 Romane Thouenon1,2
Romane Thouenon1,2 Sven Kracker1,2*†
Sven Kracker1,2*†- 1Université Paris Cité, Paris, France
- 2Laboratory of Human Lymphohematopoiesis, Imagine Institute, INSERM UMR, Paris, France
The transcription factor interferon regulatory factor 4 (IRF4) belongs to the IRF family and has several important functions for the adaptive immune response. Mutations affecting IRF family members IRF1, IRF3, IRF7, IRF8, or IRF9 have been described in patients presenting with inborn errors of immunity (IEI) highlighting the importance of these factors for the cellular host defense against mycobacterial and/or viral infections. IRF4 deficiency and haploinsufficiency have been associated with IEI. More recently, two novel IRF4 disease-causing mechanisms have been described due to the characterization of IEI patients presenting with cellular immunodeficiency associated with agammaglobulinemia. Here, we review the phenotypes and physiopathological mechanisms underlying IEI of IRF family members and, in particular, IRF4.
Introduction
The interferon regulatory factor (IRF) consists of a family of nine human transcription factors (1, 2). IRF1 was the first protein of this family identified (3) and was shown to induce the two type I interferons (IFNs) IFNα and IFNβ (4, 5). It was shown that IRF1 regulates a variety of target genes. Although both type I and type II IFNs induce IRF1 expression, IRF1-dependent responses to IFN-γ appear to be much stronger than those to type I IFNs. Subsequently, other proteins were associated with this family because of their structural and functional (as transcription factors) similarities (Supplementary Figure 1) (1, 6–9). The unique function of a particular IRF is related to a combination of cell-type-specific expression and interactions with other members of the IRF family and/or other transcription factors (8). They are expressed in different tissues with IRF1, IRF2, IRF3, and IRF6 present in almost every tissue; IRF4 and IRF8 mainly in hematopoietic cells; IRF5 in bone marrow, spleen, and lymph nodes; IRF7 in liver and spleen; and finally IRF9 in female genitals and in the respiratory tract. On the cellular level, specifically in hematopoietic cells, IRF1 and IRF2 are mainly expressed in neutrophils and monocytes. IRF3 expression is detected in all hematopoietic lineages with high expression in B and T lymphocytes. IRF4 is expressed in B and T lymphocytes as well as in dendritic cells (DCs). IRF5 is expressed in monocytes, B cells, and DCs. IRF6 expression is low in immune cells. IRF7 and IRF8 are expressed in DCs, and IRF8 expression is also detected in B cells and monocytes. IRF9 is expressed in all hematopoietic lineages with high expression in granulocytes (the human protein atlas https://www.proteinatlas.org/). All IRFs are regulated by IFNs, except IRF4 (10), whose expression is controlled by antigen receptors, Toll-like receptors, and CD40 signaling pathways (11, 12). IRF proteins contain two functional domains: an N-terminal helix-turn-helix DNA-binding domain (DBD) containing conserved tryptophan residues, and a C-terminal interferon activation domain (IAD, also known as the IRF-association domain) known to mediate protein–protein interactions (13–15). Phylogenetic analysis of IRF DBD indicated that human IRF can be divided into four subfamilies: the IRF1 subfamily (including IRF1 and IRF2), the IRF3 subfamily (including IRF3 and IRF7), the IRF4 subfamily (including IRF4, IRF8, and IRF9) and the IRF5 subfamily (including IRF5 and IRF6) (15). IRF4 contains a C-terminal flexible autoinhibitory region that binds directly to the DBD to modulate its interaction with DNA, in contrast to the other IRF4 subfamily members (Supplementary Figure 1) (13). IRF family members are known to be important for immune cell development and functions. All IRF proteins stimulate the expression of IFN-inducible genes (16–18), except IRF2 that antagonizes IRF1, hence attenuating type I IFN response (6, 19). IRF1, IRF3, IRF5, and IRF7 also enhance TLR-dependent gene induction (20–24) and IRF8 stimulates IFN-γ- and PAMP-inducible genes (25, 26). IRF4 regulation differs from that of the other members of the IRF family and the IRF4 gene has been described as a sensor of B-cell receptor or T-cell receptor signaling (27, 28). In response to BCR/TCR signaling, the IRF4 gene is expressed through the NFκB signaling pathway. Subsequently, signaling-induced changes in the IRF4 protein level control the fate of activated B and T lymphocytes (27–29).
