 Ronghua He
Ronghua He Subei Zhao2
Subei Zhao2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 October 2023
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1234535
This article is part of the Research Topic Challenges in Inflammatory Bowel Disease: Current, Future and Unmet Needs - Volume II View all 8 articles
Inflammatory bowel disease (IBD) is a chronic inflammatory disease typically involving the gastrointestinal tract but not limited to it. IBD can be subdivided into Crohn’s disease (CD) and ulcerative colitis (UC). Extraintestinal manifestations (EIMs) are observed in up to 47% of patients with IBD, with the most frequent reports of cutaneous manifestations. Among these, pyoderma gangrenosum (PG) and erythema nodosum (EN) are the two most common skin manifestations in IBD, and both are immune-related inflammatory skin diseases. The presence of cutaneous EIMs may either be concordant with intestinal disease activity or have an independent course. Despite some progress in research on EIMs, for instance, ectopic expression of gut-specific mucosal address cell adhesion molecule-1 (MAdCAM-1) and chemokine CCL25 on the vascular endothelium of the portal tract have been demonstrated in IBD-related primary sclerosing cholangitis (PSC), little is understood about the potential pathophysiological associations between IBD and cutaneous EIMs. Whether cutaneous EIMs are inflammatory events with a commonly shared genetic background or environmental risk factors with IBD but independent of IBD or are the result of an extraintestinal extension of intestinal inflammation, remains unclear. The review aims to provide an overview of the two most representative cutaneous manifestations of IBD, describe IBD’s epidemiology, clinical characteristics, and histology, and discuss the immunopathophysiology and existing treatment strategies with biologic agents, with a focus on the potential pathophysiological associations between IBD and cutaneous EIMs.
IBD is an immune-mediated chronic inflammatory disease mainly involving the gastrointestinal tract. It is caused by the interaction of multiple factors, including genetic, environmental, immune, epithelial, and microbial, making it complex (1). The global incidence and prevalence of IBD vary widely by geographic region, ranging from 0.1 to 58 cases per 100,000 person-years and 0.9 to 505 cases per 100,000 population, respectively; the highest rates have been reported in North America and Europe (2). In recent years the incidence has also increased rapidly in many newly industrializing countries (3). IBD’s peak usually occurs between the ages of 20–40 years but a moderate second peak occurs after the age of 60 years (4). In Asia, IBD is more common in males, while in the Western world, patients with IBD are predominantly females (5). Depending on the location and depth of intestinal involvement, IBD is divided into two subtypes, CD and UC (6).
Although IBD is a chronic inflammation of the gut, the manifestations of CD and UC are not limited to the gastrointestinal tract. EIMs are frequently observed in patients with IBD, with a reported frequency of 6%–47% (7). The skin, musculoskeletal, joints, eyes, and hepatobiliary tract are the organs most affected by the EIMs (8, 9). Of these, cutaneous EIMs are the most frequently reported, with an incidence of more than 17% (7). According to the different pathophysiological mechanisms, cutaneous EIMs are divided into the following four categories: (1) specific skin manifestations with the same histological features as IBD, such as metastatic CD; (2) reactive skin manifestations with similar pathophysiological mechanisms to IBD, such as PG, Sweet’s syndrome, EN, and aphthous ulcer; (3) IBD-related skin diseases, such as psoriasis, hidradenitis suppurativa, and atopic dermatitis, and (4) IBD-treatment-induced skin lesions, such as anti-tumor necrosis factor (TNF)-α-induced skin eruptions (10). A clinical cohort study by Hung et al. showed that patients with IBD are at different degrees of risk of having certain skin lesions at or after the diagnosis of IBD (11). PG and EN are the two most common skin manifestations in IBD (12) and both are immune-mediated inflammatory skin diseases. In addition to the association between cutaneous EIMs and intestinal disease, various cutaneous EIMs are linked. For example, PG and EN are related, while EN is also associated with aphthous ulcers (13). Based on the correlation between different cutaneous EIMs and between cutaneous EIMs and IBD, along with the recently confirmed efficacy of biologic agents for IBD and cutaneous EIMs, the proposed hypothesis is that there may be a potential pathogenic association between these conditions. This potential pathogenic association is not fully understood but it is speculated that damage to the gastrointestinal mucosa in patients with IBD may trigger excessive immune responses in the skin through multiple pathways. In this review, we provide an overview of the two most representative cutaneous manifestations of IBD; describe its epidemiology, clinical characteristics, and histology, and discuss the immunopathophysiology and applicable treatment options with biologic agents, with a focus on the potential pathophysiological associations between IBD and cutaneous EIMs.
Genetic, environmental, epithelial, microbial, and immune factors are involved in the occurrence and development of IBD. In IBD, gut microbiota dysregulation is associated with mucus layer disruption, epithelial cell tight junction dysregulation, defective Paneth cell number and function, and increased gut permeability, leading to increased gut microbial exposure (1). Antigen-presenting cells (APCs) continually recognize and sample these exposed microorganisms and produce abundant pro-inflammatory cytokines such as TNF-α, interleukin (IL)-1, IL-6, IL-12, and IL-23, and then migrate to mesenteric lymph nodes (MLN) or gut-associated lymphoid tissue (GALT), where they present antigens to naïve CD4+T cells. Under the influence of these pro-inflammatory cytokines, conserved pathogenic structures, or danger signals released by damaged cells, naïve CD4+T cells undergo proliferation and differentiation into effector T cell subsets comprising type 1 helper T (Th1) and type 17 helper T (Th17) cells (1). These primed and activated effector T cell subsets migrate out of the MLN or GALT and move into the bloodstream (14, 15). Under the interaction of chemokines with chemokine receptors and integrins with cell adhesion molecules, they migrate from the blood along chemotactic gradients to the lamina propria of the intestinal mucosa (14). Effector T cell subsets homing to the gut adapt the composition of their surface molecules (e.g., upregulation of integrin αEβ7) in response to the inflammatory circumstances, thereby allowing them to reside in the tissue (16). These effector T cell subsets residing in the intestinal mucosal lamina propria produce pro-inflammatory cytokines associated with prolonged intestinal mucosal inflammation and tissue damage (1, 17, 18) (Figure 1).
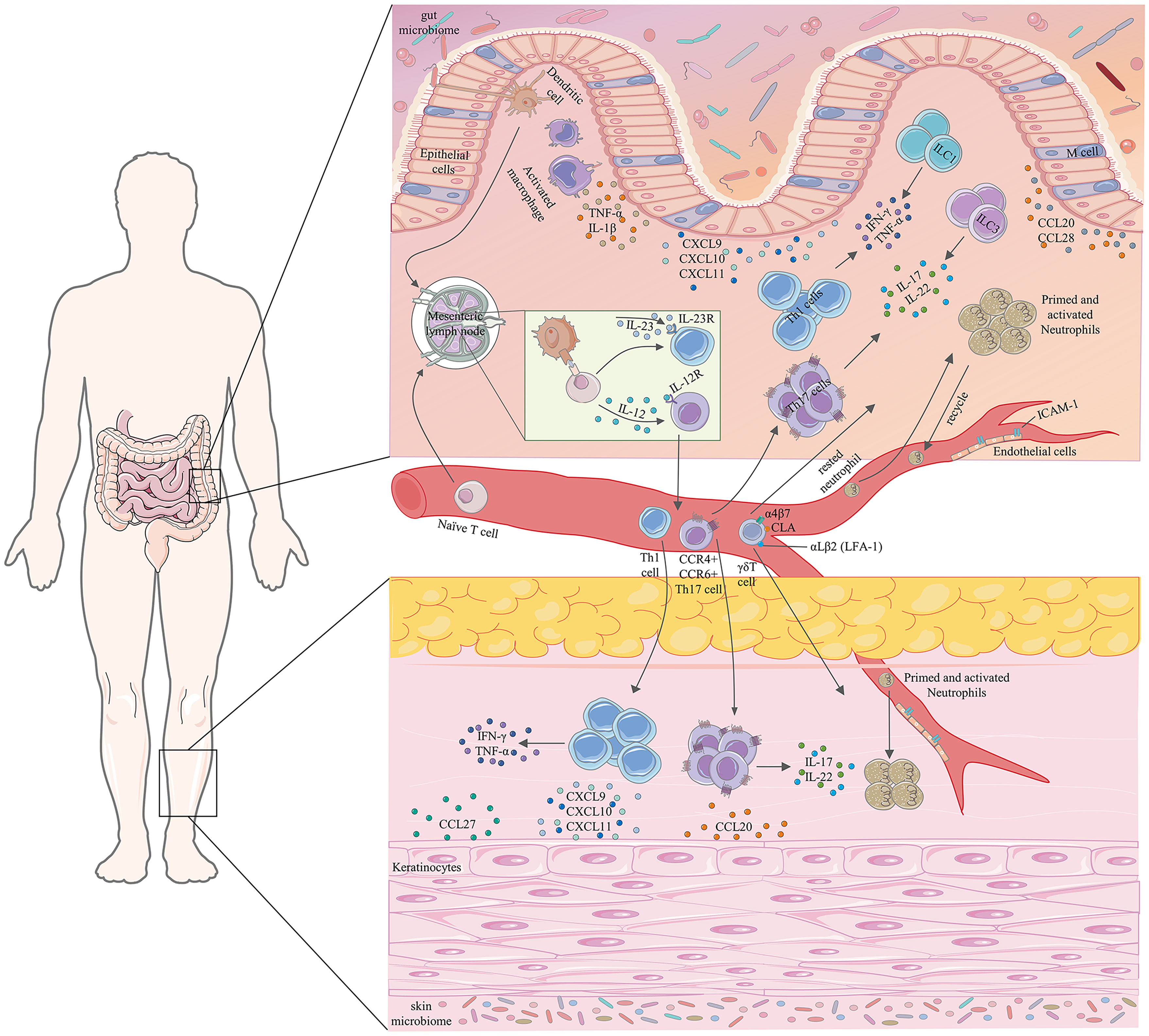
Figure 1 Partial pathophysiological associations between IBD and cutaneous EIMs. TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; IL, interleukin; CXCL, C-X-C motif chemokine ligand; ILCs, innate lymphoid cells; CLA, cutaneous lymphocyte-associated antigen; LFA-1, lymphocyte function-associated antigen-1; ICAM-1, intercellular adhesion molecule-1; Th1, type 1 helper T; Th17, type 17 helper T.
