 Jie Xing
Jie Xing Changfeng Man
Changfeng Man Yingzhao Liu
Yingzhao Liu Zhengdong Zhang
Zhengdong Zhang Huiyong Peng
Huiyong Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 August 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1224269
This article is part of the Research Topic Metabolism Regulation of T Cell Mediated-Tumor Immunity and Immunotherapy View all 5 articles
Tumor development is closely associated with a complex tumor microenvironment, which is composed of tumor cells, blood vessels, tumor stromal cells, infiltrating immune cells, and associated effector molecules. T helper type 17 (Th17) cells, which are a subset of CD4+ T cells and are renowned for their ability to combat bacterial and fungal infections and mediate inflammatory responses, exhibit context-dependent effector functions. Within the tumor microenvironment, different molecular signals regulate the proliferation, differentiation, metabolic reprogramming, and phenotypic conversion of Th17 cells. Consequently, Th17 cells exert dual effects on tumor progression and can promote or inhibit tumor growth. This review aimed to investigate the impact of various alterations in the tumor microenvironment on the antitumor and protumor effects of Th17 cells to provide valuable clues for the exploration of additional tumor immunotherapy strategies.
Malignant neoplasms represent a critical affliction in many nations and are characterized by high prevalence and mortality rates. Statistically, in over 100 countries, including China, the United States, the United Kingdom, Argentina, Spain, and South Korea, the proportion of deaths caused by cancer among noncommunicable diseases is 23.3%. As of 2020, the leading types of cancer in terms of incidence rates are breast cancer (11.7%), lung cancer (11.4%), prostate cancer (7.3%), nonmelanoma of the skin (6.2%), colon cancer (6.0%), and stomach cancer (5.6%) (1, 2). The increase in tumor incidence can be attributed to multifaceted pathogenesis, including the establishment of a tumor microenvironment that serves as a vital habitat for tumor sustenance (3–5). The tumor microenvironment is composed of diverse constituents, including neoplastic cells, vascular structures, stromal cells, infiltrating immunocytes, and assorted secreted effector molecules (6, 7). The inherent uniqueness of tumors provides novel therapeutic approaches for cancer treatment beyond established interventions such as surgical resection and chemotherapy. These approaches include tumor immunotherapy, such as chimeric antigen receptor T-cell therapy, cytokine therapy and miRNA drugs (8–10). However, neither T cell transfer therapy nor immune checkpoint therapy is universal, and they are only effective against a limited number of tumors (11, 12).
In order to address the limitations of these treatments and explore novel therapeutic approaches, extensive and in-depth research has been conducted on the intricate relationship between the tumor microenvironment and immune cells. This endeavor has even given rise to a new conceptual framework known as the ‘tumor immune microenvironment’, which is aimed at elucidating the role of immune cells and their effector molecules within the context of the tumor microenvironment (13). Immune cells within the tumor microenvironment play more than a singular role of promoting or inhibiting tumor progression. They display a contextual responsiveness, which is characterized as resistance or promotion based on dynamic changes in their surroundings. This effect transcends temporal and spatial limitations, meaning that these immune cells can manipulate tumors to vary degrees across different stages or locations within the same disease. In other words, these immune cells possess spatiotemporal adaptations (14). For instance, macrophages exhibit an antitumor M1 phenotype when exposed to high glucose and IFN-γ levels, whereas these cells acquire a protumor M2 phenotype in high lactate environments and in the presence of Colony-stimulating factor 1 (CSF1) (15–17). The polarization of neutrophils associated with tumors is influenced by the presence of transforming growth factor-β (TGF-β) in the local milieu (18, 19). Th17 cells follow a similar pattern, although they lack the clear functional categorization observed in the aforementioned cell types (20, 21).
Th17 cells are a type of CD4+ T cell that produces IL-17 and whose development is mainly regulated by transcription factors such as RORγt and STAT3, as well as various cytokines such as IL-6, IL-1β, IL-21, IL-23, and TGF-β. IL-6 and TGF-β induce RORγt transcription in CD4+ T cells, promoting the expression of IL-17 and IL-17F in Th17 cells. RORγt+ T cells act with IL-6 to upregulate IL-23R expression, facilitating the response to IL-23. This contributes to the maintenance, expansion, or further differentiation of Th17 cells. The combined signaling of TGF-β with IL-6/STAT3 and IL-21/STAT3 drives the differentiation of Th17 cells by promoting the expression of the transcription factors RORγt and RORα. STAT3 also directly binds and transactivates the promoters of IL-17 and IL-21, facilitating their expression (22–28).
Th17 cells have been shown to play an important role in various autoimmune diseases (29–31). In recent years, due to increasing research on tumors and inflammation, tumor microenvironments, and immune cells, the role of Th17 cells in tumors has been further explored. Through various studies on their role in tumors, the dual nature of Th17 cells in tumors cannot be ignored. Many scholars have suggested why Th17 cells promote or inhibit tumor growth and development, such as by promoting tumor angiogenesis to provide a rich blood supply to tumors, thereby promoting tumors, which occurs in melanoma, bladder cancer, hepatocellular carcinoma, and colorectal cancer (11, 20, 32). However, there is also evidence that Th17 cells can play an antitumor role by recruiting Cytotoxic T lymphocytes (CTLs) and Natural killer (NK) cells, such as in ovarian cancer and lung cancer (32). Most research has focuses on how downstream effectors of Th17 cells regulate tumor development. How upstream molecules affect Th17 cells in tumor microenvironments is still up for debate. Therefore, this article will focus on how the tumor microenvironment affects the dual function of Th17 cells.
