 Emily Horner
Emily Horner Janet M. Lord
Janet M. Lord Jon Hazeldine
Jon Hazeldine- 1Institute of Inflammation and Ageing, University of Birmingham, Birmingham, United Kingdom
- 2National Institute for Health Research Surgical Reconstruction and Microbiology Research Centre, Queen Elizabeth Hospital Birmingham, Birmingham, United Kingdom
Associated with the development of hospital-acquired infections, major traumatic injury results in an immediate and persistent state of systemic immunosuppression, yet the underlying mechanisms are poorly understood. Detected in the circulation in the minutes, days and weeks following injury, damage associated molecular patterns (DAMPs) are a heterogeneous collection of proteins, lipids and DNA renowned for initiating the systemic inflammatory response syndrome. Suggesting additional immunomodulatory roles in the post-trauma immune response, data are emerging implicating DAMPs as potential mediators of post-trauma immune suppression. Discussing the results of in vitro, in vivo and ex vivo studies, the purpose of this review is to summarise the emerging immune tolerising properties of cytosolic, nuclear and mitochondrial-derived DAMPs. Direct inhibition of neutrophil antimicrobial activities, the induction of endotoxin tolerance in monocytes and macrophages, and the recruitment, activation and expansion of myeloid derived suppressor cells and regulatory T cells are examples of some of the immune suppressive properties assigned to DAMPs so far. Crucially, with studies identifying the molecular mechanisms by which DAMPs promote immune suppression, therapeutic strategies that prevent and/or reverse DAMP-induced immunosuppression have been proposed. Approaches currently under consideration include the use of synthetic polymers, or the delivery of plasma proteins, to scavenge circulating DAMPs, or to treat critically-injured patients with antagonists of DAMP receptors. However, as DAMPs share signalling pathways with pathogen associated molecular patterns, and pro-inflammatory responses are essential for tissue regeneration, these approaches need to be carefully considered in order to ensure that modulating DAMP levels and/or their interaction with immune cells does not negatively impact upon anti-microbial defence and the physiological responses of tissue repair and wound healing.
1 Introduction
Accounting for an estimated 16% of the global burden of disease (1), and the leading cause of death in individuals aged ≤44 years (2), traumatic injuries are a significant cause of morbidity and mortality. However, due in part to advances in trauma medicine, most notably in the management of coagulopathy where haemorrhage-induced deaths have fallen by more than 12% since the 1980s (3), the likelihood of patients surviving the initial effects of a severe trauma is increasing (4). Consequently, it is the onset of secondary complications such as multiple organ failure (MOF), multiple organ dysfunction syndrome (MODS) and nosocomial infections that are emerging as major determinants of clinical outcomes. Indeed, when compared to those with non-eventful episodes, trauma patients who develop MOF, MODS or hospital-acquired infections (HAIs) experience increased lengths of intensive care unit stay, require prolonged mechanical ventilation, report poorer physical and psychological recovery, and are less likely to return to work (5–10). Of these secondary complications, HAIs are highly prevalent (5, 6, 11–14) and remain a significant clinical challenge, with one recent study estimating that the prevention of pneumonia amongst hospitalised trauma patients would reduce the incidence of end organ dysfunction, mortality and the need for mechanical ventilation by 22.1%, 7.8% and 6.8% respectively (13).
Two opposing and concurrent syndromes underpin the immune and inflammatory response to major trauma. With the intention of localising and eliminating endogenous stressors, and promoting wound repair, a systemic inflammatory response syndrome (SIRS) is triggered within minutes of injury (15). Associated with such physiological changes as tachycardia and tachypnea, the SIRS response is characterised by elevated levels of circulating pro-inflammatory cytokines and immune cell activation. To counteract this state of systemic hyperinflammation, the SIRS response is accompanied by a compensatory anti-inflammatory response syndrome (CARS) (16). Defined in part by raised circulating levels of anti-inflammatory cytokines, lymphocyte apoptosis and decreased cytokine production by endotoxin challenged monocytes, the concomitant CARS response aims to restore immunological homeostasis (17). Over the past decade, it has become increasingly evident that the kinetics of the simultaneous SIRS and CARS responses markedly influence the clinical trajectories of hospitalised trauma patients, with robust, dysregulated and prolonged SIRS/CARS responses associated with delayed physical recovery, organ dysfunction, an increased risk of nosocomial infections and mortality (16, 18, 19). Furthermore, failure to resolve trauma-induced inflammatory responses can lead to the development of persistent inflammation, immunosuppression and catabolism syndrome (PICS). Detected in >40% of patients with chronic critical illness, PICS has been suggested to have replaced MOF as the predominant phenotype of chronic critical illness (20–23). Characterised by lymphopenia, neutrophilia, a persistent acute phase response and ongoing protein catabolism, the development of PICS predisposes to recurrent HAIs, poor wound healing and a greater need for post-discharge rehabilitation in long-term care facilities (20–24).
Hypothesising that factors released into the circulation or present in wound fluids may modulate the local and systemic immune response to sterile injury, several groups have investigated the phenotypic and functional consequences of culturing immune cells isolated from healthy subjects or mice with plasma, serum or wound fluids obtained from their injured counterparts (25–28). Compared to control samples, plasma acquired from trauma patients at day 1 post-injury, wound fluids obtained either 3- or 6-hours post-surgery or from closed drains in injury sites, and serum collected from mice within 30 minutes of injury, were found to promote the expansion of both CD8+ T cells and CD11c+ Natural Killer (NK) cells, and to suppress the anti-microbial responses of neutrophils and monocytes (25–28). These are all features reminiscent of the trauma associated SIRS and CARS responses (15, 29–31). Whilst recognising that a multitude of factors are present in the circulation of trauma patients, the authors suggested a potential role for damage associated molecular patterns (DAMPs) in mediating the effects they observed (25, 26, 28). Detected in the circulation of major trauma patients in the minutes, hours and days following injury (30–33), DAMPs, also known as alarmins, are a heterogeneous collection of cytosolic, nuclear and mitochondrial-derived lipids, proteins and DNA. Passively released from damaged/necrotic tissue, and actively secreted by immune cells (34, 35), DAMPs are detected primarily by pathogen recognition receptors (PRRs), particularly members of the Toll-like receptor (TLR) family (36) (Table 1). To date, much attention has focussed on the immune stimulatory properties of DAMPs (69, 70), with studies showing that in vitro exposure to mitochondrial, nuclear and cytosolic-derived DAMPs can trigger ROS generation, degranulation, formation of extracellular traps and the production of pro-inflammatory cytokines by neutrophils, monocytes, macrophages and invariant NKT cells (33, 71–75). Similar pro-inflammatory responses have also been observed in animal studies, where administration of mitochondrial-derived DAMPs (mtDAMPs) has been shown to increase plasma cytokine concentrations and activate neutrophils, systemic responses that result in immune-mediated organ injury (33, 76). Importantly, these observations are aligned with the results of prospective studies of trauma patients, where elevated circulating concentrations of mitochondrial-derived DNA (mtDNA) and the nuclear-derived DAMP high mobility group box-1 (HMGB-1) are associated with the development of SIRS and MODS (76–80).
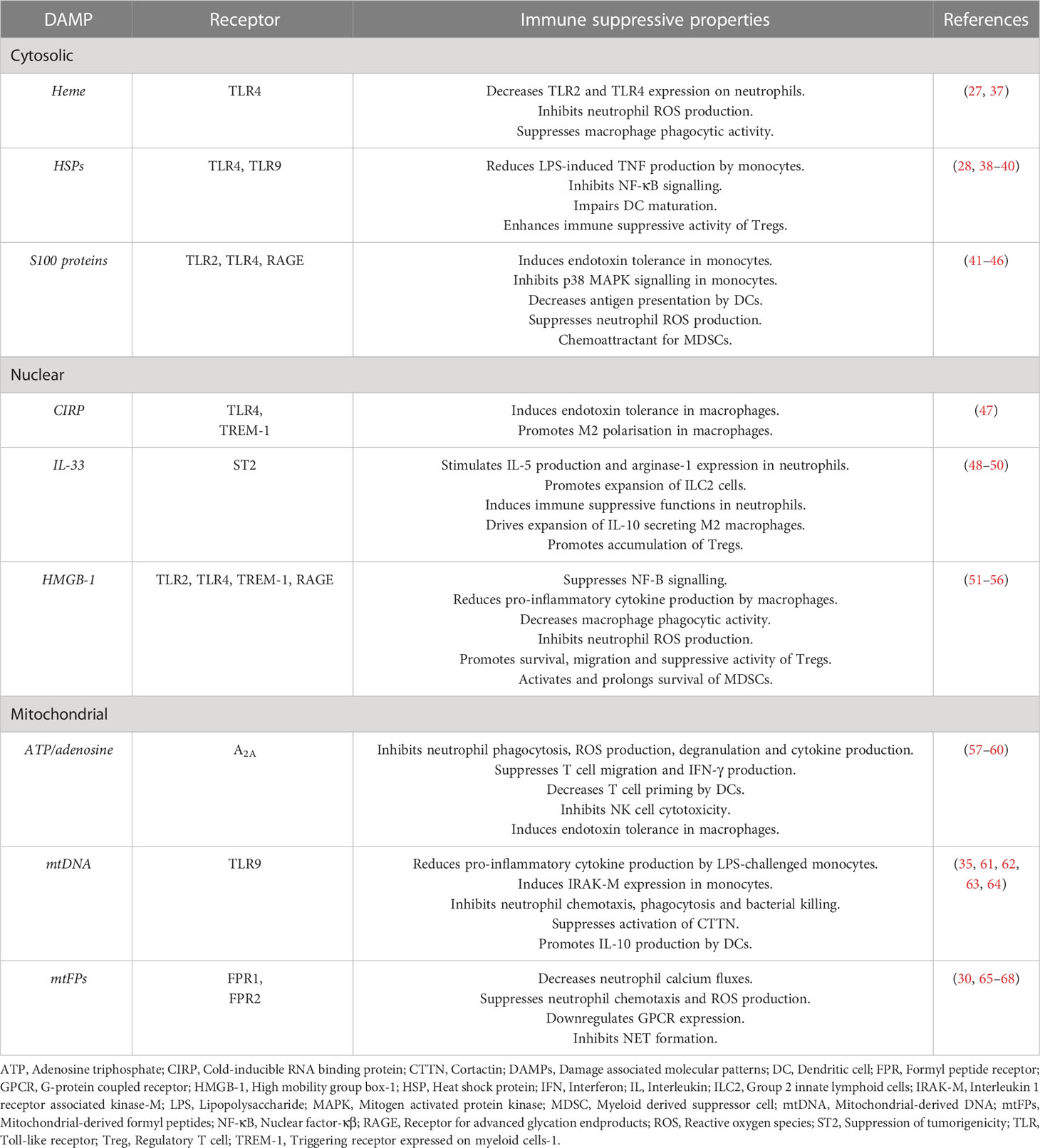
Table 1 Summary of the immune suppressive properties of DAMPs.
Interestingly, in addition to their widely accepted role as instigators of the SIRS response, data are emerging that implicates DAMPs as potential mediators of the CARS response, with in vitro studies demonstrating that neutrophils and monocytes pre-treated with cytosolic and/or mitochondrial-derived DAMPs exhibit a state of functional tolerance upon secondary stimulation with such inflammatory agonists as bacterial endotoxins and chemokines (30, 35, 41, 61, 65, 81–83) (Table 1). This DAMP-induced immune suppression may also extend to the adaptive arm of the immune system, where it has been suggested that, by promoting interleukin-4 production and T helper 2 immunity, DAMPs may suppress pro-inflammatory T helper 1 responses (81, 84). Alongside DAMPs that are passively released from damaged/necrotic tissue, a novel class of de novo synthesised DAMPs with immunosuppressive properties, termed suppressing/inhibiting inducible DAMPs (SAMPs), have recently been described. Produced by activated leukocytes, SAMPs, which include prostaglandin E2 and Annexin A1, have been suggested to promote exaggerated anti-inflammatory and hyper-resolution responses that culminate in systemic immunosuppression (82). Thus, exaggerated and/or persistent DAMP driven resolution responses may predispose the hospitalised trauma patient to the development of secondary infections (82). Supporting this idea, a study of 166 adult trauma patients found plasma levels of both mtDNA and cell-free nuclear DNA (used as a generic readout for cell damage and death) were significantly higher at hospital admission in subjects who subsequently developed an infection within 28 days (31). Moreover, the authors reported that plasma concentrations of the cytosolic DAMP, heat shock protein-70 (HSP-70), and nuclear DNA (nDNA) both correlated negatively with the expression of HLA-DR on monocytes (31), a marker of immune competence whose downregulation is associated with a higher risk of nosocomial infections in critically ill patients (85–87).
Proposed as a potential explanation for the failures of randomised clinical trials that have tested the practicalities and benefits of immunomodulatory interventions for the treatment of critically-ill patients, compartmentalisation is a concept that refers to how immune responses differ between anatomical sites within the body (88). Examples of compartmentalisation include the varied phenotypic and morphological features described for neutrophils isolated from the blood and lungs of patients with acute respiratory distress syndrome (ARDS) (89), and the different mediators that drive the phagocytic dysfunction of peripheral blood and alveolar residing neutrophils in critically ill patients (90). To date, almost all studies that have investigated DAMP-induced immunosuppression in an ex vivo setting have examined the functional and phenotypic features of circulating leukocytes. Thus, whilst one may assume that DAMPs, whose concentrations at sites of tissue damage exceed those measured in the circulation (91), would also suppress localised immune responses, there is a current paucity of information on whether DAMPs can modulate the behaviours of tissue resident immune cells. That said, with results from a small number of studies demonstrating a role for DAMPs in the impaired ex vivo responses of macrophages and T cells (47, 48, 92), and in promoting the expansion of tissue resident myeloid-derived suppressor cells (MDSCs) (93), it does appear that the release of DAMPs from damaged tissue may contribute to the immune dysfunction that develops in different tissue spaces following major trauma.
Bringing together data generated from in vitro, in vivo and ex vivo studies, the purpose of this review is to summarise our current understanding of DAMP-induced immunosuppression in the setting of sterile injury. The immune suppressive properties of cytosolic, nuclear and mitochondrial-derived DAMPs will be discussed, and where identified, their underlying mechanisms described. Furthermore, with the aim of reducing the susceptibility of critically-ill patients to HAIs, we will highlight the therapeutic strategies that are under consideration as potential approaches for preventing or reversing DAMP-induced immunosuppression.