Chemokines and adhesion molecules are crucial components of our immune system and regulate the migration and localization of immune cells in tissues. Extensive analysis of various immune cells in the innate and adaptive immune systems has revealed their different migratory capacities, which are reflected in the unique chemokine receptors expressed by the various immune cells themselves, that mediate the migration of immune cells in the direction of increasing chemokine concentration. In addition to mediating the migration of immune cells themselves, chemokine receptors can help distinguish between different cell subsets, such as Th1 cells (CXCR3+) and Th17 cells (CCR4+CCR6+) (19, 20). The ligands of CXCR3 include CXCL9 (a monokine induced by interferon (IFN)-γ), CXCL10 (IFN-induced protein-10), and CXCL11 (IFN-inducible T-cell alpha chemoattractant), which are produced by human intestinal epithelial cells, are secreted in large quantities in an inflammatory environment rich in IFN-γ and can recruit CXCR3+ cells to the inflammatory site (21, 22). Overexpression of chemokine pairs CXCR3-CXCL9, CXCL10, and CXCL11 in human IBD has been demonstrated (23, 24). They are tightly associated with Th1 cell responses (19, 25) and increased secretion of pro-inflammatory cytokine by monocytes, especially with IFN-γ, to synergistically induce the secretion of IL-12 and IL-23 (26). CCL20 is also a chemokine secreted by human intestinal epithelial cells, which can chemotactically attract Th17 cells expressing CCR6 and is closely associated with inflammation and homeostasis (27). The expression of CCR6 is a common feature of IL-17-secreting cells, such as Th17 cells and γδT cells (20, 28). Multiple pro-inflammatory cytokines (e.g., TNF-α) induce CCL20 expression, and high upregulation of the CCR6-CCL20 axis has been demonstrated in the intestinal mucosa of patients with IBD (27, 29, 30). Meanwhile, Soler et al. found that Th lymphocytes expressing CCR4 can be mediated to home to the skin (31). CCR9-CCL25 plays an indispensable role in the recruitment of intestinal immune cells. CCL25 is the only receptor for CCR9, which is expressed not only by small intestinal epithelial cells under homeostatic conditions but also by colonic cells during inflammation (32). Papadakis et al. have confirmed that the CCR9-CCL25 axis is associated with inflammation, particularly in small bowel CD (33). Interaction of CCL25 with CCR9 contributes to the homing of expressing α4β7+ T cells to intestinal sites (32). CCR9+ T cells isolated from patients with CD show significantly higher expression of IL-17 and IFN-γ after stimulation compared to healthy controls (34). In addition, expression of CXCR1 and CXCR2 is characteristic of neutrophils, which can bind to seven neutrophil attraction and chemotactic factors, CXCL1-3 and CXCL5-8, of which CXCL8 (IL-8) is the most efficient and abundantly secreted protein by the cells, inducing neutrophil infiltration from the blood into inflammatory tissues and amplifying inflammation (35). The involvement of neutrophils and CXCL8 in the pathogenesis and development of intestinal mucosal inflammation in IBD has been previously described (36).
Lymphocyte homing is a process whereby lymphocytes in the blood migrate directionally and settle in peripheral immune organs or tissues, including lymphocyte recycling and lymphocyte migration to inflammatory sites (such as intestinal mucosa and skin). The molecular basis of lymphocyte homing is the interaction between lymphocyte homing receptors expressed on the surface of lymphocytes and adhesion molecules expressed on the surface of endothelial cells, wherein T lymphocyte trafficking is the most important (37, 38). Under the binding of chemokines to their cognate receptors on lymphocytes and other soluble pro-inflammatory cytokines, the expression of integrins (αLβ2 (lymphocyte function-associated antigen-1 (LFA-1), α4β1(very late antigen-4 (VLA-4), and α4β7) on the membranes of effector T cell subsets and cell adhesion molecules (intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and MAdCAM-1) on endothelial cells are upregulated (15, 39). Firm interactions between cell adhesion molecules and integrins allow effector T cell subsets to adhere tightly to the vessel wall and eventually to transmigrate through the endothelial cells to the intestinal mucosal lamina propria (39). MAdCAM-1, a major ligand in effector lymphocytes that expresses integrin α4β7, is highly expressed in the venules of intestinal tissue in close contact with the intestinal epithelium (15), and its expression is strongly induced by TNF-α in patients with IBD (40). Since MAdCAM-1 is usually expressed only in gastrointestinal tissues, the interaction of MAdCAM-1 with α4β7 is thought to be exclusive to the migration occurring in the gastrointestinal tract (41). The interaction of αLβ2 with ICAM-1 and α4β1 with VCAM-1 contributes to the migration of intestinal effector lymphocytes (15). The expressions of ICAM-1, VCAM-1, and MAdCAM-1 are upregulated in the active site of intestinal inflammation in patients with IBD (16).
Th1 and Th17 cells are crucial central pathways in the pathogenesis of IBD, and their mediators and upstream and downstream molecules are abundantly involved in the lesion (42). The most relevant cytokines that promote the differentiation of Th1 and Th17 cells are IL-12 and IL-23, respectively, which are mainly oversecreted by activated dendritic cells and macrophages (43, 44). IL-12 is a heterodimeric cytokine composed of IL-12p35 and IL-12p40 subunits that induces upregulation of the transcription factor, T-bet, in naïve T cells and promotes differentiation of Th1 cells that produce IFN-γ, TNF-α and recruit macrophages, natural killer cells, and CD8+T cells. In the presence of IL-1, IL-6, and transforming growth factor β (TGF-β), a cytokine, IL-23, composed of IL-12p40 and IL-23p19 subunits, can induce upregulation of the master transcription factor, RORγt, and promotes differentiation of Th17 cells, which produce IL-17, IL-22, and recruit neutrophils (1, 45). Innate lymphoid cells (ILCs) lie adjacent to the intestinal mucosal barrier and the effector cytokines secreted by them can modulate the pathology of IBD; for example, group 1 ILCs and group 3 ILCs produce IFN-γ, TNF-α, IL-17, and IL-22 (46). In the intestinal mucosa, IL-17 can induce the expression of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6), various chemokines (e.g., CCL20, which attracts Th17 cells, γδT cells, and dendritic cells; CXCL8/IL-8, CXCL1, which attracts neutrophils, and monocyte chemoattractant protein (MCP-1), which attracts monocytes and macrophages), and adhesion molecule (ICAM-1) (44, 47), resulting in profound pro-inflammatory effects. High levels of IFN-γ secreted by Th1 cells may activate vascular endothelial cells, leading to the infiltration of various immune cells from the blood into the intestinal mucosa (48). Simultaneously, IFN-γ itself, as the main product of Th1 cells, not only makes the immune response biased towards Th1 phenotype but also upregulates the expression of chemokines (such as CXCL9, CXCL10) that attract Th1 cells and MHC molecules on tissues cells and APCs (48). In chronic inflammation of the intestinal tract, the pleiotropic and strongly potent pro-inflammatory cytokine, TNF-α, can induce a wide range of pro-inflammatory effects, including activation of endothelial cells, expression of inflammatory chemokines, activation of macrophage and effector T cells, necrosis of Paneth cells, and damage to epithelial cells (49, 50). Presumably, IL-17 alone appears to elicit only a mild to moderate inflammatory cellular response but synergistically with IFN-γ and/or TNF-α enhances cellular IL-6, IL-8(CXCL8) synthesis and release, along with ICAM-1 expression (51, 52). A recent study showed that TNF-α and IFN-γ also act synergistically and lead to intestinal epithelial cell death through induction of the caspase-8-JAK1/2-STAT1 module (53). All of these indicate the role of these cytokines in the core network of IBD.
Although chemokines, adhesion molecules, and cytokines play an indispensable role in maintaining immune homeostasis and lymphoid tissue development, the occurrence of aberrant homing of lymphocytes in IBD indicates that the mechanisms are imperfect. Aberrant homing of autoreactive pathogenic T cells is closely related to many autoimmune diseases (54). For example, aberrant homing of gut-directed T lymphocytes in IBD may lead to autoimmune, chronic T lymphocyte-mediated cutaneous EIMs, characterized by specific effector mechanisms of different T lymphocyte subsets and non-specific inflammation.
PG is the second most common cutaneous manifestation of IBD (55), occurring in approximately 0.5–2.6% of patients with IBD (56), and is more common in UC than in CD (57). Although PG can occur in all age groups and both males and females, it is more likely to occur in the elderly (~50 years old) and females (58, 59). Interestingly, up to 50% of patients with PG also have underlying IBD (60), which in most cases precedes the diagnosis of PG (7).
PG is an immune-related autoinflammatory neutrophilic disease with severe and debilitating clinical behavior (57, 59). PG can be induced by minor trauma and usually presents as single or multiple tender inflammatory papules or pustules that rapidly break down over the next few days into painful necrotic ulcers ranging in size from 2–20 cm, with a characteristic violaceous undermined borders and peripheral erythema, accompanied by sterile purulent discharge (59, 61) (Figure 2). However, in severe cases, necrotic ulcers may form spontaneously even in the absence of a history of trauma (59). The base of a PG ulcer usually does not extend beyond the adipose tissue underlying the dermis and is initially an oozy exudative base, which can transform into exuberant granulation tissue within a few weeks (59). Although PG can occur in any part of the skin, extensor surfaces of the legs (shins) and adjacent to postsurgical stoma areas are most commonly involved (57). PG is often associated with several other immune-mediated diseases, most commonly IBD and rheumatoid arthritis (63).
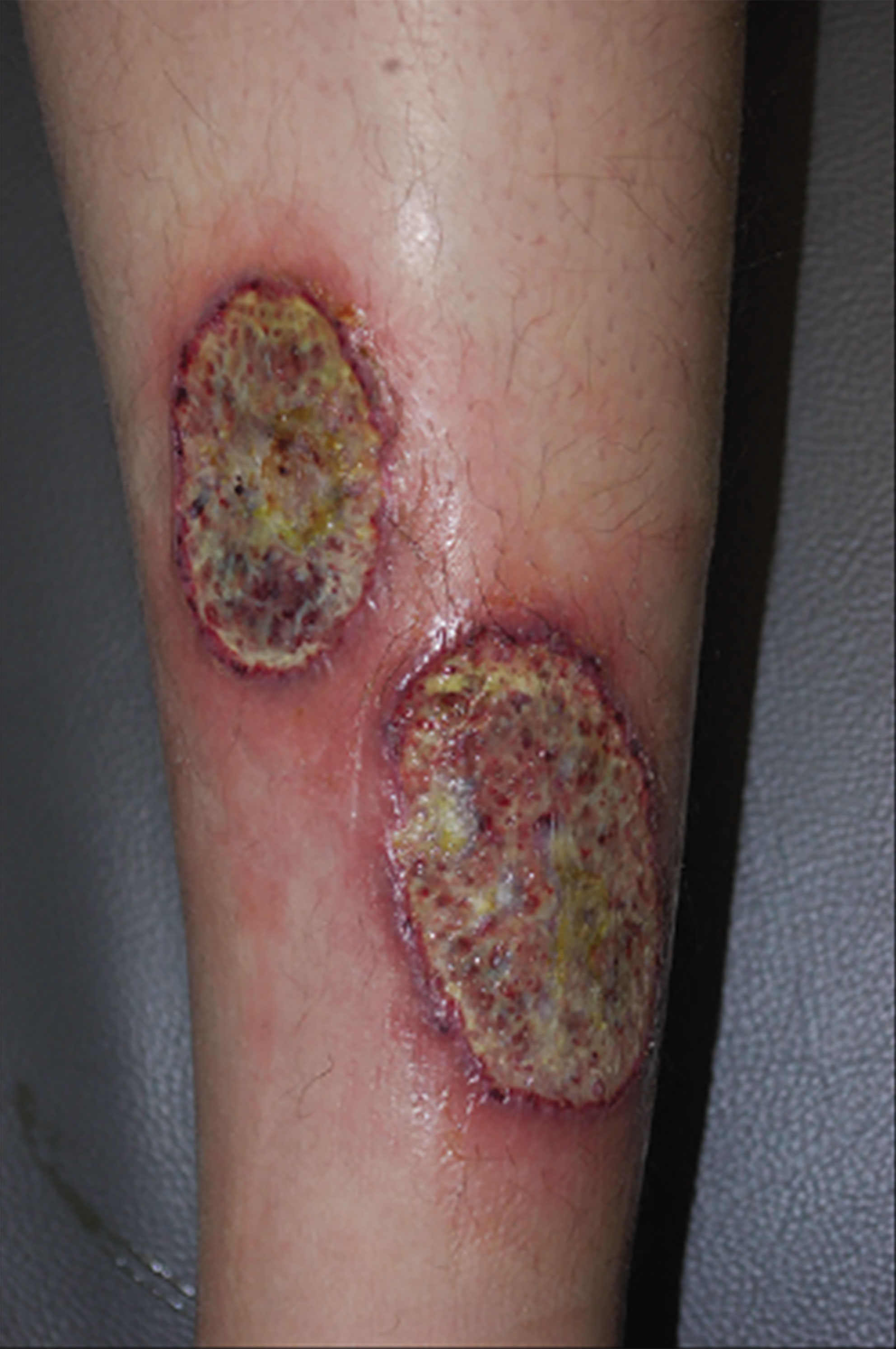
Figure 2 Ulcerative PG, the most common type, presents as a rapidly enlarged deep ulceration with erythematous borders. Reproduced with permission from (62). © 2023 Indian Journal of Dermatology, Venereology and Leprology - Published by Scientific Scholar.
