
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 July 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1232629
This article is part of the Research TopicInflammasomes in Human Diseases and MetabolismView all 8 articles
 Miguel A. Ortega1,2*†
Miguel A. Ortega1,2*† Diego De Leon-Oliva1,2†
Diego De Leon-Oliva1,2† Cielo García-Montero1,2
Cielo García-Montero1,2 Oscar Fraile-Martinez1,2
Oscar Fraile-Martinez1,2 Diego Liviu Boaru1,2Amador Velazquez de Castro1,2Miguel A. Saez1,2,3Laura Lopez-Gonzalez2,4
Diego Liviu Boaru1,2Amador Velazquez de Castro1,2Miguel A. Saez1,2,3Laura Lopez-Gonzalez2,4 Julia Bujan1,2
Julia Bujan1,2 Miguel Angel Alvarez-Mon1,2,5
Miguel Angel Alvarez-Mon1,2,5 Natalio García-Honduvilla1,2Raul Diaz-Pedrero2,4,6‡
Natalio García-Honduvilla1,2Raul Diaz-Pedrero2,4,6‡ Melchor Alvarez-Mon1,2,7‡
Melchor Alvarez-Mon1,2,7‡Inflammasomes are multiprotein signaling platforms in the cytosol that senses exogenous and endogenous danger signals and respond with the maturation and secretion of IL-1β and IL-18 and pyroptosis to induce inflammation and protect the host. The inflammasome best studied is the Nucleotide-binding oligomerization domain, leucine-rich repeat-containing family pyrin domain containing 3 (NLRP3) inflammasome. It is activated in a two-step process: the priming and the activation, leading to sensor NLRP3 oligomerization and recruitment of both adaptor ASC and executioner pro-caspase 1, which is activated by cleavage. Moreover, NLRP3 inflammasome activation is regulated by posttranslational modifications, including ubiquitination/deubiquitination, phosphorylation/dephosphorylation, acetylation/deacetylation, SUMOylation and nitrosylation, and interaction with NLPR3 protein binding partners. Moreover, the connection between it and metabolism is receiving increasing attention in this field. In this review, we present the structure, functions, activation, and regulation of NLRP3, with special emphasis on regulation by mitochondrial dysfunction-mtROS production and metabolic signals, i.e., metabolites as well as enzymes. By understanding the regulation of NLRP3 inflammasome activation, specific inhibitors can be rationally designed for the treatment and prevention of various immune- or metabolic-based diseases. Lastly, we review current NLRP3 inflammasome inhibitors and their mechanism of action.
The innate immune system plays a crucial role in protecting the body against various pathogens and maintaining tissue homeostasis. In recent years, the relationship between the innate immune system, inflammation, metabolism, and metabolic diseases has gained significant attention (1–4). It is now recognized that dysregulation of metabolic processes can profoundly impact immune responses and contribute to the development of metabolic diseases, such as obesity, type 2 diabetes, and cardiovascular disorders (5–7).
In this context, inflammasomes, multiprotein signaling platforms, have emerged as critical players linking metabolism and inflammation (8, 9). Inflammasomes are multiprotein signaling platforms that detect exogenous and endogenous danger signals in the cytosol and respond activating caspase-1 which leads to production and release of proinflammatory cytokines IL-1β and IL-18 (10). Likewise, inflammasome is capable of inducing pyroptosis (also known as inflammatory cell necrosis), a specialized form of programmed cell death that is triggered by inflammatory signaling pathways characterized by the rapid and lytic cell death accompanied by the release of pro-inflammatory cytokines and cellular contents (11). Therefore, inflammasomes are an important component of innate immunity that ensure protective responses against pathogens and tissue homeostasis (12–14). In addition to their role in detecting danger signals, inflammasomes have been implicated in metabolic dysfunction, insulin resistance, and the development of metabolic diseases. The activation of inflammasomes can disrupt insulin signaling, promote chronic low-grade inflammation, and contribute to metabolic imbalances (15–18). Moreover, inflammasomes have recently been linked to others programmed cell deaths (PCD), besides pyroptosis, including apoptosis, necroptosis, PANoptosis and ferroptosis (19–23). PCD is an essential process that preserve tissue homeostasis, eliminate damaged cells and regulate immunological responses (24, 25). Apoptosis, necroptosis, PANoptosis and ferroptosis have drawn the most interest because of their consequences in both physiological and medical conditions. Recent studies have shown the complex interaction between the inflammasome and these programmed cellular deaths, highlighting how this interaction affects inflammatory reactions and disease progression (19).
There exist different types of inflammasomes. Nucleotide-binding oligomerization domain (NOD), leucine-rich repeat-containing (LRR) (NLR) family pyrin domain containing 3 (NLRP3) inflammasome is the most studied. NLRP3 mutation have been associated with cryopyrin-associated periodic syndrome (CAPS), a group of autoinflammatory disorders characterized by recurrent fevers and systemic inflammation (26).
This review aims to provide a comprehensive overview of the current understanding of the interplay between metabolism and NLRP3 inflammasome activation. We will delve into the molecular mechanisms through which metabolic factors influence NLRP3 inflammasome signaling, emphasizing the role of mitochondrial dysfunction-mitochondrial reactive oxygen species (mtROS) production, metabolites and metabolic pathways in this process. Furthermore, we will explore the therapeutic opportunities that arise from targeting metabolic pathways to modulate NLRP3 inflammasome activation, offering potential strategies for the development of novel anti-inflammatory therapies.
Inflammasomes are composed of the sensors/receptors of the danger signals, the adaptor and the effector. There are a variety of sensors/receptors that classify the distinct inflammasomes which are activated by different stimuli: NLRP1, NLRP3, NLRC4, AIM2 or pyrin (27). The adaptor protein is the apoptosis-associated speck-like protein containing a CARD (ASC) that recruits and activates pro-caspase-1 (28). Finally, the effector is the pro-caspase-1 which is activated by cleavage into caspase-1 (29). NLRP3 is a 1036 amino acid protein that contains three domains, an amino (N)-terminal pyrin domain (PYD), a central NACHT domain and a carboxy (C)-terminal LRR. PYD (3–91) consists of six antiparallel α-helix, is a member of the death domain-fold superfamily and mediates the supramolecular complex formation, probably due to its ability of dimerization (30). The NACHT (NAIP, CIITA, HET-E, TP1) domain (131–649) is responsible for the oligomerization and presents ATPase activity. Is subdivided into FISNA (131–218), NBD (219–372), HD1 (373–434), WHD (435–541) and HD2 (542–649) domains (31). The LRR domain (650-1036) at C-terminal is involved in the activation and ligand sensing of the NLRP3 inflammasome (32, 33) and binds a NIMA-related kinase 7 (NEK7), an essential mediator of NLRP3 activation (34). ASC presents an N-terminal PYD and a C-terminal CARD which allow the interaction with NLRP3 and pro-caspase-1, respectively (35) (see Figure 1). Indeed, ASC oligomerization, by assembling into filaments, leads to a supramolecular complex formation, ASC speck, that generates numerous potential sites for caspase-1 activation and serves as an amplification signal (36–38). Caspase-1 is synthesized as an inactive zymogen called pro-caspase-1, which presents an N-terminal CARD, a central p20 catalytic subunit and a C-terminal p10 subunit (39). Pro-caspase-1 is recruited through homotypic interactions of their CARDs with CARDs of ASC. The clustering of individual pro-caspase-1 monomers within the inflammasome triggers proximity-induced dimerization, which leads to the formation of protease active sites, with two active sites per dimer. Then, pro-caspases-1 are autoprocessed by cleavage of interdomain linker, between p20 and p10, to become fully active (40).
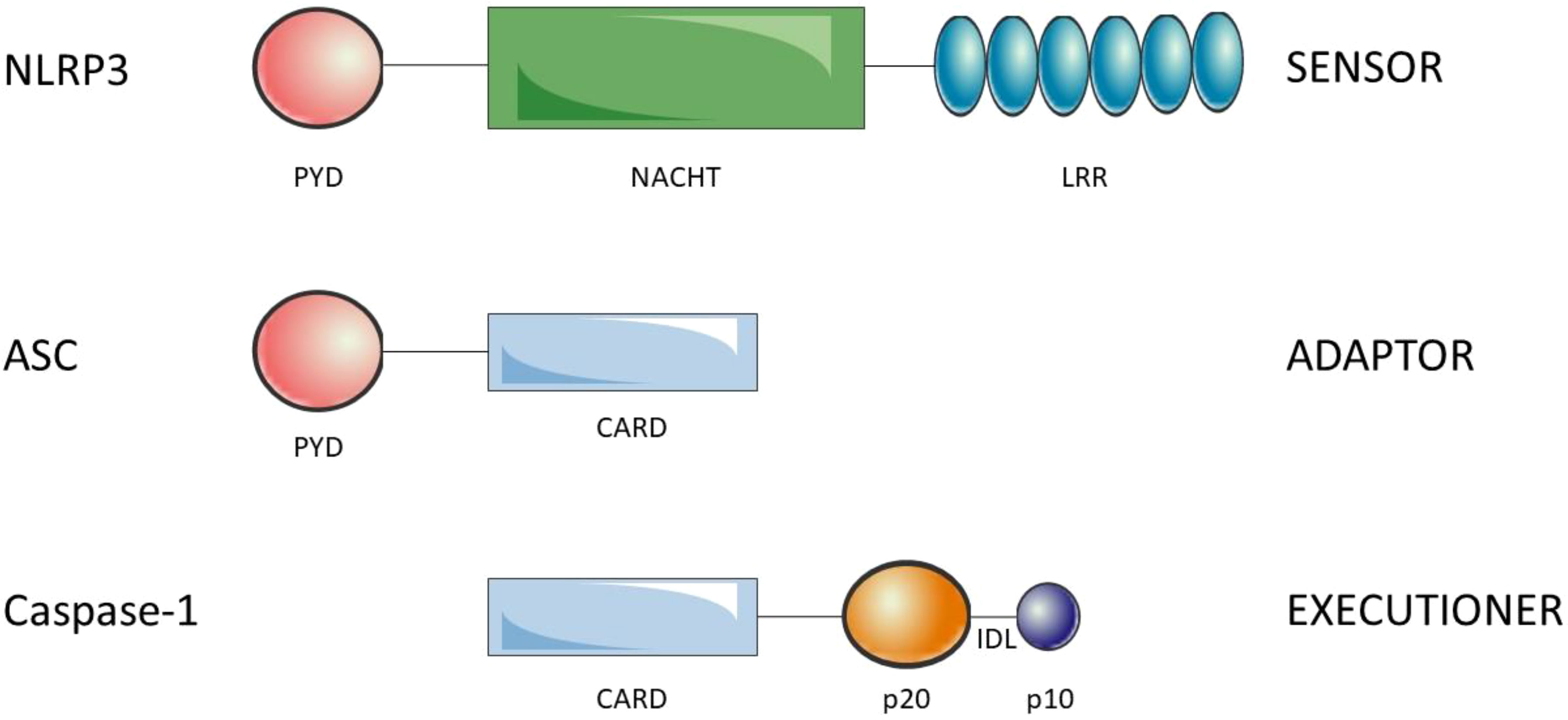
Figure 1 Structure of the components of the NLRP3 inflammasome. NLRP3 is composed of the N-ter PYD, NACHT and C-ter LRR. ASC is composed of theN-ter PYD and the C-ter CARD. Finally, caspase-1 is composed of the N-ter CARD, p20 (large catalytic subunit) and C-ter p10 (small catalytic subnit). The interdomain linker (IDL) binds p20 and p10 and is cleaved to process inactive zymogen pro-caspase-1 to active caspase-1. Oligomerization of NLRP3 and homotypic interactions PYD-PYD and CARD-CARD lead to the NLRP3 inflammasome assembly.
