 Yao Jiang
Yao Jiang Kai Hong
Kai Hong Yingchao Zhao
Yingchao Zhao Kai Xu
Kai Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 21 June 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1228200
This article is part of the Research Topic Community Series in Post-Translational Modifications of Proteins in Cancer Immunity and Immunotherapy, volume II View all 12 articles
Immune evasion is essential for carcinogenesis and cancer progression. Programmed death-ligand 1 (PD-L1), a critical immune checkpoint molecule, interacts with programmed death receptor-1 (PD-1) on immune cells to suppress anti-tumor immune responses. In the past decade, antibodies targeting PD-1/PD-L1 have tremendously altered cancer treatment paradigms. Post-translational modifications have been reported as key regulators of PD-L1 expression. Among these modifications, ubiquitination and deubiquitination are reversible processes that dynamically control protein degradation and stabilization. Deubiquitinating enzymes (DUBs) are responsible for deubiquitination and have emerged as crucial players in tumor growth, progression, and immune evasion. Recently, studies have highlighted the participation of DUBs in deubiquitinating PD-L1 and modulating its expression. Here, we review the recent developments in deubiquitination modifications of PD-L1 and focus on the underlying mechanisms and effects on anti-tumor immunity.
Immune evasion is essential for carcinogenesis and cancer progression. Cancer cells have developed multiple mechanisms to evade immune surveillance, including reducing immunogenicity, limiting antigen recognition, inducing T cell exhaustion, and expressing inhibitory immune checkpoint proteins (1). Among these checkpoint molecules, programmed death-ligand 1 (PD-L1) is one of the most critical players. PD-L1 interacts with programmed death receptor-1 (PD-1) on immune cells, including T cells, dendritic cells, macrophages, and natural killer (NK) cells to restrain anti-tumor immunity (2). Elevated PD-L1 expression has been observed in multiple cancers including cervical cancer, non-small cell lung cancer (NSCLC), and hepatocellular cancer (3). Currently, PD-1/PD-L1-targeting treatments have significantly affected cancer treatment approaches, and PD-L1 expression has emerged as an indicator for the selection of patients who are more likely to benefit from PD-1/PD-L1 inhibitors (4, 5). Therefore, exploring PD-L1 regulatory mechanisms is of great importance.
The expression of PD-L1 is modulated at various levels, including epigenetic, transcriptional, post-transcriptional, and post-translational mechanisms (3, 6–8). Deubiquitination and ubiquitination are among the most important post-translational modifications, which dynamically control protein degradation and stability, thereby influencing cellular processes. Deubiquitination, mediated by deubiquitinating enzymes (DUBs), involves the covalent cleavage of conjugated monoubiquitin or polyubiquitin chains from various substrates (9, 10). To date, approximately 100 DUBs have been discovered. Based on their structural homology, DUBs can be classified into seven categories as follows: ubiquitin-specific proteases (USPs), otubain proteases (OTUs), and JAB1/MPN/Mov34 metalloenzymes (JAMMs), ubiquitin C-terminal hydrolases, Machado-Joseph disease proteases, MIU-containing novel DUB family proteases, and Zn-finger and UFSP domain proteins. With the exception of JAMMs, all of these DUBs belong to the cysteine protease family.
DUBs have been extensively studied in a variety of cellular activities including cell proliferation, apoptosis, cell cycle control, and adaptive immune response (11). Several DUBs have been demonstrated to deubiquitinate PD-L1 and regulate its expression in cancer (7) (Figure 1). Here, we provide a comprehensive review of the recent advancements in deubiquitination modifications of PD-L1, focusing on their impact and the underlying mechanisms related to anti-tumor immunity (Table 1).
