 Lingyun Zhang1,2
Lingyun Zhang1,2 Shiyou Wei
Shiyou Wei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 September 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1238233
This article is part of the Research Topic Improving Responses to Immunotherapy in Glioblastoma Multiforme View all 10 articles
Despite tremendous efforts to exploit effective therapeutic strategies, most glioblastoma (GBM) inevitably relapse and become resistant to therapies, including radiotherapy and immunotherapy. The tumor microenvironment (TME) of recurrent GBM (rGBM) is highly immunosuppressive, dominated by tumor-associated macrophages (TAMs). TAMs consist of tissue-resident microglia and monocyte-derived macrophages (MDMs), which are essential for favoring tumor growth, invasion, angiogenesis, immune suppression, and therapeutic resistance; however, restricted by the absence of potent methods, the heterogeneity and plasticity of TAMs in rGBM remain incompletely investigated. Recent application of single-cell technologies, such as single-cell RNA-sequencing has enabled us to decipher the unforeseen diversity and dynamics of TAMs and to identify new subsets of TAMs which regulate anti-tumor immunity. Here, we first review hallmarks of the TME, progress and challenges of immunotherapy, and the biology of TAMs in the context of rGBM, including their origins, categories, and functions. Next, from a single-cell perspective, we highlight recent findings regarding the distinctions between tissue-resident microglia and MDMs, the identification and characterization of specific TAM subsets, and the dynamic alterations of TAMs during tumor progression and treatment. Last, we briefly discuss the potential of TAM-targeted strategies for combination immunotherapy in rGBM. We anticipate the comprehensive understanding of the diversity and dynamics of TAMs in rGBM will shed light on further improvement of immunotherapeutic efficacy in rGBM.
Glioblastoma (GBM) is the most prevalent and malignant type of brain tumors. Nearly 90% of GBM relapse despite the standard of care involving surgery, radiotherapy, and chemotherapy (1). Recurrent GBM (rGBM) generally differs from primary GBM in their molecular and histological characteristics, intra-tumor heterogeneity, immune microenvironment, and biological behaviors due to the therapeutic pressure and clonal selection, which contributes to the aggressiveness and therapeutic resistance of rGBM (2). Therefore, patients with GBM rapidly succumb to this disease, with a median overall survival of 12-15 months after initial diagnosis and a 5-year survival rate of less than 10% (3, 4); however, due to the lack of abundant high-quality rGBM samples, most current studies focus on primary GBM, while the biology of rGBM remains largely unknown, and practical therapeutic approaches against rGBM are lacking. Therefore, it is instrumental to understand the biology of rGBM for developing effective therapeutic strategies and improving the clinical outcome of patients with GBM.
Recently, emerging immunotherapies, including immune checkpoint blockade (ICB), vaccine, and chimeric antigen receptor (CAR) T-cell therapy, have revolutionized the therapeutic landscape of multiple types of cancers (5); however, several clinical trials have revealed disappointing therapeutic efficacy of immunotherapy in rGBM, and the underlying mechanisms remain incompletely elucidated (6–8). The tumor microenvironment (TME) is pivotal in orchestrating immune activity and modulating response to immunotherapy. GBM is a typically “cold tumor” with an immunosuppressive TME featured by the paucity of cytotoxic T cells (CTLs) and the abundance of immunosuppressive cells such as myeloid cells (8–11). Tumor-associated macrophages (TAMs) are the most predominant non-malignant cells infiltrating GBM, which consist of tissue-resident microglia and monocyte-derived macrophages (MDMs). Growing evidence has suggested pro-tumor functions of TAMs in GBM include aggravating tumor growth and metastasis, angiogenesis, immunosuppression, treatment resistance, etc (12–14). Besides, the level of TAMs is markedly increased in rGBM, which in turn is associated with poor prognosis of patients (8, 15).
TAMs are incredibly plastic and heterogeneous, exhibiting diverse phenotypes and functions when responding to the environment-specific stimuli. Recently, high-resolution methodologies [e.g., single-cell RNA-sequencing (scRNA-seq)] have been instrumental in identifying and characterizing various subsets of TAMs with distinct functions in GBM (13, 16). Interestingly, emerging evidence has revealed the dynamic alterations of TAMs during disease progression and therapeutic resistance in rGBM. Meanwhile, different TAM-targeted therapeutic approaches have been developed, showing promising potential in multiple types of cancers, including rGBM (17, 18). Therefore, an elaborated understanding of the complexity of TAMs and molecular mechanisms underlying the tumor-promoting roles of TAMs in rGBM is vital to facilitating TAM-modulating treatment in order to overcome resistance of rGBM to immunotherapy.
In this review, we first provide a concise overview of characteristics of TME and immunotherapy for patients with rGBM. We then describe the biology of TAMs in rGBM, including their origins, categories and functions. In particular, we review recent advances regarding the phenotypic and functional diversity of TAMs in rGBM at the single-cell resolution, and focus on distinctions between tissue-resident microglia and MDMs, the characterization of specific subsets, and the dynamic changes of TAMs during tumor evolution and treatment in GBM. Finally, we highlight the potential of therapeutically targeting TAM as the basis for combination immunotherapy for patients with rGBM.
In the context of cancer, various types of immune cells enter the central nervous system (CNS) by disrupting the blood-brain barrier. The TME of GBM is dominated by immunosuppressive cells, including TAMs, myeloid-derived suppressor cells (MDSC), and regulatory T cells (Treg) (9, 10). Besides, genetic alterations are associated with the immune status of GBM, and diverse immune landscapes in four molecular subtypes of GBM [neural, pro-neural (PN), classical (CL), and mesenchymal (MES)] have been documented (19, 20). For example, tumor-infiltrating CTLs were scarce in the CL subtype but abundant in the MES subtype (21). Also, a preponderance of TAMs was identified in the MES subtype (22). In addition, the IDH-1 mutation, which frequently occurrs in the PN subtype, is correlated with reduced Tregs and monocyte signatures, PD-L1 expression, and a favorable prognosis (23–25). Thus, the heterogeneity of TME that is associated with cancer genetics provides a foundation for tailoring therapies for patients with GBM.
Crucially, the TME altered by the treatment results in a unique TME for rGBM that differs from that of primary GBM. For instance, 82% of rGBM lost the expression of epidermal growth factor receptor variant III (EGFRvIII). EGFRvIII is an immunogenic mutation widely detected and constitutively activated in primary GBM, indicating that immunologic escape occured after a period of progression-free survival in rGBM (26). Recently, it was reported that CD103+ Tregs with upregulated lipid metabolism accumulated in response to ICB therapy and concurrent radiotherapy, which hindered the cytotoxic activity of CTLs in GBM (27). Additionally, rGBM exhibited an increase in the infiltration of CD68+ macrophages following anti-angiogenic therapy, suggestive of the potential role of TAMs in controlling therapeutic resistance and tumor relapse (15). More dynamic changes of TAMs during tumor progression and treatment will be reviewed in the following sessions. Taken together, the highly heterogeneous, dynamic and immunosuppressive TME is a key player contributing to anti-tumor immune evasion in rGBM.
ICB therapy could inhibit the immune checkpoint pathways such as programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) signaling, thus alleviating T cell exhaustion and enhancing CTLs-mediated tumor killing (28). Despite the therapeutic success of anti-PD-1/PD-L1 treatment in multiple types of cancers, the phase III clinical trial Checkmate 143 reported that anti-PD-1 antibody nivolumab failed to achieve survival benefits compared with bevacizumab in rGBM patients (6, 29–31). In contrast, another clinical trial conducted by Cloughesy et al. demonstrated that OS in rGBM patients treated with neoadjuvant anti-PD-1 therapy (surgery following pembrolizumab) was improved compared with adjuvant-only treatment, which was accompanied by increases in the expression levels of genes related to T cells and interferon (IFN)-γ within the tumor (32). Besides, several studies have suggested the promising anti-tumor effects of ICB-based combination therapy in pre-clinical GBM mouse models, but clinical trials are needed to determine the clinical efficacy (33–36). Overall, current evidence hints that a single ICB treatment might be insufficient to revert the immunosuppressive TME of rGBM and elicit satisfactory efficacy. Therefore, it is worth investigating an ICB-based combination treatment against rGBM.
The tumor-specific peptide vaccination provides a promising approach to trigger specific immune responses by targeting tumor-associated antigens (TAAs). rGBM possesses a broad spectrum of TAAs, including CD133, gp100, EGFRvIII, IL-13Rα2, Wilms’ tumor 1 (WT1), HER2, etc (37–40). Several clinical trials have demonstrated the survival benefit of GBM-specific peptide vaccination, but the therapeutic response was hampered by pre-treatment lymphopenia, which highlighted the necessity of more rigorous selection criteria for patient enrollment (41). On the other hand, given the crucial role of dendritic cells (DCs) in antigen presentation and activation of CTLs, DC vaccination therapy has also exhibited an encouraging effect in treating patients with rGBM (42–45); however, it is incredibly time-consuming to isolate and purify autologous DCs, making it challenging to exploit DCs-based immunotherapy for rapidly progressing rGBM.
Recently, CAR-T cell immunotherapy has presented an attractive anti-tumor method and succeeded in treatment of hematological malignancies (46–48). Several studies have demonstrated the safety and feasibility of IL-13Rα2-specific and HER2-specific CAR T cells in patients with rGBM (49–52); however, researchers reported the limited efficacy of EGFRvIII-specific CAR-T cell therapy in patients with rGBM (7). EGFRvIII was highly expressed in primary GBM but exhibited a specific loss or decreased expression in tumors resected after CAR-T cell therapy (7, 53, 54). Apart from EGFRvIII antigen escape, the adaptive immunosuppressive response was observed in the TME upon CAR-T therapy, suggested by the upregulated expression of inhibitory molecules, including PD-L1, TGF-β, IDO, and IL-10 and infiltration of Tregs (7). Currently, the durable clinical efficacy of CAT-T cell therapy in rGBM is hindered by the short lifespan of CAR-T cells, the poor infiltration of T cells in tumor tissues, tumor heterogeneity, and antigen escape, which needs to be addressed in the future (55).