NLRP3 canonical function consists in the sensing of danger signals, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), in the cytoplasm of cells as a pattern recognition receptor (PRR) of the innate immune system and respond with the activation and assembly of NLRP3 inflammasome (41). NLRP3 inflammasome activates caspase-1 which drives the cell death by pyroptosis and maturation and release of IL-1β and IL-18 (42). Non canonical functions of NLRP3 are independent of NLRP3 inflammasome and will not be discussed in this review, including TGF-β signaling and R-Smad activation in epithelial cells (43), transcriptional regulator of driving T helper type 2 (TH2) polarization in CD4+ T cells (44) and regulation of apoptosis in epithelial cells trough a conserved non-canonical platform for caspase-8 activation (45, 46).
Pyroptosis is a form of programmed cell death that is characterized by a highly inflammatory response. It is mediated by inflammatory caspases, which are caspase-1 in human and mice, caspases-4 and -5 in human and the ortholog caspase-11 in mice (47). NLRP3 inflammasome participates in the canonical and noncanonical inflammasome pyroptotic pathways. The former is driven by the recognition of dangerous signals by the NLRP3 inflammasome and the subsequent activation of caspase-1, which cleaves gasdermin D (GSDMD) at D275 and pro-IL-1β and pro-IL-18 (48, 49). The N-terminal fragment of GSDMD forms a transmembrane pore that disrupts ion and water equilibrium and secretes IL-1β and IL-18, resulting in cell death and inflammatory response (50). The latter is initiated by the recognition and binding of caspases 4/5/11 with lipopolysaccharide (LPS) of Gram-negative bacteria, and their oligomerization and activation (51). Next, GSDMD is cleaved by caspases 4/5/11 and the impairment in ion equilibrium, probably potassium efflux, activates the canonical NLRP3 inflammasome (52). GSDMD transmembrane pores are the mediators of pyroptosis and result in the release of cytosolic content, including PAMPS, DAMPS, IL-1β, IL-18, alarmins, to the extracellular space, alteration of water and ion equilibrium, cell swelling and in some cases the lysis of the cell (50). The major established physiological functions of pyroptosis cell death are host defense against microbial infections (53) and regulation of inflammation (54), and exhibits close associations with a range of conditions, including diseases of the nervous system, infectious diseases, autoimmune disorders, cardiovascular conditions, and tumorigenesis (55).
The activated form of caspase-1 cleaves pro-IL-1β and pro-IL-18 at D116 and D36, respectively (56). IL-1β and IL-18 are two members of the IL-1 family. They act through binding with their respective receptors. The secretory mechanism described are through GSDMD transmembrane pores and through membrane rupture and lysis (57). IL-1β and IL-18 play important roles in inflammation (58–60), innate and adaptive immunity (61), bone metabolism (62), central nervous system (CNS) function (63, 64) and metabolism (65–67). Due to their pleiotropic effects, both ILs are involved in several diseases (68, 69).
Despite the mechanism is less well understood, NLRP3 inflammasome is also involved in the secretion of IL-1α (70, 71), high mobility group box 1 (HMGB1) (72, 73), M1 macrophage polarization (74) and modulation of glycolysis (75).
The activation of NLRP3 inflammasome is well regulated because it is activated in a two-step process, the priming and the activation steps (76) (see Figure 2). Likewise, post-translational modifications (PTMs) and multiple NLRP3-interacting protein partners also modulate NLRP3 inflammasome activity at both phases (77). The priming step leads to the transcription of NLRP3, caspase-1, IL-1β and IL-18, despite IL-18 is constitutively expressed, and it is believed that licenses the cell to rapidly responds to activity stimuli. The priming signals are PAMPs/DAMPs detected by PRRs, such as LPS/TLR4 or muramyl dipeptide (MDP)/NOD2, and cytokines that bind to their receptors, such as tumor necrosis factor-α (TNF-α)/TNFR or IL-1β/IL-1R. These signals activate nuclear factor-κB (NF-κB) transcription, which upregulates NLRP3, pro-IL-1β and pro-caspase-1 (41, 78).
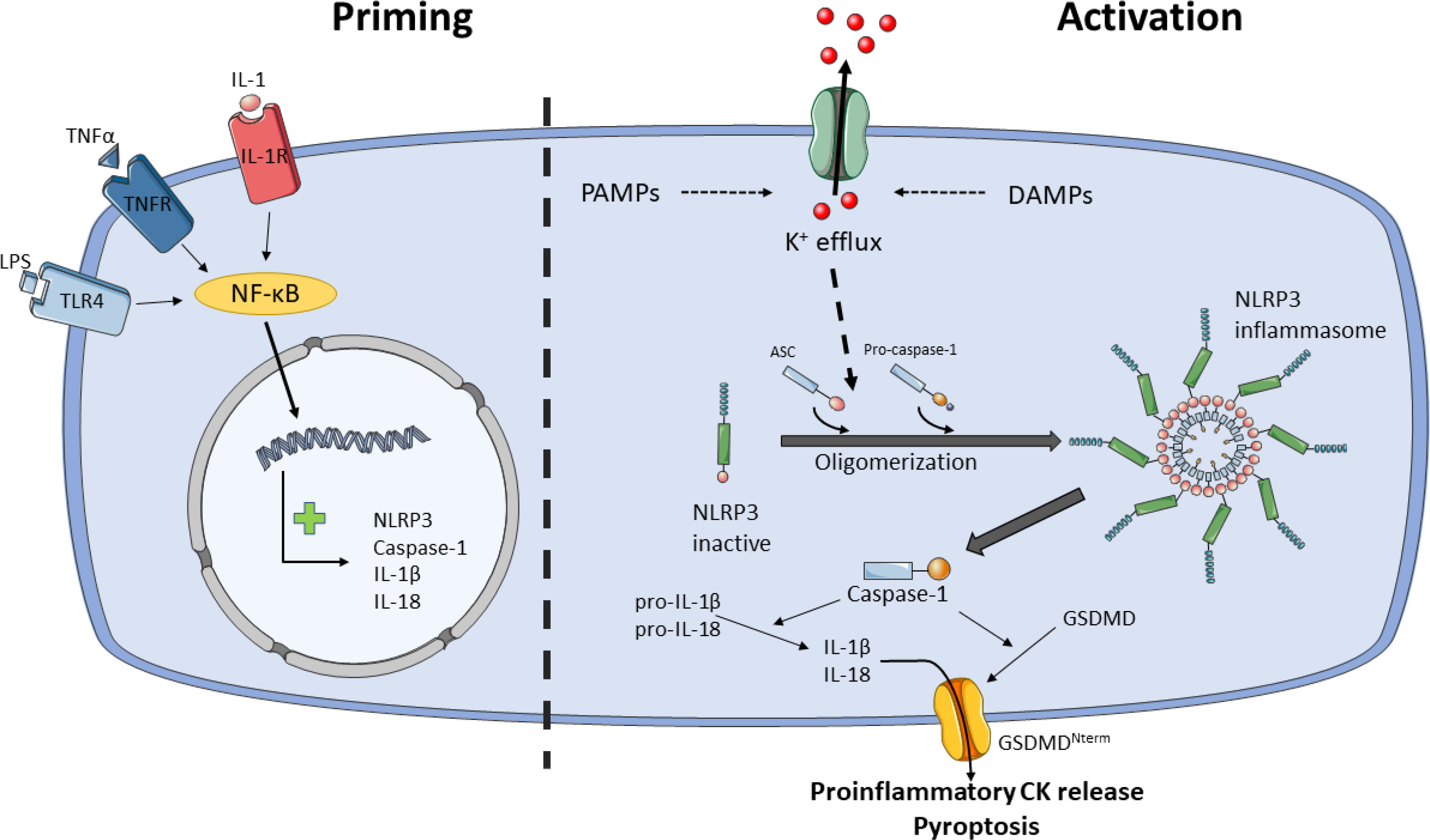
Figure 2 Model for NLRP3 activation by two-step process. Priming (step 1) licenses the cell for NLRP3 inflammasome activation. Certain PAMPs and cytokines (CK) bind to their receptors, e.g.: LPS/TLR4, TNF-α/TNFR or IL-1/IL-1R, leading to activation of NF-κB, which translocate into the nucleus and activates transcription of NLRP3, caspase-1, IL-1β and IL-18. Activation (step 2) starts with the sensing of danger signals, PAMPs and DAMPs, by NLRP3. These stimuli trigger the activation of NLRP3 inflammasome through K+ or Cl- efflux, Ca2+ flux, lysosomal disruption, mitochondrial dysfunction and reactive oxygen species (ROS) generation, metabolic signals and trans-Golgi disassembly, and it seems that K+ efflux is the convergence point for the activation of NLRP3 inflammasome. NLRP3 inflammasome activation occurs through NLRP3 and ASC oligomerization and assembly. NLRP3 inflammasome recruits and activates pro-caspase-1. Caspase-1 cleaves and activates pro-IL-1β and pro-IL-18 and GSDMD. Proinflammatory CKs are released through GSDMD pore and pyroptosis. Ultimately, NLRP3 inflammasome signals for inflammation and cell death by pyroptosis.