After being stimulated by IL-6, IL-21, TGF-β, and other factors, naive CD4+ T cells are activated, leading to the differentiation of Th17 cells through the activation of STAT3 by RORγt (33, 34). These cells play a crucial role in host defense against pathogens and inflammatory responses (35, 36).Additionally, they are involved in the development of autoimmune diseases and cancer (37, 38). Th17 cells exert different functional effects depending on the environmental conditions and the type of cytokine stimulation they receive. TGF-β and IL-6 promote the generation of nonpathogenic Th17 cells from naive CD4+ T cells, mainly due to TGF-β mediated inhibition of T-bet expression. At this stage, Th17 cells can secrete IL-9 and IL-10, and IL-10 exerts antagonistic effects on Th17 cells pathogenicity during infection or inflammation (39, 40). Additionally, research has shown that TGF-β derived from Th17 cells play an important role in maintaining Th17 cells stability and blocking their pathogenic transformation by downregulating IL-12 and IL-27 expression (41). TGF-β can also inhibit the differentiation of pathogenic Th17 cells by suppressing the extracellular-signal-regulated kinase (ERK) pathway, whereas the TGF-β superfamily cytokine Activin-A can activate the ERK pathway through Activin-A receptor ALK4 to promote the differentiation of pathogenic Th17 cells (42). IL-23 and IL-1β can also stimulate Th17 cells differentiation, but since immature T cells do not express IL-1R and IL-23R, their effects mainly occur after Th17 cells differentiation begins (43, 44). Furthermore, IL-1β inhibits the production of IL-10, indicating that Th17 cells at this stage tend to promote disease development (45). Finally, IL-23 participates in Th17 cells pathogenicity (43). The transcription factors involved in the differentiation and functional regulation of Th17 cells are numerous. Among these, the most critical factors are STAT3 and RORγt. STAT3 is activated in response to stimulation by the JAK family kinases and requires subsequent activation of RORγt to drive Th17 cells differentiation (46). RORα shares gene binding sites with RORγt, and in the presence of the latter, it can cooperatively bind to RORE or DNA to enhance the inflammatory effects of Th17 cells (47). The presence of basic leucine zipper transcription factor ATF-like (BATF) promotes the expression of STAT3 while inhibiting IL-2-dependent STAT5, thereby inhibiting the competitive binding between STAT3 and STAT5. This disruption inhibits the differentiation of T cells towards Th1 cells and Treg cells subsets and facilitates the early differentiation of Th17 cells, which is associated with the expression of IL-17, IL-21, and IL-22 (48–50). Interferon regulatory factor 4 (IRF4) synergistically upregulates RORγt and downregulates FOXP3 in collaboration with STAT3, thereby promoting the differentiation of Th17 cells. The presence of IL-6 intensifies the aforementioned response and facilitates the expression of IL-17 and IL-21 (51, 52). The expression of aryl hydrocarbon receptor (AHR) is upregulated in Th17 cells, particularly in pathogenic Th17 cells, and the levels are sustained and stable. The upregulation of AHR is associated with the presence of TGF-β1 and IL-6, which facilitates the secretion of IL-17A and IL-22 by Th17 cells (53). The transcription factors T-bet and E26 transformation specific-1 (ETS-1) positively regulate Th1 cells and Th2 cells, respectively, and can inhibit Th17 cells differentiation. In the presence of IL-23, T-bet negatively modulates IL-17 secretion by Th17 cells. Moreover, ETS-1 negatively regulates Th17 cells by activating STAT5 in an IL-2-dependent manner (49, 54, 55). The transcription factor growth factor-independent 1 transcriptional repressor (GFI1), which is associated with the Th2 cells phenotype, act as a negative regulator of Th17 cells. IL-4 is required to inhibit the expression of RORγt and IL-17, but this inhibitory effect can be disrupted by TGF-β (56). Similarly, the transcription factor FOXP3, which is involved in Treg cells regulation, can suppress Th17 cells differentiation by inhibiting RORγt in the presence of TGF-β (57).To efficiently reach the site of inflammation, Th17 cells share transport receptors with Th1 cells, Th2 cells, and Treg cells. They share the receptors CCR4, CCR5, CCR6, CXCR3, and CXCR6 with Treg cells, while they share the receptors CCR2, CCR5, CCR7, CXCR3, and CXCR6 with Th1 cells. Th2 cells share the receptors CCR4 and CCR7 (58).This receptor sharing not only improves the migration of Th17 cells but can potentially define a distinct subgroup of Th17 cells. These Th17 cells express CCR6, produce CCL20, contribute to the recruitment of Th17 cells, are closely associate with IL-17 production, and participate in the inflammatory microenvironment associated with Th17 cells responses. These receptors may serve as pathogenic molecular markers of Th17 cells and are related to FOXP3+Th17 cells (31, 59).Notably, Th17 cells expressing CCR2 also express T-bet and RUNX family transcription factor 1 (RUNX1) and can secrete IFN-γ and GM-CSF, which are implicated in the pathogenicity of Th17 cells (60, 61).Th17 cells and neutrophils can mutually recruit each other. Neutrophils chemotactically attract Th17 cells through CCL2 and CCL20, while Th17 cells chemotactically attract neutrophils by producing and releasing CXCL8 (62).The effector molecules involved in the function of Th17 cells are IL-17A, IL-17F, IL-21, IL-22, and GM-CSF. Among these, IL-17A and IL-17F primarily contribute to host defense against bacteria and participate in mucosal defense. IL-17A can inhibit the pathogenicity of Th17 cells by inducing NF-κB activation and IL-24 production (63–65). IL-21 promotes Th17 cells expansion through self-amplification by stimulating RORγt and inhibits the expression of FOXP3 (66, 67) (68), while IL-22 is involved in tissue repair and resistance to pathogens (69). In addition to genetic factors, cellular metabolism influences the pathogenicity of Th17 cells. Th17 cells promote disease progression under conditions of active glycolysis and high tricarboxylic acid (TCA) activity, while protective Th17 cells seem to rely on fatty acid oxidation for energy (70). However, Wang et al. also discovered that when CD5L, which is a member of the scavenger receptor cysteine-rich superfamily, is inhibited by IL-23 in Th17 cells, it can alter lipid metabolism pathways and subsequently affect the downregulation of RORγt expression, leading to the generation of pathogenic Th17 cells (71). Furthermore, the active polyamine metabolism pathway promotes the generation of FOXP3+ Th17 cells and contributes to disease progression through the regulation of Jumonji domain-containing protein-3 (JMJD3) (70). Under different environmental conditions, Th17 cells exhibit varying functions based on the stimuli received. The complex tumor microenvironment is likely to cause a series of functional changes in Th17 cells. In the following section, this article will delve into various aspects of how the tumor microenvironment impacts the pathogenicity of Th17 cells (Table 1).
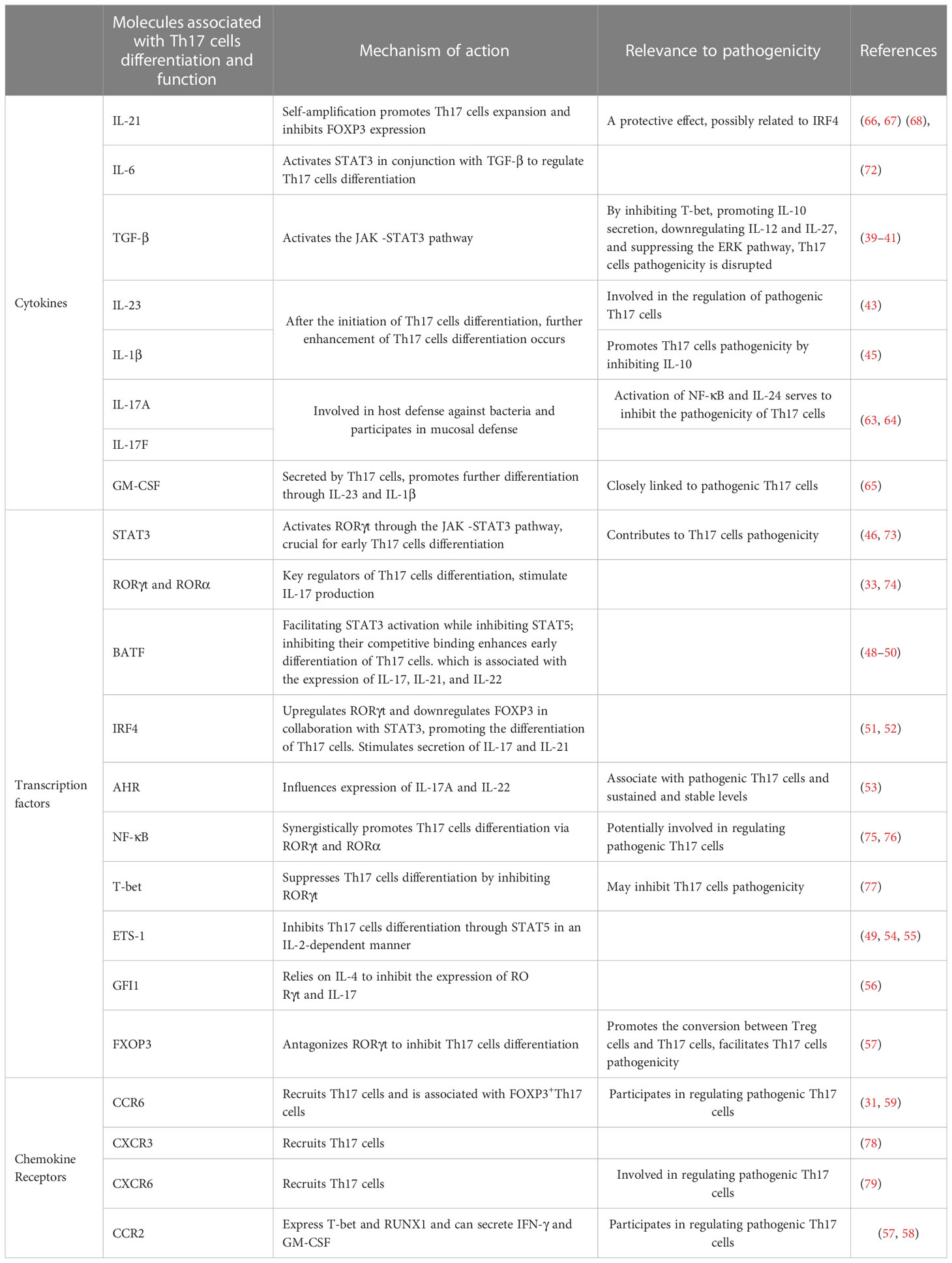
Table 1 Effectors that regulate Th17 cells differentiation, development, function, and pathogenicity.