IRF family member variants associated with cancer
The IRF protein family members were also found in the literature to be associated with different kinds of cancer. For example, IRF1, IRF2, IRF5, IRF8, and IRF9 were found to be upregulated in glioma, and their mRNA levels correlated to the tumor grade and the clinical outcome of the disease (30). Moreover, IRF1 was identified to be deleted in patients presenting with leukemia, myelodysplastic syndromes for hematopoietic malignancies (31), and esophageal and gastric cancers (32, 33). The pathophysiological role of IRF2 in cancer was mainly associated with impaired immune surveillance associated with IFN-mediated CD8 T-cell exhaustion (34). Furthermore, in colorectal cancer, loss of IRF2 expression mediated by oncogenic KRAS promotes the migration of myeloid-derived suppressor cells to the tumor microenvironment driving resistance to anti-PD-1 therapy (35). IRF8 has also been associated with the induction of T-cell exhaustion in cancer microenvironment (36, 37). IRF3 expression was linked to a better survival outcome mainly for gastrointestinal cancers (38, 39). IRF4 chromosomal translocation/mutation was mainly associated with hematopoietic cancers and found in particular in multiple myeloma (40), large B-cell lymphoma (41), and adult T-cell lymphoma-leukemia (42). The catalog of somatic mutation in cancer (COSMIC v97) contains 146 somatic missense variants identified in hematopoietic cancers affecting the whole IRF4 gene (Figure 1A). Of note, the two most frequent IRF4 mutations annotated in COSMIC v97 p.K59R (c.176A>G) and pL70V (c.208C>G) were located in IRF4’s DBD (42).
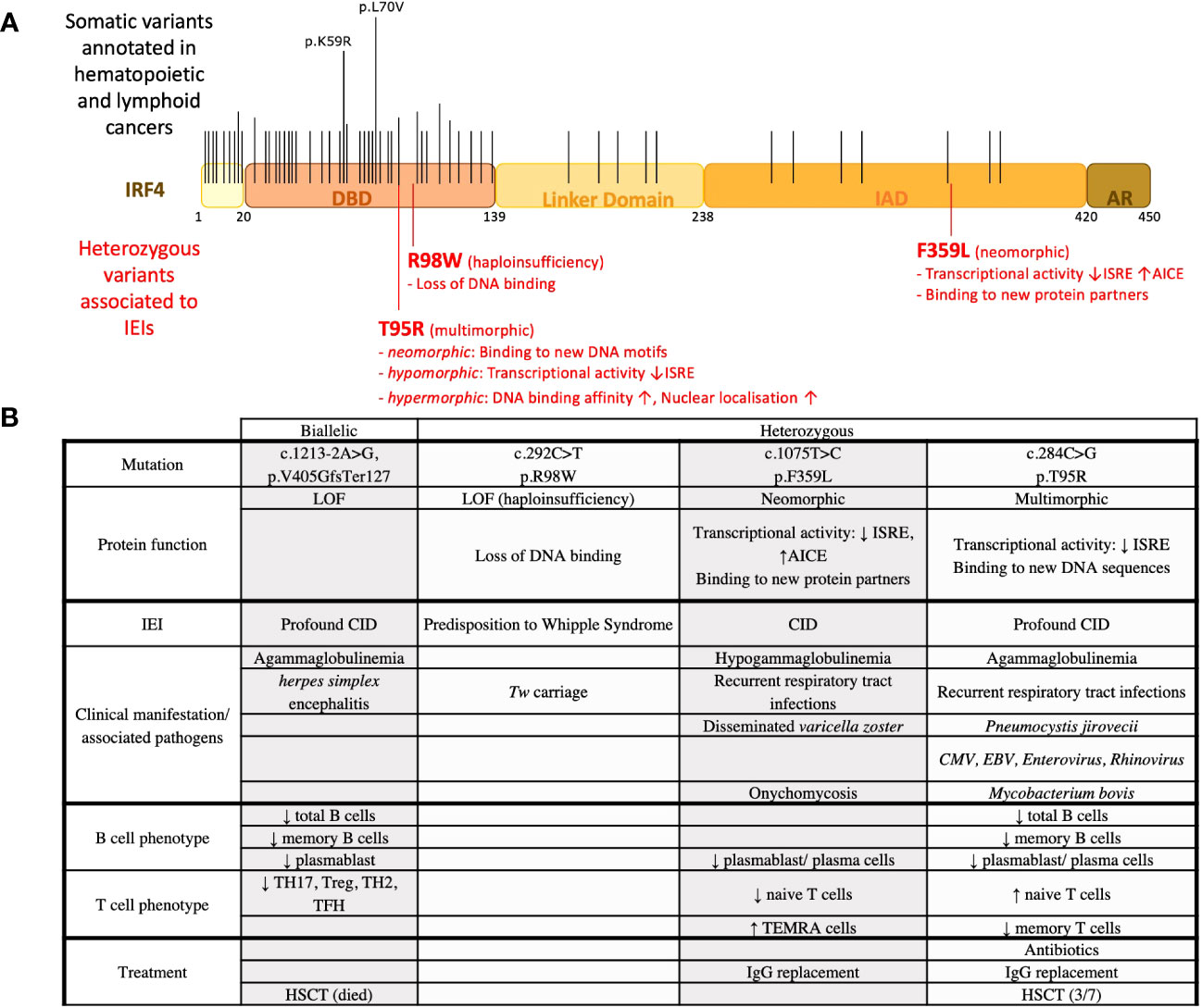
Figure 1 IRF4 variants associated with IEIs and observed in hematopoietic cancers. (A) IRF4 variants found in IEIs and hematopoietic cancers. Black lines indicate the location of recurrent somatic mutations noted in COSMICv97. Long lines indicate mutations that were found more frequently. Red lines indicate the location of heterozygous variants found in patients with IEI. (B) IRF4 variants associated with IEI. Clinical and immunological phenotypes of patients are recapitulated.
