 Toshihiko Taya
Toshihiko Taya Fumiya Teruyama
Fumiya Teruyama Satoshi Gojo
Satoshi Gojo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 September 2023
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1227467
This article is part of the Research Topic Overcoming Challenges in Microbial Immunology: 2022 View all 10 articles
Bacterial infections still impose a significant burden on humanity, even though antimicrobial agents have long since been developed. In addition to individual severe infections, the f fatality rate of sepsis remains high, and the threat of antimicrobial-resistant bacteria grows with time, putting us at inferiority. Although tremendous resources have been devoted to the development of antimicrobial agents, we have yet to recover from the lost ground we have been driven into. Looking back at the evolution of treatment for cancer, which, like infectious diseases, has the similarity that host immunity eliminates the lesion, the development of drugs to eliminate the tumor itself has shifted from a single-minded focus on drug development to the establishment of a treatment strategy in which the de-suppression of host immunity is another pillar of treatment. In infectious diseases, on the other hand, the development of therapies that strengthen and support the immune system has only just begun. Among innate immunity, the first line of defense that bacteria encounter after invading the host, the molecular mechanisms of the phagolysosome pathway, which begins with phagocytosis to fusion with lysosome, have been elucidated in detail. Bacteria have a large number of strategies to escape and survive the pathway. Although the full picture is still unfathomable, the molecular mechanisms have been elucidated for some of them, providing sufficient clues for intervention. In this article, we review the host defense mechanisms and bacterial evasion mechanisms and discuss the possibility of host-directed therapy for bacterial infection by intervening in the phagolysosome pathway.
The treatment of bacterial infections has changed dramatically with the development of antibiotics, and many lives have been saved; in the era of the COVID-19 pandemic, tuberculosis, a widespread and deadly disease, has not been conquered, but it has been set aside as an infectious disease. On the other hand, bacterial infections are still strictly a major threat to people’s lives and a major treatment target, one of which is sepsis caused by a rapid and massive bacterial load (1) and another is the emergence of antimicrobial resistance bacteria spurred by antibiotic overuse (2). Infectious diseases other than sepsis, such as tuberculosis, infectious gastroenteritis, bacterial pneumonia, and food poisoning, are limited to localized areas, and dysfunction of the tissues of the infected foci comes to the fore. Sepsis is defined as organ damage due to an inadequate host response to infection (3). In addition to the classic treatment of infusion, removal of infected lesions, and respiratory circulatory support, treatment is aimed at normalizing coagulation abnormalities to maintain organ microcirculation (4). Nevertheless, more than 11 million lives are lost annually to sepsis, making it the cause of nearly 20% of deaths worldwide (5). One of the reasons that current medical treatment for sepsis has been so hampered is that the host’s immune system forms a troublesome response to sepsis. That response is the coexistence of an excessive inflammatory response and a prolonged state of immunosuppression (6). The former, also called a cytokine storm, is characterized by an overproduction of inflammatory cytokines as the predominant phenotype (7). Much of the pathology of sepsis is associated with this unhelpfully exuberant reaction of the host, which is thought to be a common end pathway that occurs with viral infections as well as bacterial infections, and suppression of excess cytokines and regulation of their receptors is thought to reduce the disease state (8). However, the major proinflammatory cytokines IL-1 (9) and TNF-α (10) which are major inflammatory cytokines in sepsis, have failed to improve the survival of sepsis. In addition, inhibitors of Toll-like receptor (TLR) 4, which detects many bacteria and transduces intracellular signals that trigger inflammation, have also failed to improve sepsis survival (8). Despite such disappointment, transcriptome analysis of leukocytes from patient blood in sepsis revealed that up to 80% of the pathways of cellular function are altered and that inflammatory and regulatory mechanisms are simultaneously driven in the first few hours after onset (11). The setbacks in these clinical trials and the genetic approach to pathophysiology have led to a major shift in our current understanding of the pathogenesis of sepsis, in which host immunity to sepsis is a conflict between attack and suppression, far from its original goal of eliminating pathogens (12). This understanding of the pathogenesis has led to a search for therapeutic strategies that achieve homeostasis of host immune function.
For antibiotics, the invention of new drugs in the nearly 30 years since the 1940s, a golden age, has been a wonderful scientific breakthrough that has led to an overly optimistic fantasy that bacterial infections will cease to be a threat to humanity (2). However, in the half-century since the 1970s, only a few new classes of antibiotics have been invented, and in addition, we have been handicapped by the disastrous situation with multidrug-resistant bacteria. Despite advances in understanding the life cycles of bacteria and long-awaited advances in molecular biology and genetics, biology and medicine today are far behind the good old days of the past 30 years in terms of progress in the field of infectious disease treatment. In the past half century, mankind has not only used invented antibiotics in large quantities in medicine but also abused them in livestock in search of economic rewards (13). The result has been a situation in which bacteria that have acquired multidrug resistance, also known as superbugs, have become rampant. Tuberculosis remains an uncontrolled infectious disease worldwide, and is the leading cause of mortality among mono infectious diseases, with 1.4 million deaths per year (14). Currently, it is estimated that 1/4 of the earth’s population is infected with Mycobacterium tuberculosis, the majority of which is considered to be in the latent stage, but reactivation is a common occurrence (15). Although the current standard of care is to continue the four-drug combination for at least six months (16), reinfection cannon be completely prevented, and 18% of these infections are caused by multidrug-resistant organisms (14, 17). Modern medicine has devoted many resources, and with the struggles of the field, has managed to prevent the global spread of antibiotic-resistant bacteria from becoming a pandemic threat like COVID-19. Given that the doubling time of bacteria is in hours, the speed of their molecular evolution is tremendous. Considering that even if a new drug is invented, it takes a certain amount of time for its clinical development, this weasel-word is extremely at disadvantage for humans. Nevertheless, new antibiotics are being developed to win the battle, and in addition, methods to attenuate bacterial toxins and phage-based methods (18, 19).
Understanding the pathogenesis of sepsis is directly linked to drug development, which is moving toward therapies that can eliminate pathogens while balancing the active and regulatory systems of the immune system (6). One of the molecular basis of sepsis is the transformation of the energy supply system of immune cells, and various compounds related to PGC1α, which activates mitochondrial biosynthesis, are being investigated for their efficacy in the treatment of sepsis (20). In the treatment of antibiotic-resistant bacteria, the use of immune checkpoint inhibitors that block inhibitory signals in T cells, TGF-β to activate T cells, M1-like macrophage adaptive transfer, and strategies such as the administration of gelsolin, an endogenous protein, to enhance the pathogen clearance of macrophages are beginning to be explored (5). Methodologies to intervene in host immunity and promote pathogen elimination are beginning to emerge in the form of specific methods and compounds.
In this review, we focus on therapeutic strategies for infectious diseases through intervention in the host rather than approaches to the pathogens themselves. In the field of cancer therapy, the development of drugs aimed at killing the cancer cells themselves and the intervention of host immunity have made remarkable progress (21). Although immunity plays a major role in infectious diseases, the host approach has been neglected to date. Defense against microorganisms is mediated by the effector mechanisms of innate and adaptive immunity. Innate immunity is mainly responsible for defense in the early stages of infection, whereas adaptive immunity, together with innate immunity, provides a stronger and more specific response, and establishes a sustained defense posture with immune memory (22). The balance between these host immune responses and the acquisition of microbial resistance determines whether infection is established (23). The initial response of the host to bacterial infection is recognition of the bacteria by cells possessing pattern recognition receptors, release of inflammatory cytokines such as IL-1β, TNF-α, and IL-6, vasodilation, and increased vascular permeability (24). This leads to the accumulation of leukocytes, mainly neutrophils, which are non-specialized phagocytes, and more phagocytes in the infected foci. These cells phagocytose extracellular bacteria and infected cells and serve as the first line of defense in bacterial clearance (25). Inflammatory cytokines activate adaptive immunity, leading to enhanced antibody production by B cells, and opsonized bacteria are subject to phagocytosis by phagocytes, while T cells produce a variety of cytokines, including IFN-γ, to enhance bacterial killing by phagocytosis (26). Antibodies, together with activated complement, cause bacterial neutralization and lysis and play a role in the host defense system. In the early stages of infection, phagocytosis plays a central role in bacterial killing. On the other hand, bacteria that cause intracellular infection ensure their survival and replication by disabling the phagolysosomal system, which is the executor of intracellular disinfection.
Mycobacterium tuberculosis, which causes intracellular infection, can cause delayed-type hypersensitivity and tissue damage. Slow-growing Mycobacterium tuberculosis evades the killing of the phagolysosomal system and survives intracellularly, resulting in persistent stimulation of T cells and macrophages and the formation of granulomas (27). This granuloma and the solid tumor microenvironment share common features of immunosuppressive conditions such as lymphocyte exhaustion/elimination, macrophage polarization to M2-like phenotype, hypoxia, immunomodulatory cytokines such as TGF-β/IL-10, and infiltration of myeloid-derived suppressor cells (5). This similarity reminds us that host-directed therapy, which has been successful in anticancer therapy, could bear great fruit in infectious diseases. Among host immune mechanisms, the phagolysosomal system is considered to be at the center of pathogen control and an appropriate target for infection control. In order to examine the possibility of intervention in the phagolysosome system in host-directed therapy, the molecular mechanism of the pathway from phagocytosis to phagosomes reaching lysosomes is discussed from the perspective of host-pathogen interaction. Finally, the current status and future potential of drug discovery targeting the phagolysosome pathway will be discussed.
When considering host-directed therapy for bacterial infections, it is important to divide bacteria into those that cause extracellular infections and those that cause intracellular infections. Bacteria that cause extracellular infections include Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumoniae, Escherichia coli, Vibrio cholerae, Clostridium tetani, Neisseria meningitidis, and Corynebacterium diphtheriae. On the other hand, bacteria that cause intracellular infections include Mycobacterium, Listeria monocytogenes, and Legionella pneumophila (22). Microorganisms can be classified into three types: 1) obligate extracellular growth parasites, which cannot grow inside the cell but only outside, 2) facultative intracellular growth parasites, which can grow both inside and outside the cell, and 3) obligate intracellular growth parasites, which can grow only inside the cell. Obligate extracellular growth parasites are eliminated by phagocytes and have developed resistance mechanisms against phagocytosis (22). S. aureus is classically recognized as a bacterium that causes only extracellular infections such as furuncles, carbuncles, impetigo, abscesses, septicemia, necrotizing pneumonia, and biofilm formation (28). Recent studies have shown that S. aureus can survive and proliferate intracellularly, which is a major factor in pathogenesis, making it a second category of bacteria (29) (Horn). Bacteria belonging to this category have evolved the ability to neutralize the phagolysosome. The third category of bacteria includes rickettsia and chlamydia, which are dependent on the host in terms of membrane structure and metabolism, respectively, but the immune mechanisms against them are beyond the scope of this review and should be referred to the cited review (30).
Innate immunity to extracellular infections is centered on complement activation, phagocyte activation, and inflammatory responses, and the final execution mechanism of bacterial elimination depends on the phagolysosomal system. Phagocytes directly recognize bacteria via mannose and scavenger receptors and enhance phagocytosis (25). In addition, both peptidoglycan, a major membrane component of Gram-positive bacteria, and LPS, an endotoxin of Gram-negative bacteria, activate the alternative complement pathway to opsonize bacteria. Like complement, bacteria opsonized by antibodies enhance phagocytosis (26). Extracellular bacterial protein antigens cause activation of CD4+ T cells, also assisting phagocytosis. Although neutralization and lysis of bacteria by antibodies are important defense systems, phagolysosomes as the final executor of bacterial elimination are central to bactericidal activity. Bacteria that produce intracellular infections have found a microenvironment (niche) within the phagocyte that is isolated from strong adaptive immunity and have acquired mechanisms that allow them to survive and replicate there. These bacteria have evolved mechanisms to disable the phagolysosomal system within the phagocyte and hijack the phagosome to survive (27). Adaptive immunity attempts to execute bacterial clearance through activation of the phagolysosomal system by recruiting phagocytes with the CD40 ligand signal and INFγ by CD4+ T cells. In the process of escape from the phagosome, the host can trigger a mechanism by which CD8+ T cells, upon receiving the signal, eliminate the infected cell itself (22). This section on host-pathogen interactions describes the general effector function of the host’s phagolysosomal system on pathogens.
The immune system quickly detects invading bacteria in the body and timely initiates phagocytosis as the appropriate response to eliminate the threat (Figure 1) (25). Phagocytes are estimated to make up less than 1% of all cells in the body (31). The ability of these cells to adequately patrol and scavenge throughout the body is critical for defense against foreign enemies (32). Although phagocytes form a constant protrusion (33), and signals from the calcium-sensing receptor (CaSR), a G protein coupled receptor, regulate phosphatidylinositide phosphorylation plasma membrane remodeling (34), and polymerization of the branched actin network just below the plasma membrane (35). Pathogen-sensing receptors include the pattern recognition receptors (PRRs) such as TLR4 that directly bind to pathogen surface structures (24), Fcγ receptors, and complement receptors interact with antibodies or complement that opsonize pathogens, and their interactions play a role in signaling to phagosome formation (25). The pattern recognition receptors (PRRs) involved in bacterial infections are Toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), C- and C- type lectin receptors (CLRs), and absent in melanoma-2 (AIM2)-like receptors (ALRs). Ten TLRs have been identified in humans and are present in dimeric form at the plasma membrane or phagosomal membrane (36). In the plasma membrane, TLRs exist as homodimers or heterodimers and recognize lipids, proteins, lipoproteins, and other components of microorganisms. On the other hand, in phagosome membranes, they exist as homodimers and recognize microbial nucleic acids (37). TLR1- TLR2 and TLR2-TLR6 are expressed in monocytes, dendritic cells, and are involved in the recognition of triacyl lipopeptide, lipoprotein, lipopeptide, lipoteichoic acid, arabinomannan, peptidoglycan. TLR4 homodimer is expressed in macrophages and dendritic cells and binds to lipopolysaccharide. TLR4 homodimer is expressed in macrophage and dendritic cells and recognizes lipopolysaccharide. TLR5 homodimer is expressed in intestinal epithelial cells and senses flagellin (24). NOD1 in the cytoplasm of intestinal epithelial cells and macrophages recognizes γ-D-glu-meso-diaminopimelic acid in the cell wall of gram-negative bacteria, while NOD2 recognizes muramyl depeptide in the cell wall of all bacteria (38). CLRs are expressed on macrophages and dendritic cells and play a critical role in anti-fungal immunity. They include the mannose receptor, which recognizes mannose units repeated on the surface of bacteria such as mycobacterium and induces phagocytosis, and the Asian glycoprotein receptor family, which includes Dectin-2, which recognizes mannose-capped lipoarabinomannan (39). ALRs are PRRs that recognize intracellular double strand DNA and do not participate in innate immunity but are involved in apoptosis (40).