The histopathologic features of PG are not specific (Figure 3). They can be explained by the chronology of lesions and one relies on the site of the biopsy (64). Early lesions present as deep perifollicular inflammation with a dense lymphocytic infiltration (64, 65). Ulceration of the epidermis is often secondary to inflammation of the dermis (64), and their centers show sterile purulent dermatitis and panniculitis with numerous infiltrations of macrophages and multinucleated histiocytes (66). Inflammatory cell infiltration, predominantly by lymphocytes, is often reported in the surrounding skin (64, 66). Biopsies of later-stage ulcers show a dense neutrophilic infiltrate with features of abscess formation (64). As the lesion regresses, plasma cells and macrophages intrude the dermis, and eventually, the inflammation is replaced by fibrosis (66).
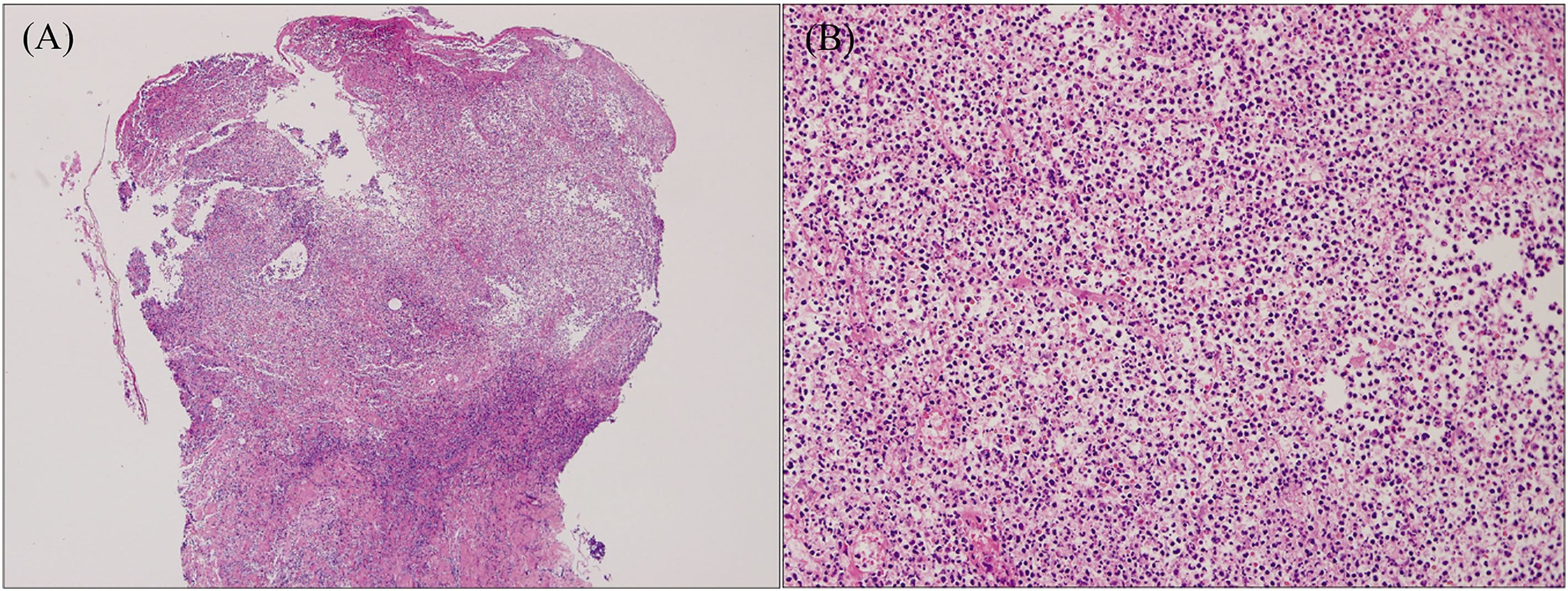
Figure 3 Histopathology of ulcerative PG. (A) Low-power magnification (H&E staining, ×125) reveals diffuse epidermal necrotic ulcer with massive neutrophilic infiltrations and dermal granulation tissue. (B) High-power magnification (H&E staining, ×2000) demonstrates that the majority of lesional cells consist of neutrophils. Reproduced with permission from (62). © 2023 Indian Journal of Dermatology, Venereology and Leprology - Published by Scientific Scholar.
The pathogenesis of PG is not fully understood but is generally considered an autoinflammatory disease involving both innate and adaptive immune responses (59). Part of the mechanism may involve an adaptive immune response mediated by Th1 cells and Th17 cells (59). Histological analysis of early PG papules has revealed that perivascular and peripilosebaceous infiltrates are dominated by dense T lymphocytes rather than neutrophils (65). Gene expression analysis of these papules has revealed overexpression of genes encoding neutrophil-attracting chemokines (e.g., CXCL8/IL-8, CXCL3, CXCL5), Th1 cell-attracting chemokines (e.g., CXCL9, CXCL10, CXCL11), and multiple cytokines (e.g., IL-17, IFN-γ, TNF-α) (65). The genes of transcription factors consistent with the Th1 phenotype are up-regulated. Particularly, there is a strong up-regulation of the Th1 cell-promoting transcription factors, namely STAT1 (signal transducer and activator of transcription-1) and STAT4, along with the down-regulation of the Th2 cell-promoting transcription factor, GATA3 (65). There appears to be an imbalance between Th17 cells and regulatory T (Treg) cells in early PG, characterized by overexpression of Th17 cell-associated cytokines and a decrease in Treg cells (65, 67). These cytokines may be important factors driving the autoimmune inflammatory response of early PG. Th1 cell-mediated immune responses are important for the profound promotion and feedback amplification of inflammation, while Th17 cell-mediated immune responses are critical for the proliferation, maturation, and recruitment of neutrophils, which may be partially mediated by its upstream molecule, IL-23 (68–70). Moreover, neutrophils are the predominant cell type within the ulcer bed and below the destruction border in cases of advanced PG (59). Therefore, T cells in PG may be preferentially polarized into Th1 and Th17 cytokine secretion profiles, which are important sources of IFN-γ, TNF-α, and IL-17 (59), and play an important role in mediating organ specificity and tissue damage and the evolution of early-late-stage lesions in PG.
The clinical behavior of PG is often debilitating and unpredictable. Despite treatment, it may persist. However, early and proactive intervention is necessary and attempts should be made to cure the lesion quickly. Since it is unclear whether PG occurs parallelly with the IBD activity (57), it is necessary to treat PG independently while treating IBD. Mild cases may be treated with local or topical treatments, including intralesional corticosteroid injections, moist dressings, and topical sodium cromoglycate 1-2% solution (59, 71) but systemic medications are usually required, including oral corticosteroids (prednisone 0.5–2 mg/kg/day, prednisolone 0.75 mg/kg/day), oral cyclosporine (3.5–5 mg/kg/day), and dapsone (50–200 mg/day) (59, 71, 72). Treatment with biologic agents has been reported to be effective in improving PG, most notably with the use of anti-TNF-α drugs. Only one randomized controlled trial for PG showed that the portion of patients reporting a clinical response was higher in the infliximab-treated (single dose of 5mg/kg) group compared to the placebo group (6/13 [46%] vs 1/17 [6%]) for the primary endpoint at week 2, and an overall complete remission rate (including 16 patients from the placebo group) of 21% (6/29) for the secondary endpoints of clinical remission at week 6 (73). A retrospective observational study by Argüelles-Arias et al. reported a cure rate of up to 92% (22/24) with infliximab and a healing time of 4–8 weeks (74), while a multicenter retrospective study by Regueiro et al. (single dose of 5mg/kg) showed cure rates as high as 100% (13/13) with complete healing times ranging from 1–30 weeks (75). Some case reports have shown that adalimumab (160/80 mg at 0 and 2 weeks, then 40 mg every other week) has a certain clinical effect on PG (76, 77). IL-12/23 antagonists may be a theoretically feasible therapeutic approach. Two recent retrospective multicenter studies have reported that ustekinumab is also clinically effective for treating patients with PG. Phillips et al. reported complete remission in all three patients with PG (median time to remission 4 months) (78), while De Risi-Pugliese et al. reported complete response in three and partial remission (> 50% reduction of symptoms) in one of four cases with PG (90 mg at 0, 4 and 8, and then every 8 weeks) at week 16 after initiating treatment (79). Although these studies suggest promising prospects for the clinical treatment of PG with biologics, large and randomized placebo-controlled clinical trials are needed to assess the efficacy and safety of these biologics for PG treatment.
EN is the most common skin manifestation of IBD (55) and appears to be more prevalent in CD than in patients with UC (61). The prevalence of EN is 2–10% in patients with UC and 5–15% in those with CD (8). Although EN can occur in any age group, both in males and females, it appears to be more common in people between the ages of 20 and 30 years and females (12, 61, 80). Typically, EN is diagnosed later than IBD, and the proportion of patients with EN before IBD diagnosis is less than 15% (7).
EN is an immune-mediated inflammatory skin disease belonging to typical septal panniculitis and is associated with different pathological processes (81, 82). The main clinical features are symmetrical, raised, red, or violet tender non-ulcerative subcutaneous nodules 1–5 cm in diameter (81) (Figure 4). Typically, EN is self-limiting, with skin lesions mostly resolving within 2–8 weeks without ulceration, scarring, or atrophy (82, 83). EN is usually symmetrically distributed over the extensor surface of the lower extremities, most commonly in the anterior tibial region, and rarely affects the face, trunk, and upper extremities (57). EN is not only associated with IBD but is seen in other immune/non-immune mediated diseases, including Behçet’s syndrome, systemic lupus erythematosus, sarcoidosis, infection, and on some medications (82, 83).