Despite the recent breakthrough of immunotherapy in a subset of patients with rGBM, there are still many obstacles in the practical application. More efforts should be made to solve the issues regarding the optimum approach, treatment timing, patient selection, and combination modalities to augment the efficacy of immunotherapies for patients with rGBM.
Microglia are the brain-resident macrophages originating from yolk sac-derived embryogenetic precursors. Under normal physiological conditions, microglia comprise 10% of the adult brain cell populations, represent the main component of brain macrophages, and play an essential role in maintaining the immune homeostasis of CNS (16, 56). Upon inflammatory stimulation, such as infection and cancer, bone marrow-derived monocytes in the peripheral blood are recruited to the tumor site and then differentiate into macrophages. Various recruitment signals have been recognized, including colony-stimulating factor-1 (CSF-1), monocyte chemoattractant protein-1 (MCP‐1), and stromal-derived factor (SDF)-1α derived from tumor cells and other cells in the TME (12, 57–60).
The term TAMs in GBM include both tissue-resident microglia and MDMs. It is challenging to distinguish or separate microglia from MDMs using conventional approaches (e.g., flow cytometry) due to the lack of specific markers (61); however, growing evidence has demonstrated the dramatic distinctions in preferential localizations and functions between these two subpopulations. For example, Chen et al. found that MDMs accounted for most TAMs in GBM and were mainly located in perivascular areas. Inversely, microglia only represented a minor TAM population, usually appearing in the peritumoral zones (58). Moreover, microglia-derived TAMs are predominant in primary GBM but are outnumbered by MDMs following recurrence, especially under hypoxia (62). Phenotypically, MDMs upregulate immunosuppressive cytokines and show an altered metabolism compared to microglial TAMs (63). More studies are needed to dissect the exact origin and specific roles of TAM populations in GBM.
Based on the polarization status and regulatory functions under inflammation, macrophages are divided into classically activated macrophages (M1, pro-inflammatory) and alternatively activated macrophages (M2, anti-inflammatory) (64). M1 macrophages can be induced by lipopolysaccharide (LPS), IFN-γ, granulocyte–macrophage colony-stimulating factor (GM-CSF) or Toll-like receptor signaling pathway. M1 macrophages spur inflammation by releasing cytokines such as IL-1α, IL-1β, IL-6, IL-12, and tumor necrosis factor (TNF)-α. On the other hand, M2 macrophages are stimulated by IL-4, IL-10, IL-13, and glucocorticoid. M2 macrophages express PD-L1 and exert immunosuppressive functions by secreting IL-10, arginase-1, TGF-β, etc (64, 65). Generally, M1 macrophages exert an anti-tumor role, whereas M2 macrophages play a pro-tumor role. Several markers distinguish the M1 from the M2 phenotype, e.g., CD80, CD86, and MHC-II for M1, CD163 and CD206 for M2, although they are not absolutely specific (66, 67). CSF-1, TGF-β1, macrophage inhibitory cytokine 1 (MIC-1), osteopontin (OPN), and Periostin produced by GBM cells recruit and polarize macrophages to a tumor-supporting M2-like phenotype (68–71). CD163 and CD206 are highly expressed in perivascular macrophages in the brain tumor cores and are associated with an immunosuppressive TME (72).
M1/M2 nomenclature is proposed mainly based on in vitro data when macrophages were stimulated with type 1 or 2 cytokines. This nomenclature remains oversimplified, albeit widely used (66). Indeed, macrophages are highly plastic and heterogeneous, with the capacity of being reprogrammed into distinct phenotypes by different microenvironmental stimuli. Besides, canonical M1 and M2 markers are co-expressed in individual cells, implying that macrophages could possess a mixed M1/M2 phenotype (63). Beyond M1/M2, the more complicated phenotypic and functional diversity of TAMs in GBM has been recently appreciated (73–75). Next, we will review the diverse roles of TAMs in regulating progression of GBM, and summarize recent advances that reveal the complexity of TAMs in GBM based on single-cell omics approaches.
TAMs are involved in tumor development and progression via releasing various factors and interacting with other cells in multiple malignancies (17, 76). In GBM, the pro-tumor roles of TAMs are well documented that implicate the importance of TAMs as a therapeutic vulnerability in GBM, involving tumor growth, invasion, angiogenesis, immunosuppression, and treatment resistance (Figure 1) (12, 18).
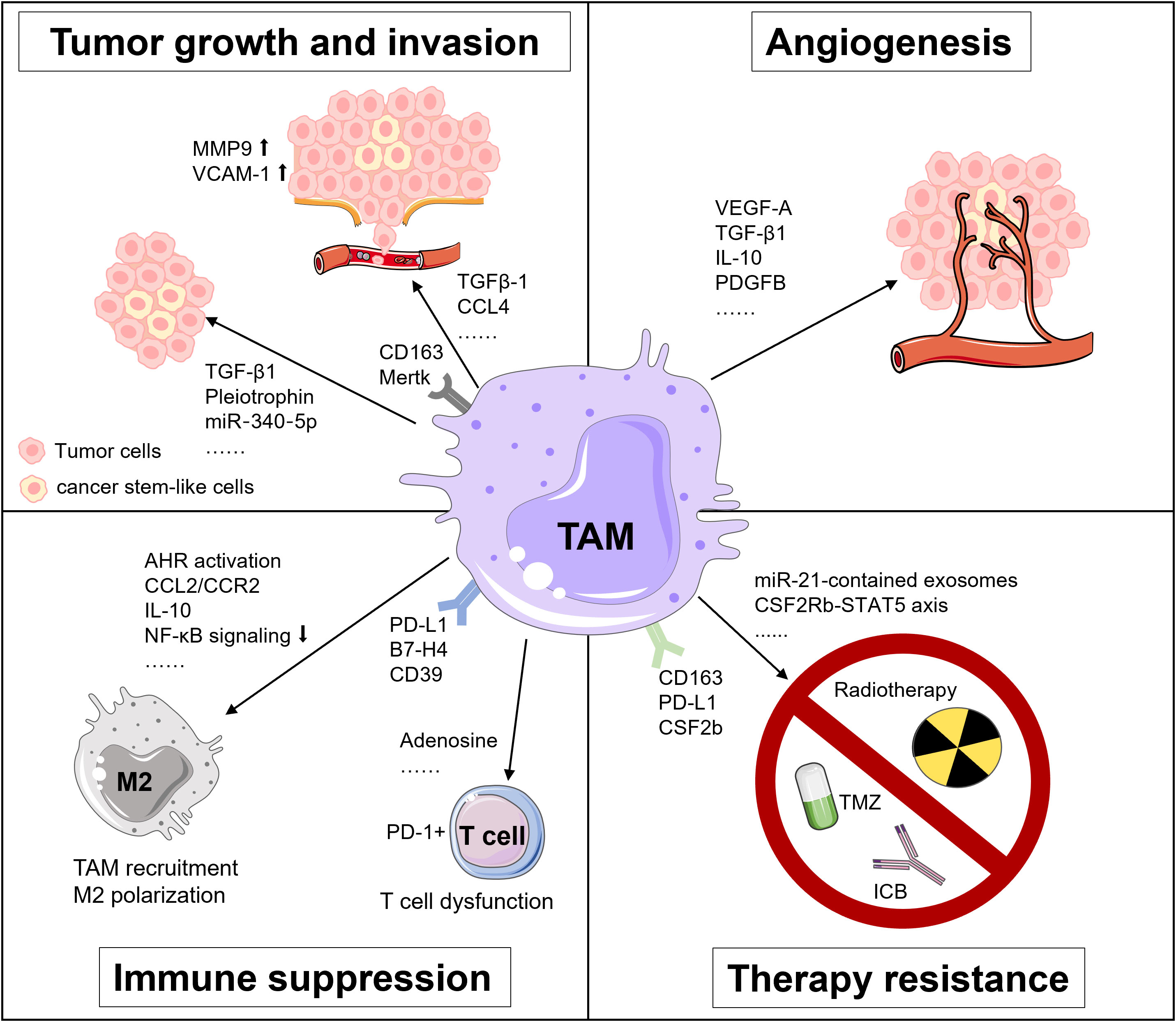
Figure 1 Tumor-supportive functions of TAMs in GBM. TAMs foster tumor growth and invasion, angiogenesis, immunosuppression and treatment resistance in GBM via multiple pathways.
The molecular interaction between TAMs and tumor cells is critical for regulating tumor growth and invasion. For instance, TAMs secrete TGF-β1 to recruit CD133+ cancer stem-like cells (CSCs) (77). Pleiotrophin (PTN) derived from CD163+ M2 macrophages binds to its receptor protein tyrosine phosphatase receptor type Z1 (PTPRZ1) on the surface of CSCs. The binding of PTN and PTPRZ1 contributes to the stemness maintenance and tumorigenic capacity of CSCs, thus accelerating the growth of GBM (78). Besides, TAMs upregulate the expression of metalloproteinase 9 (MMP-9) of CSCs via TGF-β1 and CCL4-CCR5 signaling to enhance the GBM invasiveness (79). Also, the vascular cell adhesion molecule-1 (VCAM-1)-mediated interaction between macrophages and GBM cells reinforces GBM invasion (80). Additionally, Liu et al. unveiled that a miR‐340‐5p‐macrophage feedback loop regulated tumor progression and was related to a poor prognosis for patients with GBM (81). Moreover, TAMs expressing myeloid-epithelial-reproductive tyrosine kinase (MerTK), a critical tyrosine kinase for phagocytosis function, are associated with tumor growth (82).