IRF family member variants associated with IEI
IEI affecting IRF family members present with defective control of viral infection and/or mycobacterial infections (Table 1). IRF1 autosomal recessive (AR) loss of function (LOF) variants have been identified in two related patients presenting with Mendelian susceptibility to mycobacterial diseases (MSMD). Functional characterization showed, in addition to a defect of NK, naive αβ CD8+ T and myeloid DC differentiation, the absence of IRF1 impacting IFN type II (IFNγ) production, leading to the development of MSMD (43). IRF3 autosomal dominant (AD) LOF variants have been described for a patient presenting with HSV-1 encephalitis and for another patient presenting with recurrent cutaneous herpes. This has been characterized as an impairment of the type I IFN expression after HSV-1 infection in these patients (44–47). AR LOF IRF7 variants were also found in patients presenting with severe infection by influenza virus and SARS-CoV-2-induced life-threatening induced pneumonia. The impairment of IFN type I and type III expression was observed in plasmacytoid DCs (pDCs) deficient in IRF7 and has been associated with poor clinical outcome (47, 52–54). Then, AD and AR LOF variants found in the IRF8 gene were also found in patients presenting with MSMD (55, 56, 60). AD IRF8-deficiency caused impaired development of conventional DC and Th1 cells, whereas AR IRF8 deficiency was associated with an absence of circulating monocytes and DCs, and with reduced NK cell numbers and function in some patients (55, 56, 60). IRF9 AR LOF variants were found in patients presenting with severe influenza virus infection and might result in susceptibility to infection by a wide range of viruses. Functionally, it has been associated with compromised type I and III IFN signaling, which then leads to defective control of viral infections (57–59). Finally, variants found in the IRF4 gene have also been associated with IEI, and this aspect will be developed in the following section.

Table 1 Variants affecting IRF family members found in IEI patients.
IRF4 variants associated with IEI
IRF4 complete deficiency due to uniparental isodisomy
The first IRF4 LOF variant associated with IEI reported in the literature was a case report of an IRF4 deficiency inherited by uniparental isodisomy found in a patient with profound combined immunodeficiency (50). The patient died at 2 years of age shortly after an allogeneic hematopoietic stem cell transplantation. Immunological evaluation revealed an agammaglobulinemia with an absence of memory B cells and plasma cells (in blood, bone marrow, and lymph nodes). Only a small percentage of regulatory CD4 T cells was observed and CD8 effector memory T cells were undetectable. Histological analysis of lymph node biopsies showed a loss of normal architecture characterized by a lack of follicles and the presence of necrotic areas. An increased number of pDCs was noted whereas an inadequate differentiation of monocytes towards monocyte-derived DCs was observed in vitro (based on the lack of CD1a expression). Genetic analysis identified a mutation, c.1213-2A>G, p.V405GfsTer127, in the IRF4 gene located on chromosome 6 found to be homozygous in the patient and heterozygous in the mother (Figure 1B). Results of a single-nucleotide polymorphism array suggested a maternal uniparental isodisomy of chromosome 6 (50). Because of the very complex genetics of this unique case of IRF4 deficiency, the possible involvement of other genetic factors cannot be excluded.