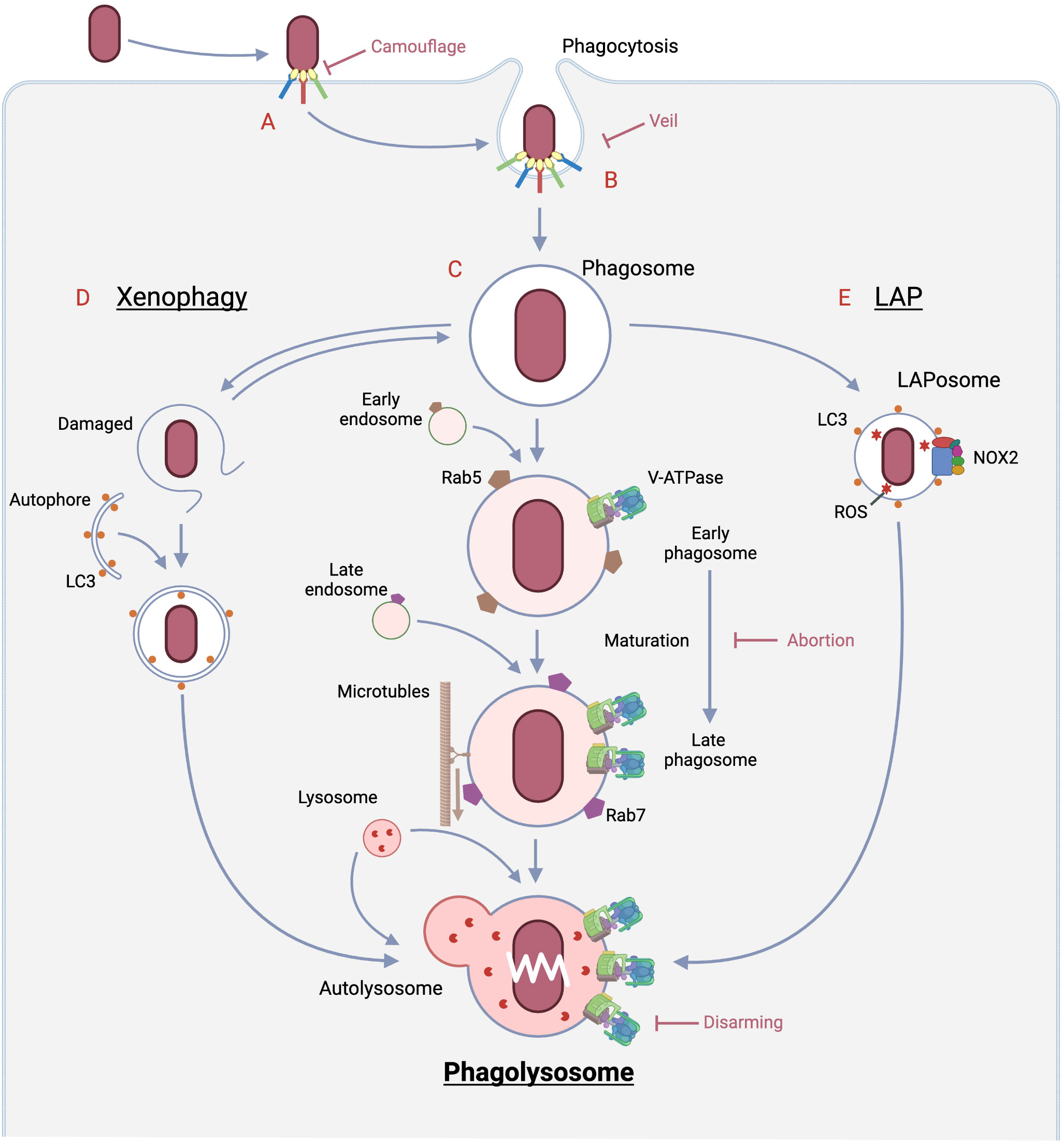
Figure 1 Pathways of bacterial eradication by phagocytes and immune evasion strategies by bacteria. A: The encounter between the bacteria and the host phagocyte is the initial site of rivalry that determines whether infection is established or not, and begins with the sensing of the bacteria by the host. Bacteria LPS is sensed by PPRs, and if opsonized by immunoglobulin and complement, by Fc or complement receptors, respectively. In the case of opsonization by complement, some bacteria evade detection by the complement receptor by mimicking the regulatory mechanism of complement. In the case of opsonization by antibodies, some bacteria prevent binding to the Fc receptor by secreting an enzyme that digests the antibody. Some bacteria alter the structure recognized by PRRs by phosphorylation or other means to escape from the PRRs. B: The process from phagocytic ruffle through cup closure and scission to phagosome formation requires dramatic changes in the cytoskeleton and in membrane phosphatidylinositides, which are regulated by many signals such as phosphorylation. Bacteria prevent phagocytosis formation by disrupting these signals through the secretion of dephosphorylases. On the other hand, there are viruses that produce substances that mimic the “Don’t eat me” signal as phagocytosis checkpoints. C: Phagolysosome pathway (Middle): Nascent phagosomes undergo fusion with early/late endosomes, hydrolase increases toward full set and V-ATPase also increases. As a result, the phagosome lumen becomes acidified and moves along the cytoskeleton toward the site of lysosome presence, where it fuses with the lysosome. The resulting phagolysosome reaches a pH near 4.6, the optimum pH for many hydrolases, to carry out complete bacterial degradation. D: Xenophagy (Left): When the phagosome is damaged by a bacterial escape mechanism, galectin, which is only exposed in the lumen, is exposed in the cytoplasm, which triggers autophagy initiation. If the cargo is a pathogen such as a bacterium, the autophagy is called xenophagy. The vesicles also eventually fuse with the lysosomes, resulting in complete digestion of the pathogen. E: LAP (Right): LAPosomes, in which LC3, which plays an important role in autophagy, engages the nascent phagosome, recruits NOX2 and produces reactive oxygen species. Reactive oxygen species are formed most efficiently in a neutral environment, and they damage pathogen-forming lipids, proteins, and nucleic acids more rapidly than phagolysosomes. The final disposition of the inclusions of this pathway is also completed by fusion with lysosomes. Created in BioRender.
The protrusion-captured target induce clustering of phagocytic receptors, and the immunoreceptor tyrosine-based activation motif (ITAM) in their intracellular domain (41) is productively phosphorylated by Src-family tyrosine kinases (SFKs), spleen tyrosine kinases (Syk) (42). As soon as the phagocytosis signal begins to amplify and a transient increase in PI (4, 5)P2 occurs, conversion occurs from PI (4, 5)P2 to PI (3–5)P3 by PI3K recruited to adaptor proteins (43). PI (3–5)P3 surges recruits phospholipase Cγ which breaks down PI (3–5)P3 to produce diacylglycerol (DAG) and inositol (1, 4, 5)-triphosphate (IP3) (44). DAG acts as a second messenger for signaling between phagocytic receptors (45), while IP3 provides calcium spike from the endoplasmic reticulum (ER) into the cytoplasm (46). These two 2nd messengers cooperate to activate small G protein Rap1, which mediates the “inside-out” response of integrin (47). PI3K activates Rho family GTPases that facilitate cytoskeletal remodeling directly and through the GEF. This activation dynamically alters the cytoskeleton to form phagocytic cups. NADPH oxidase 2 (NOX2), which is responsible for the generation of reactive oxygen species (ROS) that cause oxidative bursts, engages in the newly formed phagocytic cups (48).
On the other hand, there are systems that prevent phagocytosis, which phagocytoses pathogens and apoptotic cells, from running amok and eliminating normal cells. In cancer research, immune checkpoints have been identified as entities of T cell regulatory mechanisms (49). Immune checkpoint inhibitors such as anti-programmed cell death protein 1 (PD-1; pembrolizumab and nivolumab) (50), anti-cytotoxic T lymphocyte-associated protein 4 (CTLA-4; ipilimumab and tremelimumab) (51), anti-PD-1 ligand 1 (PD-L1: atezolizumab, avelumab and durvalumab) (52) have developed, and demonstrated to significantly improve outcome in patients suffered from devastating cancers (Figure 2A). In innate immunity, phagocytosis checkpoints recognize “Don’t eat me” signals during the phagocytosis process, and are beginning to be recognized as important new targets for cancer immunotherapy (53). The discovery was signal-regulatory protein α (SIRPα) expressed in the myeloid lineage (54). Upon the interaction of SIRPα and CD47, the intracellular domain of SIRPα, an immunoreceptor tyrosine-based inhibitory motif (ITIM) recruits SH2-containing protein tyrosine phosphatase 1 (SHP1) or SHP2, preventing myosin IIA dephosphorylation, subsequent rearrangement of the cytoskeleton, and the phagocytosis they form (55) (Figure 2B). Although PD-1 expression is an important marker of T cell exhaustion (56), it also is expressed on various immune cells, including macrophages. The interaction with PD-1 and PD-L1 provides a suppressive signal for the phagocytosis of tumor-associated macrophages (TAMs) (57). Cancer cells can escape macrophage-induced phagocytosis by expressing PD-L1. Sialic acid-binding immunoglobulin-like lectin (SIGLEC), which contains inhibitory receptor motifs (ITIMs) in its intracellular domain, is induced at the surface of macrophages and its expression confers a poor prognosis in cancer patients (58). The ligand for SIGLEC is CD24, and this interaction serves as an entity for anti-phagocytic action (59).
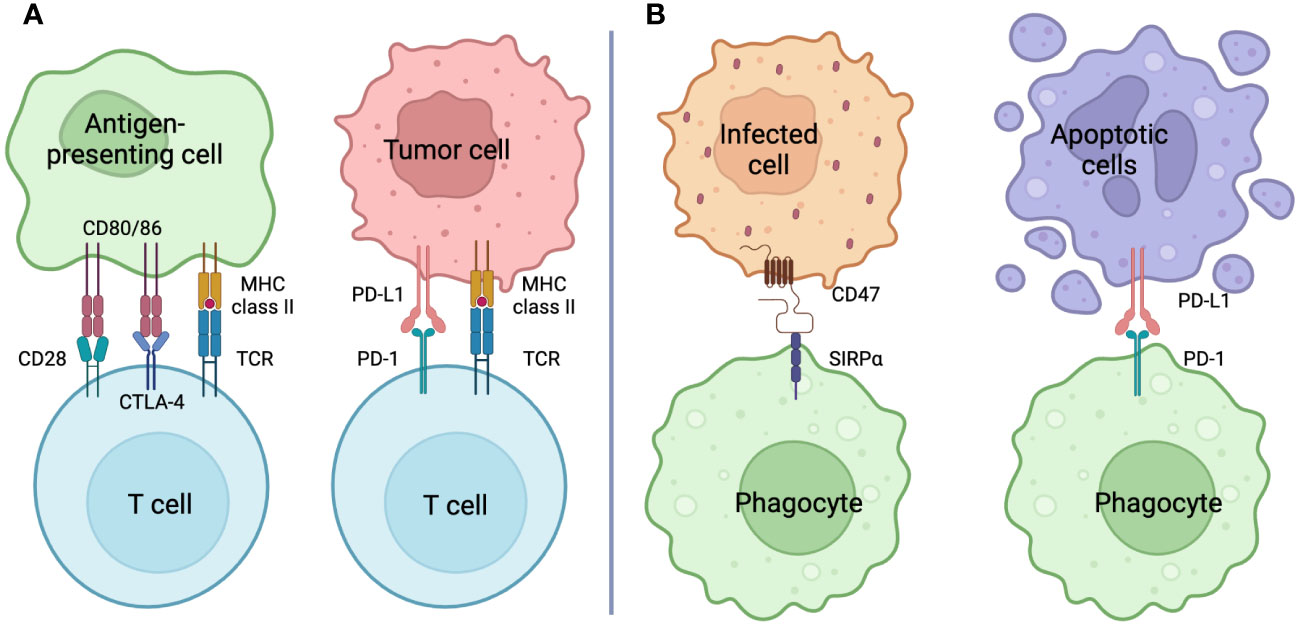
Figure 2 Checkpoint Inhibition (A) Immune checkpoint: T cells exercise T cell activation, clonal expansion, and effector function by transmitting signals via the TCR and signals from CD28 through binding to CD80/86 as costimulatory signals. On the other hand, inhibitory signals from CTLA-4 through binding to CD80/86 result in T cell deactivation. T cells expressing PD-1, which is also recognized as a T cell exhaustion marker, transmit inhibitory signals by engaging its ligand PD-L1, causing T cell deactivation similar to that of CTLA-4. Antibodies against molecules that comprise this immune checkpoint (immune checkpoint inhibitors: ICIs) shield the target antigen and cause T cell activation by releasing the T cell brake. Host-directed therapy with ICIs for cancer is an important anti-tumor strategy that works in tandem with anticancer drugs that kill the cancer cells themselves. (B) Phagocytosis checkpoint: Phagocytes, like T cells, have receptors with intracellular domains that transmit inhibitory signals to control their activity. The lignads, also called “Don’t eat me” signals, include CD47 and PD-L1, whose receptors are SIRPα and PD-1, respectively. Cells infected with pathogens show enhanced expression of CD47 as the main inducer of INF-g, while cells in apoptosis express PD-L1 and escape clearance by phagocytes. These phagocytosis checkpoint inhibitors may promote phagosytosis and, in the case of bacterial infection, may promote bacterial killing. Created in BioRender.
Non-specialized phagocytes involved in bacterial infections include epithelial cells, endothelial cells, osteoblasts, and fibroblasts. Epithelial cells, which play another major role among non-specialized phagocytes, cover the outer and luminal surfaces of the body and organs. Depending on where they are present, epithelial cell functions include absorption in the lungs and intestinal tract, secretion in the kidneys and stomach, and material transport in the trachea and oral-nasal cavity. Regardless of their location, the basic function of epithelial cells is to interact closely with the external environment and, in particular, to serve as the first line of defense in the immune system (60). Epithelial cells possess pattern recognition receptors to detect external hazards, but do not express receptors to capture opsonized pathogens as do specialized phagocytes. Therefore, phagocytosis of pathogens by epithelial cells is initiated by two following main methods. One is a trigger mechanism in which the cytoskeleton is restructured by effector molecules secreted by the bacteria, forming ruffles on the plasma membrane, and the other is a zipper mechanism in which the bacteria attach to proteins involved in cell adhesion, such as integrins and cadherins (61). After internalization, the phagolysosome system takes over for clearance.