2 Immunosuppressive properties of DAMPs
2.1 Cytosolic-derived DAMPs
2.1.1 Heme
Heme is an iron containing porphyrin synthesised in the cytosol and mitochondria of erythrocytes. Acting as the prosthetic group for such hemoproteins as haemoglobin, myoglobin and cytochrome P450, heme plays an essential role in a range of biological processes, which include oxygen transport and storage, electron transfer and cellular metabolism (94). However, when released from hemoproteins, unbound heme promotes oxidative stress, inflammation and tissue injury via its ability to catalyse the formation of highly reactive oxygen free radicals, amplify TLR-mediated immune responses and activate the complement cascade (94–98). Thus, in the setting of critical illness, links have been suggested between elevated concentrations of cell-free heme, the SIRS response and organ injury (99). To counteract its cytotoxicity, circulating levels of cell-free heme are regulated primarily by the endogenous heme-binding plasma protein hemopexin (100), with this scavenging system supported by albumin, α-2-macroglobulin and haptoglobin, three heme/haemoglobin binding plasma proteins that assist in the neutralisation, recycling and removal of free heme and iron (94, 95, 99).
Compared to healthy controls, significantly higher concentrations of free heme have been detected in plasma samples obtained from trauma patients at day 1 post-injury (27). Interestingly, insights from a murine model of non-lethal blunt liver trauma suggest a rapid emergence and clearance of circulating heme post-injury, with serial measurements across a 24-hour window (0.5, 1, 4 and 24 hours) detecting a significant trauma-induced increase in plasma heme concentrations only in samples obtained within 30 minutes of injury (27). This apparent discordance in the kinetics of heme release, and its persistence, between humans and mice could potentially reflect species differences in the scavenging of free heme. For instance, in murine models of trauma and burn wound infections, significant increases in plasma levels of hemopexin and haptoglobin have been reported (27, 101, 102), whereas in cohorts of trauma and burns patients, plasma concentrations of hemopexin, haptoglobin and α-2-macroglobulin were found to be comparable to or lower than the levels detected in healthy controls (27, 101–103). In line with these observations, a study of fifty stably resuscitated trauma haemorrhage patients found that within 2-3 hours of resuscitation, plasma hemopexin levels were significantly lower than those of free heme, resulting in a hemopexin:free heme ratio of <1 (37). In addition to the act of trauma itself, medical interventions may influence circulating heme levels, with studies reporting long-term storage of leukoreduced red blood cells resulted in heme release (37, 104, 105). Consequently, when compared to fresh red blood cells, resuscitation with stored red blood cells results in significantly elevated circulating concentrations of free heme (37).
Results of in vitro and in vivo studies have provided direct evidence of the immune suppressive properties of heme (27, 37). In a murine model of Staphylococcus aureus (S.aureus) challenge, Lee et al. recovered significantly higher bacterial loads from the bronchoalveolar lavage fluid samples of mice that had been administered heme prior to infection (27), whilst impaired bacterial clearance was reported in hemopexin knockout mice challenged with S.aureus following a liver crush injury (27). Moreover, scavenging free heme via systemic administration of hemopexin in mice, either following blunt liver trauma, or prior to resuscitation with stored red blood cells, was shown to improve the clearance rates of S.aureus and Pseudomonas aeruginosa (27, 37). At the cellular level, data points towards suppression of the innate immune response as being the mechanism by which heme treatment delays the elimination of invading pathogens, with analysis of circulating neutrophils isolated from heme treated mice revealing significantly reduced surface expression of TLR2 and TLR4 (27). Moreover, culturing neutrophils from naïve mice in the presence of serum collected from mice within 30 minutes of a liver crush injury, when circulating heme levels peak, reduced ROS production in response to S.aureus challenge (27). As well as neutrophils, heme treatment has been reported to inhibit the phagocytic activity of macrophages by 90% (37). Interestingly, this inhibitory effect appears to be mediated by a heme-induced release of HMGB-1 by activated macrophages since treating macrophages with an anti-HMGB-1 blocking antibody prior to heme exposure restored their phagocytic activity to a level comparable to that of untreated controls (37).
Due to its well-established cytotoxic, pro-inflammatory and immune amplifying properties, cell-free heme has previously been discussed as a potential therapeutic target for alleviating systemic inflammation and organ injury in critically-ill patients. Heme scavenging via hemopexin therapy and the prevention of heme-induced activation of TLR4 are two examples of strategies considered to date (101, 105). Now, with data implicating heme as a potential mediator of trauma-induced immunosuppression, the idea that modulating the circulating levels of cell-free heme could reduce patient susceptibility to HAIs has been raised (27). However, our understanding of cell-free heme in the setting of traumatic injury, and its relationship with infections, must improve to test the feasibility of such a strategy. As mentioned above, animal-based studies suggest that heme levels peak within 30 minutes of injury, with levels returning to those found in uninjured controls within 4 hours (27). Currently, our knowledge of trauma-induced heme release in humans is limited to a single study whose sample acquisition began post-hospital admission, suggesting that the levels of heme reported to date may not reflect peak concentrations (27). Moreover, no study to our knowledge has examined whether a relationship exists between circulating levels of heme, hemopexin and/or haptoglobin, and the development of infections in hospitalised trauma patients. Finally, given the differences that appear to exist between rodents and humans in how trauma impacts the levels of cell-free heme and/or its scavenger proteins (27, 101–103), using murine models to test potential therapeutic strategies for counteracting heme-induced immunosuppression in human patients may be inappropriate.
2.1.2 Heat shock proteins
A heterogeneous family of proteins, HSPs are molecular chaperones implicated in protein folding, stabilisation and transport (106). Constitutively expressed, and induced by such stressors as heat shock and UV radiation, HSPs are released into the extracellular environment passively, from necrotic tissue, or actively via the non-classic protein release pathway, where they are secreted as free proteins or packaged within extracellular vesicles (107, 108). Linked to the pathology of several chronic inflammatory conditions, HSPs are highly immunogenic, with studies demonstrating their capacity to stimulate cytokine production by monocytes and macrophages (107, 109), promote the maturation of dendritic cells (107) and activate the complement system (110).
Positively associated with injury severity scores (ISS), and indices of illness severity (e.g. sequential organ failure assessment score and simplified acute physiology score 3), significantly higher concentrations of HSP60, HSP70, HSP72 and/or HSP90 alpha have been measured in the circulation of polytrauma and traumatically brain-injured (TBI) patients when compared to healthy controls (31, 111–114). Correlating positively with systemic concentrations of interleukin (IL)-6 and IL-8, elevated levels of HSPs have been detected within minutes of injury and persist for up to ten days (31, 112). Intracellular expression of HSPs are also increased post-trauma, with an injury-induced induction of HSP27, HSP32, HSP60, HSP70 and HSP90 reported in neutrophils and/or monocytes isolated from major burns and trauma patients (115–117). In accordance with their pro-inflammatory nature, prospective studies have measured significantly higher concentrations of HSP60 at hospital admission (118) and HSP70 at 24- and 48-hours post-injury (111) in trauma patients that subsequently develop acute lung injury or MODS respectively. Interestingly, and suggesting a dual inflammatory role for HSPs, Ren et al. detected significantly higher serum concentrations of HSP70 in infected trauma patients 60-90 hours post-injury when compared to their non-infected counterparts (113), whilst a study of 166 adult trauma patients reported plasma HSP70 levels at hospital admission were negatively associated with leukocyte HLA-DR expression (31). Thus, a role for HSPs in mediating the post-injury CARS response has been proposed (28, 119), a theory supported by a body of literature that demonstrates the immune suppressive properties of HSPs.
Using TLR-induced pro-inflammatory cytokine production as a readout of anti-microbial capacity, HSPs have been shown to induce functional tolerance in innate phagocytic cells. Compared to untreated controls, primary human monocytes, or differentiated THP-1 cells, treated with HSP70 exhibit impaired tumour necrosis factor alpha (TNF) production in response to stimulation with lipopolysaccharide (LPS), S.aureus peptidoglycan or Salmonella typhimurium flagellin (28, 38, 83, 120). Mechanistically, the HSP70-induced impairment in TNF production triggered by LPS stimulation has been attributed to reduced gene transcription, with studies demonstrating HSP70 treatment inhibits activation of the nuclear factor-κβ (NF-κβ) signalling pathway and promotes enrichment of the transcriptional repressors heat shock transcription factor 1 and constitutive HSE-binding factor at the TNF promoter (38, 83). These tolerance-inducing properties of HSP70 contradict data showing that this protein can stimulate monocytes/macrophages to secrete TNF (107, 109). Addressing these contrasting observations, studies have suggested that endotoxin contamination within HSP70 preparations may explain the cytokine-inducing capacity assigned to this DAMP (38, 121). In addition to innate responses, HSPs exert suppressive effects on the adaptive arm of the immune system. Culminating in reduced T cell responses (122), HSP70 treatments have been shown to (1): enhance the immunosuppressive activity of regulatory T cells (39) (2); induce a tolerogenic phenotype in monocyte-derived dendritic cells (DC) (122) (3); impair DC maturation (40) (4); suppress the formation of T helper 1 cells (123). Thus, given the abundance of data demonstrating immune suppressive properties for HSPs, and the suggestion that LPS contamination of preparations may explain their reported pro-inflammatory nature, the idea that HSPs are “DAMPERs” rather than DAMPs appears more likely (124).
2.1.3 S100 proteins
Located within the cytoplasm of leukocytes, microglia and astrocytes, S100 proteins are a family of calcium binding proteins involved in the regulation of cellular proliferation, metabolism and division (125, 126). Detected by the PRRs TLR2 and TLR4, and the receptor for advanced glycation endproducts (RAGE), elevated levels of S100A8, S100A9 and S100B have been detected in the circulation of TBI, polytrauma and burns patients at hospital admission and persist for up to 7 days (41, 127–129). Positively correlated with ISS, percentage total body surface area (TBSA) burn and Glasgow coma scale score (41, 129–131), prospective studies have reported significantly higher levels of S100 proteins as early as day one of injury in patients who subsequently experience poor clinical outcomes such as mortality or long-term disability (41, 127, 130–134). By augmenting the SIRS responses, the anti-apoptotic (135) and pro-inflammatory properties of S100 proteins, which include the activation of neutrophils (136), the amplification of TNF production by monocytes (137) and the promotion of leukocyte transepithelial migration (138), offer potential explanations for how injury-induced elevations in S100 proteins may contribute to poor clinical trajectories.
Studies that have investigated the immune modulatory functions of S100 proteins have shown S100A8, S100A9 and calprotectin, an S100A8/A9 heterodimer, possess immune tolerising properties. In an elegant series of experiments, Austermann et al, who measured acutely and persistently elevated serum concentrations of calprotectin in polytrauma and burns patients, uncovered the molecular mechanisms by which S100A8/A9 inhibits monocyte function (41). Resulting in impaired LPS-induced TNF production, the authors demonstrated that prior exposure of monocytes to calprotectin led to heterochromatin formation at the TNF promoter (41). Underlying this epigenetic modification was a calprotectin-induced recruitment to the TNF promoter of G9a, a methyltransferase that suppresses gene transcription via demethylation of histone H3 on lysine K9 (41). In line with this finding, other studies have shown that pre-treating macrophages or monocytes with S100A8 alone can attenuate pro-inflammatory cytokine production to bacterial challenge, with this induction of tolerance a consequence of reduced activation of the P38 mitogen activated protein kinase (MAPK) signalling pathway following ligation of TLR2/TLR4 (42). In addition to inhibiting monocyte function, S100A8, S100A9 and/or calprotectin have been reported to (1): suppress T cell priming by reducing antigen presentation by DCs (43) (2); inhibit neutrophil chemotaxis (44, 139) (3); suppress LPS and phorbol 12 myristate 13 acetate (PMA)-induced ROS production by neutrophils, with this inhibition attributed to an S100A8 and S100A9 mediated production of adenosine metabolites (140). As well as direct effects on immune cells, raised concentrations of S100 proteins at sites of tissue damage may create a localised immune suppressive environment as both S100A8 homodimers and calprotectin have been shown to act as chemoattractants for MDSCs (45, 46). Through binding to carboxylated N-glycans on such cell surface receptors as RAGE, calprotectin triggers NF-κβ signalling in MDSCs, with MDSC activation resulting in the synthesis and secretion of S100A8/A9 heterodimers (45). Thus, a post-injury vicious cycle of immunosuppression could be established, in which S100 proteins from damaged tissue not only directly suppress the function of infiltrating leukocytes, but recruit MDSCs, whose subsequent activation and production of calprotectin exacerbates existing immune dysfunction and promotes additional immunosuppression.
Interestingly, in the only study to our knowledge to have investigated the relationship between circulating calprotectin and infectious episodes in hospitalised trauma patients, Joly and colleagues found that when compared to their non-infected counterparts, patients who developed a HAI exhibited a smaller increase in plasma calprotectin concentrations during the first five days of hospitalisation (141). If calprotectin exerts suppressive effects on innate and adaptive immune cells, how can a smaller increase in its circulating levels make patients prone to infection? Since calprotectin is secreted by activated neutrophils and monocytes (142, 143), the authors speculated that their findings may reflect a sign of systemic immune exhaustion and thus decreased anti-microbial activity, thereby increasing patient susceptibility to HAIs (141).
2.2 Nuclear-derived DAMPs
2.2.1 Cold-inducible RNA binding protein
A regulator of mRNA translation and cell proliferation, CIRP is an RNA chaperone protein, which in the steady state resides in the nucleus (144, 145). In times of stress, triggered by mild hypothermia, hypoxia or oxidation, CIRP is transported into the cytosol, where it is actively secreted, either via the lysosomal pathway or through gasdermin D membrane channels (145–148), or released passively into the extracellular space as a consequence of necroptosis (149).
Signalling through TLR4, triggering receptor expressed on myeloid cells-1 (TREM-1) and NF-κβ (147, 150–153), extracellular CIRP (eCIRP) has been shown to: (1) induce TNF production, ROS generation and extracellular trap formation by macrophages and monocytes (147, 151, 153–155); (2) promote ICAM-1 expression and extracellular trap generation by neutrophils (152, 156, 157); (3) stimulate differentiation of CD4+ T cells to Th1 cells (150); (4) enhance the cytotoxic profile of CD8+ T cells (150). Unsurprisingly therefore, in in vivo models of sepsis or haemorrhage, eCIRP amplifies systemic inflammatory responses that culminate in organ dysfunction, injury and increased mortality (147, 148, 150, 151, 153, 158). Interestingly, eCIRP may also exacerbate inflammation by delaying the resolution of inflammatory responses. In a series of in vitro co-culture experiments, Chen et al. demonstrated that eCIRP-induced NETs significantly inhibited macrophage efferocytosis, with this suppression a consequence of NET associated neutrophil elastase cleaving the integrins αvβ3 and αvβ5 from the macrophage surface (157).
eCIRP has been detected in the circulation of critically-ill ICU patients and in the serum of rats subjected to haemorrhagic shock and cecal ligation and puncture (47, 147). As reported for other DAMPs, innate immune cells pre-exposed to CIRP exhibit impaired functional responses upon secondary stimulation. Compared to vehicle controls, pre-treatment of peritoneal macrophages isolated from healthy mice with CIRP for 24 hours resulted in significantly reduced IL-6 and TNF production upon subsequent LPS challenge (47). This induction of tolerance was confirmed ex vivo, with macrophages obtained from mice injected with CIRP exhibiting impaired IL-6 production when stimulated with LPS in vitro (47). Mechanistically, this state of endotoxin tolerance was attributed to an eCIRP-mediated activation of STAT3, which was downstream of eCIRP binding to the IL-6 receptor (IL-6R) (47). Indeed, pharmacological inhibition of STAT3 signalling or the use of anti-IL-6R antibodies reversed eCIRP-induced macrophage endotoxin tolerance (47). Pointing towards a role in modulating macrophage polarisation, eCIRP treatment of RAW264.7 cells increased their expression of arginase-1 and CD206 (47). The up-regulation of these markers suggests eCIRP promotes polarisation of macrophages towards an M2 anti-inflammatory phenotype (47). Interestingly, besides inflammatory cytokines, eCIRP treatment of RAW264.7 cells results in secretion of HMGB-1 (147), a DAMP that also possesses endotoxin tolerising properties (51). Thus, a feedback loop of DAMP-induced immunosuppression, initiated by eCIRP and reinforced by HMGB-1, could be established at sites of tissue injury that would result in localised immunosuppression and impaired bacterial clearance.
