 Yuping Zheng
Yuping Zheng Qingrui Huang
Qingrui Huang Qiannan Li
Qiannan Li- Department of Dermatology, Shuguang Hospital Affiliated with Shanghai University of Traditional Chinese Medicine, Shanghai, China
Transient receptor potential (TRP) channels are a class of transmembrane proteins that can sense a variety of physical/chemical stimuli, participate in the pathological processes of various diseases and have attracted increasing attention from researchers. Recent studies have shown that some TRP channels are involved in the development of pathological scarification (PS) and directly participate in PS fibrosis and re-epithelialization or indirectly activate immune cells to release cytokines and neuropeptides, which is subdivided into immune inflammation, fibrosis, pruritus and mechanical forces increased. This review elaborates on the characteristics of TRP channels, the mechanism of PS and how TRP channels mediate the development of PS, summarizes the important role of TRP channels in the different pathogenesis of PS and proposes that therapeutic strategies targeting TRP will be important for the prevention and treatment of PS. TRP channels are expected to become new targets for PS, which will make further breakthroughs and provide potential pharmacological targets and directions for the in-depth study of PS.
1 Introduction
Pathological scarification (PS) is a pathological result of wound healing and is caused by inflammation and trauma. Epidemiology shows that the overall prevalence of PS is 30%-90%. The incidence of hypertrophic scar (HS) in patients with full-thickness burns is as high as 80% (1). In the United States alone, the cost of treating HS is estimated to be $400 million per year (2), which brings a huge economic burden to families and society. PS is one of the important complications of tissue damage repair, including HS and keloids. It is mainly characterized by the infiltration of inflammatory cells such as macrophages, lymphocytes, mast cells and neutrophils (3). Inflammatory mediators secreted by immune cells induce fibroblasts (FBs) fibrosis and keratinocytes (KCs) re-epithelialization, resulting in excessive deposition of a large amount of extracellular matrix (ECM) components, which is accompanied by mechanical stretching and angiogenesis and eventually develops into PS (4–6). Furthermore, recent studies have shown that oxidative stress and epigenetic regulation are involved in the occurrence of PS (7, 8). PS easily causes appearance damage and local tissue pruritus, pain, tumor-like hyperplasia or varying degrees of dysfunction, affecting the physical and mental health of patients (9). PS research has always been a challenging topic in the field of burns, plastic surgery and dermatology department (10). However, the exact pathogenesis of PS is unclear and still needs to be examined.
Recently, many studies have shown that transient receptor potential (TRP) channels are involved in many mechanisms of PS, such as participating in the development of PS fibrosis and re-epithelialization or affecting the secretion of TGF-β1 and ECM by nonimmune cells, and regulating cytokine release, cell migration and phagocytosis through immune-related mechanisms (11). Some TRP channels are involved in PS mechanical conduction, oxidative stress, epigenetics and pruritus. Therefore, the expression of TRP channels in PS deserves more attention. This review focused on specific channels in the TRP family, such as TRPV, TRPC, TRPA and TRPM, especially TRPV1-4, TRPC3, TRPC6, TRPA1 and TRPM7. These are channel proteins that play important roles in the wound healing and the development of PS.
The analysis of this information aimed to demonstrate immune inflammation and fibrosis to examine TRP channels as potential targets for inhibiting PS.
2 TRP channels classification and function
The TRP superfamily consists of nonselective cation channels with the ability to sense local environmental changes (12). In 1969, Cosens (13) found that light stimulation only caused a transient increase in intracellular Ca2+ concentrations in a Drosophila mutant with light sensing defects. Subsequently, Hardie (14) found that this was due to the lack of functional copies of genes encoding ion channels and named this type of calcium-permeable cation channel the TRP channel. According to differences in amino acid sequence homology, the mammalian TRP family is divided into six groups: ankyrin (TRPA), canonical (TRPC), melastatin (TRPM), mucolipin (TRPML), polycystin (TRPP), and vanilloid (TRPV) (15).
TRP channels are abundantly expressed in various cell types (16). For example, KCs, melanocytes, FBs and a variety of immune cells express TRP channels (17). TRP channels can be activated by external stimuli or local environmental changes (including pain, pruritus, heat, warmth or cold, odor, mechanical stimulation, and osmotic pressure changes) (18). In addition, TRP channels are critical in physiological processes such as regulating skin homeostasis, melanin synthesis, wound healing, epigenetic regulation, and pathological processes such as barrier damage, vascular stress relaxation, oxidative stress, and skin cancer caused by ultraviolet irradiation (19). There is growing evidence that the TRP family plays an important role in mediating disease fibrosis (20). These results are consistent with the mechanism of PS.
The TRPV subfamily consists of six members, which can be subdivided into heat-activated TRPV (TRPV1-4) channels and Ca2+-selective TRPV channels (TRPV5, TRPV6) (21), and these channels can be activated by different stimuli such as heat, pruritus, pain, osmotic pressure or chemical stimulation (21, 22). TRPV channels are involved in the activation and differentiation of immune cells and play an important role in activating macrophages, stimulating the type 17 immune inflammatory response and inducing neutrophil adhesion and chemotaxis (23–25). TRPV is closely related to fibrosis and is mainly involved in myofibroblasts (MFBs) differentiation and collagen deposition (26). TRPV is also an important osmotic-mechanical sensitive channel that mediates abnormal mechanical conduction into specific biochemical signals (23, 27). In addition, TRPV is related to angiogenesis (28), epigenetic regulation (29), electrolyte homeostasis (30) and the maintenance of barrier function (31).
The TRPC family can be further divided into four subgroups (TRPC1, TRPC2, TRPC4/5 and TRPC3/6/7) according to their amino acid sequence and functional homology (32). TRPC channels may mediate fibrotic diseases as mechanosensitive ion channels (33) and can sense and regulate oxidative stress responses. For example, the oxidation product OONO- upregulates the mRNA and protein expression of TRPC6 and TRPC3 in monocytes (34). In addition, TRPC channels are involved in the inflammatory response (35), mitochondrial metabolism in ageing (36), cell proliferation, wound healing (37), and angiogenesis (38). In particular, TRPC3 and TRPC6 have been shown to be crucial in the mechanical conduction of wound healing (39).
TRPA channels have been widely studied in the field of pruritus, pain and neurogenic inflammation (15). TRPA1 is the only member of the mammalian TRPA family that can be activated by cold and heat stimulation, mechanical forces, chemicals and endogenous signals associated with cell damage. TRPA1 is also an important mediator of acute and chronic itching perception. Exogenous and endogenous pruritus can produce scratching behavior by activating neuronal TRPA1 (19). In addition, TRPA1 is expressed in immune cells, KCs, melanocytes, FBs, epithelial cells and sensory neurons and plays a key role in the pathophysiology of almost all systems (40–42).
The TRPM subfamily consists of eight members (TRPM1-8), is the largest subfamily of TRP channels and has a specific structure and physical function (43). The TRPM subfamily is expressed in various organs and cells of the peripheral, central and immune systems and is vital in various biological processes, such as cold and heat stimulation, ion homeostasis, autophagy, vascular tension, epigenetics, and immune inflammation (29, 44–46). An increasing number of studies have shown that TRPM channel participates in fibrotic diseases (47, 48).
3 Cutaneous wound healing and the mechanism of PS
3.1 Physiological wound healing process
The physiological wound healing process is divided into four stages: hemostasis, inflammation, proliferation and remodeling (49). To a certain extent, this process is mediated by growth factors and regulatory molecules (50).
① Collagen and tissue factors promote the clumping of platelet aggregation in the affected area, releasing chemotactic and growth factors and eventually forming clots (51). ② The infiltration of inflammatory cells marks the beginning of the inflammatory phase of wound healing (50). Immune cells such as neutrophils, lymphocytes, mast cells and monocytes release inflammatory mediators to defend against microorganisms and remove wound pathogens and tissue fragments (52). When proinflammatory M1 macrophages transform into anti-inflammatory M2 macrophages, the wound healing process shifts to the proliferative phase (53). The hemostasis and inflammatory phases typically take 3 days (54). ③ The proliferative phase is an important stage associated with angiogenesis, KC migration, granulation tissue formation, ECM accumulation and epithelialization (55). FBs synthesize ECM and promote the formation of granulation tissue (53). FBs activation, KCs proliferation and migration, and new epithelial differentiation jointly promote wound re-epithelialization (56). Furthermore, FBs can be activated and differentiate into MFBs, which promote matrix remodeling to promote wound healing and angiogenesis (56, 57). ④ The final step is the remodeling phase, which typically lasts for weeks or even years (58). This stage mainly involves excessive tissue degradation (59). Excessive ECM is degraded, and collagen type III (COL-3) is replaced by mature collagen type I (COL-1), eventually leading to wound healing (60), as shown in Figure 1.
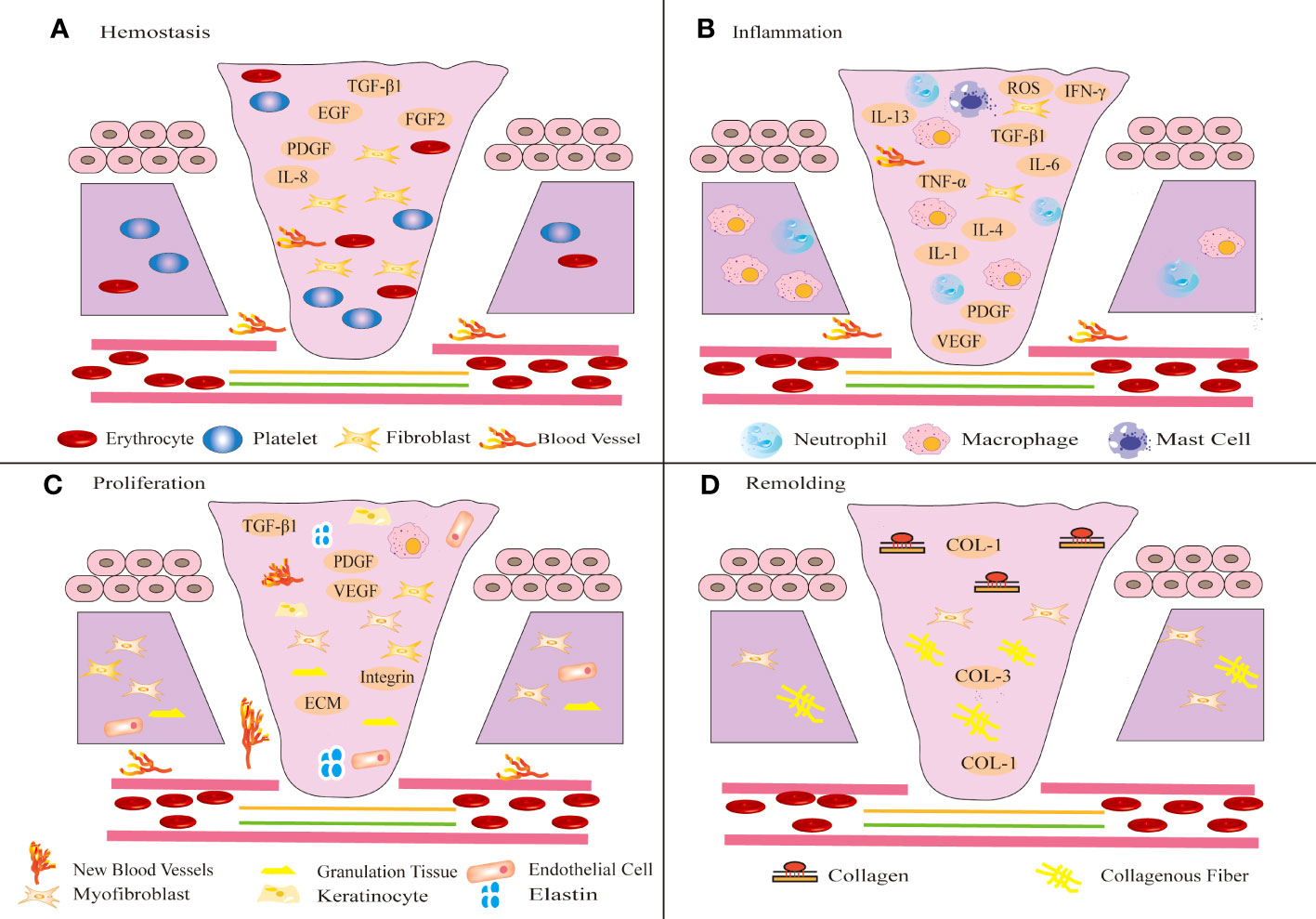
Figure 1 Schematic diagram of different stages of wound healing. Wound healing includes four stages: hemostasis (A), inflammation (B), proliferation (C) and remodeling (D). (A) After injury, platelets aggregate, release chemotactic and growth factors, and eventually form clots. (B) Shortly thereafter, immune inflammatory cells release inflammatory mediators to resist microbial invasion and remove wound pathogens and tissue fragments. (C) Subsequently, FBs migrate to the wound tissue and synthesize ECM to promote the formation of granulation tissue at the wound site. The proliferation of KCs at the wound edge promotes wound re-epithelialization and migrates down to the injured dermis. (D) In the final remodeling stage, the tensile strength of the wound increases and the wound is completely covered by the new epidermis.
TRP channels play a key role in various stages of physiological wound healing, with TRPV1-4, TRPC3, TRPC6, TRPA1, and TRPM7 being closely associated. Specifically, TRPV1, TRPV3, TRPV4, and TRPA1 are involved in the inflammatory, proliferative, and remodeling phases of physiological wound healing. For instance, TRPV1 deficiency can lead to neutrophil inflammation and NETs formation, as well as defective re-epithelialization, which can prolong wound healing (61).
TRPV3, on the other hand, can induce fibrosis through TRPV3/TSLP/Smad2/3 pathway, resulting in significantly increased expression levels of α-SMA, fibronectin, COL1A1, and TSLP (62). Additionally, the selective TRPV3 activator KS0365 has been shown to accelerate the migration of KCs and promote re-epithelialization during physiological wound healing (63). Furthermore, the presence of TRPV3 in macrophage lysosomes may play a crucial role in the inflammatory phase of PS (64).
The activation of TRPV4 has been shown to promote TGF-β1 and IL-6 induced fibrosis and inflammation (20). Conversely, the lack of TRPA1 has been found to retard macrophage infiltration, subsequent fibrotic tissue formation, and mRNA expression of α-SMA and COL-1, which may further impair fibrotic behavior in fibroblasts (65). Additionally, TRPV2, TRPC3, TRPC6, and TRPM7 have been implicated in the proliferative and remodeling phases of wound healing. Specifically, TRPV2 mediates FB differentiation and contraction by promoting TGF-β1 and α-SMA expression (66).
Inhibition of TRPC3 and TRPC6 can suppress MFBs trans-differentiation and the expression of αSMA and TGF-β1 (67, 68). Furthermore, overexpression of TRPM7 promotes fibrosis and ECM deposition in wound healing (69).
3.2 Mechanism of PS
PS can be induced by prolonged hemostasis and inflammatory phases, leading to an abnormal increase in activated cells and their accumulation at the injury site, abnormal proliferation of FBs and excessive collagen deposition during the proliferative phase; reduced degradation of ECM and excessive wound contraction during the remodeling phase. The mechanism of PS is complex, and current research is mainly related to immune inflammation, fibrosis, re-epithelialization, epigenetics, oxidative stress, and mechanical forces.
3.2.1 The mechanism by which immune cells regulate PS
Immune cells mainly prevent the invasion of pathogenic microorganisms during wound healing, and an imbalance will change the outcome of wound healing. Immune cells release cytokines or chemokines to promote fibrosis and re-epithelialization, resulting in excessive deposition of ECM and eventually leading to PS. At present, PS-related immune cells mainly include macrophages, lymphocytes, mast cells and neutrophils (Figure 2).
