 Giuseppina Granato1,2Iacopo Gesmundo1,2Francesca Pedrolli1,2Ramesh Kasarla1,2Laura Begani1,2Dana Banfi1,2Stefania Bruno2,3Tatiana Lopatina2Maria Felice Brizzi2Renzhi Cai4,5Wei Sha4Ezio Ghigo1,2Andrew V. Schally4,5,6,7Riccarda Granata1,2*
Giuseppina Granato1,2Iacopo Gesmundo1,2Francesca Pedrolli1,2Ramesh Kasarla1,2Laura Begani1,2Dana Banfi1,2Stefania Bruno2,3Tatiana Lopatina2Maria Felice Brizzi2Renzhi Cai4,5Wei Sha4Ezio Ghigo1,2Andrew V. Schally4,5,6,7Riccarda Granata1,2*- 1Department of Medical Sciences, Division of Endocrinology, Diabetes and Metabolism, University of Turin, Turin, Italy
- 2Department of Medical Sciences, University of Turin, Turin, Italy
- 3Molecular Biotechnology Center, University of Turin, Turin, Italy
- 4Endocrine, Polypeptide, and Cancer Institute, Veterans Affairs Medical Center, Miami, FL, United States
- 5South Florida VA Foundation for Research and Education, Veterans Affairs Medical Center, Miami, FL, United States
- 6Department of Medicine, Divisions of Medical/Oncology and Endocrinology, and the Department of Pathology, Miller School of Medicine, University of Miami, Miami, FL, United States
- 7Sylvester Comprehensive Cancer Center, Miller School of Medicine, University of Miami, Miami, FL, United States
COVID-19 is characterized by an excessive inflammatory response and macrophage hyperactivation, leading, in severe cases, to alveolar epithelial injury and acute respiratory distress syndrome. Recent studies have reported that SARS-CoV-2 spike (S) protein interacts with bacterial lipopolysaccharide (LPS) to boost inflammatory responses in vitro, in macrophages and peripheral blood mononuclear cells (PBMCs), and in vivo. The hypothalamic hormone growth hormone-releasing hormone (GHRH), in addition to promoting pituitary GH release, exerts many peripheral functions, acting as a growth factor in both malignant and non-malignant cells. GHRH antagonists, in turn, display potent antitumor effects and antinflammatory activities in different cell types, including lung and endothelial cells. However, to date, the antinflammatory role of GHRH antagonists in COVID-19 remains unexplored. Here, we examined the ability of GHRH antagonist MIA-602 to reduce inflammation in human THP-1-derived macrophages and PBMCs stimulated with S protein and LPS combination. Western blot and immunofluorescence analysis revealed the presence of GHRH receptor and its splice variant SV1 in both THP-1 cells and PBMCs. Exposure of THP-1 cells to S protein and LPS combination increased the mRNA levels and protein secretion of TNF-α and IL-1β, as well as IL-8 and MCP-1 gene expression, an effect hampered by MIA-602. Similarly, MIA-602 hindered TNF-α and IL-1β secretion in PBMCs and reduced MCP-1 mRNA levels. Mechanistically, MIA-602 blunted the S protein and LPS-induced activation of inflammatory pathways in THP-1 cells, such as NF-κB, STAT3, MAPK ERK1/2 and JNK. MIA-602 also attenuated oxidative stress in PBMCs, by decreasing ROS production, iNOS and COX-2 protein levels, and MMP9 activity. Finally, MIA-602 prevented the effect of S protein and LPS synergism on NF-кB nuclear translocation and activity. Overall, these findings demonstrate a novel antinflammatory role for GHRH antagonists of MIA class and suggest their potential development for the treatment of inflammatory diseases, such as COVID-19 and related comorbidities.
1 Introduction
To date, the cases of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have exceeded 758 million, with 6.8 million deaths worldwide (1). The most common symptoms of COVID-19 are fever, cough, dyspnea, and myalgia, while severe disease encompasses pulmonary and systemic hyperinflammation, leading to sepsis, multiorgan failure and acute respiratory distress syndrome (ARDS) (2, 3). SARS-CoV-2 is made up of four structural proteins, of which the spike (S) protein is responsible for cell entry by binding to angiotensin-converting enzyme 2 (ACE2) (4). Receptor engagement leads to virus entry in host cell, by either endocytosis and subsequent S2 subunit cleavage by cathepsins, or activation and cleavage of S2 at the cell surface by transmembrane protease, serine 2 (TMPRSS2) (5). Viral components and products of apoptotic and necrotic cells promote inflammatory responses in innate immune cells such as macrophages, monocytes, and neutrophils, via pattern recognition receptors (PRRs) (6). Subsequently, PRRs, including toll-like receptors (TLRs), NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs), induce activation of transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), leading to production of inflammatory cytokines/chemokines, cytokine storm and lung inflammation. Indeed, COVID patients show high levels of interleukin (IL)-1, IL-6, IL-8, tumor necrosis factor alpha (TNF-α), and monocyte chemoattractant protein (MCP)-1, among others (2, 3). In addition, SARS-CoV-2 triggers an excess of reactive oxygen species (ROS) and enhances oxidative stress, resulting in more severe disease (7).
Recent studies have demonstrated that S protein interacts with low levels of bacterial lipopolysaccharide (LPS), a known TLR4 ligand, to boost NF-κB activation and inflammatory cytokine release in vitro, in THP-1-derived macrophages and human peripheral blood mononuclear cells (PBMCs), and in vivo (8–10). Moreover, elevated levels of LPS have been observed in severe COVID-19, often associated with increased death of hospitalized patients (11, 12). Thus, identifying drugs that counteract oxidative stress and inflammation, also induced by S protein and LPS synergism, would help define novel therapeutic approaches to ameliorate hyperinflammation during COVID-19.
Growth hormone-releasing hormone (GHRH) is a hypothalamic hormone that stimulates the synthesis and release of GH from the anterior pituitary (13). In addition, several studies have demonstrated that GHRH and its agonistic analogs exert many peripheral activities, promoting cell growth in both nonmalignant and tumor cells and displaying protective effects in different cells and tissues, through binding to GHRH receptors (GHRH-Rs) (14–18). Moreover, an autocrine/paracrine stimulatory loop formed by locally produced GHRH and its receptors, particularly the splice variant 1 (SV1), promotes the growth of many cancers, a mechanism that can be blocked by GHRH antagonists. These molecules can also inhibit the autocrine/paracrine proliferative effects of tumoral insulin-like growth factor (IGF)-I and -II (19). In the past decades, many GHRH antagonists have been developed and tested in our laboratories, including the latest of Miami (MIA) and AVR class (19–22).
The potent anticancer effects of MIA antagonists have been demonstrated in several in vitro and in vivo models of human cancer, including lung and colorectal cancer, mesothelioma and pituitary tumors (22–27). These compounds, characterized by increased receptor binding affinity, also exhibited strong antinflammatory and antioxidant activities, in both malignant and non-malignant models, while displaying only weak endocrine GH inhibitory effects (22, 25, 28, 29). In fact, MIA-602 and MIA-690 alleviated LPS-induced ocular inflammation (30, 31), reduced prostatic enlargement and inflammation (32), blunted colon and lung inflammation (33, 34), inhibited inflammation in a model of sarcoidosis (35), and supported lung endothelial barrier function (36). Mechanistically, these effects included attenuation of lymphocyte and macrophage recruitment, inhibition of cytokine release and inflammatory pathways, such as MAPKs, JAK2/STAT3, NF-кB, as well as attenuation of oxidative markers, including ROS, cyclooxygenase (COX)−2, and inducible nitric oxide synthase (iNOS) (27, 28, 30, 32, 34, 36, 37). Overall, the above-mentioned studies suggest potential protective effects of GHRH antagonists against systemic hyperinflammation and sepsis-induced ARDS, main characteristics of severe COVID-19 (2); however, the role of these peptides in SARS-CoV-2-induced inflammation remains unexplored. Thus, based on the foregoing, we aimed to verify whether MIA-602 reduces inflammation in human THP-1-derived macrophages and PBMCs exposed to SARS-CoV-2 S protein and LPS synergism. Additionally, we examined the signaling pathways involved in the effects of the peptide.
2 Materials and methods
2.1 Reagents
GHRH-R antagonists MIA602 was prepared by Dr. Renzi Cai and Dr. Andrew Schally at the Veterans Affairs Medical Center, University of Miami, Miami, FL, as described previously (25). The chemical structure of MIA-602 is ([(PhAc-Ada)0-Tyr1, D-Arg2, Fpa56, Ala8, Har9, Tyr(Me)10, His11, Orn12, Abu15, His20,Orn21, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2). MIA-602 was dissolved in 100% dimethyl sulfoxide (DMSO) (Merk Life Science, Billerica, MA, USA) and diluted with appropriate incubation medium. The concentration of DMSO never exceed 0.1% (vol/vol). Trypsin, lipopolysaccharides (LPS) from Escherichia coli serotype O111:B4, bovine serum albumin (BSA), 4’,6-diamidino- 2-phenylindole (DAPI), formaldehyde solution, Triton X-100, Bicinchoninic Acid (BCA) protein assay kit, all primers for Real-Time PCR and Coomassie Brilliant Blue R-250 were obtained from (MilliporeSigma, Milan, Italy). RT-PCR, Real-Time PCR reagents and TaqMan probes were obtained from Life Technologies (Thermo Fisher Scientific, Milan, Italy). PepTivator SARS-CoV-2 Spike protein (S protein, code: 130-126-700) was purchased from Miltenyi Biotec (Bologna, Italy). Western blot reagents were purchased from Bio-Rad (Milan, Italy). Rabbit polyclonal antibodies against GHRH-R (code: ab76263) and ERK (code: ab16869) were obtained from Abcam (Cambridge, UK). Rabbit monoclonal primary antibodies against NFkB p65 (code: 8242) and P-NFkB p65 (code: 3033), glyceraldehyde-3- phosphate dehydrogenase (GAPDH), (code: 2118), P-ERK (code: 3192), SAPK/JNK (code: 9252), P-SAPK/JNK (code: 4668), STAT3 (code:30835) and P-STAT3 (code:9131) were obtained from Cell Signaling Technology (Danvers, MA, USA). Horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit-IgG secondary antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Goat normal serum and Alexa Fluor-488 goat anti-rabbit secondary antibody were obtained from Jackson Immuno-Research Europe (Newmarket, UK). All other chemicals were purchased from MilliporeSigma (Milan, Italy).