Given its pro-inflammatory properties and associations with organ dysfunction in models of systemic inflammation, eCIRP has been discussed as a potential therapeutic target for the treatment of sepsis (148, 159). Lending support to this idea, in vitro and in vivo studies have shown antagonists of CIRP signalling that include C23, which binds to the TLR4-MD2 receptor complex, and M3, which blocks the interaction of eCIRP with TREM-1, can suppress macrophage activation, reduce circulating concentrations of pro-inflammatory cytokines, attenuate organ injury and improve survival rates (151, 153, 158). With a link now emerging between eCIRP, the IL6R and immunosuppression, a call for studies to investigate whether targeting this pathway could negate eCIRP-induced immune tolerance in critically-ill patients has been made, with the suggestion that research should begin by testing small peptide antagonists that target the interaction between eCIRP and IL6R in preclinical models of immune tolerance and secondary infections (47).
2.2.2 Interleukin -33
Constitutively expressed by epithelial, endothelial and stromal cells, and induced in macrophages and DCs in times of inflammation and infection, IL-33 is a member of the IL-1 cytokine superfamily (160, 161). Residing in the nucleus, IL-33 is a dual function protein, acting as a transcriptional repressor and cytokine (160). When released into the extracellular environment, either as a result of cellular damage or active secretion, IL-33 binds to its receptor suppression of tumorigenicity (ST2) (160, 161). Expressed by such cells as mast cells, eosinophils, Th2 cells, group 2 innate lymphoid cells (ILC2) and NK cells, activation of ST2 signalling pathways results in cytokine production, degranulation and T cell polarisation (162, 163).
In a prospective study of 472 blunt trauma patients admitted to ICU, Xu and colleagues detected an early (upon hospital admission) and persistent (up to day 7) post-injury elevation in circulating IL-33 levels (49), a finding that was confirmed in a subsequent independent study of 14 polytrauma patients, in whom plasma IL-33 concentrations were significantly higher 6 to 72 hours post-injury when compared to a mixed control population of monotrauma patients and healthy controls (164). Interestingly, in burns patients, a significant injury-induced decrease in serum IL-33 levels has been reported, with this reduction evident on the day of hospital admission and persisting for up to 28 days (165). Whilst it is debatable whether circulating IL-33 levels are influenced by injury severity (49, 164, 166), differences have been observed in IL-33 concentrations between patients with varied clinical outcomes. For example, in the abovementioned study of 14 polytrauma patients, plasma IL-33 levels at hospital admission were reported to be positively associated with SIRS and Denver MOF scores at day 3 post-injury (164), whereas non-survivors of blunt trauma presented with significantly lower IL-33 concentrations 24-72 hours post-injury when compared to survivors (166). Acting as a decoy receptor and endogenous inhibitor of IL-33, elevated levels of soluble ST2 (sST2) have been detected in the circulation of trauma and burns patients in the days and weeks following injury (49, 165–167), with one prospective study measuring the highest concentrations of sST2 12-24 hours after the peak in IL-33 levels, a delay that was suggested would allow IL-33 to modulate systemic inflammatory responses (49). In respect to clinical outcomes, significantly higher concentrations of sST2 have been detected at several post-injury time points (hospital admission to day 5) in patients who develop pulmonary complications or succumb to their injuries (165, 166)
Suggesting a potential link between raised IL-33 levels and post-trauma immunosuppression, significantly higher concentrations of this DAMP have been measured in plasma samples acquired during the early (4-24 hours) and acute (day 4) post-injury phase from patients that develop HAIs when compared to those who do not (49). Mechanistically, this relationship could potentially be explained by IL-33 activating ILC2 cells and neutrophils to produce the type 2 cytokines IL-4, IL-5 and IL-13, whose plasma levels are significantly elevated in injured patients that experience HAIs (49, 168). Supporting this idea, positive associations have been reported between circulating concentrations of IL-33, IL-4, IL-5 and IL-13 in severely injured individuals (49), whilst in rodent models of trauma haemorrhage, IL-33 signalling is critical for both the post-injury expansion of ILC2 cells and the production of IL-5 by neutrophils (49). Recently, in the context of viral infections, IL-33-mediated production of IL-13 by ILC2 cells was shown to induce arginase-1 expression in neutrophils, the significance of which was demonstrated in neutrophil:T cell co-culture systems, where arginase-1 positive neutrophils significantly inhibited T cell proliferation (50). Relating these observations to traumatic injury, increased arginase-1 expression and activity has been reported in CD16+ granulocytes isolated from blunt/penetrative trauma patients for up to two weeks post-injury (169).
By inducing the expansion of IL-10 secreting M2 macrophages, and promoting the accumulation of regulatory T cells (Tregs), IL-33 has been proposed as a potential mediator of long-term immunosuppression in murine models of sepsis (48). Providing clinical relevance, significantly higher concentrations of IL-33 and IL-10, as well as increased frequencies of Tregs, have been detected in the circulation of sepsis survivors sampled 5-10 months post-diagnosis when compared to age and sex-matched controls (48). Thus, could a similar mechanism underlie the development of PICS in critically-injured subjects? With the potential to identify therapeutic targets for the treatment of this debilitating condition, future studies that prospectively measure the circulating levels of IL-33, its associated cytokines (e.g. IL-4, IL-5, IL-10 and IL-13) and the frequencies of Tregs in the months following major trauma are warranted. It is of interest that significantly elevated serum concentrations of IL-4, IL-5 and IL-13 have been reported in burns patients for up to three years post-injury (170).
2.2.3 HMGB-1
HMGB-1 is a nuclear residing DNA-binding chaperone protein that regulates gene transcription. Entering the circulation as a consequence of necrosis, pyroptosis or active secretion through a secretory vesicular pathway (171–173), HMGB-1 concentrations are significantly elevated in the minutes, hours and days following major trauma (32, 174, 175). Positively associated with injury severity, base deficit and pro-inflammatory mediators (e.g. MPO, IL-6, IL-8), elevated levels of HMGB-1 have been linked to such poor clinical outcomes as MODS, organ injury and mortality (32, 174, 175). Recognised by several PRRs, which include TLR2, TLR4, TREM-1 and RAGE, HMGB-1 can both activate and inhibit immune cell function, with these opposing traits dependent upon its redox state. The immune suppressive actions of HMGB-1 arise from the terminal oxidation of three cysteine residues located within its DNA binding domains and acidic tail (159)
Like many other DAMPs, exposure to HMGB-1 blunts the pro-inflammatory responses of monocytes. Attributed to impaired activation of NFκB, resulting from reduced phosphorylation and degradation of IκBα, HMGB-1 preconditioning decreases TNF production by differentiated THP-1 cells and bone marrow-derived macrophages challenged with the TLR agonists LPS and lipoteachoic acid (51, 52). For the induction of endotoxin tolerance, HMGB-1 signalling through the RAGE receptor appears to be critical since pre-treatment with HMGB-1 failed to impair TNF production by LPS stimulated macrophages isolated from RAGE-/- mice (51, 52). Contributing to further innate immune dysfunction, HMGB-1 reduces macrophage phagocytic activity (53), promotes the release of immature monocytes from the bone marrow (176) and inhibits neutrophil anti-microbial responses (54). Compared to vehicle controls, neutrophils treated with HMGB-1 exhibit decreased ROS production upon stimulation with PMA, an impairment that is associated with reduced phosphorylation of the NADPH oxidase subunit p40phox (54). In terms of clinical applicability, culturing neutrophils isolated from healthy controls with plasma from critically ill septic shock patients has been shown to significantly decrease their ROS production (54). Suggesting a role for HMGB-1 in mediating this effect, the addition of a neutralising anti HMGB-1 antibody to patient plasma restored neutrophil anti-microbial function (54). Whether similar mechanisms contribute to post-injury immunosuppression is unclear, but in a cohort of 88 blunt trauma patients plasma HMGB-1 levels were significantly higher within 4 hours of injury in those who later developed a HAI (168).
In a study that aimed to investigate the effect of HMGB-1 on T cell-mediated immunity following thermal injury, Huang et al. reported a profound effect of this DAMP on both CD4+ T cells and Tregs in rats (92). Compared to those isolated from sham controls, the authors found that post-burn, T cells from rodents subjected to a 30% TBSA burn exhibited reduced proliferative activity and skewed polarisation towards a Th2 phenotype, whilst Tregs produced greater amounts of IL-10 and expressed higher levels of RAGE (92). Implying a role for HMGB-1 in these responses, administration of an anti-RAGE antibody 6- and 24-hours post-burn injury reversed the burn-induced changes in both T cell function and phenotype (92). Supporting and expanding upon these results, a subsequent in vitro study found, in a TLR4 or RAGE dependent manner, that HMGB-1 treatment inhibited IFN-γ production by human CD4+ T cells and promoted the survival, migration and suppressive activity of Tregs (55).
Alongside direct modulation of immune cells, HMGB-1 may promote systemic immunosuppression post-injury by promoting the expansion of MDSCs. MDSCs suppress innate and adaptive immune responses via a number of mechanisms, which include the production of IL-10 and prostaglandin E2, the sequestration of cysteine, the breakdown of arginine and the generation of ROS (177, 178). Across several studies, HMGB-1 has been shown to promote the differentiation, survival, migration and activation of MDSCs (56). In the context of traumatic injury, elevated frequencies of MDSCs have been detected in the bone marrow, blood and spleen of mice subjected to peripheral tissue trauma, with this elevation prevented by post-injury administration of a blocking anti-HMGB-1 antibody (93). An expansion in spleen resident MDSCs has also been reported in a murine model of neurological trauma, which was accompanied by a significant increase in arginase-1 (ARG-1) expression in CD11b+ monocytes (176). A reduction in MDSC frequency in RAGE-/- mice, and the absence of the trauma-induced increase in ARG-1 expression in monocytes isolated from mice pre-treated with an anti-HMGB-1 antibody, demonstrates a role for this DAMP in mediating systemic immunosuppression (176).
Human monocytes treated with HMGB-1 secrete the peptide hormone resistin (179). Shown in vitro to inhibit both neutrophil chemotaxis and ROS generation (180–182), and to suppress TNF production by E.coli or LPS challenged monocytes (183, 184), elevated resistin concentrations have been measured in the circulation of TBI and thermally-injured patients (185–188). Furthermore, and with the potential to create a perpetual cycle of immunosuppression, monocytes treated with physiological concentrations of resistin secrete HMGB-1 and upregulate their expression of TLR4 (189). Thus, systemically, and at sites of tissue damage, HMGB-1 and resistin may combine to create an immune suppressive environment that predisposes the hospitalised trauma patient to secondary infections (Figure 1). Crucially, this cycle may be amenable to therapeutic intervention, with studies showing that in vitro hemoadsorption and nucleic acid scavenging microfiber meshes can deplete resistin from serum samples of critically-ill patients (190) and HMGB-1 from supernatants of necrotic cells (191). In the case of resistin, the correction of hyperresistinemia restored neutrophil migration and bacterial killing capacity in serum co-culture assays (190).
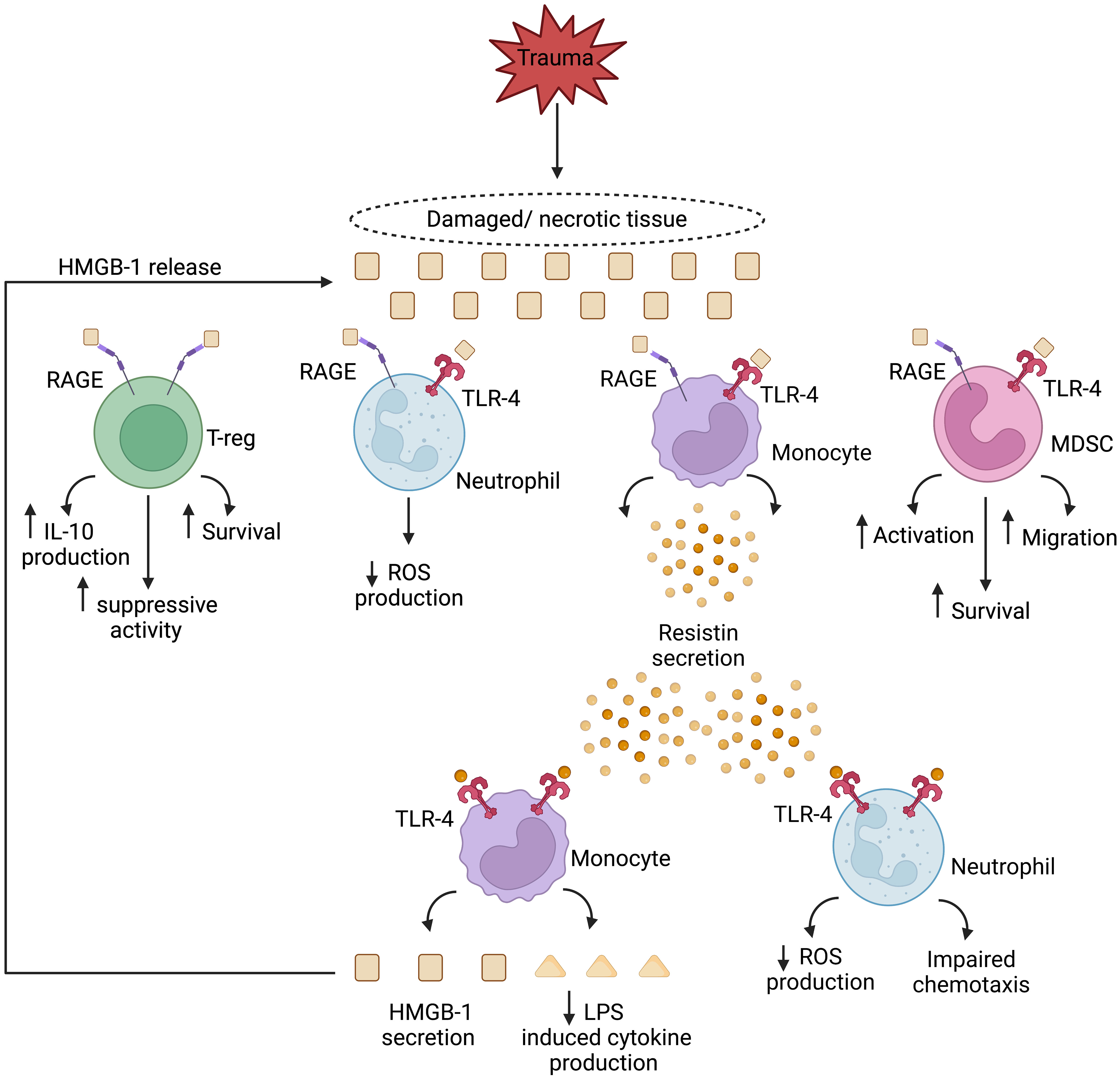
Figure 1 Theoretical model of a HMGB-1 and resistin-induced vicious cycle of immunosuppression in the setting of sterile injury. Passively released from damaged/necrotic tissue, the nuclear-derived damage associated molecular pattern high mobility group box-1 (HMGB-1) modulates the functions of innate and adaptive immune cells. Via binding to the pathogen recognition receptors toll like receptor-4 (TLR-4), and receptor for advanced glycation end products (RAGE), HMGB-1 directly inhibits neutrophil anti-microbial functions and enhances the suppressive activities of regulatory T cells and myeloid derived suppressor cells (MDSCs). Monocytes activated by HMGB-1 secrete resistin, an inducer of endotoxin tolerance, and a negative regulator of neutrophil chemotaxis and reactive oxygen species (ROS) production. In a self-perpetuating feedback loop, resistin, through TLR-4 signalling, promotes the secretion of HMGB-1 by monocytes, culminating in a localised immune suppressive environment at the site of tissue injury. IL-10, Interleukin-10; LPS, Lipopolysaccharide. Figure was generated at Biorender.com.