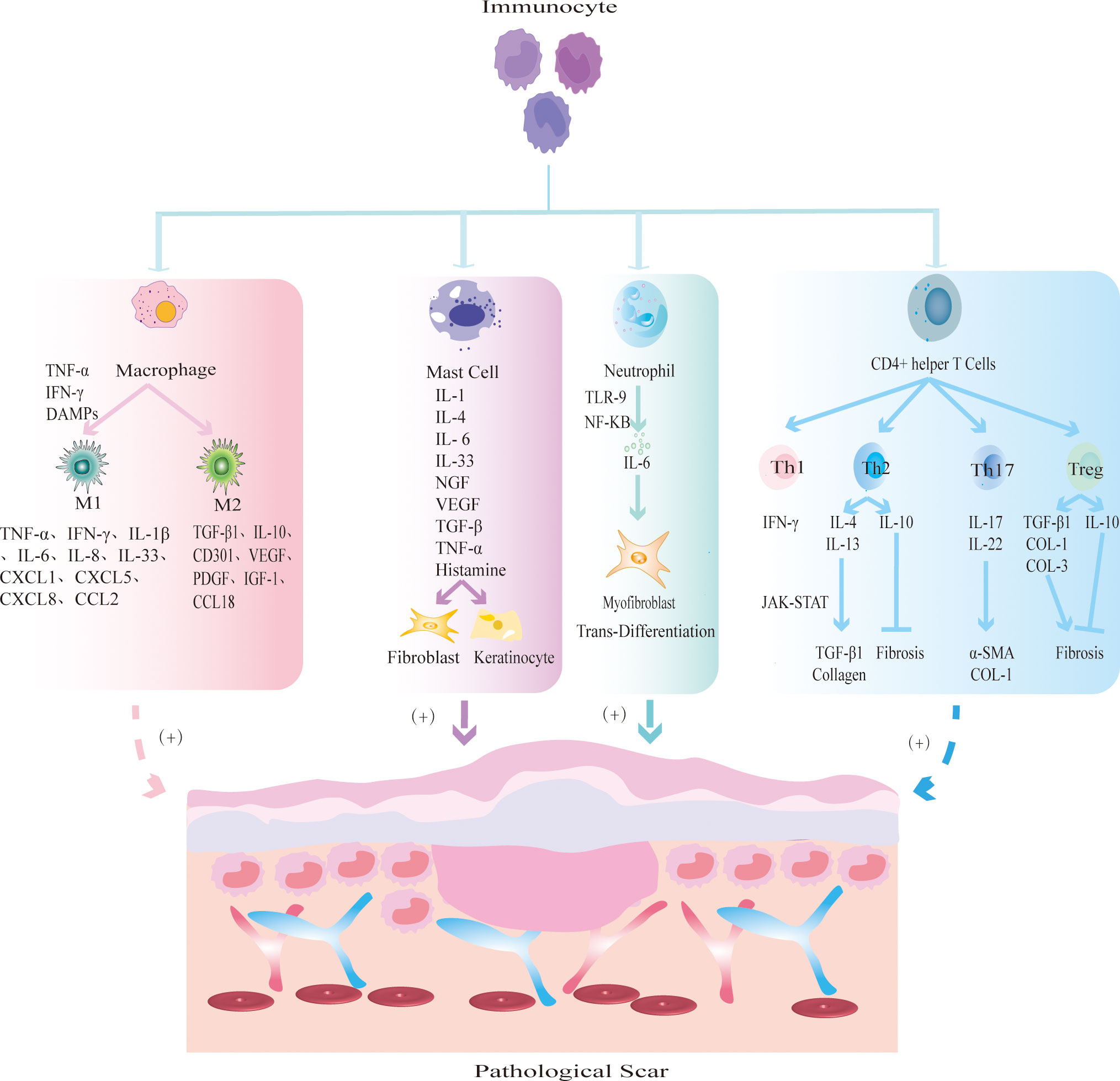
Figure 2 The mechanism by which different immune cells regulate PS. Macrophages are polarized into two main phenotypes: M1 and M2. M1 macrophages secrete TNF-α and IFN-γ to initiate inflammatory responses; M2 macrophages secrete TGF-β1 and IL-10 to promote tissue fibrosis. Mast cells release inflammatory mediators such as IL-1 and IL-4, and promote FBs fibrosis and KCs re-epithelialization. Neutrophils release IL-6 to induce MFBs transdifferentiation. CD4+ helper T cells are further divided into Th1, Th2, Th17 and Treg subsets. Th1 releases pro-inflammatory cytokine IFN-γ; Th2 cells produce anti-inflammatory factors IL-4, IL-13 to promote fibrosis and IL-10 to inhibit fibrosis; Th17 cells secrete IL-17 and IL-22 to promote the expression of α-SMA and collagen; Treg cells secrete pro-fibrotic cytokines TGF-β1, COL-1, COL-3 and anti-fibrotic cytokine IL-10.
Macrophages are the key effector cells of innate immunity and play an important protective role in clearing pathogenic microorganisms and tissue fragments, presenting antigens, and promoting wound repair (70). Studies have shown that macrophages undergo significant phenotypic and functional changes to coordinate changes in the microenvironment at different stages of wound healing (71). In different microenvironments, macrophages can be polarized into two main phenotypes: M1 and M2. During wound healing, monocytes are polarized into the M1 phenotype by microorganisms, proinflammatory Th1 cytokines, damage-associated molecular patterns (DAMPs) and lipopolysaccharide (LPS) to initiate the inflammatory response (72, 73). Furthermore, the number of M1 macrophages begins to increase at 0-2 days after injury, peaks at 7-14 days after injury, and decreases significantly at 14-28 days after injury (72). This finding indicates that M1 cells secrete many inflammatory mediators in the early stage of normal scar formation (74, 75). During the transition from the inflammatory phase to the proliferative phase of wound healing, M1 cells are transformed into the M2 phenotype by the phagocytosis of neutrophils or the change of local wound microenvironment (76, 77). However, how M1 macrophages differentiate into M2 macrophages is not clear. The anti-inflammatory M2 phenotype is mainly involved in the proliferation and remodeling phases of wound healing. The secretion of vascular growth factors, cytokines and chemokines induces the proliferation and differentiation of FBs and MFBs, the re-epithelialization of KCs, the deposition of ECM and angiogenesis (71, 78–81). M2 cells are significantly increased at 28 days after injury and returned to baseline at 56 days (80). Normal wound healing is characterized by the transition from the early inflammatory stage, which is dominated by M1 macrophages, to the recovery stage, which is dominated by M2 macrophages (71). Increased secretion of inflammatory cytokines by M1 macrophages promotes the development of inflammation, or increased secretion of cytokines by M2 macrophages promotes the development of fibrosis, which leads to the formation of PS.
Most studies on lymphocytes in PS focus on T cells. Studies have shown that there may be a decrease in CD8+ cytotoxic T cells in keloids, and the number and activity of FBs co-cultured with CD8+ cytotoxic T cells are significantly reduced (82, 83). CD4+ helper T cells can be further divided into Th1, Th2, Th17 and regulatory T (Treg) cell subsets. The dynamic balance of the proinflammatory Th1 response with the anti-inflammatory Th2 response is crucial in wound healing. Once the balance is disturbed, PS may occur. During PS, Th1 cells can produce the proinflammatory factor IFN-γ to protect against fibrosis (84, 85). Th2 cells produce IL-4 and IL-13 driven by the transcription factor GATA3, which can not only induce macrophage polarization to the M2 phenotype but also induce TGF-β1 and collagen synthesis through the JAK-STAT signaling pathway and induce pruritus (86, 87). In addition, Th2 cells can produce the anti-inflammatory mediator IL-10 to protect against fibrosis (88). Treg cells can secrete cytokines and interact with other inflammatory cells to regulate PS. The profibrotic cytokines TGF-β1, COL-3, and COL-3/COL-1, anti-fibrotic cytokine IL-10 and nuclear transcription factor Foxp3 are secreted by Treg cells, which can directly regulate PS (89). Treg cells can promote macrophage polarization to the M2 phenotype and interact with helper T cells to indirectly regulate PS (88). Th17 cells activate FBs differentiation and KCs proliferation by secreting IL-17 and IL-22 (88, 90). Overall, these studies showed that lymphocytes can induce the differentiation of FBs and KCs by releasing inflammatory mediators or participate in the development of PS by interacting with macrophages. However, there are relatively few studies, and the specific mechanism needs further study.
Mast cells are mainly involved in PS and its pruritus by releasing inflammatory mediators, promoting FBs and KCs activation and excessive collagen deposition. Mast cells release inflammatory mediators, induce degranulation, directly activate FBs fibrosis, angiogenesis and KCs re-epithelialization, recruit more immune cells to migrate to the injured site; and indirectly promote tissue repair (91, 92). Furthermore, the mast cell inhibitor DSCG can reduce the width of PS and the levels of the wound inflammatory factors IL-1α, IL-1β and CXCL1 (93). In addition, mast cells are closely related to PS pruritus. Compared with those in non-pruritus keloids, the number and degranulation of mast cells in pruritus keloids were increased (94). Therefore, number of mast cells and their storage particles are important factors affecting the PS.
Neutrophils are the first immune cells to reach the wound site and secrete various cytokines to participate in wound healing. Neutrophils kill microorganisms, remove tissue debris, and contribute to the activation of macrophages (95). However, the persistent presence of neutrophils in peripheral tissues triggers an inflammatory response. Studies have shown that neutrophil extracellular traps (NETs), which are network structures by which neutrophils kill pathogens, are highly expressed in HS and induce FBs to differentiate into MFBs through the TLR-9/NF-κB/IL-6 signaling pathway (96). It is known that the IL-6 signaling pathway is critical in the pathogenesis of PS (97, 98). At present, there is a lack of studies on the specific mechanisms by which neutrophils induce FBs differentiation and interact with other immune cells to mediate PS.
3.2.2 The mechanism by which nonimmune cells regulate PS
In addition to immune cells, many nonimmune cells are involved in the development of PS (Figure 3). The nonimmune cells involved in PS are mainly FBs, MFBs and KCs. During physiological wound healing, MFBs undergo apoptosis or revert to static FBs. When the mechanical environment around the wound changes or the internal environment is disordered, FBs are activated by cytokines and chemokines secreted by immune cells and differentiate into MFBs, which is accompanied by excessive secretion of ECM components which ultimately leads to PS (99). MFBs mainly mediate information exchange through autocrine and paracrine mechanisms. Autocrine signaling involves binding to its own receptor to trigger TGF-β1 and induce MFBs differentiation. When TGF-β1 is inhibited, it causes the dedifferentiation of MFBs (100). In addition, the paracrine pathway mainly recruits immune cells such as macrophages and neutrophils to achieve indirect communication and jointly promote the development of PS. KCs play an important role in the development of PS by inducing wound healing re-epithelialization and regulating FBs differentiation (101). During normal skin differentiation, KCs move from the basal layer of the epidermis. When the skin is damaged, these cells proliferate and migrate to the wound, promoting wound healing (102). In HS, the thicker the epidermis, the stronger the re-epithelialization of KCs (103). In addition, KCs upregulate the expression of profibrogenic molecules to accelerate FBs proliferation and collagen production (104). At present, the abnormal interaction between KCs and MFBs is one of the most widely recognized mechanisms in PS. In conclusion, the excessive differentiation of FBs into MFBs, which in turn promotes fibrosis and excessive re-epithelialization of KCs, leads to the occurrence of PS.
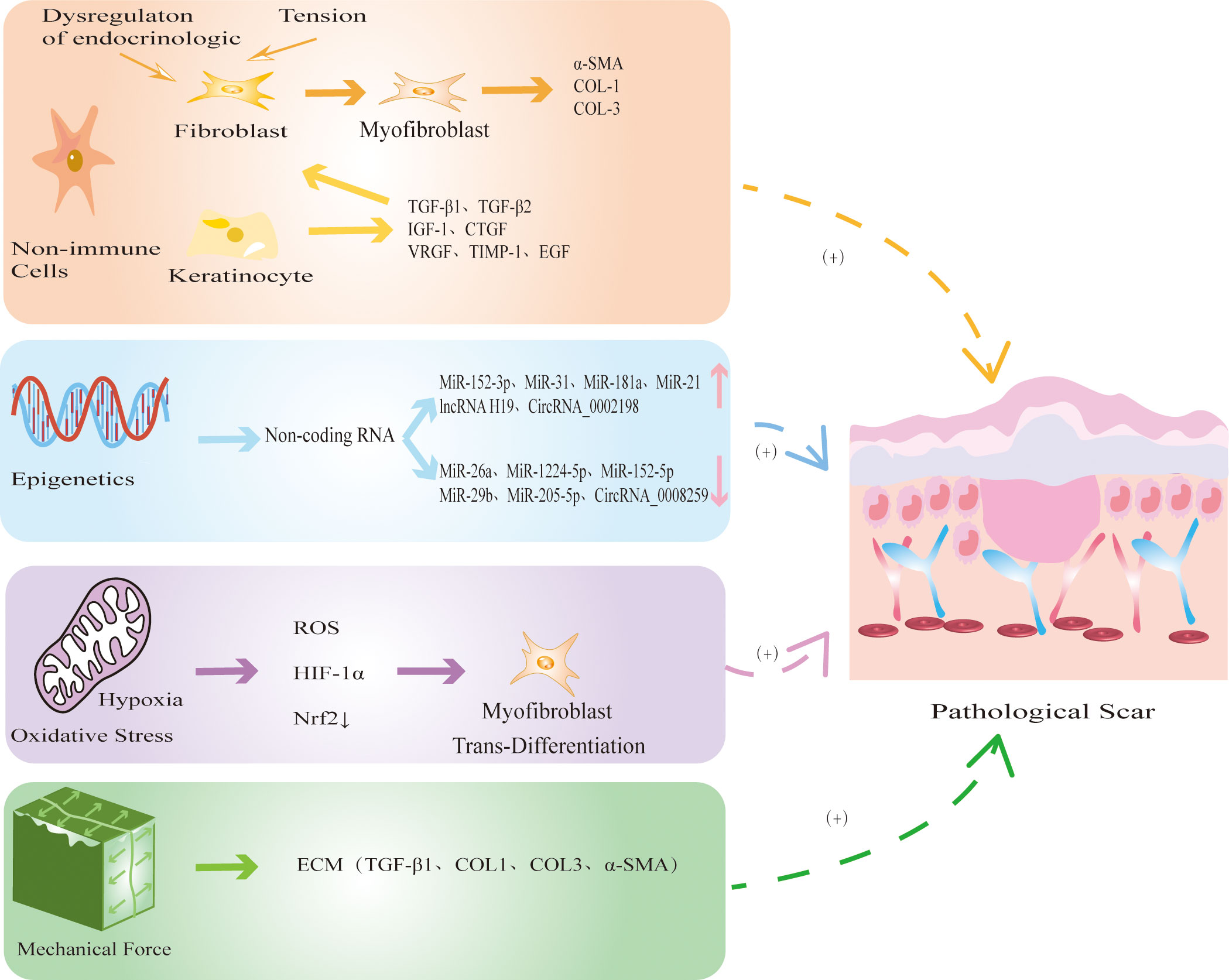
Figure 3 The mechanism by which non-immune cells and other factors regulate PS. Non-immune cells mainly include FBs, MFBs and KCs. When the mechanical environment around the wound changes or the internal environment is disordered, FBs differentiate into MFBs, accompanied by excessive secretion of COL-1, COL-3, α-SMA, eventually leading to PS. At the same time, KCs secretes TGF-β1 and TGF-β2 to promote re-epithelialization and FBs fibrosis. Epigenetic modification represented by noncoding RNAs is abnormally expressed in PS, which further promotes the fibrosis of FBs. Hypoxia releases ROS and HIF-1α, reduces the expression of antioxidant protein Nrf2, and induces MFBs transdifferentiation. Continuous mechanical stretching induces FBs to synthesize ECM, promotes collagen secretion and fiber synthesis.
