 Maciej Tota
Maciej Tota Piotr Donizy2
Piotr Donizy2 Magdalena Krajewska
Magdalena Krajewska Mariusz Kusztal
Mariusz Kusztal- 1Faculty of Medicine, Wroclaw Medical University, Wrocław, Poland
- 2Department of Clinical and Experimental Pathology, Wroclaw Medical University, Wrocław, Poland
- 3Department of Nephrology and Transplantation Medicine, Wroclaw Medical University, Wrocław, Poland
Although associations of IgA nephropathy (IgAN) and ANCA-associated vasculitis (AAV) have been described, this coexistence scarcely occurs and requires multidisciplinary management. Herein, we discuss a course of treatment introduced in a patient with two exacerbations. Furthermore, alterations in histopathological images between two kidney biopsies are presented. The applicability of traditional inflammatory markers, e.g., CRP, in monitoring disease severity in AAV and IgAN is limited. Based on our patient and current literature, we suggest ANCA testing in patients with rapidly progressing IgAN for therapeutic and prognostic purposes. As regards the therapy of IgAN associated with AAV, aggressive immunosuppressive regimens with methylprednisolone and cyclophosphamide are recommended. Alternatively, methylprednisolone with rituximab, plasma exchange, mycophenolate mofetil, and intravenous immunoglobulin (IVIG) could also be considered.
1 Introduction
IgA nephropathy (IgAN) is the most common glomerulonephritis worldwide (1). Numerous disease entities, such as liver, dermatological, gastrointestinal, autoimmune, oncological, and respiratory disorders, as well as infectious, iatrogenic (drugs), and environmental factors, are recognized as triggers for IgAN development (2). As regards the pathophysiology of IgAN, the four-hit hypothesis is suggested. Elevated levels of galactose-deficient IgA1 (hit 1.) and the production of specific anti-glycan antibodies (hit 2.) cause the formation of pathogenic IgA1-containing immune complexes (hit 3.). The subsequent accumulation causes the release of cytokines and extracellular matrix proteins (hit 4.), contributing to renal dysfunction (3).
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a condition that causes severe systemic small-vessel vasculitis and is defined by the emergence of autoantibodies against the neutrophil proteins leukocyte proteinase 3 (PR3-ANCA) or myeloperoxidase (MPO-ANCA) (4). Three major ANCA-associated vasculitides have been identified according to the 2012 Chapel Hill Consensus: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA) (5). More than 75% of patients with AAV have renal involvement, characterized by rapidly progressing glomerulonephritis. Untreated AAVs have a less than one-year survival rate. The 5-year kidney survival rate is 70–75% in patients receiving the proper immunosuppressive medication (6). Genetics, environmental factors, and reactions of the innate and adaptive immune systems all play a role in the complex origin and pathophysiology of AAV (7).
Two studies on the prevalence of AAV in IgAN showed similar results: 1.2% (4/330) and 1.5% (35/2,390) of IgAN biopsy-proven patients were ANCA-positive (8, 9). Hence, the coincidence of AAV and IgAN is rare and few reports have described it. The aim of our study was to summarize the current literature on the suggested common pathomechanism, clinical presentation, and management of AAV and IgAN. We also present a successful course of treatment in a patient with AAV and IgAN and discuss the role of traditional inflammatory markers in monitoring disease activity.
2 Case report
In January 2019, a 56-year-old caucasian man was referred to the nephrology department due to fatigue, joint and muscle pain, arterial hypertension, hemoptysis, and bloody discharge from the nose lasting for two months. Chest computed tomography (CT) performed in an outpatient setting revealed diffuse alveolar hemorrhage (DAH) with a 20% alteration of lung parenchyma. Furthermore, abdominal ultrasonography (USG) showed mild dilatation of the pelvicalyceal system in the left kidney. The family history was positive for cerebral stroke and negative for autoimmune diseases.
On admission, petechiae on the legs were observed. Laboratory studies revealed positive PR3-ANCA (>1000 RU/ml) and ANA (1:320), proteinuria (0.33–0.44 g/24 h), and microhematuria. MPO-ANCA and anti-GBM antibodies were negative. Moreover, mild inflammation (C-reactive protein (CRP): 11 mg/L; erythrocyte sedimentation rate (ESR) within the normal limit: 19 mm/h) and normal kidney function (estimated glomerular filtration rate (eGFR): 108 ml/min/1.73 m2). During the first hospitalization, 40 mg of methylprednisolone (MTPD) was introduced, resulting in a nearly complete resolution of the reported symptoms. One month later, hematuria persisted and PR3-ANCA levels were 555 RU/ml. Surprisingly, a kidney biopsy showed none of the typical morphological features of ANCA-related vasculitis but IgA nephropathy with an Oxford score MEST-C: M1, E0, S0, T0, C0 (Figures 1A, B). Such a possible negativation of proliferative lesions could have been a result of commenced immunosuppressive treatment. Therefore, MTPD pulses 4x250 mg with conversion to prednisone 40 mg/day and methotrexate (MTX) 7.5 mg were introduced. At the follow-up visit in April, the second CT demonstrated a complete resolution of previous changes. All symptoms were completely resolved. The MTX dose was increased to 10 mg/week and MTPD was tapered to 12 mg/day. Although laboratory tests and the patient’s clinical state were well controlled, a gradual increase in PR3-ANCA antibodies was noted (Figure 2A). CRP levels were within normal limits (Figure 2B).