The rapid proliferation of tumor cells accelerates the consumption of oxygen and nutrients in the TME, rendering a highly hypoxic environment for GBM, especially in its core region (83). Under the hypoxic condition, TAMs produce angiogenesis-promoting cytokines, chemokines, and growth factors, like vascular endothelial growth factor A (VEGF-A), a well-known factor for vascularization and immunosuppression in multiple cancers, including GBM (83–85). Besides, Cui et al. found that GBM-induced M2-like macrophages secreted more TGF-β1 and IL-10. These anti-inflammatory cytokines facilitated endothelial capillary proliferation and angiogenic sprouting through integrin (αvβ3) receptors and Src-PI3K-YAP signaling. Hence, dual blockade of integrin (αvβ3) and cytokine receptor (TGFβ-R1) could suppress the neovascularization of GBM induced by the TAM-endothelial interaction (86). Moreover, a recent study by Zhu et al. suggested that the expression of cat eye syndrome critical region protein 1(CECR1) was upregulated in M2 macrophages and correlated with microvascular density in GBM. Mechanistically, CECR1 mediated the crosstalk between macrophages and vascular mural cells via the PDGFB-PDGFRβ signaling axis, leading to recruitment of pericytes, migration, and tumor angiogenesis (87). Collectively, TAMs exert potent pro-angiogenic properties in GBM, implying that therapeutically targeting TAMs may present an attractive way against rGBM.
The highly immunosuppressive TME represents a hallmark of GBM, which is primarily attributed to TAMs via multiple mechanisms. For instance, decreased IKBKB expression and NF-κB signaling in TAMs support M2 polarization and correlate with defective expression of immune/inflammatory genes, resulting in immune suppression in GBM (88). Accordingly, NF-κB-targeted therapy could reverse M2 polarization, induce tumor regression and improve survival of a GBM mouse model in T cell-dependent manner (89). Besides, Takenaka and colleagues recently uncovered mechanisms by which TME controlled TAMs and T cells in GBM. Kynurenine produced by GBM cells elicited the activation of aryl hydrocarbon receptor (AHR) in TAMs, which further increased the expression of CCR2 and boosted TAM recruitment via the CCL2/CCR2 axis. Aside from that, AHR drove the expression of ectonucleotidase CD39 in TAMs and led to the dysfunction of CTLs via adenosine accumulation. Moreover, elevated expression of AHR was associated with glioma grade and unfavorable prognosis in patients with GBM (90).
Immune checkpoint molecules are critical inducers of immunosuppressive TME. Reportedly, GBM could upregulate PD-L1 expression in circulating monocytes and TAMs through the IL-10 signaling axis in an autocrine/paracrine manner. In vitro, macrophages stimulated by IL-10 induced T cell apoptosis, which could be attenuated by inhibiting IL-10 and its receptor (91). Besides, Yao et al. unveiled that CD133+ CSCs activated the expression of B7-H4 in TAMs via the IL6/JAK/STAT3 pathway. Such B7-H4-mediated crosstalk between glioma-initiating cells and TAMs was associated with a bleak prognosis of human GBM (92).
TAMs are involved in therapuetic resistance of GBM to temozolomide (TMZ), radiotherapy, and immunotherapy. For example, TAMs release oncomiR-21-contained exosomes, which upregulate the production of PDCD4, SOX2, STAT3, IL-6, and TGF-1 in GBM cells, rendering resistance of GBM to TMZ. Pacritinib, a STAT3 inhibitor, could overcome resistance to TMZ by decreasing miR-21-enriched exosomes from TAMs (93). Besides, Miyazaki et al. revealed that TMZ-resistant GBM cells produced M2-related cytokines including IL-10, IL-4, IL-13, and CSF-1, and PD-L1 expression. Upon in vivo anti-PD-L1 antibody administration, TMZ-resistant GBM tumor tissues showed abundant infiltration of CD163+ M2 macrophages. Expectedly, remarkable anti-tumor efficacy was achieved using a combination therapy of anti-PD-L1 antibody plus IPI-549, a PI3Kγ inhibitor that could skew M2 macrophages to M1 macrophages (94). Moreover, dynamic transcriptional alterations of TAMs in the irradiated and recurrent tumors have been observed in mouse and human GBM, and CSF-1R inhibition could overcome resistance of pre-clinical models to radiotherapy (95).
As for the significance of TAMs in mediating resistance of GBM to immunotherapy, Simonds et al. previously discovered the association between PD-L1+ TAMs and resistance to ICB by comparing the TME of human ICB-refractory GBM and ICB-responsive tumors using cytometry by time-of-flight (CyTOF) (96). Additionally, through integrated analyses of multi-dimensional data, Lee et al. demonstrated that neoadjuvant anti-PD-1 blockade induced conventional type 1 DC (cDC1) and activation of T cells but failed to eliminate immunosuppressive TAMs in rGBM (97). Interestingly, in the mouse model of brain metastases, pro-inflammatory activation of TAM which was mediated by the compensatory CSF2Rb–STAT5 signaling axis fostered tumor recurrence after CSF1R inhibition. Furthermore, blockade of CSF1R combined with STAT5 signaling inhibitor could sustain tumor control and rectify adaptive resistance to CSF1R inhibition (98). All of these findings highlight the potential benefit of TAM-targeted therapeutic intervention for overcoming treatment resistance in rGBM and metastatic brain tumors.
As mentioned before, owing to the plastic and heterogenous nature of TAMs, the linear M1/M2 activation theory is insufficient to explain the in vivo complexity of TAMs in GBM (66). On the other hand, despite the well-documented anti-tumor functions of M1 macrophages and pro-tumoral functions of M2 macrophages, the prognostic value of CD163+ and CD206+ M2 macrophages was controversial in different cohorts of patients with GBM, emphasizing an unmet need to decipher the exact function of specific TAM subtypes in GBM (20, 99); however, limited by conventional approaches, hurdles exist to distinguish and characterize TAM subpopulations in GBM. In the past years, the application of high-dimensional and high-resolution techniques has enabled us to decipher unprecedented macrophage subclusters in the brain under homeostasis and disease, and moved us beyond the binary M1/M2 polarization paradigm. Herein, we will review recent advances in the phenotypic and functional diversity of TAMs in GBM and provide insights into the therapeutic potential of TAM-based strategies for patients with rGBM.
Microglia and MDMs are different in the spatial distribution, enrichment extent, phenotypic and functional characteristics during disease progression in GBM (Figure 2). A study by Darmanis et al. suggested that the majority of myeloid cells within the tumor center preferentially exhibited gene signatures of macrophages, whereas microglia-related genes were mainly expressed in myeloid cells located in the surrounding space in human GBM (100). Similarly, by analyzing RNA-seq data from the human GBM cohort, Kim et al. recently confirmed that microglial genes (CX3CR1, TMEM119, and P2RY12) were mainly expressed in the periphery, while activated macrophage genes (TNF, CCL2, LYZ, CCR2, CXCR4, and SIGLEC1) were predominantly detected within the core tumor regions (74). Besides, Muller’s group has reported that differing from microglia, MDMs were usually enriched in perivascular and necrotic areas in human glioma (63).
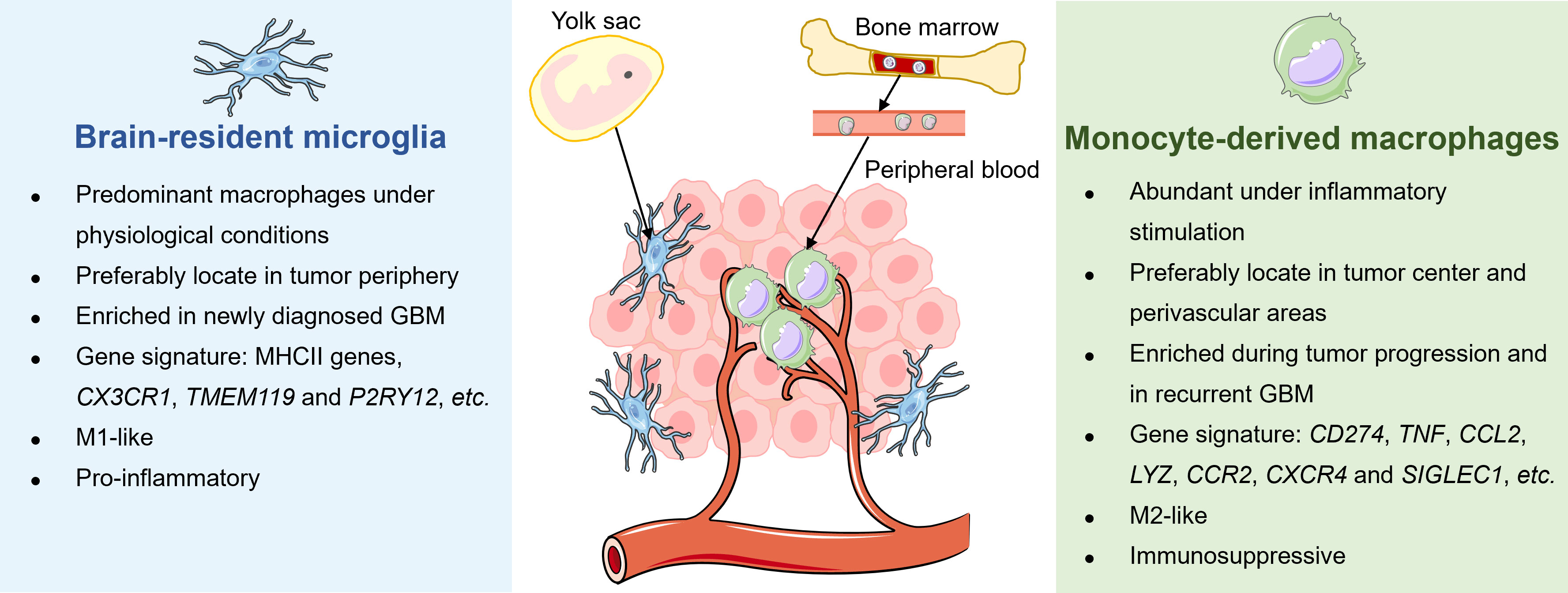
Figure 2 Distinctions between tissue-resident microglia and monocyte-derived macrophages in GBM. The tissue-resident microglia originate from the yolk sac and represent the primary macrophages in the brain under physiological conditions to maintain immune homeostasis. Under malignant conditions, microglia preferably locate in the periphery of newly diagnosed GBM, exhibiting a pro-inflammatory M1-like phenotype. Meanwhile, bone marrow-derived monocytes in circulation are recruited into the brain and differentiate into macrophages. Unlike microglia, MDMs are enriched during tumor progression, particularly in tumor core and perivascular regions of rGBM, and present immunosuppressive M2-like properties.