3.2.3 The mechanism by which other factors regulate PS
Increasing evidence shows that epigenetic modifications, represented by DNA methylation, histone modification and noncoding RNAs (ncRNAs), play a key role in the gene regulation of PS (105, 106). Many studies have focused on the expression of miRNAs in PS. The upregulation of miR-152-3p, miR-31, miR-181a, miR-21, lncRNA H19, and circRNA_0002198 (107–112) and the downregulation of miR-26a, miR-1224-5p, microRNA-152-5p, miR-29b, miR-205-5p, and circRNA_0008259 (112, 113) can promote the proliferation of FBs and the formation of collagen and ultimately induce the formation of PS (Figure 3).
There is hypoxia during PS. Compared with those in normal tissues, HIF-1α and ROS are highly expressed in PS (114, 115). Furthermore, hypoxia can induce the transformation of FBs to MFBs in keloids through the TGF-β1/SMAD3 pathway (114). In addition, Lee showed that antioxidant protein Nrf2 in keloids was significantly lower than that in normal skin tissue, and the protein levels of the oxidation product 2,4-dinitrophenylhydrazine were significantly higher than in normal skin (116). This finding suggests that oxidative stress is one of the mechanisms of PS (Figure 3).
Mechanical forces are important factors leading to PS (117, 118). TRP channels have been shown to play key roles in response to mechanical conduction, and TRPV2, TRPV4, TRPC3 and TRPC6 are potential mechanical force sensors involved in PS (3, 119). Studies have shown that continuous mechanical stretching can lead to the formation of PS by inducing FBs to synthesize ECM, indirectly activating the immune response to promote collagen secretion and fiber synthesis (120). In addition, local high mechanical forces are associated with abnormal skin fibrosis (1). These studies showed that mechanical forces can not only induce the release of inflammatory mediators by activating the immune response but also directly promote fibrosis in PS (Figure 3).
4 Modulation of TRP channels in PS
TRP channels are involved in PS (Table 1). The specific manifestations include immune inflammation, fibrosis, re-epithelialization, abnormal oxidative stress, epigenetic disorders, and excessive mechanical stretching. The factors are related to TRP channels. In addition, some TRP channels mediate PS-induced pruritus. TRP channels that are most closely related to PS are described below. Furthermore, we offer some opinions for reference in expounding the related problems.
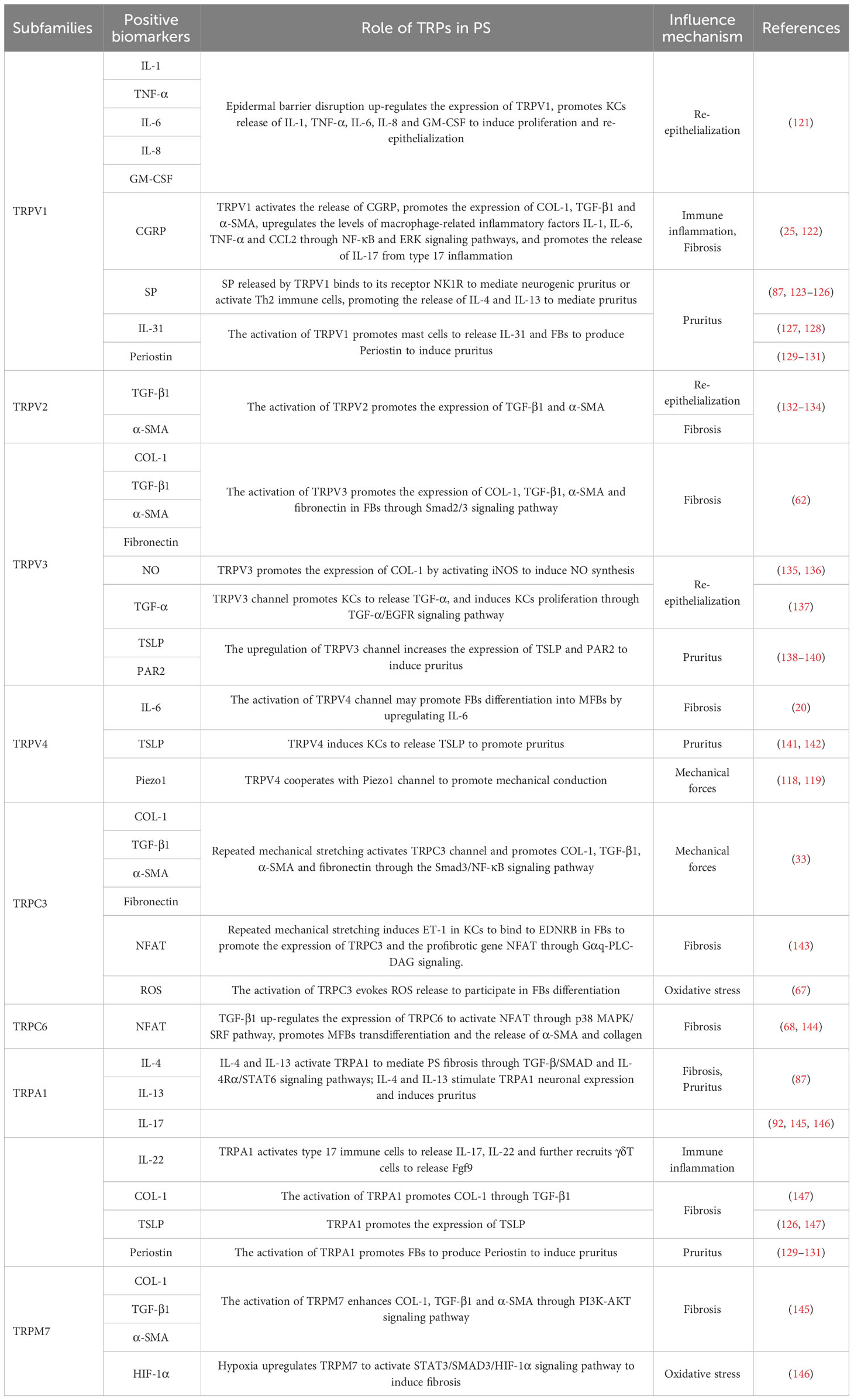
Table 1 The function of TRP channels in PS.
4.1 TRPV1
TRPV1 is not only a heat-activated capsaicin receptor but also a multimodal receptor that is activated through multiple pathways in different microenvironments and mediates PS fibrosis, re-epithelialization and inflammatory responses, as well as being involved in PS pruritic signaling (Figure 4) (26, 148, 149).
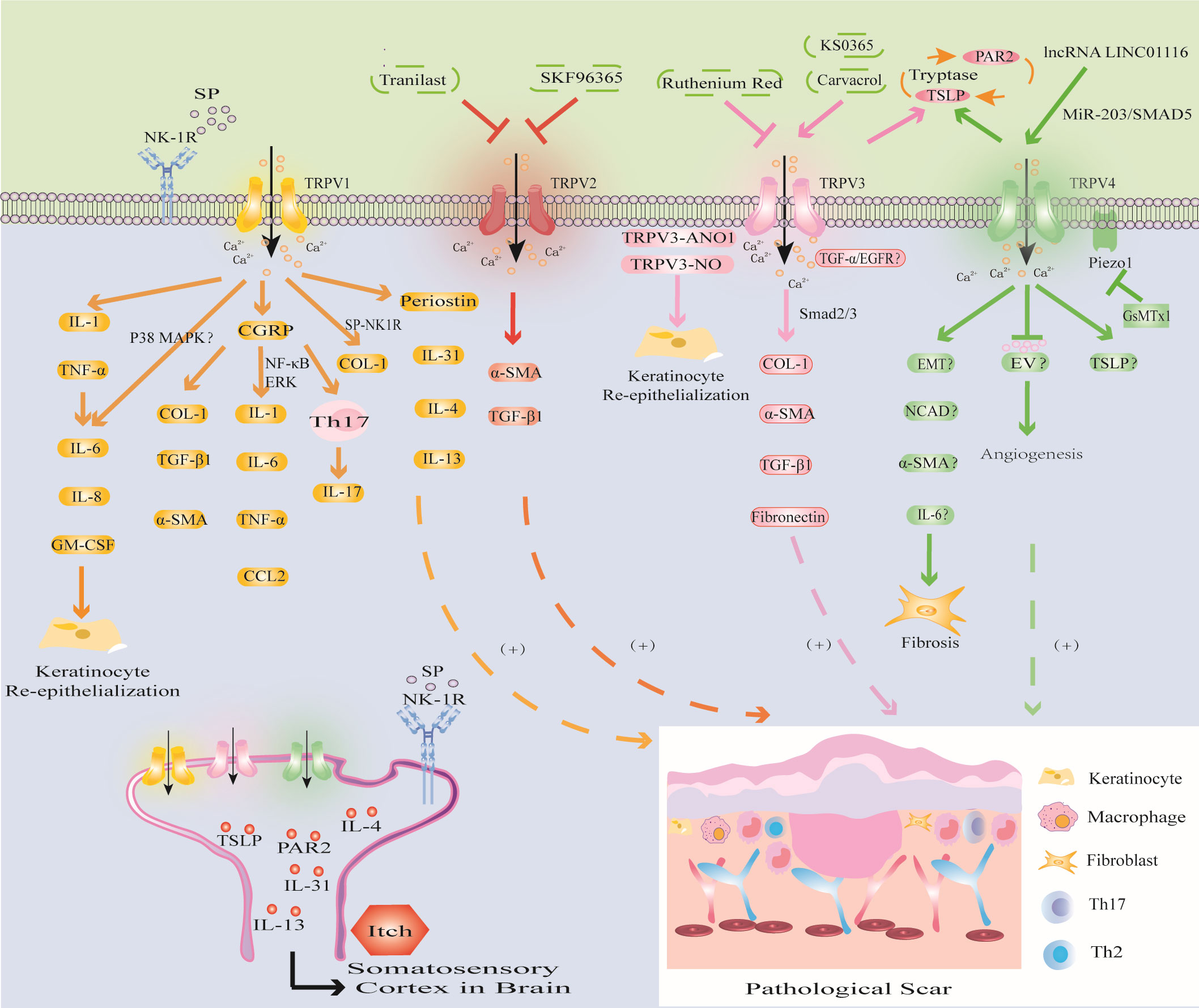
Figure 4 Modulation of TRPV channels in PS. TRPV1 channel activates IL-1 and TNF-α, triggers inflammatory response, promotes KCs proliferation and re-epithelialization; CGRP released by TRPV1 directly promotes PS fibrosis, and can induce PS by upregulating the levels of inflammatory factors through NF-κB and ERK signaling pathways; the SP released by TRPV1 activation mediates PS fibrosis and pruritus. The activation of TRPV1 promotes the expression of Periostin, IL-4, IL-13 and induces PS pruritus. (2) TRPV2 inhibitors SKF96365 and tranilast inhibited PS by downregulating TGF-β1 and α-SMA. (3) TRPV3 activator carvacrol promotes fibrosis through Smad2/3 pathway; activation of TRPV3 may interact with ANO1 and NO or promote PS through TGF-α/EGFR signaling pathway. PS pruritus may be related to the PAR2-TSLP positive feedback pathway mediated by TRPV3 and TRPV4 channels. (4) The activation of TRPV4 may promote the expression of EMT, NCAD, α-SMA, IL-6 to induce fibrosis, inhibit EV to inhibit abnormal angiogenesis, or release TSLP to induce PS pruritus and cooperate with Piezo1 channel to promote the formation of PS induced by mechanical stretching.
In HS, the expression of TRPV1 is upregulated. Studies have shown that trans-epidermal water loss (TEWL) in HS is significantly higher than that in normal skin (150). This finding suggests that there is the presence of epidermal barrier dysfunction in HS, which is consistent with the clinical manifestations of skin dryness in some HS patients. Studies have shown that the barrier function of the skin is closely related to the expression of TRPV1 in KCs. Overactivation of TRPV1 delays the recovery of epidermal barrier function (151). Further studies have shown that disrupting the water barrier leads to KCs production of IL-1 and TNF-α to trigger the inflammatory response, and upregulate IL-6, IL-8, and granulocyte/macrophage colony stimulating factor (GM-CSF), promote KCs proliferation and re-epithelialization, and ultimately induce PS (121). In addition, Capsaicin induces increases in proliferation through IL-6 upregulation and TRPV1 induces the proliferation of human corneal epithelial cells through global MAPK activation (152, 153). These studies have shown that the expression of TRPV1 is related to the barrier function and inflammatory response of HS. The loss of TRPV1 inhibited inflammatory cell invasion and expression of TGF-β1 and other proinflammatory gene expression in cultured ocular fibroblasts (154). This is due to the overexpression of TRPV1 in HS which affects KCs proliferation and differentiation (20). Therefore, interfering with TRPV1 expression in KCs may become a new therapeutic strategy for PS. In addition, studies have shown that TRPV1 activation can release calcitonin gene-related peptide (CGRP) stored in vesicles from nerve endings to mediate immune inflammation and local vasodilation (155, 156). Zhou found increased CGRP levels in both human and mouse HS tissues. Furthermore, CGRP antagonists can directly reduce the expression of COL-1, TGF-β1 and α-SMA and can downregulate the levels of the macrophage-related inflammatory factors IL-1, IL-6, TNF-α and CCL2 through the NF-κB and ERK signaling pathways. Moreover, CGRP can promote PS by inducing the Th17 immune response (122). Cohen found that IL-17 expression was closely related to the Th17 immune response in light-simulated TRPV1-Ai32 mice (25). However, this process requires TRPV1 to induce CGRP release (25). The development of macrophages, the Th17 inflammatory response and fibrosis in HS induced by CGRP may be related to the release of TRPV1 from neurons. The neuroimmune mechanism by which TRPV1 regulates PS provides a new research direction. In summary, these studies have shown that TRPV1 may promote PS by directly promoting fibrosis and re-epithelialization and indirectly inducing inflammatory stimulation.
Pruritus is the most important symptom affecting the quality of life of PS patients. TRPV1 inhibitors may be one of the effective treatment strategies. The study found that the degree of scar pruritus after TRPV1 gene knockout was significantly less than that of wild type rats (152). Further studies have shown that TRPV1 can induce pruritus by promoting the expression of the pruritus mediators IL-31 and SP (127). The level of IL-31 secreted by mast cells was increased in HS compared with that in normal tissues, and the number of mast cells was also increased (128). This finding suggests that HS pruritus may be related to TRPV1-mediated promotion of mast cell degranulation and the release of inflammatory factors. In addition, the expression of SP and TRPV1 were significantly higher in HS skin than in normal skin. Furthermore, immunofluorescence analysis showed that the distribution of TRPV1 and SP was consistent (157). As a neuropeptide, SP mediates angiogenesis, macrophage polarization, mast cell degranulation, KCs proliferation and fibrosis and is an important neuromodulator of pruritus. SP can selectively bind to its specific receptor neurokinin-1 receptor (NK-1R) to mediate neurogenic pruritus (123, 125). Further studies have shown that the SP-NK1R signaling pathway promotes FBs to secrete COL-1 and is positively correlated with SP levels (157). The mechanism of skin neurogenic pruritus mediated by the TRPV1-mediated SP-NK1R signaling pathway may provide a new therapeutic target for PS pruritus conduction. Study has confirmed that hyperbaric oxygen therapy (HBOT) can alleviate the pruritus symptoms of keloid patients by reducing the expression of TRPV1 (158), but the specific mechanism is still unclear. Recently, Hashimoto found that the new pruritic Periostin was upregulated in PS (129). Periostin is produced by TGF-β1 and histamine-stimulated FBs and induces pruritus by binding to the aVb3 integrin receptor or inducing the Th2 cytokine cascade (130). However, this process requires the activation of TRPV1 and TRPA1 (130, 131). The discovery of Hashimoto provided a new direction for researching the mechanism of Periostin-mediated pruritus in PS through the activation of TRPV1 and TRPA1. Overall, these results indicate that targeting TRPV1 channels may be a prospective therapeutic strategy for PS.
4.2 TRPV2
TRPV2 is mainly involved in PS by promoting the release of TGF-β1 from KCs and the differentiation of FBs (Figure 4).