2.2 Cell culture and treatments
The THP-1 cells (ATCC, TIB-202) and MSTO-211H human biphasic pleural mesothelioma cell line were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% fetal bovine serum (FBS), 2-ß-mercaptoethanol (0.05 mM), penicillin (100 U/ml), streptomycin (100 µg/ml) and amphotericin B (250 ng/ml) (MilliporeSigma, Milan, Italy) at 37°C in a 5% CO2 humidified atmosphere. Differentiation of THP-1 cells in macrophage-like cells was induced by adding phorbol-12-myristate-13-acetate (PMA) (MilliporeSigma, Milan, Italy) (25 nM) for 48 h, as previously described (38). Differentiated THP-1 cells were maintained in PMA-free medium for further 24 h, then stimulated with S protein (500 ng/ml), MIA-602 (MIA, 1 µM) or LPS (50 ng/ml) alone for 24 h or pretreated with LPS for 3 h then incubated up to 24 h with S protein, in the presence or absence of MIA-602, at the same concentrations. PBMCs were isolated from human peripheral blood of healthy male donors (n=10, age range 30-65 years by Histopaque®-1077 density gradient centrifugation (MilliporeSigma) following the manufacturer’s instructions. Cells were maintained in RPMI-1640 with 10% FBS, penicillin (100 U/ml), streptomycin (100 µg/ml) and amphotericin B (250 ng/ml) at 37°C in a 5% CO2 humidified atmosphere, then exposed to either LPS (10 ng/ml), S protein (500 ng/ml), MIA-602 (1 µM) alone for 24 h or pre-treated with LPS for 3 h, then for further 24 h with either S protein, MIA-602 or both, at the same concentrations. Untreated cells were used as control.
2.3 Western blotting
Protein extraction and Western blot analysis were performed as described previously (15, 25, 39). Briefly, 50 µg proteins were resolved in 10% SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 1% BSA in Tris-buffered saline with 0.1% Tween for 2 h at room temperature, membranes were incubated overnight at 4°C with the specific antibody (dilution 1:1000). Subsequently, blots were re-probed with the respective total antibodies (1:1000) or GAPDH (1:1500) for normalization. Immunoreactive proteins were visualized using horseradish peroxidase-conjugated goat anti-mouse (1:4000) and goat anti-rabbit (1:10000) secondary antibodies by enhanced chemiluminescence substrate (ECL, Bio-Rad, Milan, Italy) using ChemiDoc XRS (Bio-Rad, Milan, Italy); densitometric analysis was performed with Quantity One software (Bio-Rad, Milan, Italy). Each experiment was performed in triplicate. Densitometric analysis was carried out with Quantity One software (Bio-Rad).
2.4 Fluorescence microscopy
THP-1 and MSTO-211H cells were plated on glass coverslips in 35-mm dishes at a density of 5 x 105 cells and then treated with the different stimuli for 24 h. Next, cells were washed once with ice-cold 1X PBS, fixed with 4% paraformaldehyde for 5 min, washed again for three times and stored at 4°C until analysis. PBMCs (3 x 105 cells) were immobilized on glass microscope slides by cytospin and then fixed in 90% ethanol/acetic acid (2:1 solution) at -20°C for 5 min. After fixation, all cells were permeabilized with 0.1% Triton X-100 for 2 min, blocked in goat normal serum (dilution 1:10 in 1X PBS) for 30 min at room temperature and stained overnight at 4°C with rabbit polyclonal anti-GHRH-R (1:200) or anti-NFkB (1:200) antibody. The day after, the cells were incubated for 1 h at room temperature with Alexa Fluor 488- conjugated goat anti-rabbit secondary antibody diluted 1:300 in 1X PBS. Then, cells were washed three times and counterstained with DAPI for 15 min at room temperature. Slides were mounted using Fluoromount-G Mounting Medium (MilliporeSigma). Images were captured using a Leica DM200 fluorescent microscope and a Leica DFC340 FX camera (Leica, Solms, Germany) at 40x magnification.
2.5 Real-time PCR
Total RNA and reverse transcription to cDNA (1 µg of RNA) from THP-1 cells and PBMCs were performed as described previously (26). RNA purity and quantification were detected spectrophotometrically by 260/280 nm absorbance ratio with NanoDrop One (Thermo Fisher Scientific, Milan, Italy). For real-time PCR, reaction was performed with 50 ng cDNA, 100 nM of each primer, and the Luna Universal qPCR Master Mix (New England BioLabs, Ipswich, MA, USA) using the ABI-Prism 7300 (Applied Biosystems; Thermo Fisher Scientific, Milan, Italy). The following primer pairs [designed with the Primer 3 Software (http://www.primer3.org/)] were used: TNF-α, forward 5'-ATGAGCACTGAAAGCATGATCC-3', reverse 5'-GAGGGCTGATTAGAG AGAGGTC-3' (NM_000594.4); IL-6, forward 5'-AACCTGAACCTTCCAAAGATGG−3', reverse 5'-TCTGGCTTGTTCCTCACTACT-3' (NM_001371096.1); IL-8, forward 5'-TGGACCACACTG CGCCAACAC-3', reverse 5'-ACTTCTCCACAACCCTCTGCA-3' (NM_001354840.3); IL-1β, forward 5'-CCTGTCCTGCGTGTTGAAAGA-3' reverse 5'-GGGAACTGGGCAGACTCAAA-3' (XM_054341810.1); 18S rRNA, forward 5′-CCCATTCGAACGTCTGCCCTATC-3′, reverse 5′-TG CTGCCTTCCTTGGATGTGGTA-3′ (NR_146144.1). 18S rRNA was used as endogenous control. Real-time PCR for MCP1 was performed using commercially available primers [TaqMan Gene Expression Assays; FAM dye labeled TaqMan MGB probe: HS00234140_m1 (MCP1)]. 18S [probe: Hs03003631_g1 (18S)] was used as housekeeping gene. Relative quantification was performed using the comparative Ct (2-ΔΔCt) method.
2.6 ELISA
THP-1-derived macrophages and PBMCs were seeded in 6-well plates at the concentrations of 6 x 106 cells/well. Conditioned medium from treated cells was collected and centrifuged at 300 x g for 10 min, then stored at -80°C until analysis. TNF-α, IL-1β and IL-6 were measured using human DuoSet ELISA Kit (R&D Systems, Minneapolis, MN, USA), following the manufacturer’s instructions. Absorbance was assessed at 450 nm using the LT-4000 microplate reader (Euroclone, Milan, Italy).
2.7 ROS detection
Intracellular ROS levels were measured using the Muse Oxidative Stress Cell Kit (Luminex, Austin, TX, USA) according to the manufacturer’s instructions. Briefly, PBMCs were seeded in 24-well plate (1 x 106 cells/well) and treated with the different stimuli for 24 h. Collected cells were resuspended in Muse Oxidative Stress reagent, incubated at 37°C for 30 min and analysis was performed using Muse Cell Analyzer and Muse Cell Analyzer Software (Luminex, Austin, TX, USA).
2.8 Gelatin zymography
Gelatinolytic activity of MMP9 in PBMC conditioned medium was determined by gelatin zymography. Equal amounts of proteins (30 μg) were separated in SDS-PAGE under non reducing conditions without boiling on 10% SDS-polyacrylamide gel co-polymerized with 2 mg/ml of gelatin. After electrophoresis (35 mA for 150-210 min at 4°C), gel was washed in 2.5% Triton X-100 solution for 1 h to remove SDS and then incubated in developing buffer (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM CaCl2 and 5 µM ZnCl2) at 37°C for 48 h, which allows substrate degradation. Finally, gel was fixed in 30% methanol and 10% acetic acid for 30 min, stained with 0.1% Coomassie Brilliant Blue R-250 and destained in 50% methanol and 5% acetic acid. Proteolytic bands were visualized as clear band against a blue background. MMP9 enzymatic activity was determined by quantification of clear bands with ImageJ software (http://rsbweb.nih.gov/ij/).
2.9 Statistical analysis
Results are presented as mean ± SEM. Significance was calculated by one-way ANOVA followed by Tukey’s multiple comparison test for post-hoc analysis. Analysis was performed using GraphPad Prism 8.0 (San Diego, CA, USA). Significance was established at P <0.05.
3 Results
3.1 GHRH-R and SV1 are expressed in THP-1-derived macrophages and human PBMCs
THP-1 monocytes were first differentiated into macrophage-like cells. Light microscopy analysis showed that after treatment with PMA, the round shape non-adherent cells became adherent and acquired the typical flat, elongated, and branching macrophage morphology (Figure 1A). Because GHRH agonists and antagonists display their biological effects through binding to GHRH-R and/or its splice variant SV1, we assessed the presence of these receptors in THP-1-derived macrophages and human PBMCs. Western blot analysis, using a polyclonal antibody against a common segment of GHRH-R and SV1, showed strong expression of GHRH-R protein and low levels of SV1 in THP-1 cells and PBMCs (Figure 1B). Immunofluorescence analysis confirmed the presence of GHRH-R in both cell types (Figure 1C). MSTO-211H pleural mesothelioma cells, which express GHRH-Rs (24), were used as positive control in both experiments.