Several studies have attempted to dissect longitudinal changes of TAM composition throughout tumor evolution and recurrence in GBM. For instance, Yeo et al. reported a progression of TME from M1-like proinflammatory microglia towards an M2-like pro-tumorigenic infiltrating macrophages during tumor growth in the mouse model of GBM. Notably, a similar transition was observed in tumor biopsies derived from patients with low-grade glioma and GBM (101). Besides, Pombo Antunes et al. profiled myeloid cells in mouse and human GBM at new diagnosis and recurrence by employing scRNA-seq and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) technical platforms. The researchers found that microglia-derived TAMs were predominant in initial tumors but were outnumbered by MDMs in recurrent tumors, especially under hypoxic conditions. Although microglia and MDMs exhibited functional specialization to some extent, both of them showed a convergent angiogenic and T-cell suppressive capacity (62).
Emerging evidence has suggested additional phenotypic and functional differences between microglia and MDMs. By performing scRNA-seq of IDH-mutant human gliomas, Venteicher et al. unmasked a continuous rather than a bimodal distribution of transcriptional signatures of microglia/macrophages, underscoring the plasticity of cellular states of TAMs. Besides, the macrophage signature, but not the microglia signature, was associated with clinical grade and increased vascularity in gliomas (102). Additionally, Ochocka et al. identified distinct transcriptional programs of microglia and monocytes/macrophages in mice bearing GBM via scRNA-seq analysis. The transcriptional responses of macrophages were associated with the activation of immunosuppressive genes such as Cd274 encoding PD-L1, while microglia had higher expression levels of major histocompatibility complex II (MHC-II) genes in a sex-specific manner (103). Consistently, Muller et al. determined that MDMs tended to express immunosuppressive cytokines and to undergo metabolic reprogramming compared with microglia in human gliomas (63). Collectively, these results shed light on the dynamic alterations of TAMs with different origins in response to disease status and microenvironment stimulation in GBM.
Substantial efforts have been made to identify specific subsets of TAMs with the most promising therapeutic potential in GBM (Table 1). For instance, by integrating analyses of scRNA-seq and CyTOF data, Sankowski et al. mapped microglial states in the human brain under healthy and malignant conditions, and uncovered a disease-associated transcriptional signature in TAMs from patients with GBM. These TAMs exhibited down-regulation of the microglia core signature and concomitant up-regulation of inflammatory, metabolic, and hypoxia-related genes, including SPP1, and several type I interferon genes, APOE, and CD163. Furthermore, the top differentially expressed proteins, including HLA-DR, TREM2, APOE, CD163, and GPR56, could be detected in a TAM subset via CyTOF, providing a possibility for therapeutically targeting specific TAM states in GBM (75).
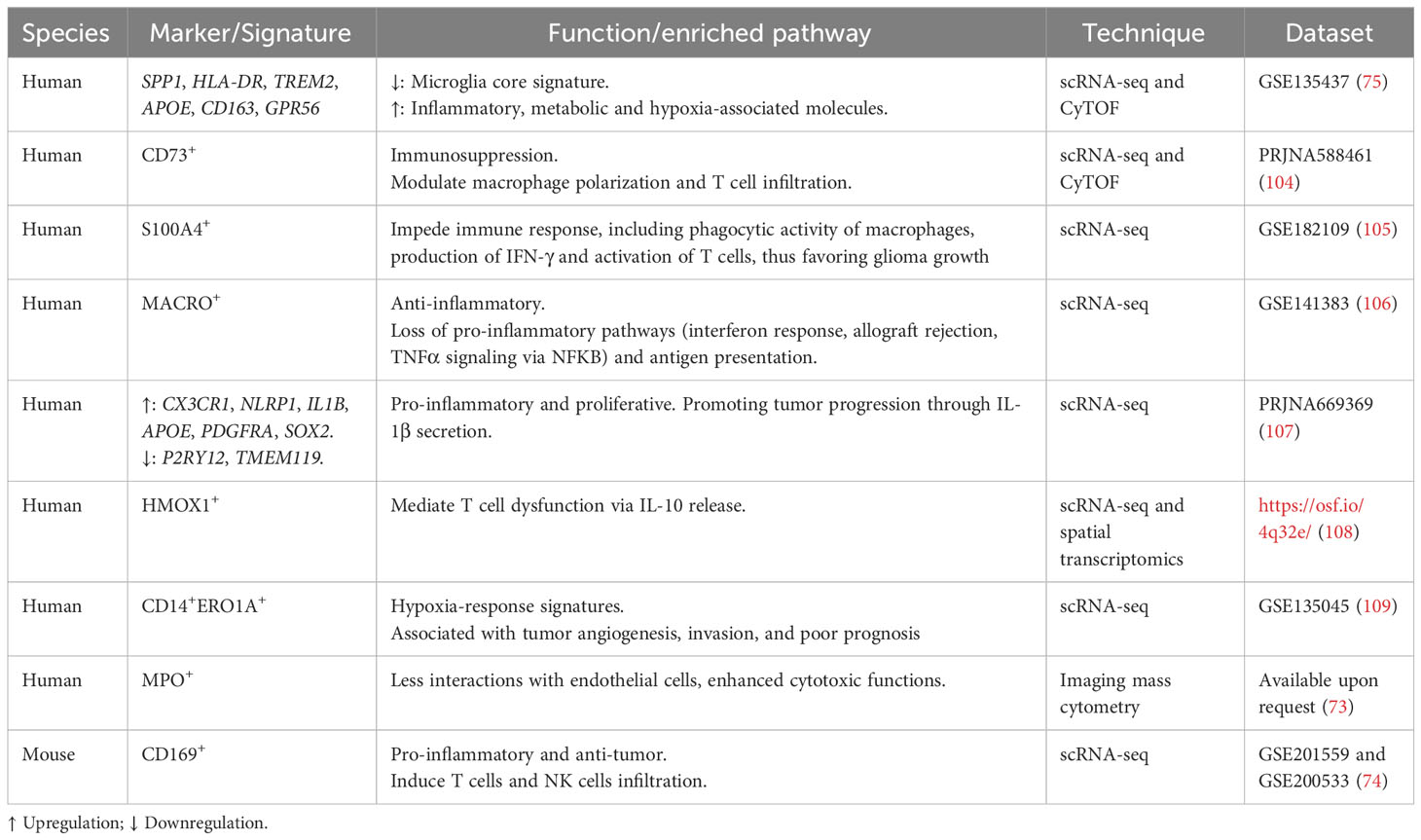
Table 1 Specific TAM subsets in GBM identified through single-cell omics technologies.
Using similar approaches, Goswami et al. determined the persistence of a unique population of CD73+ macrophages in patients with GBM upon anti-PD-1 treatment. They further demonstrated the critical function of CD73+ macrophages in conferring resistance to ICB, which was mediated by the modulation of macrophage polarization and T cell infiltration. Compared with wild-type mice, the authors detected a decrease of immunosuppressive CD206+Arg1+VISTA+PD-1+CD115+ myeloid cluster and an increase of iNOS+ myeloid cells in CD73-deficient mice. Upon the treatment of CD73-knockout mice with the combination with anti-CTLA-4 plus anti-PD-1, the tumor burden was significantly alleviated and survival was improved, along with the elevated ratio of the granzyme B+ effector CD8 T cells to the CD206+ macrophages in the TME. The study indicated that CD73 was a promising immunotherapeutic target to augment anti-tumor immune responses to the combination immunotherapy in GBM (104).
Recently, through multi-regional and -dimensional analyses at the single-cell level, Abdelfattah et al. sought to discover immune modulatory targets in GBM. scRNA-seq analysis identified that S100A4 was highly expressed in innate and adaptive immunosuppressive cells, including Tregs, exhausted T cells, and three subsets of pro-tumorigenic myeloid cells. The immunofluorescence staining confirmed the expression of S100A4 in immunosuppressive CD163+, CD206+ macrophages, and FOXP3+ T cells in human glioma. Moreover, a higher expression of S100A4 was markedly associated with a worse survival and was recognized as an independent prognostic indicator for patients with GBM. Subsequent animal experiments and functional analyses demonstrated the roles of S100A4 in impeding immune response, including phagocytic activity of macrophages, production of IFN-γ and activation of T cells, thus favoring the growth of glioma, supporting S100A4 as a promising immunotherapeutic target (105).