In new granulation tissue in the wound, KCs, FBs and macrophages at the edge of the wound promote wound re-epithelialization by secreting cytokines such as TGF-β1 and stimulate MFBs to produce α-SMA, eventually leading to PS (132, 133). In the cell collagen contraction model based on this theory, researchers found that the TRPV2 inhibitor SKF96365 or tranilast inhibited collagen contraction, while a TRPV2 inhibitor or TRPV2 knockdown using siRNA reduced TRPV2 agonist-induced Ca2+ influx in FBs. It was further confirmed that the TRPV2 inhibitors SKF96365 and tranilast could induce FBs differentiation and collagen contraction by downregulating TGF-β1 and α-SMA expression (134). These studies showed that altering FBs and KCs differentiation with drugs targeting TRPV2 channels is beneficial for preventing PS and contracture.
4.3 TRPV3
TRPV3 plays an important role in mediating KCs re-epithelialization and FBs fibrosis and promoting ECM deposition in PS. In addition, TRPV3-mediated PS pruritus may be related to the expression of PAR2 and TSLP (Figure 4).
Studies have shown that the new TRPV3 channel activator KS0365 promotes wound healing by accelerating the re-epithelialization of KCs, while the broad-spectrum channel blocker ruthenium red and siRNA-mediated TRPV3 knockdown inhibit this process (63, 159). This finding indicates that overexpression of TRPV3 channels can promote excessive wound healing leading to the formation of PS. A clinical study showed that the TRPV3 activator carvacrol could promote the expression of COL-1, α-SMA, TGF-β1 and fibronectin in FBs through the Smad2/3 pathway, thereby promoting HS fibrosis (62). In addition, NO is an important mediator involved in biological processes such as wound healing, fibrosis, inflammation and KCs differentiation (160). Cobbold found that compared with those in normal tissues, NO levels in keloids were increased, and NO produced by nitric oxide synthase (iNOS) promoted the expression of COL-1 (135). TRPV3 induces NO synthesis by activating iNOS, thereby promoting KCs re-epithelialization and facilitating wound repair (136). This finding suggests that TRPV3-induced NO overexpression leads to PS. In addition, epidermal growth factor receptor (EGFR) can promote wound healing by promoting KCs proliferation, inflammation and angiogenesis (161). Aijima found that the phosphorylation of EGFR in the oral epithelial cells of TRPV3-KO mice was inhibited, and TGF-α, which is a ligand of EGFR, was released from KCs through the activation of TRPV3 (137). This finding indicates that the TGF-α/EGFR signaling pathway plays a role in oral mucosal wound healing through TRPV3. Although the oral mucosa repairs faster than skin wounds and has fewer scars (137), the mechanism of oral mucosal repair suggests that the activation of TRPV3 may promote PS through the TGF-α/EGFR signaling pathway. Recent studies have shown that anoctamin1 (ANO1), a calcium-activated chloride channel, can promote the migration and proliferation of cancer cells (162). The interaction of TRPV3 with ANO1 promotes the proliferation of KCs during wound healing, while TRPV3 and ANO1 inhibitors inhibit the proliferation of KCs (162). The mechanism of the TRPV3-ANO1 interaction may provide a new target for PS.
PS pruritus may be related to the expression of PAR2 and TSLP, which is mediated by TRPV3 channels. The expression of PAR2 can be detected in burn scars with pruritus (138). Furthermore, inhibiting TRPV3 channels can reduce the expression of PAR2 and inhibit the itching of burn scars (139). In addition, compared with normal tissues, the expression of TRPV3 and TSLP in KCs in burn scars was upregulated, especially in burn scar tissues with pruritus (139). Further studies have shown that the synergistic effect of TSLP and PAR2 is particularly important in mediating pruritus signal transduction. TSLP triggers mast cell degranulation and the release of tryptase by binding to its receptor. Tryptase binds to PAR2 in KCs and activates TRPV3 channels to induce Ca2+ influx to promote the expression of TSLP, forming a positive feedback loop. TSLP binds to its receptor and transmits to the spinal dorsal root ganglion to induce pruritus (140). Kim found that higher levels of the TRPV3 activator carvacrol were associated with higher NRS scores of the burn scar pruritus index (138). Therefore, the expression of TRPV3 in PS may be positively correlated with the degree of pruritus, and the upregulation of TRPV3 channels may be related to the increased expression of PAR2 and TSLP and the involvement of the PAR2-TSLP positive feedback pathway. The current research results provide a reference for the function of TRPV3 in PS pruritus. Further research is expected to determine whether the combination of TRPV3 with TSLP and PAR2 inhibitors can provide a feasible solution for PS pruritus.
4.4 TRPV4
Recent literature shows that TRPV4 may be involved in PS fibrosis, angiogenesis, pruritus, mechanical conduction and epigenetic regulation (Figure 4).
Epithelial-mesenchymal transition (EMT) plays an important role in wound healing by inducing re-epithelialization and promoting MFBs contraction and the secretion of ECM (163, 164). Sharma found that TGF-β1-induced EMT-like changes in KCs were dependent on TRPV4. Furthermore, TRPV4 promoted the expression of the mesenchymal markers N-cadherin (NCAD) and α-SMA in a bleomycin-induced mouse skin fibrosis model (165). Whether the activation of TRPV4 is involved in EMT in PS and affects the levels of NCAD and α-SMA remains to be further studied. In addition, studies have shown that IL-6 deficiency in TRPV4-deficient corneal FBs decreases MFBs differentiation, resulting in delayed corneal wound closure (20). It is well known that the IL-6 signaling pathway plays an important role in the pathogenesis of PS (97, 98). However, in the alkali burn wound healing response of TRPV4-null mice, biomarker gene expression of fibrosis, collagen1a1 and α-SMA were attenuated along with macrophage release of IL-6 whereas TGF-β release was unchanged (166). Therefore, TRPV4 channel may promote the differentiation of FBs into MFBs to mediate the development of PS, but whether the release of IL-6 promotes this process needs further study. Recent studies have shown that extracellular vesicles (EVs) are involved in skin wound healing (167). Lactobacillus delbrueckii-derived EVs (LDEVs) may inhibit PS fibrosis by inhibiting the expression of collagen and α-SMA (168). Furthermore, Wnt4 in mesenchymal stromal cell-derived extracellular vesicles (MSC-EVs) stimulated the proliferation and migration of FBs and KCs in a dose-dependent manner, enhanced the production of collagen and fibronectin to accelerate the process of wound healing (169, 170). In addition, studies have shown that EV induces abnormal angiogenesis by downregulating TRPV4-mediated ERK phosphorylation and activating VEGFR2 and YAP signaling (171). This finding suggests that studying the activity and participation of EVs in PS will provide new intervention targets for exploring the mechanism of TRPV4-mediated PS. In summary, these studies provide new ideas for TRPV4-induced PS fibrosis and angiogenesis, and the related mechanisms need to be further elucidated.
TRPV4 may also be involved in PS pruritus. Yang found that TRPV4 mRNA expression was significantly increased in patients with burn scar pruritus and was positively correlated with pruritus intensity compared with that in patients without scar pruritus (141). Lee found that skin dryness relied on TRPV4 channels to induce TSLP production in KCs and promote pruritus (142). This study provides a new research direction for the mechanism of TRPV4-mediated PS pruritus.
In addition, TRPV4 is related to the mechanical conduction of PS, and immune inflammatory cells and FBs fibrosis are involved. Studies have revealed the effect of TRPV4 on the reaction of implanted foreign bodies, and in the absence of TRPV4, macrophage-induced FBs differentiation into MFBs was significantly reduced (172). This finding suggests that TRPV4-mediated mechanical conduction contributes to the accumulation of MFBs. In addition, studies have shown that TRPV4 activation by some stresses (excessive mechanical, osmotic, and chemical stimulation) induces pain through ATP release in human corneal epithelial cells (173). Therefore, the interaction between mechanical conduction, immune cells and fibrosis may be related to TRPV4. In addition, a novel mechanically activated cation channel Piezo1 is overexpressed in HS, and its inhibitor GsMTx1 can protect rats from stretch-induced HS (118). Furthermore, Piezo1 interacts with TRPV4 after activation to produce a continuous Ca2+ signal and promote mechanical conduction (119). Therefore, TRPV4 can promote PS by cooperating with the Piezo1 channel, and Piezo1 blockers may be used to treat PS. These findings provide new research targets for reducing PS mechanical contraction.
Furthermore, TRPV4 may be involved in the epigenetic regulation of PS. In liver fibrosis, TRPV4 is a direct target of miR-203 and promotes TGF-β1-induced hepatic stellate cell proliferation (174). Studies have shown that miR-203 regulates wound healing and scar formation by inhibiting Hes1 expression in epidermal stem cells (175). Furthermore, studies have shown that downregulating lncRNA LINC01116 inhibits keloid by regulating the miR-203/SMAD5 axis. Western blot analysis showed that lncRNA LINC01116 and SMAD5 were upregulated in keloids, while miR-203 expression was downregulated (176). Therefore, downregulating miR-203 expression and upregulating lncRNA LINC01116 expression may promote PS by regulating TRPV4 channel, but the specific mechanism needs further research.
4.5 TRPC3
TRPC3 induces mechanical stretch to promote the differentiation of FBs and KCs and regulates oxidative stress by promoting the production of ROS and H2O2 in PS (Figure 5).
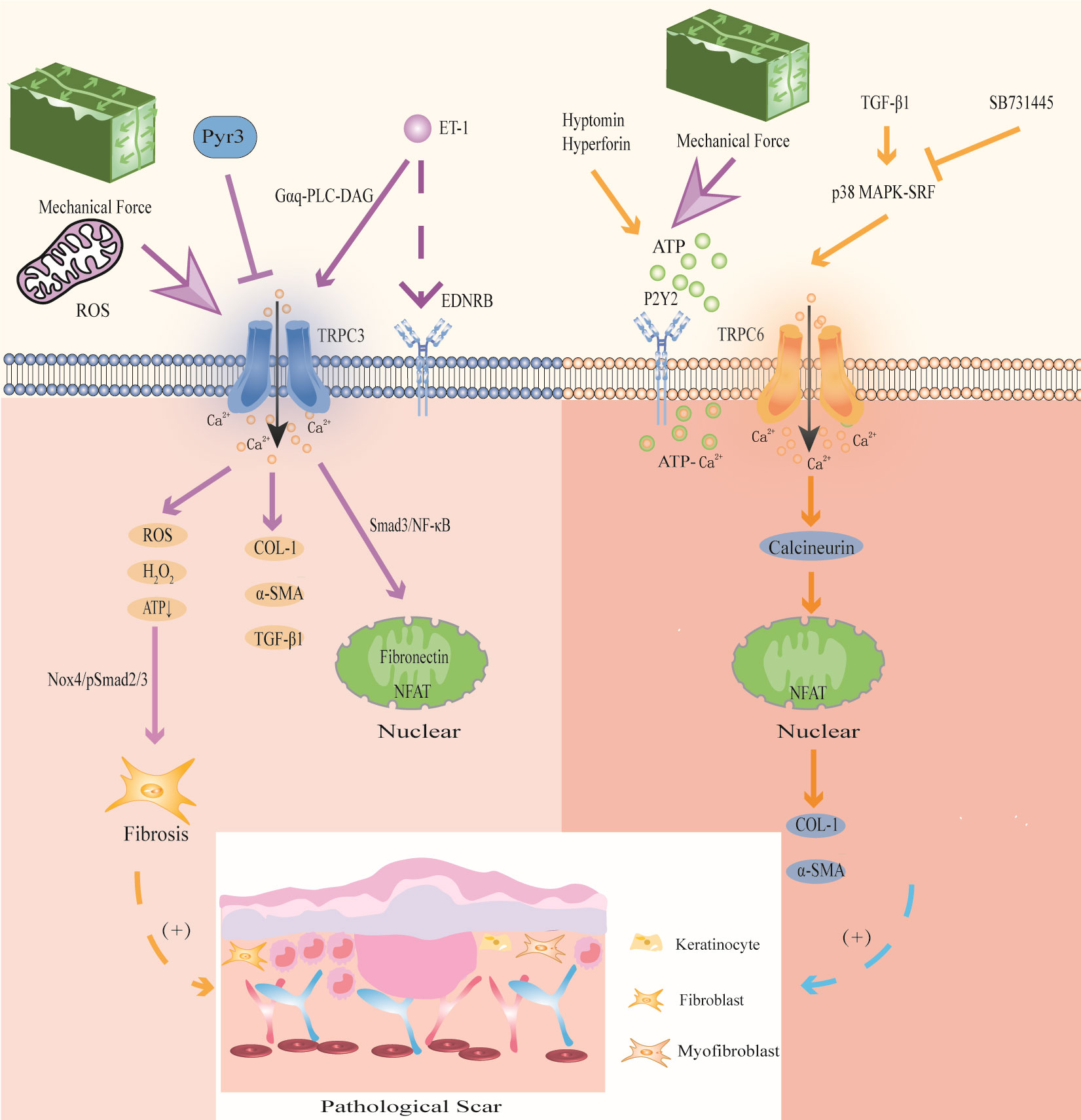
Figure 5 Modulation of TRPC3 and TRPC6 channels in PS. TRPC3 channel induces Ca2+ influx accompanied by the expression of ROS, activates Nox4/pSmad2/3 pathway, and participates in FBs differentiation; activation of TRPC3 can also directly promote the expression of COL-1, α-SMA and TGF-β1;mechanical stretching and oxidative stress activate TRPC3 channels to increase Ca2+ influx, and then activated Smad3/NF-κB migrates to the nucleus to induce fibrin expression and promote wound contraction; the signal exchange between ET-1 secreted by KCs and EDNRB activates TRPC3 channel and promotes the expression of the profibrotic gene NFAT. Mechanical stretching stimulates ATP release and activates TRPC6 channel upon binding to P2Y2 receptors, enhancing ATP- Ca2+ influx and triggering wound healing; TGF-β1 activates TRPC6 channel through p38 MAPK-SRF signaling pathway, and promotes the expression of collagen and α-SMA through TRPC6/calcineurin/NFAT signaling pathway.
Studies have shown that repeated mechanical stretching can promote the expression of TRPC3 in HS (33). TRPC3 promotes the expression of fibronectin through the Smad3/NF-κB pathway, thereby affecting FBs fibrosis in HS. In addition, TRPC3 protein and mRNA levels were positively correlated with VSS in HS patients (33). Further in vitro experiments showed that compared with that in Trpc3+/+ mice, the expression of TGF-β1, α-SMA, fibronectin and COL-1 in the granulation tissue of Trpc3-/- mice was significantly decreased. In addition, the TRPC3 inhibitor Pyr3 could significantly downregulate the expression of TGF-β1 (67). This finding indicates that mechanical stretching induces FBs fibrosis in PS by activating TRPC3 channels. In addition, the signal exchange between ET-1 secreted by KCs induced by mechanical stretch in PS and the ET-1 receptor EDNRB in FBs enhanced the expression of TRPC3 in FBs through Gαq-PLC-DAG signaling, promoting Ca2+ influx and the expression of the profibrotic gene NFAT (143).
TRPC3 is involved in hypoxia-induced PS. A study showed that ROS and H2O2 was significantly enhanced and ATP was significantly reduced in HS, while the TRPC3 inhibitor Pyr3 could decrease mitochondrial ROS and H2O2 and promote ATP production, thereby reducing the level of oxidative stress (67). Furthermore, the skin FBs of Trpc3-/- mice and Trpc3+/+ mice were used for detection in animal models. It was found that the levels of PDHE1α, a key subunit of the mitochondrial tricarboxylic acid cycle, and NOX4, a ROS-producing enzyme, in the skin FBs of Trpc3-/- mice, were significantly reduced compared to those of Trpc3+/+ mice (67). These results show that TGF-β1 upregulates TRPC3 expression and promotes PDHE1α phosphorylation during wound healing, resulting in increased mitochondrial ROS and H2O2 in FBs, decreased ATP production, activation of the Nox4/pSmad2/3 pathway, and thus participating in FBs differentiation in PS. We suggest that this is the mechanism by which TRPC3 promotes PS in the context of oxidative stress. Therefore, intervention with TRPC3 may be a new idea for the treatment of PS.