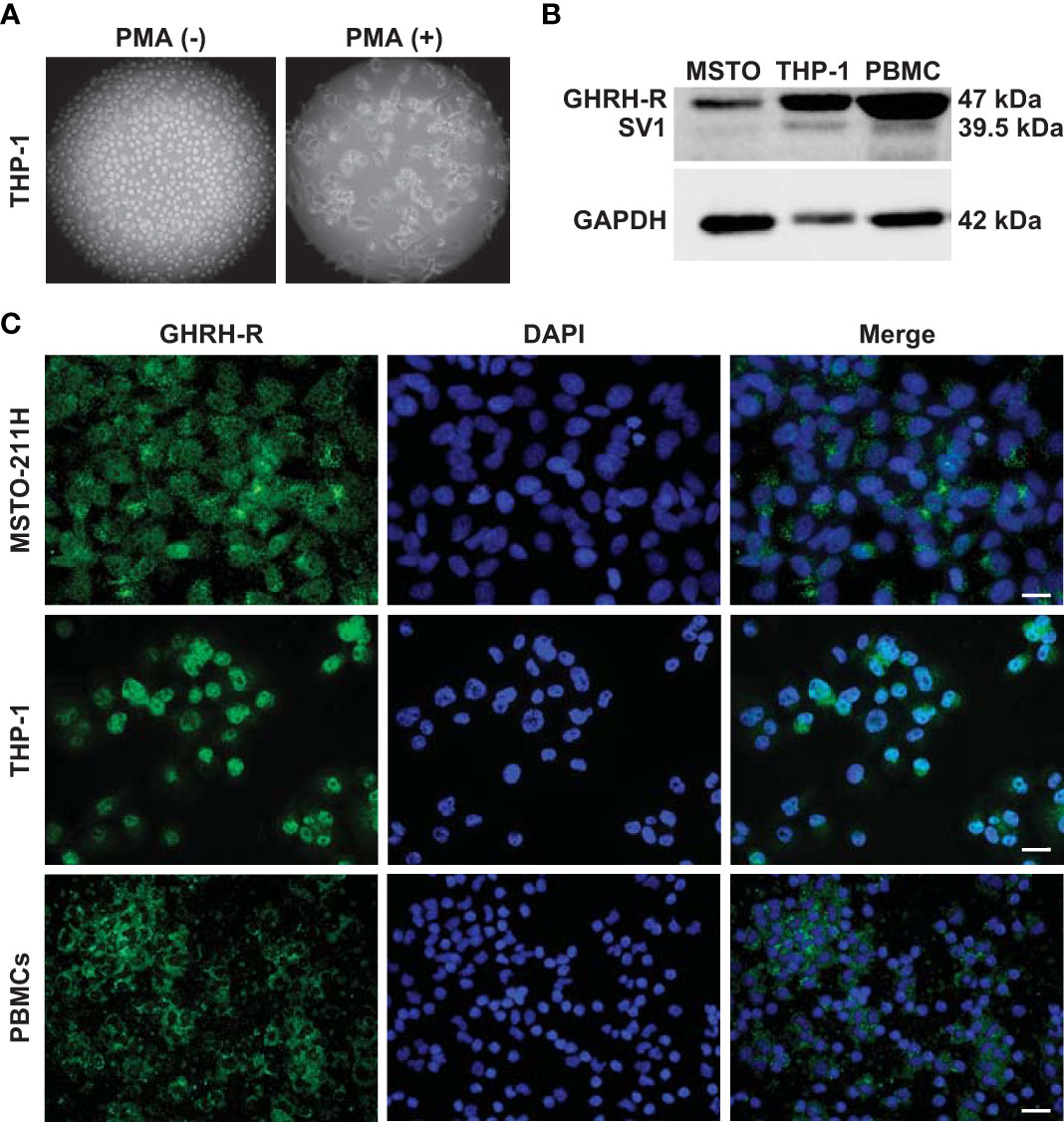
Figure 1 Expression of GHRH-R and SV1 in THP-1-derived macrophages and PBMCs. (A) Representative phase contrast microscopy images (magnification, X10) of THP-1 monocytes (left panel) differentiated into macrophage-like cells (right panel) after treatment for 48 h with PMA (25 nM). (B) Western blot analysis for GHRH-R and SV1 in THP-1-derived macrophages and PBMCs (top). MSTO-211H pleural mesothelioma cells were used as positive control. Equal protein loading was determined by reprobing with antibody to GAPDH (bottom). (C) Representative fluorescence microscopy micrographs showing GHRH-R expression (green) in THP-1-derived macrophages (middle panels) and PBMCs (bottom panels). Nuclei were stained blue with DAPI. MSTO-211H cells were used as positive control (top panels) (scale bar: 200 μm).
3.2 MIA-602 reduces cytokine and chemokine production in THP-1-derived macrophages treated with S protein and LPS combination
To evaluate the antinflammatory role of MIA-602, THP-1 cells were stimulated with S protein, either alone or with LPS. Both S protein and MIA-602 as single agents had no effect on the mRNA levels of TNF-α, IL-6 and IL-8. As expected, the treatment with LPS for 24 h strongly increased TNF-α, IL-1β, IL-6 and IL-8 levels, compared with control, an effect particularly evident for IL-6. The addition of S protein produced a further elevation in IL-1β and IL-8, compared with LPS alone, while TNF-α and IL-6 were not significantly different. MIA-602, used at 1 µM, in agreement with our previous studies (24, 26), blunted the LPS-driven increase of all cytokines, except IL-6. Furthermore, the combination of MIA-602 with S protein and LPS strongly inhibited TNF-α, IL-1β and IL-8 mRNAs, compared with S protein+LPS alone, while IL-6 was slightly, but not significantly, reduced (Figures 2A-D). Similar results were obtained for MCP-1 chemokine (also known as C-C motif chemokine ligand 2, CCL2), whose mRNA levels were inhibited by the combination of MIA-602 with S protein and LPS, compared with S protein and LPS alone (Figure 2E). In line with the mRNA results, MIA-602 attenuated the production of TNF-α and IL-1β proteins in THP-1 cells exposed to S protein and LPS, while showing no effect on IL-6. (Figures 2F-H). Overall, these results indicate the ability of MIA-602 to reduce inflammation in THP-1-derived macrophages, particularly under S protein and LPS synergism.
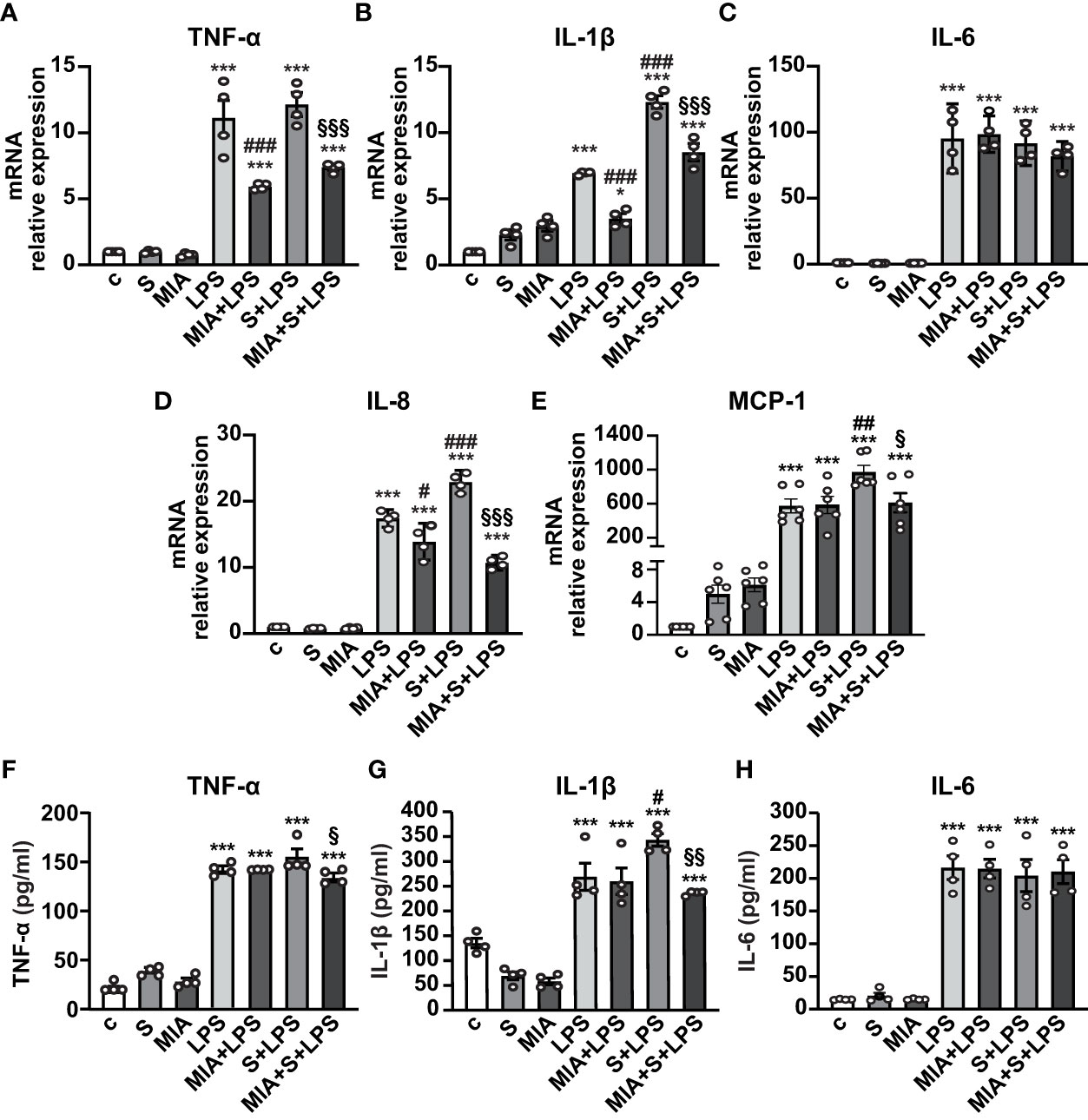
Figure 2 Inhibitory effect of MIA-602 on cytokine and chemokine expression in THP-1-derived macrophages. The cells were either untreated (c, control) or exposed to S protein (S, 500 ng/ml), MIA-602 (MIA, 1 µM) or LPS (50 ng/ml) alone for 24 h or pretreated with LPS for 3 h and then up to 24 h with S protein, in the presence or absence of MIA-602, at the same concentrations. (A–E) mRNA levels of TNF-α, IL-1β, IL-6, IL-8 and MCP-1 assessed by real time-PCR and normalized to 18S rRNA. (F–H) TNF-α, IL-1β and IL-6 secretion measured by ELISA in cell conditioned medium. Results are means ± SEM. *P < 0.05; ***P < 0.001 vs. control (c); #P < 0.05, ##P < 0.01, ###P < 0.001 vs. LPS; §P < 0.05, §§P < 0.01, §§§P < 0.001 vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 4).