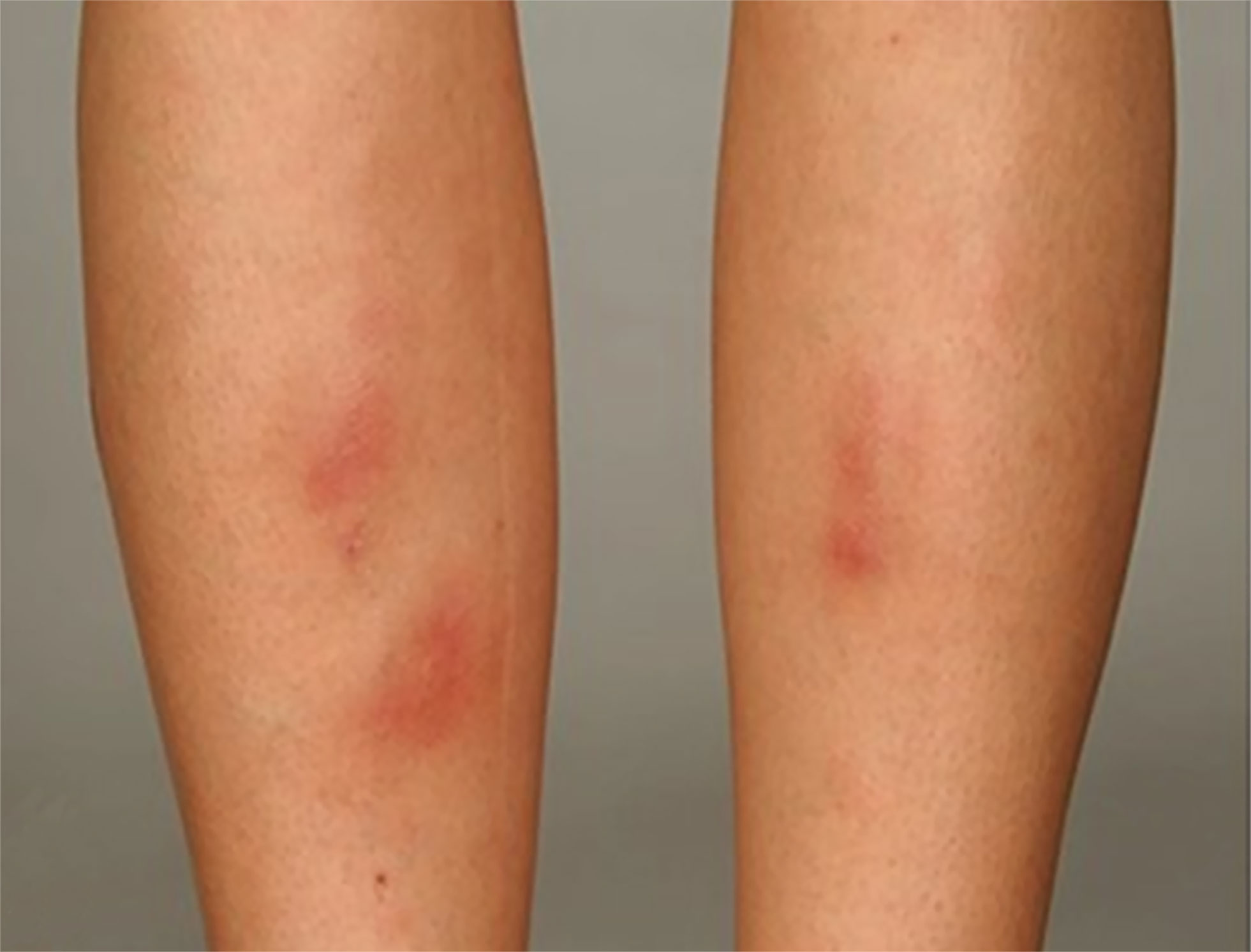
Figure 4 A typical EN shows symmetrical, raised, erythematous, tender non-ulcerative subcutaneous nodules on the tibial anterior region. Reproduced with permission from (57). © 2015 Crohn’s & Colitis Foundation of America, Inc.
Although EN is associated with the pathological processes of a variety of diseases, histopathological examination of the lesion site demonstrates typical septal panniculitis without vasculitis, regardless of the underlying etiology (83, 84). It usually changes with the phase of the disease (85). The early lesions show interlobular septal edema with mixed inflammatory cell infiltration, including lymphocytes, histiocytes, eosinophils, and numerous neutrophils (Figure 5). Inflammation is usually concentrated in the periphery of the septa. The late lesions present thickening and fibrosis of the septa infiltrated by lymphocytes, histiocytes, multinucleated giant cells, and a few neutrophils. Inflammation extends to the periphery of the fat lobules (82, 85, 86). Miescher’s radial granulomas can also be observed (84) (Figure 5). Miescher’s radial granulomas present with nodular aggregations of small histiocytes and macrophages radially arranged around star-shaped cleft-like spaces, which are typical characteristics of EN (82, 83, 86).
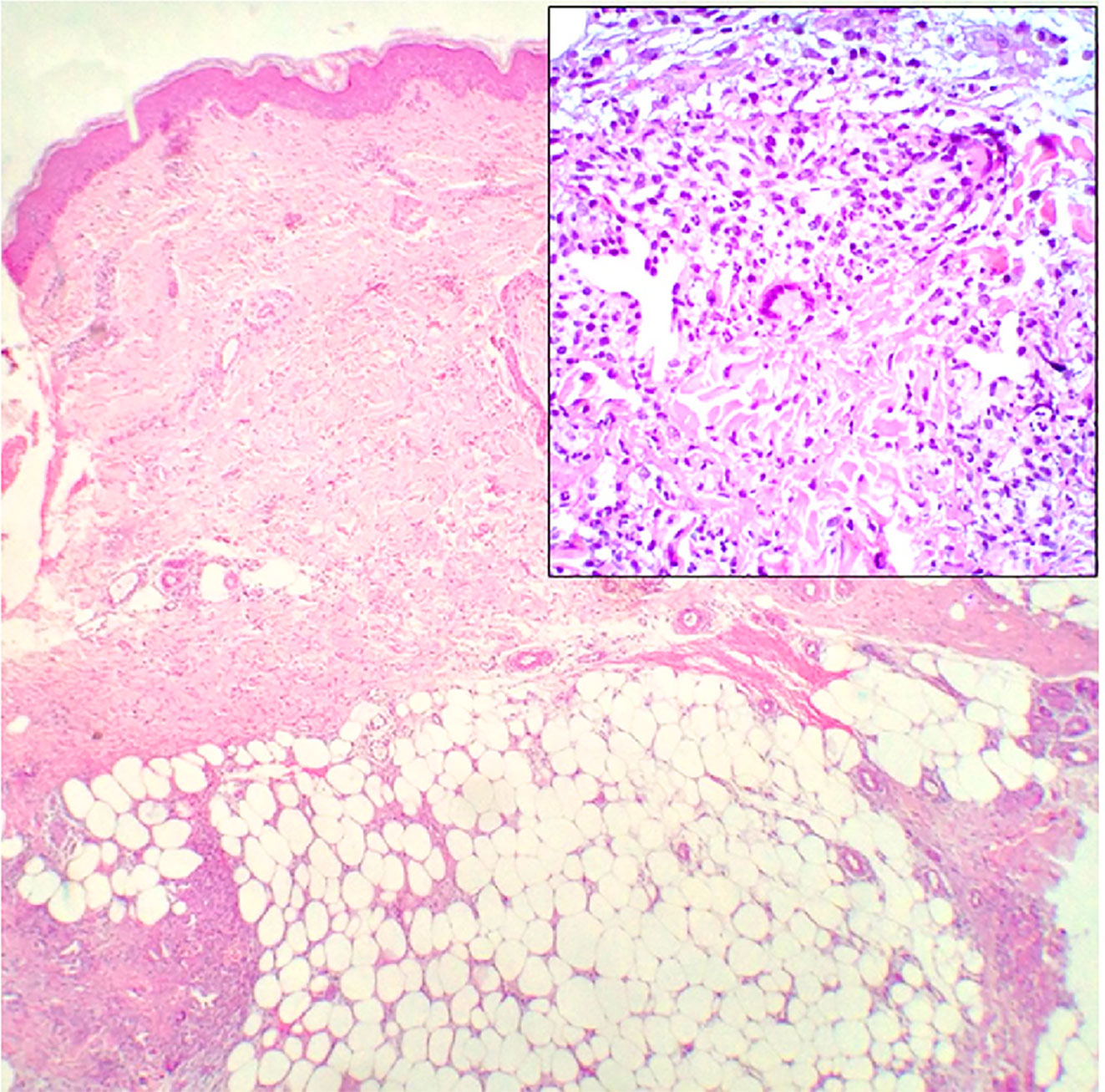
Figure 5 Histopathology of EN shows septal panniculitis and a mixed inflammatory infiltrate predominantly composed of lymphocytes, histiocytes, and neutrophils (H&E staining, ×40). Inset: Miescher’s radial granuloma (H&E staining, ×200). Reproduced with permission from (84). © 2016 The International Society of Dermatology.
The underlying pathogenesis of EN is complex and not fully understood, but part of the pathogenesis of EN is considered to involve type IV hypersensitivity reaction (71). Histopathology of the lesion site has revealed abundant infiltration of lymphocytes, histiocytes, and neutrophils in the lower dermis (86), with neutrophils usually predominating in the early lesions (55). Furthermore, a previous study found significant overexpression of genes for encoding Th1 cytokines (IL-2, IFN-γ) in cutaneous lesions and peripheral blood of patients with EN (87). Correspondingly, Simone et al. in 2016 also found elevated levels of Th1 cytokines in the lesional skin and serum of patients with EN with particularly high levels of IFN-γ and IL-12 (84). These findings provide direct evidence that a polarized Th1 immune response is present in the skin lesions of patients with EN. Simultaneously, high levels of cytokines mainly involved in neutrophil recruitment and activation, including IL-6, IL-8, TNF-α, granulocyte colony-stimulating factor, and MCP-1, are present in the skin of the lesion site and serum (84). However, Simone et al. found that IL-17 levels were low in both serum and skin samples in five patients, and that the balance between Th17 cells and Treg cells varied widely in the five enrolled patients. Therefore, they believe that IL-17 and Th17 cells play a negligible role in the pathogenesis of EN (84). Although IL-12 can induce IFN-γ production by Th1 cells, thereby enhancing the expression of chemokines to promote neutrophil recruitment and activation (88), IL-17 is also a powerful pro-neutrophil cytokine, critical for neutrophil proliferation, maturation, and chemotaxis (68). Given the primary EN, small sample size, and lack of healthy skin controls included in this study, the role of IL-17 and Th17 cells in EN secondary to IBD may be controversial. Shimizu et al. found significantly higher expression of IL-17 and TGF-β in EN secondary to Behçet’s syndrome compared to patients with primary EN (89). Moreover, another study found that STAT3, which is required for up-regulation of the master transcription factor RORγt, is not only overexpressed in IBD and PG but also significantly overexpressed in EN (90). Therefore, IL-17 and Th17 cells may be upregulated in EN secondary to IBD. Many adhesion molecules and inflammatory mediators are also involved in the development of EN. The expression of several adhesion molecules has been observed on the endothelial cells of patients with EN, such as E-selectin, P-selectin, platelet endothelial cell adhesion molecule-1, VCAM-1, and ICAM-1 (91). The formation of circulating immune complexes and deposition in and around venules of the connective tissue septa of the subcutaneous fat suggest that antigens and antibodies may also play an important role in the pathogenesis of EN (92, 93).