4.6 TRPC6
TRPC6 has great potential in mediating MFBs fibrosis and mechanical stretching in PS (Figure 5).
Genome-wide screening identified TRPC6 as an essential channel for MFBs transformation during wound healing and tissue remodeling, and TRPC6 overexpression activates MFBs differentiation (68). Further studies have shown that TGF-β1 induces TRPC6 expression through the p38 MAPK-serum response factor (SRF) signaling pathway to promote wound healing, and the p38 MAPK inhibitor SB731445 can completely block TRPC6 expression (68). Furthermore, TRPC6 is activated to induce Ca2+ influx and MFBs transdifferentiation through the TRPC6/calcineurin/NFAT signaling pathway, further promoting the expression of α-SMA and collagen (68). Calcineurin inhibitors can be used to treat keloids (144). The TRPC6/calcineurin/NFAT signaling pathway, which is a key signaling pathway that mediates MFBs transdifferentiation to promote wound healing, is expected to provide new intervention targets for PS and fibrotic diseases.
In addition, TRPC6 can promote wound healing through mechanical conduction. Mechanical stimulation induces HaCaT cells to release ATP, which acts as an autocrine mediator and binds to the P2Y2 receptor, activating TRPC6 channels in HaCaT cells, promoting Ca2+ influx, and participating in wound healing (177). Further studies have shown that the TRPC6 activators hyperforin and hypericin, which are Chinese herbal medicines that promote wound healing, can upregulate the expression of TRPC6 in HaCaT cells induced by mechanical stretching, mediate Ca2+ influx and enhance ATP-Ca2+ signaling to promote wound healing (177, 178). These studies suggest that mechanical stimulation enhances ATP-Ca2+ signaling by activating TRPC6 channels, which may be a potential mechanism by which TRPC6 participates in PS.
4.7 TRPA1
TRPA1 mainly regulates immune inflammation and fibrosis of PS. Furthermore, this channel mediates PS pruritus (Figure 6).
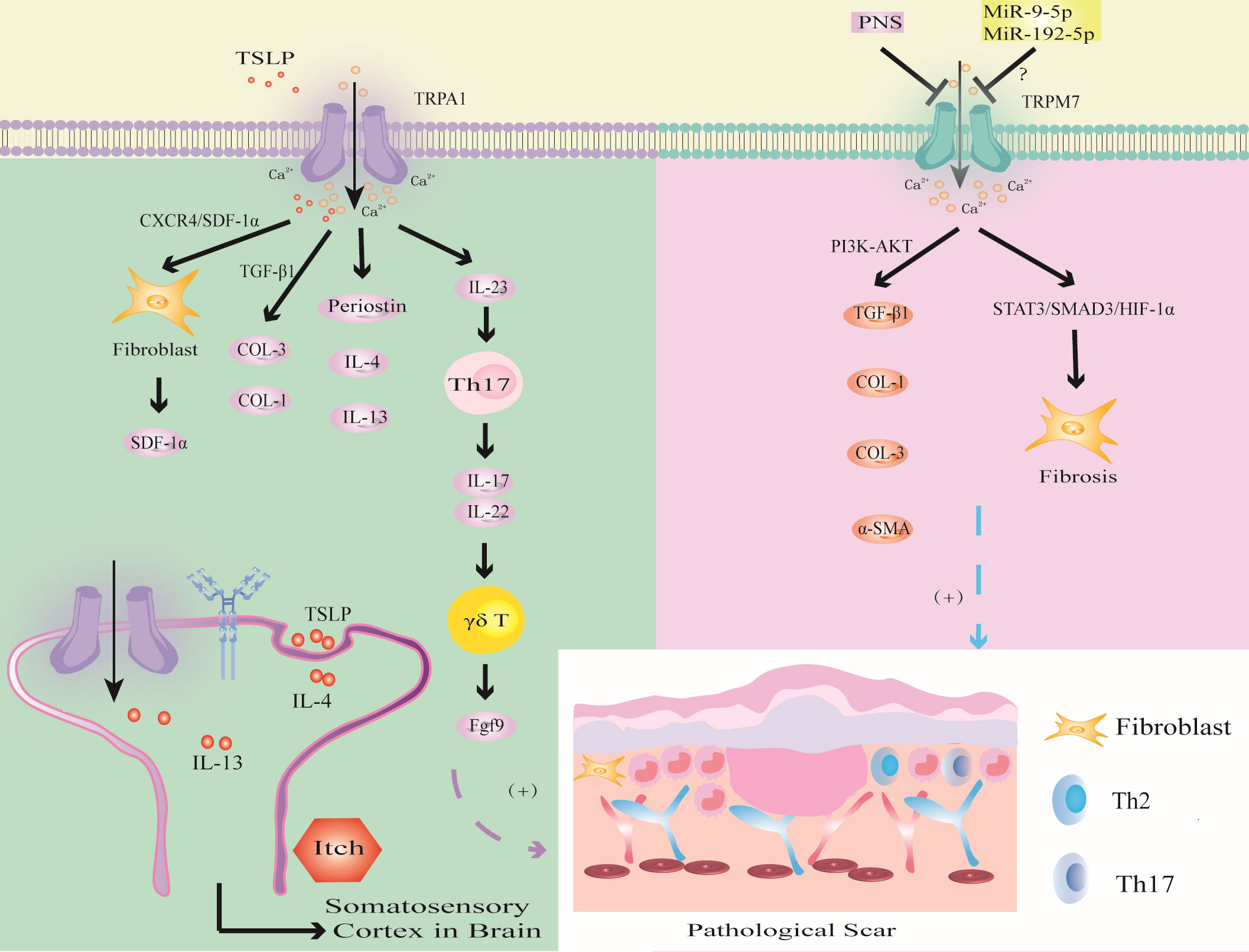
Figure 6 Modulation of TRPA1 and TRPM7 channels in PS. TSLP triggers TRPA1 channel to promote fibrosis through CXCR4/SDF-1 axis or promote the synthesis of COL-1 and COL-3 through TGF-β1; the activation of TRPV1 promotes the expression of Periostin, IL-4 and IL-13, which induced PS pruritus. TRPA1 activates immune cells to release inflammatory factors to promote wound healing. The activation of TRPM7 channel upregulates the expression of TGF-β1, COL-1, COL-3 and α-SMA through PI3K-AKT signaling pathway, or induces hypoxia through STAT3/SMAD3/HIF-1α signaling pathway. MiR-9-5p and miR-192-5p may inhibit PS by targeting TRPM7.
Murata found that TRPA1 deficiency inhibited the infiltration of FBs, T cells and the expression of α-SMA and COL-1 during wound healing (65). TSLP, which is a Th2 cytokine, was positively correlated with TRPA1 expression (126). Studies have shown that TSLP promotes the expression of SDF-1a in FBs through the CXCR4/SDF-1 axis and promotes the synthesis of COL-1 and COL-3 through TGF-β1, thereby inducing keloids (147). In addition, TRPA1-expressing neurons stimulate dendritic cells to produce IL-23, leading to skin inflammation, which in turn activates type 17 immune cells to produce IL-17 and IL-22 to further recruit γδT cells, which release fibroblast growth factor 9 (Fgf9) to promote wound healing (92, 145, 146). Excessive upregulation of Fgf9 may lead to PS. In addition, the absence of TRPA1-induced upregulation of TGFβ1-related signaling cascades inhibits chemical injury-induced corneal wound inflammation and fibrosis in mice (20). Thus, TRPA1-mediated inflammation and fibrosis play important roles in wound healing. Targeting TRPA1 may provide a new strategy for the treatment of PS (20).
Studies have shown that the expression of TRPA1 in the scar tissue of patients with burn scar pruritus is higher than that of patients without pruritus, especially in mast cells (141). Studies have shown that IL-4 and IL-13 can upregulate the transcription of TRPV1 and TRPA1 (126). IL-4 and IL-13 can not only mediate PS fibrosis through the TGF-β/SMAD and IL-4Rα/STAT6 signaling pathways but also directly stimulate neurons through IL-4 receptors to induce PS pruritus (87). This finding suggests that IL-4 and IL-13 secreted by Th2 cells may mediate PS pruritus through TRPV1 and TRPA1 neurons. These studies provide a new direction for improving PS pruritus.
4.8 TRPM7
Abnormal TRPM7 expression may be involved in PS fibrosis, oxidative stress and noncoding RNA-related gene regulation (Figure 6).
Zhi found that the expression of TRPM7 was upregulated and enhanced the expression of TGF-β1, COL-1, COL-3 and α-SMA through the PI3K-AKT signaling pathway in HS (179). Furthermore, Panax Noto ginseng saponins (PNSs) can protect against HS by inhibiting TRPM7 expression, cell migration and viability, and collagen deposition by downregulating the PI3K/AKT pathway, thus inducing apoptosis and cell cycle arrest. This finding indicates that TRPM7 can promote fibrosis and collagen deposition through the PI3K/AKT signaling pathway and ultimately induce PS.
Zhang found that the overexpression of TRPM7 regulated the BAX/Bcl-2 balance and antioxidant processes through the STAT3/SMAD3/HIF-1α signaling pathway, thereby promoting FBs migration and inducing wound healing (69). This led us to hypothesize that TRPM7 may accelerate PS through the STAT3/SMAD3/HIF-1α signaling pathway.
In addition, TRPM7 may be related to the regulation of epigenetic noncoding RNA-related genes in PS. MiR-9-5p mediates endothelial cell proliferation, migration and angiogenesis by targeting TRPM7 through the PI3K/AKT/autophagy pathway (180). Studies have shown that miR-9-5p can inhibit the expression of α-SMA and COL-1 in HS by targeting PPARβ, promote the proliferation of FBs and accelerate apoptosis (181). Therefore, we hypothesize that miR-9-5p targets PPARβ to activate TRPM7 channel through the PI3K/AKT pathway, inhibit FBs fibrosis and collagen deposition, which may be an important target for PS. In addition, studies have shown that expression of the tumor suppressor miR-192-5p can be reversed by TRPM7 overexpression, thereby promoting the proliferation, migration and invasion of cervical cancer cells (182). However, miR-192-5p derived from adipose tissue-derived mesenchymal stem cell-derived exosomes (ADSC-Exos) inhibits HS by targeting IL-17 to regulate the Smad pathway (183). Therefore, inhibiting miR-9-5p to promote the expression of TRPM7 may aggravate PS fibrosis by activating the IL-17/Smad pathway. In-depth study of TRP channels may become an important research direction to determine the mechanism of PS in the context of epigenetics.
We now acknowledge that while few studies have reported on the involvement of other TRP channels in the mechanism of PS, the similarity of the structure and function of the homologous TRP channel family leads us to speculate that other TRP channels may also be related to the mechanism of PS, which needs further research and exploration in the future. In recent years, studies have found that the expression of TRPC1 is associated with renal fibrosis. CircRNA_010383 colocalized with miR-135a, and that overexpression of circRNA_010383 increased the level of TRPC1, which is a target protein of miR-135a. Additionally, the overexpression of circRNA_010383 inhibited the high glucose-induced accumulation of ECM (184). TRPC1 may have a potential relationship with the fibrosis of PS, but further research is needed to confirm this. Additionally, the study mentions that TRPM2 ablation has been found to significantly reduce renal interstitial fibrosis by decreasing TGF-β1 levels. This reduction in TGF-β1 is accompanied by a decrease in α-SMA, connective tissue growth factor (CTGF), fibronectin, and COL-1. The study also suggests that TRPM2 ablation may protect against renal fibrosis and inflammation by impeding JNK activation regulated by TGF-β1 (185). TRPM2 may be involved in inflammation and fibrosis of PS. Additionally, TRPML channels have important functional activities in immune cells such as macrophages, dendritic cells, neutrophils, NK cells, and B lymphocytes, which may provide important ideas for exploring the mechanism of TRPML in the inflammation and immunity of PS (186).In addition, TRPP1-like proteins may form mechanosensors in primary cilia. The phosphorylation of the COOH-terminus of TRPP1 may recruit TRPP2 to the cell membrane, which can promote Id2 to enter the nucleus and induce renal cell proliferation (187). The study suggests that Id2, as a key regulator of cell proliferation and differentiation, may provide a research direction for future studies on the mechanism of TRPP channel and PS.
5 Discussion and conclusion
In recent years, it has been found that a variety of TRP channels are involved in the development of PS, especially in immune inflammation and fibrosis, which has great research and clinical values. In this review, we introduced the specific physiological and pathological functions of TRP channels and focused on their important roles in mediating PS immune inflammation, fibrosis, mechanical conduction, epigenetics, oxidative stress and pruritus. TRP channels promote tissue fibrosis and re-epithelialization by regulating the proliferation, migration, phagocytosis and cytokine release of immune cells or by directly activating FBs and KCs to promote the deposition of ECM and mediate the development of PS; mechanical stimulation which directly promotes fibrosis by activating TRP channels or recruiting immune cells to participate in fibrosis; noncoding RNA which regulates FBs fibrosis and collagen deposition by activating TRP channels; and the activation of TRP channels that can promote the release of oxidative stress-related factors such as ROS and H2O2, which in turn induce PS. Based on the latest reports, this review summarized the most novel concepts for the therapeutic drugs of PS from targeting TRP channels as follows: EVs are paracrine molecules that regulate cell signal transduction in the corneal scar and fibrosis, and studying how EVs participate in the development of PS through TRP channels may provide a promising treatment (167); Mechanical forces stimulate some TRP receptors on skin sensory fibers to release neuropeptides including SP and CGRP, which may provide direction for TRP-mediated PS neurogenic inflammation mechanism (122); and IL-31 mediates pruritus through TRPV1 and TRPA1 channels, indicating a new therapeutic target for the treatment of PS pruritus (127, 128). In addition, most of the current research on PS focuses on immunity. Immune signals enhance epithelial cell proliferation, differentiation and migration to accelerate skin repair (188), which suggests that targeting immune-epithelial communication to promote repair or reduce inflammation can be a new strategy for PS treatment. Therefore, studying the important role of TRP channels on account of the immune perspective may prompt novel targets for improved treatment of PS.
Although many studies have been carried out on immune inflammation in PS, the role of TRP channels in the regulation and treatment of PS is still in its infancy, mainly because it is a relatively new field involving wound healing and PS mechanisms, and many problems require further study. First, in response to different intensities of mechanical stretching, some mechanoreceptors in the TRP family promote wound healing. Excessive activated TRP in the wound site may lead to PS. Therefore, identifying the most suitable mechanical force triggering wound healing is particularly important to avoid the occurrence of PS. Second, the effects of various TRP channels on PS are different. For instance, the mechanism of TRP channels in the inflammatory, proliferative and remodeling stages of PS, TRP needs to be further explored. Finally, although several TRP agonists or antagonists have been developed for the treatment of PS, such as SKF96365 or tranilast, they have infrequently used in clinical practice due to the problems of drug permeability, side effects, and poor efficacy (189). Therefore, there is an urgent need for a comprehensive summary and in-depth evaluation of TRP channels to provide novel directions for identifying more selective and efficacious drug target to treat PS. This paper summarized the latest progress in understanding the role of TRP channels in PS, and the role of other TRP channels in PS needs to be further examined.
In the future, as the mechanisms of different TRP channels and their interactions with cytokines and growth factors become increasingly clear, TRP channels are expected to have further breakthroughs in the prevention and treatment of PS. In this article, we summarized the important roles of TRP channels in the pathogenesis of PS and proposed that drug development targeting TRP channels will be a critical topic for PS. Therefore, we hope to provide potential pharmacological targets and directions for future deeper understanding of PS and the evolution of new drugs by summarizing the mechanism of TRP channels in PS.