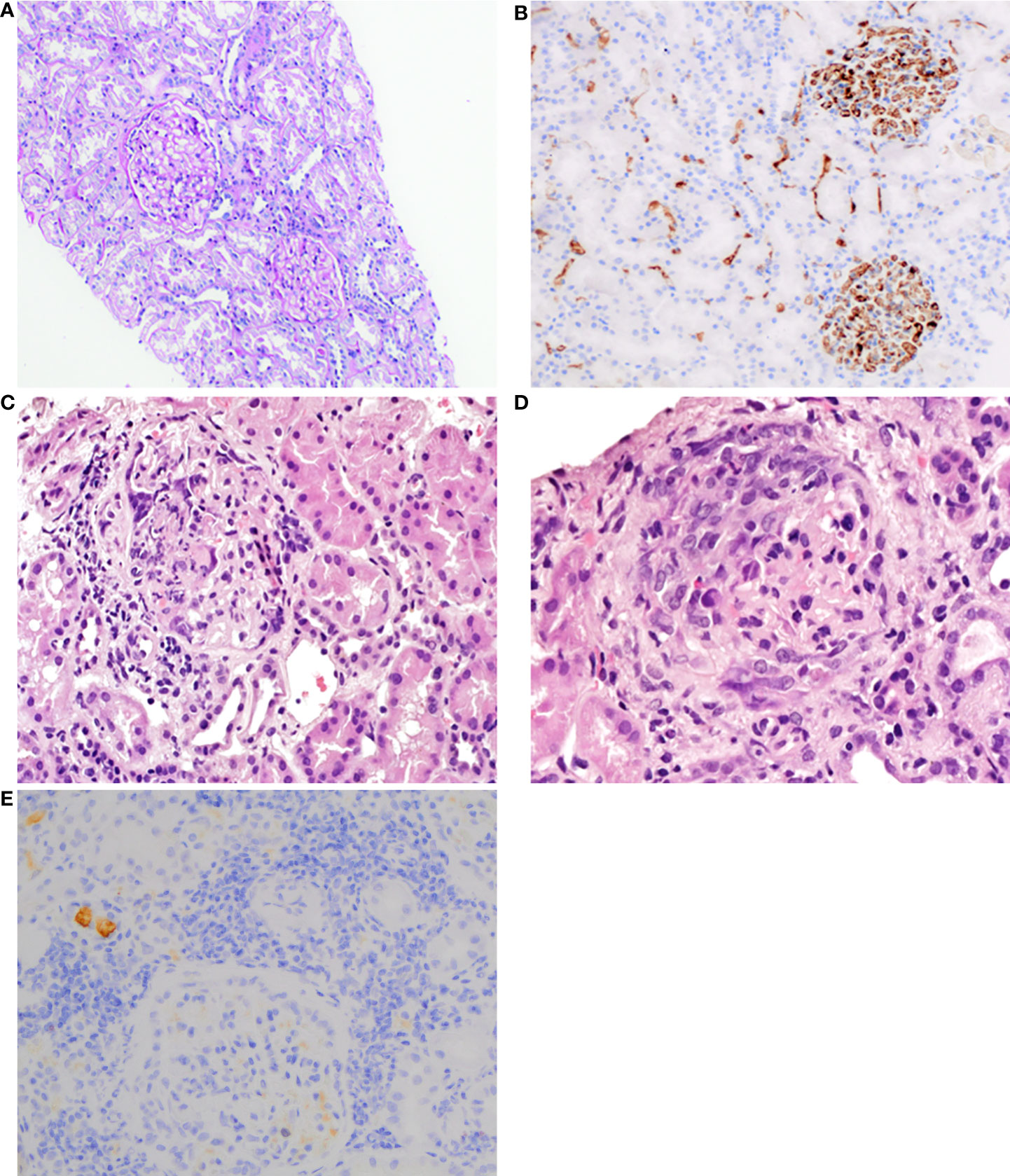
Figure 1 (A) First kidney biopsy. PAS staining of the cortex shows normal to mild mesangial hypercellularity with normal tubules and interstitium (100×). (B) First kidney biopsy. Immunohistochemistry for IgA reveals intense granular staining, predominantly mesangial (200×). (C) Second kidney biopsy. Glomerulus with fibrinoid necrosis and karyorrhexis. HE staining (400×). (D) Second kidney biopsy. Glomerulus with a cellular crescent. HE staining (600×). (E) Second kidney biopsy. Immunohistochemistry reveals a significant reduction of IgA reactivity in glomeruli (200×).