Bacterial degradation in first line innate immunity is carried out through three main pathways (Figure 1): phagolysosome, xenophagy, and LAP (Microtubule-associated proteins 1A/1B light chain 3B (LC3)-associated phagocytoisis), each of which is followed by a vesicle: phagosome, autophagosomes, and LAPosomes, respectively, through fusion with lysosomes (60). In case of the first pathway, it begins with internalization by phagosomes after pathogen recognition, during which signaling occurs in the cell, leading to phagosome maturation (25) The phagosomes are then translocated to the lysosomes. Subsequently, the pathogen is degraded by phagolysosomes that are generated by fusion with lysosomes (60). On the other hand, autophagy is triggered when the imported bacterium attempts to escape from the phagosome, causing vesicle damage (62). Autophagy to enclose pathogens is called xenophagy, whose efficacy largely depends upon lysosomal function. In the third pathway, LC3, which plays a major role in autophagy, is embedded in the phagosome membrane in the form of LC3 modified by phosphatidylethanolamine (referred to as LC3-II), resulting in the formation of the LAPosome. The LAPosome is partitioned by a single membrane like the phogosome, unlike autophagy, which has a double membrane beginning in the phagophore (63). Its major feature is the quick recruitment of NADPH and the burst of reactive oxygen species that its enzymatic activity leads to, and the maturation of the LAPosome is faster than that of conventional phagocytosis. Review of bacterial killing in LAP and xenophagy are beyond the scope of this review; see other reviews (62, 63).
Inactivation and decay of phagocytosed pathogens leading to acquired immunity requires dramatic transformation of the formed phagosomes, a process termed phagosome maturation. It is a process that many pathogens target for survival (64). This process leads to two intermediate states: early phagosome and late phagosome, and eventually to the formation of phagolysosomes. Nascent phagosome fuse with early endosomes and are responsible for sorting phagocytosed prey for reusability. The late phagosomes fused with the late endosomes create a more acidic environment in the lumen and migrate along the microtubules toward the lysosomes. The elaborate molecular mechanisms of this process have been detailed, with Rab GTPase and phosphatidylinositide playing major roles.
The newly formed phagosome has a PI (3–5)P3-rich membrane composition, and the recruitment of Rab5 GTPase to it promotes membrane fusion with early endosomes through several pathways (65) Vps34, type III PI3K, is recruited by Rab5 (66) and converts PI to PI3P, which becomes a major component of the membrane and attracts multiple effectors (67). One of them is early endosome antigen 1 (EEA1) (68), which interact with Soluble N-ethylmaleimide-Sensitive Factor Attachment Proteins (SNAPs), Syntaxin 6 (69) and Syntaxin 13 (70), to promote membrane fusion of nascent phagosomes and early endosomes (71).
The conversion to the late phagosome begins when the positive feedback loop of Rab5 is severed and replaced by Rab7 (72) PIs that make up the membrane are transformed from PI3P to PI4P by recruitment of 3- phosphatases of the myotubularin family and PtdIns4P kinase 2A (PI4K2A) (73). Rab7 forms homotypic fusion and vacuole protein sorting (HOPS), which mediate a tether between membrane with binding to Rab7, by replacing some of the components of class C core vacuole/endosome tether (CORVET) that mediate Rab5-mediated inter-vesicular tethering (74). GTP-bound active Rab7 recruits two Rab7 effectors: the Rab7-interacting lysosomal protein (RILP) and the long splice variant of the oxysterol-binding protein (OSBP)-related protein 1 (ORP1L) to move toward the microtubule-organizing center (MTOC) for a complete fusion with lysosome (75). They form a scaffold for dynein-dynactin to bridge the microtubule and are transported to the minus end along microtubules (76).
Centripetal movement brings the late phagosome and lysosome into close proximity, and Syntaxin 7 is involved, causing the phagolysosome (77). The two organelles undergo a process of tethering, docking, consolidation, and fusion, in which actin polymerization and calmodulin are involved in the tethering process, protein-protein interaction regulates docking, and Ca spiking leads to consolidation (78). In this stage, the composition of membrane PIs changes dramatically, with PI (3, 5)P2 joining the major PIs components. V-ATPase, which is responsible for intraluminal acidification, is completely incorporated in the phagolysosome membrane (79). Analysis of the immune escape mechanism of Mycobacterium tuberculosis revealed that V-ATPase also acts as a major player in membrane fusion and associates with HOPS (80).
Lysosomes are the defeaters that break down phagocytosed materials into their constituent parts and squeeze out the substances necessary for the host, and they have an arsenal of various weapons for this purpose. Against bacteria, the lysosome attempts to destroy them with reactive radicals, various digestive hydrolases, acidic milieu, and nutrient segregation as its main weapons. However, their regulation differs greatly among cell types, even among professional phagocytes (81). The control of these factors seems to depend on the roles of bactericidal action by reactive oxygen species and nitrogen, followed by complete digestion of the structure and transmission to the acquired immune system by antigen presentation to T cells. Macrophages are highly plastic cells and are classified into two activation states, M1 or classic and M2 or alternative, and the process leading to this state is defined as polarization (82). M1 M2 macrophages are induced by Th1 cytokines such as IFN-γ and TNF-α or LPS and secrete high levels of pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6 and TNF-α. -13 and secrete abundant levels of anti-inflammatory cytokines such as IL-10 and TGF-β (82). Macrophages have been shown to regulate V-ATPase and NOX2 very oppositely by polarization (83). M1 macrophages and neutrophils have low V-ATPase activity and a near-neutral lysosomal lumen but abundant production of reactive oxygen species (ROS). In contrast, M2 macrophages have high V-ATPase activity, the lysosome lumen is strongly acidic, and reactive oxygen species are not so high. However, the classification of M1/M2 macrophages is an oversimplification, as the two states are flexible and dynamically plastic, with intermediate rather than binary states, and there is a subset of regulatory macrophages in addition to activated and healing macrophages (84). It remains to be seen how polarization and its shift are regulated in infection, when the regulatory subset is committed during infection in vivo, and what are the keys that control these processes.
In lysosomes and endosomes, V-ATPase is the only machinery that consumes energy to transfer protons into the lumen, but the acidic milieu produced by V-ATPase provides an optimal pH for the intraluminal hydrolase to perform bacterial killing (Figure 3). V-ATPase also plays an extremely multifaceted role in the phagolysosomal pathway (85). For example, recycling of plasma membrane receptors taken up into the lumen (86), recovery of the mannose 6-phosphate receptor into the trans-Golgi network (87), loading of external antigens into the major histocompatibility complex (88), and endosome tethering in phagosome maturation (80). In addition, acidic milieu plays an essential role in the following processes: neurotransmitter uptake (89), maturation by degradation of prohormone (90), nutrient sensing in association with mTORC1 (91), amino acid supply to the cytoplasm (92), and macroautophagy (93, 94). In signal transduction, WNT and Notch signals require an acidic milieu in the vesicle. In the WNT pathway, the Frizzled/LRP6 complex is in close proximity to V-ATPase via prorenin receptors on the signaling endosome. LRP6 phosphorylation required for the β-catenin destruction inhibition signal is dependent on V-ATPase activity (95). In Notch signaling, the Notch receptor is internalized by ligand binding and transferred onto the signaling endosome, and the acidic environment of the vesicle causes the Notch intracellular domain to be released into the cytoplasm through S3 cleavage by the γ-secretase (96).
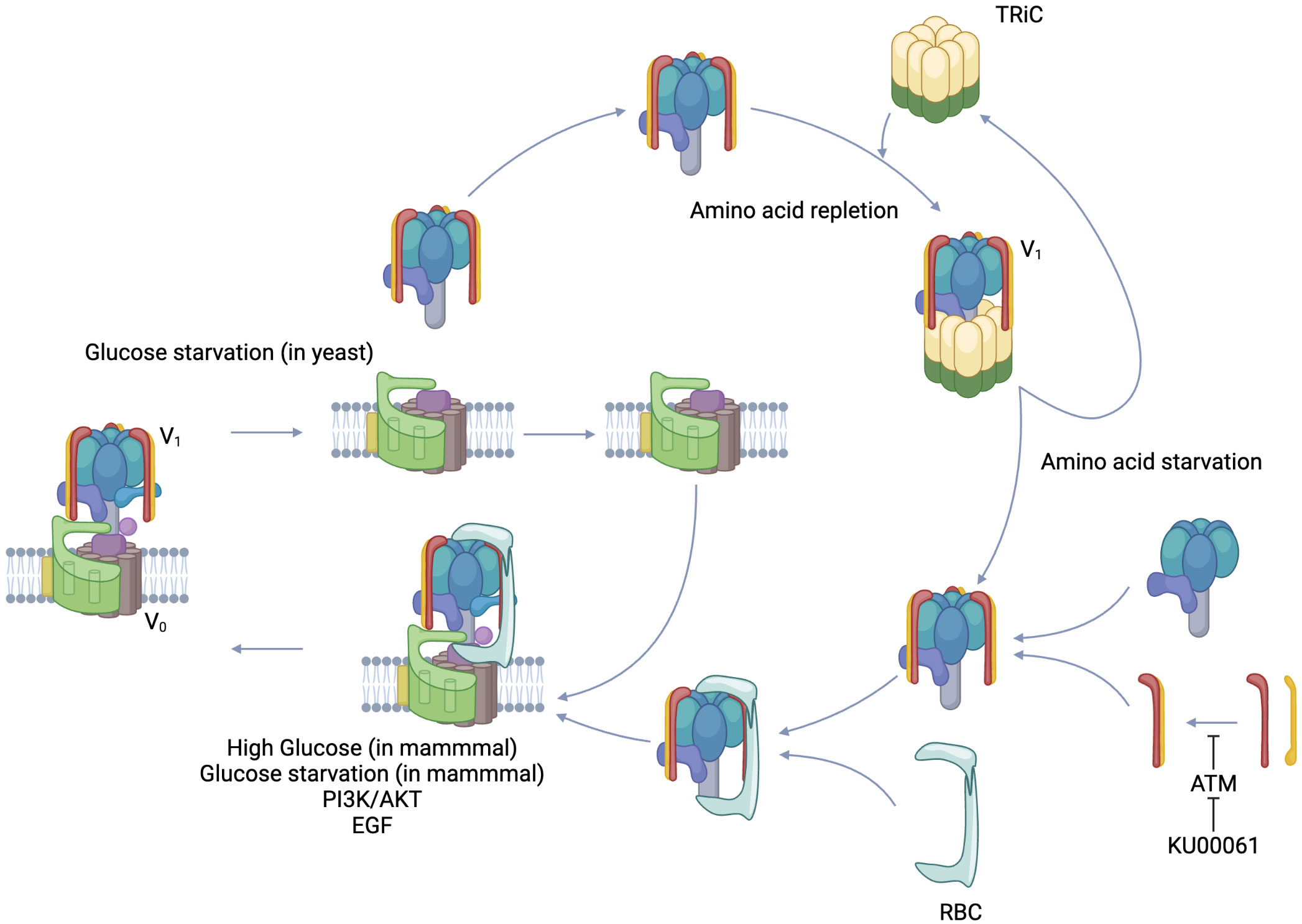
Figure 3 Regulated assembly/Reversible disassembly of V-ATPase. V-ATPase is composed of a membrane-integrated V0 complex and a V1 complex that can exist free in the cytoplasm. PI3K/AKT, EGF signal, and in mammals, glucose starvation and high glucose induce regulated assembly. On the other hand, glucose starvation induces reversible disassembly in yeast. In the case of amino acid deficiency, TRiC, which holds the V1 complex in the cytoplasm, releases the V1 complex and leans toward V-ATPase assembly. Rbc3 recruits V1 to V0 as a chaperone molecule. The dimer formation of V1E and V1G, which form the peripheral stalk of the V1 complex, is inhibited by the phosphorylation of V1G by ATM, resulting in inhibition of V-ATPase assembly. KU-60019, which inhibits ATM, promotes V-ATPase assembly. Created in BioRender.
Various hydrolases encapsulated in the lysosome have pH-dependent enzymatic activities and their acidity is precisely regulated. The methods reported for its regulation include 1. nutrition, 2. signaling, 3. cofactors, and 4. modification by enzymes. Nutrition was first reported as reversible disassembly in yeast during glucose starvation, and its biological significance is thought to be the limitation of ATP consumption under nutrient-depleted conditions (97). On the other hand, in mammals, V-ATPase assembly was shown to occur at an excess glucose concentration of 25 mM (98). Excess glucose increases glycolysis which leads to acidic environment in the cytoplasm, whereas the promotion of V-ATPase assembly is thought to be responsible for keeping the cytoplasm neutral by accumulating protons in the lysosome. It was reported that reversible disassembly in yeast is mediated by PKA, while regulated assembly in mammals is mediated by PI3K. Initially, the response of V-ATPase to glucose availability in yeast and mammalian cells appeared to be consistent, but the report that glucose starvation also promotes regulated assembly in mammalian cells showed the diversity of the regulatory mechanism (99). Glucose starvation activates AMPK, which is further enhanced by a complex with Regulator and assembled V-ATPase that provides a binding site for AMPK through AXIN (100). This AMPK activation may be directed toward improving energy supply and demand by shifting metabolism toward catabolism, one of which may be autophagy (101). Amino acids also have a significant effect on V-ATPase. In amino acid starvation, Regulator forms a tight complex with V1A of V-ATPase and eliminates mTORC1 while assembling V-ATPase (91). V-ATPase assembly leads to increased activity, and autophagy enhanced by amino acid starvation leads to amino acid acquisition by degradation of proteins brought to the lysosomes, resulting in release of amino acids into the cytoplasm to maintain homeostasis (101). With respect to signals, PI3K and its downstream AKT bring about regulated assembly (102). PI3K inhibitors do not prevent assembly (103), while AKT inhibitors prevent assembly (104). This suggests direct binding of AKT to V-ATPase (105). On the other hand, mTORC1 activity, which is downstream of PI3K/AKT, does not participate in V-ATPase assembly in the absence of amino acids (103). The AKT-mediated increase in the regulated assembly of V-ATPase, which results in decreased intraluminal pH, enhanced proteolysis, and increased cytoplasmic amino acid content, is also the mechanism by which EGF activates mTORC1 (106).