Author contributions
YPZ conceived and wrote the article. QH, YFZ, and LG collected the data. WW was responsible for the idea. HZ was responsible for conceptualization and funding. XH was responsible for project administration and funding. QL were responsible for the idea, funding, and paper revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 82205125), the Research on New Carrier Technology Development of Skin Barrier Repair Nursing Agent—Horizontal Project (Zhang Huimin), the Health Industry Clinical Research Special Project of Shanghai Municipal Health Commission (Grant No. 20224Y0224), Shanghai Science and Technology Commission (Grant No. 21ZR14640), Shanghai “Rising Stars of Medical Talent Youth Development Program for Youth Medical Talents-Specialist Program” (Grant No. 2022-38).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finnerty CC, Jeschke MG, Branski LK, Barret JP, Dziewulski P, Herndon DN. Hypertrophic scarring: the greatest unmet challenge after burn injury. Lancet (2016) 388(10052):1427–36. doi: 10.1016/s0140-6736(16)31406-4
2. Aarabi S, Longaker MT, Gurtner GC. Hypertrophic scar formation following burns and trauma: new approaches to treatment. PloS Med (2007) 4(9):e234. doi: 10.1371/journal.pmed.0040234
3. Wang ZC, Zhao WY, Cao Y, Liu YQ, Sun Q, Shi P, et al. The roles of inflammation in keloid and hypertrophic scars. Front Immunol (2020) 11:603187. doi: 10.3389/fimmu.2020.603187
4. Wang X, Liang B, Li J, Pi X, Zhang P, Zhou X, et al. Identification and characterization of four immune-related signatures in keloid. Front Immunol (2022) 13:942446. doi: 10.3389/fimmu.2022.942446
5. Yin J, Zhang S, Yang C, Wang Y, Shi B, Zheng Q, et al. Mechanotransduction in skin wound healing and scar formation: Potential therapeutic targets for controlling hypertrophic scarring. Front Immunol (2022) 13:1028410. doi: 10.3389/fimmu.2022.1028410
6. Huang C, Ogawa R. The vascular involvement in soft tissue fibrosis-lessons learned from pathological scarring. Int J Mol Sci (2020) 21(7):2542. doi: 10.3390/ijms21072542
7. Qin H, Zhang L, Li M, Liu Y, Sun S, Nie W, et al. EGR1/NOX4 pathway regulates oxidative stress and further facilitates fibrosis progression in keloids responses to TGF-β1. J Dermatol Sci (2022) 108(3):138–45. doi: 10.1016/j.jdermsci.2022.12.009
8. Amjadian S, Moradi S, Mohammadi P. The emerging therapeutic targets for scar management: genetic and epigenetic landscapes. Skin Pharmacol Physiol (2022) 35(5):247–65. doi: 10.1159/000524990
9. Partida-Sanchez S, BN D, Schwab A, Zierler S. Editorial: TRP channels in inflammation and immunity. Front Immunol (2021) 12:684172. doi: 10.3389/fimmu.2021.684172
10. Mousavizadeh SM, Torbati PM, Daryani A. The effects of kiwifruit dressing on hypertrophic scars in a rabbit ear model. J Wound Care (2021) 30(Sup9a):XVi–XVvii. doi: 10.12968/jowc.2021.30.Sup9a.XV
11. Khalil M, Alliger K, Weidinger C, Yerinde C, Wirtz S, Becker C, et al. Functional role of transient receptor potential channels in immune cells and epithelia. Front Immunol (2018) 9:174. doi: 10.3389/fimmu.2018.00174
12. Yang F, Sivils A, Cegielski V, Singh S, Chu XP. Transient receptor potential (TRP) channels in pain, neuropsychiatric disorders, and epilepsy. Int J Mol Sci (2023) 24(5):4714. doi: 10.3390/ijms24054714
13. Cosens DJ, Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature (1969) 224(5216):285–7. doi: 10.1038/224285a0
14. Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron (1992) 8(4):643–51. doi: 10.1016/0896-6273(92)90086-s
15. Zhong T, Zhang W, Guo H, Pan X, Chen X, He Q, et al. The regulatory and modulatory roles of TRP family channels in Malignant tumors and relevant therapeutic strategies. Acta Pharm Sin B (2022) 12(4):1761–80. doi: 10.1016/j.apsb.2021.11.001
16. Samanta A, Hughes TET, Moiseenkova-Bell VY. Transient receptor potential (TRP) channels. Subcell Biochem (2018) 87:141–65. doi: 10.1007/978-981-10-7757-9_6
17. Koivisto AP, Belvisi MG, Gaudet R, Szallasi A. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat Rev Drug Discovery (2022) 21(1):41–59. doi: 10.1038/s41573-021-00268-4
18. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem (2007) 76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819
19. Caterina MJ, Pang Z. TRP channels in skin biology and pathophysiology. Pharm (Basel) (2016) 9(4):77. doi: 10.3390/ph9040077
20. Okada Y, Sumioka T, Reinach PS, Miyajima M, Saika S. Roles of epithelial and mesenchymal TRP channels in mediating inflammatory fibrosis. Front Immunol (2021) 12:731674. doi: 10.3389/fimmu.2021.731674
21. Seebohm G, Schreiber JA. Beyond hot and spicy: TRPV channels and their pharmacological modulation. Cell Physiol Biochem (2021) 55(S3):108–30. doi: 10.33594/000000358
22. Rosenbaum T, Islas LD. Molecular physiology of TRPV channels: controversies and future challenges. Annu Rev Physiol (2023) 85:293–316. doi: 10.1146/annurev-physiol-030222-012349
23. Michalick L, Kuebler WM. TRPV4-A missing link between mechanosensation and immunity. Front Immunol (2020) 11:413. doi: 10.3389/fimmu.2020.00413
24. Lan Z, Chen L, Feng J, Xie Z, Liu Z, Wang F, et al. Mechanosensitive TRPV4 is required for crystal-induced inflammation. Ann Rheum Dis (2021) 80(12):1604–14. doi: 10.1136/annrheumdis-2021-220295
25. Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, et al. Cutaneous TRPV1(+) neurons trigger protective innate type 17 anticipatory immunity. Cell (2019) 178(4):919–32.e14. doi: 10.1016/j.cell.2019.06.022
26. Peng G, Tang X, Gui Y, Yang J, Ye L, Wu L, et al. Transient receptor potential vanilloid subtype 1: A potential therapeutic target for fibrotic diseases. Front Physiol (2022) 13:951980. doi: 10.3389/fphys.2022.951980
27. Kärki T, Tojkander S. TRPV protein family-from mechanosensing to cancer invasion. Biomolecules (2021) 11(7):1019. doi: 10.3390/biom11071019
28. Smani T, Gómez LJ, Regodon S, Woodard GE, Siegfried G, Khatib AM, et al. TRP channels in angiogenesis and other endothelial functions. Front Physiol (2018) 9:1731. doi: 10.3389/fphys.2018.01731
29. Ohya S, Kito H, Hatano N, Muraki K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol Ther (2016) 160:11–43. doi: 10.1016/j.pharmthera.2016.02.001
30. Blanchard MG, Kellenberger S. Effect of a temperature increase in the non-noxious range on proton-evoked ASIC and TRPV1 activity. Pflugers Arch (2011) 461(1):123–39. doi: 10.1007/s00424-010-0884-3
31. Szöllősi AG, Vasas N, Angyal Á, Kistamás K, Nánási PP, Mihály J, et al. Activation of TRPV3 regulates inflammatory actions of human epidermal keratinocytes. J Invest Dermatol (2018) 138(2):365–74. doi: 10.1016/j.jid.2017.07.852
32. Wang H, Cheng X, Tian J, Xiao Y, Tian T, Xu F, et al. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol Ther (2020) 209:107497. doi: 10.1016/j.pharmthera.2020.107497
33. Ishise H, Larson B, Hirata Y, Fujiwara T, Nishimoto S, Kub T, et al. Hypertrophic scar contracture is mediated by the TRPC3 mechanical force transducer via NFkB activation. Sci Rep (2015) 5:11620. doi: 10.1038/srep11620
34. Wuensch T, Thilo F, Krueger K, Scholze A, Ristow M, Tepel M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes (2010) 59(4):844–9. doi: 10.2337/db09-1100
35. Sun ZC, Ma SB, Chu WG, Jia D, Luo C. Canonical transient receptor potential (TRPC) channels in nociception and pathological pain. Neural Plast (2020) 2020:3764193. doi: 10.1155/2020/3764193
36. Farfariello V, Gordienko DV, Mesilmany L, Touil Y, Germain E, Fliniaux I, et al. TRPC3 shapes the ER-mitochondria Ca(2+) transfer characterizing tumour-promoting senescence. Nat Commun (2022) 13(1):956. doi: 10.1038/s41467-022-28597-x
37. Kim J, Moon SH, Shin YC, Jeon JH, Park KJ, Lee KP, et al. Erratum to: Intracellular spermine blocks TRPC4 channel via electrostatic interaction with C-terminal negative amino acids. Pflugers Arch (2016) 468(7):1297. doi: 10.1007/s00424-016-1824-7
38. Zhu Y, Gao M, Zhou T, Xie M, Mao A, Feng L, et al. The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J Biol Chem (2019) 294(1):28–37. doi: 10.1074/jbc.RA118.005392
39. Quick K, Zhao J, Eijkelkamp N, Linley JE, Rugiero F, Cox JJ, et al. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol (2012) 2(5):120068. doi: 10.1098/rsob.120068
40. Talavera K, Startek JB, Alvarez-Collazo J, Boonen B, Alpizar YA, Sanchez A, et al. MamMalian transient receptor potential TRPA1 channels: from structure to disease. Physiol Rev (2020) 100(2):725–803. doi: 10.1152/physrev.00005.2019
41. Zappia KJ, Garrison SR, Palygin O, Weyer AD, Barabas ME, Lawlor MW, et al. Mechanosensory and ATP release deficits following keratin14-cre-mediated TRPA1 deletion despite absence of TRPA1 in murine keratinocytes. PloS One (2016) 11(3):e0151602. doi: 10.1371/journal.pone.0151602
42. Oh MH, Oh SY, Lu J, Lou H, Myers AC, Zhu Z, et al. TRPA1-dependent pruritus in IL-13-induced chronic atopic dermatitis. J Immunol (2013) 191(11):5371–82. doi: 10.4049/jimmunol.1300300
43. Jimenez I, Prado Y, Marchant F, Otero C, Eltit F, Cabello-Verrugio C, et al. TRPM channels in human diseases. Cells (2020) 9(12):2604. doi: 10.3390/cells9122604
44. Najder K, Rugi M, Lebel M, Schröder J, Oster L, Schimmelpfennig S, et al. Role of the intracellular sodium homeostasis in chemotaxis of activated murine neutrophils. Front Immunol (2020) 11:2124. doi: 10.3389/fimmu.2020.02124
45. Shi R, Fu Y, Zhao D, Boczek T, Wang W, Guo F. Cell death modulation by transient receptor potential melastatin channels TRPM2 and TRPM7 and their underlying molecular mechanisms. Biochem Pharmacol (2021) 190:114664. doi: 10.1016/j.bcp.2021.114664
46. Wrighton KH. Epigenetics: the TRPM7 ion channel modifies histones. Nat Rev Mol Cell Biol (2014) 15(7):427. doi: 10.1038/nrm3824
47. Xu T, Wu BM, Yao HW, Meng XM, Huang C, Ni MM, et al. Novel insights into TRPM7 function in fibrotic diseases: a potential therapeutic target. J Cell Physiol (2015) 230(6):1163–9. doi: 10.1002/jcp.24801
48. Mirbod SM, Khanahmad H, Amerizadeh A, Amirpour A, Mirbod SM, Zaker E. Viewpoints on the role of transient receptor potential melastatin channels in cardiovascular system and disease: A systematic review. Curr Probl Cardiol (2023) 48(2):101012. doi: 10.1016/j.cpcardiol.2021.101012
49. Orlowski P, Zmigrodzka M, Tomaszewska E, Ranoszek-Soliwoda K, Pajak B, Slonska A, et al. Polyphenol-conjugated bimetallic au@AgNPs for improved wound healing. Int J Nanomed (2020) 15:4969–90. doi: 10.2147/ijn.S252027
50. Kolanthai E, Fu Y, Kumar U, Babu B, Venkatesan AK, Liechty KW, et al. Nanoparticle mediated RNA delivery for wound healing. Wiley Interdiscip Rev Nanomed Nanobiotechnol (2022) 14(2):e1741. doi: 10.1002/wnan.1741
51. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res (2010) 89(3):219–29. doi: 10.1177/0022034509359125
52. Segal AW. How neutrophils kill microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
53. Zhang T, Wang XF, Wang ZC, Lou D, Fang QQ, Hu YY, et al. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. BioMed Pharmacother (2020) 129:110287. doi: 10.1016/j.biopha.2020.110287
54. Zhang X, Shu W, Yu Q, Qu W, Wang Y, Li R. Functional biomaterials for treatment of chronic wound. Front Bioeng Biotechnol (2020) 8:516. doi: 10.3389/fbioe.2020.00516
55. Pignet AL, Schellnegger M, Hecker A, Kohlhauser M, Kotzbeck P, Kamolz LP. Resveratrol-induced signal transduction in wound healing. Int J Mol Sci (2021) 22(23):12614. doi: 10.3390/ijms222312614
56. Qiang L, Yang S, Cui YH, He YY. Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing. Autophagy (2021) 17(9):2128–43. doi: 10.1080/15548627.2020.1816342
57. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: A cellular perspective. Physiol Rev (2019) 99(1):665–706. doi: 10.1152/physrev.00067.2017
58. Wang PH, Huang BS, Horng HC, Yeh CC, Chen YJ. Wound healing. J Chin Med Assoc (2018) 81(2):94–101. doi: 10.1016/j.jcma.2017.11.002
59. Lee HJ, Jang YJ. Recent understandings of biology, prophylaxis and treatment strategies for hypertrophic scars and keloids. Int J Mol Sci (2018) 19(3):711. doi: 10.3390/ijms19030711
60. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res (2012) 49(1):35–43. doi: 10.1159/000339613
61. Ueno K, Saika S, Okada Y, Iwanishi H, Suzuki K, Yamada G, et al. Impaired healing of cutaneous wound in a Trpv1 deficient mouse. Exp Anim (2023) 72(2):224–32. doi: 10.1538/expanim.22-0124
62. Um JY, Kang SY, Kim HJ, Chung BY, Park CW, Kim HO. Transient receptor potential vanilloid-3 (TRPV3) channel induces dermal fibrosis via the TRPV3/TSLP/Smad2/3 pathways in dermal fibroblasts. J Dermatol Sci (2020) 97(2):117–24. doi: 10.1016/j.jdermsci.2019.12.011
63. Maier M, Olthoff S, Hill K, Zosel C, Magauer T, Wein LA, et al. KS0365, a novel activator of the transient receptor potential vanilloid 3 (TRPV3) channel, accelerates keratinocyte migration. Br J Pharmacol (2022) 179(24):5290–304. doi: 10.1111/bph.15937
64. Sahu RP, Goswami C. Presence of TRPV3 in macrophage lysosomes helps in skin wound healing against bacterial infection. Exp Dermatol (2023) 32(1):60–74. doi: 10.1111/exd.14683
65. Murata S, Yamanaka M, Taniguchi W, Kajioka D, Suzuki K, Yamada G, et al. Lack of transient receptor potential ankyrin 1 (TRPA1) retards cutaneous wound healing in mice: A preliminary study. Biochem Biophys Rep (2022) 31:101322. doi: 10.1016/j.bbrep.2022.101322