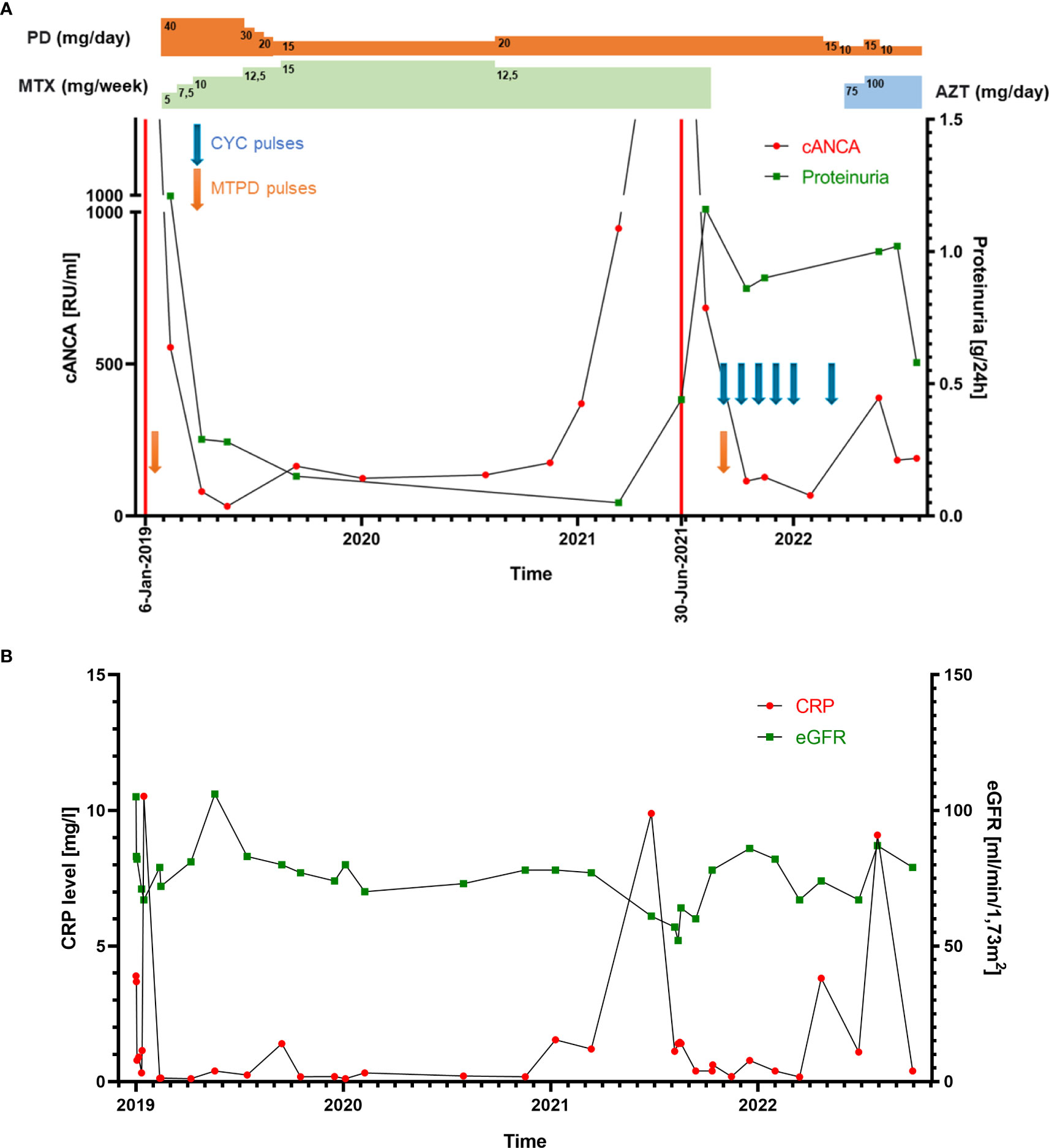
Figure 2 (A) Line graph showing the patient’s cANCA (PR3-ANCA) levels and proteinuria throughout therapy. The two exacerbations are marked with red lines (6 January 2019 and 30 June 2021), consistent with cANCA and proteinuria level elevation. PD, prednisone; MTX, methotrexate; AZT, azathioprine; CYC, cyclophosphamide; MTPD, methylprednisolone. (B) Line graph representing the patient’s CRP level and eGFR ratio changes.