Following priming step, recognition of NLRP3 activators leads to oligomerization, assembly and activation of NLRP3 inflammasome. While most PRRs detect one or two PAMPs or DAMPs, the NLRP3 inflammasome stands out for the wide variety of stimuli of different nature it recognizes, including PAMPs such as pore-forming toxins, LPS, viral RNA, fungal β-glucans or DAMPs such as ATP, cholesterol/monosodium urate crystals, alum or silica (79). Due to the diverse nature of these stimuli is unlikely that NLRP3 interacts with all of them. It is currently accepted that cellular stress induced by these stimuli trigger the activation of NLRP3 inflammasome through K+ or Cl- efflux, Ca2+ flux, lysosomal disruption, mitochondrial dysfunction and reactive oxygen species (ROS) generation, metabolic signals and trans-Golgi disassembly (79–81), and it seems that K+ efflux is the convergence point for the activation of NLRP3 inflammasome (82–84). However, other levels of regulation have been described such as epigenetic regulation by microRNAs (85, 86), subcellular localization (87), crosstalk between the gut microbiota and NLRP3 is involved in signaling in the gut-brain axis (88), exosomes (89), acute-phase proteins (90) or neuropeptide Calcitonin Gene-Related Peptide (CGRP) (91).
PTMs of NLRP3 inflammasome affect mainly the NLRP3, but also ASC and caspase-1 and promotes or inhibits NLRP3 inflammasome activity depending on the specific modified amino acid residue (77). PTMs include ubiquitination/deubiquitination, phosphorylation/dephosphorylation, acetylation/deacetylation, SUMOylation and nitrosylation (92, 93). The research of these PTMs and the signaling pathways are of great interest in the search for drugs that modulate NLRP3 inflammasome activity in different diseases in which it is deregulated.
Several NLPR3 protein binding partners modulate both priming and activation via domain-domain interactions with NLRP3, ASC or caspase-1. Positive modulators are GBP5, MARK4, HSP90, TXNIP, NEK7, DDX3X, MAVS, MFN2, PKR and MIF, while negative modulators include COPs, POPs, GNB1, SHP, PPARγ, HSP70 and RACK1 (81, 94–96).
It is worth to mention a less-understood alternative NLRP3 activation pathway in human and porcine monocytes and murine dendritic cells (DCs) that only requires signal 1 to activate NLRP3 inflammasome via TLR4-TRIF-RIPK1-FADD-CASP8 signaling upstream of NLRP3, leading to IL-1β secretion but no to pyroptosis (97, 98).
Lastly, pathogenic pollutants and cigarette smoke activate NLRP3 inflammasome via ROS and mtROS generation, contributing to chronic inflammation and fibrosis in the respiratory system, which plays a crucial role in the pathogenesis of respiratory diseases such as asthma, chronic obstructive pulmonary disease (COPD) and lung fibrosis (99). Fine particulate matter (PM2.5) is a major component of air pollution. Exposure to PM2.5 resulted in lung injury in mice, characterized by the infiltration of inflammatory cells and structural abnormalities in the alveoli and upregulation of NRP3 inflammasome (100). ROS production induced by PM2.5 is linked to ATP alteration (101). NLRP3 inflammasome is activated by PM2.5 trough lysosomal damage and cathepsin B release and induce fibrosis in mouse lung (102). Asbestos and crystalline silica also induce NLRP3 inflammasome activation through ROS production (99). BV-2 and HMC-3 microglial cell lines subjected to oxygen–glucose deprivation and reoxygenation with PM2.5 exposure show decreased cell viability, increased NLRP3 inflammasome activation and ROS production (103). Similarly, ozone-induced lung inflammation in murine model activates NLRP3 trough mtROS production (104, 105).
The role of cigarette smoke (CS) in the activation of NLRP3 inflammasome remains controversial. On the one hand, constituents of CS are involved in the activation of NLRP3 through oxidative stress, mitochondrial dysfunction MyD88/NF-κB, HMGB1, endoplasmic reticulum stress, lysosomal destabilization and TLR/NF-κB signaling pathway (106–108). On the other hand, Buscetta et al. found that CS inhibited the expression of NLRP3 at the transcriptional level, but activated caspase-1 in an NLRP3-independent manner (109). Moreover, CS increases NLRP3 ubiquitin-mediated proteasomal processing (110).
Because the aim of this review is to connect the inflammasome activity and metabolism, we will take a closer look at the regulation by mitochondrial dysfunction-ROS generation and metabolic signaling.
The mitochondria are the organelles in charge of adenosine triphosphate (ATP) generation. However, it also regulates cellular processes, including the cell death by apoptosis, through the intrinsic pathway (111). It is considered the connection between the innate immunity and metabolism (112). Moreover, mitochondrion is an important mediator of NLRP3 inflammasome activation as shown in the multiple regulatory mechanisms where it is involved and inhibition of electron transport chain prevents NLRP3 inflammasome activation (113). The best-recognized mechanisms are the increased generation of mitochondrial ROS (mtROS) during cellular stress (114), the release of oxidized mitochondrial DNA (mtDNA) into the cytosol (115) and the recruitment of NLRP3 through cardiolipin (116). Furthermore, multiple NLRP3 signaling activators lead to mitochondrial dysfunction, which is associated with the pathogenesis of metabolic diseases (99, 117, 118).
mtROS are increased during cellular stress and mediated through voltage dependent anion-selective channel (VDAC) (100). Inhibition of mitochondrial Complexes I and III induce mtROS and NLRP3 activation, independent of K+ efflux (101, 102, 114). However, Muñoz-Planillo et al. showed that ROS production is dispensable for NLRP3 activation (82) and the nuclear factor erythroid 2-related factor 2 (NRF2), a regulator of redox homeostasis, has been described as both activator (103) and inhibitor (119) of NLRP3 inflammasome.
Likewise, mtDNA binds to and activates NLRP3 inflammasome (115, 120). According to Nakahira et al, mtDNA is released into the cytoplasm through mitochondrial permeability transition (MPT) pores in a mtROS-dependent manner (101). Moreover, TLR2, TLR3 or TLR4 agonists increase mtDNA synthesis through upregulation of IRF1-CMPK2, allowing activation of the NLRP3 inflammasome (121). In line with the above mentioned, mitophagy reduces mtROS and mtDNA and thus, activation of NLRP3 inflammasome, and inhibitors of mitophagy achieve the opposite effect (114).
Lastly, when mitochondrial cardiolipin (mtCL), a unique phospholipid of the inner membrane (IMM), translocate to the outer membrane (OMM) binds the LRR domain of NLRP3 (122). Recently discovered, mtCL recruits IL-1α through a similar CL binding domain present in LC3b, a CL binding protein that triggers mitophagy (123). Both lead to NLRP3 inflammasome activation. The mitochondrial antiviral signalling protein (MAVS), in complex with mitofusin 2, recruits NLRP3 to the mitochondria during RNA viral infections. Subramanian et al. identify an N-terminus sequence in NLRP3 that associates with MAVS. Park et al. suggest that putting close NLRP3 to mtROS may promote its oligomerization and activation. However, MAVS does not seem essential for NLRP3 inflammasome activation (124, 125). Lastly, to demonstrate the importance of subcellular localization, NLRP3 associates to the endoplasmic reticulum (ER) in inactivated cells. Then, activators of NLRP3 inhibit Sirtuin2, and through acetylated α-tubulin via dynein-dependent mitochondrial transport, ASC on mitochondria is apposed to NLRP3 (126). Its assembly takes place in mitochondria-associated endoplasmic reticulum membranes (MAMs) in the perinuclear space (87, 126).
Metabolic reprogramming refers to the alterations in cellular metabolic pathways that occur in immune cells, enabling them to adapt their energy production and biosynthetic processes to support immune responses effectively (127). Indeed, Traba et al. demonstrated that nutrient levels in human subjects modulate NLRP3 inflammasome activation (128). For instance, activated macrophages undergo a metabolic switch to aerobic glycolysis (129, 130). Not only glucose metabolism, but also lipid and purine metabolism and the tricarboxylic acid (TCA) cycle can regulate the activation/inhibition of the NLRP3 inflammasome. Table 1 presents a detailed view of the principal mechanisms of activation of NLRP3 inflammasome through metabolites and metabolic pathways.