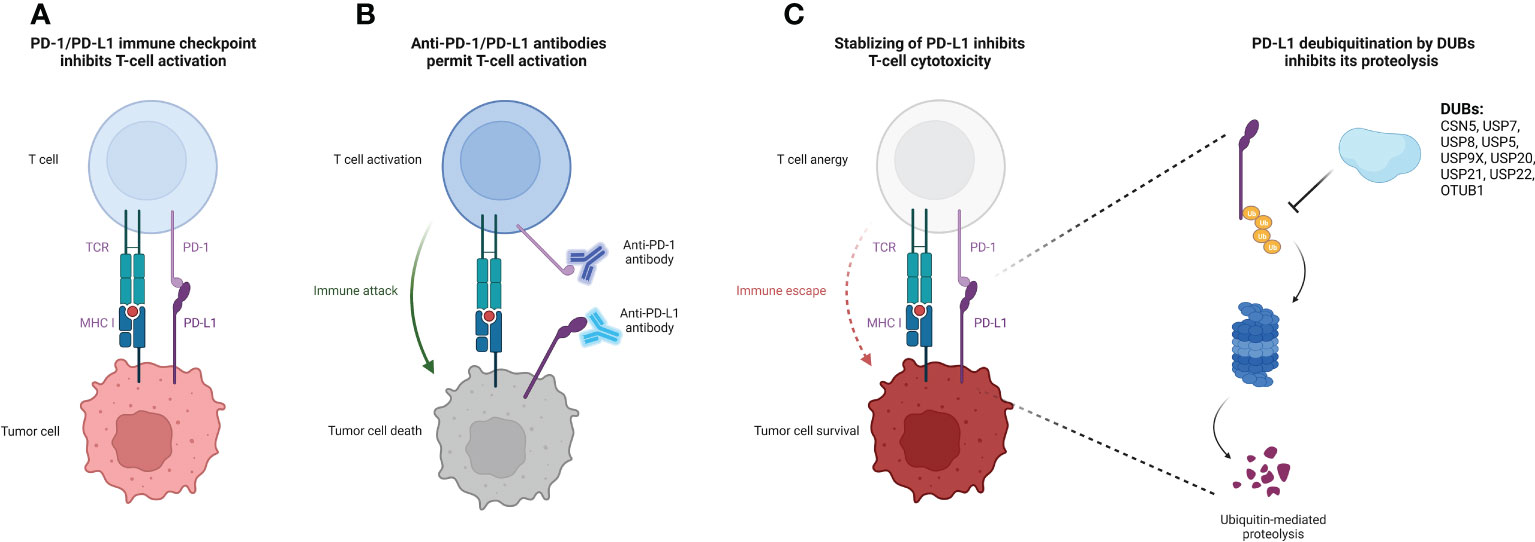
Figure 1 Deubiquitination of PD-L1 protein by DUBs causes increased PD-L1 stability and suppressed T-cell cytotoxicity. (A) Binding of PD-L1 on the tumor cell surface to their receptor PD-1 on the T cell surface releases the immune suppression signal, thereby inhibiting T cell activation and cytotoxicity. (B) The administration of specific antibodies to PD-L1/PD-1 reverses the T cell activation suppression signal, facilitating for the immune attack form of cytotoxic T cells to target tumor cells. (C) DUBs of PD-L1 stabilize PD-L1 and protect it from ubiquitin-mediated proteolysis, promoting tumor cell immune escape from T cell attack. The figure was created with Biorender.com. PD-L1, programmed death-ligand-1; DUB, deubiquitinating enzyme; MHC-I, major histocompatibility complex class I; PD-1, programmed death receptor-1; TCR, T cell receptor.
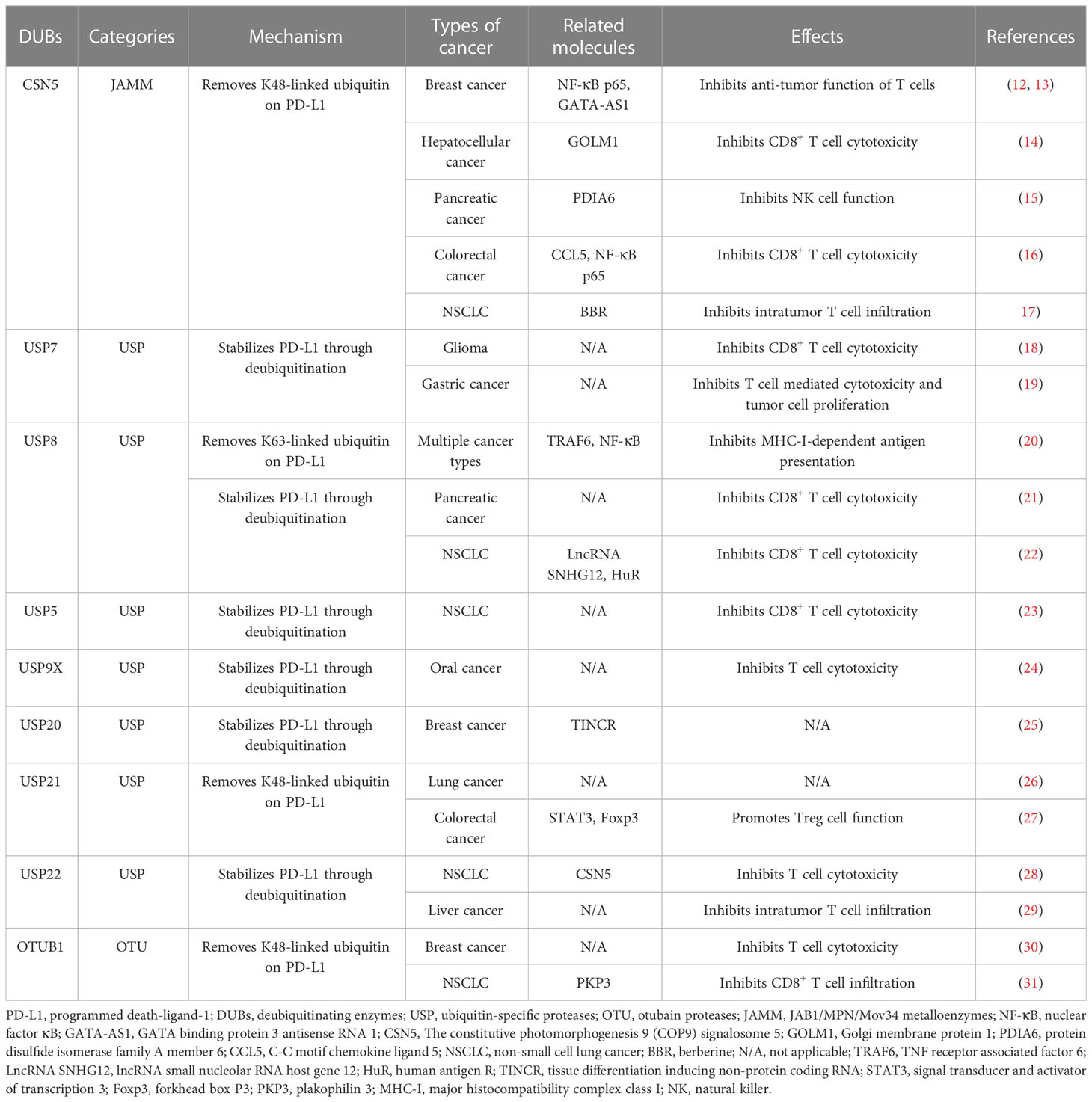
Table 1 Major deubiquitinating enzymes (DUBs) of PD-L1 and their biological effects in cancer.