In May 2021, the second exacerbation occurred. The patient presented at the ophthalmologist’s office due to redness and eye pain. Episcleritis was diagnosed and treated with loteprednol and brimonidine/timolol eye drops. In the next month, the patient developed muscle and joint pain that resolved spontaneously after a few weeks. Due to the overall clinical picture, a second kidney biopsy was performed. It revealed ANCA-associated glomerulonephritis, namely endocapillary proliferation within 8/20 of the glomeruli, with segmental fibrinoid necrosis, karyorrhexis, and prominent cellular crescents in three glomeruli. A low intensity of chronic inflammation (TIN Grade 0/1) in the interstitium with mild fibrosis was observed (2–3% of the available renal cortex) (Figures 1C, D). An immunohistochemistry examination of the kidney specimen did not show IgA deposition (Figure 1E). Chest CT scans showed irregular linear opacity with traction of the adjacent lung parenchyma seen in the right upper lobe with a size of 2.3x2.0 cm; signs corresponding to inflammation. Otherwise, normal lung parenchyma. The patient received 1 g of cyclophosphamide (CYC), and MTPD pulses 2x500 mg with conversion to prednisone 35 mg/day. In subsequent monthly follow-up visits, the patient received 800 mg of CYC (a total dose of 5000 mg; 62.5 mg/kg) (Figure 2A).
In March 2022, azathioprine (AZT) was introduced (75 mg, which was increased to 100 mg after four weeks). In October 2022, remission was achieved based on clinical and laboratory images, with a Birmingham Vasculitis Activity Score (BVAS) = 1.
3 Discussion
In recent years, MTX versus CYC for induction of AAV remission has been a matter of debate. Considering CYC adverse events, MTX was suggested in patients with non-life-threatening AAV (10). Hence, our patient was administered with MTX. However, recently published studies have shown that MTX use is associated with a higher relapse rate and late accrual damage compared to CYC (11). Thus, CYC with glucocorticoids over MTX is recommended for induction of remission of AAV according to KDIGO 2021 Guidelines. Alternatively, rituximab with glucocorticoids may be considered. In life-threatening cases, plasmapheresis is recommended. Conversely, in a no-organ-threatening course, mycophenolate mofetil may be administered (11).
Our report represents a patient with AAV and a first histopathological diagnosis unequivocal for IgAN. Traditional inflammatory marker CRP ratios in our patient were significantly elevated only during exacerbations. Thus, we conclude that CRP has a limited role in assessing disease activity. Our thesis is consistent with previous reports demonstrating weak or no correlation between CRP and BVAS (12, 13). Similarly, other inflammatory markers, e.g., ESR, procalcitonin, and calprotectin, were not applicable in monitoring patients with AAV (14, 15).
Ozcan et al. described the coexistence of primary IgAN diagnosed two years prior and a novel AAV diagnosis that may had been provoked by COVID-19 infection (Table 1). Similar to our patient, a CT scan showed an alveolar hemorrhage (16). The link between COVID-19 and AAV and IgAN has not been elucidated yet. Comparably, nonspecific upper respiratory tract infection preceded AAV and IgAN diagnosis in our patient. Growing evidence suggests that the formation of neutrophil extracellular traps (NETs) during COVID-19 infection may lead to AAV development (31, 32) (Figure 3). As concerns kidney diseases, lupus nephritis, anti-glomerular basement membrane disease, thrombotic microangiopathies, and diabetic nephritis are also suggested to share the contribution of NETs in pathogenesis (33). Such a possible link was not corroborated for IgAN and should be evaluated by future studies.
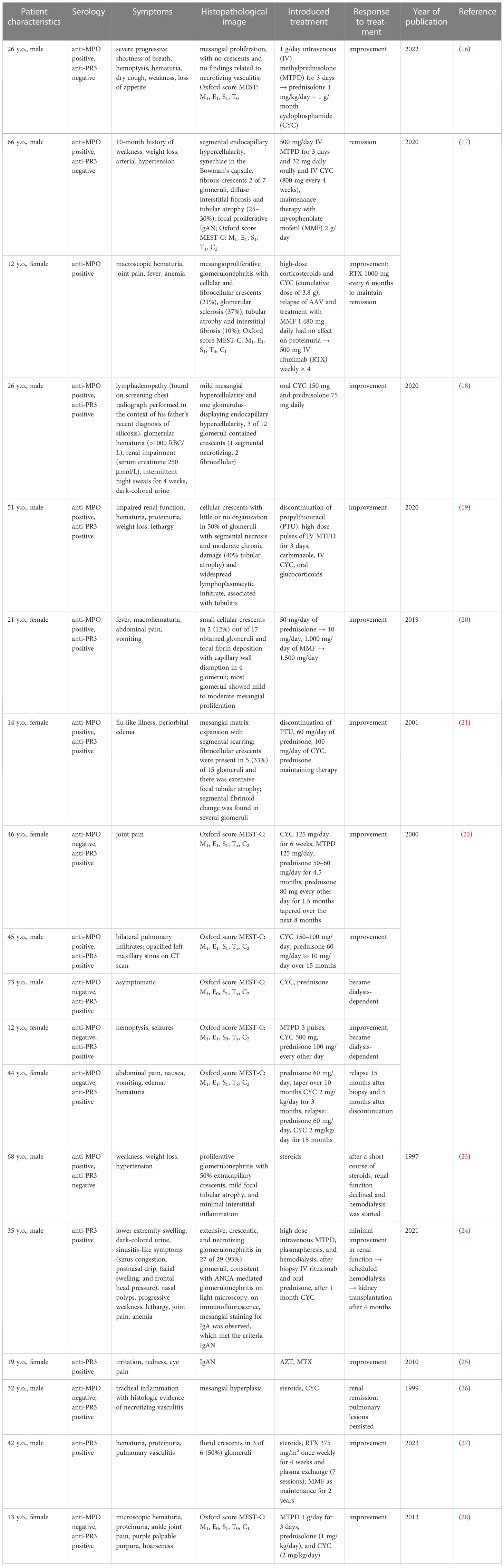
Table 1 Summary of to-date published case reports on AAV and IgAN coexistence.