In cases of EN secondary to IBD, as the skin lesions reflect the activity and onset of intestinal disease, treatment of the underlying IBD usually results in remission of the skin manifestations (55). EN is usually self-limiting, even without specific treatment for the cause, and symptomatic supportive treatment including compression bandages, limb elevation (> 30 min twice daily), analgesics, and potassium iodide (300 mg three times daily, 2-3 weeks after remission) is typically sufficient for most patients (57, 71, 85). For severe or refractory cases, systemic corticosteroids (prednisone 20 mg for 7-10 days) may be considered as the first-line therapy (57, 61, 71). Anti-TNF treatment may serve as rescue therapy, especially in patients with active IBD. An analysis from the Swiss IBD Cohort Study showed the best response rate of up to 80% (8/10) with anti-TNF-α treatment for EN (94). A multicenter, open-label, Phase 3b study by Löfberg et al. showed that adalimumab (160/80 mg at 0 and 2 weeks, then 40 mg every other week) reduced the incidence of EN from 2.4% (23/945) at baseline to 0.4% (4/942) on the clinical endpoints at 20 weeks (95). Additionally, several case reports have highlighted the benefits of adalimumab (40 mg every 14 days) and infliximab (5 mg/kg at 0, 2, and 6 weeks) in the treatment of severe or refractory EN (96, 97). Recent studies have found that monoclonal antibodies against the IL-12/23p40 subunit may exert some clinical effects for the treatment of EN. A multicenter case series by Phillips et al. reported four complete remissions and one partial remission in five patients with EN treated with ustekinumab (78). A prospective multicenter study of 221 patients with CD (the initial intravenous infusion with ustekinumab based on weight [260 mg < 55 kg, 390 mg between 55 and 85 kg, 520 mg > 85 kg], 90 mg subcutaneously at 8 weeks, then 90 mg every 8 or 12 weeks) reported data on EN. Two patients had EN at baseline and 50% (1/2) of patients presented with the primary outcome of corticosteroid-free clinical remission. Three patients developed new-onset EN during follow-up, with a response rate of 66.7% (2/3) (98).
The underlying pathophysiological mechanisms of IBD and cutaneous EIMs are complex, diverse, and not fully understood. EIMs reportedly occur at a frequency of 6–47% in the lifetime of patients with IBD, with the skin being among the most commonly affected organs (7). There may be a common pathogenic association between IBD and cutaneous EIMs. Further, two different theories have been proposed to explain this potential pathogenic association. One is that EIMs are only independent inflammatory events that share a genetic background or environmental risk factors with IBD, and the other is that EIMs may be the result of an extraintestinal extension of intestinal inflammation (99). These mechanisms are not mutually exclusive and often multiple pathological mechanisms may interact to promote the occurrence and progression of skin EIMs. Understanding the potential association between IBD and cutaneous EIMs is critical for identifying targeted treatment strategies in the future.
Aberrant homing of gut-specific lymphocytes in IBD may contribute to extraintestinal skin manifestations of IBD (100). MAdCAM-1 is typically expressed exclusively in the gut, and its interaction with the α4β7 integrin selectively transports gut-tropic T cells to the intestinal mucosa (41). The interaction of the gut-expressed chemokine, CCL25, with its corresponding chemokine receptor, CCR9, also contributes to lymphocyte homing to the gut site (32). Thus, the ectopic expression of gut-specific adhesion molecules and chemokines may facilitate the transport of inflammatory T lymphocytes to extraintestinal sites. In PSC, ectopic expression of MAdCAM-1 and CCL25 on the vascular endothelium of the portal tract has been demonstrated (101, 102). However, apart from PSC, evidence that this condition can occur in organs or tissues other than the liver, especially in the skin, is scarce. Vavricka et al. analyzed 31 biopsy samples from EN and PG and showed that gut-specific MAdCAM-1 was not overexpressed in the skin (90). Despite the lack of gut-specific expression of MAdCAM-1 in the skin, a recent large real-life experience cohort study found that vedolizumab (an anti-human α4β7 integrin antibody) resulted in complete remission rates of up to 75% in the treatment of cutaneous EIMs (103). It is unclear whether this is a direct effect of vedolizumab on cutaneous EIMs or an indirect effect through the control of intestinal disease activity. Lymphocytes may also co-express gut-specific and skin-specific homing molecules, which may allow the tropism of lymphocytes in IBD toward skin sites (104). For example, Th17 cells can simultaneously express CCR4 and CCR6, which can mediate Th lymphocyte chemotaxis to skin sites and intestinal lymphoid tissues, respectively (27, 31). γδT cells can co-express integrin α4β7 and skin-homing molecules like cutaneous lymphocyte-associated antigen (CLA) (105) (Figure 1).
The up-regulation of non-gut-specific chemokines and adhesion molecules at inflammatory sites may also mediate the transport of T lymphocytes to extraintestinal sites. Both intestinal epithelial cells and skin keratinocytes secrete CCL20, a chemokine strongly upregulated by inflammatory stimulation, which attracts the recruitment of Th17 cells expressing its cognate receptor, CCR6 (27, 106) (Figure 1). CCL27 expressed by skin keratinocytes and CCL28 expressed by intestinal mucosal epithelial cells have high homology, and both chemotactically recruit T lymphocytes expressing CCR10 (Figure 1). Thus, inflammatory T lymphocytes derived from the gut and expressing CCR10 may be recruited to skin sites (107). Simultaneously, non-specific chemokines CXCL9, CXCL10, and CXCL11 are produced at the site of inflammation, which may attract Th1 cells and γδT cells from the intestinal site of patients with IBD expressing the receptor CXCR3 to the skin site (21, 105) (Figure 1). In addition, non-specific ligand and receptor interactions between lymphocytes and endothelial cells may also contribute to the transport of lymphocytes to extraintestinal sites. Human circulating γδT cells express LFA-1 (αLβ2), which primarily recognizes ICAM-1 expressed by endothelial cells and which further increases under inflammatory conditions and tissue stress (105) (Figure 1). Thus, in the context of a genetic predisposition of patients with IBD, low-grade inflammation, injury or mechanical stress of skin against external hazards, toxic substances, and pathogens may lead to increased concentrations and recruitment of Th1, Th17, and γδT cells in the gut at the diseased skin (99). In conclusion, specific and non-specific lymphocyte-endothelial cell interactions may induce ectopic homing of intestinal lymphocytes to skin sites, leading to the development of cutaneous EIMs.
Th1 and Th17 pathways and the TNF pathway play an important role in the occurrence and development of IBD. In IBD, exposure to pathogenic microorganisms, antigen presentation by APCs, and interaction of APCs with Th cells lead to macrophage activation. Activated macrophages produce cytokines with profound pro-inflammatory effects, including TNF-α, IL-12, and IL-23 (1). TNF-α binds to death receptors of the TNF receptor superfamily resulting in intracellular signaling (108), while IL-12 and IL-23 act by binding to IL-12 receptors (consisting of IL-12Rβ1 and IL-12Rβ2) and IL-23R (consisting of IL-12Rβ1 and a novel IL-23R), respectively (109, 110). The p40 subunit shared by IL-12/23 binds to the transmembrane IL-12Rβ1 on the cell surface (109, 111). A monoclonal antibody (ustekinumab) directed against the p40 subunit shared by IL-12 and IL-23, as well as a monoclonal antibody against TNF-α, are effective in the treatment of CD and UC (112–114). Therefore, inhibition of Th1 and Th17 signaling pathways and the TNF-α pathway has become a successful strategy for the treatment of IBD. Recent studies have shown that anti-TNF-α is effective in the treatment of cutaneous EIMs (94, 115), and ustekinumab is also effective in the control of cutaneous EIMs (116). An analysis based on the Swiss IBD Cohort Study showed that response rates to anti-TNF-α therapy were 80% (8/10) for EN and 50% (6/12) for PG (94). A prospective multicenter study of 221 patients with CD treated with ustekinumab reported data on EN. Two patients had EN at baseline and 50% of patients showed clinical remission. Three patients developed new EN during follow-up, with a response rate of 66.7% (98). Another retrospective multicenter study evaluated the efficacy of ustekinumab in neutrophilic dermatosis associated with CD. Four of the seven CD patients included had PG, with three complete healings and one partial response defined as more than 50% improvement of the skin lesions (79). These data suggest a common Th1 and Th17 pathway- and TNF-α pathway- based pathogenic association between IBD and cutaneous EIMs, and between different cutaneous EIMs (Figure 1). In fact, in early PG studies a large number of Th1 and Th17 cell infiltrates as well as high levels of TNF-α, IFN-γ, IL-12, IL-17, and IL-23 at the diseased skin have been reported (59). These cytokine profiles are similar to the results observed in PG-associated diseases such as IBD (1), suggesting that the Th1 and Th17 pathways, as well as the TNF-α pathway, underlie commonly, associating together these diseases. There is growing evidence that the Th1 and Th17 pathways, as well as the TNF-α pathway, are involved in the pathogenesis of cutaneous EIMs. However, it is unclear whether this is the result of an extension of intestinal disease activity to the skin or a widespread pro-inflammatory cytokine imbalance in genetically predisposed patients. However, the detection of high levels of TNF-α, Th1 cytokines, and Th17 cytokines in the sera of patients with IBD seems to confirm the latter possibility. These high levels of pro-inflammatory cytokines associated with IBD may lead to the occurrence of cutaneous EIMs by activating immune cells at the skin site.
Both the gut and skin are active, complex immune and neuroendocrine organs that are constantly exposed to the external environment and have a broad microbial community (117, 118). Many studies have associated gastrointestinal health with skin homeostasis and dysregulation and have proposed the concept of a gut-skin axis. There is evidence of a bi-directional interaction between the gut and the skin, which is mainly achieved through immuno-crosslinking (118, 119). Polkowska-Pruszynska et al. discussed the role of dysregulation of the gut microbiota in the pathogenesis of a range of autoimmune inflammatory skin diseases, including psoriasis, hidradenitis suppurativa, atopic dermatitis, and alopecia areata (119). Intestinal dysbacteriosis is associated with some less common but more serious skin diseases, such as EN and PG (120). Although specific skin disorders are associated with gut health and the balance of the microbiome within the gut, the exact mechanisms by which gut microbes affect the skin have not been fully elucidated. The cross-reactivity of antigens (molecular mimicry) plays an essential role in the pathogenesis of autoimmune diseases (121). Autoimmune reactions can occur when foreign antigens (from bacteria or viruses) have sequence and/or structural similarities to self-antigens (121). Molecular mimicry has typically been characterized for T cells and at the antibody level (121). Antigens from the gut microbiota are thought to be key targets for gut effector T cells (119). Similar pathogenic microbial communities between the gut and skin may exist (99) which produce similar microbial antigens. Thus, in the context of IBD, cells at the skin site may be reactivated by similar antigens or host cross-antigens of the gut and skin microbiota, thereby presenting skin EIMs. Mice with reduced or absent gut microbiota have significantly lower levels of γδT cells, Th17 cells, and Th1 cells both locally and systemically, and imiquimod can hardly induce the development of skin inflammation (122). Gut microbes drive psoriasis-like skin inflammation in mice by inducing stronger γδT cell, Th17 cell, and Th1 cell responses (122), which are likely to be cross-reactive bacterial antigens, providing evidence for a direct association between gut microbiota, cross-reactive autoantigens, and skin inflammation. Presently, whether modulating the gut microbiota can effectively target the treatment of cutaneous EIMs is uncertain. Satta et al. have shown that the use of probiotics is significantly inversely associated with the occurrence of skin lesions in patients with IBD (123).
Circulating autoantibodies can extend the intestinal immune inflammatory response to extraintestinal sites, and immune complex-mediated inflammation can contribute to cutaneous EIMs (124). Earlier studies have demonstrated the presence of autoantibodies which are reactive to colonic proteins in patients with IBD (125, 126), and share certain unique epitopes between the human colon and skin, as evidenced by the presence of monoclonal antibodies (127). However, the exact role of these autoantibodies or immune complexes in promoting the development of cutaneous EIMs in patients with IBD is unclear.