The constitutive photomorphogenesis 9 signalosome 5 (CSN5) contains a conserved JAMM motif, which belongs to the JAMM subfamily (32). On one hand, CSN5 could directly interact with a variety of molecules, including c-Jun and p53, thereby influencing tumor proliferation (12). On the other hand, as a deubiquitinase, CSN5 deubiquitinates PD-L1, promoting tumor progression and immune escape (12). One study demonstrated that the activation of nuclear factor κB (NF-κB), further transactivates CSN5, leading to PD-L1 deubiquitination and stabilization (12). Moreover, PD-L1 and CSN5 expression levels are positively correlated in breast cancer (12). The application of a CSN5 inhibitor, curcumin, results in PD-L1 destabilization, increases the cytotoxic activity of T cells, and synergizes with anti-cytotoxic T-lymphocyte associated protein 4 antibodies (12).
Multiple mechanisms regulate CSN5-mediated PD-L1 deubiquitination. Golgi membrane protein 1 upregulates the expression of PD-L1 in hepatocellular cancer cells through the CSN5-mediated deubiquitination of PD-L1, leading to the suppression of CD8+ T cells (14). Ma et al. reported that protein disulfide isomerase family A member 6 interacts with CSN5 and promotes the deubiquitination of PD-L1 in pancreatic cancer cells (15). In colorectal cancer, macrophages-derived C-C motif chemokine ligand 5 (CCL5) promotes the activation of NF-κB p65 activation, which binds to the CSN5 promoter, increases CSN5 expression, and upregulates PD-L1 protein level (16). In triple negative breast cancer, long non-coding RNA GATA binding protein 3 antisense RNA 1 stabilized PD-L1 via the miR-676-3p/CSN5 axis (13). Interestingly, berberine, an established anti-inflammatory drug, interacts with CSN5 and inhibits CSN5/PD-L1 interaction, resulting in PD-L1 ubiquitination (17).
USP7 is a deubiquitinase that contains a USP domain (33), and is aberrantly expressed in several human cancers. USP7 mediates cell cycle control, tumor growth, chemoresistance, and tumor immunity by regulating multiple cellular signaling pathways, including the p53 and Wnt pathways (33, 34).
Regulatory T cells (Tregs) suppress the activity of effector T cells and promote immune escape (35). Moreover, Tip60, a histone acetyltransferase, promotes the acetylation and dimerization of the key transcription factor, forkhead box P3 (Foxp3), and regulates the activity of Tregs (35). A previous study discovered that USP7 directly deubiquitinates Foxp3 and stabilizes it. Further, USP7 depletion disrupts the immunosuppressive functions of Tregs in vivo (36). Another investigation revealed that USP7 is a deubiquitinase of both Tip60 and Foxp3, which enhances Tregs functions by increasing protein abundance (37).
Emerging studies have demonstrated that USP7 expression correlates with PD-L1 levels in cancer (18, 38). One recent investigation observed the overexpression of USP7 and PD-L1 proteins in glioma (18). USP7 mediates the deubiquitination of PD-L1, leading to increased PD-L1 expression. Abrogated USP7 expression promotes CD8+ T cell proliferation, elevates tumor necrosis factor (TNF) alpha and interferon gamma (IFN-γ) levels, and inhibits glioma cell immune evasion, which can be reversed by PD-L1 overexpression (18). Similarly, USP7 expression is upregulated and positively associated with PD-L1 in gastric cancer. Silencing USP7 decreases PD-L1 expression on cell surfaces, and augments the T cell-mediated killing of cancer cells (19). However, the regulatory relationship between USP7 and PD-L1 appears to be context-dependent. A negative association between the USP7 and PD-L1 expression in lung adenocarcinoma was revealed using The Cancer Genome Atlas data (38). In addition, the targeted inhibition of USP7 significantly increases PD-L1 protein levels in both cancer cells and the tumor microenvironment. Furthermore, abrogated USP7 expression inhibits the M2 macrophages transformation and their function, and promotes IFN-γ+CD8+ T cells infiltration, augmenting anti-tumor immunity. Additionally, a USP7 inhibitor, P5091, has shown a synergistic anti-tumor effect with a PD-1 inhibitor in vivo (38, 39).