2.3 MtDAMPs
2.3.1 ATP
ATP is released non-specifically from necrotic cells, or actively by phagocytes, endothelial cells and apoptotic cells in response to inflammatory challenge, hypoxia or oxidative stress, with its secretion mediated by vesicular exocytosis or through pore-forming channels such as connexins and pannexins (192). Murine models of sepsis have reported elevated plasma concentrations of ATP within two hours of cecal ligation and puncture (193), whilst resuscitation protocols may exacerbate direct trauma-induced increases in circulating ATP levels since in vitro exposure to hypertonic saline induces ATP secretion from neutrophils and T cells (194, 195).
Although not reported by all studies (196, 197), the current literature points towards ATP as a DAMP with potent pro-inflammatory properties and an initiator of the SIRS response. For example, signalling through the P2 family of purinergic receptors, ATP (1): promotes inflammasome-mediated secretion of IL-1β by LPS primed macrophages (198) (2); triggers TNF, IL-6 and IL-1α production by macrophages (199); (3) induces T cell activation and IL-2 production (200); (4) augments fMLP-induced superoxide production by neutrophils (194); (5) stimulates neutrophil degranulation, aggregation and adherence to endothelium (201). Moreover, in a murine model of LPS-induced shock, systemic administration of the ATP hydrolase apyrase reduced the cytokine storm, immune cell infiltration into peripheral organs and apoptotic cell death that was observed in vehicle-treated littermates (202). Removal of ATP has therefore been proposed as a potential strategy for dampening the SIRS response and its associated toxicity in the setting of sterile inflammation (202).
In the extracellular environment, ATP levels are regulated by the ectonucleotidases CD39 and CD73. Expressed by various cell types that include endothelial cells, monocytes, lymphocytes, neutrophils and Tregs, CD39 converts ATP and ADP into AMP, which is subsequently degraded into adenosine by CD73 (203, 204). A potent anti-inflammatory molecule, adenosine inhibits neutrophil phagocytosis, ROS production, degranulation and cytokine production (57), suppresses T cell migration, adhesion and IFN-γ production (58), downregulates antigen presentation and T cell priming by DCs (59), reduces TLR-induced pro-inflammatory cytokine production by macrophages (205) and decreases NK cell cytotoxicity (58). Mechanistically, this suppression is mediated through the A2 receptor family, and in particular the A2A subclass, whose signalling pathways result in the production of cyclic AMP and activation of protein kinase A (57, 59). Interestingly, a recent study reported a trend towards an increased frequency of CD8+ CD39+ T cells in the circulation of trauma patients at days 0/1 post-injury when compared to control patients undergoing elective surgery (206). Immune phenotyping revealed these T cells exhibited signs of immune exhaustion, with increased expression of the markers PD-1 and TIM-3 (206). These changes were accompanied by a trend towards reduced granzyme B content, suggesting potentially reduced cytotoxic capacity (206). Prominent in trauma patients with SIRS, it was speculated that an abundance of immunologically exhausted CD8+ CD39+ PD-1+ Tim-3+ T cells may contribute to the state of systemic immune dysfunction that increases the susceptibility of trauma patients to infection (206).
Data are emerging that implies potential interplay between ATP, mtDNA, HSPs and heme metabolism in mediating post-trauma immunosuppression. Suggesting that recognition of mtDNA may enhance T cell mediated conversion of ATP/ADP to its immune suppressive metabolite adenosine, a small cohort study of 11 trauma patients reported a positive association between plasma mtDNA levels and the frequency of CD8+CD39+ T cells heckler (206). In a series of in vitro experiments, Haschemi et al. demonstrated a positive feedback loop between adenosine and HSP32 in suppressing LPS-induced TNF production by macrophages (60). Through activation of the A2A receptor, the authors found adenosine induced expression of heme oxygenase (HSP32) in macrophages (60). Degradation of heme by HSP32 resulted in the production of carbon monoxide, which increased the sensitivity of macrophages to the anti-inflammatory effects of adenosine by increasing surface expression of the A2A receptor (60). Relating these findings to trauma, monocytes isolated from injured patients with severe SIRS have been shown to exhibit increased expression of HSP32 and impaired TNF production following bacterial stimulation (115).
2.3.2 MtDNA
In a seminal study published in 2010, Zhang and colleagues provided mechanistic insights into how sterile traumatic injury results in systemic inflammation and organ dysfunction (33). Passively released from necrotic/damaged tissues, and actively secreted by immune cells (35, 207, 208), they showed that mtDNA, whose levels are acutely and persistently elevated post-trauma (31, 33, 76, 77, 209) is a potent activator of neutrophils, with exposure to this DAMP triggering IL-8 production and activation of MAPKs (33). Subsequent studies have confirmed these observations and demonstrated that mtDNA also induces NET formation and neutrophil degranulation in a TLR9 dependent manner (72, 210, 211). Linking the immunostimulatory properties of mtDNA to systemic inflammation and organ dysfunction, injection of mtDNA into rodents results in hepatic inflammation (210), whilst in models of traumatic injury and shock, plasma mtDNA levels positively associate with lung MPO levels and circulating urea and IL-6 concentrations (76). Providing clinical relevance to these findings, prospective studies of critically-injured subjects have shown plasma mtDNA levels in the early and acute post-injury phase is an independent predictor of SIRS (77, 212). Further, mtDNA load was significantly higher in non-survivors versus survivors as well as patients who develop such secondary complications as ARDS and MODS (76, 78, 213). Interestingly, circulating cell-free mtDNA (cfmtDNA) exists in two forms, a low molecular weight form and a larger form, with the latter comprising 95% of cfmtDNA (214, 215). Comparing these two fractions in plasma samples from 25 major trauma patients, Briggs et al. demonstrated that it was the levels of low molecular weight cfmtDNA that were positively associated with poor clinical outcomes such as ICU length of stay, ventilator use and duration of MOF (216). When interpreting their findings, the authors suggested that the larger forms of cfmtDNA are membrane encapsulated and therefore unable to bind to TLR9 and trigger immune responses (216).
Alongside its pro-inflammatory properties, data are emerging that demonstrates mtDNA is a potent immune suppressor. Treatment of human monocytes with mtDNA inhibits, in a TLR9 dependent manner, their production of IL-1β, IL-6 and TNF upon subsequent stimulation with LPS (61, 62). This mtDNA-induced reduction in cytokine production was attributed to impaired activation of NF-κB signalling and the induction of IRAK-M, a negative regulator of TLR signalling (61). In the setting of sterile neurological trauma, circulating levels of mtDNA are significantly elevated in patients who develop HAIs, with ex vivo studies showing that pre-treatment of monocytes isolated from HCs with sera from these patients reduced LPS-induced TNF production, an impairment that was reversed when monocytes were treated with the TLR9 antagonist ODN prior to serum incubation (62). In addition to monocytes, mtDNA induces functional tolerance in neutrophils, with prior exposure to this DAMP inhibiting neutrophil chemotaxis, phagocytosis and bacterial killing (35, 63). A critical step in each of these defence mechanisms is reorganisation of actin filaments, a process that involves activation of such cytoskeletal proteins as the F-actin binding protein cortactin (CTTN). Through activation of G protein coupled receptor kinase 2 (GRK2), mtDNA treatment suppresses CTTN activation by promoting its deacetylation by histone deacetylases (HDACs) (63). Coinciding with elevated plasma mtDNA levels, activated GRK2 and reduced CTTN acetylation have been detected in neutrophils isolated from trauma patients (63). Demonstrating the potential relevance of these observations to clinical outcomes, GRK2 activity and CTTN acetylation, at days 1-6 post-injury, were increased and decreased respectively in neutrophils of trauma patients who later developed HAIs (63).
With potential implications for the efficiency of adaptive immune responses, mtDNA negatively impacts upon DC function. Actively engulfed from the extracellular environment, elevated cytoplasmic mtDNA levels have been measured in DCs isolated from septic mice and in bone marrow-derived DCs challenged with LPS and mtDNA in vitro (64). Through activation of the cGAS-stimulator of interferon genes (STING) pathway, JAK-1/2 and STAT3 signalling, the accumulation of cytoplasmic mtDNA inhibited LPS-induced expression of the co-stimulatory molecules CD40, CD80 and CD86 (64). Accompanied by reduced production of IL-12p70 and enhanced synthesis of IL-10, these phenotypic changes were associated with an impaired ability of DCs to promote the proliferation of CD4+ T cells in co-culture systems (64). Bearing unmethylated CpG islands, mtDNA is a ligand for the surface expressed and endosomal residing PRR TLR9. In murine models of thermal injury, CD4+ T cells cultured with TLR9 activated DCs isolated from burn-injured mice were found to secrete markedly lower levels of Th1 and Th17 cytokines when compared to T cells cultured with DCs from sham controls (217). This shift in T cell polarisation was associated with increased production of IL-10 and reduced secretion of IL-6, IL-12p70 and TNF by TLR9 challenged DCs post-burn (217).
As a DAMP that possesses both immune stimulatory and inhibitory properties, mtDNA and/or its associated signalling pathways have been discussed as possible therapeutic targets for the treatment of post-traumatic SIRS, ARDS and immunosuppression (35, 63, 76, 78, 213). Focussing on the latter, preclinical studies have demonstrated the potential immune enhancing effects of GRK2 and HDAC inhibitors (63). Administered 30 minutes after a liver crush injury and tracheal inoculation of S.aureus, a combined therapy of valproate, a HDAC inhibitor, and the GRK2 inhibitor paroxetine restored levels of bacterial clearance to those recorded in uninjured controls and increased survival rates (63). Importantly, for its potential translation into the hospital setting, the treatment regimen was not associated with any evidence of organ injury or failure (63).
2.3.3 Mitochondrial-derived N-formylated peptides
Mitochondria synthesise 13 formylated peptides that share molecular similarities with the formyl peptides derived from bacteria. All but one of these formylated peptides, namely Cox1, have been detected in the circulation of trauma patients, with concentrations of ND6, a subunit of mitochondrial NADH dehydrogenase, significantly elevated within 1-hour of injury and remaining so for up to 72 hours (30, 66).
Detected by surface expressed formyl peptide receptor (FPR) 1 and FPR2 (67), mtFPs are neutrophil chemoattractants, with the proteins that most closely resemble bacterial-derived formylated peptides, namely ND3, ND4, ND5 and ND6 possessing the greatest chemotactic potency (66). Data generated by several studies have demonstrated that neutrophils pre-treated with ND6 exhibit reduced calcium fluxes, chemotactic responses and ROS production upon secondary stimulation with other GPCR agonists such as IL-8, leukotriene B4 (LTB4) and fMLP (30, 65–68). Contributing to this heterologous receptor desensitisation and suppression in anti-microbial functions is an ND6-mediated downregulation of GPCRs, for which activation of GRK2 is a potential underlying mechanistic explanation (63, 65, 66, 218).
Associated with reduced bacterial clearance, a peripheral fracture injury in animal models has been shown to reduce neutrophil recruitment to the lung following a pulmonary contusion (219). As femoral fracture reamings are a rich source of mtDAMPs (91), it has been proposed that the increased susceptibility of trauma patients to HAIs may arise in part from mtDAMPs impairing host resistance by attracting neutrophils towards distal injury sites and away from regions of pathogenic challenge (219, 220). Supporting this hypothesis, intra-peritoneal injection of mitochondrial debris was shown to markedly attenuate neutrophil migration to the lung following a pulmonary contusion (220). A role for mtFPs in mediating this effect is suggested by the results of another animal study in which administration of the FPR1 receptor antagonist cyclosporin H, alongside mtDAMPs, significantly improved bacterial clearance in the lung following intra-tracheal injection of S.aureus (218).
Implicated in the capture, neutralisation and elimination of pathogens, the generation of NETs is significantly reduced post-injury (30, 219). In a series of in vitro and ex vivo experiments, we attributed this impairment to a mtFP-induced dysfunction in neutrophil metabolism (30). Compared to those isolated from healthy controls, neutrophils obtained from trauma patients, as early as 1-hour post-injury, exhibited reduced glucose uptake and breakdown, two prerequisites for NET formation (30). Replicating these impairments in vitro, we showed that treatment of control neutrophils with whole mtDAMP preparations, but not purified mtDNA, inhibited PMA-induced NET production, a defect that was associated with reduced aerobic glycolysis and a mtFP dependent activation of AMP-activated protein kinase, a negative regulator of NET formation (30).