66. Ishii T, Uchida K, Hata S, Hatta M, Kita T, Miyake Y, et al. TRPV2 channel inhibitors attenuate fibroblast differentiation and contraction mediated by keratinocyte-derived TGF-beta1 in an in vitro wound healing model of rats. J Dermatol Sci (2018) 90(3):332–42. doi: 10.1016/j.jdermsci.2018.03.003
67. Xia W, Wang Q, Lu Y, Hu Y, Zhang X, Zhang J, et al. Transient receptor potential channel canonical type 3 deficiency antagonizes myofibroblast transdifferentiation in vivo. BioMed Res Int (2020) 2020:1202189. doi: 10.1155/2020/1202189
68. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell (2012) 23(4):705–15. doi: 10.1016/j.devcel.2012.08.017
69. Zhang H, Li H, Wang H, Lei S, Yan L. Overexpression of TRPM7 promotes the therapeutic effect of curcumin in wound healing through the STAT3/SMAD3 signaling pathway in human fibroblasts. Burns (2023) 49(4):889–900. doi: 10.1016/j.burns.2022.06.016
70. Moretti L, Stalfort J, Barker TH, Abebayehu D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J Biol Chem (2022) 298(2):101530. doi: 10.1016/j.jbc.2021.101530
71. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci (2017) 18(7):1545. doi: 10.3390/ijms18071545
72. Chen L, Wang J, Li S, Yu Z, Liu B, Song B, et al. The clinical dynamic changes of macrophage phenotype and function in different stages of human wound healing and hypertrophic scar formation. Int Wound J (2019) 16(2):360–9. doi: 10.1111/iwj.13041
73. Sim SL, Kumari S, Kaur S, Khosrotehrani K. Macrophages in skin wounds: functions and therapeutic potential. Biomolecules (2022) 12(11):1659. doi: 10.3390/biom12111659
74. van den Broek LJ, van der Veer WM, de Jong EH, Gibbs S, Niessen FB. Suppressed inflammatory gene expression during human hypertrophic scar compared to normotrophic scar formation. Exp Dermatol (2015) 24(8):623–9. doi: 10.1111/exd.12739
75. Jung BK, Roh TS, Roh H, Lee JH, Yun CO, Lee WJ. Effect of mortalin on scar formation in human dermal fibroblasts and a rat incisional scar model. Int J Mol Sci (2022) 23(14):7918. doi: 10.3390/ijms23147918
76. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev (2016) 273(1):357–70. doi: 10.1111/imr.12453
77. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol (2015) 185(10):2596–606. doi: 10.1016/j.ajpath.2015.06.001
78. Pavlou S, Lindsay J, Ingram R, Xu H, Chen M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol (2018) 19(1):24. doi: 10.1186/s12865-018-0261-0
79. Smigiel KS, Parks WC. Macrophages, wound healing, and fibrosis: recent insights. Curr Rheumatol Rep (2018) 20(4):17. doi: 10.1007/s11926-018-0725-5
80. Xu X, Gu S, Huang X, Ren J, Gu Y, Wei C, et al. The role of macrophages in the formation of hypertrophic scars and keloids. Burns Trauma (2020) 8:tkaa006. doi: 10.1093/burnst/tkaa006
81. Feng Y, Sun ZL, Liu SY, Wu JJ, Zhao BH, Lv GZ, et al. Direct and indirect roles of macrophages in hypertrophic scar formation. Front Physiol (2019) 10:1101. doi: 10.3389/fphys.2019.01101
82. Xu H, Zhu Z, Hu J, Sun J, Wo Y, Wang , et al. Downregulated cytotoxic CD8(+) T-cell identifies with the NKG2A-soluble HLA-E axis as a predictive biomarker and potential therapeutic target in keloids. Cell Mol Immunol (2022) 19(4):527–39. doi: 10.1038/s41423-021-00834-1
83. Shan M, Liu H, Hao Y, Song K, Feng C, Wang Y. The role of CD28 and CD8(+) T cells in keloid development. Int J Mol Sci (2022) 23(16):8862. doi: 10.3390/ijms23168862
84. Ley K. M1 means kill; M2 means heal. J Immunol (2017) 199(7):2191–3. doi: 10.4049/jimmunol.1701135
85. Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol (2015) 36(12):763–77. doi: 10.1016/j.it.2015.10.003
86. Knipper JA, Willenborg S, Brinckmann J, Bloch W, Maaß T, Wagener R, et al. Interleukin-4 receptor α Signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity (2015) 43(4):803–16. doi: 10.1016/j.immuni.2015.09.005
87. Nguyen JK, Austin E, Huang A, MaMalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res (2020) 312(2):81–92. doi: 10.1007/s00403-019-01972-3
88. Short WD, Wang X, Keswani SG. The role of T lymphocytes in cutaneous scarring. Adv Wound Care (New Rochelle) (2022) 11(3):121–31. doi: 10.1089/wound.2021.0059
89. Chen Y, Jin Q, Fu X, Qiao J, Niu F. Connection between T regulatory cell enrichment and collagen deposition in keloid. Exp Cell Res (2019) 383(2):111549. doi: 10.1016/j.yexcr.2019.111549
90. Zaharie RD, Popa C, Schlanger D, Vălean D, Zaharie F. The role of IL-22 in wound healing. Potential implications in clinical practice. Int J Mol Sci (2022) 23(7):3693. doi: 10.3390/ijms23073693
91. Komi DEA, Khomtchouk K, Santa Maria PL. A review of the contribution of mast cells in wound healing: involved molecular and cellular mechanisms. Clin Rev Allergy Immunol (2020) 58(3):298–312. doi: 10.1007/s12016-019-08729-w
92. Chesko DM, Wilgus TA. Immune cells in cutaneous wound healing: A review of functional data from animal models. Int J Mol Sci (2022) 23(5):2444. doi: 10.3390/ijms23052444
93. Chen L, Schrementi ME, Ranzer MJ, Wilgus TA, DiPietro LA. Blockade of mast cell activation reduces cutaneous scar formation. PloS One (2014) 9(1):e85226. doi: 10.1371/journal.pone.0085226
94. Lee SS, Yosipovitch G, Chan YH, Goh CL. Pruritus, pain, and small nerve fiber function in keloids: a controlled study. J Am Acad Dermatol (2004) 51(6):1002–6. doi: 10.1016/j.jaad.2004.07.054
95. Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regener (2010) 18(2):139–53. doi: 10.1111/j.1524-475X.2009.00553.x
96. Shao Y, Guo Z, Yang Y, Liu L, Huang J, Chen Y, et al. Neutrophil extracellular traps contribute to myofibroblast differentiation and scar hyperplasia through the Toll-like receptor 9/nuclear factor Kappa-B/interleukin-6 pathway. Burns Trauma (2022) 10:tkac044. doi: 10.1093/burnst/tkac044
97. Chen ZH, Yu B, Ye QF, Wang YF. [Research advances on interleukin-6 in hypertrophic scar formation]. Zhonghua Shao Shang Za Zhi (2022) 38(9):874–7. doi: 10.3760/cma.j.cn501120-20210331-00111
98. Kurachi I, Kurita E, Takushima A, Suga H. Human CD206+ Macrophages Show Antifibrotic Effects on Human Fibroblasts through an IL-6-Dependent Mechanism in vitro. Plast Reconstr Surg (2021) 147(2):231e–9e. doi: 10.1097/prs.0000000000007563
99. Zhou B, Tu T, Gao Z, Wu X, Wang W, Liu W. Impaired collagen fibril assembly in keloids with enhanced expression of lumican and collagen V. Arch Biochem Biophys (2021) 697:108676. doi: 10.1016/j.abb.2020.108676
100. Kosla J, Dvorakova M, Dvorak M, Cermak V. Effective myofibroblast dedifferentiation by concomitant inhibition of TGF-β signaling and perturbation of MAPK signaling. Eur J Cell Biol (2013) 92(12):363–73. doi: 10.1016/j.ejcb.2013.10.013
101. Russo B, Brembilla NC, Chizzolini C. Interplay between keratinocytes and fibroblasts: A systematic review providing a new angle for understanding skin fibrotic disorders. Front Immunol (2020) 11:648. doi: 10.3389/fimmu.2020.00648
102. Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, et al. Epithelialization in wound healing: A comprehensive review. Adv Wound Care (New Rochelle) (2014) 3(7):445–64. doi: 10.1089/wound.2013.0473
103. Yang S, Sun Y, Geng Z, Ma K, Sun X, Fu X. AbnorMalities in the basement membrane structure promote basal keratinocytes in the epidermis of hypertrophic scars to adopt a proliferative phenotype. Int J Mol Med (2016) 37(5):1263–73. doi: 10.3892/ijmm.2016.2519
104. Li B, Gao C, Diao JS, Wang DL, Chu FF, Li Y, et al. Aberrant Notch signalling contributes to hypertrophic scar formation by modulating the phenotype of keratinocytes. Exp Dermatol (2016) 25(2):137–42. doi: 10.1111/exd.12897
105. Nyika DT, Khumalo NP, Bayat A. Genetics and epigenetics of keloids. Adv Wound Care (New Rochelle) (2022) 11(4):192–201. doi: 10.1089/wound.2021.0094
106. Stevenson AW, Melton PE, Moses EK, Wallace HJ, Wood FM, Rea S, et al. A methylome and transcriptome analysis of normal human scar cells reveals a role for FOXF2 in scar maintenance. J Invest Dermatol (2022) 142(5):1489–98.e12. doi: 10.1016/j.jid.2021.08.445
107. Wang R, Bai Z, Wen X, Du H, Zhou L, Tang Z, et al. MiR-152-3p regulates cell proliferation, invasion and extracellular matrix expression through by targeting FOXF1 in keloid fibroblasts. Life Sci (2019) 234:116779. doi: 10.1016/j.lfs.2019.116779
108. Zhang J, Xu D, Li N, Li Y, He Y, Hu X, et al. Downregulation of microRNA-31 inhibits proliferation and induces apoptosis by targeting HIF1AN in human keloid. Oncotarget (2017) 8(43):74623–34. doi: 10.18632/oncotarget.20284
109. Rang Z, Wang ZY, Pang QY, Wang YW, Yang G, Cui F. MiR-181a targets PHLPP2 to augment AKT signaling and regulate proliferation and apoptosis in human keloid fibroblasts. Cell Physiol Biochem (2016) 40(3-4):796–806. doi: 10.1159/000453139
110. Li Y, Zhang J, Lei Y, Lyu L, Zuo R, Chen T. MicroRNA-21 in skin fibrosis: potential for diagnosis and treatment. Mol Diagn Ther (2017) 21(6):633–42. doi: 10.1007/s40291-017-0294-8
111. Wang Z, Feng C, Song K, Qi Z, Huang W, Wang Y. lncRNA-H19/miR-29a axis affected the viability and apoptosis of keloid fibroblasts through acting upon COL1A1 signaling. J Cell Biochem (2020) 121(11):4364–76. doi: 10.1002/jcb.29649
112. Zhang Z, Yu K, Liu O, Xiong Y, Yang X, Wang S, et al. Expression profile and bioinformatics analyses of circular RNAs in keloid and normal dermal fibroblasts. Exp Cell Res (2020) 388(1):111799. doi: 10.1016/j.yexcr.2019.111799
113. An G, Liang S, Sheng C, Liu Y, Yao W. Upregulation of microRNA-205 suppresses vascular endothelial growth factor expression-mediated PI3K/Akt signaling transduction in human keloid fibroblasts. Exp Biol Med (Maywood) (2017) 242(3):275–85. doi: 10.1177/1535370216669839
114. Zhao B, Guan H, Liu JQ, Zheng Z, Zhou Q, Zhang J, et al. Hypoxia drives the transition of human dermal fibroblasts to a myofibroblast-like phenotype via the TGF-β1/Smad3 pathway. Int J Mol Med (2017) 39(1):153–9. doi: 10.3892/ijmm.2016.2816
115. De Felice B, Garbi C, Santoriello M, Santillo A, Wilson RR. Differential apoptosis markers in human keloids and hypertrophic scars fibroblasts. Mol Cell Biochem (2009) 327(1-2):191–201. doi: 10.1007/s11010-009-0057-x
116. Lee YJ, Kwon SB, Kim CH, Cho HD, Nam HS, Lee SH, et al. Oxidative damage and nuclear factor erythroid 2-related factor 2 protein expression in normal skin and keloid tissue. Ann Dermatol (2015) 27(5):507–16. doi: 10.5021/ad.2015.27.5.507
117. Harn HI, Ogawa R, Hsu CK, Hughes MW, Tang MJ, Chuong CM. The tension biology of wound healing. Exp Dermatol (2019) 28(4):464–71. doi: 10.1111/exd.13460
118. He J, Fang B, Shan S, Xie Y, Wang C, Zhang Y, et al. Mechanical stretch promotes hypertrophic scar formation through mechanically activated cation channel Piezo1. Cell Death Dis (2021) 12(3):226. doi: 10.1038/s41419-021-03481-6
119. Peng S, Poole DP, Veldhuis NA. Mini-review: Dissecting receptor-mediated stimulation of TRPV4 in nociceptive and inflammatory pathways. Neurosci Lett (2022) 770:136377. doi: 10.1016/j.neulet.2021.136377
120. Wang Z, Huang X, Zan T, Li Q, Li H. A modified scar model with controlled tension on secondary wound healing in mice. Burns Trauma (2020) 8:tkaa013. doi: 10.1093/burnst/tkaa013
121. Kloeters O, Schierle C, Tandara A, Mustoe TA. The use of a semiocclusive dressing reduces epidermal inflammatory cytokine expression and mitigates dermal proliferation and inflammation in a rat incisional model. Wound Repair Regener (2008) 16(4):568–75. doi: 10.1111/j.1524-475X.2008.00404.x
122. Zhou Y, Hua T, Weng X, Ma D, Li X. Calcitonin gene-related peptide alleviates hypertrophic scar formation by inhibiting the inflammation. Arch Dermatol Res (2022) 314(1):53–60. doi: 10.1007/s00403-020-02179-7
123. Chiang RS, Borovikova AA, King K, Banyard DA, Lalezari S, Toranto JD, et al. Current concepts related to hypertrophic scarring in burn injuries. Wound Repair Regener (2016) 24(3):466–77. doi: 10.1111/wrr.12432
124. Dong J, Feng F, Xu G, Zhang H, Hong L, Yang J. Elevated SP/NK-1R in esophageal carcinoma promotes esophageal carcinoma cell proliferation and migration. Gene (2015) 560(2):205–10. doi: 10.1016/j.gene.2015.02.002
125. Leal EC, Carvalho E, Tellechea A, Kafanas A, Tecilazich F, Kearney C, et al. Substance P promotes wound healing in diabetes by modulating inflammation and macrophage phenotype. Am J Pathol (2015) 185(6):1638–48. doi: 10.1016/j.ajpath.2015.02.011
126. Meng J, Li Y, Fischer MJM, Steinhoff M, Chen W, Wang J. Th2 modulation of transient receptor potential channels: an unmet therapeutic intervention for atopic dermatitis. Front Immunol (2021) 12:696784. doi: 10.3389/fimmu.2021.696784
127. Tang L, Gao J, Cao X, Chen L, Wang H, Ding H. TRPV1 mediates itch-associated scratching and skin barrier dysfunction in DNFB-induced atopic dermatitis mice. Exp Dermatol (2022) 31(3):398–405. doi: 10.1111/exd.14464
128. Lee MY, Shin E, Kim H, Kwak IS, Choi Y. Interleukin-31, interleukin-31RA, and OSMR expression levels in post-burn hypertrophic scars. J Pathol Transl Med (2018) 52(5):307–13. doi: 10.4132/jptm.2018.08.03
129. Yin SL, Qin ZL, Yang X. Role of periostin in skin wound healing and pathologic scar formation. Chin Med J (Engl) (2020) 133(18):2236–8. doi: 10.1097/cm9.0000000000000949