Additionally, Chen et al. integrated analyses of newly generated and published single-cell RNA-seq data, which identified a tumor-supportive subcluster of TAMs characterized by the scavenger receptor MARCO, almost exclusively expressed in IDH1-wild-type (IDH-WT) GBM. Moreover, MACRO was reportedly detrimental in melanoma and non-small cell lung cancer. The expression of MARCO in bulk tumors was also associated with a disappointing prognosis and mesenchymal subtype in the GBM cohort. Further analysis observed the loss of pro-inflammatory pathways (interferon response, allograft rejection, and TNFα signaling via NFKB) and antigen presentation in MACRO+ TAMs, supporting its anti-inflammatory phenotype. Altogether, the study revealed a novel TAM subpopulation driving the progression of GBM and implied a potential strategy for MACRO+ TAM-targeted therapy (106).
Increasing evidence has also elucidated how TAMs orchestrate the immunosuppressive TME through various crosstalk with their neighboring components. Through scRNA-seq analysis of human GBM, Liu et al. identified a unique pro-inflammatory and proliferative subpopulation of microglia, marked by upregulated expression of CX3CR1, NLRP1, IL1B, APOE, PDGFRA, and SOX2. The microglia were activated by TGF-β1 derived from SETD2-mut/IDH-WT tumor cells, and accelerated tumor progression via secreting IL-1β. Notably, depletion of TGF-β1/TGF-β RI successfully reduced the pro-inflammatory and proliferative microglia and restrained tumor growth (107). Through integrative analysis of single-cell and spatial transcriptomics data of human GBM, Ravi et al. revealed that a subset of HMOX1+ microglia and macrophages released IL-10 and mediated T cell dysfunction, thus fostering an immunosuppressive TME (108). Additionally, a specific CD14+ERO1A+ TAM cluster with detrimental prognostic value in human primary GBM has been identified, which showed a gene signature enriched in hypoxia-response, invasion and extracellular matrix organization. The CD14+ERO1A+ TAM cluster, together with two hypoxia-dependent MES-like tumor cells expressed VEGFA, indicating their contribution to the induction of angiogenesis in GBM via interacting with endothelial cells (109).
Contrary to the well-known tumor-supportive functions of TAMs, studies also showed that several subsets of macrophages favor anti-tumor immunity against GBM. For example, through the analysis of scRNA-seq data, Kim et al. unraveled that CD169+ TAMs were IFN-responsive macrophages, which produced pro-inflammatory chemokines, hence inducing the infiltration of T cells and NK cells in human and mouse gliomas. Mechanistically, CD169+ TAMs originated from CCR2+ blood monocytes, and IFN-γ derived from NK cells was critical for recruiting CD169+ macrophages into gliomas. CD169 boosted the phagocytosis capacity of macrophages through ligands of apoptotic tumor cells and ignited antigen-specific T cell responses. Moreover, the clearance of CD169+ TAMs impaired anti-tumor responses mediated by T cells and shortened the survival of mice bearing glioma (74). Recently, Karimi et al. characterized the immune landscape of primary and metastatic human brain tumors at the single-cell level by applying imaging mass cytometry. Specifically, they identified a unique subpopulation of myeloperoxidase (MPO)-positive neutrophil-like macrophages, which was related to reduced interactions with endothelial cells, enhanced cytotoxic functions and survival benefit for patients with GBM (73).
The remarkably distinct and even inverse roles of TAMs in orchestrating GBM progression reflect their plasticity and heterogeneity. Therefore, it is necessary to identify specific targets and mechanisms for tailoring TAMs-modulating therapy regimens. Still, the vast diversity of TAMs in GBM remains incompletely illustrated, highlighting an urgent demand for more investigations that utilize high-dimension and high-resolution approaches and platforms.
Several studies have investigated alterations of TAMs during tumor evolution in GBM at the single-cell level (Table 2). For instance, Rajendran et al. not only revealed an immune-activated feature displayed by TAM clusters in low-grade murine glioma but also demonstrated an immunosuppressive property in murine high-grade murine glioma, accompanied by the restriction of T cell trafficking and activation. They further identified high expression of CD74 and its binding partner, macrophage migration inhibition factor (MIF) in distinct TAM populations, which was subsequently validated in human samples and supported the CD74-MIF axis as a potential target for TAMs (11). As mentioned above, the preponderance of TAMs underwent a transition from M1-like proinflammatory microglia to M2-like pro-tumorigenic macrophages during GBM progression, which was conserved in human and mouse. The transition was concurrent with a disruption of the blood-brain barrier and an explosive growth of malignant cells (101).
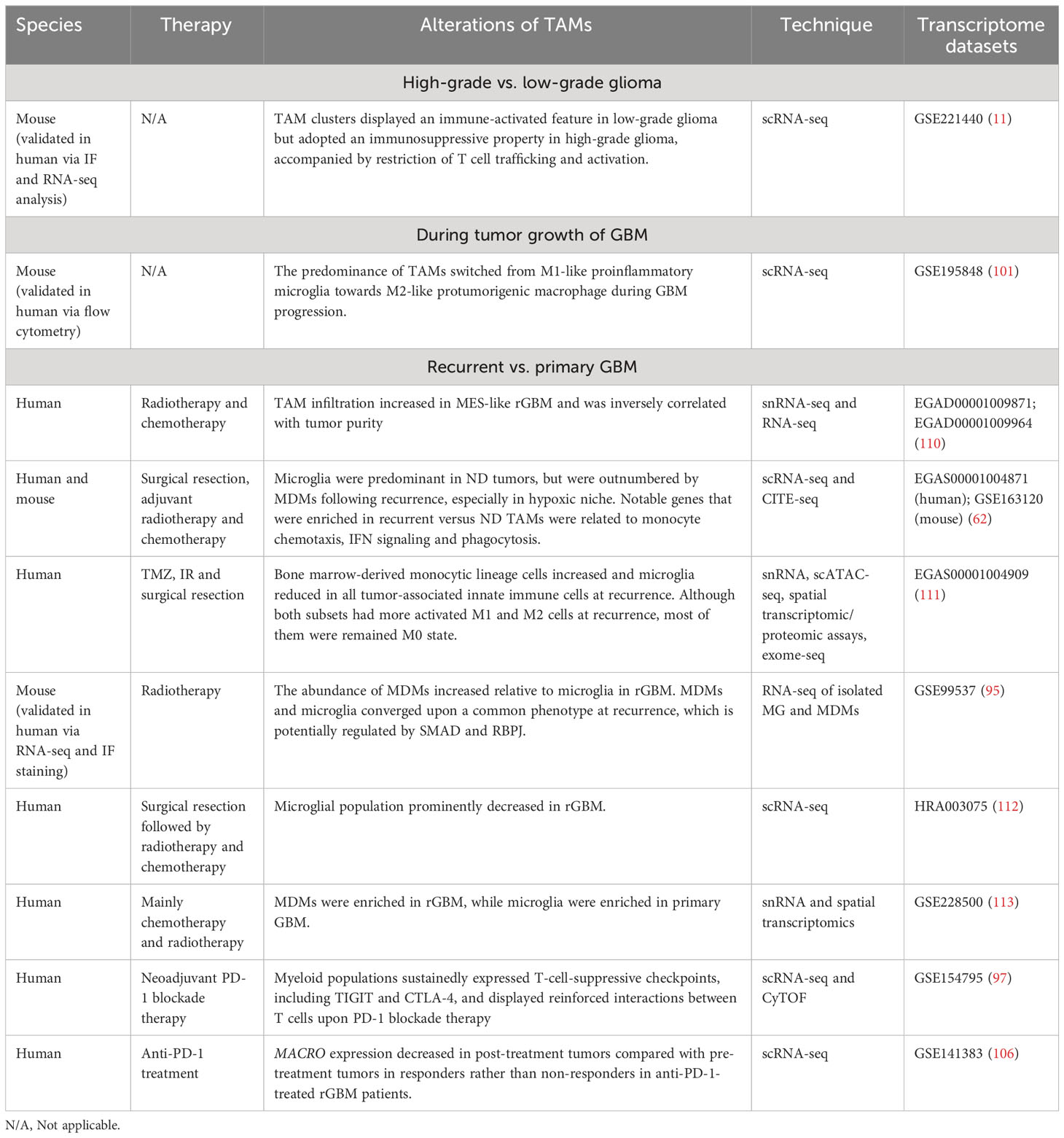
Table 2 The dynamic alterations of TAMs during disease progression and treatment in GBM at the single-cell resolution.
Hoogstrate et al. conducted a large-scale transcriptome analysis of paired primary-recurrent GBM resections of patients following standard therapy and suggested that rGBM preferentially progressed to MES-like subtype (110). Macrophages are known to be recruited by GBM stem cells and induce the MES-like state of GBM cells (110, 114, 115). Consistently, Hoogstrate et al.’s study identified significant increase of TAM infiltration in MES-like rGBM, which was inversely correlated with tumor purity, supporting the essential role of TAMs in favoring MES-like GBM progression at recurrence (110).
As mentioned above, Pombo Antunes et al. compared the immune landscape of newly diagnosed (ND) GBM versus rGBM following surgery, adjuvant radiotherapy and chemotherapy through scRNA-seq and CITE-seq analyses. The TME of ND GBM mainly consisted of TAMs (82–97%), followed by T cells (2–20%), while rGBM displayed a more diverse immune compartment including increased T cells, NK, B cells and monocytes. Microglia formed the major TAM fraction in ND GBM, but MDMs outcompeted microglia in rGBM, especially in the hypoxic tumor niche. TAMs in recurrent versus ND GBM displayed higher expression of genes related to monocyte chemotaxis, IFN signaling, and phagocytosis (62). Additionally, a single-cell multi-omics analysis conducted by Wang et al. demonstrated an increase of bone marrow-derived monocytic lineage cells and a reduction of microglia in all tumor-associated innate immune cells at recurrence upon standard-of-care therapy including TMZ, IR and surgical resection. Although both subsets had more activated M1 and M2 macrohages at recurrence, most of them were classified as M0 state without expressing either program above. It may be attributed to the oversimplification of M1/M2 paradigm, and a continuous modal for macrophage classification is demanded (111).