As a DAMP that suppresses a range of neutrophil anti-microbial functions, studies have investigated whether mtFPs may represent a potential therapeutic target to combat post-trauma immunosuppression. Several studies have shown that pre-treatment of neutrophils with FPR1 inhibitors not only prevents mtFP-induced impairments in calcium mobilisation and chemotactic responses (65, 66, 68) but also reduces ND6-mediated downregulation of GPCRs (65, 218). Another strategy under consideration is the intra-tracheal instillation of neutrophils, which aims to overcome the ability of mtFPs generated at regions of tissue injury to direct neutrophils away from sites of pathogenic challenge. In preclinical models of pseudo fracture and bacterial inoculation, intra-tracheal instillation of exogenous bone marrow derived murine neutrophils or neutrophils isolated from human blood samples has been shown to prevent the establishment of and/or treat pneumonia by reversing the mtDAMP-induced impairment in bacterial clearance from the lungs (68, 221, 222). Encouragingly, no adverse effects in respect of organ damage have been detected post-instillation (221, 222). Furthermore, and relevant to its potential translation to the clinic, preliminary data suggest that cryopreserved neutrophils are just as effective as freshly isolated neutrophils in promoting bacterial clearance from the lung in the presence of mtDAMPs (222).
3 Conclusions
The detection of DAMPs in blood samples obtained from critically-injured patients in the pre-hospital setting offers a mechanistic explanation for the simultaneous induction of the SIRS and CARS response that occurs within minutes of injury (15, 31). Indeed, alongside their renowned pro-inflammatory properties, cytosolic, nuclear and mitochondrial-derived DAMPs are now recognised as potent immune suppressors, impairing the anti-microbial functions of innate and adaptive immune cells either directly or by driving the expansion and differentiation of Tregs and MDSCs. Exacerbating both the local and systemic immune suppressive environments created by the instantaneous release of DAMPs from injured tissues, are secondary insults such as fluid resuscitation, surgical interventions and transfusions with aged blood cell products, which either induce the release of endogenous DAMPs or are themselves a source of exogenous DAMPs. Furthermore, alongside pre-existing DAMPs, a novel class of suppressive inducible DAMPs, termed SAMPs, have recently been described and suggested to activate hyper-resolution responses within minutes of the SIRS response being initiated (82).
Currently, very few prospective studies have investigated whether post-trauma elevations in circulating DAMPs are associated with and/or predictive of the development of HAIs. However, studies in other cohorts of critically-ill patients suggest such a relationship may exist (65, 223). If translatable to trauma patients, then the question arises of whether DAMPs and/or their associated signalling pathways represent feasible therapeutic targets for the prevention and/or treatment of secondary infections post-injury. Scavenging circulating DAMPs via the use of microfiber meshes or polymers, or inhibiting ligand-receptor interactions through delivery of receptor antagonists, are strategies under consideration for treating the secondary complications associated with the post-traumatic SIRS response that may also be suitable for combating the immune suppressive actions of DAMPs (Figure 2) (76, 191). However, the timing of such treatments would need to be carefully considered as it must not be forgotten that the purpose of DAMP release is to alert the body to tissue damage. Thus, a fine balance will exist for achieving an optimal immune response, with excessive or inappropriate removal of DAMPs, or inhibition of immune cell signalling, having the potential to negatively impact upon the DAMP-induced initiation of inflammatory responses that are critical for the regenerative processes of tissue repair and healing.
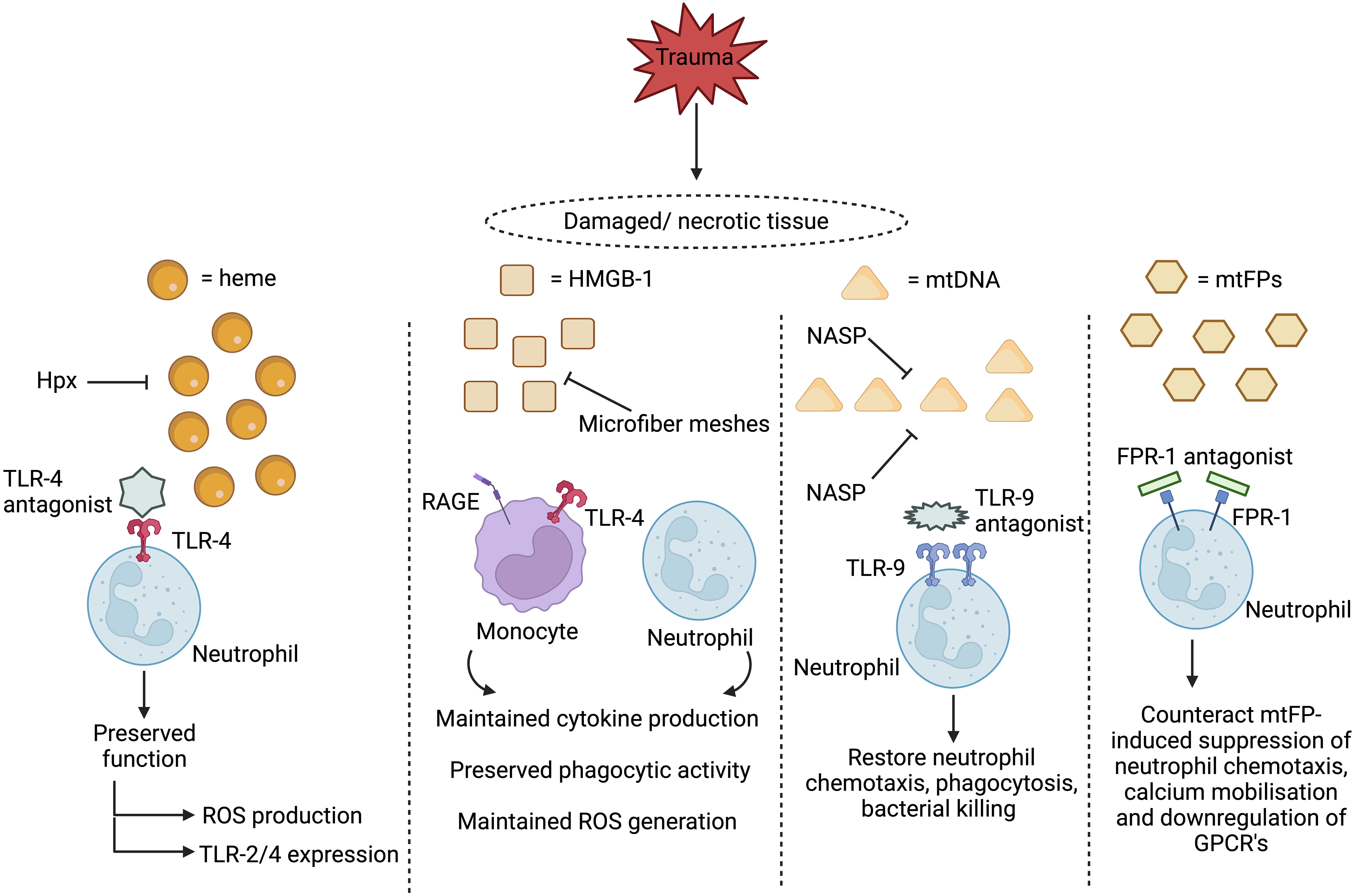
Figure 2 Proposed therapeutic strategies for treating damage associated molecular pattern (DAMP)-induced immunosuppression. Two therapeutic approaches under consideration for preventing and/or reversing DAMP-induced immunosuppression are to scavenge circulating DAMPs or to inhibit their activation of immune cells. Restoring circulating levels of hemopexin (Hpx), a heme scavenging protein whose plasma concentrations are reduced post-injury, may help combat heme-induced suppression of neutrophil anti-microbial functions, whilst the use of microfiber meshes or nucleic acid scavenging polymers (NASPs) to reduce the systemic load of circulating mtDNA and HMGB-1 could alleviate the post-trauma impairments reported in monocyte and neutrophil function. Showing promise in in vitro studies, blocking DAMP-induced activation of neutrophils through the use of toll-like receptor-9 (TLR-9) or formyl peptide receptor-1 (FPR-1) antagonists prevents the induction of functional tolerance to subsequent secondary stimulation with inflammatory agonists or microbial-derived proteins. HMGB-1, High mobility group box-1, mtDNA, Mitochondrial-derived DNA; mtFPs, Mitochondrial-derived N-formylated peptides; RAGE, Receptor for advanced glycation end products; ROS, Reactive oxygen species; TLR, Toll-like receptor. Figure was were generated at Biorender.com.
Author contributions
EH and JH conceived and wrote the manuscript. JL critically appraised and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
JH is supported by a Career Development Award from the UKRI-Medical Research Council. JH and JL are members of the National Institute for Health Research Surgical Reconstruction and Microbiology Research Centre.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed here are those of the authors and not necessarily those of the NIHR, NHS or Department of Health and Social Care.
References
1. Krug EG, Sharma GK, Lozano R. The global burden of injuries. Am J Public Health (2000) 90:523–6. doi: 10.2105/AJPH.90.4.523
2. The Trauma Audit and Research Network. Performance comparison: trauma care . Available at: https://www.tarn.ac.uk/Content.aspx?ca=15 (Accessed 10th June 2023).
3. Pfeifer R, Tarkin IS, Rocos B, Pape HC. Patterns of mortality and causes of death in polytrauma patients–has anything changed? Injury (2009) 40:907–11. doi: 10.1016/J.INJURY.2009.05.006
4. Van Breugel JMM, Niemeyer MJS, Houwert RM, Groenwold RHH, Leenen LPH, Van Wessem KJP. Global changes in mortality rates in polytrauma patients admitted to the ICU-a systematic review. World J Emerg Surg (2020) 15. doi: 10.1186/S13017-020-00330-3
5. Hessels AJ, Kuo YH, Ahmed N. Epidemiology and impact of healthcare-associated infections in trauma patients: A national data analysis. Surg Infect (Larchmt) (2020) 21:871–6. doi: 10.1089/SUR.2019.294
6. Glance LG, Stone PW, Mukamel DB, Dick AW. Increases in mortality, length of stay, and cost associated with hospital-acquired infections in trauma patients. Arch Surg (2011) 146:794–801. doi: 10.1001/ARCHSURG.2011.41
7. Kesinger MR, Kumar RG, Wagner AK, Puyana JC, Peitzman AP, Billiar TR, et al. Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg (2015) 78:396–402. doi: 10.1097/TA.0000000000000526
8. Czaja AS, Rivara FP, Wang J, Koepsell T, Nathens AB, Jurkovich GJ, et al. Late outcomes of trauma patients with infections during index hospitalization. J Trauma (2009) 67:805–14. doi: 10.1097/TA.0B013E318185E1FB
9. Manson J, Cole E, De’Ath HD, Vulliamy P, Meier U, Pennington D, et al. Early changes within the lymphocyte population are associated with the development of multiple organ dysfunction syndrome in trauma patients. Crit Care (2016) 20. doi: 10.1186/S13054-016-1341-2
10. Cabrera CP, Manson J, Shepherd JM, Torrance HD, Watson D, Longhi MP, et al. Signatures of inflammation and impending multiple organ dysfunction in the hyperacute phase of trauma: A prospective cohort study. PloS Med (2017) 14. doi: 10.1371/JOURNAL.PMED.1002352
11. Van Wessem KJP, Hietbrink F, Leenen LPH. Attenuation of MODS-related and ARDS-related mortality makes infectious complications a remaining challenge in the severely injured. Trauma Surg Acute Care Open (2020) 5. doi: 10.1136/TSACO-2019-000398
12. Yadollahi M, Kashkooe A, Feyzi M, Bornapour S. Risk factors of mortality in nosocomial infected traumatic patients in a trauma referral center in south of Iran. Chin J Traumatol (2018) 21:267–72. doi: 10.1016/J.CJTEE.2018.03.002
13. Lee PT, Krecko LK, Savage S, O’Rourke AP, Jung HS, Ingraham A, et al. Which hospital-acquired conditions matter the most in trauma? An evidence-based approach for prioritizing trauma program improvement. J Trauma Acute Care Surg (2022) 93:446–52. doi: 10.1097/TA.0000000000003645
14. Cole E, Davenport R, Willett K, Brohi K. The burden of infection in severely injured trauma patients and the relationship with admission shock severity. J Trauma Acute Care Surg (2014) 76:730–5. doi: 10.1097/TA.0B013E31829FDBD7
15. Hazeldine J, Naumann DN, TOman E, Davies D, Bishop JRB, Su Z, et al. Prehospital immune responses and development of multiple organ dysfunction syndrome following traumatic injury: A prospective cohort study. PloS Med (2017) 14. doi: 10.1371/JOURNAL.PMED.1002338
16. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208:2581–90. doi: 10.1084/JEM.20111354
17. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med (2008) 29:617–25. doi: 10.1016/J.CCM.2008.06.010
18. Orr SK, Butler KL, Hayden D, Tompkins RG, Serhan CN, Irimia D. Gene expression of proresolving lipid mediator pathways is associated with clinical outcomes in trauma patients. Crit Care Med (2015) 43:2642–50. doi: 10.1097/CCM.0000000000001312
19. Yang YW, Wu CH, Tsai HT, Chen YR, Chang YP, Han YY, et al. Dynamics of immune responses are inconsistent when trauma patients are grouped by injury severity score and clinical outcomes. Sci Rep (2023) 13. doi: 10.1038/S41598-023-27969-7
20. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg (2012) 72:1491–501. doi: 10.1097/TA.0B013E318256E000
21. Vanzant EL, Lopez CM, Ozrazgat-Baslanti T, Ungaro R, Davis R, Cuenca AG, et al. Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg (2014) 76:21–30. doi: 10.1097/TA.0B013E3182AB1AB5
22. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin (2017) 33:245–58. doi: 10.1016/J.CCC.2016.12.001
23. Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, et al. Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front Immunol (2018) 9:1511. doi: 10.3389/FIMMU.2018.01511
24. Hesselink L, Hoepelman RJ, Spijkerman R, de Groot MCH, van Wessem KJP, Koenderman L, et al. Persistent inflammation, immunosuppression and catabolism syndrome (PICS) after polytrauma: A rare syndrome with major consequences. J Clin Med (2020) 9. doi: 10.3390/JCM9010191
25. Cahill LA, Guo F, Nguyen J, Zhang F, Seshadri A, Keegan J, et al. Circulating factors in trauma plasma activate specific human immune cell subsets. Injury (2020) 51:819–29. doi: 10.1016/J.INJURY.2020.03.009
26. Kim HI, Park J, Konecna B, Huang W, Riça I, Gallo D, et al. Plasma and wound fluids from trauma patients suppress neutrophil extracellular respiratory burst. J Trauma Acute Care Surg (2022) 92:330–8. doi: 10.1097/TA.0000000000003461
27. Lee GR, Gallo D, de Souza RWA, Tiwari-Heckler S, Csizmadia E, Harbison JD, et al. Trauma-induced heme release increases susceptibility to bacterial infection. JCI Insight (2021) 6. doi: 10.1172/JCI.INSIGHT.150813
28. Flohé SB, Bangen JM, Flohé S, Agrawal H, Bergmann K, SChade FU. Origin of immunomodulation after soft tissue trauma: potential involvement of extracellular heat-shock proteins. Shock (2007) 27:494–502. doi: 10.1097/SHK.0B013E31802DEC51
29. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, et al. Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: A prospective observational cohort study. Ann Surg (2017) 265:1241–9. doi: 10.1097/SLA.0000000000001807
30. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic injury and exposure to mitochondrial-derived damage associated molecular patterns suppresses neutrophil extracellular trap formation. Front Immunol (2019) 10:685. doi: 10.3389/FIMMU.2019.00685
31. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med (2016) 42:551–61. doi: 10.1007/S00134-015-4205-3
32. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care (2009) 13. doi: 10.1186/CC8152
33. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/NATURE08780
34. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis (2021) 26:152–62. doi: 10.1007/S10495-021-01663-3
35. Konecna B, Park J, Kwon WY, Vlkova B, Zhang Q, Huang W, et al. Monocyte exocytosis of mitochondrial danger-associated molecular patterns in sepsis suppresses neutrophil chemotaxis. J Trauma Acute Care Surg (2021) 90:46–53. doi: 10.1097/TA.0000000000002973
36. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/S41577-019-0215-7
37. Wagener BM, Hu PJ, Oh JY, Evans CA, Richter JR, Honavar J, et al. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: A preclinical experimental study. PloS Med (2018) 15. doi: 10.1371/JOURNAL.PMED.1002522
38. Ferat-Osorio E, Sánchez-Anaya A, Gutiérrez-Mendoza M, Boscó-Gárate I, Wong-Baeza I, Pastelin-Palacios R, et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J Inflammation (Lond) (2014) 11. doi: 10.1186/1476-9255-11-19
39. Wachstein J, Tischer S, Figueiredo C, Limbourg A, Falk C, Immenschuh S, et al. HSP70 enhances immunosuppressive function of CD4(+)CD25(+)FoxP3(+) T regulatory cells and cytotoxicity in CD4(+)CD25(-) T cells. PloS One (2012) 7. doi: 10.1371/JOURNAL.PONE.0051747