IRF4 haploinsufficiency associated with Whipple’s disease with incomplete penetrance
Whipple’s disease (WD) is an intestinal inflammatory disease due to infection by the bacteria Tropheryma whipplei (Tw). The vast majority of infected individuals clear Tw primary infections; however, a minority become chronic asymptomatic carriers, of whom a very small proportion develop WD (61, 62). IRF4 haploinsufficiency associated with WD has been reported due to the study of four related patients diagnosed with WD with a mean age of diagnosis of 58 years (51). The four patients from a large multiplex family were found carriers of a heterozygous missense IRF4 variant (c.292 C>T, p.R98W). Eight other relatives were heterozygous for this variant. Interestingly, five of them were reported to be Tw carriers with a mean age of 55 years. The familial segregation of the IRF4R98W allele was therefore consistent with an autosomal dominant pattern of WD inheritance with an age-dependent incomplete penetrance. Of note, chronic Tw carriage also followed an autosomal dominant pattern. Immunological abnormalities were not reported. However, it is unclear to which extent in-depth immunophenotyping was performed for symptomatic and asymptomatic IRF4R98W allele carriers. Functional characterization of the IRF4R98W variant showed that this IRF4 variant has lost its ability to bind to known IRF4 DNA motifs like interferon-stimulated response elements (ISREs). In addition, a higher level of IRF4 protein in the cytoplasm was observed in cells from IRF4 R98W allele carriers than in wild-type homozygotes. Thus, as a mechanism, haploinsufficiency not due to loss of expression but due to lack of activity of the IRF4R98W protein variant in the nucleus has been proposed (51) (Figures 1A, B). Whether IRF4 has a function in the cytoplasm and whether IRF4R98W protein interferes with it remain to be elucidated.
IRF4 autosomal dominant variant with neomorphic functions
A heterozygous IRF4 variant with neomorphic functions has been discovered due to the study of three patients from a multigenerational non-consanguineous family. All three patients suffered from recurrent respiratory tract infections associated with hypogammaglobulinemia and reduced circulating plasmablasts/plasma cells. The patients presented with early (teenage-onset) hair graying and skin depigmentation. This is possibly associated with the reported IRF4 expression in melanocytes and its role in the MITF/Tyr signaling pathway (63). Of note, IRF4 single-nucleotide polymorphisms (SNPs) have been associated with a light skin and hair color (49). For the T-cell compartment, they presented with low naïve T-cell counts and high effector memory counts for both CD4 and CD8 T cells. Genetic analysis of these patients revealed a germline heterozygous missense IRF4 gene variant (c.1075 T>C, p.F359L) affecting the IRF4 protein’s IAD (64). IRF4 is known to bind to erythroblast-transformation-specific (ETS) interferon composite elements (EICEs) as an PU-1/IRF4 heterodimer and to AP-1-IRF composite elements (AICEs) as an AP-1 (composed of BATF and JUN)/IRF4 heterodimer (27, 65, 66). At high nuclear protein level, IRF4 is known to bind to ISRE, allowing the formation of the IRF4 homodimer (14) and the expression of pro-plasmablast genes as XBP1 and PRDM1 (67). A combination of different functional analysis including luciferase reporter assays, chromatin immunoprecipitation sequencing, and transcriptomic analysis indicated that the IRF4F359L variant deregulated IRF4 transcriptional activity on AICE and ISRE sites. On AICE sites, an increased transcriptional activity mediated by the IRF4F359L variant was observed. In contrast, the IRF4F359L variant supressed transcription on ISRE sites. Proteomic analysis discovered the transcriptional repressor ETV6 as a novel protein binding partner for IRF4F359L. Notably, ETV6 has already been shown to be a negative regulator of IRF8 transcriptional activity on ISRE sequences (68). As the transcriptional activity mediated by IRF4F359L on ISRE sites in ETV6-knockdown cells was comparable to the one of IRF4 wild-type expressing cells, it is reasonable to suggest that, on ISRE sites, the presence of IRF4F359L allows the recruitment of ETV6, resulting in the suppression of IRF4-triggered transcriptional activity. One other protein interacting with IRF4F359L is JUN-B. The family of JUN proteins is composed of c-JUN, JUN-B, and JUN-D, and they differ in their binding affinity to AP-1 (69). For T-cell differentiation, JUN-B has been mainly associated with the generation of regulatory T cell and Th17 cells (70–72). Thus, the high terminal effector counts for CD4 and CD8 T cells could be associated with an increased activity of IRF4F359L (possibly in conjunction with JUN-B) on AICE sequence. Of note, IRF4F359L appears to function in heterochromatin organization as evidenced by diffuse nuclear H3K9 trimethylation immunofluorescent signals in patients’ derived B-EBV cells. This might be associated with a loss of IRF4F359L binding to PC4 (64), a complex component reported to function in heterochromatin organization in activated B lymphocytes (48, 73) (Figures 1A, B). Taken together, deregulation of IRF4 transcriptional activity causing an AD IEI can be associated with new protein interaction(s) mediated by an IRF4 neomorphic variant.