3.3 MIA-602 inhibits NF-κB, JAK-STAT and MAPK inflammatory pathways in THP-1-derived macrophages treated with S protein and LPS combination
The mechanisms involved in the antinflammatory effects of MIA-602 were next evaluated in THP-1 cells treated with S protein and LPS. Western blot analysis showed that the phosphorylation of the NF-кB subunit p65 was increased by LPS, an effect further potentiated by S protein. MIA-602 had no effect with LPS alone, however, the peptide strongly attenuated the S protein+LPS-induced increase in p65 phosphorylation (Figure 3A). Similarly, MIA-602 reduced the phosphorylation of STAT3 (Figure 3B), MAPK ERK1/2 and JNK (Figures 3C, D), that was enhanced by cotreatment of S protein with LPS, compared with LPS alone. Collectively, these findings indicate that MIA-602 inhibits the inflammatory pathways activated by the combination of S protein and LPS.
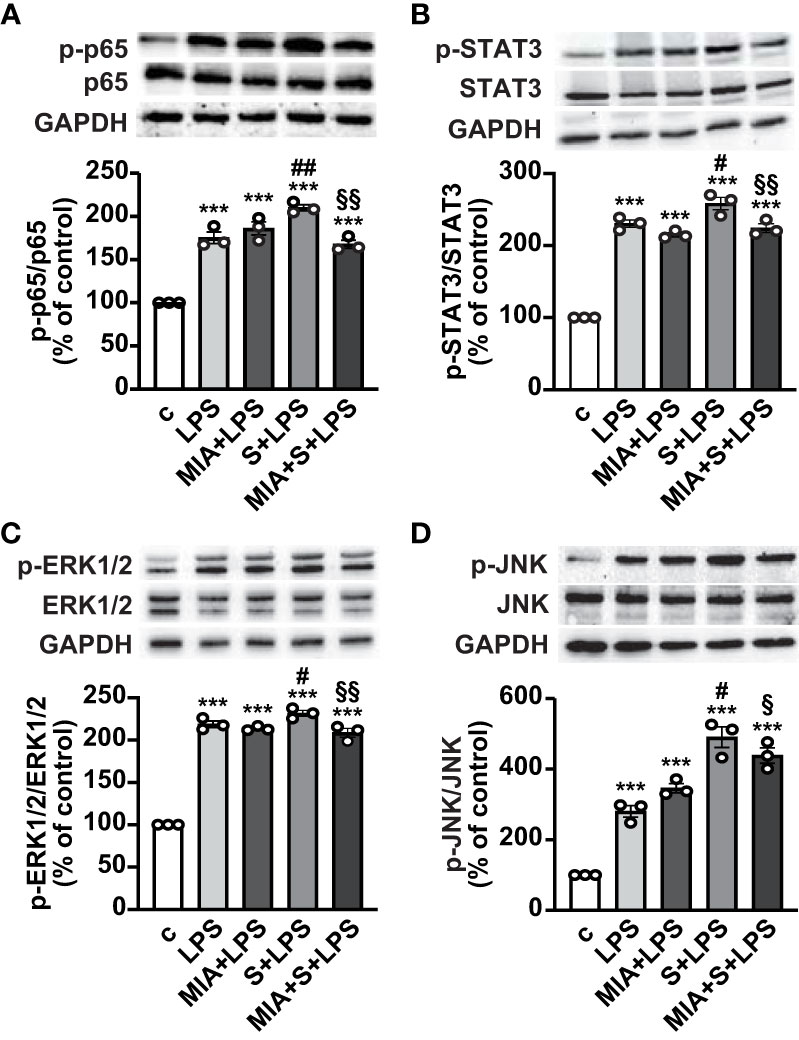
Figure 3 Inhibitory effect of MIA-602 on LPS+S protein-induced activation of inflammatory pathways in THP-1-derived macrophages. Representative Western blots for phosphorylated (p)-p65 (A), p-STAT3 (B), p-ERK1/2 (C) and p-JNK (D) assessed by Western blot in THP-1 cells either untreated (c, control) or pretreated for 3 h with LPS (50 ng/ml), then up to 24 h with either S protein (500 ng/ml), MIA-602 (1 µM) or both (top panel). Blots were reprobed with non-phosphorylated antibodies for normalization (middle panel) or GAPDH, used as internal control (bottom panel). Graphs show the densitometric analysis of phosphorylated proteins normalized to total proteins and reported as percentage of control (means ± SEM). ***P < 0.001 vs. c; #P < 0.05, ##P < 0.01 vs. LPS; §P < 0.05 and §§P < 0.01 vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 3).
3.4 MIA-602 reduces inflammatory responses in PBMCs exposed to S protein and LPS combination
We next asked whether MIA-602 could inhibit the release of inflammatory cytokines and chemokines in human PBMCs stimulated with S protein and LPS for 24 h. ELISA results showed that S protein alone produced no response, while a robust increase of TNF-α and IL-1β secretion was observed with LPS as single agent, an effect further enhanced by S protein and LPS synergism and counteracted by the addition of MIA-602 (Figures 4A, B). By contrast, while LPS alone increased IL-6, the addition of either S protein or the combination of S protein, LPS and MIA-602 showed no effect (Figure 4C). As for THP-1 cells, MIA-602 reduced MCP-1 mRNA levels, that were elevated by S protein and LPS combination (Figure 4D). These results indicate that, except for IL-6, MIA-602 reduces inflammation in PBMCs stimulated with S protein and LPS.
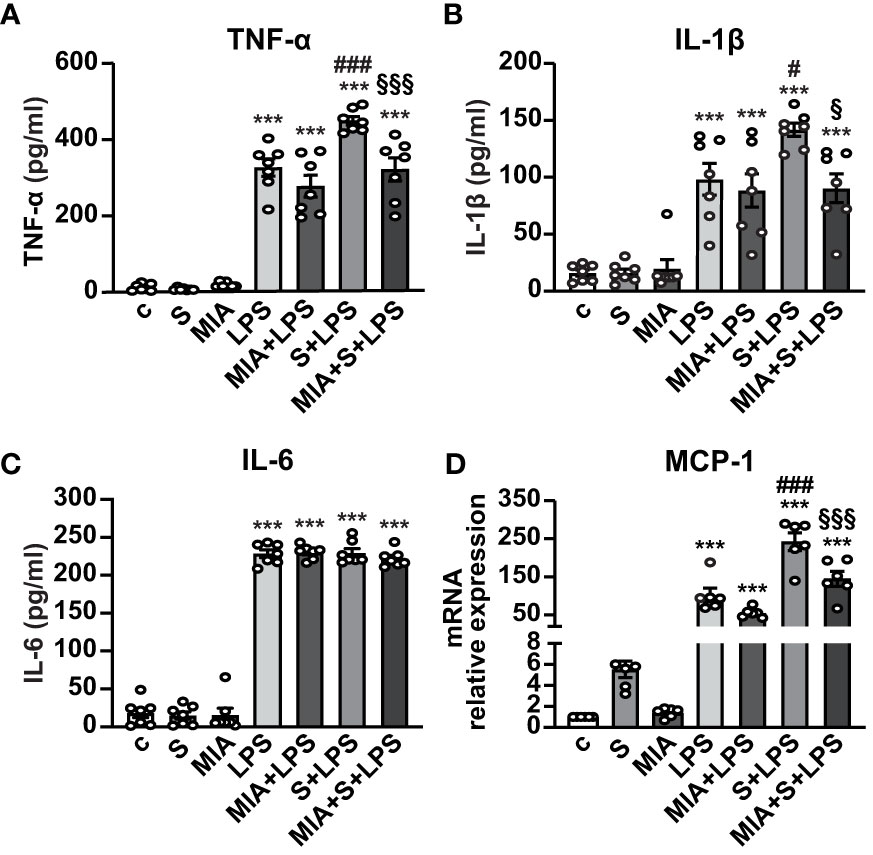
Figure 4 Inhibitory activity of MIA-602 on release and expression of inflammatory cytokines and chemokines in human PBMCs. The cells were either untreated (c, control) or pretreated for 3 h with LPS (10 ng/ml), then up to 24 h with either S protein (500 ng/ml), MIA-602 (1 µM) or both. TNF-α (A), IL-1β (B) and IL-6 (C) secretion measured by ELISA in cell conditioned medium. (D) MCP-1 mRNA expression assessed by real time-PCR. Results are means ± SEM. ***P < 0.001 vs. c; #P < 0.05 and ###P < 0.001 vs. LPS; §P < 0.05 and §§§P < 0.001 vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 7 for A-C, n = 6 for D).
3.5 MIA-602 reduces oxidative stress induced by S protein and LPS combination
In addition to inflammation, SARS-CoV-2 infection and its complications have been associated with dysregulated redox balance and increased oxidative stress (7, 40). To explore whether MIA-602 reduces oxidative stress induced by S protein and LPS combination, we first examined ROS production in PBMCs by flow cytometry. As expected, LPS alone elevated ROS levels, an effect further enhanced by S protein. MIA-602 was ineffective in cells exposed to LPS alone, however, it reduced ROS production when added to S protein+LPS combination (Figures 5A, B). Similar findings were obtained for COX-2 and iNOS proteins, whose increase, promoted by S protein and LPS synergism, was blunted by MIA-602 (Figures 5C, D).