Increasing evidence suggests that USP8 expression is upregulated, which stabilizes multiple oncogenes, in various cancers (22, 40). Additionally, USP8 is involved in T cell development and homeostasis. It is also essential for thymocyte maturation, proliferation, and the suppressive function of Treg cells on γδ T cells. Mechanistically, USP8 interacts with Gads and 14-3-3β, forming a complex with the T cell receptor (TCR)−CD28 cluster upon stimulation. Subsequently, USP8 is degraded via a caspase-dependent pathway, leading to the downregulation of interleukin-7 receptor subunit alpha (IL-7Rα) levels through the Forkhead box protein O1−IL-7Rα axis (41). Another study demonstrated that USP8 deubiquitinates and increases the expression of the type II transforming growth factor-β receptor (TβRII) in tumor-derived extracellular vesicles (TEVs). The inhibition of USP8 reduces the abundance of TβRII+ circulating TEVs and prevents CD8+ T cell exhaustion (42).
In a screening study performed by Xiong et al., it was revealed that DUBs-IN-2, a USP8 inhibitor, significantly increases PD-L1 protein levels in multiple cancer cell lines (20). Furthermore, a negative association between USP8 and PD-L1 was confirmed in lung squamous cancer tissues. Mechanistically, USP8 specifically removes K63-linked ubiquitination, but promotes the K48-linked ubiquitination of PD-L1, which finally promotes PD-L1 degradation. Furthermore, by deubiquitinating the K63-linked modification of TNF receptor associated factor 6 (TRAF6), USP8 up-regulates the expression of most genes in the major histocompatibility complex class I pathways, which limits the NF-κB signaling pathway and inhibits the immune response and antigen presentation. A USP8 inhibitor synergizes with anti-PD-1/PD-L1 treatments, dramatically inhibits tumor growth, and improves survival rates in mouse colon cancer models (20). Conversely, a recent study showed that the expression levels of USP8 and PD-L1 are positively correlated in pancreatic cancer. USP8 deficiency decreases PD-L1 protein abundance by promoting PD-L1 ubiquitination-mediated degradation. Moreover, a combined strategy comprising a USP8 inhibitor and PD-L1 inhibitor decreases tumor growth and enhances CD8+ T cell mediated killing of cancer cells (21).
USP5 belongs to the USP subfamily and can specifically recognize unconjugated polyubiquitin and cleave ubiquitin linkages. USP5 participates in multiple cellular procedures, including inflammatory responses (43, 44). The NLR family pyrin domain-containing 3 (NLRP3) inflammasome is critical for defense against microbial pathogens, and its dysregulation is implicated in various inflammatory diseases. Notably, USP5 is involved in regulating NLRP3 inflammasome activity, unrelated to its DUBs function. Mechanistically, USP5 acts as a pivotal scaffold protein recruiting a specific E3 ligase to NLRP3, promoting its ubiquitination and autophagic degradation. In addition, in alum-induced peritonitis mouse models, the overexpression of USP5 reduces interleukin 1 beta (IL-β) levels and polymorphonuclear infiltration (45). Furthermore, recent studies have demonstrated that USP5 directly deubiquitinates and stabilizes PD-L1. In NSCLC tissues, elevated USP5 expression correlates with PD-L1 expression, indicating of unfavorable clinical outcomes. Moreover, the inhibition of USP5 suppresses tumor growth in vivo by downregulating PD-L1 expression (23).
USP 9, X-linked (USP9X) is a positive regulator of the TCR signaling pathway. Silencing of USP9X in vivo suppresses T-cell growth, cytokine production, and the differentiation of T helper (Th) cells, without affecting T-cell survival and the development of specific T-cell populations in the thymus. Moreover, USP9X knockdown in human and mouse T-cell lines attenuates the TCR signaling-mediated activation of NF-κB through the deubiquitination of Bcl10 (46). USP9X knockout also results in a proliferation defect in both CD4+and CD8+ T cells, impairs the development of T cells in the thymus, and downregulates proximal TCR signaling. In vivo studies demonstrated that the T cell-specific knockout of Usp9x elevates PD-1-expressing T cell populations, leading to the incidence of specific autoimmune disease (47). In B lymphocytes, USP9X is necessary for the kinase activity of protein kinase C beta after B cell antigen receptor-dependent activation (48). In a model of sepsis with liver injury, USP9X promotes CD8+ T cells dysfunction in the liver through the inhibition of autophagy, which can be reversed by the conditional depletion of mechanistic target of rapamycin (49). Moreover, USP9X directly binds PD-L1, and USP9X reduces PD-L1 ubiquitination and increases its protein abundance. Additionally, a positive association was found between USP9X and PD-L1 expression in oral cancer (24).