130. Hashimoto T, Mishra SK, Olivry T, Yosipovitch G. Periostin, an emerging player in itch sensation. J Invest Dermatol (2021) 141(10):2338–43. doi: 10.1016/j.jid.2021.03.009
131. Hashimoto T, Nattkemper LA, Kim HS, Kursewicz CD, Fowler E, Shah SM, et al. Dermal periostin: A new player in itch of prurigo nodularis. Acta Derm Venereol (2021) 101(1):adv00375. doi: 10.2340/00015555-3702
132. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol (2002) 3(5):349–63. doi: 10.1038/nrm809
133. Yang L, Chan T, Demare J, Iwashina T, Ghahary A, Scott PG, et al. Healing of burn wounds in transgenic mice overexpressing transforming growth factor-beta 1 in the epidermis. Am J Pathol (2001) 159(6):2147–57. doi: 10.1016/s0002-9440(10)63066-0
134. Ishii T, Uchida K, Hata S, Hatta M, Kita T, Miyake Y, et al. TRPV2 channel inhibitors attenuate fibroblast differentiation and contraction mediated by keratinocyte-derived TGF-β1 in an in vitro wound healing model of rats. J Dermatol Sci (2018) 90(3):332–42. doi: 10.1016/j.jdermsci.2018.03.003
135. Cobbold CA. The role of nitric oxide in the formation of keloid and hypertrophic lesions. Med Hypotheses (2001) 57(4):497–502. doi: 10.1054/mehy.2001.1373
136. Miyamoto T, Petrus MJ, Dubin AE, Patapoutian A. TRPV3 regulates nitric oxide synthase-independent nitric oxide synthesis in the skin. Nat Commun (2011) 2:369. doi: 10.1038/ncomms1371
137. Aijima R, Wang B, Takao T, Mihara H, Kashio M, Ohsaki Y, et al. The thermosensitive TRPV3 channel contributes to rapid wound healing in oral epithelia. FASEB J (2015) 29(1):182–92. doi: 10.1096/fj.14-251314
138. Kim HO, Jin Cheol K, Yu Gyeong K, In Suk K. Itching caused by TRPV3 (Transient receptor potential vanilloid-3) activator application to skin of burn patients. Medicina (Kaunas) (2020) 56(11):560. doi: 10.3390/medicina56110560
139. Park CW, Kim HJ, Choi YW, Chung BY, Woo SY, Song DK, et al. TRPV3 channel in keratinocytes in scars with post-burn pruritus. Int J Mol Sci (2017) 18(11):2425. doi: 10.3390/ijms18112425
140. Ikoma A, Steinhoff M, Ständer S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat Rev Neurosci (2006) 7(7):535–47. doi: 10.1038/nrn1950
141. Yang YS, Cho SI, Choi MG, Choi YH, Kwak IS, Park CW, et al. Increased expression of three types of transient receptor potential channels (TRPA1, TRPV4 and TRPV3) in burn scars with post-burn pruritus. Acta Derm Venereol (2015) 95(1):20–4. doi: 10.2340/00015555-1858
142. Lee WJ, Shim WS. Cutaneous neuroimmune interactions of TSLP and TRPV4 play pivotal roles in dry skin-induced pruritus. Front Immunol (2021) 12:772941. doi: 10.3389/fimmu.2021.772941
143. Kawai K, Ishise H, Kubo T, Larson B, Fujiwara T, Nishimoto S, et al. Stretching promotes wound contraction through enhanced expression of endothelin receptor B and TRPC3 in fibroblasts. Plast Reconstr Surg Glob Open (2023) 11(4):e4954. doi: 10.1097/gox.0000000000004954
144. Memariani H, Memariani M, Moravvej H, Shahidi-Dadras M. Emerging and Novel Therapies for Keloids: A compendious review. Sultan Qaboos Univ Med J (2021) 21(1):e22–33. doi: 10.18295/squmj.2021.21.01.004
145. Wei JJ, Kim HS, Spencer CA, Brennan-Crispi D, Zheng Y, Johnson NM, et al. Activation of TRPA1 nociceptor promotes systemic adult mamMalian skin regeneration. Sci Immunol (2020) 5(50):eaba5683. doi: 10.1126/sciimmunol.aba5683
146. Li S, Hu M, Lorenz HP. Treatment of full-thickness skin wounds with blood-derived CD34(+) precursor cells enhances healing with hair follicle regeneration. Adv Wound Care (New Rochelle) (2020) 9(5):264–76. doi: 10.1089/wound.2019.0974
147. Shin JU, Kim SH, Kim H, Noh JY, Jin S, Park CO, et al. TSLP is a potential initiator of collagen synthesis and an activator of CXCR4/SDF-1 axis in keloid pathogenesis. J Invest Dermatol (2016) 136(2):507–15. doi: 10.1016/j.jid.2015.11.008
148. Nilius B, Voets T. TRP channels: a TR(I)P through a world of multifunctional cation channels. Pflugers Arch (2005) 451(1):1–10. doi: 10.1007/s00424-005-1462-y
149. Li F, Wang F. TRPV1 in pain and itch. Adv Exp Med Biol (2021) 1349:249–73. doi: 10.1007/978-981-16-4254-8_12
150. Dolivo D, Rodrigues A, Sun L, Hou C, Li Y, Chung E, et al. Simvastatin cream alleviates dermal fibrosis in a rabbit ear hypertrophic scar model. J Cosmet Dermatol (2023) 22(2):534–41. doi: 10.1111/jocd.15142
151. Denda M, Sokabe T, Fukumi-Tominaga T, Tominaga M. Effects of skin surface temperature on epidermal permeability barrier homeostasis. J Invest Dermatol (2007) 127(3):654–9. doi: 10.1038/sj.jid.5700590
152. Sumioka T, Okada Y, Reinach PS, Shirai K, Miyajima M, Yamanaka O, et al. Impairment of corneal epithelial wound healing in a TRPV1-deficient mouse. Invest Ophthalmol Vis Sci (2014) 55(5):3295–302. doi: 10.1167/iovs.13-13077
153. Yang H, Wang Z, Capo-Aponte JE, Zhang F, Pan Z, Reinach PS. Epidermal growth factor receptor transactivation by the cannabinoid receptor (CB1) and transient receptor potential vanilloid 1 (TRPV1) induces differential responses in corneal epithelial cells. Exp Eye Res (2010) 91(3):462–71. doi: 10.1016/j.exer.2010.06.022
154. Okada Y, Reinach PS, Shirai K, Kitano A, Kao WW, Flanders KC, et al. TRPV1 involvement in inflammatory tissue fibrosis in mice. Am J Pathol (2011) 178(6):2654–64. doi: 10.1016/j.ajpath.2011.02.043
155. Pinho-Ribeiro FA, Baddal B, Haarsma R, O'Seaghdha M, Yang NJ, Blake KJ, et al. Blocking neuronal signaling to immune cells treats streptococcal invasive infection. Cell (2018) 173(5):1083–97.e22. doi: 10.1016/j.cell.2018.04.006
156. Spekker E, Körtési T, Vécsei L. TRP channels: recent development in translational research and potential therapeutic targets in migraine. Int J Mol Sci (2022) 24(1):700. doi: 10.3390/ijms24010700
157. Zhang S, Li K, Yu Z, Chai J, Zhang Z, Zhang Y, et al. Dramatic effect of botulinum toxin type A on hypertrophic scar: A promising therapeutic drug and its mechanism through the SP-NK1R pathway in cutaneous neurogenic inflammation. Front Med (Lausanne) (2022) 9:820817. doi: 10.3389/fmed.2022.820817
158. Li WB, Liu S, Zhang MZ, Liu H, Dong XH, Hao Y, et al. Hyperbaric oxygen therapy relieved pruritus and pain of keloid patients. Am J Transl Res (2020) 12(2):574–82.
159. Moore CL. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun (1971) 42(2):298–305. doi: 10.1016/0006-291x(71)90102-1
160. Cals-Grierson MM, Ormerod AD. Nitric oxide function in the skin. Nitric Oxide (2004) 10(4):179–93. doi: 10.1016/j.niox.2004.04.005
161. Repertinger SK, Campagnaro E, Fuhrman J, El-Abaseri T, Yuspa SH, Hansen LA. EGFR enhances early healing after cutaneous incisional wounding. J Invest Dermatol (2004) 123(5):982–9. doi: 10.1111/j.0022-202X.2004.23478.x
162. Yamanoi Y, Lei J, Takayama Y, Hosogi S, Marunaka Y, Tominaga M. TRPV3-ANO1 interaction positively regulates wound healing in keratinocytes. Commun Biol (2023) 6(1):88. doi: 10.1038/s42003-023-04482-1
163. Shu DY, Lovicu FJ. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog Retin Eye Res (2017) 60:44–65. doi: 10.1016/j.preteyeres.2017.08.001
164. El Ayadi A, Jay JW, Prasai A. Current approaches targeting the wound healing phases to attenuate fibrosis and scarring. Int J Mol Sci (2020) 21(3):1105. doi: 10.3390/ijms21031105
165. Sharma S, Goswami R, Zhang DX, Rahaman SO. TRPV4 regulates matrix stiffness and TGFβ1-induced epithelial-mesenchymal transition. J Cell Mol Med (2019) 23(2):761–74. doi: 10.1111/jcmm.13972
166. Okada Y, Shirai K, Miyajima M, Reinach PS, Yamanaka O, Sumioka T, et al. Loss of TRPV4 function suppresses inflammatory fibrosis induced by alkali-burning mouse corneas. PloS One (2016) 11(12):e0167200. doi: 10.1371/journal.pone.0167200
167. Yeung V, Boychev N, Farhat W, Ntentakis DP, Hutcheon AEK, Ross AE, et al. Extracellular vesicles in corneal fibrosis/scarring. Int J Mol Sci (2022) 23(11):5921. doi: 10.3390/ijms23115921
168. Han F, Wang K, Shen K, Wang J, Han S, Hu D, et al. Extracellular vesicles from Lactobacillus druckerii inhibit hypertrophic scar fibrosis. J Nanobiotechnol (2023) 21(1):113. doi: 10.1186/s12951-023-01861-y
169. An Y, Lin S, Tan X, Zhu S, Nie F, Zhen Y, et al. Exosomes from adipose-derived stem cells and application to skin wound healing. Cell Prolif (2021) 54(3):e12993. doi: 10.1111/cpr.12993
170. Hade MD, Suire CN, Mossell J, Suo Z. Extracellular vesicles: Emerging frontiers in wound healing. Med Res Rev (2022) 42(6):2102–25. doi: 10.1002/med.21918
171. Guarino B, Katari V, Adapala R, Bhavnani N, Dougherty J, Khan M, et al. Tumor-derived extracellular vesicles induce abnormal angiogenesis via TRPV4 downregulation and subsequent activation of YAP and VEGFR2. Front Bioeng Biotechnol (2021) 9:790489. doi: 10.3389/fbioe.2021.790489
172. Goswami R, Arya RK, Sharma S, Dutta B, Stamov DR, Zhu X, et al. Mechanosensing by TRPV4 mediates stiffness-induced foreign body response and giant cell formation. Sci Signal (2021) 14(707):eabd4077. doi: 10.1126/scisignal.abd4077
173. Lapajne L, Lakk M, Yarishkin O, Gubeljak L, Hawlina M, Krizaj D. Polymodal sensory transduction in mouse corneal epithelial cells. Invest Ophthalmol Vis Sci (2020) 61(4):2. doi: 10.1167/iovs.61.4.2
174. Zhan L, Yang Y, Ma TT, Huang C, Meng XM, Zhang L, et al. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol Cell Biochem (2015) 402(1-2):9–22. doi: 10.1007/s11010-014-2298-6
175. Zhou Z, Shu B, Xu Y, Liu J, Wang P, Chen L, et al. microRNA-203 modulates wound healing and scar formation via suppressing hes1 expression in epidermal stem cells. Cell Physiol Biochem (2018) 49(6):2333–47. doi: 10.1159/000493834
176. Yuan W, Sun H, Yu L. Long non-coding RNA LINC01116 accelerates the progression of keloid formation by regulating miR-203/SMAD5 axis. Burns (2021) 47(3):665–75. doi: 10.1016/j.burns.2020.07.027
177. Takada H, Furuya K, Sokabe M. Mechanosensitive ATP release from hemichannels and Ca2+ influx through TRPC6 accelerate wound closure in keratinocytes. J Cell Sci (2014) 127(Pt 19):4159–71. doi: 10.1242/jcs.147314
178. Takada H, Yonekawa J, Matsumoto M, Furuya K, Sokabe M. Hyperforin/HP-β-cyclodextrin enhances mechanosensitive ca(2+) signaling in haCaT keratinocytes and in atopic skin ex vivo which accelerates wound healing. BioMed Res Int (2017) 2017:8701801. doi: 10.1155/2017/8701801
179. Zhi Y, Wang H, Huang B, Yan G, Yan LZ, Zhang W, et al. Panax Notoginseng Saponins suppresses TRPM7 via the PI3K/AKT pathway to inhibit hypertrophic scar formation in vitro. Burns (2021) 47(4):894–905. doi: 10.1016/j.burns.2020.10.003
180. Zhou DM, Sun LL, Zhu J, Chen B, Li XQ, Li WD. MiR-9 promotes angiogenesis of endothelial progenitor cell to facilitate thrombi recanalization via targeting TRPM7 through PI3K/Akt/autophagy pathway. J Cell Mol Med (2020) 24(8):4624–32. doi: 10.1111/jcmm.15124
181. Chai CY, Tai IC, Zhou R, Song J, Zhang C, Sun S. MicroRNA-9-5p inhibits proliferation and induces apoptosis of human hypertrophic scar fibroblasts through targeting peroxisome proliferator-activated receptor β. Biol Open (2020) 9(12):bio051904. doi: 10.1242/bio.051904
182. Dong RF, Zhuang YJ, Wang Y, Zhang ZY, Xu XZ, Mao YR, et al. Tumor suppressor miR-192-5p targets TRPM7 and inhibits proliferation and invasion in cervical cancer. Kaohsiung J Med Sci (2021) 37(8):699–708. doi: 10.1002/kjm2.12398
183. Li Y, Zhang J, Shi J, Liu K, Wang X, Jia Y, et al. Exosomes derived from human adipose mesenchymal stem cells attenuate hypertrophic scar fibrosis by miR-192-5p/IL-17RA/Smad axis. Stem Cell Res Ther (2021) 12(1):221. doi: 10.1186/s13287-021-02290-0
184. Peng F, Gong W, Li S, Yin B, Zhao C, Liu W, et al. circRNA_010383 acts as a sponge for miR-135a, and its downregulated expression contributes to renal fibrosis in diabetic nephropathy. Diabetes (2021) 70(2):603–15. doi: 10.2337/db20-0203
185. Wang Y, Chen L, Wang K, Da Y, Zhou M, Yan H, et al. Suppression of TRPM2 reduces renal fibrosis and inflammation through blocking TGF-beta1-regulated JNK activation. BioMed Pharmacother (2019) 120:109556. doi: 10.1016/j.biopha.2019.109556
186. Spix B, Chao YK, Abrahamian C, Chen CC, Grimm C. TRPML cation channels in inflammation and immunity. Front Immunol (2020) 11:225. doi: 10.3389/fimmu.2020.00225
187. Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol (2005) 7(12):1202–12. doi: 10.1038/ncb1326
188. Guenin-Mace L, Konieczny P, Naik S. Immune-epithelial cross talk in regeneration and repair. Annu Rev Immunol (2023) 41:207–28. doi: 10.1146/annurev-immunol-101721-062818
Keywords: transient receptor potential channels, pathological scarification, inflammatory, fibrosis, re-epithelialization
Citation: Zheng Y, Huang Q, Zhang Y, Geng L, Wang W, Zhang H, He X and Li Q (2023) Multimodal roles of transient receptor potential channel activation in inducing pathological tissue scarification. Front. Immunol. 14:1237992. doi: 10.3389/fimmu.2023.1237992
Received: 10 June 2023; Accepted: 15 August 2023;
Published: 29 August 2023.
Edited by:
Wang Xiqiao, Shanghai Jiao Tong University, ChinaReviewed by:
Peter S. Reinach, Wenzhou Medical University, ChinaYunsheng Chen, Shanghai Jiao Tong University, China
Copyright © 2023 Zheng, Huang, Zhang, Geng, Wang, Zhang, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang He, aGVyb3hpYW5nQGhvdG1haWwuY29t; Qiannan Li, bGlxaWFubmFueWVzQDE2My5jb20=