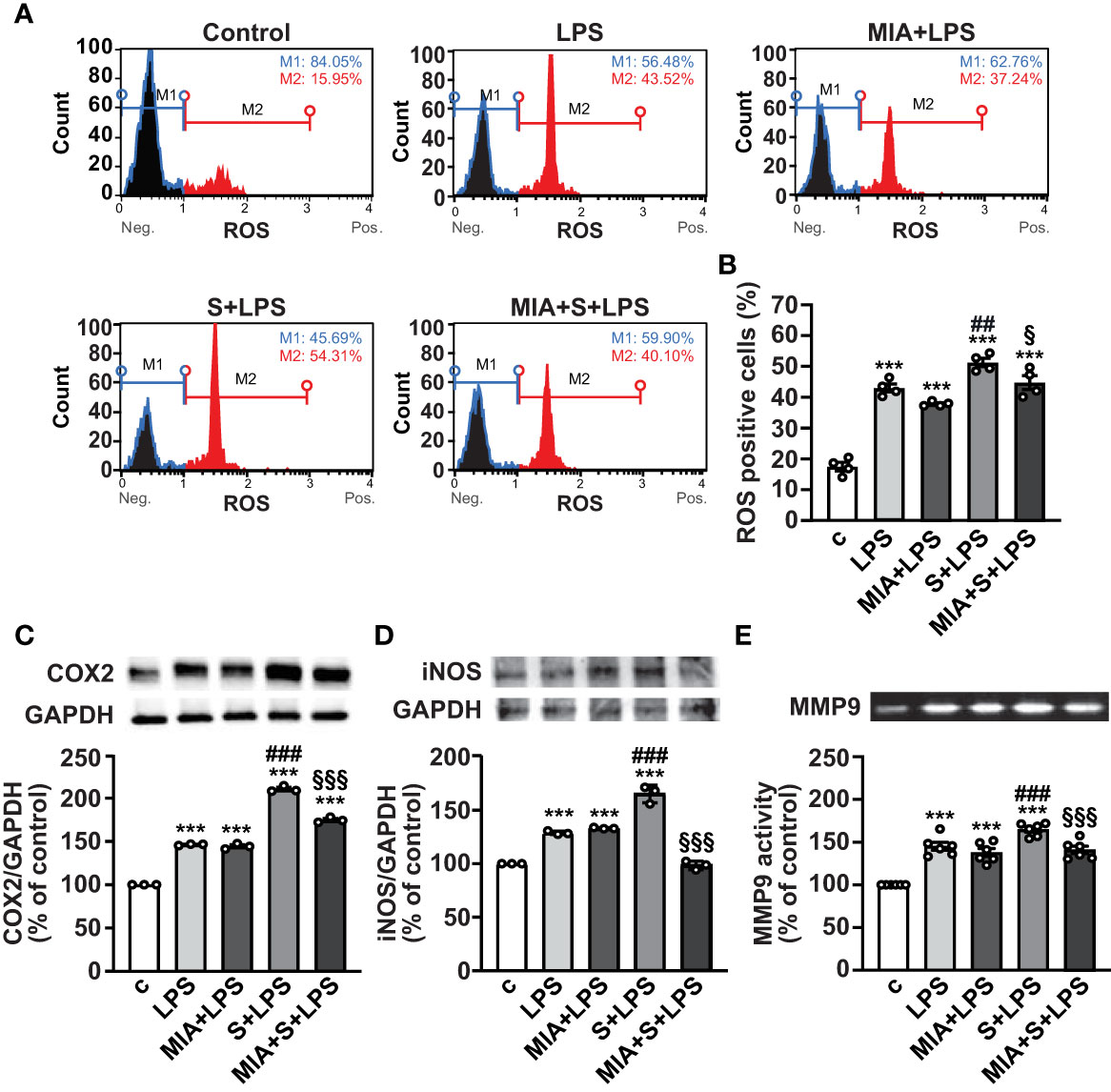
Figure 5 MIA-602-induced protection against oxidative stress in PBMCs. The cells were either untreated (c, control) or pretreated for 3 h with LPS (10 ng/ml), then up to 24 h with either S protein (500 ng/ml), MIA-602 (1 µM) or both. (A) Representative images of ROS production assessed by flow cytometry using the Muse® Oxidative Stress Kit. M1: ROS negative cells, M2: ROS-positive cells. (B) Graph showing the percentage of ROS positive cells after the different treatments. Results are means ± SEM. ***P < 0.001 vs. control (c); ##P < 0.01 vs. LPS; §P < 0.05, vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 4). COX-2 (C) and iNOS (D) protein levels assessed by Western blot (top panels). Blots were reprobed with antibody for GAPDH for normalization (bottom panels). (E) Representative gelatin zymography of MMP9 activity in PBMC conditioned medium. Graph values show the densitometric analysis of MMP9, reported as percentage of control. Results are means ± SEM. ***P < 0.001 vs. control (c); ###P < 0.001 vs. LPS; §§§P < 0.001 vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 6).
MMPs have an essential proinflammatory role in lung diseases, and elevated levels of MMP9 have been reported in patients with severe COVID-19, showing positive correlation with the risk of death (41, 42). Gelatin zymography analysis in PBMC conditioned medium showed that the combination of S protein and LPS enhanced the LPS-mediated increase of MMP9, an effect opposed by MIA-602 (Figure 5E). Overall, these data indicate that MIA-602 reduces oxidative stress and gelatinase activity induced by S protein and LPS combination in PBMCs.
3.6 MIA-602 attenuates NF-кB nuclear translocation and activity in PBMCs stimulated with S protein and LPS combination
Nuclear translocation of NF-κB is necessary for its activation and transcription of inflammatory mediators (43); thus, we next explored the role of MIA-602 on NF-κB activity in PBMCs. Figure 6A shows that nuclear localization of NF-κB subunit p65 was markedly enhanced by S protein and LPS combination, compared with untreated cells, an effect abrogated by MIA-602 (Figure 6A). Western blot analysis revealed that MIA-602, although increasing LPS-driven p65 phosphorylation, exerted a strong inhibitory effect when combined with S protein and LPS, compared with S protein+LPS alone (Figure 6B). These results indicate that, along with its inhibitory function in THP-1 cells, MIA-602 attenuates NF-κB activity in PBMCs.
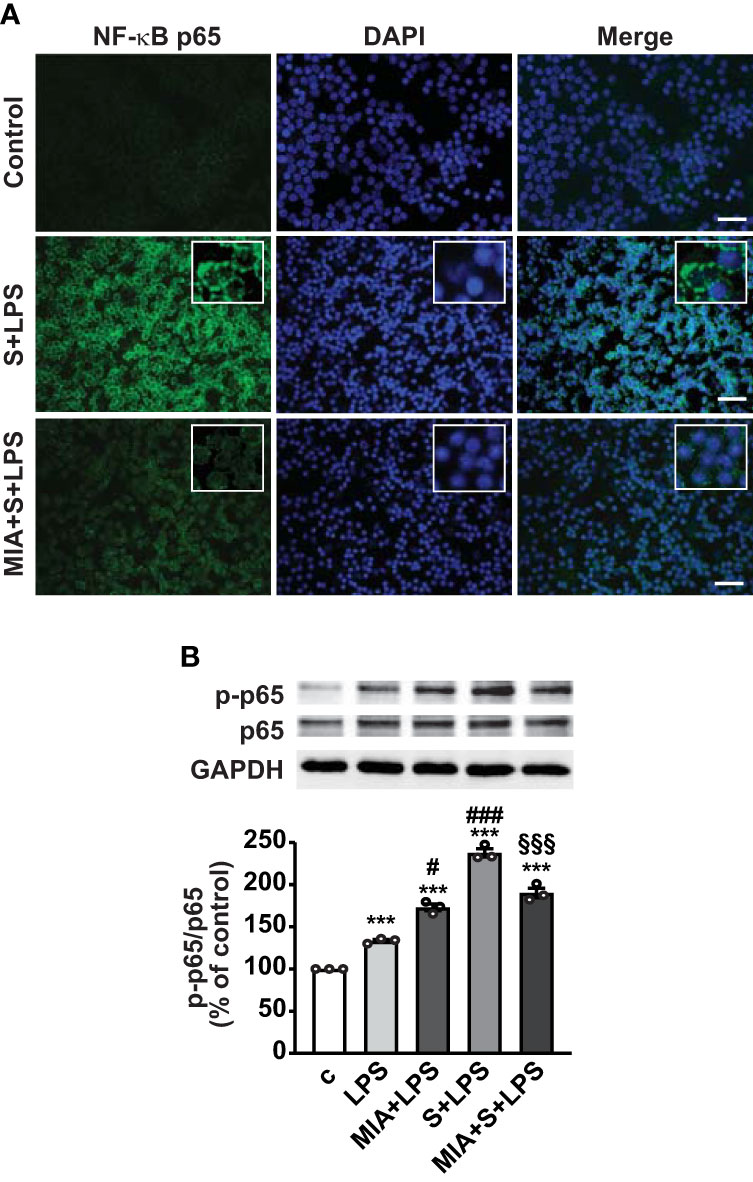
Figure 6 Inhibitory effect of MIA-602 on LPS+S protein-induced translocation and activation of NF-кB in PBMCs. The cells were either untreated (c, control) or pretreated for 3 h with LPS (10 ng/ml), then up to 24 h with S protein (500 ng/ml) or S protein+MIA-602 (1 µM). (A) Representative fluorescence microscopy images showing cells stained for NF-кB p65 (green). Nuclei were counterstained with DAPI (blue). Insets show higher magnification (scale bar: 200 μm). (B) Representative Western blot for phosphorylated (p)-p65. Blots were reprobed with non-phosphorylated p65 for normalization. Graph shows the densitometric analysis, reported as percentage of control (means ± SEM). ***P < 0.001 vs. c; #P < 0.05, ###P < 0.001 vs. LPS, and §§§P < 0.01 vs. LPS+S protein by one-way ANOVA and Tukey’s post-hoc test (n = 3).