Tumor cells, tumor-infiltrating lymphocytes, and other immune or stromal cells within the tumor microenvironment compete for limited nutrients, and certain metabolic byproducts generated within the tumor microenvironment can inhibit antitumor immunity. Therefore, understanding the various metabolic conditions within the tumor microenvironment and their impact on Th17 cells is crucial for harnessing metabolic substances within the microenvironment to regulate tumors. In colorectal cancer, elevated levels of reactive oxygen species (ROS) disrupt glycolysis and PKM2-dependent phosphorylation of STAT3, thereby inhibiting the differentiation of Th17 cells. Activation of the PRAK-NRF2-ROS axis could inhibit ROS production and promote the differentiation of Th17 cells, leading to the induction of an antitumor effect (80). However, pathogenic Th17 cells are also present in colorectal cancer. The active glycolytic pathway is inhibited, thereby enhancing the inflammatory response and facilitating the disease progression. The origin of these Th17 cells is likely associated with the transdifferentiation of Treg cells (81). The active glycolytic environment plays a role in the antitumor effect of Th17 cells, but it falls short in sustaining their long-term antitumor efficacy. Studies have indicated that Th17 cells can secrete IL-17 and low levels of IFN-γ under glycolytic and oxidative phosphorylation (OXPHOS) metabolic conditions. In the tumor microenvironment, OXPHOS-mediated induction of BCL-XL and inhibition of BIM contribute to Th17 cells survival and prevent apoptosis. This fosters the stemness of Th17 cells and plays a crucial role in maintaining their antitumor capacity. Additionally, OXPHOS can enhance the use of glutamine by Th17 cells, promoting their differentiation (82). Wyatt et al. also supported this viewpoint in their investigation of antitumor Th17 cells. They found that the antitumor activity of Th17 cells was significantly enhanced by weak inducible T cell co-stimulator (ICOS) signaling or low TCR signaling. Additionally, these cells rely on glycolysis and oxidative phosphorylation, along with a memory cell phenotype (83). High levels of insulin-like growth factor 1 (IGF1) and insulin in obesity downregulate Glycogen synthase kinase 3 beta (GSK3B) activity by phosphorylating its serine residue, thereby altering the IL-17 signaling pathway, including upregulation of CXCL1, CCL20, and IL-6 expression. Melatonin can inhibit the upregulation of IL-17 by enhancing GSK3B activity. Preliminary validation has been conducted in human prostate cancer by Ge, D. et al. and there is evidence suggesting that IL-17 promotes prostate cancer progression (84, 85). This finding suggests that melatonin can exert its antitumor effects by inhibiting IL-17, but direct evidence is still required for confirmation. Apart from glucose and lipid metabolism, Filip-Psurska et al. constructed a mouse model of breast cancer and proposed that vitamin D and its metabolites could mediate the expression of osteopontin through VDR, thereby recruiting Th17 cells and promoting tumor progression. Osteopontin can serve as a tumor-associated inflammatory mediator, facilitating tumor metastasis, and its association with Th17 cells has been demonstrated in autoimmune diseases (86, 87). Considering the significant impact of metabolism on the plasticity of Th17 cells, the emerging trend in cancer treatment is the development of suitable antitumor Th17 cells for adoptive cell therapy (88). By constructing Th17/Th1 cells, which is a group of cells that possesses the stem cell characteristics of Th17 cells and the tumor-suppressive IFN-γ secretion abilities of Th1 cells, these composite cells exhibit a metabolic state characterized by suppressed glycolysis and enhanced glutamine breakdown and exert high expression of histone deacetylase sirtuin-1 (SIRT1) and Forkhead box O1 (FOXO1) under the conditions of a CD38 inhibitor and increased nicotinamide adenine dinucleotide+ (NAD+). Their antitumor effect is significantly enhanced compared to that of Th1 cells, as confirmed in mouse melanoma and prostate cancer. During the screening of composite cells, it was discovered that both IL-1β and TGF-β could stimulate the production of IFN-γ in composite cells for tumor suppression. However, IL-1β was more effective than TGF-β in this regard. This is because TGF-β induces the expression of CD73, which converts ATP to adenosine and inhibits the production of IFN-γ, thus counteracting some of the antitumor effect. Additionally, IL-1β is associated with more active glycolytic metabolism. Although IL-1β-induced Th17 cells have a favorable effect, they lack long-term stem cell properties. On the other hand, the presence of TGF-β can enhance their stem cell characteristics. Therefore, after adjusting the concentration of TGF-β, it was found that a low concentration of TGF-β in combination with IL-1β yields the optimal antitumor effect and stimulated Th17 cells (89–91) (Figure 1).