Cofactors and assembly chaperones such as Rabconnectin3 (Rbc3), TRiC, and mEAK7 and enzymes such as ATM have been reported. In yeast, RAVE (Regulator of H+-ATPase of Vacuolar and Endosomal membranes) regulates luminal acidity of lysosomes and endosomes via V-ATPase assembly, and in mammals Rbc3 is functionally equivalent to RAVE (107). Rbc3 is a heterodimer composed of Rbc3α and Rbcβ (108). The former is composed of either of two isoforms, DMXL1, or DMXL2 (109), and the latter is formed by WDR7. The combination varies among tissues and intracellular organelles (107). Rav1, which is a subunit of the yeast RAVE, recruits free V1C in the cytoplasm and contributes to V-ATPase assembly (110). DMXL1 and DMXL2 are homologs of Rav1, and the amino acid sequence in which Rav1 interacts with V1C is also conserved in DMXL (111). Functionally, silencing of any of the components of Rbc3 reduced the acidity in the vesicles (112). Knockout of WDR7 attenuated V-ATPase assembly (113). The regulation of Rbc3 is still to be elucidated, but one clue is calcium dynamics. CAB2.2, a transmembrane calcium channel, binds to DMXL (114), and CAPS1, which is involved in endoplasmic reticulum acidification through calcium dynamics, also binds to Rbc3 (115). TRiC holds the V1 component in the amino acid-replete cytoplasm, while releasing it for V-ATPase assembly in the presence of amino acid deprivation (116). A regulatory mechanism of TRiC could the phosphorylation of a subunit constituting TRiC. Phosphorylation of CCT2, a component of TRiC, modulates its function (117). mTORC1 signal may modify TRiC components to stabilize the TRiC/V1 component complex. mEAK7 engages V1A, B, and E in the N-terminal domain and binds to V1D in the C-terminal domain, but does not contribute to luminal acidification and affects mTOR signaling (118). Although ataxia telangiectasia mutated (ATM) was initially identified as a protein involved in the DNA damage response, it was recently reported to phosphorylate V1G and prevent the interaction with V1E, resulting in inhibition of the formation of a peripheral stalk (119).
Among the microbicidal effects induced by bacterial phagocytosis, the production of reactive radicals in the lumen is mainly mediated by NOX2 of the NADPH oxidase (NOX) family and inducible nitric oxide synthase (iNOS), which are triggered most rapidly after pathogen entry. The former produces reactive oxygen species (ROS) most prominently in neutrophils, while the latter produces reactive nitrogen species (RNS) mainly in macrophages (64). The superoxide anion (O2-.) produced by NOX2 leads to the production of ROS represented by hydrogen peroxide (H2O2), hydroxyl radical (OH*), and hypochlorous acid (HOCl). This process is called a respiratory or oxidative burst because of the surge in oxygen uptake and glucose consumption unresponsive to cyanide (120).
The activated NOX2 complex transfers electrons from the cytoplasmic NADPH into the lumen of the phagosome, and the resulting charge imbalance is resolved by the voltage-gated proton channel Hv1, indicating that the activity of the NOX2 complex requires this channel to be activated (121). Since this ion channel does not consume energy, it is thought to function only when the pH of the phagosome lumen is near neutral, indicating that the respiratory burst of phagocytes induced by NOX2 complex activation occurs only in a very narrow range near neutral (122). The superoxide anion (O2-) generated by the NOX2 complex can utilize electrons from further NOX2 complexes and hydrogen from the phagosome lumen and Hv1 to induce superoxide reductase (SOR) or three types of superoxide dismutases (SODs) to produce hydrogen peroxide, leading to the formation of additional hydroxy radicals (123). These reactive radicals carry out their microbicidal action by disrupting structures containing DNA, Fe-S clusters, hemes, sulfhydryls, thioethers, and alkenes (124). ROS from the NOX2 complex are not prominent in macrophages, but in macrophages that swallowed the pathogen in defense against S. aureus infection, mitochondria-derived vesicles, which contain abundant hydrogen peroxide, fuse with phagosomes to provide reactive radicals, which are lacking in bacterial killing (125).
It is mainly the inducible nitric oxide synthase (iNOS) that produces reactive nitrogen species (RNS) that cooperate with ROS in pathogen killing (64). Nitrogen oxide is made of cytoplasmic L-arginine and oxygen, which undergo various catalytic reactions to produce nitrogen dioxide, peroxynitrite, dinitrogen trioxide, and dinitrosyl iron. Unlike NOX2, the regulatory mechanism occurs at the transcriptional regulation, and de novo protein synthesis is required for RNS production (126). Activation signals for iNOS include the extracellular proinflammatory cytokine interferon gamma (IFNγ) and the intracellular signaling molecule NF-κB (127).. It had been thought that iNOS is not recruited to the phagosome and remains in the cytoplasm; therefore, the RNS produced reaches the phagosome lumen by diffusion (128). In research on Mycobacterium spp., it was revealed that iNOS is recruited to phagosomes through binding with the scaffolding protein EBP50, while the bacillus attenuates the recruitment (129).
Iron, alone or incorporated into Fe-S clusters or heme, is essential for respiration, amino acid metabolism, and nucleic acid synthesis, not only in eukaryotes but also in prokaryotes. Excess iron leads to the ROS formation, while catalases and peroxidases that relieve oxidative stress require heme as a cofactor (130). The innate immune system has acquired the tactic of making iron unavailable to pathogens so that they can feed on the pathogens they have taken in (131). Lactoferrin is structurally very similar to transferrin, and it strongly binds to divalent iron ions even in the highly acidic environment of the lumen of the lysosome and exhibits antimicrobial action as an iron chelating agent (132). Iron is absorbed by bacteria via siderophore from the environment. Siderocalin (neutrophil gelatinase-associated lipocalin (GAL)), which inhibits siderophores, has been shown to effectively function, especially in sepsis caused by E. coli (133) and Mycobacterium spp (134).. Natural resistance-associated macrophage protein-1 (Nramp1/Slc11a1), in the membrane of phagosomes and functions as a divalent metal-proton symporter, has been implicated in the defense of intravesicular pathogens (IPs). Nramp1 starves IPs such as Mycobacteria, Salmonella typhimurium, and Leishmania domovanii by removing Fe2+, Co2+, and Mn2+ from the phagosome (135).
Pathogens have various strategies to evade immunity and survive. In particular, the pathway from phagocytosis to digestion in the phagocyte, the first line of defense of innate immunity that the pathogen encounters, is the most important site that must be neutralized (64). Numerous molecular mechanisms have been described that allow pathogens to disarm the phagosome pathway and thereby acquire the microenvironment in which to survive and proliferate (Figure 4; Tables 1–3) (136).
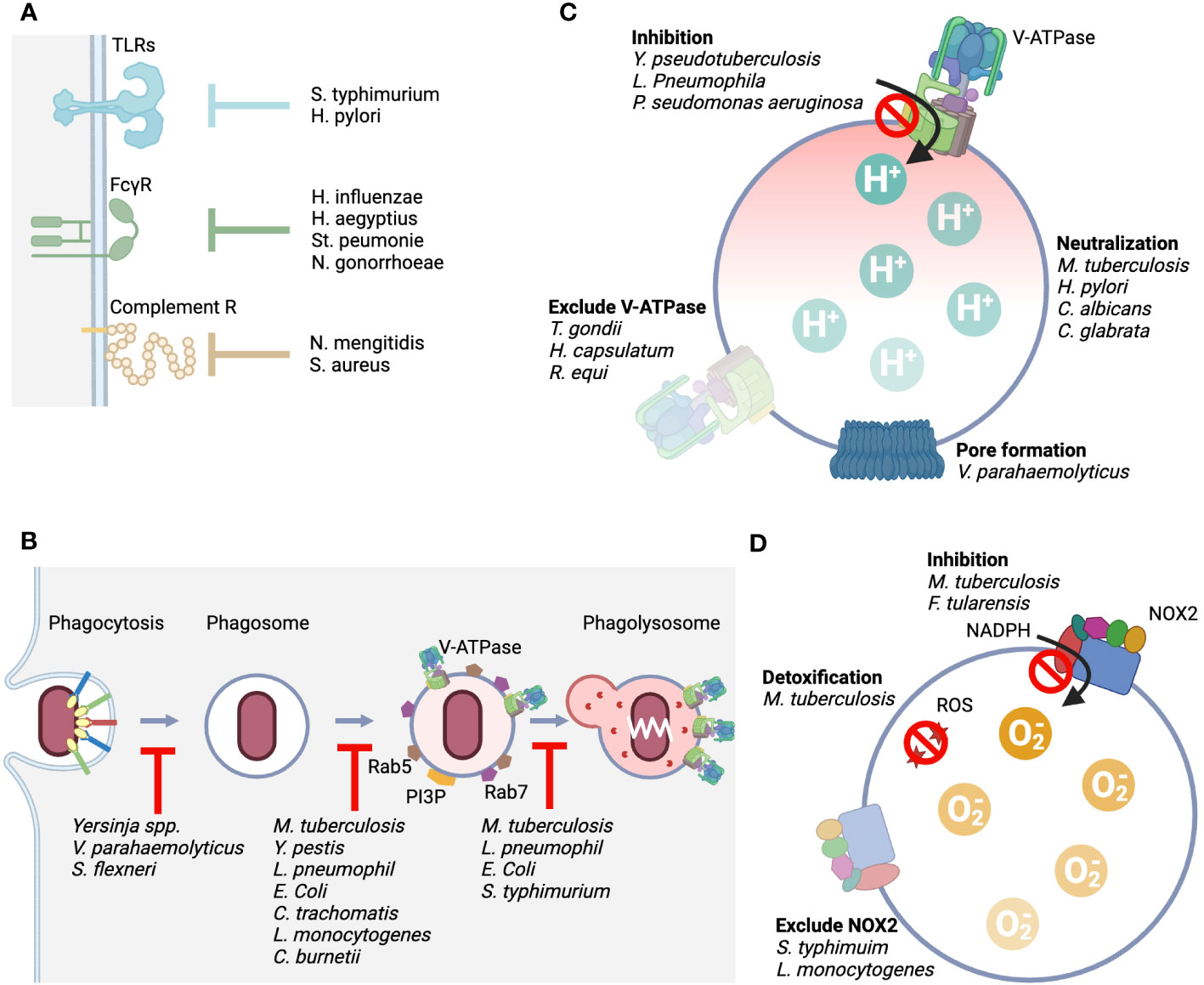
Figure 4 Rpresentative mechanisms by which pathogens evade from the host phagolysosomal system. (A) Representative mechanisms by which the host senses pathogens include TLRs, FcγRs, and complement receptors, and we show bacteria that possess mechanisms that counteract this sensing. (B). It shows bacteria with an evade mechanism by inhibiting the process of phagocytosis, phagosome maturation, and fusion with lysosomes. (C). The point of action at which the function of V-ATPase is attacked and the bacteria that carry it out. Four typical mechanisms are shown. Direct inhibition of pump function, elimination of the pump from the membrane, loss of proton concentration differences by generating pores, and production of enzymes that alkalinize acidified intralumens. (D). The point of action at which the function of NOX2 is attacked and the bacteria that carry it out. Three typical mechanisms are shown: direct inhibition of the mechanism that produces superoxide, elimination of the complex from the membrane, and detoxification of the produced reactive oxygen species.
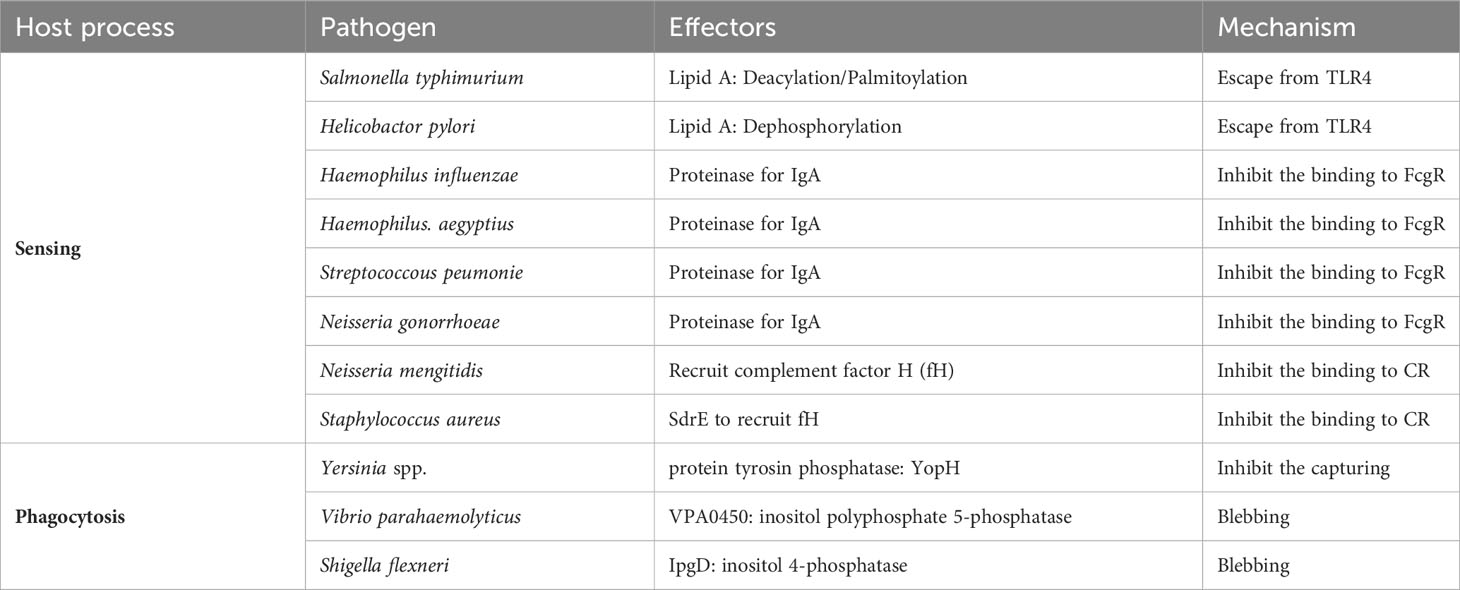
Table 1 Immune evasion strategies for processes from sensing to internalization.
