibit Fc receptor-mediated signaling in monocytes. Eur J Immunol (1998) 28(11):3423–34. doi: 10.1002/(SICI)1521-4141(199811)28:11<3423::AID-IMMU3423>3.0.CO;2-2
43. Kang X, Kim J, Deng M, John S, Chen H, Wu G, et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle (2016) 15(1):25–40. doi: 10.1080/15384101.2015.1121324
44. Bellon T, Kitzig F, Sayos J, Lopez-Botet M. Mutational analysis of immunoreceptor tyrosine-based inhibition motifs of the Ig-like transcript 2 (CD85j) leukocyte receptor. J Immunol (2002) 168(7):3351–9. doi: 10.4049/jimmunol.168.7.3351
45. Sayos J, Martinez-Barriocanal A, Kitzig F, Bellon T, Lopez-Botet M. Recruitment of C-terminal Src kinase by the leukocyte inhibitory receptor CD85j. Biochem Biophys Res Commun (2004) 324(2):640–7. doi: 10.1016/j.bbrc.2004.09.097
46. Ketroussi F, Giuliani M, Bahri R, Azzarone B, Charpentier B, Durrbach A. Lymphocyte cell-cycle inhibition by HLA-G is mediated by phosphatase SHP-2 and acts on the mTOR pathway. PloS One (2011) 6(8):e22776. doi: 10.1371/journal.pone.0022776
47. Li C, Houser BL, Nicotra ML, Strominger JL. HLA-G homodimer-induced cytokine secretion through HLA-G receptors on human decidual macrophages and natural killer cells. Proc Natl Acad Sci U.S.A. (2009) 106(14):5767–72. doi: 10.1073/pnas.0901173106
48. Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A, et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol (2011) 186(5):2990–7. doi: 10.4049/jimmunol.1003078
49. Lepin EJ, Bastin JM, Allan DS, Roncador G, Braud VM, Mason DY, et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol (2000) 30(12):3552–61. doi: 10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L
50. Navarro F, Llano M, Bellon T, Colonna M, Geraghty DE, Lopez-Botet M. The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur J Immunol (1999) 29(1):277–83. doi: 10.1002/(SICI)1521-4141(199901)29:01<277::AID-IMMU277>3.0.CO;2-4
51. Dulberger CL, McMurtrey CP, Holzemer A, Neu KE, Liu V, Steinbach AM, et al. Human leukocyte antigen F presents peptides and regulates immunity through interactions with NK cell receptors. Immunity (2017) 46(6):1018–29. doi: 10.1016/j.immuni.2017.06.002
52. Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity (1999) 11(5):603–13. doi: 10.1016/S1074-7613(00)80135-1
53. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U.S.A. (2003) 100(15):8856–61. doi: 10.1073/pnas.1431057100
54. Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H, et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem (2006) 281(15):10439–47. doi: 10.1074/jbc.M512305200
55. Held W, Mariuzza RA. Cis interactions of immunoreceptors with MHC and non-MHC ligands. Nat Rev Immunol (2008) 8(4):269–78. doi: 10.1038/nri2278
56. Nam G, Shi Y, Ryu M, Wang Q, Song H, Liu J, et al. Crystal structures of the two membrane-proximal Ig-like domains (D3D4) of LILRB1/B2: alternative models for their involvement in peptide-HLA binding. Protein Cell (2013) 4(10):761–70. doi: 10.1007/s13238-013-3908-x
57. Wang Q, Song H, Cheng H, Qi J, Nam G, Tan S, et al. Structures of the four Ig-like domain LILRB2 and the four-domain LILRB1 and HLA-G1 complex. Cell Mol Immunol (2020) 17(9):966–75. doi: 10.1038/s41423-019-0258-5
58. Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y, et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol (2008) 181(7):4742–51. doi: 10.4049/jimmunol.181.7.4742
59. Hudson LE, Allen RL. Leukocyte ig-like receptors - A model for MHC class I disease associations. Front Immunol (2016) 7:281. doi: 10.3389/fimmu.2016.00281
60. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci U.S.A. (2006) 103(44):16412–7. doi: 10.1073/pnas.0605228103