Neutrophils are the major circulating white blood cells in the human body. In healthy individuals, circulating neutrophils are in the “dormant state”. When neutrophils are exposed to infection and with subsequent release of inflammatory cytokines or other signals, these are primed, activated, and recruited to the site of infection, producing and releasing large amounts of antibacterial peptides, proteases, and reactive oxygen species (ROS), all of which contribute to host defense. Physiologically primed and activated neutrophils can be deprimed and deactivated and can return to circulation as quiescent neutrophils or eventually die by apoptosis. However, in inflammatory diseases, neutrophils can be highly primed and persistently activated, leading to tissue damage and excessive inflammatory responses (128). Circulating neutrophils in patients with IBD present morphological evidence of activation (129) and secrete higher levels of TNF-α and IL-1β (130). Primed neutrophils may fail to deprimed and then re-enter the bloodstream in a primed state (128) (Figure 1). Thus, in IBD, these neutrophils activated in the gut may be re-activated in the skin, resulting in skin damage and inflammation. The two most common cutaneous EIMs in IBD, PG and EN, present a large amount of neutrophil infiltration at the lesion site (55, 59). Magro et al. showed clonal neutrophil infiltration that is not associated with myeloproliferative disease in both PG and Sweet’s syndrome (131). A recent study by Jatana et al. demonstrated inflammatory skin-gut crosstalk is mediated by IL-1β-primed neutrophils in a murine model of PG with intestinal inflammation (132).
In addition to the above mechanisms, some pathological associations exist. In IBD, systemic inflammatory cytokines, leakage of microbial products into circulation due to increased intestinal permeability, or changes in metabolites of the intestinal microbiota can all lead to altered innate immunity (99). For example, the impaired pro-inflammatory cytokine secretion by macrophages in patients with CD results in a reduced recruitment of neutrophils to the skin, causing reduced or delayed bacterial clearance from the skin site (133, 134), and ultimately, resulting in inflammation or damage at the skin site. Short-chain fatty acids (SCFAs) produced by gut commensal microbes facilitate the generation of peripheral anti-inflammatory Treg cells (135). Several studies have confirmed that compared to healthy subjects, the abundance of SCFA-producing bacteria in the feces of patients with IBD decreases significantly (136, 137), further inhibiting the generation of peripheral Treg cells and causing the disruption of the balance between pro- and anti-inflammatory in the skin. Additionally, intestinal inflammation promotes the differentiation of fibroblast subsets in the intestinal stroma into preadipocytes with innate antimicrobial activity (138). In IBD, preadipocytes mature into “creeping fat” with independent pro-inflammatory functions, and this process depends on the presence of hyaluronan (HA) in the extracellular matrix (138, 139). Skin inflammation degrades the matrix composed of extracellular HA. The degraded HA fragments enter the intestine through the circulatory system, thereby exacerbating this reactive fat maturation process. Therefore, skin inflammation exacerbates intestinal inflammation through HA (138).
In summary, extraintestinal skin manifestations of IBD are common and its manifestations range from mild and self-limiting (e.g., EN) to severe and debilitating courses (e.g., PG). The presence of cutaneous EIMs significantly affects morbidity and mortality due to IBD. Control of intestinal inflammation is effective for cutaneous EIMs associated with intestinal disease activity (e.g., EN), and multiple biologic agents are efficacious in treating cutaneous EIMs that develop independently of intestinal disease activity (e.g., PG). Clinicians may consider this treatment option to manage cutaneous EIMs. However, prospective and controlled studies with larger sample sizes are needed to confirm the efficacy and safety of various biologics in cutaneous EIMs. Common genetic backgrounds and risk factors are increasingly being recognized. Although some parts of the pathogenic mechanisms remain controversial, they are not mutually exclusive and may contribute to different skin EIMs to different degrees and be the result of multiple pathological mechanisms acting together. Therefore, elucidating the potential pathological associations between IBD and cutaneous EIMs is expected to help identify more new therapeutic targets in the future.
RH wrote the first draft of the manuscript. RH and SZ prepared the figures and table. RH, SZ, and XW conceived the central idea and finalized the paper. SZ, MC, YC, JM, and JL prepared the material and collected the data. XW revised the manuscript critically for intellectual content and supported the funding. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Jilin Province [grant number 20210101275JC].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med (2020) 383:2652–64. doi: 10.1056/NEJMra2002697
2. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet (2017) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
3. Windsor JW, Kaplan GG. Evolving epidemiology of IBD. Curr Gastroenterol Rep (2019) 21:40. doi: 10.1007/s11894-019-0705-6
4. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol (2015) 12:205–17. doi: 10.1038/nrgastro.2015.34
5. Mak WY, Zhao M, Ng SC, Burisch J. The epidemiology of inflammatory bowel disease: East meets west. J Gastroenterol Hepatol (2020) 35:380–89. doi: 10.1111/jgh.14872
6. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature (2020) 578:527–39. doi: 10.1038/s41586-020-2025-2
7. Vavricka SR, Rogler G, Gantenbein C, Spoerri M, Prinz Vavricka M, Navarini AA, et al. Chronological order of appearance of extraintestinal manifestations relative to the time of IBD diagnosis in the swiss inflammatory bowel disease cohort. Inflammation Bowel Dis (2015) 21:1794–800. doi: 10.1097/MIB.0000000000000429
8. Rogler G, Singh A, Kavanaugh A, Rubin DT. Extraintestinal manifestations of inflammatory bowel disease: current concepts, treatment, and implications for disease management. Gastroenterology (2021) 161:1118–32. doi: 10.1053/j.gastro.2021.07.042
9. Marotto D, Atzeni F, Ardizzone S, Monteleone G, Giorgi V, Sarzi-Puttini P. Extra-intestinal manifestations of inflammatory bowel diseases. Pharmacol Res (2020) 161:105206. doi: 10.1016/j.phrs.2020.105206
10. Gordon H, Burisch J, Ellul P, Karmiris K, Katsanos K, Allocca M, et al. ECCO guidelines on extraintestinal manifestations in inflammatory bowel disease. J Crohns Colitis (2023). doi: 10.1093/ecco-jcc/jjad108
11. Hung Y-T, Le P-H, Kuo C-J, Tang Y-C, Chiou M-J, Chiu C-T, et al. The temporal relationships and associations between cutaneous manifestations and inflammatory bowel disease: A nationwide population-based cohort study. J Clin Med (2021) 10:1311. doi: 10.3390/jcm10061311
12. Farhi D, Cosnes J, Zizi N, Chosidow O, Seksik P, Beaugerie L, et al. Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: a cohort study of 2402 patients. Med (Baltimore) (2008) 87:281–93. doi: 10.1097/MD.0b013e318187cc9c
13. Vavricka SR, Brun L, Ballabeni P, Pittet V, Prinz Vavricka BM, Zeitz J, et al. Frequency and risk factors for extraintestinal manifestations in the Swiss inflammatory bowel disease cohort. Am J Gastroenterol (2011) 106:110–19. doi: 10.1038/ajg.2010.343
14. Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn's disease and ulcerative colitis. Inflammopharmacology (2012) 20:1–18. doi: 10.1007/s10787-011-0104-6
15. Veny M, Fernández-Clotet A, Panés J. Controlling leukocyte trafficking in IBD. Pharmacol Res (2020) 159:105050. doi: 10.1016/j.phrs.2020.105050
16. Wiendl M, Becker E, Müller TM, Voskens CJ, Neurath MF, Zundler S. Targeting immune cell trafficking - insights from research models and implications for future IBD therapy. Front Immunol (2021) 12:656452. doi: 10.3389/fimmu.2021.656452
17. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol (2019) 20:970–79. doi: 10.1038/s41590-019-0415-0
18. Nakase H. Treatment of inflammatory bowel disease from the immunological perspective. Immunol Med (2020) 43:79–86. doi: 10.1080/25785826.2020.1751934
19. Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med (1998) 187:129–34. doi: 10.1084/jem.187.1.129
20. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol (2007) 8:639–46. doi: 10.1038/ni1467
21. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89:207–15. doi: 10.1038/icb.2010.158
22. Shibahara T, Wilcox JN, Couse T, Madara JL. Characterization of epithelial chemoattractants for human intestinal intraepithelial lymphocytes. Gastroenterology (2001) 120:60–70. doi: 10.1053/gast.2001.20904
23. Ostvik AE, Granlund AVB, Bugge M, Nilsen NJ, Torp SH, Waldum HL, et al. Enhanced expression of CXCL10 in inflammatory bowel disease: potential role of mucosal Toll-like receptor 3 stimulation. Inflammation Bowel Dis (2013) 19:265–74. doi: 10.1002/ibd.23034
24. Schroepf S, Kappler R, Brand S, Prell C, Lohse P, Glas J, et al. Strong overexpression of CXCR3 axis components in childhood inflammatory bowel disease. Inflammation Bowel Dis (2010) 16:1882–90. doi: 10.1002/ibd.21312
25. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
26. Zhao Q, Kim T, Pang J, Sun W, Yang X, Wang J, et al. A novel function of CXCL10 in mediating monocyte production of proinflammatory cytokines. J Leukoc Biol (2017) 102:1271–80. doi: 10.1189/jlb.5A0717-302
27. Lee AYS, Eri R, Lyons AB, Grimm MC, Korner H. CC chemokine ligand 20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: odd couple or axis of evil? Front Immunol (2013) 4:194. doi: 10.3389/fimmu.2013.00194
28. Schirmer L, Rothhammer V, Hemmer B, Korn T. Enriched CD161high CCR6+ γδ T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol (2013) 70:345–51. doi: 10.1001/2013.jamaneurol.409
29. Kaser A, Ludwiczek O, Holzmann S, Moschen AR, Weiss G, Enrich B, et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol (2004) 24:74–85. doi: 10.1023/B:JOCI.0000018066.46279.6b
30. Marafini I, Monteleone I, Dinallo V, Di Fusco D, De Simone V, Laudisi F, et al. CCL20 is negatively regulated by TGF-β1 in intestinal epithelial cells and reduced in crohn's disease patients with a successful response to mongersen, a smad7 antisense oligonucleotide. J Crohns Colitis (2017) 11:603–09. doi: 10.1093/ecco-jcc/jjw191
31. Soler D, Humphreys TL, Spinola SM, Campbell JJ. CCR4 versus CCR10 in human cutaneous TH lymphocyte trafficking. Blood (2003) 101:1677–82. doi: 10.1182/blood-2002-07-2348
32. Trivedi PJ, Bruns T, Ward S, Mai M, Schmidt C, Hirschfield GM, et al. Intestinal CCL25 expression is increased in colitis and correlates with inflammatory activity. J Autoimmun (2016) 68:98–104. doi: 10.1016/j.jaut.2016.01.001
33. Papadakis KA, Prehn J, Moreno ST, Cheng L, Kouroumalis EA, Deem R, et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn's disease. Gastroenterology (2001) 121:246–54. doi: 10.1053/gast.2001.27154
34. Saruta M, Yu QT, Avanesyan A, Fleshner PR, Targan SR, Papadakis KA. Phenotype and effector function of CC chemokine receptor 9-expressing lymphocytes in small intestinal Crohn's disease. J Immunol (2007) 178:3293–300. doi: 10.4049/jimmunol.178.5.3293
35. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol (2023) 20:217–51. doi: 10.1038/s41423-023-00974-6
36. Zhou GX, Liu ZJ. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J Dig Dis (2017) 18:495–503. doi: 10.1111/1751-2980.12540
37. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science (1996) 272:60–6. doi: 10.1126/science.272.5258.60
38. von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med (2000) 343:1020–34. doi: 10.1056/NEJM200010053431407
39. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
40. Biancheri P, Di Sabatino A, Rovedatti L, Giuffrida P, Calarota SA, Vetrano S, et al. Effect of tumor necrosis factor-α blockade on mucosal addressin cell-adhesion molecule-1 in Crohn's disease. Inflammation Bowel Dis (2013) 19:259–64. doi: 10.1097/MIB.0b013e31828100a4
41. Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol (1999) 72:209–53. doi: 10.1016/S0065-2776(08)60022-X
42. Nakase H, Sato N, Mizuno N, Ikawa Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun Rev (2022) 21:103017. doi: 10.1016/j.autrev.2021.103017
43. Dong C. Cytokine regulation and function in T cells. Annu Rev Immunol (2021) 39:51–76. doi: 10.1146/annurev-immunol-061020-053702
44. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature (2008) 453:1051–57. doi: 10.1038/nature07036
45. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol (2018) 15:458–69. doi: 10.1038/s41423-018-0004-4
46. Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. Int J Mol Sci (2021) 22:7618. doi: 10.3390/ijms22147618
47. Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukoc Biol (2002) 71:1–8.
48. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi: 10.1189/jlb.0603252
49. Jang D-I, Lee AH, Shin H-Y, Song H-R, Park J-H, Kang T-B, et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α Inhibitors in therapeutics. Int J Mol Sci (2021) 22:2719. doi: 10.3390/ijms22052719
50. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14:329–42. doi: 10.1038/nri3661
51. Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a "fine-tuning cytokine" in inflammation processes. Arthritis Rheum (2001) 44:2176–84. doi: 10.1002/1529-0131(200109)44:9<2176::AID-ART371>3.0.CO;2-4
52. Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol (1999) 162:494–502. doi: 10.4049/jimmunol.162.1.494
53. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis (2021) 12:864. doi: 10.1038/s41419-021-04151-3
54. Comerford I, Kara EE, McKenzie DR, McColl SR. Advances in understanding the pathogenesis of autoimmune disorders: focus on chemokines and lymphocyte trafficking. Br J Haematol (2014) 164:329–41. doi: 10.1111/bjh.12616
55. Marzano AV, Borghi A, Stadnicki A, Crosti C, Cugno M. Cutaneous manifestations in patients with inflammatory bowel diseases: pathophysiology, clinical features, and therapy. Inflammation Bowel Dis (2014) 20:213–27. doi: 10.1097/01.MIB.0000436959.62286.f9
56. States V, O'Brien S, Rai JP, Roberts HL, Paas M, Feagins K, et al. Pyoderma gangrenosum in inflammatory bowel disease: A systematic review and meta-analysis. Dig Dis Sci (2020) 65:2675–85. doi: 10.1007/s10620-019-05999-4
57. Vavricka SR, Schoepfer A, Scharl M, Lakatos PL, Navarini A, Rogler G. Extraintestinal manifestations of inflammatory bowel disease. Inflammation Bowel Dis (2015) 21:1982–92. doi: 10.1097/MIB.0000000000000392
58. Xu A, Balgobind A, Strunk A, Garg A, Alloo A. Prevalence estimates for pyoderma gangrenosum in the United States: An age- and sex-adjusted population analysis. J Am Acad Dermatol (2020) 83:425–29. doi: 10.1016/j.jaad.2019.08.001
59. Maverakis E, Marzano AV, Le ST, Callen JP, Brüggen M-C, Guenova E, et al. Pyoderma gangrenosum. Nat Rev Dis Primers (2020) 6:81. doi: 10.1038/s41572-020-0213-x
60. Rothfuss KS, Stange EF, Herrlinger KR. Extraintestinal manifestations and complications in inflammatory bowel diseases. World J Gastroenterol (2006) 12:4819–31. doi: 10.3748/wjg.v12.i30.4819
61. Greuter T, Navarini A, Vavricka SR. Skin manifestations of inflammatory bowel disease. Clin Rev Allergy Immunol (2017) 53:413–27. doi: 10.1007/s12016-017-8617-4
62. Kim YJ, Lee KH, Won CH, Chang SE, Lee MW, Choi JH, et al. Clinicopathological features and prognosis of pyoderma gangrenosum in Korea: A single centre, retrospective, observational study over 20 years. Indian J Dermatol Venereol Leprol (2023) 89:25–34. doi: 10.25259/IJDVL_968_20
63. Kridin K, Cohen AD, Amber KT. Underlying systemic diseases in pyoderma gangrenosum: A systematic review and meta-analysis. Am J Clin Dermatol (2018) 19:479–87. doi: 10.1007/s40257-018-0356-7
64. Brooklyn T, Dunnill G, Probert C. Diagnosis and treatment of pyoderma gangrenosum. BMJ (2006) 333:181–84. doi: 10.1136/bmj.333.7560.181
65. Wang EA, Steel A, Luxardi G, Mitra A, Patel F, Cheng MY, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: A hypothesis based on molecular and clinicopathologic studies. Front Immunol (2017) 8:1980. doi: 10.3389/fimmu.2017.01980
66. Wollina U. Clinical management of pyoderma gangrenosum. Am J Clin Dermatol (2002) 3:149–58. doi: 10.2165/00128071-200203030-00002
67. Caproni M, Antiga E, Volpi W, Verdelli A, Venegoni L, Quaglino P, et al. The Treg/Th17 cell ratio is reduced in the skin lesions of patients with pyoderma gangrenosum. Br J Dermatol (2015) 173:275–78. doi: 10.1111/bjd.13670
68. Bettelli E, Kuchroo VK. IL-12- and IL-23-induced T helper cell subsets: birds of the same feather flock together. J Exp Med (2005) 201:169–71. doi: 10.1084/jem.20042279
69. Guenova E, Teske A, Fehrenbacher B, Hoerber S, Adamczyk A, Schaller M, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol (2011) 147:1203–5. doi: 10.1001/archdermatol.2011.168
70. Antiga E, Maglie R, Volpi W, Bianchi B, Berti E, Marzano AV, et al. T helper type 1-related molecules as well as interleukin-15 are hyperexpressed in the skin lesions of patients with pyoderma gangrenosum. Clin Exp Immunol (2017) 189:383–91. doi: 10.1111/cei.12989
71. Pagani K, Lukac D, Bhukhan A, McGee JS. Cutaneous manifestations of inflammatory bowel disease: A basic overview. Am J Clin Dermatol (2022) 23:481–97. doi: 10.1007/s40257-022-00689-w
72. Din RS, Tsiaras WG, Li DG, Mostaghimi A. Efficacy of systemic dapsone treatment for pyoderma gangrenosum: A retrospective review. J Drugs Dermatol (2018) 17:1058–60.
73. Brooklyn TN, Dunnill MGS, Shetty A, Bowden JJ, Williams JDL, Griffiths CEM, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut (2006) 55:505–09. doi: 10.1136/gut.2005.074815
74. Argüelles-Arias F, Castro-Laria L, Lobatón T, Aguas-Peris M, Rojas-Feria M, Barreiro-de Acosta M, et al. Characteristics and treatment of pyoderma gangrenosum in inflammatory bowel disease. Dig Dis Sci (2013) 58:2949–54. doi: 10.1007/s10620-013-2762-2
75. Regueiro M, Valentine J, Plevy S, Fleisher MR, Lichtenstein GR. Infliximab for treatment of pyoderma gangrenosum associated with inflammatory bowel disease. Am J Gastroenterol (2003) 98:1821–26. doi: 10.1111/j.1572-0241.2003.07581.x
76. Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn's disease. Inflammation Bowel Dis (2009) 15:803–06. doi: 10.1002/ibd.20748
77. Sagami S, Ueno Y, Tanaka S, Nagai K, Hayashi R, Chayama K. Successful use of adalimumab for treating pyoderma gangrenosum with ulcerative colitis under corticosteroid-tapering conditions. Intern Med (2015) 54:2167–72. doi: 10.2169/internalmedicine.54.4853
78. Phillips FM, Verstockt B, Sebastian S, Ribaldone D, Vavricka S, Katsanos K, et al. Inflammatory cutaneous lesions in inflammatory bowel disease treated with vedolizumab or ustekinumab: an ECCO CONFER multicentre case series. J Crohns Colitis (2020) 14:1488–93. doi: 10.1093/ecco-jcc/jjaa078
79. de Risi-Pugliese T, Seksik P, Bouaziz J-D, Chasset F, Moguelet P, Gornet J-M, et al. Ustekinumab treatment for neutrophilic dermatoses associated with Crohn's disease: A multicenter retrospective study. J Am Acad Dermatol (2019) 80:781–84. doi: 10.1016/j.jaad.2018.06.065
80. Schwartz RA, Nervi SJ. Erythema nodosum: a sign of systemic disease. Am Fam Physician (2007) 75:695–700.
81. Antonelli E, Bassotti G, Tramontana M, Hansel K, Stingeni L, Ardizzone S, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med (2021) 10. doi: 10.3390/jcm10020364
82. Leung AKC, Leong KF, Lam JM. Erythema nodosum. World J Pediatr (2018) 14:548–54. doi: 10.1007/s12519-018-0191-1
83. Requena L, Yus ES. Erythema nodosum. Dermatol Clin (2008) 26:425–38. doi: 10.1016/j.det.2008.05.014
84. De Simone C, Caldarola G, Scaldaferri F, Petito V, Perino F, Arena V, et al. Clinical, histopathological, and immunological evaluation of a series of patients with erythema nodosum. Int J Dermatol (2016) 55:e289–94. doi: 10.1111/ijd.13212
85. Blake T, Manahan M, Rodins K. Erythema nodosum - a review of an uncommon panniculitis. Dermatol Online J (2014) 20:22376. doi: 10.5070/D3204022376
86. Pérez-Garza DM, Chavez-Alvarez S, Ocampo-Candiani J, Gomez-Flores M. Erythema nodosum: A practical approach and diagnostic algorithm. Am J Clin Dermatol (2021) 22:367–78. doi: 10.1007/s40257-021-00592-w
87. Llorente L, Richaud-Patin Y, Alvarado C, Reyes E, Alcocer-Varela J, Orozco-Topete R. Elevated Th1 cytokine mRNA in skin biopsies and peripheral circulation in patients with erythema nodosum. Eur Cytokine Netw (1997) 8:67–71.
88. Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol (2010) 162:100–07. doi: 10.1111/j.1365-2249.2010.04201.x
89. Shimizu J, Izumi T, Arimitsu N, Fujiwara N, Ueda Y, Wakisaka S, et al. Skewed TGFβ/Smad signalling pathway in T cells in patients with Behçet's disease. Clin Exp Rheumatol (2012) 30:S35–9.