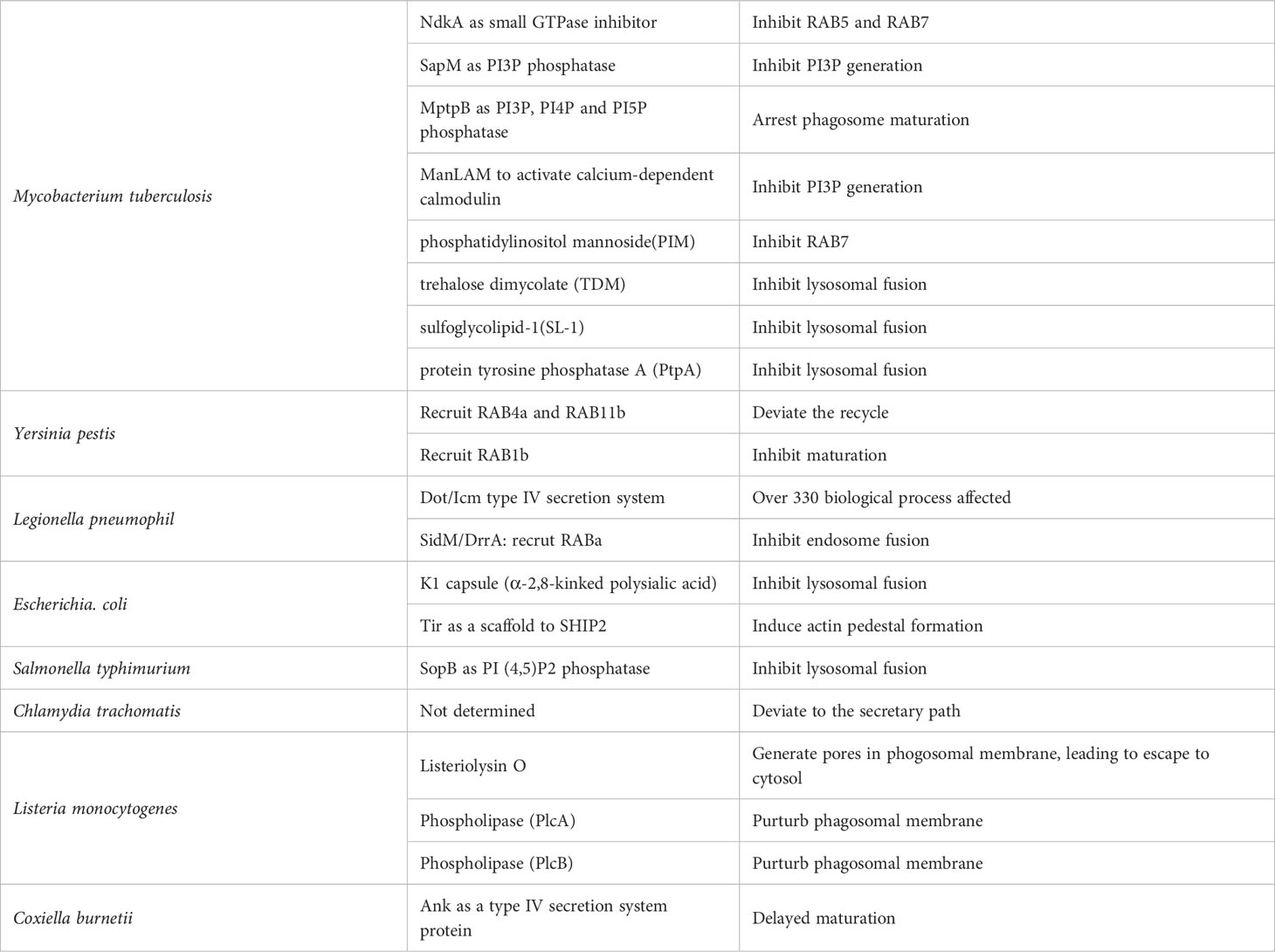
Table 2 Immune evasion strategies to prevent phagosome maturation.
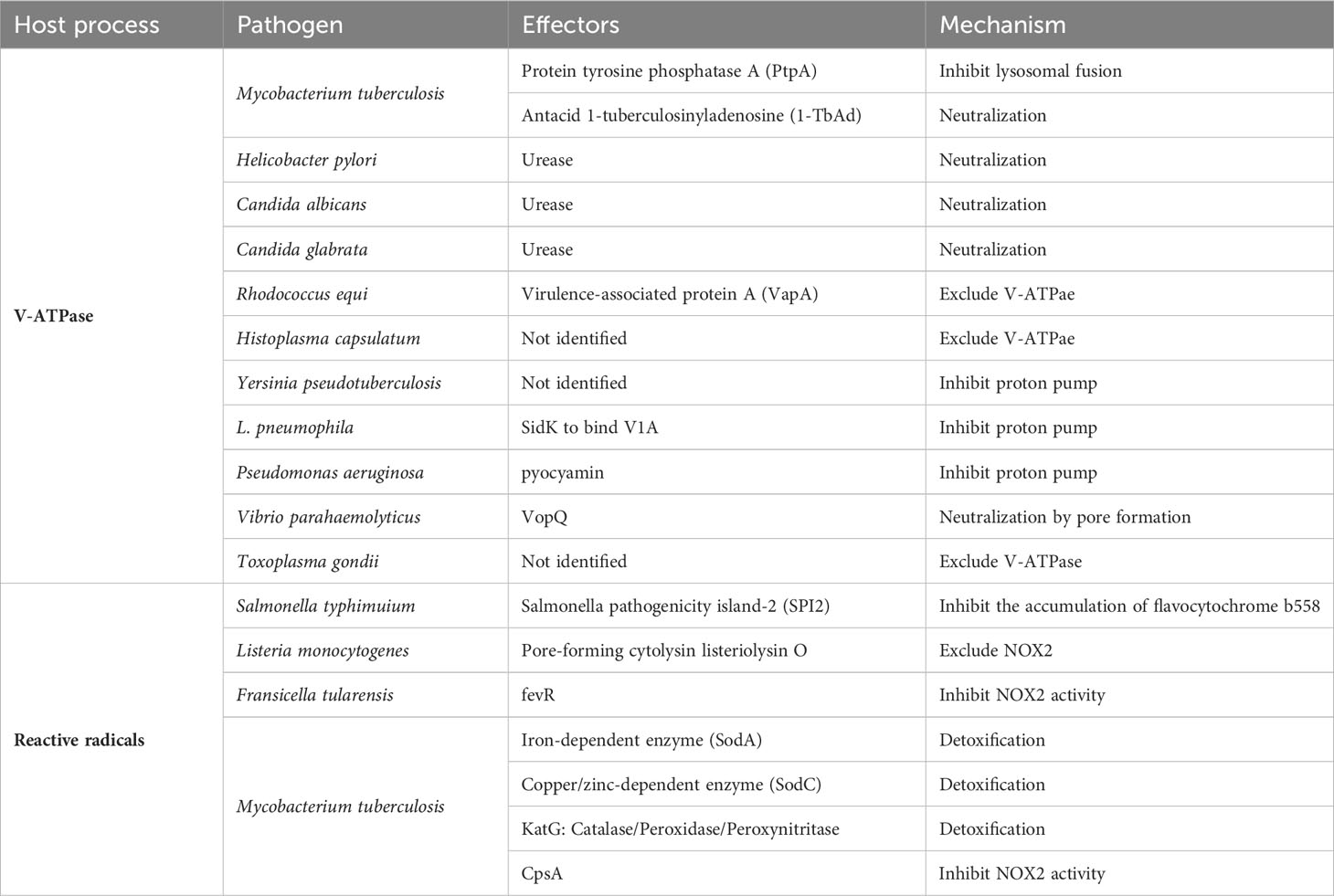
Table 3 Immune evasion strategies to neutralize bacterial destruction mechanisms.
Salmonella typhimurium interferes with TLR4 recognition and signaling through deacylation and palmitoylation of lipid A present on their surfaces (137). Helicobacter pylori dephosphorylates lipid A to escape TLR4 (138). Haemophilus influenzae, H. aegyptius, Streptococcus peumonie, and Neisseria gonorrhoeae secrete proteases that selectively cleave immunoglobulin A, which is responsible for opsonization (139, 140). Complement also plays a role in opsonization, but some bacteria exploit the elaborate activation pathway of complement. Neisseria mengitidis is a bacterium that avoids phagocytosis by mimicking the host complement factor H (fH), the regulatory substance of complement, on its own surface (141). Staphylococcus aureus also recruits fH to its surface by secreting a substance called SdrE (142). YopH, a protein tyrosine phosphatase produced by Yersinia spp., dephosphorylates host phosphotyrosine proteins and prevents phagocytosis (143). Vibrio parahaemolyticus secretes an inositol polyphosphate 5-phosphatase, VPA0450, which disrupts host cell membrane integrity and causes blebbing (144). This tactic is also used by Shigella flexneri, which secretes IpgD, an inositol 4-phosphatase, as its virulence factor, causing plasma membrane blebbing by converting PI (4, 5)P2 to PI (5)P (145). Although not a bacterium, m128L encoded by Myxoma virus has a high homology to CD47, which is known to inhibit host phagocytosis as a “don’t eat me” signal (53). The “don’t eat me” signal is essential for establishing the lethal infection through inhibition of host phagocytosis (146). Although there are no reports of cases in which the bacteria themselves encode CD47 mimic, enhanced expression of CD47 in cells infected with S. typhi and Borrelia burgdorferi has been reported (147). The upregulation of CD47 expression occurs through signaling from PRRs and is also enhanced by inflammatory cytokines, suggesting that the CD47-SIRPα axis may work to suppress excessive inflammatory responses. Bacteria make good use of this host regulation mechanism to aid immune evasion. On the other hand, M. tuberculosis, a phagocytosis-dependent intracellular parasite, is unique in that it does not enhance CD47 expression, unlike other bacteria.
Although we are far from having a complete picture of the diverse strategies by which bacteria neutralize host immune attack within the cell, learning the molecular mechanisms is the first thing we must do to win the war against bacteria. The strategy of bacteria that acquire permissive niches intracellularly as intracellular pathogens (IPs) (148) provides a great clue for the construction of host-directed therapeutics (HDTs) (27). M. tuberculosis, a leading IP, has long been a major scourge to mankind due to its high prevalence and high mortality rate on a global scale (149), and is one of the most carefully investigated (150). M. tuberculosis is thought to have evolved in such a way to struggle with the host immune system that an exhaustive list of protein and lipid effectors produced by the bacillus has been compiled (151). In addition to M. tuberculosis, other potential IPs include Rickettsia rickettsia, Chlamydia trachomatis, Legionella peumophila, Coxiella burnetiid, Brucella abortus, and Salmonella enterica; Cryptococcus neoformans and Aspergillus fumigatus among fungi. Candida albicans uses other intracellular organelles such as mitochondria as a habitat. The mechanisms by which they evade the phagosome pathway have been intensively studied, and detailed molecular mechanisms have been elucidated (152).
M. tuberculosis has spun out more than a dozen countermeasures in the phagosome maturation stage alone. First, it secretes NdkA to repress the small GTPases RAB5 and RAB7, which are central regulatory molecules in phagosome maturation (153). In addition, SapM produced by the bacillus acts as a PI3P phosphatase and inhibits the formation of PI3P, which is essential for the maturation of membrane composition (154). Glycolipids on the surface of the bacillus also greatly influence this phagosome maturation. Mannose-capped lipoarabinomannane (ManLAM) suppresses PI3P generation via the calcium-dependent calmodulin pathway and prevents phagosome maturation (155). In addition, phosphatidylinositol mannoside (PIM) present in the envelope supports the retention of early endosome RAB proteins such as RAB5, RAB22A, and RAB14 and prevents the recruitment of late endosome RAB proteins such as RAB7 (156). With respect to fusion with the lysosome, trehalose dimycolate (TDM) (157) and sulfoglycolipid-1 (SL-1) (158) as lipid effectors prevent the fusion process between the lysosome and the phagosome. Furthermore, M. tuberculosis secretes toxins that inhibit the Ca/Calmodulin-PI3K cascade and attempts to survive as IP through a strategy of inhibiting the fusion of phagosomes and lysosomes (159). In phagosomes harboring M. tuberculosis, protein tyrosine phosphatase A (PtpA) secreted by the bacillus binds to V-ATPase V1H and inhibits the association of V-ATPase and HOPS, as well as dephosphorylates vacuolar protein sorting 33B (VPS33B), which forms HOPS, and thus loses its function as a fusion machinery (80).
Yersinia pestis targets organelle trafficking and recruits Rab4a early in infection and Rab11b late in infection to prevent phagosome maturation and inhibit acidification in the lumen (160). These small GTPases are involved in endosome recycling, and the Yersinia-containing vacuole mimics this process. In addition, Y. pestis recruits Rab1b to phagosomes to inhibit phagosome acidification by suppressing lysosome fusion (161). Legionella pneumophila has evolved a defect in organelle trafficking: intracellular multiplication (Dot/Icm) type IV secretion system to make the phagosome of alveolar macrophages a proliferative niche (162). This system provides more than 330 effector proteins that interfere with host biological processes to assist in bacterial replication and survival (163). Among them, the system is involved in the recruitment of Rab1 like Y. pestis (164) and provides Sid1/DrrA, which is involved in the regulation of Rab1 (165). E.coli K1 has a K1 capsule composed of α-2,8-kinked polysialic acid on its surface that inhibits the fusion of phagosomes and lysosomes. Salmonella-containing phagosomes also inhibits the fusion of phagosomes and lysosomes (166). Salmonella secretes phosphoinositide phosphatase to maintain PI3P levels in the membrane, thereby preventing phagosome maturation and fusion with the lysosome and ensuring its survival (167). Chlamydia trachomatis avoids the fusion of its internalized phagosomes with endosomes and directs them to the secretory pathway to avoid an acidic environment (168).
It was shown 30 years ago that M. tuberculosis, when it reaches the phagolysosome, excludes V-ATPase from its membrane and maintains the lumen at a pH of 6.3 or higher (169). The entity responsible for excluding V-ATPase from the phagosome was PtpA (80). In addition, antacid 1-tuberculosinyladenosine (1-TbAd), which neutralizes acidification of the lumen, is secreted by the bachillus (170). H. pylori, which can live in highly acidic stomachs, has evolved various genes to adapt to the acidic environment. One of the effectors is urease, which produces ammonium ions that allow the pathogen survive in the harsh acid environment of the stomach (171). Candida albicans and C. glabrata use amino acids in their lysosomes to produce ammonium ions to neutralize the intraluminal pH (172, 173). Mycobacteria spp. have the same strategy (174). Research has been conducted to create more effective vaccine that lacks urease (175). a single bacterium has multiple defense mechanisms against the host offense of lysosomal acidification. In addition to Mycobacterium spp., Rhodococcus spp (176, 177). and Histoplasma capsulatum (178) were reported to exclude V-ATPases from phagosomes that contain them.
Some bacteria secrete substances that directly inhibit V-ATPase. Y. pseudotuberculosis directly inhibits the activity of the proton pump without affecting the expression level of the protein component of V-ATPase, thereby causing lysosomal deacidification (179). SidK produced by L. pneumophila physically binds to V-ATPase V1A and inhibits its proton transport (180). Structural analysis of the binding of SidK to V-ATPase showed that the two α-helical bundles at the N-terminus of SidK bind to V1A and markedly reduce the flexibility of its subunit (181). Pseudomonas aeruginosa secretes pyocyamin, which is a potent inhibitor of V-ATPase (182). Toxoplasma gondii survives by eliminating all components involved in membrane fusion with endosomes, resulting in non-acidic vacuole (183, 184). Vibrio parahaemolyticus secretes VopQ, a type III effector protein, which is incorporated into the lysosome membrane as a channel for the free passage of protons, and the pH in the cytoplasm and lysosome lumen is balanced (185).