In another study, by analysing RNA-seq data of isolated microglia and MDMs in pre- and post-treatment murine gliomas, Akkari and colleagues identified stage-dependent transcriptional reprogramming of these two TAM subpopulations in irradiated murine glioma. In line with previous studies, the results confirmed the increased abundance of MDMs relative to microglia in rGBM compared with primary GBM (62, 111–113). MDMs and microglia maintained their ontogeny-based identities and converged upon a common phenotype at recurrence, which is potentially regulated by SMAD and RBPJ. Notably, recurrence-specific transcriptional changes of TAMs were also observed in human rGBM. Inhibition of CSF1-R could counteract the recurrence-induced gene signature alterations in TAMs, enhance the efficacy of radiation therapy and delay tumor regrowth in pre-clinical mouse models (95). Collectively, the findings disclosed the dynamics and plasticity of individual TAM populations during radiation treatment and provided novel insight into improving the treatment landscape in GBM.
Additionally, a study by Lee et al. also mapped the landscape of infiltrating immune cells in GBM, with a particular focus on alterations in TME following neoadjuvant PD-1 blockade therapy. By exploiting high-dimensional proteomics, scRNA-seq and quantitative multiplex immunofluorescence (mIF), the authors determined increased activation and infiltration of T cells and cDC1 after ICB treatment; however, TAMs and monocytes maintained the dominance of tumor-infiltrating immune cells upon anti-PD-1 therapy. Although the interferon-mediated T-cell chemotactic factors (such as CXCL9, CXCL10, and CXCL11) were secreted in myeloid populations after PD-1 blockade, these cells sustainedly expressed T cell-suppressive checkpoints, including TIGIT and CTLA-4. Furthermore, the analysis of scRNA-seq data recognized reinforced interactions between T cells and myeloid cells through TIGIT- and CTLA-4-related signaling after PD-1 blockade therapy, which could impede optimal and durable activation of CTLs. Therefore, additional strategies targeting TIGIT and/or CTLA-4 may enhance the strength and durability of CTL-mediated anti-tumor response of GBM to immunotherapy (97).
Besides, in the above-described study, decreased expression of MACRO was observed in post-treatment tumors compared with pre-treatment tumors in responders rather than non-responders in a longitudinal cohort of patients with rGBM treated with anti-PD-1. However, there were no apparent changes in expression of MARCO after treatment in another longitudinal cohort of patients of GBM with standard therapy. These findings suggested that MACRO was altered upon treatment in an immunotherapy-specific and response-dependent manner (106).
More investigations are required to delineate the dynamics and plasticity of TAMs during treatment and determine the mechanisms by which TAMs modulate therapy outcomes, thus providing translational relevance for enhancing therapeutic efficacy in GBM.
The strong tumor-promoting activity of TAMs has highlighted its promising potential as a therapeutic target against rGBM. Multiple TAM-targeted approaches have been explored in preclinical and clinical settings for patients with rGBM, mainly including: i). reduction of the recruitment of TAMs into tumors; ii). elimination of TAMs within tumors; iii). reprogramming of TAMs. Since these strategies have been reviewed recently, we provide a concise summary here (13, 14, 17, 18, 116).
The inhibition of TAM infiltration could be realized by directly blocking signalings between chemokines and their receptors. For example, CCL2 derived from tumor cells recruit CCR2+ myeloid cells; hence CCR2 antagonist could directly reduce TAM infiltration and improve the efficacy of ICB in murine GBM (117). As mentioned above, kynurenine produced by GBM cells led to AHR activation, further promoting CCR2 expression and enhancing TAM recruitment. In this case, AHR antagonist effectively suppressed GBM growth via reducing CCL2/CCR2-mediated TAM infiltration (90). Besides CCL2/CCR2 axis, other chemoattractant-receptor interactions have also shown therapeutic potential in GBM, such as lysyl oxidase (LOX)/β1 integrin, OPN/αvβ5 integrin, and slit guidance ligand 2 (SLIT2)/Roundabout 1 and 2 (ROBO1/2) (71, 118, 119).
CSF-1R is expressed on macrophages and critical for regulating the survival, proliferation, differentiation, and polarization of TAMs by binding with its ligands CSF-1 and IL-34 (120, 121). Targeting CSF-1R using antibodies or small molecule inhibitors has represent a powerful strategy to deplete TAMs and induce TAM repolarization in various types of cancers, including GBM (122–124). It is worth noting that monotherapy of targeting CSF-1R was insufficient to elicit satisfactory efficacy, and combination therapy with immunotherapy or radiation demonstrated better clinical outcome, which highlight the necessity of combination strategy in clinical exploration (125, 126). Besides, along with our extended understanding of the complexity of TME and the heterogeneity of TAM subpopulations, we should realize that the unbiased depletion of the whole TAM cluster may not be an optimal option, because it is likely to eliminate beneficial TAM subpopulations and influence other TME components. Therefore, more efforts should be made to identify specific tumor-supporting subtypes of TAMs (e.g., CD73+, MACRO+, and HMOX1+ TAMs) and develop targeted therapy across ravious scenarios.
In spite of the detrimental function of TAM subsets, macrophage play an essential role in phagocytosis and antigen presentation, which is beneficial for the activation of anti-tumor immunity (127). Therefore, rather than macrophage clearance and recruitment inhibition, another attractive strategy is to reprogram/re-deucate TAMs, i.e., reprogram immunosuppressive TAMs to immune-supportive TAMs by restoring their phagocytic and antigen presenting capacities (76). To achieve reprogramming of TAMs in GBM, multiple approaches have been developed. For instance, the blockade of phagocytosis checkpoint pairs, e.g., CD47/SIRPα, CD24/Siglec-10 could augment the phagocytic ability of TAMs (128–132). To unleash the immune-stimulatory capacity of TAMs, blockade of CSF1/CSF1R, stimulation of CD40/CD40L, as well as inhibition of PI3Kγ, IL-6, SLIT2, monoacylglycerol lipase (MAGL) have shown promising targetable potential and are worth further investigation (94, 119, 124, 133–135).
Notably, no single TAM-targeted agent has been successful in clinical trials for patients with GBM. Given the complexity of the TME and the close interplay between TAMs, tumor cells, and other non-malignant cells, combination therapy emerges as an attractive option. For instance, in a mouse model of GBM, the SDF-1α inhibitor in combination with VEGF blockade was more efficient in suppressing TAM recruitment, reducing tumor vasculature and improving survival compared with monotherapy of VEGF blockade (136). As discussed in the above, CSF1-R blockade in combination with radiotherapy substantially inhibited tumor growth and prolonged survival by reversing transcriptional changes of TAM induced by radiation in pre-clinical glioma models, thus overcoming resistance to radiotherapy (95). Similarly, a triple combination of oncolytic virus expressing IL-12, and anti-PD-1 plus anti-CTLA-4 antibodies synergistically cured pre-clinical murine GBM via increasing M1-like polarization and the ratio of effector T cells to Tregs (137). Further investigation regarding combination therapy regimens in the clinical setting is dispensable for strengthening the efficacy of immunotherapies for patients with rGBM.
rGBM has been characterized by a highly immunosuppressive TME and an extremely low response to immunotherapy. TAMs, originating from microglia and peripheral monocytes, represent the dominant non-malignant cells in the TME of rGBM. TAMs exert various tumor-supportive functions, contributing to tumor growth and invasion, angiogenesis, immune evasion, and treatment resistance. More importantly, TAMs are plastic and heterogeneous, displaying more complicated phenotypes beyond the binary M1/M2 polarization. Recently, single-cell omics methodologies have enabled us to characterize the dynamics and diversity of TAMs in the TME of GBM at the single-cell resolution. Microglia and MDMs show different spatial distribution and exhibit distinctive transcriptional alterations across disease stages. Besides, specific subsets of TAMs with different functions have been determined in the context of rGBM, e.g., the pro-tumor MACRO+, CD73+, HMOX1+, and S100A4+ macrophages, and anti-tumor CD169+ and MPO+ macrophages, further underscoring the complexity of TAMs. Moreover, several studies have interrogated the dynamic changes of TAMs responding to treatment and microenvironmental stimulation, providing novel insights into how TAMs modulate therapeutic response and resistance. Therefore, harnessing TAMs via different approaches may be feasible in treating rGBM, and TAM-based combination therapy regimens have started to show a promising potential.
Still, dynamics and diversities of TAMs in the context of rGBM, and underlying molecular mechanisms remain incompletely clarified, which warrants further investigations and integrated analyses of high-dimensional and high-resolution data, such as spatial scRNA-seq, single-cell proteomics, and single-cell sequencing assay for transposase-accessible chromatin (scATAC-seq). More studies in the near future should focus on i). distinguishing microglia from MDMs; ii). identifying specific tumor-supportive and tumor-suppressive TAM subclusters; iii). delineating the stage- and therapy-specific reprogramming of TAMs in longitudinal cohorts; iv). dissecting the cellular crosstalk between TAMs and other cells; v). exploring rationale-based combination therapy modalities in clinical trials targeting TAMs. Ultimately, comprehensively understanding TAMs and their interplay with other cells will be instrumental for optimal immunomodulation and enhanced immunotherapeutic efficacy for patients with rGBM.
GZ and SW conceived and designed the study. LZ wrote the original draft. YJ, GZ and SW revised the manuscript. All authors contributed to the article and approved the submitted version.