90. Vavricka SR, Galván JA, Dawson H, Soltermann A, Biedermann L, Scharl M, et al. Expression patterns of TNFα, MAdCAM1, and STAT3 in intestinal and skin manifestations of inflammatory bowel disease. J Crohns Colitis (2018) 12:347–54. doi: 10.1093/ecco-jcc/jjx158
91. Demirkesen C, Tüzüner N, Senocak M, Türkmen I, Aki H, Kepil N, et al. Comparative study of adhesion molecule expression in nodular lesions of Behçet syndrome and other forms of panniculitis. Am J Clin Pathol (2008) 130:28–33. doi: 10.1309/3YJG2AWU06H0NX8G
92. Jones JV, Cumming RH, Asplin CM. Evidence for circulating immune complexes in erythema nodosum and early sarcoidosis. Ann N Y Acad Sci (1976) 278:212–19. doi: 10.1111/j.1749-6632.1976.tb47032.x
93. Requena L, Sánchez Yus E. Erythema nodosum. Semin Cutan Med Surg (2007) 26:114–25. doi: 10.1016/j.sder.2007.02.009
94. Vavricka SR, Gubler M, Gantenbein C, Spoerri M, Froehlich F, Seibold F, et al. Anti-TNF treatment for extraintestinal manifestations of inflammatory bowel disease in the swiss IBD cohort study. Inflammation Bowel Dis (2017) 23:1174–81. doi: 10.1097/MIB.0000000000001109
95. Löfberg R, Louis EV, Reinisch W, Robinson AM, Kron M, Camez A, et al. Adalimumab produces clinical remission and reduces extraintestinal manifestations in Crohn's disease: results from CARE. Inflammation Bowel Dis (2012) 18:1–9. doi: 10.1002/ibd.21663
96. Ortego-Centeno N, Callejas-Rubio JL, Sanchez-Cano D, Caballero-Morales T. Refractory chronic erythema nodosum successfully treated with adalimumab. J Eur Acad Dermatol Venereol (2007) 21:408–10. doi: 10.1111/j.1468-3083.2006.01893.x
97. Clayton TH, Walker BP, Stables GI. Treatment of chronic erythema nodosum with infliximab. Clin Exp Dermatol (2006) 31:823–24. doi: 10.1111/j.1365-2230.2006.02221.x
98. Biemans VBC, van der Meulen-de Jong AE, van der Woude CJ, Löwenberg M, Dijkstra G, Oldenburg B, et al. Ustekinumab for crohn's disease: results of the ICC registry, a nationwide prospective observational cohort study. J Crohns Colitis (2020) 14:33–45. doi: 10.1093/ecco-jcc/jjz119
99. Hedin CRH, Vavricka SR, Stagg AJ, Schoepfer A, Raine T, Puig L, et al. The pathogenesis of extraintestinal manifestations: implications for IBD research, diagnosis, and therapy. J Crohns Colitis (2019) 13:541–54. doi: 10.1093/ecco-jcc/jjy191
100. Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol (2006) 6:244–51. doi: 10.1038/nri1784
101. Grant AJ, Lalor PF, Hübscher SG, Briskin M, Adams DH. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology (2001) 33:1065–72. doi: 10.1053/jhep.2001.24231
102. Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hübscher SG, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med (2004) 200:1511–17. doi: 10.1084/jem.20041035
103. Tadbiri S, Peyrin-Biroulet L, Serrero M, Filippi J, Pariente B, Roblin X, et al. Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther (2018) 47:485–93. doi: 10.1111/apt.14419
104. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet (2002) 359:150–57. doi: 10.1016/S0140-6736(02)07374-9
105. Mann ER, McCarthy NE, Peake STC, Milestone AN, Al-Hassi HO, Bernardo D, et al. Skin- and gut-homing molecules on human circulating γδ T cells and their dysregulation in inflammatory bowel disease. Clin Exp Immunol (2012) 170:122–30. doi: 10.1111/j.1365-2249.2012.04649.x
106. Furue M, Furue K, Tsuji G, Nakahara T. Interleukin-17A and keratinocytes in psoriasis. Int J Mol Sci (2020) 21:1275. doi: 10.3390/ijms21041275
107. Xiong N, Fu Y, Hu S, Xia M, Yang J. CCR10 and its ligands in regulation of epithelial immunity and diseases. Protein Cell (2012) 3:571–80. doi: 10.1007/s13238-012-2927-3
108. Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech (2000) 50:184–95. doi: 10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H
109. Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, et al. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A (1996) 93:14002–07. doi: 10.1073/pnas.93.24.14002
110. Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev (2004) 202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x
111. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol (2002) 168:5699–708. doi: 10.4049/jimmunol.168.11.5699
112. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for crohn's disease. N Engl J Med (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
113. Sands BE, Sandborn WJ, Panaccione R, O'Brien CD, Zhang H, Johanns J, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med (2019) 381:1201–14. doi: 10.1056/NEJMoa1900750
114. Jeong DY, Kim S, Son MJ, Son CY, Kim JY, Kronbichler A, et al. Induction and maintenance treatment of inflammatory bowel disease: A comprehensive review. Autoimmun Rev (2019) 18:439–54. doi: 10.1016/j.autrev.2019.03.002
115. Peyrin-Biroulet L, Van Assche G, Gómez-Ulloa D, García-Álvarez L, Lara N, Black CM, et al. Systematic review of tumor necrosis factor antagonists in extraintestinal manifestations in inflammatory bowel disease. Clin Gastroenterol Hepatol (2017) 15:25–36.e27. doi: 10.1016/j.cgh.2016.06.025
116. Guillo L, D'Amico F, Danese S, Peyrin-Biroulet L. Ustekinumab for extra-intestinal manifestations of inflammatory bowel disease: A systematic literature review. J Crohns Colitis (2021) 15:1236–43. doi: 10.1093/ecco-jcc/jjaa260
117. Coates M, Lee MJ, Norton D, MacLeod AS. The skin and intestinal microbiota and their specific innate immune systems. Front Immunol (2019) 10:2950. doi: 10.3389/fimmu.2019.02950
118. Salem I, Ramser A, Isham N, Ghannoum MA. The gut microbiome as a major regulator of the gut-skin axis. Front Microbiol (2018) 9:1459. doi: 10.3389/fmicb.2018.01459
119. Polkowska-Pruszyńska B, Gerkowicz A, Krasowska D. The gut microbiome alterations in allergic and inflammatory skin diseases - an update. J Eur Acad Dermatol Venereol (2020) 34:455–64. doi: 10.1111/jdv.15951
120. Szántó M, Dózsa A, Antal D, Szabó K, Kemény L, Bai P. Targeting the gut-skin axis-Probiotics as new tools for skin disorder management? Exp Dermatol (2019) 28:1210–18. doi: 10.1111/exd.14016
121. Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol (2012) 42:102–11. doi: 10.1007/s12016-011-8294-7
122. Zákostelská Z, Málková J, Klimešová K, Rossmann P, Hornová M, Novosádová I, et al. Intestinal microbiota promotes psoriasis-like skin inflammation by enhancing th17 response. PLoS One (2016) 11:e0159539. doi: 10.1371/journal.pone.0159539
123. Satta R, Pes GM, Rocchi C, Pes MC, Dore MP. Is probiotic use beneficial for skin lesions in patients with inflammatory bowel disease? J Dermatolog Treat (2019) 30:612–16. doi: 10.1080/09546634.2018.1527998
124. Fernández Bussy R, Crisci CD, Fiorenza G, Bertoya N. Evaluation of circulating immune complexes in cutaneous diseases associated with immune disorders. Allergol Immunopathol (Madr) (1990) 18:47–52.
125. Biancone L, Mandal A, Yang H, Dasgupta T, Paoluzi AO, Marcheggiano A, et al. Production of immunoglobulin G and G1 antibodies to cytoskeletal protein by lamina propria cells in ulcerative colitis. Gastroenterology (1995) 109:3–12. doi: 10.1016/0016-5085(95)90263-5
126. Geng X, Biancone L, Dai HH, Lin JJ, Yoshizaki N, Dasgupta A, et al. Tropomyosin isoforms in intestinal mucosa: production of autoantibodies to tropomyosin isoforms in ulcerative colitis. Gastroenterology (1998) 114:912–22. doi: 10.1016/S0016-5085(98)70310-5
127. Das KM, Vecchi M, Sakamaki S. A shared and unique epitope(s) on human colon, skin, and biliary epithelium detected by a monoclonal antibody. Gastroenterology (1990) 98:464–69. doi: 10.1016/0016-5085(90)90839-S
128. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie J-C, Gougerot-Pocidalo M-A, Dang PM-C. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev (2016) 273:180–93. doi: 10.1111/imr.12447
129. McCarthy DA, Rampton DS, Liu YC. Peripheral blood neutrophils in inflammatory bowel disease: morphological evidence of in vivo activation in active disease. Clin Exp Immunol (1991) 86:489–93. doi: 10.1111/j.1365-2249.1991.tb02958.x
130. Nikolaus S, Bauditz J, Gionchetti P, Witt C, Lochs H, Schreiber S. Increased secretion of pro-inflammatory cytokines by circulating polymorphonuclear neutrophils and regulation by interleukin 10 during intestinal inflammation. Gut (1998) 42:470–76. doi: 10.1136/gut.42.4.470
131. Magro CM, Kiani B, Li J, Crowson AN. Clonality in the setting of Sweet's syndrome and pyoderma gangrenosum is not limited to underlying myeloproliferative disease. J Cutan Pathol (2007) 34:526–34. doi: 10.1111/j.1600-0560.2006.00654.x
132. Jatana S, Ponti AK, Johnson EE, Rebert NA, Smith JL, Fulmer CG, et al. A novel murine model of pyoderma gangrenosum reveals that inflammatory skin-gut crosstalk is mediated by IL-1β-primed neutrophils. Front Immunol (2023) 14:1148893. doi: 10.3389/fimmu.2023.1148893
133. Smith AM, Rahman FZ, Hayee BH, Graham SJ, Marks DJB, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med (2009) 206:1883–97. doi: 10.1084/jem.20091233
134. Marks DJB, Harbord MWN, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, et al. Defective acute inflammation in Crohn's disease: a clinical investigation. Lancet (2006) 367:668–78. doi: 10.1016/S0140-6736(06)68265-2
135. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–55. doi: 10.1038/nature12726
136. Eeckhaut V, Machiels K, Perrier C, Romero C, Maes S, Flahou B, et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut (2013) 62:1745–52. doi: 10.1136/gutjnl-2012-303611
137. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
138. Dokoshi T, Seidman JS, Cavagnero KJ, Li F, Liggins MC, Taylor BC, et al. Skin inflammation activates intestinal stromal fibroblasts and promotes colitis. J Clin Invest (2021) 131:e147614. doi: 10.1172/JCI147614
Keywords: inflammatory bowel disease, Crohn’s disease, ulcerative colitis, extraintestinal manifestations (EIMs), cutaneous manifestations, pathophysiological associations
Citation: He R, Zhao S, Cui M, Chen Y, Ma J, Li J and Wang X (2023) Cutaneous manifestations of inflammatory bowel disease: basic characteristics, therapy, and potential pathophysiological associations. Front. Immunol. 14:1234535. doi: 10.3389/fimmu.2023.1234535
Received: 04 June 2023; Accepted: 16 October 2023;
Published: 26 October 2023.
Edited by:
Giorgia Bodini, San Martino Hospital (IRCCS), ItalyReviewed by:
Maria Manuela Estevinho, Centro Hospitalar de Vila Nova de Gaia, PortugalCopyright © 2023 He, Zhao, Cui, Chen, Ma, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodong Wang, d2FuZ3hpYW9kb25nQGpsdS5lZHUuY24=
†ORCID: He Ronghua, orcid.org/0000-0001-5748-8885
Wang Xiaodong, orcid.org/0000-0001-5924-4649
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.