Salmonella typhimurium inhibits the accumulation of flavocytochrome b558 by releasing Salmonella pathogenicity island-2 (SPI2), a member of the type III secretion system (186). Listeria monocytogenes also eliminates the NOX2 membrane component by secreting the pore-forming cytolysin listeriolysin O (187). Fransicella tularensis not only excludes flavocytochrome b558 but also directly inhibits the activity of the NOX2 complex by releasing a regulatory factor called fevR (188). M. tuberculosis has also taken multiple countermeasures against reactive radicals, including two types of SODs that process ROS: iron-dependent enzyme (SodA) (189) and copper/zinc-dependent enzyme (SodC) (190) and both contribute significantly to the virulence of the pathogen. In addition, the bacillus secretes KatG, which serves as a peroxidase and peroxynitritase, to metabolizes reactive radicals produced by the phagocyte oxidative burst (191). When the bacillus is preyed upon by the LAPosome, it secretes CpsA as an effector and inhibits the activity of NOX2 (192).
Gram-negative rods such as E. coli, Salmonella spp., and Klebsiella pneumoniae restore the host-inhibited function of their own siderophores, which are responsible for iron absorption, by producing a protein called iroA (193). The thick waxy cell walls of Mycobacterium spp. provide excellent protection against severe environmental and host invasion, but are not conducive to the exchange of nutrients and metabolites necessary for growth and survival with the outside world. Mycobacterium attempts to secure iron by producing mycobactin as a siderophore (194). The host interferes with mycobactin, as it did against siderospheres of gram-negative rods, but Mycobacterium secretes Esx-3 of the type VII secretion system (Esx-1-5) to support mycobactin and iron absorption (195). Aspergillus fumigatus produces HapX in iron deficiency to suppress iron-consuming pathways such as host heme synthesis and respiration, including the TCA circuit, and to increase the production of iron-absorbing siderophores (196).
Against bacterial infections, tremendous resources have been devoted to the development of antimicrobial agents that kill the bacteria themselves. Cancer therapy has long since moved beyond the days when drugs were developed to kill cancer cells themselves, and HDTs have become a major pillar of cancer treatment. The development of HDTs for bacterial infections has just begun, and the development of HDTs for the phagolysosome pathway, the first line of innate immunity, has lagged further behind. The molecular mechanisms of the phagolysosome and bacterial evasion strategies described thus far provide major clues to HDT. In the following part, we would like to describe the current status of drug discovery that intervenes in the phagolysosome pathway.
The application of immune checkpoint blockade to infectious diseases has been investigated in the context of the interrelationship between innate and adaptive immunity, rather than between infected cells and innate immunity. Interventions on the PD1-PD-L1 axis are effective in animal studies against infectious diseases such as malaria, toxoplasma, leishmania, and Listeria (197). The results of a phase 1/2 trial of nivolumab in sepsis have overcome safety concerns, including the development of autoimmune disease (198). In cancer therapy, although macrophages have been intensively studied as targets for intervention in various aspects, phagocytosis that macrophages execute is recognized as a promising drug discovery (199). The findings of the phagocytosis checkpoint may provide clues to the treatment of infectious diseases (53). The inhibition of the PD-1-PD-L1 axis as a phagocytosis checkpoint enhances phagocytosis in liver Kupffer cells and prevent bacterial infection (200). A study using CD47 KO mice also reported that E. coli pneumonia showed better recovery compared to wild type (201).
It was reported that monocytes from imatinib-treated patients with leukemia showed an increased production of V0a3 and V0c and had more acidic lysosomes. Sera from those patients, which are added to the cell culture of macrophages, enforced more acidic lysosomes and of M. tuberculosis (202). One compound was reported to promote regulated assembly of V-ATPase, KU-60119, which was identified as an ATM inhibitor that inhibited assembly of V1E and V1G through phosphorylation of the latter (119). ZLN005 was originally recognized as a peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) activator (203). By administering ZLN005 to the cecum perforation ligation (CPL) model of sepsis and analyzing intraperitoneal cells, ZLN005 was shown to be a transcription factor EB (TFEB) activator involved in lysosome biogenesis as well as a lysosomal acidifier (204). The compound significantly improved overall survival and drastically reduced intraperitoneal bacterial load in mice at only 2 h after administration of ZLN005 in in vivo sepsis model. These results indicate that lysosomal acidification is a therapeutic target for sepsis.
When considering the pathogenesis of sepsis (205), in most cases the organism of origin is not known at the onset of the disease. When the pathogen is unknown, the choice of treatment, especially antimicrobial agents, is highly dependent on the experience and ability of the clinician. In contrast, the existence of host-directed therapies that are independent of the organisms causing the disease can first reduce the bacterial load. Even if not eradication, significant improvement in survival may be achieved if circulatory collapse and multiorgan damage can be avoided by reducing the excessive bacterial load to a controllable level, which has been beyond the reach of current initial therapy. When addressing infections caused by resistant bacteria (206), countering bacterial interference with lysosomes, which play a crucial role in bacterial elimination, emerges as a significant alternative to traditional disinfection methods.
The disadvantage of augmenting the phagolysosomal system is that overactivation of autophagy is a concern, since the lysosomal system is the final point of autophagy, which forms the basis of cellular function (62). There is also concern that digestion in lysosomes may be problematic by causing energy depletion in the cell, as significant energy expenditure is thought to occur during digestion in lysosomes (20). These are all reasons to believe that if host-directed therapeutics were to be discovered, the method of administration would need to be carefully set up.
Bacterial infections are a major public health threat no less than malignancies, neurodegenerative diseases, and cardiovascular and metabolic diseases. The development of antibiotics with the goal of disinfection shines as the most significant achievement of 20th century medicine (2). Now, the development of new antibiotics against the emergence of resistant bacteria is taking a backseat to bacterial evolution. Anticancer therapy has seen a breakthrough with the establishment of host-directed therapy in parallel with the development of therapeutic agents aimed at eliminating cancer cells (207). Host immunity plays a major role in the pathogenesis of both cancer and infectious diseases (197). However, host-directed therapy for infectious diseases is still in its infancy (208). In both innate and adaptive immunity, the phagolysosomal system plays a central role in bacterial clearance against bacterial infections (22). Bacteria have evolved to achieve evasion of this phagolysosome system by any means necessary (64, 183). Due to rapid advances in antibiotic technology, the host has not yet evolved to acquire an effector mechanism to counter the evasion mechanism exhibited by resistant bacteria. On the other hand, detailed molecular biological analyses of this system have been performed (26, 101, 209), and the time is ripe for the development of drugs with a point of action in this system. Possible targets of action are numerous, including phagocytosis, phagosome maturation, fusion with lysosomes, lysosome acidification, and lysosome quality control. This system is a fundamental cellular system and is also heavily involved in neurodegenerative diseases (93) and embodies the development strategy of organelle drug discovery rather than the framework of disease-by-disease drug discovery. The development of host-directed therapeutics for bacterial infections has the potential to revolutionize the drug discovery system. It is hoped that the development of HDT together with a new class of antimicrobial agents, will work like two wheels on a cart, dramatically increasing the life-saving rate of sepsis and creating a treatment strategy that is not afraid of superbugs.
SG designed the concept of this review. SG and TT wrote and edited the manuscript and the table. FT prepared figures. All authors contributed to the article and approved the submitted version.
The authors declare that this review was supported by funding from Remiges Ventures, Inc. (Tokyo, Japan). The funder was not involved in the concept of this review; writing of this article; or decision to submit it for publication.
FT is an employee of Kowa Corporation, but Kowa Corporation is not involved in any aspect of this study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395:200–11. doi: 10.1016/s0140-6736(19)32989-7
2. Walesch S, Birkelbach J, Jezequel G, Haeckl FPJ, Hegemann JD, Hesterkamp T, et al. Fighting antibiotic resistance-strategies and (pre)clinical developments to find new antibacterials. EMBO Rep (2023) 24:e56033. doi: 10.15252/embr.202256033
3. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
4. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
5. McCulloch TR, Wells TJ, Souza-Fonseca-Guimaraes F. Towards efficient immunotherapy for bacterial infection. Trends Microbiol (2022) 30:158–69. doi: 10.1016/j.tim.2021.05.005
6. Liu D, Huang SY, Sun JH, Zhang HC, Cai QL, Gao C, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil Med Res (2022) 9:56. doi: 10.1186/s40779-022-00422-y
7. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
8. Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA (2011) 306:2614–5. doi: 10.1001/jama.2011.1853
9. Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. Jama (1994) 271:1836–43. doi: 10.1001/jama.1994.03510470040032
10. Clark MA, Plank LD, Connolly AB, Streat SJ, Hill AA, Gupta R, et al. Effect of a chimeric antibody to tumor necrosis factor-alpha on cytokine and physiologic responses in patients with severe sepsis–a randomized, clinical trial. Crit Care Med (1998) 26:1650–9. doi: 10.1097/00003246-199810000-00016
11. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208:2581–90. doi: 10.1084/jem.20111354
12. Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis (2012) 12:503–5. doi: 10.1016/s1473-3099(12)70136-6
13. Akram F, Imtiaz M, Haq IU. Emergent crisis of antibiotic resistance: A silent pandemic threat to 21(st) century. Microb Pathog (2023) 174:105923. doi: 10.1016/j.micpath.2022.105923
14. Organization, W. H. WHO consolidated guidelines on tuberculosis: tuberculosis preventive treatment. Geneva: World Health Organization (2020).
15. Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol (2018) 16:202–13. doi: 10.1038/nrmicro.2018.8
16. Ginsberg AM, Spigelman M. Challenges in tuberculosis drug research and development. Nat Med (2007) 13:290–4. doi: 10.1038/nm0307-290
17. Allue-Guardia A, Garcia JI, Torrelles JB. Evolution of drug-resistant mycobacterium tuberculosis strains and their adaptation to the human lung environment. Front Microbiol (2021) 12:612675. doi: 10.3389/fmicb.2021.612675
18. Theuretzbacher U, Piddock LJV. Non-traditional antibacterial therapeutic options and challenges. Cell Host Microbe (2019) 26:61–72. doi: 10.1016/j.chom.2019.06.004
19. Ghosh C, Sarkar P, Issa R, Haldar J. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol (2019) 27:323–38. doi: 10.1016/j.tim.2018.12.010
20. McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, et al. The metabolic basis of immune dysfunction following sepsis and trauma. Front Immunol (2020) 11:1043. doi: 10.3389/fimmu.2020.01043
21. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
23. Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell (2006) 124:767–82. doi: 10.1016/j.cell.2006.01.034
24. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6:291. doi: 10.1038/s41392-021-00687-0
25. Levin R, Grinstein S, Canton J. The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev (2016) 273:156–79. doi: 10.1111/imr.12439
26. Uribe-Querol E, Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol (2020) 11:1066. doi: 10.3389/fimmu.2020.01066
27. Chandra P, Grigsby SJ, Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol (2022) 20:750–66. doi: 10.1038/s41579-022-00763-4
28. Patel H, Rawat S. A genetic regulatory see-saw of biofilm and virulence in MRSA pathogenesis. Front Microbiol (2023) 14:1204428. doi: 10.3389/fmicb.2023.1204428
29. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside job: Staphylococcus aureus host-pathogen interactions. Int J Med Microbiol (2018) 308:607–24. doi: 10.1016/j.ijmm.2017.11.009
30. McClure EE, Chavez ASO, Shaw DK, Carlyon JA, Ganta RR, Noh SM, et al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol (2017) 15:544–58. doi: 10.1038/nrmicro.2017.59
31. Andrew AM. NANOMEDICINE, VOLUME 1: BASIC CAPABILITIES Vol. 18. Freitas RA Jr, editor. Robotica (2000) p. 687–9. Landes Bioscience, Austin, Texas: CRC Press, 1999.