IRF4 autosomal dominant variant with multimorphic functions
Seven patients were found around the world all presenting with early-onset (<1 year of age) recurrent sinopulmonary infections associated with agammaglobulinemia and a lack of circulating plasma cells even though circulating B cells were found (74). Six out of the seven patients suffered from Pneumocystis jirovecii, causing pneumonia. In addition, clinical complications associated with Mycobacterium bovis, cytomegalovirus, and EBV infection were reported for some of the patients. For both CD4 and CD8 T cells, patients presented with high naive counts associated with low central, effector, or terminal effector memory cells. Genetic analysis of these patients led to the identification of the very same heterozygous germline mutation affecting the DBD of the IRF4 protein (c.284C>G, p.T95R). Functional analysis showed that transcriptional activity of the IRF4T95R was largely reduced on ISRE sites. This low transcription activity on ISRE sites was functionally associated with a failure of patients’ B cells to differentiate into plasmablast/plasma cells and to secrete Ig. The generation of class-switched and memory B cells was reduced. T cells from the patients failed to differentiate into TH17 and TFH cells and showed a reduced IL-2 and IFNγ secretion. Knock-in mice with the identical T95R substitution recapitulated the immunodeficient phenotype observed in these patients. Interestingly, Irf4T95R knock-in mice indicated an expansion of nonspecific germinal center B cells at the expense of antigen-specific germinal center B cells. Consistent with an increased affinity for DNA, the subcellular localization of IRF4T95R was altered, with an increased nuclear-to-cytoplasmic ratio compared with IRF4 wild type (74). CHIP-Seq analysis performed on patients’ derived B-EBV cells in conjunction with a deep learning tool (ExplaiNN) (75) unveiled IRF4T95R-altered DNA binding specificity. ExplaiNN identified a noncanonical GATA-containing ISRE motif and various noncanonical AICE motifs that appeared important for IRF4T95R binding but detrimental to IRF4 WT-specific binding. This altered DNA binding capacity of IRF4T95R can have a functional impact on the expression of new target genes containing these non-canonical AICE sites, as shown for CXCL13 (74). As such, IRF4T95R presents with a simultaneous multimorphic combination of loss, gain, and new functions for IRF4. Three out of the seven patients have undergone allogenic hematopoietic stem cell transplantation; however, reported follow-up time was less than 8 months for all them. Of note, the T95R variant has been identified as somatic mutation in adult T-cell leukemia implicating multimorphic IRF4 function also in lymphomagenesis (Figures 1A, B). Additional somatic mutation affecting the IRF4 DBD was already associated with lymphomagenesis, possibly suggesting that other germline variants affecting IRF4’s DBD might also cause IEI. Whether these IRF4 DBD variants present with a multimorphic IRF4 pathophysiology as described for IRF4T95R remains to be determined.
Conclusion
Genetic characterization of IEI is essential for an exact diagnosis and accurate treatment of patients. The use of next-generation sequencing for genetic exploration/diagnosis of IEI facilitates the discovery of novel genetic lesions (either heterozygous or biallelic) underlying IEI. Moreover, the understanding of pathophysiological mechanisms underlying IEI is evolving. Several genetic lesions in transcription factors of the IRF family are associated with IEIs, and within this family, IRF4 mutations can cause IEIs both at the heterozygous and biallelic state. Autosomal dominant IRF4 gene-associated pathophysiological mechanisms include haploinsufficiency, neomorphic, and multimorphic (a simultaneous combination of loss, gain, and new functions for IRF4), showcasing the complexity underlying IRF4 deregulation. Studying the pathophysiology of autosomal dominant IRF4 protein variants and their impact on transcriptome, epigenome, and chromatin structure organization will provide important new insights into IRF4 function and might have implications for treatments of IEI and possibly even cancer. In conclusion, the continuously increasing genetic delineation of IEI, besides its essential role in clinics, allows the description of new unsuspected physiological mechanisms, enriching our knowledge on immune responses.
Author contributions
RT and SK contributed to conception and design of the study. RT wrote the first draft of the manuscript. SK wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The work was funded by the French state [via the Agence Nationale de la Recherche’s “Investissments d’avenir” program (ANR-10-IAHU-01) and grant ANR-19-CE17-0012-01 (ANR-AID)]. SK is a Centre National de la Recherche Scientifique staff researcher.
Acknowledgments
The authors would like to thank Dr. Anne Durandy for reviewing and commenting on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1236889/full#supplementary-material
Supplementary Figure 1 | Comparison of IRF family protein structure. Schematic representation of the protein structure with indication of the DNA binding domain (DBD), the linker domain, the interferon association domain (IAD), and the autoinhibitory region.