40. Motta A, Schmitz C, Rodrigues L, Ribeiro F, Teixeira C, Detanico T, et al. Mycobacterium tuberculosis heat-shock protein 70 impairs maturation of dendritic cells from bone marrow precursors, induces interleukin-10 production and inhibits T-cell proliferation in vitro. Immunology (2007) 121:462–72. doi: 10.1111/J.1365-2567.2007.02564.X
41. Austermann J, Friesenhagen J, Fassl SK, Ortkras T, Burgmann J, Barczyk-Kahlert K, et al. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep (2014) 9:2112–23. doi: 10.1016/J.CELREP.2014.11.020
42. Coveney AP, Wang W, Kelly J, Liu JH, Blankson S, Di W, et al. Myeloid-related protein 8 induces self-tolerance and cross-tolerance to bacterial infection via TLR4- and TLR2-mediated signal pathways. Sci Rep (2015) 5. doi: 10.1038/SREP13694
43. Shimizu K, Libby P, Rocha VZ, Folco EJ, Shubiki R, Grabie N, et al. Loss of myeloid related protein-8/14 exacerbates cardiac allograft rejection. Circulation (2011) 124:2920–32. doi: 10.1161/CIRCULATIONAHA.110.009910
44. Sroussi HY, Berline J, Palefsky JM. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J Leukoc Biol (2007) 81:818–24. doi: 10.1189/JLB.0706433
45. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi: 10.4049/JIMMUNOL.181.7.4666
46. Vrakas CN, O’Sullivan RM, Evans SE, Ingram DMA, Jones CB, Phuong T, et al. The Measure of DAMPs and a role for S100A8 in recruiting suppressor cells in breast cancer lung metastasis. Immunol Invest (2015) 44:174–88. doi: 10.3109/08820139.2014.952818
47. Zhou M, Aziz M, Denning NL, Yen HT, Ma G, Wang P. Extracellular CIRP induces macrophage endotoxin tolerance through IL-6R-mediated STAT3 activation. JCI Insight (2020) 5. doi: 10.1172/JCI.INSIGHT.133715
48. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun (2017) 8. doi: 10.1038/NCOMMS14919
49. Xu J, Guardado J, Hoffman R, Xu H, Namas R, Vodovotz Y, et al. IL33-mediated ILC2 activation and neutrophil IL5 production in the lung response after severe trauma: A reverse translation study from a human cohort to a mouse trauma model. PloS Med (2017) 14. doi: 10.1371/JOURNAL.PMED.1002365
50. Liang Y, Yi P, Yuan DMK, Jie Z, Kwota Z, Soong L, et al. IL-33 induces immunosuppressive neutrophils via a type 2 innate lymphoid cell/IL-13/STAT6 axis and protects the liver against injury in LCMV infection-induced viral hepatitis. Cell Mol Immunol (2019) 16:126–37. doi: 10.1038/CMI.2017.147
51. Aneja RK, Tsung A, Sjodin H, Gefter JV, Delude RL, Billiar TR, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol (2008) 84:1326–34. doi: 10.1189/JLB.0108030
52. Robert SM, Sjodin H, Fink MP, Aneja RK. Preconditioning with high mobility group box 1 (HMGB1) induces lipoteichoic acid (LTA) tolerance. J Immunother (2010) 33:663–71. doi: 10.1097/CJI.0B013E3181DCD111
53. Wang Y, Zhang W, Xu Y, Wu D, Gao Z, Zhou J, et al. Extracellular HMGB1 impairs macrophage-mediated efferocytosis by suppressing the rab43-controlled cell surface transport of CD91. Front Immunol (2022) 13:767630. doi: 10.3389/FIMMU.2022.767630
54. Grégoire M, Tadié J-M, Uhel F, Gacouin A, Piau C, Bone N, et al. Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol (2017) 101:1281–7. doi: 10.1189/JLB.5HI0316-128RR
55. Wild CA, Bergmann C, Fritz G, Schuler P, Hoffmann TK, Lotfi R, et al. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol (2012) 24:485–94. doi: 10.1093/INTIMM/DXS051
56. Jin S, Yang Z, Hao X, Tang W, Ma W, Zong H. Roles of HMGB1 in regulating myeloid-derived suppressor cells in the tumor microenvironment. biomark Res (2020) 8. doi: 10.1186/S40364-020-00201-8
57. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol (2012) 32:856–64. doi: 10.1161/ATVBAHA.111.226845
58. Feng LL, Cai YQ, Zhu MC, Xing LJ, Wang X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int (2020) 20. doi: 10.1186/S12935-020-01195-X
59. Silva-Vilches C, Ring S, Mahnke K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front Immunol (2018) 9:2581. doi: 10.3389/FIMMU.2018.02581
60. Haschemi A, Wagner O, Marculescu R, Wegiel B, Robson SC, Gagliani N, et al. Cross-regulation of carbon monoxide and the adenosine A2a receptor in macrophages. J Immunol (2007) 178:5921–9. doi: 10.4049/JIMMUNOL.178.9.5921
61. Fernández-Ruiz I, Arnalich F, Cubillos-Zapata C, Hernández-Jiménez E, Moreno-González R, Toledano V, et al. Mitochondrial DAMPs induce endotoxin tolerance in human monocytes: an observation in patients with myocardial infarction. PloS One (2014) 9. doi: 10.1371/JOURNAL.PONE.0095073
62. Hernández-Jiménez E, Gutierrez-Fernández M, Cubillos-Zapata C, Otero-Ortega L, Rodríguez-Frutos B, Toledano V, et al. Circulating monocytes exhibit an endotoxin tolerance status after acute ischemic stroke: mitochondrial DNA as a putative explanation for poststroke infections. J Immunol (2017) 198:2038–46. doi: 10.4049/JIMMUNOL.1601594
63. Kim HI, Park J, Gallo D, Shankar S, Konecna B, Han Y, et al. Danger signals activate G-protein receptor kinases suppressing neutrophil function and predisposing to infection after tissue trauma. Ann Surg (2023). doi: 10.1097/SLA.0000000000005898
64. Tu Q, Li Y, Zhu J, Guo L, Liu C, Liu L, et al. Mitochondrial DNA mediates immunoparalysis of dendritic cells in sepsis via STING signalling. Cell Prolif (2022) 55. doi: 10.1111/CPR.13328
65. Kwon WY, Suh GJ, Jung YS, Park SM, Oh S, Kim SH, et al. Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/PNAS.2018538118/-/DCSUPPLEMENTAL
66. Kaczmarek E, Hauser CJ, Kwon WY, Rica I, Chen L, Sandler N, et al. A subset of five human mitochondrial formyl peptides mimics bacterial peptides and functionally deactivates human neutrophils. J Trauma Acute Care Surg (2018) 85:936–43. doi: 10.1097/TA.0000000000001971
67. Gabl M, Sundqvist M, Holdfeldt A, Lind S, Mårtensson J, Christenson K, et al. Mitocryptides from human mitochondrial DNA-encoded proteins activate neutrophil formyl peptide receptors: receptor preference and signaling properties. J Immunol (2018) 200:3269–82. doi: 10.4049/JIMMUNOL.1701719
68. Zhang Q, Kwon WY, Vlková B, Riça I, Kaczmarek E, Park J, et al. Direct airway instillation of neutrophils overcomes chemotactic deficits induced by injury. Shock (2021) 56:119–24. doi: 10.1097/SHK.0000000000001691
69. Pugin J. How tissue injury alarms the immune system and causes a systemic inflammatory response syndrome. Ann Intensive Care (2012) 2. doi: 10.1186/2110-5820-2-27
70. Comish PB, Carlson D, Kang R, Tang D. Damage-associated molecular patterns and the systemic immune consequences of severe thermal injury. J Immunol (2020) 205:1189–97. doi: 10.4049/JIMMUNOL.2000439
71. Schwacha MG, Rani M, Zhang Q, Nunez-Cantu O, Cap AP. Mitochondrial damage-associated molecular patterns activate γδ T-cells. Innate Immun (2014) 20:261–8. doi: 10.1177/1753425913488969
72. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PloS One (2015) 10. doi: 10.1371/JOURNAL.PONE.0120549
73. Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury (2015) 46:975–84. doi: 10.1016/J.INJURY.2015.03.028
74. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U.S.A. (2010) 107:11942–7. doi: 10.1073/PNAS.1003893107
75. Schwacha MG, Rani M, Nicholson SE, Lewis AM, Holloway TL, Sordo S, et al. Dermal γδ T-cells can be activated by mitochondrial damage-associated molecular patterns. PloS One (2016) 11. doi: 10.1371/JOURNAL.PONE.0158993
76. Aswani A, Manson J, Itagaki K, Chiazza F, Collino M, Wupeng WL, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol (2018) 9:891. doi: 10.3389/FIMMU.2018.00891
77. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PloS One (2013) 8. doi: 10.1371/JOURNAL.PONE.0072834
78. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg (2013) 258:591–6. doi: 10.1097/SLA.0B013E3182A4EA46
79. McIlroy DJ, Minahan K, Keely S, Lott N, Hansbro P, Smith DW, et al. Reduced deoxyribonuclease enzyme activity in response to high postinjury mitochondrial DNA concentration provides a therapeutic target for Systemic Inflammatory Response Syndrome. J Trauma Acute Care Surg (2018) 85:354–8. doi: 10.1097/TA.0000000000001919
80. Wang XW, Karki A, Zhao XJ, Xiang XY, Lu ZQ. High plasma levels of high mobility group box 1 is associated with the risk of sepsis in severe blunt chest trauma patients: a prospective cohort study. J Cardiothorac Surg (2014) 9. doi: 10.1186/S13019-014-0133-5
81. Vourc’h M, Roquilly A, Asehnoune K. Trauma-induced damage-associated molecular patterns-mediated remote organ injury and immunosuppression in the acutely ill patient. Front Immunol (2018) 9:1330. doi: 10.3389/FIMMU.2018.01330
82. Relja B, Land WG. Damage-associated molecular patterns in trauma. Eur J Trauma Emerg Surg (2020) 46:751–75. doi: 10.1007/S00068-019-01235-W
83. Aneja R, Odoms K, Dunsmore K, Shanley TP, Wong HR. Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J Immunol (2006) 177:7184–92. doi: 10.4049/JIMMUNOL.177.10.7184
84. Westhaver LP, Nersesian S, Nelson A, MacLean LK, Carter EB, Rowter D, et al. Mitochondrial damage-associated molecular patterns trigger arginase-dependent lymphocyte immunoregulation. Cell Rep (2022) 39. doi: 10.1016/J.CELREP.2022.110847
85. Bidar F, Bodinier M, Venet F, Lukaszewicz AC, Brengel-Pesce K, Conti F, et al. Concomitant assessment of monocyte HLA-DR expression and ex vivo TNF-α Release as markers of adverse outcome after various injuries-insights from the REALISM study. J Clin Med (2021) 11. doi: 10.3390/JCM11010096
86. de Roquetaillade C, Dupuis C, Faivre V, Lukaszewicz AC, Brumpt C, Payen D. Monitoring of circulating monocyte HLA-DR expression in a large cohort of intensive care patients: relation with secondary infections. Ann Intensive Care (2022) 12. doi: 10.1186/S13613-022-01010-Y
87. Cheron A, Floccard B, Allaouchiche B, Guignant C, Poitevin F, Malcus C, et al. Lack of recovery in monocyte human leukocyte antigen-DR expression is independently associated with the development of sepsis after major trauma. Crit Care (2010) 14. doi: 10.1186/CC9331
88. Conway Morris A, Rynne J, Shankar-Hari M. Compartmentalisation of immune responses in critical illness: does it matter? Intensive Care Med (2022) 48:1617–20. doi: 10.1007/S00134-022-06871-2
89. Juss JK, House D, Amour A, Begg M, Herre J, Storisteanu DML, et al. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3-kinase inhibition. Am J Respir Crit Care Med (2016) 194:961–73. doi: 10.1164/RCCM.201509-1818OC
90. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, et al. C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med (2009) 180:19–28. doi: 10.1164/RCCM.200812-1928OC
91. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma (2010) 24:534–8. doi: 10.1097/BOT.0B013E3181EC4991
92. Huang LF, Yao YM, Zhang LT, Dong N, Yu Y, Sheng ZY. The effect of high-mobility group box 1 protein on activity of regulatory T cells after thermal injury in rats. Shock (2009) 31:322–9. doi: 10.1097/SHK.0B013E3181834070
93. Ruan X, Darwiche SS, Cai C, Scott MJ, Pape HC, Billiar TR. Anti-HMGB1 monoclonal antibody ameliorates immunosuppression after peripheral tissue trauma: attenuated T-lymphocyte response and increased splenic CD11b (+) Gr-1 (+) myeloid-derived suppressor cells require HMGB1. Mediators Inflammation (2015) 2015. doi: 10.1155/2015/458626
94. Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol (2014) 5:61. doi: 10.3389/FPHAR.2014.00061
95. Vissa M, Larkin SK, Vichinsky EP, Kuypers FA, Soupene E. Assessment of total and unbound cell-free heme in plasma of patients with sickle cell disease. Exp Biol Med (Maywood) (2023). doi: 10.1177/15353702231157920
96. Lin T, Kwak YH, Sammy F, He P, Thundivalappil S, Sun G, et al. Synergistic inflammation is induced by blood degradation products with microbial Toll-like receptor agonists and is blocked by hemopexin. J Infect Dis (2010) 202:624–32. doi: 10.1086/654929
97. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourão-Sa D, Fortes GB, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem (2010) 285:32844–51. doi: 10.1074/JBC.M110.146076
98. Roumenina LT, Rayes J, Lacroix-Desmazes S, Dimitrov JD. Heme: modulator of plasma systems in hemolytic diseases. Trends Mol Med (2016) 22:200–13. doi: 10.1016/J.MOLMED.2016.01.004
99. Ryter SW. Significance of heme and heme degradation in the pathogenesis of acute lung and inflammatory disorders. Int J Mol Sci (2021) 22. doi: 10.3390/IJMS22115509
100. Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood (2005) 106:2572–9. doi: 10.1182/BLOOD-2005-03-1185
101. Lin T, Maita D, Thundivalappil SR, Riley FE, Hambsch J, Van Marter LJ, et al. Hemopexin in severe inflammation and infection: mouse models and human diseases. Crit Care (2015) 19. doi: 10.1186/S13054-015-0885-X
102. Maile R, Willis ML, Herring LE, Prevatte A, Mahung C, Cairns B, et al. Burn injury induces proinflammatory plasma extracellular vesicles that associate with length of hospital stay in women: CRP and SAA1 as potential prognostic indicators. Int J Mol Sci (2021) 22. doi: 10.3390/IJMS221810083
103. Dépret F, Dunyach C, De Tymowski C, Chaussard M, Bataille A, Ferry A, et al. Undetectable haptoglobin is associated with major adverse kidney events in critically ill burn patients. Crit Care (2017) 21. doi: 10.1186/S13054-017-1837-4
104. Oh JY, Stapley R, Harper V, Marques MB, Patel RP. Predicting storage-dependent damage to red blood cells using nitrite oxidation kinetics, peroxiredoxin-2 oxidation, and hemoglobin and free heme measurements. Transfusion (Paris) (2015) 55:2967–78. doi: 10.1111/TRF.13248
105. Stapley R, Rodriguez C, Oh JY, Honavar J, Brandon A, Wagener BM, et al. Red blood cell washing, nitrite therapy, and antiheme therapies prevent stored red blood cell toxicity after trauma-hemorrhage. Free Radic Biol Med (2015) 85:207–18. doi: 10.1016/J.FREERADBIOMED.2015.04.025
106. Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, et al. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm (Beijing) (2022) 3. doi: 10.1002/MCO2.161
107. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol (2000) 12:1539–46. doi: 10.1093/INTIMM/12.11.1539
108. De Maio A, Vazquez D. Extracellular heat shock proteins: a new location, a new function. Shock (2013) 40:239–46. doi: 10.1097/SHK.0B013E3182A185AB
109. Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med (2000) 6:435–42. doi: 10.1038/74697
110. Prohaszka Z, Singh M, Nagy K, Kiss E, Lakos G, Duba J Fust G. Heat shock protein 70 is a potent activator of the human complement system. Cell Stress Chaperones (2002) 7:17–22. doi: 10.1379/1466-1268
111. Guisasola MC, Ortiz A, Chana F, Alonso B, Vaquero J. Early inflammatory response in polytraumatized patients: Cytokines and heat shock proteins. A pilot study. Orthop Traumatol Surg Res (2015) 101:607–11. doi: 10.1016/J.OTSR.2015.03.014
112. Vardas K, Apostolou K, Briassouli E, Goukos D, Psarra K, Botoula E, et al. Early response roles for prolactin cortisol and circulating and cellular levels of heat shock proteins 72 and 90α in severe sepsis and SIRS. BioMed Res Int (2014) 2014. doi: 10.1155/2014/803561
113. Ren B, Zou G, Huang Y, Xu G, Xu F, He J, et al. Serum levels of HSP70 and other DAMP proteins can aid in patient diagnosis after traumatic injury. Cell Stress Chaperones (2016) 21:677–86. doi: 10.1007/S12192-016-0694-4
114. Pittet JF, Lee H, Morabito D, Howard MB, Welch WJ, Mackersie RC. Serum levels of Hsp 72 measured early after trauma correlate with survival. J Trauma (2002) 52:611–7. doi: 10.1097/00005373-200204000-00001
115. Mohri T, Ogura H, Koh T, Fujita K, Sumi Y, Yoshiya K, et al. Enhanced expression of intracellular heme oxygenase-1 in deactivated monocytes from patients with severe systemic inflammatory response syndrome. J Trauma (2006) 61:616–23. doi: 10.1097/01.TA.0000238228.67894.D7
116. Ogura H, Hashiguchi N, Tanaka H, Koh T, Noborio M, Nakamori Y, et al. Long-term enhanced expression of heat shock proteins and decelerated apoptosis in polymorphonuclear leukocytes from major burn patients. J Burn Care Rehabil (2002) 23:103–9. doi: 10.1097/00004630-200203000-00006
117. Hashiguchi N, Ogura H, Tanaka H, Koh T, Aoki M, Shiozaki T, et al. Enhanced expression of heat shock proteins in leukocytes from trauma patients. J Trauma (2001) 50:102–7. doi: 10.1097/00005373-200101000-00018
118. Pespeni M, Mackersie RC, Lee H, Morabito D, Hodnett M, Howard M, et al. Serum levels of Hsp60 correlate with the development of acute lung injury after trauma. J Surg Res (2005) 126:41–7. doi: 10.1016/J.JSS.2005.01.012
119. Guisasola MC, Alonso B, Bravo B, Vaquero J, Chana F. An overview of cytokines and heat shock response in polytraumatized patients. Cell Stress Chaperones (2018) 23:483–9. doi: 10.1007/S12192-017-0859-9
120. Bangen JM, SChade FU, Flohé SB. Diverse regulatory activity of human heat shock proteins 60 and 70 on endotoxin-induced inflammation. Biochem Biophys Res Commun (2007) 359:709–15. doi: 10.1016/J.BBRC.2007.05.167
121. Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem (2003) 278:174–9. doi: 10.1074/JBC.M208742200
122. Stocki P, Wang XN, Dickinson AM. Inducible heat shock protein 70 reduces T cell responses and stimulatory capacity of monocyte-derived dendritic cells. J Biol Chem (2012) 287:12387–94. doi: 10.1074/JBC.M111.307579
123. Tukaj S, Mantej J, Sobala M, Potrykus K, Sitko K. Autologous extracellular Hsp70 exerts a dual role in rheumatoid arthritis. Cell Stress Chaperones (2020) 25:1105–10. doi: 10.1007/S12192-020-01114-Z
124. Broere F, van der Zee R, Van Eden W. Heat shock proteins are no DAMPs, rather “DAMPERs. ” Nat Rev Immunol (2011) 11:565. doi: 10.1038/NRI2873-C1
125. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med (2013) 13:24–57.
126. Steiner J, Marquardt N, Pauls I, Schiltz K, Rahmoune H, Bahn S, et al. Human CD8(+) T cells and NK cells express and secrete S100B upon stimulation. Brain Behav Immun (2011) 25:1233–41. doi: 10.1016/J.BBI.2011.03.015
127. Goyal A, Failla MD, Niyonkuru C, Amin K, Fabio A, Berger RP, et al. S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J Neurotrauma (2013) 30:946–57. doi: 10.1089/NEU.2012.2579
128. Andersen RE, Hansson LO, Nilsson O, Dijlai-Merzoug R, Settergren G. High serum S100B levels for trauma patients without head injuries. Neurosurgery (2001) 48:1255–60. doi: 10.1097/00006123-200106000-00012
129. Thelin EP, Zibung E, Riddez L, Nordenvall C. Assessing bicycle-related trauma using the biomarker S100B reveals a correlation with total injury severity. Eur J Trauma Emerg Surg (2016) 42:617–25. doi: 10.1007/S00068-015-0583-Z
130. Yang Y, Shen L, Xu M, Chen L, Lu W, Wang W. Serum calprotectin as a prognostic predictor in severe traumatic brain injury. Clin Chim Acta (2021) 520:101–7. doi: 10.1016/J.CCA.2021.06.009
131. Pfortmueller CA, Drexel C, Krähenmann-Müller S, Leichtle AB, Fiedler GM, Lindner G, et al. S-100 B concentrations are a predictor of decreased survival in patients with major trauma, independently of head injury. PloS One (2016) 11. doi: 10.1371/JOURNAL.PONE.0152822
132. Vos PE, Jacobs B, Andriessen TMJC, Lamers KJB, Borm GF, Beems T, et al. GFAP and S100B are biomarkers of traumatic brain injury: an observational cohort study. Neurology (2010) 75:1786–93. doi: 10.1212/WNL.0B013E3181FD62D2
133. Raabe A, Grolms C, Sorge O, Zimmermann M, Seifert V. Serum S-100B protein in severe head injury. Neurosurgery (1999) 45:477–83. doi: 10.1097/00006123-199909000-00012
134. Golden N, Mahadewa TGB, Aryanti C, Widyadharma IPE. S100B serum level as a mortality predictor for traumatic brain injury: A meta-analysis. Open Access Maced J Med Sci (2018) 6:2239–44. doi: 10.3889/OAMJMS.2018.432
135. Kim DH, Gu A, Lee JS, Yang EJ, Kashif A, Hong MH, et al. Suppressive effects of S100A8 and S100A9 on neutrophil apoptosis by cytokine release of human bronchial epithelial cells in asthma. Int J Med Sci (2020) 17:498–509. doi: 10.7150/IJMS.37833
136. Sprenkeler EGG, Zandstra J, van Kleef ND, Goetschalckx I, Verstegen B, Aarts CEM, et al. S100A8/A9 is a marker for the release of neutrophil extracellular traps and induces neutrophil activation. Cells (2022) 11. doi: 10.3390/CELLS11020236
137. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, Van Zoelen MAD, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13:1042–9. doi: 10.1038/NM1638
138. Raquil M-A, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol (2008) 180:3366–74. doi: 10.4049/JIMMUNOL.180.5.3366
139. Sroussi HY, Berline J, Dazin P, Green P, Palefsky JM. S100A8 triggers oxidation-sensitive repulsion of neutrophils. J Dent Res (2006) 85:829–33. doi: 10.1177/154405910608500910
140. Sroussi HY, Lu Y, Zhang QL, Villines D, Marucha PT. S100A8 and S100A9 inhibit neutrophil oxidative metabolism in-vitro: involvement of adenosine metabolites. Free Radic Res (2010) 44:389–96. doi: 10.3109/10715760903431434
141. Joly P, Marshall JC, Tessier PA, Massé C, Page N, Frenette AJ, et al. S100A8/A9 and sRAGE kinetic after polytrauma; an explorative observational study. Scand J Trauma Resusc Emerg Med (2017) 25. doi: 10.1186/S13049-017-0455-0
142. Tardif MR, Chapeton-Montes JA, Posvandzic A, Pagé N, Gilbert C, Tessier PA. Secretion of S100A8, S100A9, and S100A12 by neutrophils involves reactive oxygen species and potassium efflux. J Immunol Res (2015) 2015. doi: 10.1155/2015/296149
143. Diercks BP, Hauschildt I, Stäb F, Wenck H, Döring O, Peters N. IL-10 promotes secretion of S100A8/A9 from human monocytes trough an inclusion in plasma membranes. Scand J Immunol (2013) 77:169–70. doi: 10.1111/SJI.12015
144. Roilo M, Kullmann MK, Hengst L. Cold-inducible RNA-binding protein (CIRP) induces translation of the cell-cycle inhibitor p27Kip1. Nucleic Acids Res (2018) 46:3198–210. doi: 10.1093/NAR/GKX1317
145. De Leeuw F, Zhang T, Wauquier C, Huez G, Kruys V, Gueydan C. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp Cell Res (2007) 313:4130–44. doi: 10.1016/J.YEXCR.2007.09.017
146. Tan C, Reilly B, Jha A, Murao A, Lee Y, Brenner M, et al. Active Release of eCIRP via Gasdermin D Channels to Induce Inflammation in Sepsis. J Immunol (2022) 208:2184–95. doi: 10.4049/JIMMUNOL.2101004
147. Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med (2013) 19:1489–95. doi: 10.1038/NM.3368
148. Han J, Zhang Y, Ge P, Dakal TC, Wen H, Tang S, et al. Exosome-derived CIRP: An amplifier of inflammatory diseases. Front Immunol (2023) 14:1066721. doi: 10.3389/FIMMU.2023.1066721
149. Reilly B, Tan C, Murao A, Nofi C, Jha A, Aziz M, et al. Necroptosis-mediated eCIRP release in sepsis. J Inflammation Res (2022) 15:4047–59. doi: 10.2147/JIR.S370615
150. Bolognese AC, Sharma A, Yang WL, Nicastro J, Coppa GF, Wang P. Cold-inducible RNA-binding protein activates splenic T cells during sepsis in a TLR4-dependent manner. Cell Mol Immunol (2018) 15:38–47. doi: 10.1038/CMI.2016.43
151. Zhang F, Brenner M, Yang WL, Wang P. A cold-inducible RNA-binding protein (CIRP)-derived peptide attenuates inflammation and organ injury in septic mice. Sci Rep (2018) 8. doi: 10.1038/S41598-017-13139-Z
152. Ode Y, Aziz M, Wang P. CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol (2018) 103:693–707. doi: 10.1002/JLB.3A0817-327RR
153. Denning NL, Aziz M, Murao A, Gurien SD, Ochani M, Prince JM, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight (2020) 5. doi: 10.1172/JCI.INSIGHT.134172
154. Lee Y, Reilly B, Tan C, Wang P, Aziz M. Extracellular CIRP induces macrophage extracellular trap formation via gasdermin D activation. Front Immunol (2021) 12:780210. doi: 10.3389/FIMMU.2021.780210
155. Li Z, Fan E K, Liu J J, Scott M, Li Y, Li S, et al. Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis (2017) 8. doi: 10.1038/CDDIS.2017.187
156. Ode Y, Aziz M, Jin H, Arif A, Nicastro JG, Wang P. Cold-inducible RNA-binding protein induces neutrophil extracellular traps in the lungs during sepsis. Sci Rep (2019) 9. doi: 10.1038/S41598-019-42762-1
157. Chen K, Murao A, Arif A, Takizawa S, Jin H, Jiang J, et al. Inhibition of efferocytosis by extracellular CIRP-induced neutrophil extracellular traps. J Immunol (2021) 206:797–806. doi: 10.4049/JIMMUNOL.2000091
158. Zhang F, Yang WL, Brenner M, Wang P. Attenuation of hemorrhage-associated lung injury by adjuvant treatment with C23, an oligopeptide derived from cold-inducible RNA-binding protein. J Trauma Acute Care Surg (2017) 83:690–7. doi: 10.1097/TA.0000000000001566
159. Zhou M, Aziz M, Wang P. Damage-associated molecular patterns as double-edged swords in sepsis. Antioxid Redox Signal (2021) 35:1308–23. doi: 10.1089/ARS.2021.0008
160. Cayrol C, Girard JP. Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine (2022) 156. doi: 10.1016/J.CYTO.2022.155891
161. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol (2014) 31:31–7. doi: 10.1016/J.COI.2014.09.004
162. Hirsiger S, Simmen HP, Werner CML, Wanner GA, Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflammation (2012) 2012. doi: 10.1155/2012/315941
163. Qi L, Zhang Q, Miao Y, Kang W, Tian Z, Xu D, et al. Interleukin-33 activates and recruits natural killer cells to inhibit pulmonary metastatic cancer development. Int J Cancer (2020) 146:1421–34. doi: 10.1002/IJC.32779
164. Haupt J, Krysiak N, Unger M, Bogner-Flatz V, Biberthaler P, Hanschen M, et al. The potential of adipokines in identifying multiple trauma patients at risk of developing multiple organ dysfunction syndrome. Eur J Med Res (2021) 26. doi: 10.1186/S40001-021-00511-Z
165. Hacker S, Dieplinger B, Werba G, Nickl S, Roth GA, Krenn CG, et al. Increased serum concentrations of soluble ST2 predict mortality after burn injury. Clin Chem Lab Med (2018) 56:2079–87. doi: 10.1515/CCLM-2018-0042
166. Billiar IM, Guardado J, Abdul-Malak O, Vodovotz Y, Billiar TR, Namas RA. Elevations in circulating sST2 levels are associated with in-hospital mortality and adverse clinical outcomes after blunt trauma. J Surg Res (2019) 244:23–33. doi: 10.1016/J.JSS.2019.05.057
167. Brunner M, Krenn C, Roth G, Moser B, Dworschak M, Jensen-Jarolim E, et al. Increased levels of soluble ST2 protein and IgG1 production in patients with sepsis and trauma. Intensive Care Med (2004) 30:1468–73. doi: 10.1007/S00134-004-2184-X
168. Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, et al. Temporal patterns of circulating inflammation biomarker networks differentiate susceptibility to nosocomial infection following blunt trauma in humans. Ann Surg (2016) 263:191–8. doi: 10.1097/SLA.0000000000001001
169. Bryk JA, Popovic PJ, Zenati MS, Munera V, Pribis JP, Ochoa JB. Nature of myeloid cells expressing arginase 1 in peripheral blood after trauma. J Trauma (2010) 68:843–52. doi: 10.1097/TA.0B013E3181B026E4
170. Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PloS One (2011) 6. doi: 10.1371/JOURNAL.PONE.0021245
171. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418:191–5. doi: 10.1038/NATURE00858
172. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun (2020) 11. doi: 10.1038/S41467-020-18443-3
173. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med (2022) 54:91–102. doi: 10.1038/S12276-022-00736-W
174. Yang Z, Simovic MO, Edsall PR, Liu B, Cancio TS, Batchinsky AI, et al. HMGB1 inhibition to ameliorate organ failure and increase survival in trauma. Biomolecules (2022) 12. doi: 10.3390/BIOM12010101
175. Fei J, Yu HJ, Zhou J, Huang XK, Liang H, Jiang Y. Study on high mobility group-1 protein in patients with multiple trauma. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue (2005) 17:273–5.
176. Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci (2015) 35:583–98. doi: 10.1523/JNEUROSCI.2439-14.2015
177. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol (2018) 200:422–31. doi: 10.4049/JIMMUNOL.1701019
178. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/S41577-020-00490-Y
179. Sundén-Cullberg J, Nyström T, Lee ML, Mullins GE, Tokics L, Andersson J, et al. Pronounced elevation of resistin correlates with severity of disease in severe sepsis and septic shock. Crit Care Med (2007) 35:1536–42. doi: 10.1097/01.CCM.0000266536.14736.03
180. Cohen G, Ilic D, Raupachova J, Hörl WH. Resistin inhibits essential functions of polymorphonuclear leukocytes. J Immunol (2008) 181:3761–8. doi: 10.4049/JIMMUNOL.181.6.3761
181. Singbartl K, Miller L, Ruiz-Velasco V, Kellum JA. Reversal of acute kidney injury-induced neutrophil dysfunction: A critical role for resistin. Crit Care Med (2016) 44:e492–501. doi: 10.1097/CCM.0000000000001472
182. Miller L, Singbartl K, Chroneos ZC, Ruiz-Velasco V, Lang CH, Bonavia A. Resistin directly inhibits bacterial killing in neutrophils. Intensive Care Med Exp (2019) 7. doi: 10.1186/S40635-019-0257-Y
183. Nieto JC, Perea L, Soriano G, Zamora C, Cantó E, Medina A, et al. Ascitic fluid regulates the local innate immune response of patients with cirrhosis. J Leukoc Biol (2018) 104:833–41. doi: 10.1002/JLB.3A0218-072R
184. Jang JC, Li J, Gambini L, Batugedara HM, Sati S, Lazar MA, et al. (2017) 114:E10399–408. doi: 10.1073/PNAS.1716015114
185. Onishi S, Matsuura H, Osuka A, Matsumoto H, Ebihara T, Ogura H. Resistin forms a network with inflammatory cytokines and is associated with prognosis in major burns. Burns (2022) 48:1680–9. doi: 10.1016/J.BURNS.2021.10.009
186. Duffy SL, Lagrone L, Herndon DN, Mileski WJ. Resistin and postburn insulin dysfunction. J Trauma (2009) 66:250–4. doi: 10.1097/TA.0B013E31815EBAD4
187. Wade CE, Mora AG, Shields BA, Pidcoke HF, Baer LA, Chung KK, et al. Signals from fat after injury: plasma adipokines and ghrelin concentrations in the severely burned. Cytokine (2013) 61:78–83. doi: 10.1016/J.CYTO.2012.08.031
188. Dong XQ, Bin YS, FL Z, QW LV, GH Z, Bin HH. Resistin is associated with mortality in patients with traumatic brain injury. Crit Care (2010) 14. doi: 10.1186/CC9307
189. Zhang DQ, Deng Y, jie ZL, min L, Qi Y, Wang J, et al. Elevated resistin levels may regulate high mobility group box 1 expression in Guillain-Barré syndrome. J Neuroimmunol (2019) 330:59–66. doi: 10.1016/J.JNEUROIM.2019.02.011
190. Bonavia A, Miller L, Kellum JA, Singbartl K. Hemoadsorption corrects hyperresistinemia and restores anti-bacterial neutrophil function. Intensive Care Med Exp (2017) 5. doi: 10.1186/S40635-017-0150-5
191. Lee J, Jackman JG, Kwun J, Manook M, Moreno A, Elster EA, et al. Nucleic acid scavenging microfiber mesh inhibits trauma-induced inflammation and thrombosis. Biomaterials (2017) 120:94–102. doi: 10.1016/J.BIOMATERIALS.2016.12.024
192. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci (2018) 19. doi: 10.3390/IJMS19041222
193. Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, et al. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock (2014) 42:142–7. doi: 10.1097/SHK.0000000000000180
194. Chen Y, Shukla A, Namiki S, Insel PA, Junger WG. A putative osmoreceptor system that controls neutrophil function through the release of ATP, its conversion to adenosine, and activation of A2 adenosine and P2 receptors. J Leukoc Biol (2004) 76:245–53. doi: 10.1189/JLB.0204066
195. Woehrle T, Yip L, Manohar M, Sumi Y, Yao Y, Chen Y, et al. Hypertonic stress regulates T cell function via pannexin-1 hemichannels and P2X receptors. J Leukoc Biol (2010) 88:1181–9. doi: 10.1189/JLB.0410211
196. Haskó G, Kuhel DG, Salzman AL, Szabó C. ATP suppression of interleukin-12 and tumour necrosis factor-alpha release from macrophages. Br J Pharmacol (2000) 129:909–14. doi: 10.1038/SJ.BJP.0703134
197. Li X, Kondo Y, Bao Y, Staudenmaier L, Lee A, Zhang J, et al. Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med (2017) 45:e97–e104. doi: 10.1097/CCM.0000000000002052
198. Zha QB, Wei HX, Li CG, Liang YD, Xu LH, Bai WJ, et al. ATP-induced inflammasome activation and pyroptosis is regulated by AMP-activated protein kinase in macrophages. Front Immunol (2016) 7:597. doi: 10.3389/FIMMU.2016.00597
199. He Y, Taylor N, Fourgeaud L, Bhattacharya A. The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflamm (2017) 14. doi: 10.1186/S12974-017-0904-8
200. Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, et al. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J (2009) 23:1685–93. doi: 10.1096/FJ.08-126458
201. Fredholm BB. Purines and neutrophil leukocytes. Gen Pharmacol (1997) 28:345–50. doi: 10.1016/S0306-3623(96)00169-3
202. Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis (2014) 5. doi: 10.1038/CDDIS.2014.70
203. Pulte ED, Broekman MJ, Olson KE, Drosopoulos JHF, Kizer JR, Islam N, et al. CD39/NTPDase-1 activity and expression in normal leukocytes. Thromb Res (2007) 121:309–17. doi: 10.1016/J.THROMRES.2007.04.008
204. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol (2013) 171:1–7. doi: 10.1111/J.1365-2249.2012.04623.X
205. Cohen HB, Briggs KT, Marino JP, Ravid K, Robson SC, Mosser DM. TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood (2013) 122:1935–45. doi: 10.1182/BLOOD-2013-04-496216
206. Tiwari-Heckler S, Lee GR, Harbison J, Ledderose C, Csizmadia E, Melton D, et al. Extracellular mitochondria drive CD8 T cell dysfunction in trauma by upregulating CD39. Thorax (2023) 78:151–9. doi: 10.1136/THORAXJNL-2021-218047
207. Thurairajah K, Briggs GD, Balogh ZJ. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur J Trauma Emerg Surg (2018) 44:325–34. doi: 10.1007/S00068-018-0954-3
208. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/CDD.2009.96
209. Lam NYL, Rainer TH, Chiu RWK, Joynt GM, Lo YMD. Plasma mitochondrial DNA concentrations after trauma. Clin Chem (2004) 50:213–6. doi: 10.1373/CLINCHEM.2003.025783
210. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock (2010) 34:55–9. doi: 10.1097/SHK.0B013E3181CD8C08
211. Mallavia B, Liu F, Lefrançais E, Cleary SJ, Kwaan N, Tian JJ, et al. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am J Respir Cell Mol Biol (2020) 62:364–72. doi: 10.1165/RCMB.2019-0140OC
212. Zhang J, Chen X, Liu Z, Wang X, Ren J, Sun T. Association between plasma mitochondrial DNA and sterile systemic inflammatory response syndrome in patients with acute blunt traumatic injury. Int J Clin Exp Med (2017) 10:3254–62.
213. Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, et al. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest (2020) 157:67–76. doi: 10.1016/J.CHEST.2019.09.028
214. Chiu RWK, Chan LYS, Lam NYL, Tsui NBY, Ng EKO, Rainer TH, et al. Quantitative analysis of circulating mitochondrial DNA in plasma. Clin Chem (2003) 49:719–26. doi: 10.1373/49.5.719
215. Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, et al. Blood contains circulating cell-free respiratory competent mitochondria. . FASEB J (2020) 34:3616–30. doi: 10.1096/FJ.201901917RR
216. Briggs GD, Gelzinnis S, Meakes S, King KL, Balogh ZJ. Not all cell-free mitochondrial dna is equal in trauma patients. Shock (2022) 58:231–5. doi: 10.1097/SHK.0000000000001969
217. Shen H, de Almeida PE, Kang KH, Yao P, Chan CW. Burn injury triggered dysfunction in dendritic cell response to TLR9 activation and resulted in skewed T cell functions. PloS One (2012) 7. doi: 10.1371/JOURNAL.PONE.0050238
218. Itagaki K, Kaczmarek E, Kwon WYMPD, Chen L, Vlková B, Zhang Q, et al. Formyl peptide receptor-1 blockade prevents receptor regulation by mitochondrial danger-associated molecular patterns and preserves neutrophil function after trauma. Crit Care Med (2020) 48:E123–32. doi: 10.1097/CCM.0000000000004094
219. Li H, Itagaki K, Sandler N, Gallo D, Galenkamp A, Kaczmarek E, et al. Mitochondrial damage-associated molecular patterns from fractures suppress pulmonary immune responses via formyl peptide receptors 1 and 2. J Trauma Acute Care Surg (2015) 78:272–81. doi: 10.1097/TA.0000000000000509
220. Zhao C, Itagaki K, Gupta A, Odom S, Sandler N, Hauser CJ. Mitochondrial damage-associated molecular patterns released by abdominal trauma suppress pulmonary immune responses. J Trauma Acute Care Surg (2014) 76:1222–7. doi: 10.1097/TA.0000000000000220
221. Itagaki K, Riça I, Zhang J, Gallo D, DePrato M, Otterbein LE, et al. Intratracheal instillation of neutrophils rescues bacterial overgrowth initiated by trauma damage-associated molecular patterns. J Trauma Acute Care Surg (2017) 82:853–60. doi: 10.1097/TA.0000000000001413
222. Macáková K, Kaczmarek E, Itagaki K. Can neutrophils prevent nosocomial pneumonia after serious injury? Int J Mol Sci (2023) 24. doi: 10.3390/IJMS24087627
Keywords: critical care, damage associated molecular patterns, immunosuppression, innate immunity, trauma
Citation: Horner E, Lord JM and Hazeldine J (2023) The immune suppressive properties of damage associated molecular patterns in the setting of sterile traumatic injury. Front. Immunol. 14:1239683. doi: 10.3389/fimmu.2023.1239683
Received: 13 June 2023; Accepted: 31 July 2023;
Published: 15 August 2023.
Edited by:
Christoph Thiemermann, Queen Mary University of London, United KingdomReviewed by:
Marcin Filip Osuchowski, Ludwig Boltzmann Institute for Experimental and Clinical Traumatology, AustriaTomasz Skirecki, Medical Centre for Postgraduate Education, Poland
Copyright © 2023 Horner, Lord and Hazeldine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon Hazeldine, ai5oYXplbGRpbmVAYmhhbS5hYy51aw==