32. Devosse T, Guillabert A, D’Haene N, Berton A, De Nadai P, Noel S, et al. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol (2009) 182:4974–84. doi: 10.4049/jimmunol.0803128
33. Bohdanowicz M, Schlam D, Hermansson M, Rizzuti D, Fairn GD, Ueyama T, et al. Phosphatidic acid is required for the constitutive ruffling and macropinocytosis of phagocytes. Mol Biol Cell (2013) 24:1700–12. doi: 10.1091/mbc.E12-11-0789
34. Canton J, Schlam D, Breuer C, Gutschow M, Glogauer M, Grinstein S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun (2016) 7:11284. doi: 10.1038/ncomms11284
35. Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol (2010) 11:237–51. doi: 10.1038/nrm2867
36. Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW. Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genomics (2008) 9:62. doi: 10.1186/1471-2164-9-62
37. Chuenchor W, Jin T, Ravilious G, Xiao TS. Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr Opin Immunol (2014) 26:14–20. doi: 10.1016/j.coi.2013.10.009
38. Pashenkov MV, Dagil YA, Pinegin BV. NOD1 and NOD2: Molecular targets in prevention and treatment of infectious diseases. Int Immunopharmacol (2018) 54:385–400. doi: 10.1016/j.intimp.2017.11.036
39. Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol (2015) 32:21–7. doi: 10.1016/j.coi.2014.12.002
40. Feng S, Chen T, Lei G, Hou F, Jiang J, Huang Q, et al. Absent in melanoma 2 inflammasome is required for host defence against Streptococcus pneumoniae infection. Innate Immun (2019) 25:412–9. doi: 10.1177/1753425919860252
41. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol (2014) 14:94–108. doi: 10.1038/nri3582
42. Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, et al. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J Exp Med (2000) 191:669–82. doi: 10.1084/jem.191.4.669
43. Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem (1999) 274:1240–7. doi: 10.1074/jbc.274.3.1240
44. Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J (1998) 17:414–22. doi: 10.1093/emboj/17.2.414
45. Fällman M, Lew DP, Stendahl O, Andersson T. Receptor-mediated phagocytosis in human neutrophils is associated with increased formation of inositol phosphates and diacylglycerol. Elevation in cytosolic free calcium and formation of inositol phosphates can be dissociated from accumulation of diacylglycerol. J Clin Invest (1989) 84:886–91. doi: 10.1172/jci114249
46. Hishikawa T, Cheung JY, Yelamarty RV, Knutson DW. Calcium transients during Fc receptor-mediated and nonspecific phagocytosis by murine peritoneal macrophages. J Cell Biol (1991) 115:59–66. doi: 10.1083/jcb.115.1.59
47. Bos JL, de Bruyn K, Enserink J, Kuiperij B, Rangarajan S, Rehmann H, et al. The role of Rap1 in integrin-mediated cell adhesion. Biochem Soc Trans (2003) 31:83–6. doi: 10.1042/bst0310083
48. Nauseef WM. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr Opin Immunol (2019) 60:130–40. doi: 10.1016/j.coi.2019.05.006
49. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
50. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
51. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
52. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
53. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
54. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature (1997) 386:181–6. doi: 10.1038/386181a0
55. Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol (2008) 180:989–1003. doi: 10.1083/jcb.200708043
56. Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell (2018) 33:547–62. doi: 10.1016/j.ccell.2018.03.012
57. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
58. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell (2019) 35:588–602.e510. doi: 10.1016/j.ccell.2019.02.009
59. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
60. Gunther J, Seyfert HM. The first line of defence: insights into mechanisms and relevance of phagocytosis in epithelial cells. Semin Immunopathol (2018) 40:555–65. doi: 10.1007/s00281-018-0701-1
61. Ribet D, Cossart P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect (2015) 17:173–83. doi: 10.1016/j.micinf.2015.01.004
62. Bah A, Vergne I. Macrophage autophagy and bacterial infections. Front Immunol (2017) 8:1483. doi: 10.3389/fimmu.2017.01483
63. Upadhyay S, Philips JA. LC3-associated phagocytosis: host defense and microbial response. Curr Opin Immunol (2019) 60:81–90. doi: 10.1016/j.coi.2019.04.012
64. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol (2009) 7:355–66. doi: 10.1038/nrmicro2128
65. Roberts RL, Barbieri MA, Ullrich J, Stahl PD. Dynamics of rab5 activation in endocytosis and phagocytosis. J Leukoc Biol (2000) 68:627–32. doi: 10.1189/jlb.68.5.627
66. Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol (2001) 155:19–25. doi: 10.1083/jcb.200107069
67. Lemmon MA. Phosphoinositide recognition domains. Traffic (2003) 4:201–13. doi: 10.1034/j.1600-0854.2004.00071.x
68. Lawe DC, Chawla A, Merithew E, Dumas J, Carrington W, Fogarty K, et al. Sequential roles for phosphatidylinositol 3-phosphate and Rab5 in tethering and fusion of early endosomes via their interaction with EEA1. J Biol Chem (2002) 277:8611–7. doi: 10.1074/jbc.M109239200
69. Simonsen A, Gaullier JM, D’Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. J Biol Chem (1999) 274:28857–60. doi: 10.1074/jbc.274.41.28857
70. McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell (1999) 98:377–86. doi: 10.1016/s0092-8674(00)81966-2
71. Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature (1999) 397:621–5. doi: 10.1038/17618
72. Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell (2010) 141:497–508. doi: 10.1016/j.cell.2010.03.011
73. Jeschke A, Zehethofer N, Lindner B, Krupp J, Schwudke D, Haneburger I, et al. Phosphatidylinositol 4-phosphate and phosphatidylinositol 3-phosphate regulate phagolysosome biogenesis. Proc Natl Acad Sci USA (2015) 112:4636–41. doi: 10.1073/pnas.1423456112
74. Bröcker C, Kuhlee A, Gatsogiannis C, Balderhaar HJ, Hönscher C, Engelbrecht-Vandré S, et al. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci USA (2012) 109:1991–6. doi: 10.1073/pnas.1117797109
75. Araki N. Role of microtubules and myosins in Fc gamma receptor-mediated phagocytosis. Front Biosci (2006) 11:1479–90. doi: 10.2741/1897
76. van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N, et al. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J Cell Sci (2013) 126:3462–74. doi: 10.1242/jcs.129270
77. Collins RF, Schreiber AD, Grinstein S, Trimble WS. Syntaxins 13 and 7 function at distinct steps during phagocytosis. J Immunol (2002) 169:3250–6. doi: 10.4049/jimmunol.169.6.3250
78. Stockinger W, Zhang SC, Trivedi V, Jarzylo LA, Shieh EC, Lane WS, et al. Differential requirements for actin polymerization, calmodulin, and Ca2+ define distinct stages of lysosome/phagosome targeting. Mol Biol Cell (2006) 17:1697–710. doi: 10.1091/mbc.e05-12-1140
79. Kissing S, Saftig P, Haas A. Vacuolar ATPase in phago(lyso)some biology. Int J Med Microbiol (2018) 308:58–67. doi: 10.1016/j.ijmm.2017.08.007
80. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci USA (2011) 108:19371–6. doi: 10.1073/pnas.1109201108
81. Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol (2011) 90:271–84. doi: 10.1189/jlb.0810457
82. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
83. Canton J. Phagosome maturation in polarized macrophages. J Leukoc Biol (2014) 96:729–38. doi: 10.1189/jlb.1MR0114-021R
84. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi: 10.1038/nri2448
85. Collins MP, Forgac M. Regulation of V-ATPase assembly in nutrient sensing and function of V-ATPases in breast cancer metastasis. Front Physiol (2018) 9:902. doi: 10.3389/fphys.2018.00902
86. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol (2007) 8:917–29. doi: 10.1038/nrm2272
87. Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol (2003) 4:202–12. doi: 10.1038/nrm1050
88. ten Broeke T, Wubbolts R, Stoorvogel W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol (2013) 5:a016873. doi: 10.1101/cshperspect.a016873
89. Farsi Z, Preobraschenski J, van den Bogaart G, Riedel D, Jahn R, Woehler A. Single-vesicle imaging reveals different transport mechanisms between glutamatergic and GABAergic vesicles. Science (2016) 351:981–4. doi: 10.1126/science.aad8142
90. Rhodes CJ, Lucas CA, Mutkoski RL, Orci L, Halban PA. Stimulation by ATP of proinsulin to insulin conversion in isolated rat pancreatic islet secretory granules. Association with the ATP-dependent proton pump. J Biol Chem (1987) 262:10712–7. doi: 10.1016/S0021-9258(18)61022-1
91. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science (2011) 334:678–83. doi: 10.1126/science.1207056
92. Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol (2015) 77:57–80. doi: 10.1146/annurev-physiol-021014-071649
93. Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nat Neurosci (2022) 25:688–701. doi: 10.1038/s41593-022-01084-8
94. Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev (2016) 32:75–88. doi: 10.1016/j.arr.2016.05.004
95. Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science (2010) 327:459–63. doi: 10.1126/science.1179802
96. Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development (2010) 137:1825–32. doi: 10.1242/dev.045484
97. Kane PM. Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J Biol Chem (1995) 270:17025–32. doi: 10.1016/s0021-9258(17)46944-4
98. Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol Cell Biol (2005) 25:575–89. doi: 10.1128/MCB.25.2.575-589.2005
99. McGuire CM, Forgac M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J Biol Chem (2018) 293:9113–23. doi: 10.1074/jbc.RA117.001327
100. Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab (2014) 20:526–40. doi: 10.1016/j.cmet.2014.06.014
101. Stransky L, Cotter K, Forgac M. The function of V-ATPases in cancer. Physiol Rev (2016) 96:1071–91. doi: 10.1152/physrev.00035.2015
102. Liberman R, Bond S, Shainheit MG, Stadecker MJ, Forgac M. Regulated assembly of vacuolar ATPase is increased during cluster disruption-induced maturation of dendritic cells through a phosphatidylinositol 3-kinase/mTOR-dependent pathway. J Biol Chem (2014) 289:1355–63. doi: 10.1074/jbc.M113.524561
103. Stransky LA, Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem (2015) 290:27360–9. doi: 10.1074/jbc.M115.659128
104. Collins MP, Stransky LA, Forgac M. AKT Ser/Thr kinase increases V-ATPase-dependent lysosomal acidification in response to amino acid starvation in mammalian cells. J Biol Chem (2020) 295:9433–44. doi: 10.1074/jbc.RA120.013223
105. Soliman M, Seo JY, Kim DS, Kim JY, Park JG, Alfajaro MM, et al. Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PloS Pathog (2018) 14:e1006820. doi: 10.1371/journal.ppat.1006820
106. Xu Y, Parmar A, Roux E, Balbis A, Dumas V, Chevalier S, et al. Epidermal growth factor-induced vacuolar (H+)-atpase assembly: a role in signaling via mTORC1 activation. J Biol Chem (2012) 287:26409–22. doi: 10.1074/jbc.M112.352229
107. Jaskolka MC, Winkley SR, Kane PM. RAVE and rabconnectin-3 complexes as signal dependent regulators of organelle acidification. Front Cell Dev Biol (2021) 9:698190. doi: 10.3389/fcell.2021.698190
108. Kawabe H, Sakisaka T, Yasumi M, Shingai T, Izumi G, Nagano F, et al. A novel rabconnectin-3-binding protein that directly binds a GDP/GTP exchange protein for Rab3A small G protein implicated in Ca(2+)-dependent exocytosis of neurotransmitter. Genes Cells (2003) 8:537–46. doi: 10.1046/j.1365-2443.2003.00655.x
109. Kraemer C, Enklaar T, Zabel B, Schmidt ER. Mapping and structure of DMXL1, a human homologue of the DmX gene from Drosophila melanogaster coding for a WD repeat protein. Genomics (2000) 64:97–101. doi: 10.1006/geno.1999.6050
110. Seol JH, Shevchenko A, Shevchenko A, Deshaies RJ. Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat Cell Biol (2001) 3:384–91. doi: 10.1038/35070067
111. Jaskolka MC, Kane PM. Interaction between the yeast RAVE complex and Vph1-containing V(o) sectors is a central glucose-sensitive interaction required for V-ATPase reassembly. J Biol Chem (2020) 295:2259–69. doi: 10.1074/jbc.RA119.011522
112. Merkulova M, Paunescu TG, Azroyan A, Marshansky V, Breton S, Brown D. Mapping the H(+) (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci Rep (2015) 5:14827. doi: 10.1038/srep14827
113. Li B, Clohisey SM, Chia BS, Wang B, Cui A, Eisenhaure T, et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat Commun (2020) 11:164. doi: 10.1038/s41467-019-13965-x
114. Gandini MA, Souza IA, Fan J, Li K, Wang D, Zamponi GW. Interactions of Rabconnectin-3 with Cav2 calcium channels. Mol Brain (2019) 12:62. doi: 10.1186/s13041-019-0483-y
115. Crummy E, Mani M, Thellman JC, Martin TFJ. The priming factor CAPS1 regulates dense-core vesicle acidification by interacting with rabconnectin3beta/WDR7 in neuroendocrine cells. J Biol Chem (2019) 294:9402–15. doi: 10.1074/jbc.RA119.007504
116. Ratto E, Chowdhury SR, Siefert NS, Schneider M, Wittmann M, Helm D, et al. Direct control of lysosomal catabolic activity by mTORC1 through regulation of V-ATPase assembly. Nat Commun (2022) 13:4848. doi: 10.1038/s41467-022-32515-6
117. Abe Y, Yoon SO, Kubota K, Mendoza MC, Gygi SP, Blenis J. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J Biol Chem (2009) 284:14939–48. doi: 10.1074/jbc.M900097200
118. Wang R, Qin Y, Xie XS, Li X. Molecular basis of mEAK7-mediated human V-ATPase regulation. Nat Commun (2022) 13:3272. doi: 10.1038/s41467-022-30899-z
119. Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol (2017) 13:616–23. doi: 10.1038/nchembio.2342
120. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev (2016) 273:180–93. doi: 10.1111/imr.12447
121. Okochi Y, Sasaki M, Iwasaki H, Okamura Y. Voltage-gated proton channel is expressed on phagosomes. Biochem Biophys Res Commun (2009) 382:274–9. doi: 10.1016/j.bbrc.2009.03.036
122. DeCoursey TE. The intimate and controversial relationship between voltage-gated proton channels and the phagocyte NADPH oxidase. Immunol Rev (2016) 273:194–218. doi: 10.1111/imr.12437
123. Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller AF, Teixeira M, et al. Superoxide dismutases and superoxide reductases. Chem Rev (2014) 114:3854–918. doi: 10.1021/cr4005296
124. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA (2000) 97:8841–8. doi: 10.1073/pnas.97.16.8841
125. Abuaita BH, Schultz TL, O’Riordan MX. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized staphylococcus aureus. Cell Host Microbe (2018) 24:625–636.e625. doi: 10.1016/j.chom.2018.10.005
126. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol (2004) 2:820–32. doi: 10.1038/nrmicro1004
127. Nathan C, Xie QW. Regulation of biosynthesis of nitric oxide. J Biol Chem (1994) 269:13725–8. doi: 10.1016/s0021-9258(17)36703-0
128. Webb JL, Harvey MW, Holden DW, Evans TJ. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect Immun (2001) 69:6391–400. doi: 10.1128/iai.69.10.6391-6400.2001
129. Davis AS, Vergne I, Master SS, Kyei GB, Chua J, Deretic V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PloS Pathog (2007) 3:e186. doi: 10.1371/journal.ppat.0030186
130. Kaplan CD, Kaplan J. Iron acquisition and transcriptional regulation. Chem Rev (2009) 109:4536–52. doi: 10.1021/cr9001676
131. Ganz T. Iron in innate immunity: starve the invaders. Curr Opin Immunol (2009) 21:63–7. doi: 10.1016/j.coi.2009.01.011
132. Gallin JI, Zarember K. Lessons about the pathogenesis and management of aspergillosis from studies in chronic granulomatous disease. Trans Am Clin Climatol Assoc (2007) 118:175–85.
133. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature (2004) 432:917–21. doi: 10.1038/nature03104
134. Saiga H, Nishimura J, Kuwata H, Okuyama M, Matsumoto S, Sato S, et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J Immunol (2008) 181:8521–7. doi: 10.4049/jimmunol.181.12.8521
135. Cellier MF, Courville P, Campion C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect (2007) 9:1662–70. doi: 10.1016/j.micinf.2007.09.006
136. Pandey S, Kant S, Khawary M, Tripathi D. Macrophages in microbial pathogenesis: commonalities of defense evasion mechanisms. Infect Immun (2022) 90:e0029121. doi: 10.1128/IAI.00291-21
137. Kawasaki K, Ernst RK, Miller SI. Deacylation and palmitoylation of lipid A by Salmonellae outer membrane enzymes modulate host signaling through Toll-like receptor 4. J Endotoxin Res (2004) 10:439–44. doi: 10.1179/096805104225006264
138. Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PloS Pathog (2011) 7:e1002454. doi: 10.1371/journal.ppat.1002454
139. Kilian M, Mestecky J, Schrohenloher RE. Pathogenic species of the genus haemophilus and streptococcus pneumoniae produce immunoglobulin A1 protease. Infection Immun (1979) 26:143–9. doi: 10.1128/iai.26.1.143-149.1979
140. Blake MS, Swanson J. Studies on gonococcus infection. XVI. Purification of Neisseria gonorrhoeae immunoglobulin A1 protease. Infection Immun (1978) 22:350–8. doi: 10.1128/iai.22.2.350-358.1978
141. Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature (2009) 458:890–3. doi: 10.1038/nature07769
142. Sharp JA, Echague CG, Hair PS, Ward MD, Nyalwidhe JO, Geoghegan JA, et al. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PloS One (2012) 7:e38407. doi: 10.1371/journal.pone.0038407
143. Andersson K, Carballeira N, Magnusson K-E, Persson C, Stendahl O, Wolf-Watz H, et al. YopH of Yersinia pseudotuberculosis interrupts early phosphotyrosine signalling associated with phagocytosis. Mol Microbiol (1996) 20:1057–69. doi: 10.1111/j.1365-2958.1996.tb02546.x
144. Broberg CA, Zhang L, Gonzalez H, Laskowski-Arce MA, Orth K. A vibrio effector protein is an inositol phosphatase and disrupts host cell membrane integrity. Science (2010) 329:1660–2. doi: 10.1126/science.1192850
145. Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, et al. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J (2002) 21:5069–78. doi: 10.1093/emboj/cdf522
146. Cameron CM, Barrett JW, Mann M, Lucas A, McFadden G. Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation. vivo. Virol (2005) 337:55–67. doi: 10.1016/j.virol.2005.03.037
147. Tal MC, Torrez Dulgeroff LB, Myers L, Cham LB, Mayer-Barber KD, Bohrer AC, et al. Upregulation of CD47 is a host checkpoint response to pathogen recognition. mBio (2020) 11:e01293-20. doi: 10.1128/mBio.01293-20
148. Anand I, Choi W, Isberg RR. The vacuole guard hypothesis: how intravacuolar pathogens fight to maintain the integrity of their beloved home. Curr Opin Microbiol (2020) 54:51–8. doi: 10.1016/j.mib.2020.01.008
149. Bagcchi S. WHO’s global tuberculosis report 2022. Lancet Microbe (2023) 4:e20. doi: 10.1016/s2666-5247(22)00359-7
150. Upadhyay S, Mittal E, Philips JA. Tuberculosis and the art of macrophage manipulation. Pathog Dis (2018) 76. doi: 10.1093/femspd/fty037
151. Augenstreich J, Briken V. Host cell targets of released lipid and secreted protein effectors of mycobacterium tuberculosis. Front Cell Infect Microbiol (2020) 10:595029. doi: 10.3389/fcimb.2020.595029
152. Gioseffi A, Edelmann MJ, Kima PE. Intravacuolar pathogens hijack host extracellular vesicle biogenesis to secrete virulence factors. Front Immunol (2021) 12:662944. doi: 10.3389/fimmu.2021.662944
153. Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PloS One (2010) 5:e8769. doi: 10.1371/journal.pone.0008769
154. Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA (2005) 102:4033–8. doi: 10.1073/pnas.0409716102
155. Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci USA (2003) 100:5437–42. doi: 10.1073/pnas.0737613100
156. Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell (2004) 15:751–60. doi: 10.1091/mbc.e03-05-0307
157. Indrigo J, Hunter RL, Actor JK. Cord factor trehalose 6,6’-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiol (Reading) (2003) 149:2049–59. doi: 10.1099/mic.0.26226-0
158. Passemar C, Arbués A, Malaga W, Mercier I, Moreau F, Lepourry L, et al. Multiple deletions in the polyketide synthase gene repertoire of Mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host-pathogen interactions. Cell Microbiol (2014) 16:195–213. doi: 10.1111/cmi.12214
159. Vergne I, Chua J, Deretic V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med (2003) 198:653–9. doi: 10.1084/jem.20030527
160. Connor MG, Pulsifer AR, Chung D, Rouchka EC, Ceresa BK, Lawrenz MB. Yersinia pestis Targets the Host Endosome Recycling Pathway during the Biogenesis of the Yersinia-Containing Vacuole To Avoid Killing by Macrophages. mBio (2018) 9:e01800-17. doi: 10.1128/mBio.01800-17
161. Connor MG, Pulsifer AR, Price CT, Abu Kwaik Y, Lawrenz MB. Yersinia pestis requires host rab1b for survival in macrophages. PloS Pathog (2015) 11:e1005241. doi: 10.1371/journal.ppat.1005241
162. Isberg RR, O’Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol (2009) 7:13–24. doi: 10.1038/nrmicro1967
163. Ensminger AW. Legionella pneumophila, armed to the hilt: justifying the largest arsenal of effectors in the bacterial world. Curr Opin Microbiol (2016) 29:74–80. doi: 10.1016/j.mib.2015.11.002
164. Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, et al. Legionella effectors that promote nonlytic release from protozoa. Science (2004) 303:1358–61. doi: 10.1126/science.1094226
165. Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature (2007) 450:365–9. doi: 10.1038/nature06336
166. Kim KJ, Elliott SJ, Di Cello F, Stins MF, Kim KS. The K1 capsule modulates trafficking of E. coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell Microbiol (2003) 5:245–52. doi: 10.1046/j.1462-5822.2003.t01-1-00271.x
167. Hernandez LD, Hueffer K, Wenk MR, Galan JE. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science (2004) 304:1805–7. doi: 10.1126/science.1098188
168. Haas A. The phagosome: compartment with a license to kill. Traffic (2007) 8:311–30. doi: 10.1111/j.1600-0854.2006.00531.x
169. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science (1994) 263:678–81. doi: 10.1126/science.8303277
170. Buter J, Cheng TY, Ghanem M, Grootemaat AE, Raman S, Feng X, et al. Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat Chem Biol (2019) 15:889–99. doi: 10.1038/s41589-019-0336-0
171. Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. Acid-adaptive genes of Helicobacter pylori. Infect Immun (2003) 71:5921–39. doi: 10.1128/IAI.71.10.5921-5939.2003
172. Danhof HA, Lorenz MC. The candida albicans ATO gene family promotes neutralization of the macrophage phagolysosome. Infect Immun (2015) 83:4416–26. doi: 10.1128/IAI.00984-15
173. Kasper L, Seider K, Gerwien F, Allert S, Brunke S, Schwarzmuller T, et al. Identification of Candida glabrata genes involved in pH modulation and modification of the phagosomal environment in macrophages. PloS One (2014) 9:e96015. doi: 10.1371/journal.pone.0096015
174. Clemens DL, Lee BY, Horwitz MA. Purification, characterization, and genetic analysis of Mycobacterium tuberculosis urease, a potentially critical determinant of host-pathogen interaction. J Bacteriol (1995) 177:5644–52. doi: 10.1128/jb.177.19.5644-5652.1995
175. Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J Clin Invest (2005) 115:2472–9. doi: 10.1172/JCI24617
176. Fernandez-Mora E, Polidori M, Luhrmann A, Schaible UE, Haas A. Maturation of Rhodococcus equi-containing vacuoles is arrested after completion of the early endosome stage. Traffic (2005) 6:635–53. doi: 10.1111/j.1600-0854.2005.00304.x
177. von Bargen K, Scraba M, Krämer I, Ketterer M, Nehls C, Krokowski S, et al. Virulence-associated protein A from Rhodococcus equi is an intercompartmental pH-neutralising virulence factor. Cell Microbiol (2019) 21:e12958. doi: 10.1111/cmi.12958
178. Strasser JE, Newman SL, Ciraolo GM, Morris RE, Howell ML, Dean GE. Regulation of the macrophage vacuolar ATPase and phagosome-lysosome fusion by Histoplasma capsulatum. J Immunol (1999) 162:6148–54. doi: 10.4049/jimmunol.162.10.6148
179. Tsukano H, Kura F, Inoue S, Sato S, Izumiya H, Yasuda T, et al. Yersinia pseudotuberculosis blocks the phagosomal acidification of B10.A mouse macrophages through the inhibition of vacuolar H(+)-ATPase activity. Microb Pathog (1999) 27:253–63. doi: 10.1006/mpat.1999.0303
180. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PloS Pathog (2010) 6:e1000822. doi: 10.1371/journal.ppat.1000822
181. Zhao J, Beyrakhova K, Liu Y, Alvarez CP, Bueler SA, Xu L, et al. Molecular basis for the binding and modulation of V-ATPase by a bacterial effector protein. PloS Pathog (2017) 13:e1006394. doi: 10.1371/journal.ppat.1006394
182. Kong F, Young L, Chen Y, Ran H, Meyers M, Joseph P, et al. Pseudomonas aeruginosa pyocyanin inactivates lung epithelial vacuolar ATPase-dependent cystic fibrosis transmembrane conductance regulator expression and localization. Cell Microbiol (2006) 8:1121–33. doi: 10.1111/j.1462-5822.2006.00696.x
183. Smith LM, May RC. Mechanisms of microbial escape from phagocyte killing. Biochem Soc Trans (2013) 41:475–90. doi: 10.1042/BST20130014
184. Sibley LD, Weidner E, Krahenbuhl JL. Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature (1985) 315:416–9. doi: 10.1038/315416a0
185. Sreelatha A, Bennett TL, Zheng H, Jiang QX, Orth K, Starai VJ. Vibrio effector protein, VopQ, forms a lysosomal gated channel that disrupts host ion homeostasis and autophagic flux. Proc Natl Acad Sci USA (2013) 110:11559–64. doi: 10.1073/pnas.1307032110
186. Gallois A, Klein JR, Allen LA, Jones BD, Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J Immunol (2001) 166:5741–8. doi: 10.4049/jimmunol.166.9.5741
187. Lam GY, Fattouh R, Muise AM, Grinstein S, Higgins DE, Brumell JH. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxidase to promote Listeria monocytogenes infection. Cell Host Microbe (2011) 10:627–34. doi: 10.1016/j.chom.2011.11.005
188. McCaffrey RL, Schwartz JT, Lindemann SR, Moreland JG, Buchan BW, Jones BD, et al. Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J Leukoc Biol (2010) 88:791–805. doi: 10.1189/jlb.1209811
189. Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeStefano MS, Tham KT, et al. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med (2001) 164:2213–9. doi: 10.1164/ajrccm.164.12.2106093
190. Dussurget O, Stewart G, Neyrolles O, Pescher P, Young D, Marchal G. Role of Mycobacterium tuberculosis copper-zinc superoxide dismutase. Infect Immun (2001) 69:529–33. doi: 10.1128/iai.69.1.529-533.2001
191. Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol (2004) 52:1291–302. doi: 10.1111/j.1365-2958.2004.04078.x
192. Koster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci USA (2017) 114:E8711–20. doi: 10.1073/pnas.1707792114
193. Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, et al. The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proc Natl Acad Sci USA (2006) 103:16502–7. doi: 10.1073/pnas.0604636103
194. Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol (2000) 54:881–941. doi: 10.1146/annurev.micro.54.1.881
195. Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, Siddiqi N, et al. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA (2009) 106:18792–7. doi: 10.1073/pnas.0900589106
196. Schrettl M, Beckmann N, Varga J, Heinekamp T, Jacobsen ID, Jochl C, et al. HapX-mediated adaption to iron starvation is crucial for virulence of Aspergillus fumigatus. PloS Pathog (2010) 6:e1001124. doi: 10.1371/journal.ppat.1001124
197. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol (2018) 18:91–104. doi: 10.1038/nri.2017.112
198. Watanabe E, Nishida O, Kakihana Y, Odani M, Okamura T, Harada T, et al. Pharmacokinetics, pharmacodynamics, and safety of nivolumab in patients with sepsis-induced immunosuppression: A multicenter, open-label phase 1/2 study. Shock (2020) 53:686–94. doi: 10.1097/shk.0000000000001443
199. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
200. Triantafyllou E, Gudd CL, Mawhin MA, Husbyn HC, Trovato FM, Siggins MK, et al. PD-1 blockade improves Kupffer cell bacterial clearance in acute liver injury. J Clin Invest (2021) 131:e140196. doi: 10.1172/JCI140196
201. Su X, Johansen M, Looney MR, Brown EJ, Matthay MA. CD47 deficiency protects mice from lipopolysaccharide-induced acute lung injury and Escherichia coli pneumonia. J Immunol (2008) 180:6947–53. doi: 10.4049/jimmunol.180.10.6947
202. Bruns H, Stegelmann F, Fabri M, Dohner K, van Zandbergen G, Wagner M, et al. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J Immunol (2012) 189:4069–78. doi: 10.4049/jimmunol.1201538
203. Zhang LN, Zhou HY, Fu YY, Li YY, Wu F, Gu M, et al. Novel small-molecule PGC-1α transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes (2013) 62:1297–307. doi: 10.2337/db12-0703
204. Suzuki Y, Kami D, Taya T, Sano A, Ogata T, Matoba S, et al. ZLN005 improves the survival of polymicrobial sepsis by increasing the bacterial killing via inducing lysosomal acidification and biogenesis in phagocytes. Front Immunol (2023) 14:1089905. doi: 10.3389/fimmu.2023.1089905
205. Vincent JL. Current sepsis therapeutics. EBioMedicine (2022) 86:104318. doi: 10.1016/j.ebiom.2022.104318
206. Moo CL, Yang SK, Yusoff K, Ajat M, Thomas W, Abushelaibi A, et al. Mechanisms of antimicrobial resistance (AMR) and alternative approaches to overcome AMR. Curr Drug Discovery Technol (2020) 17:430–47. doi: 10.2174/1570163816666190304122219
207. Chamoto K, Yaguchi T, Tajima M, Honjo T. Insights from a 30-year journey: function, regulation and therapeutic modulation of PD1. Nat Rev Immunol (2023). doi: 10.1038/s41577-023-00867-9
208. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discovery (2018) 17:35–56. doi: 10.1038/nrd.2017.162
Keywords: bacterial infection, immune evasion, host-directed therapy, sepsis, antimicrobial resistance, phagocytosis, lysosome, V-ATPase
Citation: Taya T, Teruyama F and Gojo S (2023) Host-directed therapy for bacterial infections -Modulation of the phagolysosome pathway-. Front. Immunol. 14:1227467. doi: 10.3389/fimmu.2023.1227467
Received: 23 May 2023; Accepted: 11 September 2023;
Published: 29 September 2023.
Edited by:
Nathella Pavan Kumar, National Institute of Research in Tuberculosis (ICMR), IndiaReviewed by:
Nadia Berkova, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceCopyright © 2023 Taya, Teruyama and Gojo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Gojo, Z29qb3NAa290by5rcHUtbS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.