This research is supported by grants from China Postdoctoral Science Foundation (No. 2022M720103), Postdoctoral Program of Sichuan Province (No. TB2022018), and the Fundamental Research Funds for the Central Universities (No. 2023SCU12052).
The authors thank all subjects for their participation in the research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin (2020) 70:299–312. doi: 10.3322/caac.21613
2. Campos B, Olsen LR, Urup T, Poulsen HS. A comprehensive profile of recurrent glioblastoma. Oncogene (2016) 35:5819–25. doi: 10.1038/onc.2016.85
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
4. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
5. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol (2020) 17:807–21. doi: 10.1038/s41423-020-0488-6
6. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
7. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
8. de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol (2020) 22:539–49. doi: 10.1093/neuonc/noz185
9. Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH. Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res (2019) 25:4202–10. doi: 10.1158/1078-0432.CCR-18-1627
10. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
11. Rajendran S, Hu Y, Canella A, Peterson C, Gross A, Cam M, et al. Single-cell RNA sequencing reveals immunosuppressive myeloid cell diversity during Malignant progression in a murine model of glioma. Cell Rep (2023) 42:112197. doi: 10.1016/j.celrep.2023.112197
12. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2016) 19:20–7. doi: 10.1038/nn.4185
13. Khan F, Pang L, Dunterman M, Lesniak MS, Heimberger AB, Chen P. Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J Clin Invest. (2023) 133:e163446. doi: 10.1172/JCI163446
14. Wu M, Shi Y, Zhu L, Chen L, Zhao X, Xu C. Macrophages in glioblastoma development and therapy: A double-edged sword. Life (Basel). (2022) 12:1225. doi: 10.3390/life12081225
15. Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol (2013) 15:1079–87. doi: 10.1093/neuonc/not082
16. Ochocka N, Kaminska B. Microglia diversity in healthy and diseased brain: insights from single-cell omics. Int J Mol Sci (2021) 22:3027. doi: 10.3390/ijms22063027
17. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
18. Andersen JK, Miletic H, Hossain JA. Tumor-associated macrophages in gliomas-basic insights and treatment opportunities. Cancers (Basel). (2022) 14:1319. doi: 10.3390/cancers14051319
19. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnorMalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
20. Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S, et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun (2019) 7:203. doi: 10.1186/s40478-019-0803-6
21. Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res (2013) 19:4951–60. doi: 10.1158/1078-0432.CCR-13-0551
22. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell (2019) 178:835–49 e21. doi: 10.1016/j.cell.2019.06.024
23. Rahman M, Kresak J, Yang C, Huang J, Hiser W, Kubilis P, et al. Analysis of immunobiologic markers in primary and recurrent glioblastoma. J Neurooncol. (2018) 137:249–57. doi: 10.1007/s11060-017-2732-1
24. Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol (2017) 19:1460–8. doi: 10.1093/neuonc/nox054
25. Myung JK, Cho HJ, Park CK, Kim SK, Phi JH, Park SH. IDH1 mutation of gliomas with long-term survival analysis. Oncol Rep (2012) 28:1639–44. doi: 10.3892/or.2012.1994
26. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28:4722–9. doi: 10.1200/JCO.2010.28.6963
27. van Hooren L, Handgraaf SM, Kloosterman DJ, Karimi E, van Mil L, Gassama AA, et al. CD103(+) regulatory T cells underlie resistance to radio-immunotherapy and impair CD8(+) T cell activation in glioblastoma. Nat Cancer (2023) 4:665–671. doi: 10.1038/s43018-023-00547-6
28. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
29. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
30. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
31. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372:311–9. doi: 10.1056/NEJMoa1411087
32. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med (2019) 25:477–86. doi: 10.1038/s41591-018-0337-7
33. Puigdelloses M, Garcia-Moure M, Labiano S, Laspidea V, Gonzalez-Huarriz M, Zalacain M, et al. CD137 and PD-L1 targeting with immunovirotherapy induces a potent and durable antitumor immune response in glioblastoma models. J Immunother Cancer. (2021) 9:1. doi: 10.1136/jitc-2021-002644
34. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
35. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res (2016) 4:124–35. doi: 10.1158/2326-6066.CIR-15-0151
36. Maghrouni A, Givari M, Jalili-Nik M, Mollazadeh H, Bibak B, Sadeghi MM, et al. Targeting the PD-1/PD-L1 pathway in glioblastoma multiforme: Preclinical evidence and clinical interventions. Int Immunopharmacol. (2021) 93:107403. doi: 10.1016/j.intimp.2021.107403
37. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. (2007) 81:139–48. doi: 10.1007/s11060-006-9220-3
38. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget (2015) 6:171–84. doi: 10.18632/oncotarget.2767
39. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res (2010) 16:474–85. doi: 10.1158/1078-0432.CCR-09-1322
40. Izumoto S, Tsuboi A, Oka Y, Suzuki T, Hashiba T, Kagawa N, et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg (2008) 108:963–71. doi: 10.3171/JNS/2008/108/5/0963
41. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol (2014) 16:274–9. doi: 10.1093/neuonc/not203
42. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent Malignant glioma. J Neurosurg (2015) 123:989–97. doi: 10.3171/2015.1.JNS141554
43. De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res (2008) 14:3098–104. doi: 10.1158/1078-0432.CCR-07-4875
44. Shah AH, Bregy A, Heros DO, Komotar RJ, Goldberg J. Dendritic cell vaccine for recurrent high-grade gliomas in pediatric and adult subjects: clinical trial protocol. Neurosurgery (2013) 73:863–7. doi: 10.1227/NEU.0000000000000107
45. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with alpha-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent Malignant glioma. J Clin Oncol (2011) 29:330–6. doi: 10.1200/JCO.2010.30.7744
46. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
47. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
48. Almasbak H, Aarvak T, Vemuri MC. CAR T cell therapy: A game changer in cancer treatment. J Immunol Res (2016) 2016:5474602. doi: 10.1155/2016/5474602
49. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
50. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
51. Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR, et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol (2022) 24:1318–30. doi: 10.1093/neuonc/noac024
52. Migliorini D, Dietrich PY, Stupp R, Linette GP, Posey AD Jr., June CH. CAR T-cell therapies in glioblastoma: A first look. Clin Cancer Res (2018) 24:535–40. doi: 10.1158/1078-0432.CCR-17-2871
53. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci (2009) 16:748–54. doi: 10.1016/j.jocn.2008.12.005
54. van den Bent MJ, Gao Y, Kerkhof M, Kros JM, Gorlia T, van Zwieten K, et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol (2015) 17:935–41. doi: 10.1093/neuonc/nov013
55. Wang X, Lu J, Guo G, Yu J. Immunotherapy for recurrent glioblastoma: practical insights and challenging prospects. Cell Death Dis (2021) 12:299. doi: 10.1038/s41419-021-03568-0
56. Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res (1999) 117:145–52. doi: 10.1016/s0165-3806(99)00113-3
57. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol (2003) 54:388–92. doi: 10.1002/ana.10679
58. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
59. Wang SC, Hong JH, Hsueh C, Chiang CS. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab Invest. (2012) 92:151–62. doi: 10.1038/labinvest.2011.128
60. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol (2016) 18:797–806. doi: 10.1093/neuonc/nov272
61. Muller A, Brandenburg S, Turkowski K, Muller S, Vajkoczy P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer. (2015) 137:278–88. doi: 10.1002/ijc.29379
62. Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci (2021) 24:595–610. doi: 10.1038/s41593-020-00789-y
63. Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol (2017) 18:234. doi: 10.1186/s13059-017-1362-4
64. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci (2014) 10:520–9. doi: 10.7150/ijbs.8879
65. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol (2011) 11:750–61. doi: 10.1038/nri3088
66. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
67. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
68. Alterman RL, Stanley ER. Colony stimulating factor-1 expression in human glioma. Mol Chem Neuropathol. (1994) 21:177–88. doi: 10.1007/BF02815350
69. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes Malignant growth. Nat Cell Biol (2015) 17:170–82. doi: 10.1038/ncb3090
70. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
71. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. (2019) 129:137–49. doi: 10.1172/JCI121266
72. Noorani I, Sidlauskas K, Pellow S, Savage R, Norman JL, Chatelet DS, et al. Clinical impact of anti-inflammatory microglia and macrophage phenotypes at glioblastoma margins. Brain Commun (2023) 5:fcad176. doi: 10.1093/braincomms/fcad176
73. Karimi E, Yu MW, Maritan SM, Perus LJM, Rezanejad M, Sorin M, et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature (2023) 614:555–63. doi: 10.1038/s41586-022-05680-3
74. Kim HJ, Park JH, Kim HC, Kim CW, Kang I, Lee HK. Blood monocyte-derived CD169(+) macrophages contribute to antitumor immunity against glioblastoma. Nat Commun (2022) 13:6211. doi: 10.1038/s41467-022-34001-5
75. Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci (2019) 22:2098–110. doi: 10.1038/s41593-019-0532-y
76. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther (2021) 6:127. doi: 10.1038/s41392-021-00506-6
77. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol (2012) 189:444–53. doi: 10.4049/jimmunol.1103248
78. Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun (2017) 8:15080. doi: 10.1038/ncomms15080