4 Discussion
The systemic hyperinflammatory response induced by SARS-CoV-2 has been recognized as a major cause of severe COVID-19, with hyperactivation of macrophages and other immune cells to release cytokines and chemokines, which eventually lead to multiorgan failure and ARDS (6). Restraining the uncontrolled release of cytokines is one of the effective ways to counteract the detrimental consequences of COVID-19 (2, 3).
In this work, we show that GHRH antagonist MIA-602 reduces inflammation induced by the combination of SARS-CoV-2 S protein and LPS in THP-1-derived macrophages and human PBMCs. MIA-602 ameliorated the production of cytokines and chemokines, blunted the activation of inflammatory pathways and reduced oxidative stress, main hallmarks of COVID-19.
THP-1 acute monocytic leukemia cell line is commonly used as a substitute for human monocytes, with similar host response and cytokine profile. Additionally, activation with PMA can induce their differentiation into macrophage-like cells, mimicking primary human macrophages in different features (38). However, due to their distinct origin, the two cell models may not completely overlap in terms of response to stimuli (44), which prompted us to also use human PBMCs.
The expression of both GHRH-R and SV1 has been demonstrated in a variety of cells and tissues (45, 46), as well as in normal and diseased human lung (37). Here, we first detected the presence of both pituitary GHRH-R and, although to a lesser extent, of SV1, in THP-1 cells and PBMCs, suggesting that GHRH antagonist may exert its antinflammatory and antioxidant effects by binding to both receptors.
Previous reports have demonstrated the interaction between S protein and LPS, boosting NF-κB activity and inflammatory responses in vitro, in THP-1-derived macrophages and PBMCs, and in vivo (8–10, 47). Interestingly, patients with metabolic syndrome have high blood levels of bacterial LPS due to gut dysbiosis and are at a higher risk of developing severe COVID-19 (48). Moreover, SARS-CoV-2 infection has been associated with systemic spread of bacteria and microbial products, associated with increased monocyte activation, higher levels of cytokines and chemokines and worse outcomes of the disease, including death (11, 12). These and other data suggest a pathogenetic link between endotoxemia and severe SARS-CoV-2 infection, particularly in patients with comorbidities. Accordingly, we found that the combination of S protein and LPS increased the expression and release of inflammatory cytokines and chemokines and activated inflammatory and oxidative pathways in THP-1 cells and human PBMCs, compared to LPS alone. These effects were counteracted by the addition of MIA-602, which acted as an antinflammatory compound. Interestingly, we could not detect any proinflammatory activity induced by S protein as single agent. This is in line with recent studies demonstrating that S protein-induced cytokine/chemokine production in human primary macrophages is mainly due to endotoxin (i.e., LPS) contamination, while LPS-free recombinant glycosylated S protein has no effect (47, 49). Furthermore, it has been shown that S protein alone, even at high concentrations (10 μg/ml), is inactive on IL-1β secretion in human macrophages from SARS-CoV-2 naïve individuals (50). However, others have demonstrated the ability of S protein alone to trigger inflammation in human and mouse macrophages, although it must be noted that the LPS-induced contamination had not been taken into consideration (51, 52).
The antinflammatory behavior of GHRH antagonists in both cancer and other diseases is well known (19, 28, 30, 32, 35, 36). Indeed, MIA-602, but not GHRH agonist MR-409, reduced the infiltration of macrophages and leucocytes and the production of TNF-α, IL-1β and MCP-1 in a preclinical model of LPS-induced uveitis (31). Similarly, MIA-602, but not MR-409, inhibited LPS-induced elevation of IL-1β, IL-6 and iNOS genes in vitro, in ciliary and iris epithelial cells (53). GHRH antagonists also reduced prostate size and expression of inflammatory mediators, such as IL-1β, NF-κB p65, and COX-2, in rat prostatic hyperplasia (54). Here, MIA-602 attenuated the expression and release of TNF-α, IL-1β, IL-8 and MCP-1 but not IL-6, unlike previous findings where MIA-602 was found to decrease TNF-α and IL-6 production ex vivo in mouse prefrontal cortex and colon specimens treated with LPS or dextran sulfate sodium (DSS), respectively (34, 55). Interestingly, we also observed that only LPS as single agent was able to elevate IL-6 levels, whereas S protein and LPS synergism produced no further increase, in agreement with a previous study by Petruk et al. (8). It should be also mentioned that, although IL-6 is one of the main cytokines involved in COVID-19 inflammatory response, clinical trials with IL-6 receptor antagonists gave conflicting outcomes, and the effectiveness of IL-6 targeting approach remains to be established (56, 57).
Growing evidence suggests that the endothelium may represent an important therapeutic target in COVID-19. In fact, endothelial dysfunction has been implicated in the pathogenesis and severity of the disease through both direct viral effects and increased inflammatory responses. Moreover, the development of pulmonary disease is associated with excess vascular permeability, hypercoagulation, arterial and venous embolism and endothelial injury, which also contribute to systemic symptoms (58). STAT3 is a master transcription factor of inflammatory genes, involved in the antiviral response in COVID-19 (6). Consistent with our present findings in THP-1 cells, MIA-602 was previously found to suppress STAT3 and MAPK ERK1/2 in bovine pulmonary arterial cells and to support vascular barrier integrity (36). In addition, MIA-602 inhibited the phosphorylation and expression of STAT3 and its downstream inflammatory pathways in LPS-treated human ciliary epithelial cells (30). Interestingly, GHRH agonist JI-34 also attenuated LPS- and pneumolysin (LPY)-induced endothelial dysfunction in human lung microvascular endothelial cells (HL-MVEC), while having no effect on inflammation (59), suggesting that GHRH agonists and antagonists may elicit similar protective effects in specific settings. However, it has been demonstrated that the antinflammatory and antioxidative activities of MIA-690 in vivo are greater than those of the GHRH agonist MR-409, likely due to inhibition of IGF-I or stronger direct effects of the antagonists (55).
Of note, a recent study showed that adult individuals with untreated isolated GH deficiency (IGHD) due to a GHRH-R gene mutation, characterized by severe short stature, extremely low GH and undetectable IGF-I levels, cope better with SARS-CoV-2 infection than those with normal GH (60). Additionally, macrophages from individuals with the same type of IGHD were found to be less prone to Leishmania infection compared to those of normal subjects (61). The authors of these studies suggested that the immunological benefits could be related to IGF-I deficiency, resulting in reduced cell replication, lower DNA damage, as well as decreased IGF binding protein, and insulin. In line with the above studies, we suggest that MIA-602 may reduce inflammation in THP-1-derived macrophages and PBMCs in part by attenuating the autocrine-paracrine activities of locally produced IGF-I and related inflammatory pathways (19, 62).
Here, MIA-602, besides decreasing cytokine levels, also blunted NF-κB activity, which is implicated in the pathogenesis of severe COVID-19 (63). Activation of NF-κB by inducers such as bacterial LPS, ROS, cytokines and viral DNA and RNA, promotes the expression of a wide range of cytokines, chemokines, and oxidative stress components like iNOS and COX-2 (3, 41). Accordingly, our results showed the ability of MIA-602 to also reduce oxidative stress, by attenuating ROS production, as well as iNOS and COX-2 protein levels in PBMCs exposed to S protein and LPS synergism. This is consistent with the antioxidative effects of GHRH antagonists, including MIA-602, demonstrated in different cell types and models of inflammation and cancer (24, 25, 30, 32, 34, 54, 55, 64). Interestingly, enhanced ROS production has been linked to reduced antiviral host responses and increased virus-induced inflammation and apoptosis, with activation of MAPK pathway and elevation of inflammatory cytokines (7). Our results also demonstrate that MIA-602 reduces MMP9 activity in PBMCs exposed to S protein and LPS combination, in agreement with the previously observed inhibitory effect of the peptide in human pleural mesothelioma cell lines, inflamed prostate epithelial cells and LPS-stimulated ciliary epithelial cells (24, 25, 30, 32). Noteworthy, beyond being a key mediator of tumor growth, metastasis and angiogenesis, by degrading extracellular matrix components, MMP9 has an important role in lung physiopathology and inflammatory responses (65), and its elevation has been associated with increased risk of death in patients with severe COVID-19 (41, 42).
In conclusion, in addition to corroborating the previously described antinflammatory and antioxidative functions, our findings indicate that the effects of GHRH antagonists could be extended beyond conditions such as cancer and inflammation. In fact, to the best of our knowledge, the novelty of this study consists in the fact that: i) MIA-602 acts in macrophage-like cells and PBMCs, key players in the immune system and inflammatory responses, ii) MIA-602 attenuates inflammation and oxidative stress induced by the combination of a viral protein with a bacterial component, and iii) MIA-602 inhibits signaling pathways involved in the pathogenesis and progression of severe COVID-19. Overall, our results suggest that the development of GHRH antagonists may lead to new therapies against COVID-19 and related comorbidities, including lung diseases and sepsis-induced ARDS.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Experiments using human PBMCs were conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethic Committee of A.O.U. Città della Salute e della Scienza di Torino, Turin, Italy (CS21255 - Protocol #0050416, May 16, 2019). Participants provided written informed consent to participate in this study.
Author contributions
RG and GG contributed to conception and design of the study. GG, IG, SB, FP, RK, LB, DB and TL performed the experiments and analyzed the data. RC, WS and AS provided MIA-602. MB, EG and AS revised the paper. RG supervised the work and wrote the final manuscript. All authors contributed to the revision of the manuscript and approved the submitted version.