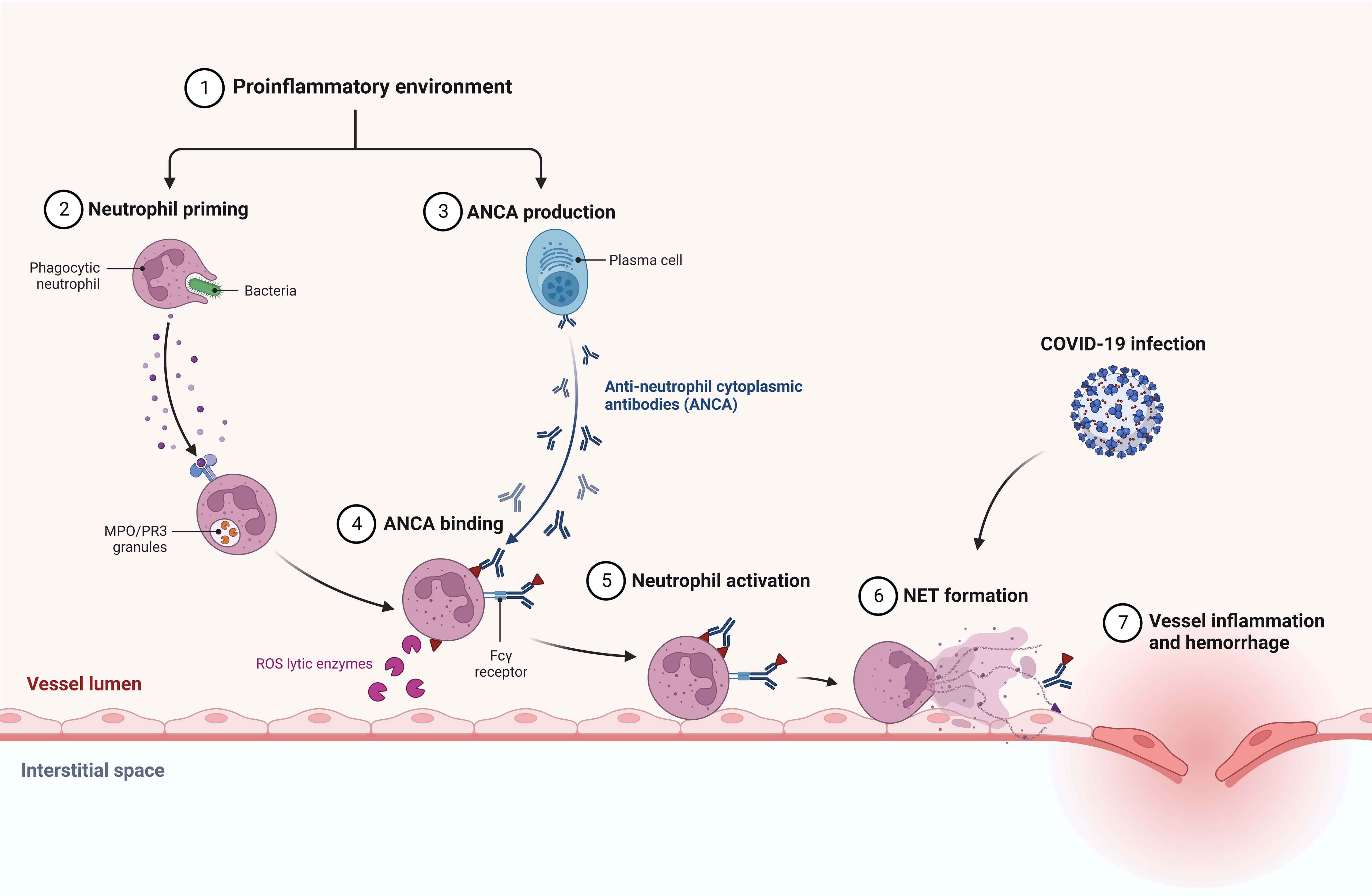
Figure 3 Suggested association between COVID-19 infection and AAV development via neutrophil extracellular traps (NETs) formation.