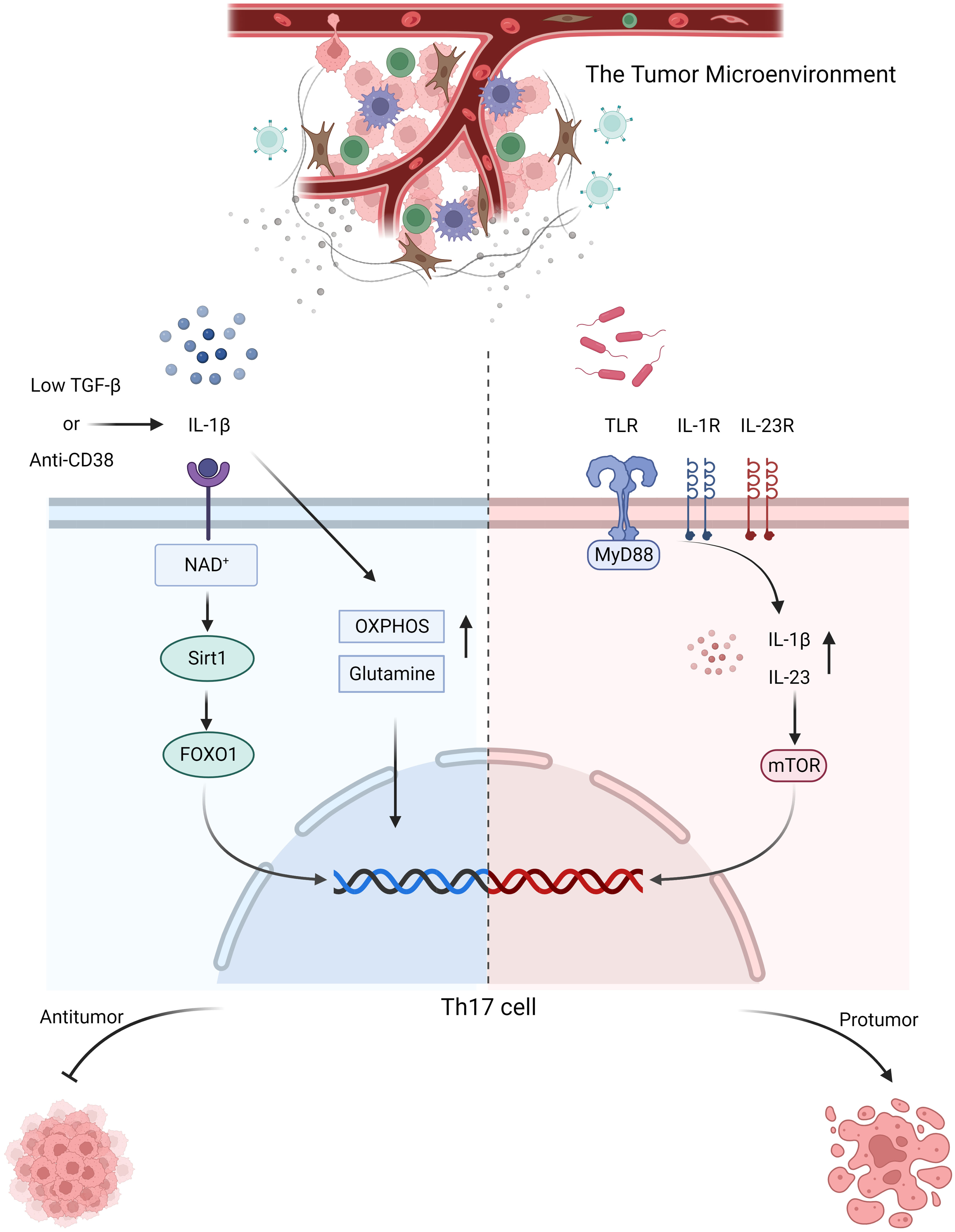
Figure 1 In response to stimulation with anti-CD38 or low-dose TGF-β combined with IL-1β, Th17 cells activate NAD+-histone deacetylase SIRT1-FOXO1 axis and undergo metabolic reprogramming, adopting a more active glutamine and OXPHOS metabolic pathway to exert their antitumor effects. In colorectal cancer, microbial signals emitted by commensal microorganisms are received by MyD88 through toll-like receptor, which sequentially activates IL-1R and IL-23R on the surface of CD4+ T cells, thereby inducing the production of IL-1β and IL-23. Consequently, mTOR expression is upregulated, promoting the proliferation and differentiation of Th17 cells, which in turn contribute to tumor promotion.
Apart from the metabolic microenvironment’s influence on Th17 cells, tumor-associated antigens can impact Th17 cells. This was demonstrated by van der Bruggen et al., who discovered that T cells could recognize the human melanoma antigen encoded by MAGE-1 (92). Tumor antigens have emerged as novel targets for antitumor immune therapy (93). However, the number of tumor antigens that can be recognized by T cells is limited, which is one of the limitations of immune checkpoint therapy (94). Jiao et al. suggested that the poor treatment efficacy of prostate cancer may be related to the number of tumor antigens and tumor-infiltrating T cells. Sufficient antigens are required to activate T cells to exert antitumor effects, but in reality, only a few antitumor T cells are activated, resulting in poor treatment effects (95). However, tumor antigens may not be scarce in number but are rather hidden by tumors. Therefore, the authors proposed the use of combination therapy to release these hidden antigens. The process of bone resorption that occurs in prostate cancer bone metastasis releases large amount of TGF-β and IL-6, which can activate Th17 cells. However, this process also reduces the activation of Th1 cells, which are the main antitumor cells. Although it is still unclear whether Th17 cells inhibit or support tumors in this context, blocking the appearance of Th17 cells can awaken the hidden antigens that activate Th1 cells, and combined with exogenous Th1 cells, should be effective in antitumor therapy.
The stimulation of Th17 cells differentiation by MyD88 can be divided into two parts. First, Th17 cells polarization is achieved through IL-1, followed by activation of the mTOR signaling pathway via IL-23, which further promoted Th17 cells differentiation. Research has indicated that the symbiotic microbiota in mouse models of colon cancer is recognized by Toll-like receptors, which stimulate MyD88, resulting in upregulated expression of IL-23 and promotion of IL-17 secretion by Th17 cells, thus facilitating tumor progression. This association may be linked to mTOR. In other words, microbial signals are received by MyD88 and subsequently activate IL-1R and IL-23R on the surface of CD4+ T cells. This activation leads to the production of IL-1β and IL-23, which, in turn, upregulate mTOR expression, facilitating the proliferation and differentiation of Th17 cells. These protumorigenic effects of Th17 cells can be inhibited by antibiotic therapy. Similarly, antibiotic treatment has been used on pancreatic cancer, although direct evidence is lacking, it is highly likely that the microbiota-mediated protumorigenic effects of Th17 cells on pancreatic cancer are also related to MyD88 (96–98) (Figure 1). The same tumorigenic effect was also confirmed in a study by Dmitrieva-Posocco, Oxana et al. The increase in tumor-promoting gut bacteria in the mouse model of colorectal cancer induced by the APC gene upregulated IL-1R1 expression on T cells, which in turn stimulated the production of IL-17 and IL-22, thereby promoting tumor progression (99). However, there is also evidence suggesting that Th17 cells exert antitumor effects on murine colorectal cancer. The presence of the gut microbiota has been shown to inhibit IL-6 and IL-1, thereby suppressing the differentiation of Th17 cells and promoting tumor progression. This finding contradicts the findings of Wang et al. and Dmitrieva-Posocco, Oxana et al. The author’s viewpoint attributes this contradiction to differences in the methods of constructing colorectal cancer models, animal models, and the cytokine milieu within the tumor microenvironment. It also underscores the reciprocal interaction between the tumor microenvironment and Th17 cells. Therefore, when studying the role of Th17 cells in tumors, it is essential to consider the composition of various components within the tumor microenvironment and their impact on the proliferation and differentiation of Th17 cells (100). The overexpression of miR-149-3p has been shown to ameliorate Th17 cells infiltration and colitis induced by Enterotoxigenic Bacteroidesfragilis, thereby inhibiting the progression to colorectal cancer. This finding not only suggests the impact of the microbe-associated inflammatory response mediated by Th17 cells on cancer progression but also highlights the significant role of Th17 cells in the transition from precancerous lesions (colitis) to colorectal cancer (101). Similarly, the presence of Th17 cells has been observed in the progression from colorectal adenoma to colorectal cancer; however, it remains unclear whether microbes are involved in this process (102). The presence of Helicobacter pylori in gastric cancer also promotes the development of Th17 cells, contributing to a tumorigenic effect (103). These findings indicate that the microbiota contribute to the regulation of Th17 cells in the tumor microenvironment, and to a large extent, they are associated with tumor-promoting effects.