61. Arnold V, Cummings JS, Moreno-Nieves UY, Didier C, Gilbert A, Barre-Sinoussi F, et al. S100A9 protein is a novel ligand for the CD85j receptor and its interaction is implicated in the control of HIV-1 replication by NK cells. Retrovirology (2013) 10:122. doi: 10.1186/1742-4690-10-122
62. Yang Z, Bjorkman PJ. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U.S.A. (2008) 105(29):10095–100. doi: 10.1073/pnas.0804551105
63. Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T, et al. Paired Ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J Immunol (2007) 178(7):4250–9. doi: 10.4049/jimmunol.178.7.4250
64. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature (2017) 552(7683):101–5. doi: 10.1038/nature24994
65. Carenza C, Calcaterra F, Oriolo F, Di Vito C, Ubezio M, Della Porta MG, et al. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front Immunol (2019) 10:1325. doi: 10.3389/fimmu.2019.01325
66. Lewis Marffy AL, McCarthy AJ. Leukocyte immunoglobulin-like receptors (LILRs) on human neutrophils: Modulators of infection and immunity. Front Immunol (2020) 11:857. doi: 10.3389/fimmu.2020.00857
67. Tedla N, Lee CW, Borges L, Geczy CL, Arm JP. Differential expression of leukocyte immunoglobulin-like receptors on cord-blood-derived human mast cell progenitors and mature mast cells. J Leukoc Biol (2008) 83(2):334–43. doi: 10.1189/jlb.0507314
68. Lesport E, Baudhuin J, Sousa S, LeMaoult J, Zamborlini A, Rouas-Freiss N, et al. Inhibition of human gamma delta [corrected] T-cell antitumoral activity through HLA-G: implications for immunotherapy of cancer. Cell Mol Life Sci (2011) 68(20):3385–99. doi: 10.1007/s00018-011-0632-7
69. Tenca C, Merlo A, Merck E, Bates EE, Saverino D, Simone R, et al. CD85j (leukocyte Ig-like receptor-1/Ig-like transcript 2) inhibits human osteoclast-associated receptor-mediated activation of human dendritic cells. J Immunol (2005) 174(11):6757–63. doi: 10.4049/jimmunol.174.11.6757
70. Young NT, Waller EC, Patel R, Roghanian A, Austyn JM, Trowsdale J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood (2008) 111(6):3090–6. doi: 10.1182/blood-2007-05-089771
71. Merlo A, Tenca C, Fais F, Battini L, Ciccone E, Grossi CE, et al. Inhibitory receptors CD85j, LAIR-1, and CD152 down-regulate immunoglobulin and cytokine production by human B lymphocytes. Clin Diagn Lab Immunol (2005) 12(6):705–12. doi: 10.1128/CDLI.12.6.705-712.2005
72. Naji A, Menier C, Morandi F, Agaugue S, Maki G, Ferretti E, et al. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol (2014) 192(4):1536–46. doi: 10.4049/jimmunol.1300438
73. Monsivais-Urenda A, Gomez-Martin D, Santana-de-Anda K, Cruz-Martinez J, Alcocer-Varela J, Gonzalez-Amaro R. Defective expression and function of the ILT2/CD85j regulatory receptor in dendritic cells from patients with systemic lupus erythematosus. Hum Immunol (2013) 74(9):1088–96. doi: 10.1016/j.humimm.2013.05.006
74. Doniz-Padilla L, Paniagua AE, Sandoval-Correa P, Monsivais-Urenda A, Leskela S, Marazuela M, et al. Analysis of expression and function of the inhibitory receptor ILT2 in lymphocytes from patients with autoimmune thyroid disease. Eur J Endocrinol (2011) 165(1):129–36. doi: 10.1530/EJE-11-0109
75. Kuroki K, Tsuchiya N, Shiroishi M, Rasubala L, Yamashita Y, Matsuta K, et al. Extensive polymorphisms of LILRB1 (ILT2, LIR1) and their association with HLA-DRB1 shared epitope negative rheumatoid arthritis. Hum Mol Genet (2005) 14(16):2469–80. doi: 10.1093/hmg/ddi247
76. Zeller T, Lutz S, Munnich IA, Windisch R, Hilger P, Herold T, et al. Dual checkpoint blockade of CD47 and LILRB1 enhances CD20 antibody-dependent phagocytosis of lymphoma cells by macrophages. Front Immunol (2022) 13:929339. doi: 10.3389/fimmu.2022.929339
77. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21(11):799–820. doi: 10.1038/s41573-022-00520-5
78. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
79. Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest (2018) 128(12):5647–62. doi: 10.1172/JCI97570
80. Ju XS, Hacker C, Scherer B, Redecke V, Berger T, Schuler G, et al. Immunoglobulin-like transcripts ILT2, ILT3 and ILT7 are expressed by human dendritic cells and down-regulated following activation. Gene (2004) 331:159–64. doi: 10.1016/j.gene.2004.02.018
81. Khanolkar RC, Kalogeropoulos M, Lawrie A, Roghanian A, Vickers MA, Young NT. Leukocyte Ig-Like receptor B1 restrains dendritic cell function through increased expression of the NF-kappaB regulator ABIN1/TNIP1. J Leukoc Biol (2016) 100(4):737–46. doi: 10.1189/jlb.1A0915-420RRR
82. Lamar DL, Weyand CM, Goronzy JJ. Promoter choice and translational repression determine cell type-specific cell surface density of the inhibitory receptor CD85j expressed on different hematopoietic lineages. Blood (2010) 115(16):3278–86. doi: 10.1182/blood-2009-09-243493
83. Davidson CL, Li NL, Burshtyn DN. LILRB1 polymorphism and surface phenotypes of natural killer cells. Hum Immunol (2010) 71(10):942–9. doi: 10.1016/j.humimm.2010.06.015
84. Morandi F, Ferretti E, Castriconi R, Dondero A, Petretto A, Bottino C, et al. Soluble HLA-G dampens CD94/NKG2A expression and function and differentially modulates chemotaxis and cytokine and chemokine secretion in CD56bright and CD56dim NK cells. Blood (2011) 118(22):5840–50. doi: 10.1182/blood-2011-05-352393
85. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
86. Beziat V, Descours B, Parizot C, Debre P, Vieillard V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PloS One (2010) 5(8):e11966. doi: 10.1371/journal.pone.0011966
87. Peppa D, Pedroza-Pacheco I, Pellegrino P, Williams I, Maini MK, Borrow P. Adaptive reconfiguration of natural killer cells in HIV-1 infection. Front Immunol (2018) 9:474. doi: 10.3389/fimmu.2018.00474
88. Muntasell A, Pupuleku A, Cisneros E, Vera A, Moraru M, Vilches C, et al. Relationship of NKG2C copy number with the distribution of distinct cytomegalovirus-induced adaptive NK cell subsets. J Immunol (2016) 196(9):3818–27. doi: 10.4049/jimmunol.1502438
89. Boudreau JE, Hsu KC. Natural killer cell education in human health and disease. Curr Opin Immunol (2018) 50:102–11. doi: 10.1016/j.coi.2017.11.003
90. Leijonhufvud C, Reger R, Segerberg F, Theorell J, Schlums H, Bryceson YT, et al. LIR-1 educates expanded human NK cells and defines a unique antitumor NK cell subset with potent antibody-dependent cellular cytotoxicity. Clin Transl Immunol (2021) 10(10):e1346. doi: 10.1002/cti2.1346
91. Favier B, Lemaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the NK-cell activating synapse. FASEB J (2010) 24(3):689–99. doi: 10.1096/fj.09-135194
92. Chen H, Chen Y, Deng M, John S, Gui X, Kansagra A, et al. Antagonistic anti-LILRB1 monoclonal antibody regulates antitumor functions of natural killer cells. J Immunother Cancer (2020) 8(2):e000515. doi: 10.1136/jitc-2019-000515
93. Morel E, Bellon T. HLA class I molecules regulate IFN-gamma production induced in NK cells by target cells, viral products, or immature dendritic cells through the inhibitory receptor ILT2/CD85j. J Immunol (2008) 181(4):2368–81. doi: 10.4049/jimmunol.181.4.2368
94. Jacquier A, Dumont C, Carosella ED, Rouas-Freiss N, LeMaoult J. Cytometry-based analysis of HLA-G functions according to ILT2 expression. Hum Immunol (2020) 81(4):168–77. doi: 10.1016/j.humimm.2020.02.001