Consequently, one can infer that infections could be triggers of AAV and IgAN development. Rathman et al. assessed 270 patients with AAV to determine whether there is an association with prior respiratory tract infection. Surprisingly, the authors found that those infections are positively associated with MPO-ANCA but not PR3-ANCA vasculitis (34). Apart from NETs formation, infectious diseases could predispose to AAV via molecular mimicry, superantigens, and proinflammatory signaling. Infectious factors reduce immune functions and may cause persistent or recurrent autoimmunity in individuals who are genetically predisposed and/or in the context of environmental exposure (35).
Chebotareva et al. were the first to describe an effective therapy for ANCA-positive IgAN with rituximab (RTX) (17). A 19-year-old patient presented with AAV glomerulonephritis exacerbation 1 and 7 years after diagnosis. During the second exacerbation, a kidney biopsy revealed IgAN and the patient was administered RTX. Four weekly infusions of 500 mg RTX led to the remission of glomerulonephritis, which was maintained with mycophenolate mofetil (MMF).
Rao et al. first reported a patient with AAV and IgAN linked to silica exposure (18). The authors suggested that exposure to silica caused immune dysregulation that resulted in IgAN and AAV development. It is recommended to consider silica exposure as a putative trigger for autoimmune diseases.
Drugs may also trigger AAV and IgAN (2, 36). Galante et al. described the coexistence of AAV and IgAN triggered by propylthiouracil (19). According to the authors’ hypothesis, propylthiouracil-induced AAV resulted in an infiltration of IgG4-rich cells in the tubulointerstitial tissue as well as epiphenomena of anti-GBM antibody production. It is unknown whether AAV occurred first or was a result of mesangial IgA deposition.
Another study showed that, in comparison to ANCA-negative IgAN patients, ANCA-positive IgAN patients present a more severe clinical and histological image. Surprisingly, the renal prognosis was worse in ANCA-negative crescentic IgAN patients than in ANCA-positive individuals after aggressive short-term immunosuppressive therapy (37). Nonetheless, due to the relatively small sample size (20 patients), the treatment outcomes should be corroborated by future studies.
Although we were able to find several eligible reports on IgAN and AAV in the current literature, we do believe that coexistence could be more prevalent and current knowledge is limited. We hypothesize that underestimation could be a result of the paucity of research on IgAN and AAV due to researchers’ low priority for case reports, publishing policies that focus on original articles, economic issues, and underdiagnosis of IgAN. An IgAN diagnosis is made on the basis of a biopsy examination followed by immunofluorescence or immunohistochemical staining; such a procedure is not ordinarily performed.
4 Conclusions
Our patient with AAV presented symptoms from multiple organ systems. As regards renal manifestation, biopsy-proven IgAN preceded AAV glomerulonephritis diagnosis. Differences in the histological picture between the two performed biopsies were remarkable. CRP and other traditional inflammatory markers were elevated only during exacerbations. Thus, these were not applicable to monitor disease activity.
Emerging evidence demonstrates that overlapping AAV with IgAN results in a more severe clinical picture, rapidly decreasing kidney function, and various extrarenal manifestations. Thus, clinicians should acknowledge the possibility of ANCA-positivity in rapidly progressing IgAN. Whether there is causality between AAV and IgAN remains unknown. Future studies should be conducted to evaluate the putative common pathophysiology of these diseases.
Since the immunosuppressive protocol utilized in AAV was successful in patients with AAV and IgAN, ANCA testing may be beneficial for patients who have rapidly progressing IgAN for therapeutic and predictive purposes. As regards the management of IgAN associated with AAV, immunosuppressive treatment with methylprednisolone and cyclophosphamide is recommended. Alternatively, methylprednisolone with rituximab, plasma exchange, mycophenolate mofetil, and intravenous immunoglobulin (IVIG) could also be considered.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceptualization: MT and MKu. Investigation: MT and MB. Resources: MT, PD, and MKu. Data curation: MT and PD. Writing—original draft preparation: MT, PD, MB, and MKu. Writing—review and editing: MT, PD, MKu, and MKr. Visualization: MT and PD. Supervision: MKu, PD, and MKr. Funding acquisition: MKr. All authors contributed to manuscript revision and read and approved the submitted version.
Funding
This study was supported by the Wroclaw Medical University statutory funds (SUBZ.C160.23.046).
Acknowledgments
Figure 3 was adapted from “Anti-neutrophil Cytoplasmic Antibody (ANCA)-associated Vasculitis” at BioRender.com (Accessed on 4 April 2023). Retrieved from https://app.biorender.com/biorender-templates.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med (1987) 64:709–27.