Whether it is the tumor metabolic microenvironment, tumor antigens, or the microbiota, their impact on Th17 cells pathogenicity relies on signaling pathways and the involvement of effector molecules. The study of tumors and Th17 cells have revealed numerous intersections between the two, indicating that tumors attract Th17 cells while creating a suitable microenvironment for themselves. In their investigation of different stages of breast cancer, Avalos-Navarro et al. discovered that Macrophage migration inhibitory factor (MIF) promoted the production of TGF-β, IL-6, and IL-1 while also participating in the activation of signaling pathways such as NF-kB, STAT3, PI3K/AKT, and MAPK (ERK1/2), thus contributing to Th17 cells generation. Moreover, they demonstrated a positive correlation between MIF and tumor progression mediated by Th17 cells (104). In their investigations on breast cancer and lung cancer in humans and mice, Voigt et al. revealed that IL-22 facilitated tumor progression, and Th17 cells were identified as one of the primary cellular sources. NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation within the tumor microenvironment can induce the activation of IL-1β, which subsequently activates RORγt to promote the secretion of IL-22 by Th17 cells, thereby promoting disease advancement (105). This finding indicates that tumors exploit signaling pathways that are advantageous to themselves, substituting the normal signals sent by the body to Th17 cells and thereby using Th17 cells to promote tumor growth. The study by Fabre et al. in skin cancer supports this possibility, as overexpression of STAT3 promoted tumor progression by stimulating the angiogenic effects of IL-17+ Th17 cells (106). Additionally, research by Xu, Z. S. revealed that Th17 cells facilitated the progression from colitis to colorectal cancer. Family with sequence similarity 64, member A (FAM64A), which is a positive regulator of STAT3, enhances IL-6-mediated STAT3 activation, promoting Th17 cells differentiation and facilitating inflammation-induced carcinogenesis (107). The expression of β-catenin in T cells is elevated in colorectal polyps, leading to the polarization of T cells towards Th17 cells through the activation of RORγt. This process promotes the occurrence of intestinal inflammation and the progression to tumor formation. Additionally, stable expression of β-catenin downregulates FOXP3 expression in Treg cells while upregulating RORγt, facilitating the conversion of Treg cells and exacerbating inflammation to promote disease progression (108). Apart from transcription factors, the presence of certain cytokines also regulates the function of Th17 cells. The study by Fabre et al. highlighted the similar roles of tumor-associated cytokines, such as IL-6. Skin cancer cells are characterized by their ability to secrete IL-6. Blocking IL-6 leads to tumor regression, as the protumor activity of IL-17 secreted by Th17 cells is regulated by IL-6 and STAT3 (109). Similarly, within the colorectal cancer microenvironment, DC cells with liver kinase B1 (LKB1) deficiency can promote tumor progression by activating the IL-6-STAT3 axis and facilitating Th17 cells differentiation (110). In KRAS-mutant lung cancer, inhibiting MEK increases the expression of TGF-β, IL-6, and IL-23, promoting the differentiation of Th17 cells and the secretion of IL-17 and IL-22, thereby enhancing tumor resistance, invasiveness, and metastasis. The presence of IL-17 increases tumor resistance by upregulating CD38, while IL-22 is associated with EMT, strengthening tumor invasiveness and metastasis. Targeted therapies can provide more effective control over tumor progression (111). Th17 cells also showed antitumor effects. Muranski et al. established a novel mouse model of melanoma called TRP-1 TCR transgenic mice. They observed that Th17 cells and Th1 cells exerted antitumor effects, and Th17 cells demonstrated superior efficacy compared to Th1 cells. This could be attributed to the increased secretion of IFN-γ and the sustained presence of Th17 cells. This aligns with the aforementioned notion of antitumor Th17 cells in the context of the microbiota and metabolism. These perspectives delineate some of the defining characteristics of antitumor Th17 cells: their persistence, stemness properties and capacity to secrete IFN-γ. Additionally, it reaffirms the influence of the tumor microenvironment on Th17 cells (112). Moreover, this subset of Th17 cells has already been defined. They exhibit abundant expression of BCL2 and BCL-XL, increased levels of stem cell-related genes, elevated expression of cell cycle proteins, and reduced CDK inhibitors. These cells also exhibit upregulated Wnt/β-catenin-associated gene expression and inhibit apoptosis through the HIF/Notch/BCL2 axis. Furthermore, they can induce the production of CXCL9 and CXCL10 by tumors to recruit Th1 cells for antitumor responses (113, 114). Knochelmann et al. further validated novel characteristics of antitumor Th17 cells using the same mouse model. The antitumor efficacy of Th17 cells was optimal on the fourth day of in vitro expansion, and was characterized by a memory cell phenotype (CD44hi CD62Llo). These cells exhibited high levels of CD25, ICOS, and OX40 expression. The presence of IL-6 further contributed to the sustained and durable antitumor capacity of Th17 cells (115). Su et al. proposed that within the tumor microenvironment of melanoma (as well as ovarian cancer, breast cancer, and colon cancer), tumor cells and tumor-associated fibroblasts secrete monocyte chemoattractant protein-1 (MCP-1) and RANTES, which mediate the migration of Th17 cells into the tumor microenvironment. Tumor cells and stromal cells further promote the generation and expansion of Th17 cells by secreting IL-1, IL-6, IL-23, and TGF-β. Inflammatory signals induced by TLRs and Nod2 also contribute to the proliferation and generation of Th17 cells. Initially, these factors facilitate the proliferation of Th17 cells mediated by tumor cells and tumor-associated fibroblasts and subsequently enhance the recruitment of Th17 cells by upregulating MCP-1 and regulated upon activation normal T cell expressed and secreted factor (RANTES) levels. Finally, these signals promote the recruitment and differentiation of Th17 cells by dendritic cells, monocytes, PBMCs, and other immune cells. The aforementioned factors that regulate Th17 cells have been shown to promote disease progression in vitro, but further validation is required in vivo (116). A similar mechanism of Th17 cells recruitment can be observed in malignant pleural effusion, where these cells also exert antitumor effects. Tumor cell-secreted CCL20 and CCL22 in the pleural effusion act on the corresponding receptors CCR4 and CCR6 on Th17 cells and recruit them (117). Additionally, in invasive bladder cancer, the recruitment of Th17 cells through chemokines is involved in exerting antitumor effects (118). These findings suggest that directly recruited Th17 cells can play an antitumor role within the tumor microenvironment. However, there are exceptions, as demonstrated in non-small cell lung cancer, in which CCL20 promotes tumor progression by recruiting Th17 cells (119). Apart from the effects of cytokines secreted by tumors themselves, immune cells in the tumor microenvironment also affect Th17 cells. Research by Li et al. has shown that the level of IL-6 was increased in tertiary lymphoid structures adjacent to hepatocellular carcinoma, which promoted the differentiation of Th17 cells and provided an active antitumor immune microenvironment. Moreover, the expression of CCR7 is high in peritumoral tissue, which may help stabilize Th17 cells in the tumor microenvironment (120). Furthermore, inhibiting secretion of immunosuppressive molecule IL-10 by B cells can also enhance the protumor effect of IL-17+ Th17 cells (121). Dendritic cells secrete IL-6, TGF-β, and IL-23 to induce polarization of Th17 cells (122).