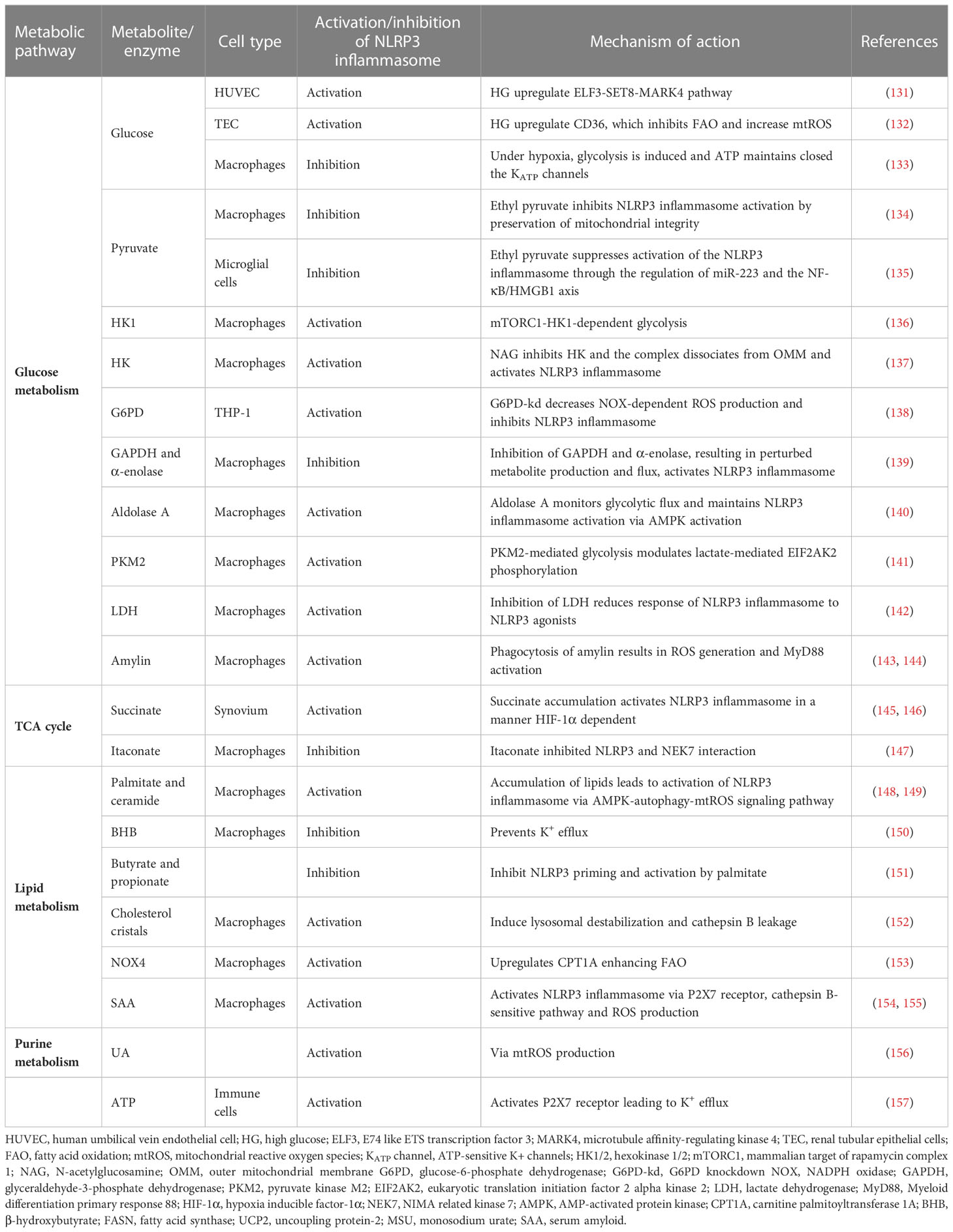
Table 1 Summary of the metabolic signals regulating the NLRP3 inflammasome activation.
First, glucose has been shown to regulate the NLRP3 inflammasome. High glucose (HG) in endothelial cells increases expression of E74 like ETS transcription factor 3 (ELF3) and decreases SET8, a methyltransferase, leading to up-regulation of microtubule affinity regulatory kinase 4 (MARK4) (131). MARK4 activates the NLRP3 inflammasome through a microtubule-dependent mechanism (158). In addition, human renal proximal tubular cells under HG conditions increase CD36 levels, leading to a metabolic shift from fatty acid oxidation to glycolysis, which promotes mtROS production and NLRP3 inflammasome activation (132). However, Watanabe et al. proposed a model of macrophages subjected to hypoxia, in which, under HG conditions, hypoxia induces glycolysis, resulting in intracellular ATP generation and subsequent closure of KATP channels, with the NLRP3 inflammasome remaining inactive (133). On the other hand, hypoxia and glucose deprivation cause a decrease in ATP levels, the opening of KATP and, therefore, the activation of the NLRP3 inflammasome. In this model, mtROS played no significant role in NLRP3 inflammasome activation. Thus, it appears that the NLRP3 inflammasome can recognize the energetic state of the cell and respond.
Pyruvate is a key intermediary metabolite, the end product of glycolysis that can be transformed into acetyl-CoA, lactate or alanine. In experiments, ethyl pyruvate prevents NLRP3 inflammasome activation while preserving mitochondrial integrity. Pyruvate inhibits ATP- and nigericin-induced accumulation of electron-dense mitochondria, maintains mitochondrial membrane potential, and prevents ATP- or nigericin-triggered release of mtDNA into the cytoplasm (134). In microglial cells, ethyl pyruvate demonstrates strong efficacy in suppressing NLRP3 inflammasome activation via regulation of miR-223 and the NF-κB/HMGB1 axis (135).
Hexokinase 1 (HK1) catalyzes glucose entry into glycolysis in the cytosol. In macrophages, NLRP3 inflammasome activation is regulated by mTORC1-induced up-regulation of HK1-dependent glycolysis (136). Furthermore, Wolf et al. propose that hexokinase is an innate immune receptor that when inhibited by N-acetylglucosamine dissociates from the outer mitochondrial membrane and activates the NLRP3 inflammasome (137).
Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the entry of glucose into the pentose phosphate pathway. One of its main functions is to control the redox state of the cell (159). Yen et al. found that in G6PD-deficient individuals, increased susceptibility to pathogens is attributed in part to altered NOX/p38 MAPK/AP-1 redox signaling, leading to negative effects on both inflammasome activation and bactericidal response (138). Inhibition of the glycolytic enzymes GAPDH and α-enolase by the small molecule GB111-NH2, which disrupts glycolytic flux, leads to NLRP3 inflammasome activation (139). Finally, the catalytic enzymes aldolase A, pyruvate kinase M2 (PKM2), lactate dehydrogenase (LDH), and the peptide hormone amylin (secreted by pancreatic β-cells) participate in the activation of the NLRP3 inflammasome in macrophages (140–144).
Regarding TCA, there are two intermediates that lead to opposite effects in terms of NLRP3 inflammasome activation. On the one hand, hypoxic induction of TGF-β1 in the synovium of rheumatoid arthritis (RA) rats resulted in elevated succinate accumulation. This was caused by inhibition of succinate dehydrogenase (SDH) activation and led to NLRP3 inflammasome activation, which was dependent on HIF-1α induction. Moreover, citrate-derived itaconate has gained attention as an anti-inflammatory modulator in macrophages (160). Hooftman et al. found that itaconate modifies a specific cysteine (C548) in NLRP3 and inhibits its activation by interfering with the binding partner of NEK7 (147). Taken together, dysregulations in the TCA cycle, the central metabolic pathway for carbohydrates, lipids, and proteins, are potential activators of the NLRP3 inflammasome.
Lipid metabolism plays a crucial role in a variety of cellular processes, such as energy production, membrane synthesis and signaling pathways. New research has revealed a significant connection between lipid metabolism and NLRP3 inflammasome activation/inhibition. First, the accumulation of fatty acids palmitate and ceramide activate the NLRP3 inflammasome through the AMPK-autophagy-mtROS signaling pathway (148, 149). In contrast, the ketone metabolite β-hydroxybutyrate (BHB) serves as an alternative source of ATP, favoring mammalian survival during periods of energy deficit. BHB blocks the NLRP3 inflammasome by inhibiting K+ efflux (150). In addition, a recent study indicates that butyrate and propionate have inhibitory effects on the priming and activation of the NLRP3 inflammasome by palmitate (151). This inhibition leads to a significant reduction in pro-IL-1β levels, whereas the effect on inflammasome activation itself is relatively modest.
Cholesterol crystals are a common feature of atherosclerotic plaques (161). Cholesterol crystals are phagocytosed by macrophages and stimulate NLRP3 inflammasome activity, probably through lysosomal destabilization (152). Regarding fatty acid oxidation (FAO), deficiency of NADPH oxidase 4 (NOX4) leads to lower expression of carnitine palmitoyltransferase 1A (CPT1A), a key mitochondrial fatty acid transporter. The result is lower FAO and reduced activation of the NLRP3 inflammasome (153). Finally, serum amyloid A (SAA) is an acute-phase protein that increases in serum during inflammation and has a pathogenic role in amyloid A amyloidosis. SAA induces activation of the NLRP3 inflammasome in macrophages through the P2X7 receptor, dependent on cathepsin B activity, but not through lysosomal destabilization, and by generating ROS (154, 155).
The recognition of the pivotal role of inflammasomes in the development and progression of metabolic diseases opens up exciting possibilities for therapeutic interventions (9, 162, 163). Targeting inflammasome pathways and modulating their activity holds promise for mitigating inflammation, improving metabolic function, and potentially treating metabolic diseases.
Developing small-molecule inhibitors that specifically target NLRP3 inflammasome components, such as NLRP3 or caspase-1, represents a promising avenue (41). Inhibitors block the activation of the inflammasome complex, thereby reducing the release of proinflammatory cytokines and alleviating inflammation associated with metabolic diseases (164–166). It is important to note that many therapeutic approaches targeting inflammasomes are still in the preclinical or early clinical stages of development. Further research is needed to elucidate the safety, efficacy, and long-term effects of these interventions in human populations. Additionally, considering the complex network of inflammasome-related pathways, combination therapies targeting multiple components may hold even greater potential for effectively managing inflammation and metabolic dysfunction. In the search for potential inhibitors, a broad range of targets can be tested due to the complex signaling cascade that governs NLRP3 inflammasome activation and its functions. For example, TLR4, TNFR and NF-κB signaling of the priming step, activators such as K+/Cl- efflux, P2X7 and mtROS, effectors such as GSDMD, interaction binding partners such as NEK7 and, obviously, the NLRP3 inflammasome components NLRP3 receptor, ASC and caspase-1. Currently, the ATPase activity of the NACHT domain in NLRP3 is the target of the majority of NLRP3 inflammasome inhibitors under clinical and preclinical research (see Table 2). Despite the existence of drugs targeting IL-1, i.e., canakinumab and anakinra, there is a need in the search of specific inhibitors that target specifically members of the NLRP3 inflammasome. These may intervene at an upstream point in the inflammatory cascade, potentially providing more comprehensive control over the inflammatory response. Moreover, directly inhibiting IL-1β signaling may affect the immune system more broadly and may have side effects. A more focused strategy, however, may be to specifically block the NLRP3 inflammasome, which lowers the chance of systemic immunosuppression while still producing the desired anti-inflammatory benefits.