Funding
This study was supported by the Italian Ministry of University and Research (MUR) PRIN 2017 (2017HRTZYA to RG and 2017S55RXB_002 to EG). University of Turin Grant Ex-60% 2019 to RG and 2020 to IG. MUR project “Dipartimenti di Eccellenza 2018-2022” to the Department of Medical Sciences, University of Turin (project N. D15D18000410001). The work in the laboratory of AS was supported by the Medical Research Service of the Department of Veterans Affairs and by the University of Miami Miller School of Medicine.
Acknowledgments
We wish to thank Dr. Robert M. Jackson (University of Miami Miller School of Medicine) and Dr. Luis A. Gonzales (South Florida VA Foundation for Research and Education, Miami, FL) for authorizing the use of GHRH analogs; we also thank Dr. Irving Vidaurre (South Florida VA Foundation for Research and Education, Miami, FL) for assistance in providing the peptides.
Conflict of interest
AS, WS and RC are co-inventors on the patents for GHRH agonists, assigned to the University of Miami and Veterans Affairs Medical Center.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. WHO. Who Coronavirus (Covid-19) Dashboard (2023). Available at: https://covid19.who.int/.
2. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of covid-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
3. Rabaan AA, Al-Ahmed SH, Muhammad J, Khan A, Sule AA, Tirupathi R, et al. Role of inflammatory cytokines in covid-19 patients: A review on molecular mechanisms, immune functions, immunopathology and immunomodulatory drugs to counter cytokine storm. Vaccines (Basel) (2021) 9(5):1–23. doi: 10.3390/vaccines9050436
4. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. Sars-cov-2 cell entry depends on Ace2 and Tmprss2 and is blocked by a clinically proven protease inhibitor. Cell (2020) 181(2):271–80 e8. doi: 10.1016/j.cell.2020.02.052
5. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of Sars-cov-2 entry into cells. Nat Rev Mol Cell Biol (2022) 23(1):3–20. doi: 10.1038/s41580-021-00418-x
6. Merad M, Martin JC. Pathological inflammation in patients with covid-19: A key role for monocytes and macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
7. Gain C, Song S, Angtuaco T, Satta S, Kelesidis T. The role of oxidative stress in the pathogenesis of infections with coronaviruses. Front Microbiol (2022) 13:1111930. doi: 10.3389/fmicb.2022.1111930
8. Petruk G, Puthia M, Petrlova J, Samsudin F, Stromdahl AC, Cerps S, et al. Sars-cov-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol (2020) 12(12):916–32. doi: 10.1093/jmcb/mjaa067
9. Puthia M, Tanner L, Petruk G, Schmidtchen A. Experimental model of pulmonary inflammation induced by Sars-cov-2 spike protein and endotoxin. ACS Pharmacol Transl Sci (2022) 5(3):141–8. doi: 10.1021/acsptsci.1c00219
10. Samsudin F, Raghuvamsi P, Petruk G, Puthia M, Petrlova J, MacAry P, et al. Sars-cov-2 spike protein as a bacterial lipopolysaccharide delivery system in an overzealous inflammatory cascade. J Mol Cell Biol (2023) 14(9):1–14. doi: 10.1093/jmcb/mjac058
11. Sirivongrangson P, Kulvichit W, Payungporn S, Pisitkun T, Chindamporn A, Peerapornratana S, et al. Endotoxemia and circulating bacteriome in severe covid-19 patients. Intensive Care Med Exp (2020) 8(1):72. doi: 10.1186/s40635-020-00362-8
12. Teixeira PC, Dorneles GP, Santana Filho PC, da Silva IM, Schipper LL, Postiga IAL, et al. Increased Lps levels coexist with systemic inflammation and result in monocyte activation in severe covid-19 patients. Int Immunopharmacol (2021) 100:108125. doi: 10.1016/j.intimp.2021.108125
13. Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science (1982) 218(4572):585–7. doi: 10.1126/science.6812220
14. Schally AV, Zhang X, Cai R, Hare JM, Granata R, Bartoli M. Actions and potential therapeutic applications of growth hormone-releasing hormone agonists. Endocrinology (2019) 160(7):1600–12. doi: 10.1210/en.2019-00111
15. Boido M, Gesmundo I, Caretto A, Pedrolli F, Schellino R, Leone S, et al. Agonist of growth hormone-releasing hormone improves the disease features of spinal muscular atrophy mice. Proc Natl Acad Sci U.S.A. (2023) 120(2):e2216814120. doi: 10.1073/pnas.2216814120
16. Gesmundo I, Miragoli M, Carullo P, Trovato L, Larcher V, Di Pasquale E, et al. Growth hormone-releasing hormone attenuates cardiac hypertrophy and improves heart function in pressure overload-induced heart failure. Proc Natl Acad Sci U.S.A. (2017) 114(45):12033–8. doi: 10.1073/pnas.1712612114
17. Granata R. Peripheral activities of growth hormone-releasing hormone. J Endocrinol Invest (2016) 39(7):721–7. doi: 10.1007/s40618-016-0440-x
18. Kanashiro-Takeuchi RM, Tziomalos K, Takeuchi LM, Treuer AV, Lamirault G, Dulce R, et al. Cardioprotective effects of growth hormone-releasing hormone agonist after myocardial infarction. Proc Natl Acad Sci U.S.A. (2010) 107(6):2604–9. doi: 10.1073/pnas.0914138107
19. Schally AV, Varga JL, Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab (2008) 4(1):33–43. doi: 10.1038/ncpendmet0677
20. Varga JL, Schally AV, Csernus VJ, Zarandi M, Halmos G, Groot K, et al. Synthesis and biological evaluation of antagonists of growth hormone-releasing hormone with high and protracted in vivo activities. Proc Natl Acad Sci U.S.A. (1999) 96(2):692–7. doi: 10.1073/pnas.96.2.692
21. Cai R, Zhang X, Wang H, Cui T, Halmos G, Sha W, et al. Synthesis of potent antagonists of receptors for growth hormone-releasing hormone with antitumor and anti-inflammatory activity. Peptides (2022) 150:170716. doi: 10.1016/j.peptides.2021.170716
22. Zarandi M, Cai R, Kovacs M, Popovics P, Szalontay L, Cui T, et al. Synthesis and structure-activity studies on novel analogs of human growth hormone releasing hormone (Ghrh) with enhanced inhibitory activities on tumor growth. Peptides (2017) 89:60–70. doi: 10.1016/j.peptides.2017.01.009
23. Wang H, Zhang X, Vidaurre I, Cai R, Sha W, Schally AV. Inhibition of experimental small-cell and non-small-cell lung cancers by novel antagonists of growth hormone-releasing hormone. Int J Cancer (2018) 142(11):2394–404. doi: 10.1002/ijc.31308
24. Villanova T, Gesmundo I, Audrito V, Vitale N, Silvagno F, Musuraca C, et al. Antagonists of growth hormone-releasing hormone (Ghrh) inhibit the growth of human Malignant pleural mesothelioma. Proc Natl Acad Sci U.S.A. (2019) 116(6):2226–31. doi: 10.1073/pnas.1818865116
25. Gesmundo I, Pedrolli F, Vitale N, Bertoldo A, Orlando G, Banfi D, et al. Antagonist of growth hormone-releasing hormone potentiates the antitumor effect of pemetrexed and cisplatin in pleural mesothelioma. Int J Mol Sci (2022) 23(19):1–16. doi: 10.3390/ijms231911248
26. Gesmundo I, Granato G, Fuentes-Fayos AC, Alvarez CV, Dieguez C, Zatelli MC, et al. Antagonists of growth hormone-releasing hormone inhibit the growth of pituitary adenoma cells by hampering oncogenic pathways and promoting apoptotic signaling. Cancers (Basel) (2021) 13(16):1–20. doi: 10.3390/cancers13163950
27. Recinella L, Chiavaroli A, Veschi S, Di Valerio V, Lattanzio R, Orlando G, et al. Antagonist of growth hormone-releasing hormone mia-690 attenuates the progression and inhibits growth of colorectal cancer in mice. BioMed Pharmacother (2022) 146:112554. doi: 10.1016/j.biopha.2021.112554
28. Barabutis N, Akhter MS, Kubra KT, Jackson K. Growth hormone-releasing hormone in endothelial inflammation. Endocrinology (2022) 164(2):1–11. doi: 10.1210/endocr/bqac209
29. Recinella L, Chiavaroli A, Orlando G, Ferrante C, Veschi S, Cama A, et al. Effects of growth hormone-releasing hormone receptor antagonist Mia-602 in mice with emotional disorders: A potential treatment for Ptsd. Mol Psychiatry (2021) 26(12):7465–74. doi: 10.1038/s41380-021-01228-5
30. Liang WC, Ren JL, Yu QX, Li J, Ng TK, Chu WK, et al. Signaling mechanisms of growth hormone-releasing hormone receptor in Lps-Induced Acute Ocular inflammation. Proc Natl Acad Sci U.S.A. (2020) 117(11):6067–74. doi: 10.1073/pnas.1904532117
31. Qin YJ, Chan SO, Chong KK, Li BF, Ng TK, Yip YW, et al. Antagonist of Gh-releasing hormone receptors alleviates experimental ocular inflammation. Proc Natl Acad Sci U.S.A. (2014) 111(51):18303–8. doi: 10.1073/pnas.1421815112
32. Popovics P, Cai R, Sha W, Rick FG, Schally AV. Growth hormone-releasing hormone antagonists reduce prostatic enlargement and inflammation in carrageenan-induced chronic prostatitis. Prostate (2018) 78(13):970–80. doi: 10.1002/pros.23655
33. Zhang C, Cai R, Lazerson A, Delcroix G, Wangpaichitr M, Mirsaeidi M, et al. Growth hormone-releasing hormone receptor antagonist modulates lung inflammation and fibrosis due to Bleomycin. Lung (2019) 197(5):541–9. doi: 10.1007/s00408-019-00257-w