95. Riteau B, Menier C, Khalil-Daher I, Martinozzi S, Pla M, Dausset J, et al. HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol (2001) 13(2):193–201. doi: 10.1093/intimm/13.2.193
96. Villa-Alvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol (2018) 9:2917. doi: 10.3389/fimmu.2018.02917
97. Vivier E, Anfossi N. Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat Rev Immunol (2004) 4(3):190–8. doi: 10.1038/nri1306
98. Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, et al. T cell subset-specific susceptibility to aging. Clin Immunol (2008) 127(1):107–18. doi: 10.1016/j.clim.2007.12.002
99. Gustafson CE, Qi Q, Hutter-Saunders J, Gupta S, Jadhav R, Newell E, et al. Immune checkpoint function of CD85j in CD8 T cell differentiation and aging. Front Immunol (2017) 8:692. doi: 10.3389/fimmu.2017.00692
100. Young NT, Uhrberg M, Phillips JH, Lanier LL, Parham P. Differential expression of leukocyte receptor complex-encoded Ig-like receptors correlates with the transition from effector to memory CTL. J Immunol (2001) 166(6):3933–41. doi: 10.4049/jimmunol.166.6.3933
101. Anfossi N, Doisne JM, Peyrat MA, Ugolini S, Bonnaud O, Bossy D, et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol (2004) 173(12):7223–9. doi: 10.4049/jimmunol.173.12.7223
102. Kim A, Han CJ, Driver I, Olow A, Sewell AK, Zhang Z, et al. LILRB1 blockade enhances bispecific T cell engager antibody-induced tumor cell killing by effector CD8(+) T cells. J Immunol (2019) 203(4):1076–87. doi: 10.4049/jimmunol.1801472
103. Dumont C, Jacquier A, Verine J, Noel F, Goujon A, Wu CL, et al. CD8(+)PD-1(-)ILT2(+) T cells are an intratumoral cytotoxic population selectively inhibited by the immune-checkpoint HLA-G. Cancer Immunol Res (2019) 7(10):1619–32. doi: 10.1158/2326-6066.CIR-18-0764
104. Dietrich J, Cella M, Colonna M. Ig-like transcript 2 (ILT2)/leukocyte Ig-like receptor 1 (LIR1) inhibits TCR signaling and actin cytoskeleton reorganization. J Immunol (2001) 166(4):2514–21. doi: 10.4049/jimmunol.166.4.2514
105. Saverino D, Fabbi M, Ghiotto F, Merlo A, Bruno S, Zarcone D, et al. The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J Immunol (2000) 165(7):3742–55. doi: 10.4049/jimmunol.165.7.3742
106. Saverino D, Merlo A, Bruno S, Pistoia V, Grossi CE, Ciccone E. Dual effect of CD85/leukocyte Ig-like receptor-1/Ig-like transcript 2 and CD152 (CTLA-4) on cytokine production by antigen-stimulated human T cells. J Immunol (2002) 168(1):207–15. doi: 10.4049/jimmunol.168.1.207
107. HoWangYin KY, Caumartin J, Favier B, Daouya M, Yaghi L, Carosella ED, et al. Proper regrafting of Ig-like transcript 2 after trogocytosis allows a functional cell-cell transfer of sensitivity. J Immunol (2011) 186(4):2210–8. doi: 10.4049/jimmunol.1000547
108. Trichet V, Benezech C, Dousset C, Gesnel MC, Bonneville M, Breathnach R. Complex interplay of activating and inhibitory signals received by Vgamma9Vdelta2 T cells revealed by target cell beta2-microglobulin knockdown. J Immunol (2006) 177(9):6129–36. doi: 10.4049/jimmunol.177.9.6129
109. Campoli M, Ferrone S. HLA antigen changes in Malignant cells: epigenetic mechanisms and biologic significance. Oncogene (2008) 27(45):5869–85. doi: 10.1038/onc.2008.273
110. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
111. Godal R, Bachanova V, Gleason M, McCullar V, Yun GH, Cooley S, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol Blood Marrow Transplant (2010) 16(5):612–21. doi: 10.1016/j.bbmt.2010.01.019
112. Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of macrophage plasticity in the tumor environment: Manipulating activation state to improve outcomes. Front Immunol (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
113. Mandel I, Haves Ziv D, Goldshtein I, Peretz T, Alishekevitz D, Fridman Dror A, et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J Immunother Cancer (2022) 10(9):e004859. doi: 10.1136/jitc-2022-004859
114. Martin-Villa JM, Vaquero-Yuste C, Molina-Alejandre M, Juarez I, Suarez-Trujillo F, Lopez-Nares A, et al. HLA-G: Too much or too little? Role in cancer and autoimmune disease. Front Immunol (2022) 13:796054. doi: 10.3389/fimmu.2022.796054
115. Lefebvre S, Antoine M, Uzan S, McMaster M, Dausset J, Carosella ED, et al. Specific activation of the non-classical class I histocompatibility HLA-G antigen and expression of the ILT2 inhibitory receptor in human breast cancer. J Pathol (2002) 196(3):266–74. doi: 10.1002/path.1039
116. Erikci AA, Karagoz B, Ozyurt M, Ozturk A, Kilic S, Bilgi O. HLA-G expression in B chronic lymphocytic leukemia: a new prognostic marker? Hematology (2009) 14(2):101–5. doi: 10.1179/102453309X385197
117. Menier C, Riteau B, Carosella ED, Rouas-Freiss N. MICA triggering signal for NK cell tumor lysis is counteracted by HLA-G1-mediated inhibitory signal. Int J Cancer (2002) 100(1):63–70. doi: 10.1002/ijc.10460
118. LeMaoult J, Zafaranloo K, Le Danff C, Carosella ED. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J (2005) 19(6):662–4. doi: 10.1096/fj.04-1617fje
119. Zhang Y, Wang H, Xu X, Liu H, Hao T, Yin S, et al. Poor prognosis and therapeutic responses in LILRB1-expressing M2 macrophages-enriched gastric cancer patients. Front Oncol (2021) 11:668707. doi: 10.3389/fonc.2021.668707
120. Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P, et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res (2016) 76(8):2153–65. doi: 10.1158/0008-5472.CAN-15-1965
121. Lorenzo-Herrero S, Sordo-Bahamonde C, Martinez-Perez A, Corte-Torres MD, Fernandez-Vega I, Solis-Hernandez MP, et al. Immunoglobulin-like transcript 2 blockade restores antitumor immune responses in glioblastoma. Cancer Sci (2023) 114(1):48–62. doi: 10.1111/cas.15575
122. Nunez SY, Ziblat A, Secchiari F, Torres NI, Sierra JM, Raffo Iraolagoitia XL, et al. Human M2 macrophages limit NK cell effector functions through secretion of TGF-beta and engagement of CD85j. J Immunol (2018) 200(3):1008–15. doi: 10.4049/jimmunol.1700737
123. Roberti MP, Julia EP, Rocca YS, Amat M, Bravo AI, Loza J, et al. Overexpression of CD85j in TNBC patients inhibits Cetuximab-mediated NK-cell ADCC but can be restored with CD85j functional blockade. Eur J Immunol (2015) 45(5):1560–9. doi: 10.1002/eji.201445353
124. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121(9):3609–22. doi: 10.1172/JCI45816
125. Vittrant B, Bergeron A, Molina OE, Leclercq M, Legare XP, Hovington H, et al. Immune-focused multi-omics analysis of prostate cancer: leukocyte Ig-Like receptors are associated with disease progression. Oncoimmunology (2020) 9(1):1851950. doi: 10.1080/2162402X.2020.1851950
126. Xu X, Yin S, Wang Y, Zhu Q, Zheng G, Lu Y, et al. LILRB1(+) immune cell infiltration identifies immunosuppressive microenvironment and dismal outcomes of patients with ovarian cancer. Int Immunopharmacol (2023) 119:110162. doi: 10.1016/j.intimp.2023.110162
127. Kang X, Lu Z, Cui C, Deng M, Fan Y, Dong B, et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat Cell Biol (2015) 17(5):665–77. doi: 10.1038/ncb3158
128. Urosevic M, Kamarashev J, Burg G, Dummer R. Primary cutaneous CD8+ and CD56+ T-cell lymphomas express HLA-G and killer-cell inhibitory ligand, ILT2. Blood (2004) 103(5):1796–8. doi: 10.1182/blood-2003-10-3372