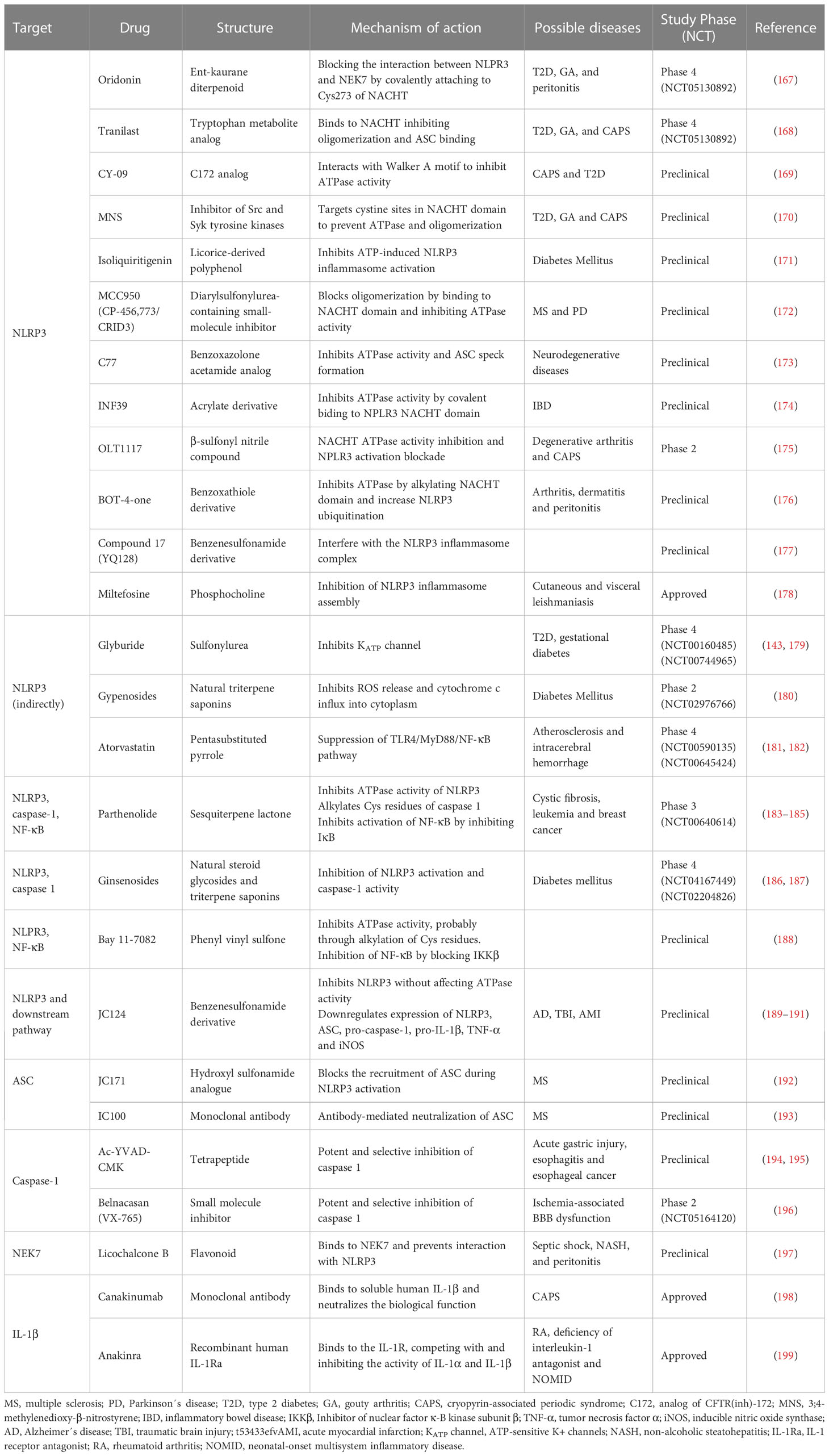
Table 2 Potential inhibitors of NLRP3 inflammasome and their targets.
The interplay between the innate immune system, inflammation, metabolism, and metabolic diseases has revealed a complex and intertwined relationship. Inflammasomes, particularly the NLRP3 inflammasome, have emerged as critical mediators linking these processes together. The activation of inflammasomes triggers the release of proinflammatory cytokines IL-1β and IL-18 and the cell death by pyroptosis. The activation of NLRP3 inflammasome is under complex regulation involving multiple signaling networks. Mitochondrial dysfunction-mtROS production as well as metabolic signaling link the bioenergetics and metabolism status of the cell with cell death by pyroptosis, inflammation and innate immune functions through NLRP3 inflammasome activation.
Future studies should focus on elucidating the specific mechanisms by which inflammasomes contribute to metabolic dysfunction, identifying potential therapeutic targets within the inflammasome pathway, and developing interventions to modulate inflammasome activation and subsequent inflammation in metabolic diseases, and ultimately, improve patient outcomes and quality of life.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The study (FIS-PI21/01244) was supported by the Instituto de Salud Carlos III (grant no. Estatal de I + D + I 2020–2027) and co-financed by the European Development Regional Fund “A way to achieve Europe”, as well as P2022/BMD-7321 (Comunidad de Madrid) and ProACapital, Halekulani S.L. and MJR. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol (2016) 12:15–20. doi: 10.1038/NRENDO.2015.189
2. Cronkite DA, Strutt TM. The regulation of inflammation by innate and adaptive lymphocytes. J Immunol Res (2018) 2018. doi: 10.1155/2018/1467538
3. Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol (2010) 184:4062. doi: 10.4049/JIMMUNOL.0903002
4. Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis (2015) 6:e1887–7. doi: 10.1038/cddis.2015.246
5. Chi H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell Mol Immunol (2022) 19:299–302. doi: 10.1038/s41423-022-00840-x
6. Saitoh S, van Wijk K, Nakajima O. Crosstalk between metabolic disorders and immune cells. Int J Mol Sci (2021) 22:505–21. doi: 10.3390/IJMS221810017
7. Zmora N, Bashiardes S, Levy M, Elinav E. The role of the immune system in metabolic health and disease. Cell Metab (2017). doi: 10.1016/j.cmet.2017.02.006
8. Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. Inflammasomes and metabolic disease. Annu Rev Physiol (2014) 76:57–78. doi: 10.1146/ANNUREV-PHYSIOL-021113-170324
9. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol (2011) 32:373. doi: 10.1016/J.IT.2011.05.004
10. Guo H, Callaway JB, Ting JP. Inflammasomes : mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
11. Pan Y, Cai W, Huang J, Cheng A, Wang M, Yin Z, et al. Pyroptosis in development, inflammation and disease. Front Immunol (2022) 13:991044/BIBTEX. doi: 10.3389/FIMMU.2022.991044/BIBTEX
12. Rathinam VAK, Chan FKM. Inflammasome, inflammation, and tissue homeostasis. Trends Mol Med (2018) 24:304–18. doi: 10.1016/J.MOLMED.2018.01.004
13. Rathinam VAK, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell (2016) 165:792–800. doi: 10.1016/J.CELL.2016.03.046
14. Fraile-Martínez Ó, García-Montero C, Pekarek L, Saz JV, Alvarez-Mon MÁ, Barrena-Blázquez S, et al. Decreased survival in patients with pancreatic cancer may be associated with an increase in histopathological expression of inflammasome marker NLRP3. Histol Histopathol (2023) 18617. doi: 10.14670/HH-18-617
15. Wani K, Alharthi H, Alghamdi A, Sabico S, Al-Daghri NM. Role of NLRP3 inflammasome activation in obesity-mediated metabolic disorders. Int J Environ Res Public Health (2021) 18:1–21. doi: 10.3390/IJERPH18020511
16. Rocha M, Apostolova N, Diaz-rua R, Muntane J, Victor VM. Mitochondria and T2D : role of autophagy , ER stress , and in fl ammasome. Trends Endocrinol Metab (2020) 31:725–41. doi: 10.1016/j.tem.2020.03.004
17. Litwiniuk A, Bik W, Kalisz M, Baranowska-bik A. Inflammasome NLRP3 potentially links obesity-associated low-grade systemic inflammation and insulin resistance with Alzheimer‘s disease. Int J Mol Sci (2021) 22(11):5603. doi: 10.3390/ijms22115603
18. Lu S, Li Y, Qian Z, Zhao T, Feng Z, Weng X, et al. Role of the inflammasome in insulin resistance and type 2 diabetes mellitus. Front Immunol (2023) 14:1052756. doi: 10.3389/fimmu.2023.1052756
19. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
20. Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 Inflammasome/Pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol (2019) 9:406/BIBTEX. doi: 10.3389/FCIMB.2019.00406/BIBTEX
21. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:238/BIBTEX. doi: 10.3389/FCIMB.2020.00238/BIBTEX
22. Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev (2020) 297:26–38. doi: 10.1111/IMR.12909
23. Chen Y, Fang ZM, Yi X, Wei X, Jiang DS. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis (2023) 14:1–13. doi: 10.1038/s41419-023-05716-0
24. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell (2011) 147:742–58. doi: 10.1016/J.CELL.2011.10.033
25. Gudipaty SA, Conner CM, Rosenblatt J, Montell DJ. Unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu Rev Cell Dev Biol (2018) 34:311. doi: 10.1146/ANNUREV-CELLBIO-100616-060748
26. Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol (2019) 39:277–86. doi: 10.1007/S10875-019-00638-Z
27. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
28. Koizumi M, Watanabe T, Masumoto J, Sunago K. Apoptosis - associated speck - like protein containing a CARD regulates the growth of pancreatic ductal adenocarcinoma. Sci Rep (2021) 11:22351. doi: 10.1038/s41598-021-01465-2
29. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol (2009) 10:241–7. doi: 10.1038/ni.1703
30. Chu LH, Gangopadhyay A, Dorfleutner A, Stehlik C. An updated view on the structure and function of PYRIN domains. Apoptosis (2015) 20:157. doi: 10.1007/S10495-014-1065-1
31. Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, et al. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature (2022) 604:184–9. doi: 10.1038/s41586-022-04467-w
32. Niu T, De Rosny C, Chautard S, Rey A, Patoli D, Groslambert M, et al. NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-26142-w
33. Duan Y, Wang J, Cai J, Kelley N, He Y. The leucine-rich repeat (LRR) domain of NLRP3 is required for NLRP3 inflammasome activation in macrophages. J Biol Chem (2022) 298. doi: 10.1016/j.jbc.2022.102717
34. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530:354–7. doi: 10.1038/nature16959
35. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
36. Dick MS, Sborgi L, Ru S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasome. Nat Commun (2016) 7:1–12. doi: 10.1038/ncomms11929
37. Hoss F, Rodriguez-Alcazar JF, Latz E. Assembly and regulation of ASC specks. Cell Mol Life Sci (2017) 74:1211–29. doi: 10.1007/s00018-016-2396-6
38. Nambayan RJT, Sandin SI, Quint DA, Satyadi DM, De Alba E. The inflammasome adapter ASC assembles into filaments with integral participation of its two death domains , PYD and CARD. J Biol Chem (2019) 294:439–52. doi: 10.1074/jbc.RA118.004407
39. Wilson KP, Black JAF, Thomson JA, Kim EE, Griffith JP, Navia MA, et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature (1994) 370:270–5. doi: 10.1038/370270A0
40. Ross C, Chan AH, Von Pein JB, Maddugoda MP, Boucher D, Schroder K. Inflammatory Caspases : toward a unified model for caspase activation by inflammasomes. Annu Rev Immunol (2022) 40:249–69. doi: 10.1146/annurev-immunol-101220-030653
41. Zhan X, Li Q, Xu G, Xiao X, Bai Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front Immunol (2023) 13:1109938. doi: 10.3389/fimmu.2022.1109938
42. Blevins HM, Xu Y, Biby S, Zhang S. The NLRP3 inflammasome Pathway : a review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front Aging Neurosci (2022) 14:1–27. doi: 10.3389/fnagi.2022.879021
43. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol (2013) 190:1239–49. doi: 10.4049/JIMMUNOL.1201959
44. Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, Boidot R, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol (2015) 16:859–70. doi: 10.1038/ni.3202