34. Recinella L, Chiavaroli A, Di Valerio V, Veschi S, Orlando G, Ferrante C, et al. Protective effects of growth hormone-releasing hormone analogs in Dss-induced colitis in mice. Sci Rep (2021) 11(1):2530. doi: 10.1038/s41598-021-81778-4
35. Zhang C, Tian R, Dreifus EM, Hashemi Shahraki A, Holt G, Cai R, et al. Activity of the growth hormone-releasing hormone antagonist Mia602 and its underlying mechanisms of action in sarcoidosis-like granuloma. Clin Transl Immunol (2021) 10(7):e1310. doi: 10.1002/cti2.1310
36. Uddin MA, Akhter MS, Singh SS, Kubra KT, Schally AV, Jois S, et al. Ghrh antagonists support lung endothelial barrier function. Tissue Barriers (2019) 7(4):1669989. doi: 10.1080/21688370.2019.1669989
37. Zhang C, Cui T, Cai R, Wangpaichitr M, Mirsaeidi M, Schally AV, et al. Growth hormone-releasing hormone in lung physiology and pulmonary disease. Cells (2020) 9(10):1–14. doi: 10.3390/cells9102331
38. Lund ME, To J, O’Brien BA, Donnelly S. The choice of Phorbol 12-Myristate 13-acetate differentiation protocol influences the response of Thp-1 macrophages to a pro-inflammatory stimulus. J Immunol Methods (2016) 430:64–70. doi: 10.1016/j.jim.2016.01.012
39. Gesmundo I, Pardini B, Gargantini E, Gamba G, Birolo G, Fanciulli A, et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic beta cells. JCI Insight (2021) 6(5):1–17. doi: 10.1172/jci.insight.141962
40. Badawy MA, Yasseen BA, El-Messiery RM, Abdel-Rahman EA, Elkhodiry AA, Kamel AG, et al. Neutrophil-mediated oxidative stress and albumin structural damage predict covid-19-associated mortality. Elife (2021) 10:1–22. doi: 10.7554/eLife.69417
41. Abers MS, Delmonte OM, Ricotta EE, Fintzi J, Fink DL, de Jesus AAA, et al. An immune-based biomarker signature is associated with mortality in covid-19 patients. JCI Insight (2021) 6(1):1–20. doi: 10.1172/jci.insight.144455
42. D’Avila-Mesquita C, Couto AES, Campos LCB, Vasconcelos TF, Michelon-Barbosa J, Corsi CAC, et al. Mmp-2 and Mmp-9 levels in plasma are altered and associated with mortality in covid-19 patients. BioMed Pharmacother (2021) 142:112067. doi: 10.1016/j.biopha.2021.112067
43. Liu T, Zhang L, Joo D, Sun SC. Nf-Kappab signaling in inflammation. Signal Transduct Target Ther (2017) 2:17023–. doi: 10.1038/sigtrans.2017.23
44. Tedesco S, De Majo F, Kim J, Trenti A, Trevisi L, Fadini GP, et al. Convenience versus biological significance: are Pma-differentiated Thp-1 cells a reliable substitute for blood-derived macrophages when studying in vitro polarization? Front Pharmacol (2018) 9:71. doi: 10.3389/fphar.2018.00071
45. Havt A, Schally AV, Halmos G, Varga JL, Toller GL, Horvath JE, et al. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc Natl Acad Sci U.S.A. (2005) 102(48):17424–9. doi: 10.1073/pnas.0506844102
46. Matsubara S, Sato M, Mizobuchi M, Niimi M, Takahara J. Differential gene expression of growth Hormone (Gh)-Releasing Hormone (Grh) and Grh receptor in various rat tissues. Endocrinology (1995) 136(9):4147–50. doi: 10.1210/endo.136.9.7649123
47. Cinquegrani G, Spigoni V, Iannozzi NT, Parello V, Bonadonna RC, Dei Cas A. Sars-cov-2 spike protein is not pro-inflammatory in human primary macrophages: endotoxin contamination and lack of protein glycosylation as possible confounders. Cell Biol Toxicol (2022) 38(4):667–78. doi: 10.1007/s10565-021-09693-y
48. Cai Q, Chen F, Wang T, Luo F, Liu X, Wu Q, et al. Obesity and covid-19 severity in a designated hospital in Shenzhen, China. Diabetes Care (2020) 43(7):1392–8. doi: 10.2337/dc20-0576
49. Ouyang W, Xie T, Fang H, Gao C, Stantchev T, Clouse KA, et al. Variable induction of pro-inflammatory cytokines by commercial Sars cov-2 spike protein reagents: potential impacts of Lps on in vitro modeling and pathogenic mechanisms in vivo. Int J Mol Sci (2021) 22(14):1–15. doi: 10.3390/ijms22147540
50. Theobald SJ, Simonis A, GeorgOmanolis T, Kreer C, Zehner M, Eisfeld HS, et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in covid-19. EMBO Mol Med (2021) 13(8):e14150. doi: 10.15252/emmm.202114150
51. Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. Sars-cov-2 spike protein induces inflammation via tlr2-dependent activation of the Nf-Kappab pathway. Elife (2021) 10:1–26. doi: 10.7554/eLife.68563
52. Umar S, Palasiewicz K, Meyer A, Kumar P, Prabhakar BS, Volin MV, et al. Inhibition of Irak4 dysregulates Sars-cov-2 spike protein-induced macrophage inflammatory and glycolytic reprogramming. Cell Mol Life Sci (2022) 79(6):301. doi: 10.1007/s00018-022-04329-8
53. Ren JL, Yu QX, Ma D, Liang WC, Leung PY, Ng TK, et al. Growth hormone-releasing hormone receptor mediates cytokine production in Ciliary and Iris epithelial cells during Lps-induced ocular inflammation. Exp Eye Res (2019) 181:277–84. doi: 10.1016/j.exer.2019.02.021
54. Rick FG, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, et al. Antagonists of Growth hormone-releasing hormone (Ghrh) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci U.S.A. (2011) 108(9):3755–60. doi: 10.1073/pnas.1018086108
55. Recinella L, Chiavaroli A, Orlando G, Ferrante C, Marconi GD, Gesmundo I, et al. Antinflammatory, antioxidant, and behavioral effects induced by administration of growth hormone-releasing hormone analogs in mice. Sci Rep (2020) 10(1):732. doi: 10.1038/s41598-019-57292-z
56. Rubin EJ, Longo DL, Baden LR. Interleukin-6 receptor inhibition in covid-19 - cooling the inflammatory soup. N Engl J Med (2021) 384(16):1564–5. doi: 10.1056/NEJMe2103108
57. Writing Committee for the R-CAPI, AM H, LR B, Lorenzi E, Murthy S, McQuilten Z, et al. Long-term (180-day) outcomes in critically ill patients with covid-19 in the remap-cap randomized clinical trial. JAMA (2023) 329(1):39–51. doi: 10.1001/jama.2022.23257
58. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in covid-19. Nat Rev Nephrol (2021) 17(1):46–64. doi: 10.1038/s41581-020-00357-4
59. Czikora I, Sridhar S, Gorshkov B, Alieva IB, Kasa A, Gonzales J, et al. Protective effect of growth hormone-releasing hormone agonist in bacterial toxin-induced pulmonary barrier dysfunction. Front Physiol (2014) 5:259. doi: 10.3389/fphys.2014.00259
60. Melo MA, Borges LP, Salvatori R, Souza DRV, Santos-Junior HT, de RNJM, et al. Individuals with isolated congenital Gh deficiency due to a Ghrh receptor gene mutation appear to cope better with Sars-cov-2 infection than controls. Endocrine (2021) 72(2):349–55. doi: 10.1007/s12020-021-02728-8
61. Barrios MR, Campos VC, Peres NTA, de Oliveira LL, Cazzaniga RA, Santos MB, et al. Macrophages from subjects with isolated Gh/Igf-I deficiency due to a Ghrh receptor gene mutation are less prone to infection by Leishmania Amazonensis. Front Cell Infect Microbiol (2019) 9:311. doi: 10.3389/fcimb.2019.00311
62. Oberlin D, Fellbaum C, Eppler E. Insulin-like growth factor I messenger Rna and protein are expressed in the human lymph node and distinctly confined to subtypes of macrophages, antigen-presenting cells, lymphocytes and endothelial cells. Immunology (2009) 128(3):342–50. doi: 10.1111/j.1365-2567.2009.03136.x
63. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The role and therapeutic potential of Nf-Kappa-B pathway in severe covid-19 patients. Inflammopharmacology (2021) 29(1):91–100. doi: 10.1007/s10787-020-00773-9
64. Akhter MS, Barabutis N. Suppression of reactive oxygen species in endothelial cells by an antagonist of growth hormone-releasing hormone. J Biochem Mol Toxicol (2021) 35(10):e22879. doi: 10.1002/jbt.22879
Keywords: GHRH, GHRH antagonist, SARS-CoV-2 spike protein, macrophages, peripheral blood mononuclear cells (PBMC), inflammation
Citation: Granato G, Gesmundo I, Pedrolli F, Kasarla R, Begani L, Banfi D, Bruno S, Lopatina T, Brizzi MF, Cai R, Sha W, Ghigo E, Schally AV and Granata R (2023) Growth hormone-releasing hormone antagonist MIA-602 inhibits inflammation induced by SARS-CoV-2 spike protein and bacterial lipopolysaccharide synergism in macrophages and human peripheral blood mononuclear cells. Front. Immunol. 14:1231363. doi: 10.3389/fimmu.2023.1231363
Received: 30 May 2023; Accepted: 24 July 2023;
Published: 15 August 2023.
Edited by:
Michael V. Volin, Midwestern University, United StatesReviewed by:
Mohamed Mahdi, University of Debrecen, HungaryManuel H. Aguiar-oliveira, Federal University of Sergipe, Brazil
Copyright © 2023 Granato, Gesmundo, Pedrolli, Kasarla, Begani, Banfi, Bruno, Lopatina, Brizzi, Cai, Sha, Ghigo, Schally and Granata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccarda Granata, riccarda.granata@unito.it