In addition to directly impacting Th17 cells to exert their antitumor effects, effector molecules in the tumor microenvironment induce the conversion of Th17 cells into Tfh cells, Th2 cells, Treg cells, and Th1 cells. In this context, Th17 cells can simultaneously possess the characteristics and functions of Th17 cells and converted cells (32, 123). This process of lineage conversion from one cell type to another is referred to as transdifferentiation (124). TGF-β can influence the reciprocal conversion between Th17 cells and Treg cells, leading to the coexpression of FOXP3 and RORγt in Treg cells and Th17 cells, which acquire dual functional capacities. The presence of AHR can also facilitate this conversion (125, 126). In breast cancer, TGF-β, PGE2 and blocking glycolysis facilitate the conversion of Th17 cells to Treg cells. These cells exhibit the phenotype of IL-17A+ FOXP3+ and possess the characteristics of Th17 cells, such as CD25 and CCR4 expression, as well as characteristics of Treg cells, such as CD161 and CD49d expression, thus promoting disease progression (127). Similarly, Roy et al. observed this phenomenon in breast cancer and proposed a Th17reg cells subset characterized by high expression of CD39 and CD73. The key mediator of this conversion is believed to be the GFI1/HDAC1 axis. Soheilifar et al. also identified a population of IL-17-secreting Treg cells whose differentiation into IL-17-secreting Treg cells was promoted by the overexpression of mir-182-5p and mir-182-3p. Marion Thibaudin discovered a subset of Th17 cells with high CD25 expression that accumulates in human breast cancer. Notably, these cell types also express CD39 and CD73, suggesting that the three aforementioned subsets are likely Th17reg cells (128–130). CD73 and CD39 have been shown to be signaling molecules that can reshape the immunosuppressive tumor microenvironment. These factors inhibit effector immune cells and activate immunoregulatory cells by dephosphorylating extracellular ATP to generate adenosine. These factors are express by not only Treg cells, but they are also expressed on various cell types within the tumor microenvironment. The expression of CD73 and CD39 on Th17 cells may be related to IL-6-STAT3 and TGF-β-GFI1, and GFI1 exerts inhibitory effects on the differentiation of Th17 cells and the expression of nucleases in Th17 cells. TGF-β can disrupt of these inhibitory mechanisms. IL-6, which is unrelated to GFI1, positively regulates the expression of CD73 and CD39 through STAT3 (131).This strongly indicates that the conversion of Th17 cells to Treg cells expressing CD73 and CD39 promotes tumor progression (132). The phenotypic conversion between Th17 cells and Treg cells in gastric cancer may arise from interactions among mesenchymal stem cells (MSCs), tumor-associated fibroblasts (PCCs), and gastric cancer stem cells (CSCs). The interplay between MSCs and CSCs prompts the secretion of TGF-β by MSCs, leading to the Th17 cells-to-Treg cells conversion, which is regulated by Notch signaling. In pancreatic cancer, the presence of Th17/Th1 cells phenotype has been identified. RIP1i-mediated macrophage reprogramming leads to the phenotypic reshaping of CD4+ T cells, resulting in high co-expression of IFN-γ, IL-17, T-bet, and RORγt. These cells lose the protumor capabilities associated with IL-17+ Th17 cells and acquire the antitumor capabilities of IFN-γ-producing Th1 cells, thus representing Th17.1 cells (133). In fact, the construction of Th17.1 cells has long been pursued for tumor therapy, as mentioned previously. These cells possess the characteristics described earlier, including a memory phenotype and the expression of IL-17 and IFN-γ. Furthermore, Muranski, P. et al. proposed that this cell population also expresses Tcf7 and β-catenin, providing evidence for the memory phenotype of Th17.1 cells and suggesting their superior antitumor capabilities compared to Th1 cells (134).In asthma, there is evidence of the conversion of Th17 cells into Th2 cells (135). Schmitt’s research indicates that in an inflammatory environment associated with autoimmune diseases, Th17 cells can acquire phenotypic characteristics of Tfh cells under the influence of TGF-β and other cytokines (136). This finding suggests the occurrence of transdifferentiation. However, this particular differentiation type has not yet been identified in tumors (Figure 2). In summary, the heterogeneity of Th17 cells is likely one of the reasons for their dual roles in tumor regulation (Table 2).
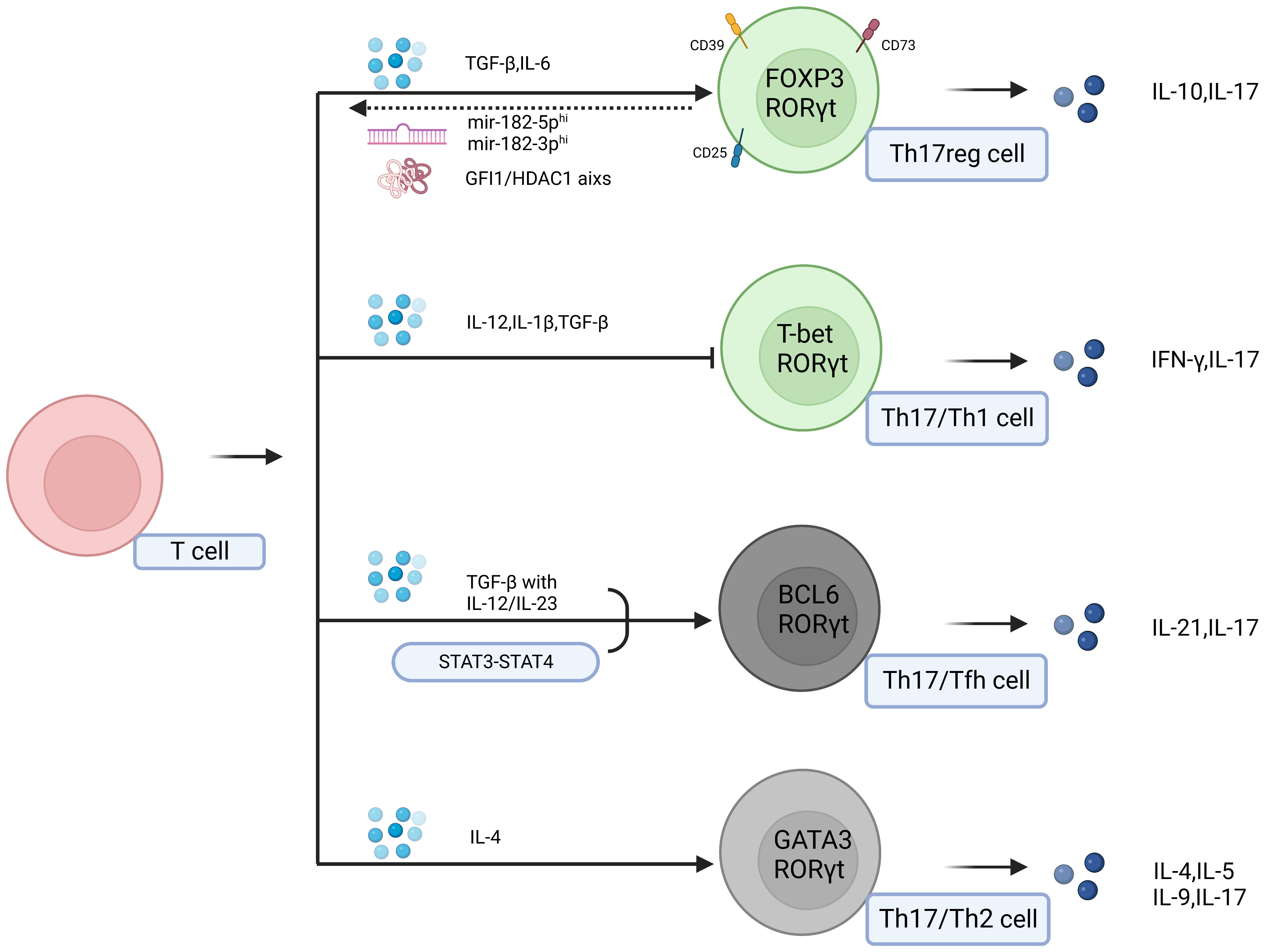
Figure 2 Effector molecules in the tumor microenvironment can induce the transdifferentiation of Th17 cells into Tfh cells, Th2 cells, Treg cells, and Th1 cells. In this context, Th17 cells can exhibit the characteristics and functions of Th17 cells as well as those of the transdifferentiated cells. The two populations of grey cells represent undiscovered transdifferentiation types within the tumor.