45. Chung H, Vilaysane A, Lau A, Stahl M, Morampudi V, Platnich JM, et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ (2016) 23:1331–46. doi: 10.1038/cdd.2016.14
46. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ (2013) 20:1149–60. doi: 10.1038/cdd.2013.37
47. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and gasdermin-InducedPyroptosis. Annu Rev Immunol (2020) 38:567. doi: 10.1146/ANNUREV-IMMUNOL-073119-095439
48. Sun Q, Scott MJ. Caspase-1 as a multifunctional inflammatory mediator: noncytokine maturation roles. J Leukoc Biol (2016) 100:961. doi: 10.1189/JLB.3MR0516-224R
49. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell (2020) 180:941–55. doi: 10.1016/j.cell.2020.02.002
50. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin d in pyroptosis. Acta Pharm Sin B (2021) 11:2768–82. doi: 10.1016/J.APSB.2021.02.006
51. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol (2020) 204:3063–9. doi: 10.4049/JIMMUNOL.2000373
52. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45:2918–26. doi: 10.1002/EJI.201545655
53. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev (2015) 265:130. doi: 10.1111/IMR.12287
54. Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T, et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol (2022) 19:971–92. doi: 10.1038/s41423-022-00905-x
55. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6:1–21. doi: 10.1038/s41392-021-00507-5
56. Afonina IS, Mu C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
57. Tapia VS, Daniels MJD, Palazón-Riquelme P, Dewhurst M, Luheshi NM, Rivers-Auty J, et al. The three cytokines IL-1β, IL-18, and IL-1α share related but distinct secretory routes. J Biol Chem (2019) 294:8325–35. doi: 10.1074/jbc.RA119.008009
58. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity (2019) 50:778–95. doi: 10.1016/j.immuni.2019.03.012
59. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
60. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289/BIBTEX. doi: 10.3389/FIMMU.2013.00289/BIBTEX
61. Pyrillou K, Burzynski LC, Clarke MCH. Alternative pathways of IL-1 activation , and its role in health and disease. Front Immunol (2020) 11:613170. doi: 10.3389/fimmu.2020.613170
62. Lee Y, Fujikado N, Manaka H, Yasuda H, Iwakura Y. IL-1 plays an important role in the bone metabolism under physiological conditions. Int Immunol (2010) 22:805–16. doi: 10.1093/intimm/dxq431
63. Hewett SJ, Jackman NA, Claycomb RJ. Interleukin-1β in central nervous system injury and repair. Eur J Neurodegener Dis (2012) 1:195–211.
64. Alboni S, Cervia D, Sugama S, Conti B. Interleukin 18 in the CNS. J Neuroinflamm (2010) 7:1–12. doi: 10.1186/1742-2094-7-9/FIGURES/1
65. Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology (2007) 148:241–51. doi: 10.1210/EN.2006-0692
66. Dror E, Dalmas E, Meier DT, Wueest S, Thévenet J, Thienel C, et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol (2017) 18(3):283–92. doi: 10.1038/ni.3659
67. Somm E, Jornayvaz FR. Interleukin-18 in metabolism: from mice physiology to human diseases. Front Endocrinol (Lausanne) (2022) 13:971745. doi: 10.3389/fendo.2022.971745
68. Kaneko N, Kurata M, Yamamoto T, Morikawa S, Masumoto J. The role of interleukin-1 in general pathology. Inflammation Regener (2019) 39:1–16. doi: 10.1186/S41232-019-0101-5
69. Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev (2018) 281:138. doi: 10.1111/IMR.12616
70. Groß O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity (2012) 36:388–400. doi: 10.1016/j.immuni.2012.01.018
71. Yazdi AS, Drexler SK. Regulation of interleukin 1α secretion by inflammasomes. Ann Rheum Dis (2013) 72 Supplem:96–9. doi: 10.1136/annrheumdis-2012-202252
72. Vande Walle L, Kanneganti T, Lamkanfi M. HMGB1 release by inflammasomes. Virulence (2011) 2:162–5. doi: 10.4161/viru.2.2.15480
73. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med (2022) 54:91–102. doi: 10.1038/s12276-022-00736-w
74. Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1 β production in inflammatory root resorption. J Clin Periodontol (2020) 47:451–60. doi: 10.1111/jcpe.13258
75. Finucane OM, Sugrue J, Rubio-Araiz A, Guillot-Sestier MV, Lynch MA. The NLRP3 inflammasome modulates glycolysis by increasing PFKFB3 in an IL-1β-dependent manner in macrophages. Sci Rep (2019) 9. doi: 10.1038/S41598-019-40619-1
76. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10:1–11. doi: 10.1038/s41419-019-1413-8
77. Xia J, Jiang S, Dong S, Liao Y, Zhou Y. The role of post-translational modifications in regulation of NLRP3 inflammasome activation. Int J Mol Sci (2023) 24. doi: 10.3390/ijms24076126
78. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:1–22. doi: 10.1038/s41421-020-0167-x
79. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
80. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
81. Paik S, Kim JK, Silwal P. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol (2021) 18:1141–60. doi: 10.1038/s41423-021-00670-3
82. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
83. Xu Z, Chen ZM, Wu X, Zhang L, Cao Y, Zhou P. Distinct molecular mechanisms underlying potassium efflux for NLRP3 inflammasome activation. Front Immunol (2020) 11:609441/BIBTEX. doi: 10.3389/FIMMU.2020.609441/BIBTEX
84. Tapia-Abellán A, Angosto-Bazarra D, Alarcón-Vila C, Baños MC, Hafner-Bratkovič I, Oliva B, et al. Sensing low intracellular potassium by NLRP3 results in a stable open structure that promotes inflammasome activation. Sci Adv (2021) 7:4468–83. doi: 10.1126/SCIADV.ABF4468
85. Yi YS. MicroRNA-mediated epigenetic regulation of inflammasomes in inflammatory responses and immunopathologies. Semin Cell Dev Biol (2022). doi: 10.1016/j.semcdb.2022.11.006
86. Saad N, Duroux-Richard I, Touitou I, Jeziorski E, Apparailly F. MicroRNAs in inflammasomopathies. Immunol Lett (2023) 256-257:48–54. doi: 10.1016/j.imlet.2023.04.001
87. Akbal A, Dernst A, Lovotti M, Mangan MSJ, McManus RM, Latz E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell Mol Immunol (2022) 19:1201–14. doi: 10.1038/s41423-022-00922-w
88. Yang D, Wang Z, Chen Y, Guo Q, Dong Y. Interactions between gut microbes and NLRP3 inflammasome in the gut-brain axis. Comput Struct Biotechnol J (2023) 21:2215–27. doi: 10.1016/j.csbj.2023.03.017
89. Li Z, Chen X, Tao J, Shi A, Zhang J, Yu P. Exosomes regulate NLRP3 inflammasome in diseases. Front Cell Dev Biol (2022) 9:802509. doi: 10.3389/fcell.2021.802509
90. Richter K, Amati AL, Padberg W, Grau V. Negative regulation of ATP-induced inflammasome activation and cytokine secretion by acute-phase proteins: a mini review. Front Pharmacol (2022) 13:981276. doi: 10.3389/fphar.2022.981276
91. Zhu F, Yu D, Qin X, Qian Y, Ma J, Li W, et al. The neuropeptide CGRP enters the macrophage cytosol to suppress the NLRP3 inflammasome during pulmonary infection. Cell Mol Immunol (2023) 20:264–76. doi: 10.1038/s41423-022-00968-w
92. McKee CM, Coll RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol (2020) 108:937–52. doi: 10.1002/JLB.3MR0720-513R
93. Seok JK, Kang HC, Cho YY, Lee HS, Lee JY. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front Immunol (2021) 11:618231/BIBTEX. doi: 10.3389/FIMMU.2020.618231/BIBTEX
94. Leu S-Y, Tsang Y-L, Ho L-C, Yang C-C, Shao A-N, Chang C-Y, et al. NLRP3 inflammasome activation, metabolic danger signals and protein binding partners. J Endocrinol (2023) 257:2. doi: 10.1530/joe-22-0184
95. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20. doi: 10.3390/IJMS20133328
96. Indramohan M, Stehlik C, Dorfleutner A. COPs and POPs patrol inflammasome activation. J Mol Biol (2018) 430:153–73. doi: 10.1016/j.jmb.2017.10.004
97. Gaidt MM, Ebert TS, Chauhan D, Cooper MA, Graf T, Gaidt MM, et al. Human monocytes engage an alternative inflammasome pathway human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
98. He Y, Franchi L, Núñez G. TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and In vivo. J Immunol (2013) 190:334–9. doi: 10.4049/jimmunol.1202737
99. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta (2017) 1863:1066. doi: 10.1016/J.BBADIS.2016.11.010
100. Zhao J, Li J, Li G, Chen M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Front Cardiovasc Med (2022) 9:1059576. doi: 10.3389/fcvm.2022.1059576
101. Nakahira K, Haspel JA, Rathinam VAK, Lee S, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi: 10.1038/ni.1980