129. Nikolova M, Musette P, Bagot M, Boumsell L, Bensussan A. Engagement of ILT2/CD85j in Sezary syndrome cells inhibits their CD3/TCR signaling. Blood (2002) 100(3):1019–25. doi: 10.1182/blood-2001-12-0303
130. Kamarashev J, Burg G, Mingari MC, Kempf W, Hofbauer G, Dummer R. Differential expression of cytotoxic molecules and killer cell inhibitory receptors in CD8+ and CD56+ cutaneous lymphomas. Am J Pathol (2001) 158(5):1593–8. doi: 10.1016/S0002-9440(10)64114-4
131. Harly C, Peyrat MA, Netzer S, Dechanet-Merville J, Bonneville M, Scotet E. Up-regulation of cytolytic functions of human Vdelta2-gamma T lymphocytes through engagement of ILT2 expressed by tumor target cells. Blood (2011) 117(10):2864–73. doi: 10.1182/blood-2010-09-309781
132. Naji A, Menier C, Maki G, Carosella ED, Rouas-Freiss N. Neoplastic B-cell growth is impaired by HLA-G/ILT2 interaction. Leukemia (2012) 26(8):1889–92. doi: 10.1038/leu.2012.62
133. Lozano E, Diaz T, Mena MP, Sune G, Calvo X, Calderon M, et al. Loss of the immune checkpoint CD85j/LILRB1 on Malignant plasma cells contributes to immune escape in multiple myeloma. J Immunol (2018) 200(8):2581–91. doi: 10.4049/jimmunol.1701622
134. Zhang Y, Lu N, Xue Y, Zhang M, Li Y, Si Y, et al. Expression of immunoglobulin-like transcript (ILT)2 and ILT3 in human gastric cancer and its clinical significance. Mol Med Rep (2012) 5(4):910–6. doi: 10.3892/mmr.2012.744
135. Tronik-Le Roux D, Sautreuil M, Bentriou M, Verine J, Palma MB, Daouya M, et al. Comprehensive landscape of immune-checkpoints uncovered in clear cell renal cell carcinoma reveals new and emerging therapeutic targets. Cancer Immunol Immunother (2020) 69(7):1237–52. doi: 10.1007/s00262-020-02530-x
136. Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods (2014) 65(1):114–26. doi: 10.1016/j.ymeth.2013.06.035
137. Tam SH, McCarthy SG, Armstrong AA, SOmani S, Wu SJ, Liu X, et al. Functional, biophysical, and structural characterization of human igG1 and igG4 fc variants with ablated immune functionality. Antibodies (Basel) (2017) 6(3):12. doi: 10.3390/antib6030012
138. Mondal K, Song C, Tian J, Ho C, Rivera LB, Huang J, et al. Preclinical evaluation of NGM707, a novel anti-ILT2/anti-ILT4 dual antagonist monoclonal antibody. Cancer Res (2021) 81(13(13 (Supplement):LB156. doi: 10.1158/1538-7445.AM2021-LB156
139. Strohl WR. Current progress in innovative engineered antibodies. Protein Cell (2018) 9(1):86–120. doi: 10.1007/s13238-017-0457-8
140. Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, Ho JM, et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med (2012) 209(10):1883–99. doi: 10.1084/jem.20120502
141. Belaunzaran OM, Rafiei A, Kumar A, Gualandi M, Westphal M, Vogt L, et al. IosH2 exerts potent anti-tumor activity by blocking LILRB1/2 and KIR3DL1 receptor signaling. J Immunother Cancer (2021) 9(Suppl 2):A906. doi: 10.1136/jitc-2021-SITC2021.865
142. Peluso MO, Adam A, Armet CM, Zhang L, O'Connor RW, Lee BH, et al. The Fully human anti-CD47 antibody SRF231 exerts dual-mechanism antitumor activity via engagement of the activating receptor CD32a. J Immunother Cancer (2020) 8(1):e000413. doi: 10.1136/jitc-2019-000413
143. Udartseva O, Pupecka-Swider M, Garcia-Broncano P, Guo N, Campbell S, Klakus E, et al. AGEN1571 is a novel high-affinity ILT2 antagonist antibody that promotes adaptive and innate immune responses. Cancer Res (2022) 82(12_Supplement):2906. doi: 10.1158/1538-7445.AM2022-2906