2. Tota M, Baron V, Musial K, Derrough B, Konieczny A, Krajewska M, et al. Secondary IgA nephropathy and IgA-associated nephropathy: A systematic review of case reports. J Clin Med (2023) 12:2726. doi: 10.3390/JCM12072726
3. Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol (2011) 22:1795–803. doi: 10.1681/ASN.2011050464
4. Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, et al. ANCA-associated vasculitis. Nat Rev Dis Prim (2020) 6:71. doi: 10.1038/S41572-020-0204-Y
5. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum (2013) 65:1–11. doi: 10.1002/ART.37715
6. Turkmen K, Ozer H, Tesar V. An update on dıagnosıs and treatment of ANCA assocıated renal vasculıtıs. Int Urol Nephrol (2023). doi: 10.1007/S11255-023-03565-6
7. Geetha D, Jefferson JA. ANCA-associated vasculitis: Core curriculum 2020. Am J Kidney Dis (2020) 75:124–37. doi: 10.1053/J.AJKD.2019.04.031
8. Ștefan G, Terinte-Balcan G, Stancu S, Zugravu A, Gherghiceanu M, Mircescu G. IgA nephropathy with serum ANCA positivity: case series and literature review. Rheumatol Int (2021) 41:1347–55. doi: 10.1007/S00296-021-04888-2
9. Xie L, He J, Liu X, Tang S, Wang W, Li F, et al. Clinical value of systemic symptoms in IgA nephropathy with ANCA positivity. Clin Rheumatol (2018) 37:1953–61. doi: 10.1007/S10067-017-3931-Z
10. Reinhold-Keller E, De Groot K. Use of methotrexate in ANCA-associated vasculitides. Clin Exp Rheumatol (2010) 28:S178–82.
11. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int (2021) 100:S1–S276. doi: 10.1016/j.kint.2021.05.021
12. Suppiah R, Mukhtyar C, Flossmann O, Alberici F, Baslund B, Batra R, et al. A cross-sectional study of the Birmingham Vasculitis Activity Score version 3 in systemic vasculitis. Rheumatol (Oxf) (2011) 50:899–905. doi: 10.1093/RHEUMATOLOGY/KEQ400
13. Morris AD, Rowbottom AW, Martin FL, Woywodt A, Dhaygude AP. Biomarkers in ANCA-associated vasculitis: Potential pitfalls and future prospects. Kidney360 (2021) 2:586. doi: 10.34067/KID.0006432020
14. Willeke P, Kümpers P, Schlüter B, Limani A, Becker H, Schotte H. Platelet counts as a biomarker in ANCA-associated vasculitis. Scand J Rheumatol (2015) 44:302–8. doi: 10.3109/03009742.2015.1006247
15. Valenzuela LM, Draibe J, Ramos MQ, Oliveras XF, Melilli E, Garrit JMC, et al. Calprotectin as a smoldering activity detection tool and renal prognosis biomarker in ANCA associated vasculitis. PloS One (2018) 13:e0205982. doi: 10.1371/JOURNAL.PONE.0205982
16. Ozcan S, Sonmez O, Karaca C, Ozdede A, Seyahi N. ANCA-associated vasculitis flare might be provoked by COVID-19 infection: a case report and a review of the literature. Clin Kidney J (2022) 15:1987. doi: 10.1093/CKJ/SFAC186
17. Chebotareva N, Kamyshova E, Bulanov N, Lysenko L, Moiseev S. Antineutrophil cytoplasmic autoantibody (ANCA) positive immunoglobulin A (IgA) nephropathy: Case reports and review of literature. Egypt Rheumatol (2020) 42:251–4. doi: 10.1016/J.EJR.2020.06.002
18. Rao N, Bendall A, Lanteri M. ANCA vasculitis and IgA nephropathy linked to silica exposure. Occup Med (Lond) (2020) 70:445–8. doi: 10.1093/OCCMED/KQAA122
19. Galante JR, Daruwalla CP, Roberts ISD, Haynes R, Storey BC, Bottomley MJ. An unusual presentation of propylthiouracil-induced anti-MPO and PR3 positive ANCA vasculitis with associated anti-GBM antibodies, IgA nephropathy and an IgG4 interstitial infiltrate: a case report. BMC Nephrol (2020) 21:295. doi: 10.1186/S12882-020-01964-W
20. Yasukawa M, Kitagawa S, Togashi R, Asakawa S, Nagura M, Arai S, et al. A patient with MPO-ANCA-positive igA nephropathy diagnosed with the clinical onset of macrohematuria. Intern Med (2019) 58:2051–6. doi: 10.2169/INTERNALMEDICINE.2475-18
21. Winters MJ, Hurley RM, Lirenman DS. ANCA-positive glomerulonephritis and IgA nephropathy in a patient on propylthiouracil. Pediatr Nephrol (2002) 17:257–60. doi: 10.1007/S00467-001-0807-9