USP20 has been linked to antiviral response, metabolic disease, neuroinflammation, and tumor progression (50–52). Tax is a viral oncoprotein which persistently activates NF-κB signaling and causes adult T cell leukemia. Through deubiquitination of TRAF6 and Tax, USP20 suppresses activation of NF-κB signaling and inhibits proliferation of leukemia cells (53).
A recent study demonstrated that USP20 interacts with PD-L1 and deubiquitinates it, which can be regulated by a long non-coding RNA, tissue differentiation inducing non-protein coding RNA (TINCR). Mechanistically, LncRNA TINCR acts as a competing endogenous RNA, which promotes stability of USP20 mRNA and upregulates the expression of PD-L1 in breast cancer (25).
Numerous studies have implicated USP21 in regulating cancer cell stemness, tumor growth, and metastasis (26, 54). In regard to immune regulation, Yang et al. have demonstrated a direct interaction between USP21 and PD-L1, whereby USP21 removes polyubiquitin chains from PD-L1, leading to its stabilization. Notably, the expression of USP21 is upregulated in lung cancer tissues, showing a positive association with PD-L1 protein levels (26). Additionally, USP21 has a role in regulating Treg cell functions (27). Li et al. demonstrated that USP21 suppresses the transformation of Th1-like Treg cells by deubiquitinating and stabilizing Foxp3. Mouse models of Usp21 depletion in Tregs exhibit spontaneous T cell activation and the expanded transformation of Tregs toward the Th1-like phenotype (55). Furthermore, emerging evidence suggests that USP21 suppresses antiviral responses in various immune cell types, including mouse embryonic fibroblasts and bone marrow-derived dendritic cells. This is achieved through the binding and deubiquitination of retinoic acid-inducible gene-I, which restricts type-I interferon production and antiviral immune defense (56). Notably, Usp21-knockout mice display enhanced resistance to vesicular stomatitis virus infection with increased production of interferons (56).
USP22, of which expression levels are elevated in various cancer types, is correlated with disease progression and an unfavorable prognosis (57, 58). This DUB plays a critical role in regulating PD-L1 stability. On one hand, USP22 directly deubiquitinates PD-L1. On the other hand, USP22 interacts with CSN5 and deubiquitinates it, thereby facilitating the interaction between PD-L1 and CSN5 (28). Moreover, USP22 and PD-L1 protein levels are positively correlated in NSCLC samples. The inhibition of USP22 enhances the cytotoxicity of T cells and reduces tumor growth (28). Another study revealed that USP22 interacts with the C terminus of PD-L1 and deubiquitinates it. In mouse models of hepatocellular carcinoma, knockout of Usp22 increases the infiltration of tumor-infiltrating lymphocytes, augments anti-tumor immunity, and synergizes with anti-PD-L1 treatments and chemotherapy (29). Furthermore, USP22 plays a part in regulating the tumor microenvironment. The knockout of USP22 in pancreatic ductal adenocarcinoma cells results in reduced myeloid cells infiltration and increased tumor infiltration of NK cells and T cells, leading to a synergistic response with combined immunotherapy (59). USP22 is also involved in regulating invariant NK T (iNKT) cells. USP22 suppression inhibits the development of iNKT cells, and attenuates iNKT1 and iNKT17 cell differentiation, while favoring iNKT2 polarization (60).
OTUB1, a member of the OTU superfamily, exhibits a preference for deubiquitinating K-48 and K-63 ubiquitin chains. Its involvement in cancer development and progression has also been observed (61). Extensive research has highlighted the role of OTUB1 in modulating immune cell responses. Depletion of OTUB1 activates NK cells and CD8+ T cells, leading to increased tumor infiltration of NK cells, DCs and T cells. Additionally, OTUB1 depletion enhances the cytokine production and the proliferation of CD4+ T cells (62, 63).
Moreover, OTUB1 has been reported to specifically interact with of PD-L1, wherein it removes the K48-linked ubiquitin chain from PD-L1 and stabilizes it. Functionally, OTUB1 depletion decreases PD-L1 expression, and increases the cytotoxicity of human peripheral blood mononuclear cells against tumor cells. The expression of OTUB1 is positively correlated with PD-L1 expression in breast cancer samples. Furthermore, OTUB1 depletion increases CD8+ T cell infiltration, elevates serum IFN-γ, and augments anti-tumor immune responses in mouse models (30). Liu et al. reported that circIGF2BP3 acts competitively to upregulate plakophilin 3 (PKP3) expression, which further stabilizes OTUB1 mRNA. CircIGF2BP3/PKP3 suppression synergized with anti-PD-1 treatment in mouse models of lung cancer (31).
Over the past few decades, the application of anti-PD-1/PD-L1 treatments has significantly improved the clinical prognoses of patients with cancer. Nevertheless, the clinical response to single-agent anti-PD-1/PD-L1 antibody therapy is limited to only a subset of patients (64, 65). Combinatorial treatments comprising anti-PD-1/PD-L1 antibodies with antiangiogenic drugs, chemotherapy, and targeted therapy have resulted in more promising clinical outcomes (66). As described above, several DUBs are participated in deubiquitination modifications of PD-L1, and regulated its expression. Thus, developing small-molecule inhibitors targeting these DUBs and the combination therapy represent an attractive therapeutic strategy.