References
1. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol (2008) 26(1):535–84. doi: 10.1146/annurev.immunol.26.021607.090400
2. Paun A, Pitha PM. The IRF family, revisited. Biochimie (2007) 89(6–7):744–53. doi: 10.1016/j.biochi.2007.01.014
3. Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell (1988) 54(6):903–13. doi: 10.1016/S0092-8674(88)91307-4
4. Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science (1996) 274(5293):1739–44. doi: 10.1126/science.274.5293.1739
5. Fujita T, Kimura Y, Miyamoto M, Barsoumian EL, Taniguchi T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature (1989) 337(6204):270–2. doi: 10.1038/337270a0
6. Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, et al. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell (1989) 58(4):729–39. doi: 10.1016/0092-8674(89)90107-4
7. Mamane Y, Heylbroeck C, Génin P, Algarté M, Servant MJ, LePage C, et al. Interferon regulatory factors: the next generation. Gene (1999) 237(1):1–14. doi: 10.1016/S0378-1119(99)00262-0
8. Taniguchi T, Tanaka N, Ogasawara K, Taki S, Sato M, Takaoka A. Transcription factor IRF-1 and its family members in the regulation of host defense. Cold Spring Harb Symp Quant Biol (1999) 64:465–72. doi: 10.1101/sqb.1999.64.465
9. Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors. Oncoimmunology (2012) 1(8):1376–86. doi: 10.4161/onci.22475
10. Nam S, Lim JS. Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch Pharm Res (2016) 39(11):1548–55. doi: 10.1007/s12272-016-0854-1
11. De Silva NS, Simonetti G, Heise N, Klein U. The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol Rev (2012) 247(1):73–92. doi: 10.1111/j.1600-065X.2012.01113.x
12. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. (2005) 102(44):15989–94. doi: 10.1073/pnas.0508327102
13. Remesh SG, Santosh V, Escalante CR. Structural studies of IRF4 reveal a flexible autoinhibitory region and a compact linker domain. J Biol Chem (2015) 290(46):27779–90. doi: 10.1074/jbc.M115.678789
14. Sundararaj S, Seneviratne S, Williams SJ, Enders A, Casarotto MG. Structural determinants of the IRF4/DNA homodimeric complex. Nucleic Acids Res (2021) 49(4):2255–65. doi: 10.1093/nar/gkaa1287
15. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct inhibition of IRF-dependent transcriptional regulatory mechanisms associated with disease. Front Immunol (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
16. Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity (2000) 13(4):539–48. doi: 10.1016/S1074-7613(00)00053-4
17. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi: 10.1038/nature03464
18. Yanai H, Chen HM, Inuzuka T, Kondo S, Mak TW, Takaoka A, et al. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci U S A. (2007) 104(9):3402–7. doi: 10.1073/pnas.0611559104
19. Ren G, Cui K, Zhang Z, Zhao K. Division of labor between IRF1 and IRF2 in regulating different stages of transcriptional activation in cellular antiviral activities. Cell Biosci (2015) 5:17. doi: 10.1186/s13578-015-0007-0
20. Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. (2004) 101(31):11416–21. doi: 10.1073/pnas.0403555101
21. Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol (2006) 7(1):40–8. doi: 10.1038/ni1282
22. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature (2001) 413(6851):78–83. doi: 10.1038/35092578
23. Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol (2003) 4(11):1144–50. doi: 10.1038/ni986
24. Colonna M. TLR pathways and IFN-regulatory factors: to each its own. Eur J Immunol (2007) 37(2):306–9. doi: 10.1002/eji.200637009
25. Masumi A, Tamaoki S, Wang IM, Ozato K, Komuro K. IRF-8/ICSBP and IRF-1 cooperatively stimulate mouse IL-12 promoter activity in macrophages. FEBS Lett (2002) 531(2):348–53. doi: 10.1016/S0014-5793(02)03556-1
26. Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol Baltim Md 1950 (2000) 165(1):271–9. doi: 10.4049/jimmunol.165.1.271
27. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
28. Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, et al. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity (2017) 47(3):481–497.e7. doi: 10.1016/j.immuni.2017.09.001
29. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14(11):1155–65. doi: 10.1038/ni.2710
30. Lei J, Zhou MH, Zhang FC, Wu K, Liu SW, Niu HQ. Interferon regulatory factor transcript levels correlate with clinical outcomes in human glioma. Aging (2021) 13(8):12086–98. doi: 10.18632/aging.202915
31. Willman CL, Sever CE, Pallavicini MG, Harada H, Tanaka N, Slovak ML, et al. Deletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science (1993) 259(5097):968–71. doi: 10.1126/science.8438156
32. Ogasawara S, Tamura G, Maesawa C, Suzuki Y, Ishida K, Satoh N, et al. Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology (1996) 110(1):52–7. doi: 10.1053/gast.1996.v110.pm8536888
33. Tamura G, Ogasawara S, Nishizuka S, Sakata K, Maesawa C, Suzuki Y, et al. Two distinct regions of deletion on the long arm of chromosome 5 in differentiated adenocarcinomas of the stomach. Cancer Res (1996) 56(3):612–5.