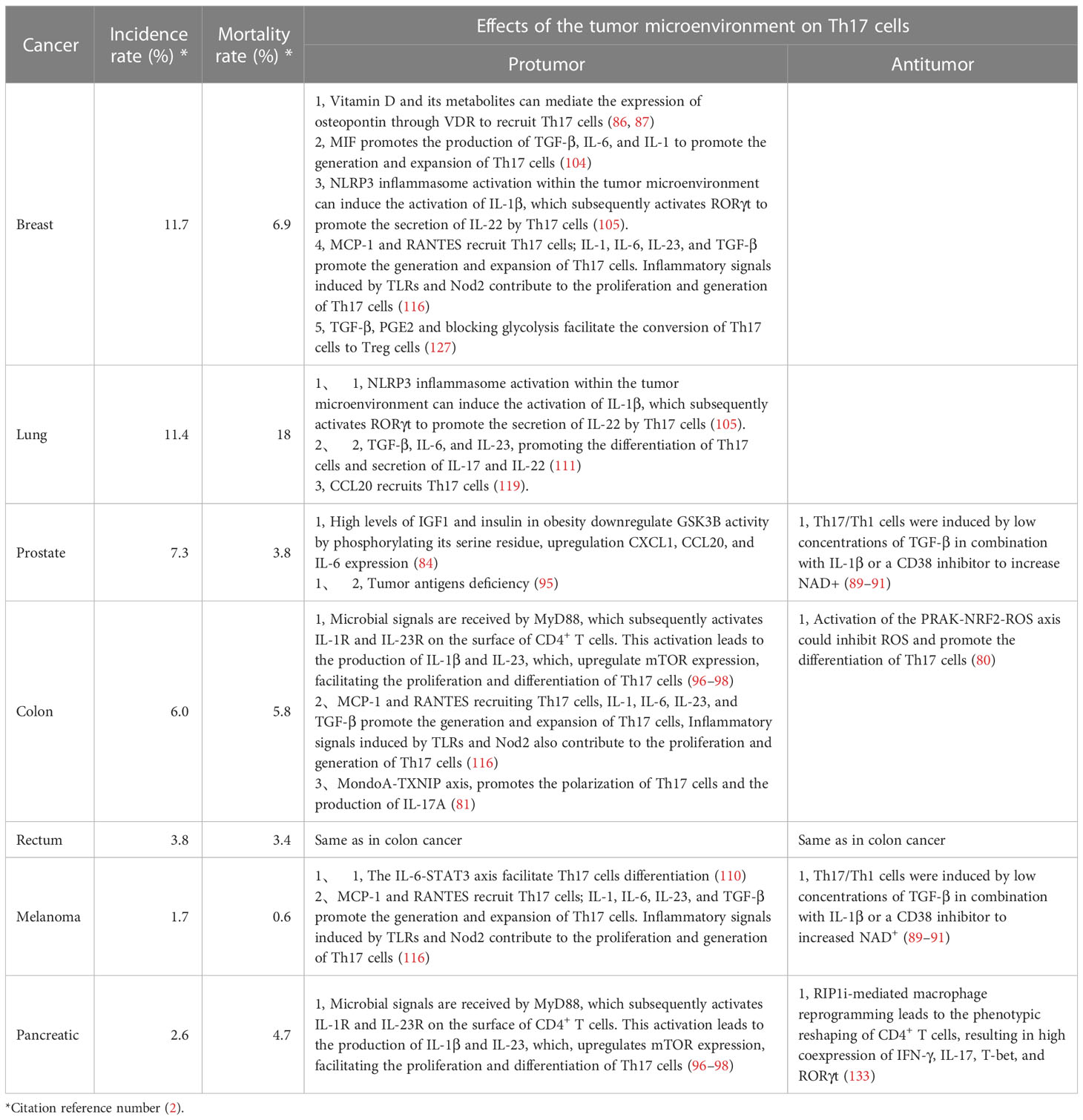
Table 2 Tumor information and the impact of the tumor microenvironment on Th17 cells.
Hanahan summarized eight fundamental hallmarks of tumors (3, 137). These eight hallmarks have resulted in a more structured study of tumor development. For example, IL-17 secretion can induce epithelial-mesenchymal transition (EMT) in lung cancer cells, promoting their migration and metastatic dissemination, which corresponds to the activation of invasion and metastasis (138). Additionally, IL-17 can promote tumor angiogenesis by coordinating with tumor fibroblasts to induce various angiogenic factors, including vascular endothelial growth factor, prostaglandin E2, and nitric oxide. corresponding to the tumor-induced vascular system (139, 140). Furthermore, apart from tumor promotion, IL-17 exhibits antitumor characteristics that correspond to the aforementioned features, such as indirectly attracting CD8+ T cells and NK cells to eliminate tumor cells (141). Th17 cells are involved in various stages of tumor development and exert antitumor and protumor effects. Therefore, the factors governing these divergent regulatory functions of Th17 cells must be considered, which constitutes the focal point of this review.