However, despite the importance of these reported DUBs, there are still questions remain to be elucidated. One such question is determining which specific DUB plays the predominant role in regulating PD-L1 expression within a particular type of cancer. Second, although some studies have demonstrated synergistic efficacy of DUBs inhibitors with anti-PD-1/PD-L1 treatments, further exploration through clinical trials is needed to validate these findings and assess their potential for clinical application. In recent years, multiple selective inhibitors of DUBs, including inhibitors of USP7, USP8, and USP9X, have been developed (11). Accordingly, further investigation of these inhibitors in clinical trials is required.
All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (NSFC) 81902854 (YJ), 81974463 (YZ), and 81702687 (KX).
We thank Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi: 10.1158/1078-0432.CCR-14-1860
2. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol (2016) 27(3):409–16. doi: 10.1093/annonc/mdv615
3. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol (2022) 19(5):287–305. doi: 10.1038/s41571-022-00601-9
4. Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol (2019) 37(17):1470–8. doi: 10.1200/JCO.18.01265
5. Mok T, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet (2019) 393(10183):1819–30. doi: 10.1016/S0140-6736(18)32409-7
6. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
7. Dai X, Gao Y, Wei W. Post-translational regulations of PD-L1 and PD-1: mechanisms and opportunities for combined immunotherapy. Semin Cancer Biol (2022) 85:246–52. doi: 10.1016/j.semcancer.2021.04.002
8. Hu X, Wang J, Chu M, Liu Y, Wang ZW, Zhu X. Emerging role of ubiquitination in the regulation of PD-1/PD-L1 in cancer immunotherapy. Mol Ther (2021) 29(3):908–19. doi: 10.1016/j.ymthe.2020.12.032
9. Schauer NJ, Magin RS, Liu X, Doherty LM, Buhrlage SJ. Advances in discovering deubiquitinating enzyme (DUB) inhibitors. J Med Chem (2020) 63(6):2731–50. doi: 10.1021/acs.jmedchem.9b01138
10. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol (2019) 20(6):338–52. doi: 10.1038/s41580-019-0099-1
11. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discovery (2018) 17(1):57–78. doi: 10.1038/nrd.2017.152
12. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30(6):925–39. doi: 10.1016/j.ccell.2016.10.010
13. Zhang M, Wang N, Song P, Fu Y, Ren Y, Li Z, et al. LncRNA GATA3-AS1 facilitates tumour progression and immune escape in triple-negative breast cancer through destabilization of GATA3 but stabilization of PD-L1. Cell Prolif (2020) 53(9):e12855. doi: 10.1111/cpr.12855
14. Chen J, Lin Z, Liu L, Zhang R, Geng Y, Fan M, et al. GOLM1 exacerbates CD8+ T cell suppression in hepatocellular carcinoma by promoting exosomal PD-L1 transport into tumor-associated macrophages. Signal Transduct Target Ther (2021) 6(1):397. doi: 10.1038/s41392-021-00784-0
15. Ma Y, Xia P, Wang Z, Xu J, Zhang L, Jiang Y. PDIA6 promotes pancreatic cancer progression and immune escape through CSN5-mediated deubiquitination of β-catenin and PD-L1. Neoplasia (2021) 23(9):912–28. doi: 10.1016/j.neo.2021.07.004
16. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ (2020) 27(6):1765–81. doi: 10.1038/s41418-019-0460-0
17. Liu Y, Liu X, Zhang N, Yin M, Dong J, Zeng Q, et al. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm Sin B (2020) 10(12):2299–312. doi: 10.1016/j.apsb.2020.06.014
18. Li B, Wang B. USP7 enables immune escape of glioma cells by regulating PD-L1 expression. Immunol Invest (2022) 51(7):1921–37. doi: 10.1080/08820139.2022.2083972
19. Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B (2021) 11(3):694–707. doi: 10.1016/j.apsb.2020.11.005
20. Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun (2022) 13(1):1700. doi: 10.1038/s41467-022-29401-6
21. Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ (2023) 30(2):560–75. doi: 10.1038/s41418-022-01102-z
22. Huang Y, Xia L, Tan X, Zhang J, Zeng W, Tan B, et al. Molecular mechanism of lncRNA SNHG12 in immune escape of non-small cell lung cancer through the HuR/PD-L1/USP8 axis. Cell Mol Biol Lett (2022) 27(1):43. doi: 10.1186/s11658-022-00343-7
23. Pan J, Qiao Y, Chen C, Zang H, Zhang X, Qi F, et al. USP5 facilitates non-small cell lung cancer progression through stabilization of PD-L1. Cell Death Dis (2021) 12(11):1051. doi: 10.1038/s41419-021-04356-6