34. Lukhele S, Rabbo DA, Guo M, Shen J, Elsaesser HJ, Quevedo R, et al. The transcription factor IRF2 drives interferon-mediated CD8+ T cell exhaustion to restrict anti-tumor immunity. Immunity (2022) 55(12):2369–2385.e10. doi: 10.1016/j.immuni.2022.10.020
35. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell (2019) 35(4):559–572.e7. doi: 10.1016/j.ccell.2019.02.008
36. Nixon BG, Kuo F, Ji L, Liu M, Capistrano K, Do M, et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity (2022) 55(11):2044–2058.e5. doi: 10.1016/j.immuni.2022.10.002
37. Fu K, Hui B, Wang Q, Lu C, Shi W, Zhang Z, et al. Single-cell RNA sequencing of immune cells in gastric cancer patients. Aging (2020) 12(3):2747–63. doi: 10.18632/aging.102774
38. Jiao S, Guan J, Chen M, Wang W, Li C, Wang Y, et al. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J Exp Med (2018) 215(2):699–718. doi: 10.1084/jem.20171116
39. Tian M, Wang X, Sun J, Lin W, Chen L, Liu S, et al. IRF3 prevents colorectal tumorigenesis via inhibiting the nuclear translocation of β-catenin. Nat Commun (2020) 11(1):5762. doi: 10.1038/s41467-020-19627-7
40. Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, et al. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet (1997) 17(2):226–30. doi: 10.1038/ng1097-226
41. Ramis-Zaldivar JE, Gonzalez-Farré B, Balagué O, Celis V, Nadeu F, Salmerón-Villalobos J, et al. Distinct molecular profile of IRF4-rearranged large B-cell lymphoma. Blood (2020) 135(4):274–86. doi: 10.1182/blood.2019002699
42. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga JI, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet (2015) 47(11):1304–15. doi: 10.1038/ng.3415
43. Rosain J, Neehus AL, Manry J, Yang R, Le Pen J, Daher W, et al. Human IRF1 governs macrophagic IFN-γ immunity to mycobacteria. Cell (2023) 186(3):621–645.e33. doi: 10.1016/j.cell.2022.12.038
44. Andersen LL, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med (2015) 212(9):1371–9. doi: 10.1084/jem.20142274
45. Lévy R, Bastard P, Lanternier F, Lecuit M, Zhang SY, Casanova JL. IFN-α2a therapy in two patients with inborn errors of TLR3 and IRF3 infected with SARS-coV-2. J Clin Immunol (2021) 41(1):26–7. doi: 10.1007/s10875-020-00933-0
46. Thomsen MM, Jørgensen SE, Storgaard M, Kristensen LS, Gjedsted J, Christiansen M, et al. Identification of an IRF3 variant and defective antiviral interferon responses in a patient with severe influenza. Eur J Immunol (2019) 49(11):2111–4. doi: 10.1002/eji.201848083
47. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
48. Das C, Hizume K, Batta K, Kumar BRP, Gadad SS, Ganguly S, et al. Transcriptional coactivator PC4, a chromatin-associated protein, induces chromatin condensation. Mol Cell Biol (2006) 26(22):8303–15. doi: 10.1128/MCB.00887-06
49. Han J, Kraft P, Nan H, Guo Q, Chen C, Qureshi A, et al. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PloS Genet (2008) 4(5):e1000074. doi: 10.1371/journal.pgen.1000074
50. Bravo García-Morato M, Aracil Santos FJ, Briones AC, Blázquez Moreno A, Del Pozo Maté Á, Domínguez-Soto Á, et al. New human combined immunodeficiency caused by interferon regulatory factor 4 (IRF4) deficiency inherited by uniparental isodisomy. J Allergy Clin Immunol (2018) 141(5):1924–1927.e18. doi: 10.1016/j.jaci.2017.12.995
51. Guérin A, Kerner G, Marr N, Markle JG, Fenollar F, Wong N, et al. IRF4 haploinsufficiency in a family with Whipple’s disease. eLife (2018) 7:e32340s. doi: 10.7554/eLife.32340
52. Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science (2015) 348(6233):448–53. doi: 10.1126/science.aaa1578
53. Campbell TM, Liu Z, Zhang Q, Moncada-Velez M, Covill LE, Zhang P, et al. Respiratory viral infections in otherwise healthy humans with inherited IRF7 deficiency. J Exp Med (2022) 219(7):e20220202. doi: 10.1084/jem.20220202
54. Thomsen MM, Jørgensen SE, Gad HH, Storgaard M, Gjedsted J, Christiansen M, et al. Defective interferon priming and impaired antiviral responses in a patient with an IRF7 variant and severe influenza. Med Microbiol Immunol (Berl). (2019) 208(6):869–76. doi: 10.1007/s00430-019-00623-8