144. Naing A, Wang JS, Sharma MR, Sommerhalder D, Gandhi L, Oh D-Y, et al. First-in-human study of NGM707, an ILT2/ILT4 dual antagonist antibody in advanced or metastatic solid tumors: Preliminary monotherapy dose escalation data. Immunooncol Technol (2022) 16(1(1 (suppl.):174P. doi: 10.1016/j.iotech.2022.100286
145. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
146. Kamber RA, Nishiga Y, Morton B, Banuelos AM, Barkal AA, Vences-Catalan F, et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature (2021) 597(7877):549–54. doi: 10.1038/s41586-021-03879-4
147. Li D, Xiong W, Wang Y, Feng J, He Y, Du J, et al. SLAMF3 and SLAMF4 are immune checkpoints that constrain macrophage phagocytosis of hematopoietic tumors. Sci Immunol (2022) 7(67):eabj5501. doi: 10.1126/sciimmunol.abj5501
148. Cockram TOJ, Dundee JM, Popescu AS, Brown GC. The phagocytic code regulating phagocytosis of mamMalian cells. Front Immunol (2021) 12:629979. doi: 10.3389/fimmu.2021.629979
149. Yeboah M, Papagregoriou C, Jones DC, Chan HTC, Hu G, McPartlan JS, et al. LILRB3 (ILT5) is a myeloid cell checkpoint that elicits profound immunomodulation. JCI Insight (2020) 5(18):e141593. doi: 10.1172/jci.insight.141593
150. Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med (2021) 218(7):e20201811. doi: 10.1084/jem.20201811
151. Siu LL, Wang D, Hilton J, Geva R, Rasco D, Perets R, et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res (2022) 28(1):57–70. doi: 10.1158/1078-0432.CCR-21-2160
152. Papadopoulos KP, Lakhani NJ, Yap TA, Naumovski AL, Brown KS, Umiker B, et al. Phase 1, first-in-human trial of JTX-8064, an anti-LILRB2/ILT4 monoclonal antibody, as monotherapy and in combination with anti-PD-1 in adult patients with advanced solid tumors (INNATE). J Clin Oncol (2021) 39(15_suppl):TPS2672. doi: 10.1200/JCO.2021.39.15_suppl.TPS2672
153. Taylor MH, Patel MR, Powderly JD, Woodard P, Chung L, Tian H, et al. A first-in-human phase 1 trial of IO-108, an antagonist antibody targeting LILRB2 (ILT4), as monotherapy and in combination with pembrolizumab in adult patients with advanced relapsed or refractory solid tumors: Dose escalation study. Cancer Res (2023) 83(8 Suppl):CT040. doi: 10.1158/1538-7445.AM2023-CT040
154. Naing A, Powderly J, Pelster M, Spira A, Schneider R, Woodard P, et al. A phase 1 trial of IO-202, an antagonist antibody targeting myeloid checkpoint LILRB4 (ILT3), as monotherapy and in combination with pembrolizumab in adult patients with advanced relapsed or refractory solid tumors. J Immunother Cancer (2022) 10(Suppl 2):A780. doi: 10.1136/jitc-2022-SITC2022.0747
155. DiNardo C, Pollyea D, Aribi A, Jonas B, Jeyakumar D, Roboz G, et al. A first-in-human phase 1 study of IO-202 (Anti-LILRB4 mab) in acute myeloid leukemia (AML) with monocytic differentiation and chronic myelomonocytic leukemia (CMML) patients. HemaSphere (2023) 7(S3):925. doi: 10.1097/01.HS9.0000969052.60533.5a
Keywords: LILRB1 (ILT2), macrophages, cancer, phagocytosis, immune checkpoint blockade, antibody therapy, NK cells, T cells
Citation: Zeller T, Münnich IA, Windisch R, Hilger P, Schewe DM, Humpe A and Kellner C (2023) Perspectives of targeting LILRB1 in innate and adaptive immune checkpoint therapy of cancer. Front. Immunol. 14:1240275. doi: 10.3389/fimmu.2023.1240275
Received: 14 June 2023; Accepted: 08 August 2023;
Published: 13 September 2023.
Edited by:
Alexandre P. A. Theocharides, University Hospital Zürich, SwitzerlandReviewed by:
Chengcheng “Alec” Zhang, University of Texas Southwestern Medical Center, United StatesCopyright © 2023 Zeller, Münnich, Windisch, Hilger, Schewe, Humpe and Kellner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Kellner, Y2hyaXN0aWFuLmtlbGxuZXJAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.