102. Groß CJ, Mishra R, Schneider KS, Schneider S, Perocchi F, Wettmarshausen J, et al. Activation by small molecules targeting article K + efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity (2016) 45:761–73. doi: 10.1016/j.immuni.2016.08.010
103. Zhao C, Gillette DD, Li X, Zhang Z, Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem (2014) 289:17020–9. doi: 10.1074/JBC.M114.563114
104. Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic Res (2019) 53:780–90. doi: 10.1080/10715762.2019.1630735
105. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res (2018) 19:1–12. doi: 10.1186/S12931-018-0931-8/TABLES/1
106. White A, Wang Z, Wang X, King M, Guo C, Mantsounga C, et al. NLRP3 inflammasome activation in cigarette smoke priming for pseudomonas aeruginosa-induced acute lung injury. Redox Biol (2022) 57:102467. doi: 10.1016/J.REDOX.2022.102467
107. Mehta S, Dhawan V. Cigarette smoke and the NLRP3 inflammasome. In: Patel VB, Preedy VR editors. Handbook of Substance Misuse and Addictions. Springer, Cham. (2022). doi: 10.1007/978-3-030-67928-6_35
108. Ma Y, Long Y, Chen Y. Roles of inflammasome in cigarette smoke-related diseases and physiopathological disorders: mechanisms and therapeutic opportunities. Front Immunol (2021) 12:720049/FULL. doi: 10.3389/FIMMU.2021.720049/FULL
109. Buscetta M, Di Vincenzo S, Miele M, Badami E, Pace E, Cipollina C. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J (2020) 34:1819–32. doi: 10.1096/FJ.201901239R
110. Han SH, Jerome JA, Gregory AD, Mallampalli RK. Cigarette smoke destabilizes NLRP3 protein by promoting its ubiquitination. Respir Res (2017) 18:1–8. doi: 10.1186/S12931-016-0485-6/FIGURES/4
111. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
112. Bahat A, MacVicar T, Langer T. Metabolism and innate immunity meet at the mitochondria. Front Cell Dev Biol (2021) 9:720490/BIBTEX. doi: 10.3389/FCELL.2021.720490/BIBTEX
113. Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol (2022) 23:692–704. doi: 10.1038/s41590-022-01185-3
114. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–6. doi: 10.1038/NATURE09663
115. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/J.IMMUNI.2012.01.009
116. Cassel SL, Elliott E, Iyer SS, Sutterwala F. Cardiolipin provides a platform for caspase-1 activation and NLRP3 inflammasome assembly. J Allergy Clin Immunol (2016) 137:AB72. doi: 10.1016/j.jaci.2015.12.244
117. Advani D, Sharma S, Tripathi R, Gupta R, Jaiswal A, Ambasta RK, et al. Mitochondrial dysfunction in metabolic disorders. In de Oliveira MR editor. Mitochondrial Dysfunction and Nanotherapeutics. Academic Press. (2021), 91–137. doi: 10.1016/B978-0-323-85666-9.00015-2
118. Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta Mol basis Dis (2020) 1866. doi: 10.1016/J.BBADIS.2020.165838
119. Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, et al. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res (2018) 336:32–9. doi: 10.1016/J.BBR.2017.06.027
120. Qiu Y, Huang Y, Chen M, Yang Y, Li X, Zhang W. Mitochondrial DNA in NLRP3 inflammasome activation. Int Immunopharmacol (2022) 108:108719. doi: 10.1016/J.INTIMP.2022.108719
121. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
122. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi: 10.1016/J.IMMUNI.2013.08.001
123. Dagvadorj J, Mikulska-Ruminska K, Tumurkhuu G, Ratsimandresy RA, Carriere J, Andres AM, et al. Recruitment of pro-IL-1α to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc Natl Acad Sci U.S.A. (2021) 118:e2015632118. doi: 10.1073/PNAS.2015632118/SUPPL_FILE/PNAS.2015632118.SM02.MOV
124. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adapter MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348. doi: 10.1016/J.CELL.2013.02.054
125. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol (2013) 191:4358–66. doi: 10.4049/JIMMUNOL.1301170
126. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14:454–60. doi: 10.1038/ni.2550
127. Hu C, Xuan Y, Zhang X, Liu Y, Yang S, Yang K. Immune cell metabolism and metabolic reprogramming. Mol Biol Rep (2022) 49:9783–95. doi: 10.1007/S11033-022-07474-2
128. Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest (2015) 125:4592–600. doi: 10.1172/JCI83260
129. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic reprogramming in macrophage responses. biomark Res (2021) 9:1–17. doi: 10.1186/S40364-020-00251-Y/TABLES/3
130. Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25:771–84. doi: 10.1038/cr.2015.68
131. Wang J, Shen X, Liu J, Chen W, Wu F, Wu W, et al. High glucose mediates NLRP3 inflammasome activation via upregulation of ELF3 expression. Cell Death Dis (2020) 11:1–14. doi: 10.1038/s41419-020-2598-6
132. Hou Y, Wang Q, Han B, Chen Y, Qiao X, Wang L. CD36 promotes NLRP3 inflammasome activation via the mtROS pathway in renal tubular epithelial cells of diabetic kidneys. Cell Death Dis (2021) 12:1–16. doi: 10.1038/s41419-021-03813-6
133. Watanabe S, Usui-Kawanishi F, Karasawa T, Kimura H, Kamata R, Komada T, et al. Glucose regulates hypoxia-induced NLRP3 inflammasome activation in macrophages. J Cell Physiol (2020) 235:7554–66. doi: 10.1002/JCP.29659
134. Li S, Liang F, Kwan K, Tang Y, Wang X, Tang Y, et al. Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol Med (2018) 24:1–11. doi: 10.1186/S10020-018-0006-9/FIGURES/7
135. Olcum M, Tufekci KU, Durur DY, Tastan B, Gokbayrak IN, Genc K, et al. Ethyl pyruvate attenuates microglial nlrp3 inflammasome activation via inhibition of HMGB1/NF-κB/miR-223 signaling. Antioxidants (2021) 10:1–14. doi: 10.3390/antiox10050745
136. Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep (2015) 12:102–15. doi: 10.1016/J.CELREP.2015.05.046
137. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell (2016) 166:624–36. doi: 10.1016/J.CELL.2016.05.076
138. Yen WC, Wu YH, Wu CC, Lin HR, Stern A, Chen SH, et al. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol (2020) 28:101363. doi: 10.1016/J.REDOX.2019.101363
139. Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife (2016) 5:1–32. doi: 10.7554/eLife.13663
140. Bai D, Du J, Bu X, Cao W, Sun T, Zhao J, et al. ALDOA maintains NLRP3 inflammasome activation by controlling AMPK activation. Autophagy (2021) 18:1673–93. doi: 10.1080/15548627.2021.1997051
141. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun (2016) 7:1–13. doi: 10.1038/ncomms13280
142. Lin HC, Chen YJ, Wei YH, Lin HA, Chen CC, Liu TF, et al. Lactic acid fermentation is required for NLRP3 inflammasome activation. Front Immunol (2021) 12:630380/BIBTEX. doi: 10.3389/FIMMU.2021.630380/BIBTEX
143. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol (2010) 11:897–904. doi: 10.1038/ni.1935
144. Westwell-Roper C, Dai DL, Soukhatcheva G, Potter KJ, van Rooijen N, Ehses JA, et al. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol (2011) 187:2755–65. doi: 10.4049/JIMMUNOL.1002854
145. Huang JJ, Xia J, Huang LL, Li YC. HIF−1α promotes NLRP3 inflammasome activation in bleomycin−induced acute lung injury. Mol Med Rep (2019) 20:3424–32. doi: 10.3892/MMR.2019.10575
146. Li Y, Zheng JY, Liu JQ, Yang J, Liu Y, Wang C, et al. Succinate/NLRP3 inflammasome induces synovial fibroblast activation: therapeutical effects of clematichinenoside AR on arthritis. Front Immunol (2016) 7:532. doi: 10.3389/FIMMU.2016.00532
147. Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab (2020) 32:468. doi: 10.1016/J.CMET.2020.07.016
148. Camell CD, Nguyen KY, Jurczak MJ, Christian BE, Shulman GI, Shadel GS, et al. Macrophage-specific de Novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem (2015) 290:29402–13. doi: 10.1074/jbc.M115.680199
149. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12:408–15. doi: 10.1038/ni.2022
150. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat Med (2015) 21:263–9. doi: 10.1038/nm.3804
151. Truax AD, Chen L, Tam JW, Cheng N, Guo H, Koblansky AA, et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe (2018) 24:364–78.e6. doi: 10.1016/J.CHOM.2018.08.009
152. Rajamüaki K, Lappalainen J, Öörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PloS One (2010) 5. doi: 10.1371/JOURNAL.PONE.0011765
153. Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med (2016) 22:1002–12. doi: 10.1038/nm.4153
154. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, et al. Serum amyloid a activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin b-sensitive pathway. J Immunol (2011) 186:6119–28. doi: 10.4049/JIMMUNOL.1002843
155. Shridas P, De Beer MC, Webb NR. High-density lipoprotein inhibits serum amyloid a–mediated reactive oxygen species generation and NLRP3 inflammasome activation. J Biol Chem (2018) 293:13257–69. doi: 10.1074/JBC.RA118.002428/ATTACHMENT/082496E9-03F5-4F5F-82A5-E20CA936DE03/MMC1.PDF
156. Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep (2017) 7:1–14. doi: 10.1038/srep39884
157. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
158. Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun (2017) 8:1–13. doi: 10.1038/ncomms15986
159. Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL, Stern A, et al. The redox role of G6PD in cell growth, cell death, and cancer. Cells (2019) 8:1–29. doi: 10.3390/cells8091055
160. Williams NC, O’Neill LAJ. A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol (2018) 9:141. doi: 10.3389/fimmu.2018.00141
161. Baumer Y, Mehta NN, Dey AK, Powell-Wiley TM, Boisvert WA. Cholesterol crystals and atherosclerosis. Eur Heart J (2020) 41:2236–9. doi: 10.1093/eurheartj/ehaa505
162. Robbins GR, Wen H, Ting JPY. Inflammasomes and metabolic disorders: old genes in modern diseases. Mol Cell (2014) 54:297–308. doi: 10.1016/J.MOLCEL.2014.03.029
163. Wen H, Ting JP, Neill LAJO. A role for the NLRP3 inflammasome in metabolic diseases [[/amp]]mdash; did warburg miss inflammation? Nat Immunol (2012) 13:352–7. doi: 10.1038/ni.2228
164. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262/BIBTEX. doi: 10.3389/FPHAR.2015.00262/BIBTEX
165. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci (2022) 43:653–68. doi: 10.1016/J.TIPS.2022.04.003
166. Zhang X, Wang Z, Zheng Y, Yu Q, Zeng M, Bai L, et al. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases (Review). Int J Mol Med (2023) 51. doi: 10.3892/ijmm.2023.5238
167. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun (2018) 9:1–12. doi: 10.1038/s41467-018-04947-6
168. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10. doi: 10.15252/EMMM.201708689
169. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214:3219–38. doi: 10.1084/JEM.20171419
170. He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Núñez G. 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem (2014) 289:1142–50. doi: 10.1074/JBC.M113.515080
171. Honda H, Nagai Y, Matsunaga T, Okamoto N, Watanabe Y, Tsuneyama K, et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J Leukoc Biol (2014) 96:1087–100. doi: 10.1189/JLB.3A0114-005RR
172. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
173. Sebastian-Valverde M, Wu H, Al Rahim M, Sanchez R, Kumar K, De Vita RJ, et al. Discovery and characterization of small-molecule inhibitors of NLRP3 and NLRC4 inflammasomes. J Biol Chem (2021) 296:100597. doi: 10.1016/J.JBC.2021.100597
174. Cocco M, Pellegrini C, Martínez-Banaclocha H, Giorgis M, Marini E, Costale A, et al. Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem (2017) 60:3656–71. doi: 10.1021/ACS.JMEDCHEM.6B01624
175. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U.S.A. (2018) 115:E1530–9. doi: 10.1073/PNAS.1716095115
176. Shim DW, Shin WY, Yu SH, Kim BH, Ye SK, Koppula S, et al. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci Rep (2017) 7:1–12. doi: 10.1038/s41598-017-15314-8
177. Jiang Y, He L, Green J, Blevins H, Guo C, Patel SH, et al. Discovery of second-generation NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem (2019) 62:9718–31. doi: 10.1021/ACS.JMEDCHEM.9B01155
178. Iacano AJ, Lewis H, Hazen JE, Andro H, Smith JD, Gulshan K. Miltefosine increases macrophage cholesterol release and inhibits NLRP3-inflammasome assembly and IL-1β release. Sci Rep (2019) 9:11128. doi: 10.1038/S41598-019-47610-W
179. Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol (2009) 187:61–70. doi: 10.1083/jcb.200903124
180. Zhang H, Chen X, Zong B, Yuan H, Wang Z, Wei Y, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med (2018) 22:4437–48. doi: 10.1111/JCMM.13743
181. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB signaling in PMA-stimulated THP-1 monocytes. BioMed Pharmacother (2016) 82:167–72. doi: 10.1016/J.BIOPHA.2016.04.043
182. Chen D, Sui L, Chen C, Liu S, Sun X, Guan J. Atorvastatin suppresses NLRP3 inflammasome activation in intracerebral hemorrhage via TLR4- and MyD88-dependent pathways. Aging (Albany NY) (2022) 14:462–76. doi: 10.18632/AGING.203824
183. Saadane A, Masters S, DiDonato J, Li J. Berger M. parthenolide inhibits IκB kinase, NF-κB activation, and inflammatory response in cystic fibrosis cells and mice. Am J Respir Cell Mol Biol (2007) 36:728–36. doi: 10.1165/rcmb.2006-0323OC
184. Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood (2007) 110:4427–35. doi: 10.1182/blood-2007-05-090621
185. D’Anneo A, Carlisi D, Lauricella M, Puleio R, Martinez R, Di Bella S, et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. a soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis (2013) 4:1–14. doi: 10.1038/cddis.2013.415
186. Liu P, Zhang Z, Cai Y, Yang Y, Yuan J, Chen Q. Inhibition of the pyroptosis-associated inflammasome pathway: the important potential mechanism of ginsenosides in ameliorating diabetes and its complications. Eur J Med Chem (2023) 253:115336. doi: 10.1016/j.ejmech.2023.115336
187. Yi YS. Roles of ginsenosides in inflammasome activation. J Ginseng Res (2019) 43:172–8. doi: 10.1016/j.jgr.2017.11.005
188. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem (2010) 285:9792–802. doi: 10.1074/jbc.M109.082305
189. Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflamm (2019) 16. doi: 10.1186/S12974-019-1471-Y
190. Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem (2018) 61:5412. doi: 10.1021/ACS.JMEDCHEM.8B00733
191. Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of alzheimer’s disease. Mol Neurobiol (2018) 55:1977. doi: 10.1007/S12035-017-0467-9
192. Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-Chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci (2017) 8:2194–201. doi: 10.1021/acschemneuro.7b00124
193. Desu HL, Plastini M, Illiano P, Bramlett HM, Dietrich WD, De Rivero Vaccari JP, et al. IC100: a novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J Neuroinflamm (2020) 17:1–10. doi: 10.1186/s12974-020-01826-0
194. Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther (2007) 321:509–16. doi: 10.1124/JPET.106.111344
195. Zhang F, Wang L, Wang JJ, Luo PF, Wang XT, Xia ZF. The caspase-1 inhibitor AC-YVAD-CMK attenuates acute gastric injury in mice: involvement of silencing NLRP3 inflammasome activities. Sci Rep (2016) 6. doi: 10.1038/SREP24166
196. Liang Y, Song P, Chen W, Xie X, Luo R, Su J, et al. Inhibition of caspase-1 ameliorates ischemia-associated blood-brain barrier dysfunction and integrity by suppressing pyroptosis activation. Front Cell Neurosci (2021) 14:540669. doi: 10.3389/FNCEL.2020.540669
197. Li Q, Feng H, Wang H, Wang Y, Mou W, Xu G, et al. Licochalcone b specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep (2022) 23. doi: 10.15252/EMBR.202153499
198. Kuemmerle Deschner JB, Haug I. Canakinumab in patients with cryopyrin-associated periodic syndrome: an update for clinicians. Ther Adv Musculoskelet Dis (2013) 5:315. doi: 10.1177/1759720X13502629
Keywords: NLRP3 inflammasome, pyroptosis, mitochondrial dysfunction, mtROS, metabolic regulation
Citation: Ortega MA, De Leon-Oliva D, García-Montero C, Fraile-Martinez O, Boaru DL, de Castro AV, Saez MA, Lopez-Gonzalez L, Bujan J, Alvarez-Mon MA, García-Honduvilla N, Diaz-Pedrero R and Alvarez-Mon M (2023) Reframing the link between metabolism and NLRP3 inflammasome: therapeutic opportunities. Front. Immunol. 14:1232629. doi: 10.3389/fimmu.2023.1232629
Received: 31 May 2023; Accepted: 04 July 2023;
Published: 20 July 2023.
Edited by:
Bernhard RYFFEL, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Sarah Huot-Marchand, UMR7355 Immunologie et Neurogénétique Expérimentales et Moléculaires (INEM), FranceCopyright © 2023 Ortega, De Leon-Oliva, García-Montero, Fraile-Martinez, Boaru, de Castro, Saez, Lopez-Gonzalez, Bujan, Alvarez-Mon, García-Honduvilla, Diaz-Pedrero and Alvarez-Mon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Ortega, bWlndWVsLmFuZ2VsLm9ydGVnYTkyQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.