55. Salem S, Gros P. Genetic determinants of susceptibility to Mycobacterial infections: IRF8, a new kid on the block. Adv Exp Med Biol (2013) 783:45–80. doi: 10.1007/978-1-4614-6111-1_3
56. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med (2011) 365(2):127–38. doi: 10.1056/NEJMoa1100066
57. Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med (2018) 215(10):2567–85. doi: 10.1084/jem.20180628
58. Lévy R, Zhang P, Bastard P, Dorgham K, Melki I, Hadchouel A, et al. Monoclonal antibody-mediated neutralization of SARS-CoV-2 in an IRF9-deficient child. Proc Natl Acad Sci U S A. (2021) 118(45):e2114390118. doi: 10.1073/pnas.2114390118
59. Bravo García-Morato M, Calvo Apalategi A, Bravo-Gallego LY, Blázquez Moreno A, Simón-Fuentes M, Garmendia JV, et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J Allergy Clin Immunol (2019) 144(1):309–312.e10. doi: 10.1016/j.jaci.2019.02.019
60. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
61. Dobbins WO. Is there an immune deficit in Whipple’s disease? Dig Dis Sci (1981) 26(3):247–52. doi: 10.1007/BF01391638
62. Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med (2007) 356(1):55–66. doi: 10.1056/NEJMra062477
63. Praetorius C, Grill C, Stacey SN, Metcalf AM, Gorkin DU, Robinson KC, et al. A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell (2013) 155(5):1022–33. doi: 10.1016/j.cell.2013.10.022
64. Thouenon R, Chentout L, Moreno-Corona N, Poggi L, Lombardi EP, Hoareau B, et al. A neomorphic mutation in the interferon activation domain of IRF4 causes a dominant primary immunodeficiency. J Exp Med (2023) 220(6):e20221292. doi: 10.1084/jem.20221292
65. Brass AL, Zhu AQ, Singh H. Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimers. EMBO J (1999) 18(4):977–91. doi: 10.1093/emboj/18.4.977
66. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature (2012) 490(7421):543–6. doi: 10.1038/nature11530
67. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity (2006) 25(2):225–36. doi: 10.1016/j.immuni.2006.07.009
68. Kuwata T, Gongora C, Kanno Y, Sakaguchi K, Tamura T, Kanno T, et al. Gamma interferon triggers interaction between ICSBP (IRF-8) and TEL, recruiting the histone deacetylase HDAC3 to the interferon-responsive element. Mol Cell Biol (2002) 22(21):7439–48. doi: 10.1128/MCB.22.21.7439-7448.2002
69. Ryseck RP, Bravo R. c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: effect of FOS proteins. Oncogene (1991) 4):533–42.
70. Wheaton JD, Ciofani M. JunB controls intestinal effector programs in regulatory T cells. Front Immunol (2020) 11:444. doi: 10.3389/fimmu.2020.00444
71. Carr TM, Wheaton JD, Houtz GM, Ciofani M. JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation. Nat Commun (2017) 8(1):301. doi: 10.1038/s41467-017-00380-3
72. Yamazaki S, Tanaka Y, Araki H, Kohda A, Sanematsu F, Arasaki T, et al. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci Rep (2017) 7(1):17402. doi: 10.1038/s41598-017-17597-3
73. Ochiai K, Yamaoka M, Swaminathan A, Shima H, Hiura H, Matsumoto M, et al. Chromatin protein PC4 orchestrates B cell differentiation by collaborating with IKAROS and IRF4. Cell Rep (2020) 33(12):108517. doi: 10.1016/j.celrep.2020.108517
74. IRF4 International Consortium, Fornes O, Jia A, Kuehn HS, Min Q, Pannicke U, et al. A multimorphic mutation in IRF4 causes human autosomal dominant combined immunodeficiency. Sci Immunol (2023) 8(79):eade7953. doi: 10.1126/sciimmunol.ade7953
Keywords: interferon regulatory factor family, IRF4, inborn errors of immunity, cancer, primary antibody deficiency
Citation: Thouenon R and Kracker S (2023) Human inborn errors of immunity associated with IRF4. Front. Immunol. 14:1236889. doi: 10.3389/fimmu.2023.1236889
Received: 08 June 2023; Accepted: 05 September 2023;
Published: 22 September 2023.
Edited by:
Peter Daniel Burrows, University of Alabama at Birmingham, United StatesReviewed by:
Carlos Rodríguez-Gallego, University Hospital of Gran Canaria Dr. Negrin, SpainCopyright © 2023 Thouenon and Kracker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Kracker, c3Zlbi5rcmFja2VyQGluc2VybS5mcg==
†ORCID: Sven Kracker, orcid.org/0000-0003-4543-8236