22. Haas M, Jafri J, Bartosh SM, Karp SL, Adler SG, Meehan SM. ANCA-associated crescentic glomerulonephritis with mesangial IgA deposits. Am J Kidney Dis (2000) 36:709–18. doi: 10.1053/AJKD.2000.17615
23. Martin SJ, Audrain MAP, Baranger T, Moreau A, Dantal J, Testa A, et al. Recurrence of immunoglobulin A nephropathy with immunoglobulin A antineutrophil cytoplasmic antibodies following renal transplantation. Am J Kidney Dis (1997) 29:125–31. doi: 10.1016/S0272-6386(97)90019-6
24. Zeidan BS, Hernandez A, Desai P. Overlap of granulomatosis with polyangiitis and IgA nephropathy. Cureus (2021) 13:e18672. doi: 10.7759/CUREUS.18672
25. Leone JE, Kern A, Williamson JD, Colandreo RM. Autoimmune dysfunction and subsequent renal insufficiency in a collegiate female athlete: A case report. J Athl Train (2010) 45:645. doi: 10.4085/1062-6050-45.6.645
26. Vrtovsnik F, Queffeulou G, Skhiri H, Nochy D, Walker F, Hayem G, et al. Simultaneous IgA nephropathy and Wegener’s granulomatosis–overlap or coincidence (the role of renal biopsy). Nephrol Dial Transplant (1999) 14:1266–7. doi: 10.1093/NDT/14.5.1266
27. Londrino F, Giuliani G, Santostefano M, Baraldi O, Mattiotti M, Mangiulli M, et al. IgA nephropathy and granulomatosis with polyangiitis-overlap: a rare coexistence of two glomerular nephropathies with remission after steroids and rituximab. G Ital di Nefrol (2023) 40.
28. Fukuhara D, Kurayama R, Ito Y, Komagata Y, Arimura Y, Yan K. Granulomatosis with polyangiitis associated with IgA nephropathy. CEN Case Rep (2013) 2:204–8. doi: 10.1007/S13730-013-0065-2
29. Javadi Parvaneh V, Shirzani A, Rahmani K, Shiari R. Pediatric granulomatosis with polyangiitis mimicking IgA vasculitis: A case report. Clin Med Insights Arthritis Musculoskelet Disord (2020) 13. doi: 10.1177/1179544120967371
30. Kataoka H, Moriyama T, Manabe S, Kawachi K, Ushio Y, Watanabe S, et al. Maximum glomerular diameter and oxford MEST-C score in IgA nephropathy: The significance of time-series changes in pseudo-R2 values in relation to renal outcomes. J Clin Med (2019) 8:2105. doi: 10.3390/JCM8122105
31. Janiuk K, Jabłońska E, Garley M. Significance of NETs formation in COVID-19. Cells (2021) 10:1–12. doi: 10.3390/CELLS10010151
32. Misra DP, Thomas KN, Gasparyan AY, Zimba O. Mechanisms of thrombosis in ANCA-associated vasculitis. Clin Rheumatol (2021) 40:4807–15. doi: 10.1007/S10067-021-05790-9
33. Nakazawa D, Marschner JA, Platen L, Anders HJ. Extracellular traps in kidney disease. Kidney Int (2018) 94:1087–98. doi: 10.1016/J.KINT.2018.08.035
34. Rathmann J, Stamatis P, Jönsson G, Englund M, Segelmark M, Jayne D, et al. Infection is associated with increased risk of MPO- but not PR3-ANCA-associated vasculitis. Rheumatol (Oxf) (2022) 61:4817–26. doi: 10.1093/RHEUMATOLOGY/KEAC163
35. Flint J, Morgan MD, Savage COS. Pathogenesis of ANCA-associated vasculitis. Rheum Dis Clin North Am (2010) 36:463. doi: 10.1016/J.RDC.2010.05.006
36. Weng CH, Liu ZC, Guo LMCALS. Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Chin Med J (Engl) (2019) 132:2848. doi: 10.1097/CM9.0000000000000539
Keywords: IgA nephropathy, ANCA-associated vasculitis, granulomatosis with polyangiitis, GPA, PR-3 ANCA, kidney biopsy, treatment, CRP
Citation: Tota M, Donizy P, Byrska M, Krajewska M and Kusztal M (2023) An unsuspected histopathological finding —concomitant IgA nephropathy in a patient with ANCA-associated vasculitis: a case report and literature review. Front. Immunol. 14:1227878. doi: 10.3389/fimmu.2023.1227878
Received: 23 May 2023; Accepted: 21 July 2023;
Published: 15 August 2023.
Edited by:
Marie-Agnes Dragon-Durey, Université Paris Cité, FranceReviewed by:
Marie-Alexandra Alyanakian, Assistance Publique Hopitaux De Paris, FranceMelchior Chabannes, Centre Hospitalier Universitaire de Besançon, France
Copyright © 2023 Tota, Donizy, Byrska, Krajewska and Kusztal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Krajewska, bWFnZGFsZW5hLmtyYWpld3NrYUB1bXcuZWR1LnBs