24. Jingjing W, Wenzheng G, Donghua W, Guangyu H, Aiping Z, Wenjuan W. Deubiquitination and stabilization of programmed cell death ligand 1 by ubiquitin-specific peptidase 9, X-linked in oral squamous cell carcinoma. Cancer Med (2018) 7(8):4004–11. doi: 10.1002/cam4.1675
25. Wang Q, Li G, Ma X, Liu L, Liu J, Yin Y, et al. LncRNA TINCR impairs the efficacy of immunotherapy against breast cancer by recruiting DNMT1 and downregulating MiR-199a-5p via the STAT1-TINCR-USP20-PD-L1 axis. Cell Death Dis (2023) 14(2):76. doi: 10.1038/s41419-023-05609-2
26. Yang S, Yan H, Wu Y, Shan B, Zhou D, Liu X, et al. Deubiquitination and stabilization of PD-L1 by USP21. Am J Transl Res (2021) 13(11):12763–74. doi: 10.21203/rs.3.rs-137970/v1
27. Deng B, Yang B, Chen J, Wang S, Zhang W, Guo Y, et al. Gallic Acid induces T-helper-1-like treg cells and strengthens immune checkpoint blockade efficacy. J Immunother Cancer (2022) 10(7): e004037. doi: 10.1136/jitc-2021-004037
28. Wang Y, Sun Q, Mu N, Sun X, Wang Y, Fan S, et al. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun Signal (2020) 18(1):112. doi: 10.1186/s12964-020-00612-y
29. Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res (2019) 7(10):1580–90. doi: 10.1158/2326-6066.CIR-18-0910
30. Zhu D, Xu R, Huang X, Tang Z, Tian Y, Zhang J, et al. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ (2021) 28(6):1773–89. doi: 10.1038/s41418-020-00700-z
31. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N6-methyladenosine-modified circIGF2BP3 inhibits CD8+ T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer (2021) 20(1):105. doi: 10.1186/s12943-021-01398-4
32. Asare Y, Ommer M, Azombo FA, Alampour-Rajabi S, Sternkopf M, Sanati M, et al. Inhibition of atherogenesis by the COP9 signalosome subunit 5. vivo Proc Natl Acad Sci U.S.A. (2017) 114(13):E2766–2766E2775. doi: 10.1073/pnas.1618411114
33. Al-Eidan A, Wang Y, Skipp P, Ewing RM. The USP7 protein interaction network and its roles in tumorigenesis. Genes Dis (2022) 9(1):41–50. doi: 10.1016/j.gendis.2020.10.004
34. Grunblatt E, Wu N, Zhang H, Liu X, Norton JP, Ohol Y, et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev (2020) 34(17-18):1210–26. doi: 10.1101/gad.340133.120
35. Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z, et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep (2014) 7(5):1471–80. doi: 10.1016/j.celrep.2014.04.021
36. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases treg-cell-suppressive capacity. Immunity (2013) 39(2):259–71. doi: 10.1016/j.immuni.2013.05.018
37. Wang L, Kumar S, Dahiya S, Wang F, Wu J, Newick K, et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. EBioMedicine (2016) 13:99–112. doi: 10.1016/j.ebiom.2016.10.018
38. Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming tumor-associated macrophages in lung cancer. Theranostics (2020) 10(20):9332–47. doi: 10.7150/thno.47137
39. Fu C, Zhu X, Xu P, Li Y. Pharmacological inhibition of USP7 promotes antitumor immunity and contributes to colon cancer therapy. Onco Targets Ther (2019) 12:609–17. doi: 10.2147/OTT.S182806
40. Islam MT, Chen F, Chen H. The oncogenic role of ubiquitin specific peptidase (USP8) and its signaling pathways targeting for cancer therapeutics. Arch Biochem Biophys (2021) 701:108811. doi: 10.1016/j.abb.2021.108811
41. Dufner A, Kisser A, Niendorf S, Basters A, Reissig S, Schönle A, et al. The ubiquitin-specific protease USP8 is critical for the development and homeostasis of T cells. Nat Immunol (2015) 16(9):950–60. doi: 10.1038/ni.3230
42. Xie F, Zhou X, Li H, Su P, Liu S, Li R, et al. USP8 promotes cancer progression and extracellular vesicle-mediated CD8+ T cell exhaustion by deubiquitinating the TGF-β receptor TβRII. EMBO J (2022) 41(16):e108791. doi: 10.15252/embj.2021108791
43. Liu Q, Wu Y, Qin Y, Hu J, Xie W, Qin FX, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv (2018) 4(5):eaar2824. doi: 10.1126/sciadv.aar2824
44. Ning F, Xin H, Liu J, Lv C, Xu X, Wang M, et al. Structure and function of USP5: insight into physiological and pathophysiological roles. Pharmacol Res (2020) 157:104557. doi: 10.1016/j.phrs.2019.104557
45. Cai B, Zhao J, Zhang Y, Liu Y, Ma C, Yi F, et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy (2022) 18(5):990–1004. doi: 10.1080/15548627.2021.1965426
46. Park Y, Jin HS, Liu YC. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc Natl Acad Sci USA (2013) 110(23):9433–8. doi: 10.1073/pnas.1221925110
47. Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med (2014) 211(10):1947–55. doi: 10.1084/jem.20140860
48. Naik E, Dixit VM. Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCβ kinase activity. J Immunol (2016) 196(8):3438–51. doi: 10.4049/jimmunol.1403165
49. Sheng L, Chen J, Tong Y, Zhang Y, Feng Q, Tang Z. USP9x promotes CD8 (+) T-cell dysfunction in association with autophagy inhibition in septic liver injury. Acta Biochim Biophys Sin (Shanghai) (2022) 54(12):1–10. doi: 10.3724/abbs.2022174
50. Qin B, Zhou L, Wang F, Wang Y. Ubiquitin-specific protease 20 in human disease: emerging role and therapeutic implications. Biochem Pharmacol (2022) 206:115352. doi: 10.1016/j.bcp.2022.115352
51. Li W, Shen M, Jiang YZ, Zhang R, Zheng H, Wei Y, et al. Deubiquitinase USP20 promotes breast cancer metastasis by stabilizing SNAI2. Genes Dev (2020) 34(19-20):1310–5. doi: 10.1101/gad.339804.120
52. Lu XY, Shi XJ, Hu A, Wang JQ, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature (2020) 588(7838):479–84. doi: 10.1038/s41586-020-2928-y
53. Yasunaga J, Lin FC, Lu X, Jeang KT. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-kappaB signaling. J Virol (2011) 85(13):6212–9. doi: 10.1128/JVI.00079-11
54. Guo Q, Shi D, Lin L, Li H, Wei Y, Li B, et al. De-ubiquitinating enzymes USP21 regulate MAPK1 expression by binding to transcription factor GATA3 to regulate tumor growth and cell stemness of gastric cancer. Front Cell Dev Biol (2021) 9:641981. doi: 10.3389/fcell.2021.641981
55. Li Y, Lu Y, Wang S, Han Z, Zhu F, Ni Y, et al. USP21 prevents the generation of T-helper-1-like treg cells. Nat Commun (2016) 7:13559. doi: 10.1038/ncomms13559
56. Fan Y, Mao R, Yu Y, Liu S, Shi Z, Cheng J, et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med (2014) 211(2):313–28. doi: 10.1084/jem.20122844
57. Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res (2014) 74(1):272–86. doi: 10.1158/0008-5472.CAN-13-1954
58. Piao S, Liu Y, Hu J, Guo F, Ma J, Sun Y, et al. USP22 is useful as a novel molecular marker for predicting disease progression and patient prognosis of oral squamous cell carcinoma. PloS One (2012) 7(8):e4254. doi: 10.1371/journal.pone.0042540
59. Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, et al. Tumor cell-intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol Res (2020) 8(3):282–91. doi: 10.1158/2326-6066.CIR-19-0661
60. Zhang Y, Wang Y, Gao B, Sun Y, Cao L, Genardi SM, et al. USP22 controls iNKT immunity through MED1 suppression of histone H2A monoubiquitination. J Exp Med (2020) 217(5): e20182218. doi: 10.1084/jem.20182218
61. Liao Y, Yang M, Wang K, Wang Y, Zhong B, Jiang N. Deubiquitinating enzyme OTUB1 in immunity and cancer: good player or bad actor. Cancer Lett (2022) 526:248–58. doi: 10.1016/j.canlet.2021.12.002
62. Mulas F, Wang X, Song S, Nishanth G, Yi W, Brunn A, et al. The deubiquitinase OTUB1 augments NF-κB-dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell Mol Immunol (2021) 18(6):1512–27. doi: 10.1038/s41423-020-0362-6
63. Stempin CC, Rojas Marquez JD, Ana Y, Cerban FM. GRAIL and otubain-1 are related to T cell hyporesponsiveness during trypanosoma cruzi infection. PloS Negl Trop Dis (2017) 11(1):e0005307. doi: 10.1371/journal.pntd.0005307
64. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv (2020) 6(38): eabd2712. doi: 10.1126/sciadv.abd2712
65. Lucibello G, Mograbi B, Milano G, Hofman P, Brest P. PD-L1 regulation revisited: impact on immunotherapeutic strategies. Trends Mol Med (2021) 27(9):868–81. doi: 10.1016/j.molmed.2021.06.005
Keywords: deubiquitinating enzymes, deubiquitination, cancer immunotherapy, post-translational modification, programmed death-ligand-1 (PD-L1)
Citation: Jiang Y, Hong K, Zhao Y and Xu K (2023) Emerging role of deubiquitination modifications of programmed death-ligand 1 in cancer immunotherapy. Front. Immunol. 14:1228200. doi: 10.3389/fimmu.2023.1228200
Received: 24 May 2023; Accepted: 09 June 2023;
Published: 21 June 2023.
Edited by:
Xiangpeng Dai, Jilin University, ChinaReviewed by:
Jiateng Zhong, Xinxiang Medical University, ChinaCopyright © 2023 Jiang, Hong, Zhao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingchao Zhao, eWluZ2NoYW96aGFvQGljbG91ZC5jb20=; Kai Xu, a3h1dG9uZ2ppQGhvdG1haWwuY29t
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.