79. Wang Y, Liu T, Yang N, Xu S, Li X, Wang D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol Rep (2016) 36:3522–8. doi: 10.3892/or.2016.5171
80. Zheng Y, Yang W, Aldape K, He J, Lu Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J Biol Chem (2013) 288:31488–95. doi: 10.1074/jbc.M113.499020
81. Liu Y, Li X, Zhang Y, Wang H, Rong X, Peng J, et al. An miR-340-5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene (2019) 38:7399–415. doi: 10.1038/s41388-019-0952-x
82. Wu J, Frady LN, Bash RE, Cohen SM, Schorzman AN, Su YT, et al. MerTK as a therapeutic target in glioblastoma. Neuro Oncol (2018) 20:92–102. doi: 10.1093/neuonc/nox111
83. Tamura R, Ohara K, Sasaki H, Morimoto Y, Kosugi K, Yoshida K, et al. Difference in immunosuppressive cells between peritumoral area and tumor core in glioblastoma. World Neurosurg (2018) 120:e601–e10. doi: 10.1016/j.wneu.2018.08.133
84. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol (2014) 5:75. doi: 10.3389/fphys.2014.00075
85. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol (2019) 37:2. doi: 10.1007/s12032-019-1329-2
86. Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials (2018) 161:164–78. doi: 10.1016/j.biomaterials.2018.01.053
87. Zhu C, Chrifi I, Mustafa D, van der Weiden M, Leenen PJM, Duncker DJ, et al. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in Malignant glioma. Oncogene (2017) 36:5356–68. doi: 10.1038/onc.2017.145
88. Mieczkowski J, Kocyk M, Nauman P, Gabrusiewicz K, Sielska M, Przanowski P, et al. Down-regulation of IKKbeta expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget (2015) 6:33077–90. doi: 10.18632/oncotarget.5310
89. Barberi T, Martin A, Suresh R, Barakat DJ, Harris-Bookman S, Drake CG, et al. Absence of host NF-kappaB p50 induces murine glioblastoma tumor regression, increases survival, and decreases T-cell induction of tumor-associated macrophage M2 polarization. Cancer Immunol Immunother. (2018) 67:1491–503. doi: 10.1007/s00262-018-2184-2
90. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci (2019) 22:729–40. doi: 10.1038/s41593-019-0370-y
91. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res (2013) 19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314
92. Yao Y, Ye H, Qi Z, Mo L, Yue Q, Baral A, et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin Cancer Res (2016) 22:2778–90. doi: 10.1158/1078-0432.CCR-15-0858
93. Chuang HY, Su YK, Liu HW, Chen CH, Chiu SC, Cho DY, et al. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR-21-Enriched Exosomes from M2 Glioblastoma-Associated Macrophages. J Clin Med (2019) 8:959. doi: 10.3390/jcm8070959
94. Miyazaki T, Ishikawa E, Matsuda M, Sugii N, Kohzuki H, Akutsu H, et al. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kgamma inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol (2020) 37:41–9. doi: 10.1007/s10014-020-00357-z
95. Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci Transl Med (2020) 12:552. doi: 10.1126/scitranslmed.aaw7843
96. Simonds EF, Lu ED, Badillo O, Karimi S, Liu EV, Tamaki W, et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer. (2021) 9:e002181. doi: 10.1136/jitc-2020-002181
97. Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun (2021) 12:6938. doi: 10.1038/s41467-021-26940-2
98. Klemm F, Mockl A, Salamero-Boix A, Alekseeva T, Schaffer A, Schulz M, et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat Cancer. (2021) 2:1086–101. doi: 10.1038/s43018-021-00254-0
99. Zeiner PS, Preusse C, Golebiewska A, Zinke J, Iriondo A, Muller A, et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol (2019) 29:513–29. doi: 10.1111/bpa.12690
100. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep (2017) 21:1399–410. doi: 10.1016/j.celrep.2017.10.030
101. Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L, et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat Immunol (2022) 23:971–84. doi: 10.1038/s41590-022-01215-0
102. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (2017) 355:eaai8478. doi: 10.1126/science.aai8478
103. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun (2021) 12:1151. doi: 10.1038/s41467-021-21407-w
104. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med (2020) 26:39–46. doi: 10.1038/s41591-019-0694-x
105. Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun (2022) 13:767. doi: 10.1038/s41467-022-28372-y
106. Chen AX, Gartrell RD, Zhao J, Upadhyayula PS, Zhao W, Yuan J, et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med (2021) 13:88. doi: 10.1186/s13073-021-00906-x
107. Liu H, Sun Y, Zhang Q, Jin W, Gordon RE, Zhang Y, et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep (2021) 36:109718. doi: 10.1016/j.celrep.2021.109718
108. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kuckelhaus J, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun (2022) 13:925. doi: 10.1038/s41467-022-28523-1
109. Xiao Y, Wang Z, Zhao M, Deng Y, Yang M, Su G, et al. Single-cell transcriptomics revealed subtype-specific tumor immune microenvironments in human glioblastomas. Front Immunol (2022) 13:914236. doi: 10.3389/fimmu.2022.914236
110. Hoogstrate Y, Draaisma K, Ghisai SA, van Hijfte L, Barin N, de Heer I, et al. Transcriptome analysis reveals tumor microenvironment changes in glioblastoma. Cancer Cell (2023) 41:678–92 e7. doi: 10.1016/j.ccell.2023.02.019
111. Wang L, Jung J, Babikir H, Shamardani K, Jain S, Feng X, et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat Cancer. (2022) 3:1534–52. doi: 10.1038/s43018-022-00475-x
112. Wu H, Guo C, Wang C, Xu J, Zheng S, Duan J, et al. Single-cell RNA sequencing reveals tumor heterogeneity, microenvironment, and drug-resistance mechanisms of recurrent glioblastoma. Cancer Sci (2023) 114:2609–21. doi: 10.1111/cas.15773
113. Al-Dalahmah O, Argenziano MG, Kannan A, Mahajan A, Furnari J, Paryani F, et al. Re-convolving the compositional landscape of primary and recurrent glioblastoma reveals prognostic and targetable tissue states. Nat Commun (2023) 14:2586. doi: 10.1038/s41467-023-38186-1
114. Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell (2021) 39:779–92 e11. doi: 10.1016/j.ccell.2021.05.002
115. Buonfiglioli A, Hambardzumyan D. Macrophages and microglia: the cerberus of glioblastoma. Acta Neuropathol Commun (2021) 9:54. doi: 10.1186/s40478-021-01156-z
116. Wang G, Zhong K, Wang Z, Zhang Z, Tang X, Tong A, et al. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front Immunol (2022) 13:964898. doi: 10.3389/fimmu.2022.964898
117. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A. (2020) 117:1129–38. doi: 10.1073/pnas.1910856117
118. Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell (2019) 35:868–84 e6. doi: 10.1016/j.ccell.2019.05.003
119. Geraldo LH, Xu Y, Jacob L, Pibouin-Fragner L, Rao R, Maissa N, et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J Clin Invest. (2021) 131:e141083. doi: 10.1172/JCI141083
120. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
121. Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans (2016) 44:333–41. doi: 10.1042/BST20150245
122. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol (2014) 5:489. doi: 10.3389/fimmu.2014.00489
123. Barca C, Foray C, Hermann S, Herrlinger U, Remory I, Laoui D, et al. The colony stimulating factor-1 receptor (CSF-1R)-mediated regulation of microglia/macrophages as a target for neurological disorders (Glioma, stroke). Front Immunol (2021) 12:787307. doi: 10.3389/fimmu.2021.787307
124. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene (2017) 36:6049–58. doi: 10.1038/onc.2017.261
125. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol (2016) 18:557–64. doi: 10.1093/neuonc/nov245
126. Przystal JM, Becker H, Canjuga D, Tsiami F, Anderle N, Keller AL, et al. Targeting CSF1R alone or in combination with PD1 in experimental glioma. Cancers (Basel). (2021) 13:2400. doi: 10.3390/cancers13102400
127. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
128. Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J, et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther (2023) 8:104. doi: 10.1038/s41392-023-01365-z
129. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPalpha immune checkpoint. Immunity (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
130. Li F, Lv B, Liu Y, Hua T, Han J, Sun C, et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology (2018) 7:e1391973. doi: 10.1080/2162402X.2017.1391973
131. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
132. Wu H, Liu J, Wang Z, Yuan W, Chen L. Prospects of antibodies targeting CD47 or CD24 in the treatment of glioblastoma. CNS Neurosci Ther (2021) 27:1105–17. doi: 10.1111/cns.13714
133. Chonan M, Saito R, Shoji T, Shibahara I, Kanamori M, Sonoda Y, et al. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol (2015) 17:1453–62. doi: 10.1093/neuonc/nov090
134. Yang F, He Z, Duan H, Zhang D, Li J, Yang H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun (2021) 12:3424. doi: 10.1038/s41467-021-23832-3
135. Yin J, Kim SS, Choi E, Oh YT, Lin W, Kim TH, et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat Commun (2020) 11:2978. doi: 10.1038/s41467-020-16789-2
136. Deng L, Stafford JH, Liu SC, Chernikova SB, Merchant M, Recht L, et al. SDF-1 blockade enhances anti-VEGF therapy of glioblastoma and can be monitored by MRI. Neoplasia (2017) 19:1–7. doi: 10.1016/j.neo.2016.11.010
Keywords: recurrent glioblastoma, tumor microenvironment, immunotherapy, microglia, monocyte-derived macrophages, single-cell
Citation: Zhang L, Jiang Y, Zhang G and Wei S (2023) The diversity and dynamics of tumor-associated macrophages in recurrent glioblastoma. Front. Immunol. 14:1238233. doi: 10.3389/fimmu.2023.1238233
Received: 11 June 2023; Accepted: 21 August 2023;
Published: 04 September 2023.
Edited by:
Riccardo Dolcetti, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Dalia Haydar, Children’s National Hospital, United StatesCopyright © 2023 Zhang, Jiang, Zhang and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gao Zhang, Z2FvemhhbmdAaGt1Lmhr; Shiyou Wei, d2Vpc2hpeW91c2N1QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.