Th17 cells are inflammatory cells that plays a significant role in the context of inflammation and tumors. Exploring the signaling pathways shared among Th17 cells, tumors, and inflammation reveals an overlapping network of molecules and cytokines (142). Remarkably, Th17 cells tend to exert tumor-promoting effects, which are often mediated by overlapping signaling molecules or cytokines. This is because Th17 cells lack precise recognition capabilities and instead rely on the environment. Leveraging this understanding, we can strategically reshape the tumor microenvironment to enhance the antitumor capacity of Th17 cells. For instance, anti-PD-1 therapy upregulates the solute carrier family 11 member 1 (SLC11A1) gene in the tumor microenvironment, promoting the differentiation of Th17 cells. The levels of Th17 cells are significantly associated with improved overall survival (OS), suggesting that Th17 cells may serve as prognostic indicators for anti-PD-1 therapy (97, 143). Additionally, the microbiome’s involvement in intestinal tumors coordinates with the sustained presence of chronic inflammation, thus promoting tumor development by inducing the differentiation of Th17 cells (144). In fact, there are therapeutic approaches targeting these pathways in tumor therapy. For instance, modulation signaling pathways by manipulation of noncoding RNA expression, such as through overexpression or knockdown, can impact the function of Th17 cells and other immune cell subsets. Additionally, antibiotic treatment targets microbial infections can also combat tumors (101). However, these findings do not fully elucidate the additional aspects of the antitumor effect mediated by Th17 cells. In addition to inflammation, the plasticity of Th17 cells themselves may play a role in regulating tumor behavior (123, 145).There may be two distinct states of Th17 cells: classical Th17 cells and transdifferentiated Th17 cells, which are characterized by different phenotype traits such as Th17reg cells and Th1/Th17 cells. The existence of these diverse forms may be associated with the presence of TGF-β. Currently, chemokine recruitment of Th17 cells primarily involves classical Th17 cells, which subsequently undergo transformation in various directions in response to interactions with the microenvironment through processing and modification. It has been shown that the transdifferentiation of Th17 cells to Th1 cells and Th17 cells to Treg cells are reversible processes (146, 147). Additionally, metabolically, transdifferentiated Th17 cells differ from classical Th17 cells. For example, Th17reg cells may require lipid metabolism for activation, similar to Treg cells (148). However, clear evidence to support this notion is currently lacking. If proven, targeting cellular metabolic pathways to alter the transdifferentiation of immune cells could become a novel approach for tumor therapy. In summary, the assessment of the role of Th17 cells in the tumor microenvironment may require comprehensive consideration of the following factors: 1)cellular metabolism, including glycolysis and oxidative phosphorylation; 2) the level of signaling factors such as TGF-β, IL-6, and IL-23 in the tumor microenvironment, as well as the expression of STAT3, T-bet, FOXP3, IL-17, IL-22, IFN-γ, CD39, CD73 and chemotactic factors with cellular specificity in Th17 cells; and 3) the presence of a memory phenotype or long-lived characteristics in Th17 cells, as indicated by the expression of CD44hi, CD62Llo, and the involvement of stem cell-related genes such as Wnt/β-catenin and Tcf7. In conclusion, considering these factors comprehensively will facilitate more precise characterization of the effector functions of Th17 cells in tumors and enable more effective antitumor therapies.
JX, and CM drafted the manuscript and drawn the tables and figures. ZZ conducted the project guidance. YL and HP designed the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (Grant No. 81800698), Jiangsu Provincial Medical Key Discipline Cultivation Unit (Grant No. JSDW202241), Zhenjiang Sixth Phase 169 Project Training Fund Support Project (2021-169GG-28), Zhenjiang Science and Technology Planning Project (Grant Nos. SH2022032, SH2023006 and SH2023008), and Zhenjiang First People’s Hospital Support Project (Grant No.Y2023005-S).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (2018) 392:1736–88. doi: 10.1016/s0140-6736(18)32203-7
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
4. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
5. DeGregori J. Connecting cancer to its causes requires incorporation of effects on tissue microenvironments. Cancer Res (2017) 77:6065–8. doi: 10.1158/0008-5472.CAN-17-1207
6. Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer (2020) 6:605–18. doi: 10.1016/j.trecan.2020.02.022
7. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
8. Xing Y, Ruan G, Ni H, Qin H, Chen S, Gu X, et al. Tumor immune microenvironment and its related miRNAs in tumor progression. Front Immunol (2021) 12:624725. doi: 10.3389/fimmu.2021.624725
9. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
10. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
11. Pulendran B, Davis MM. The science and medicine of human immunology. Science (2020) 369(6511):eaay4014. doi: 10.1126/science.aay4014
12. Lv B, Wang Y, Ma D, Cheng W, Liu J, Yong T, et al. Immunotherapy: reshape the tumor immune microenvironment. Front Immunol (2022) 13:844142. doi: 10.3389/fimmu.2022.844142
13. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
14. Roquilly A, Mintern JD, Villadangos JA. Spatiotemporal adaptations of macrophage and dendritic cell development and function. Annu Rev Immunol (2022) 40:525–57. doi: 10.1146/annurev-immunol-101320-031931
15. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
16. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
17. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22(13):6995. doi: 10.3390/ijms22136995
18. Furumaya C, Martinez-Sanz P, Bouti P, Kuijpers TW, Matlung HL. Plasticity in pro- and anti-tumor activity of neutrophils: shifting the balance. Front Immunol (2020) 11:2100. doi: 10.3389/fimmu.2020.02100
19. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
20. Feng R, Cui Z, Liu Z, Zhang Y. Upregulated microRNA-132 in T helper 17 cells activates hepatic stellate cells to promote hepatocellular carcinoma cell migration in vitro. Scand J Immunol (2021) 93:e13007. doi: 10.1111/sji.13007
21. Vitiello GA, Miller G. Targeting the interleukin-17 immune axis for cancer immunotherapy. J Exp Med (2020) 217(1):e20190456. doi: 10.1084/jem.20190456
22. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
23. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
24. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
25. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8:967–74. doi: 10.1038/ni1488
26. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
27. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203:2271–9. doi: 10.1084/jem.20061308
28. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448:484–7. doi: 10.1038/nature05970
29. Hisada R, Yoshida N, Umeda M, Burbano C, Bhargava R, Scherlinger M, et al. The deacetylase SIRT2 contributes to autoimmune disease pathogenesis by modulating IL-17A and IL-2 transcription. Cell Mol Immunol (2022) 19:738–50. doi: 10.1038/s41423-022-00874-1
30. Xiao F, Lin X, Tian J, Wang X, Chen Q, Rui K, et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjogren’s syndrome. Cell Mol Immunol (2017) 14:924–34. doi: 10.1038/cmi.2017.8
31. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
32. Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol (2013) 182:10–20. doi: 10.1016/j.ajpath.2012.08.041
33. Jiang P, Zheng C, Xiang Y, Malik S, Su D, Xu G, et al. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor Rev (2023) 69:28–42. doi: 10.1016/j.cytogfr.2022.07.005
34. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol (2019) 41:283–97. doi: 10.1007/s00281-019-00733-8
35. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14:275–81. doi: 10.1038/nm1710
36. Bacher P, Hohnstein T, Beerbaum E, Rocker M, Blango MG, Kaufmann S, et al. Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell (2019) 176:1340–1355.e1315. doi: 10.1016/j.cell.2019.01.041
37. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol (2018) 15:458–69. doi: 10.1038/s41423-018-0004-4
38. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, Jadidi-Niaragh F, et al. The paradox of Th17 cell functions in tumor immunity. Cell Immunol (2017) 322:15–25. doi: 10.1016/j.cellimm.2017.10.015
39. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467:967–71. doi: 10.1038/nature09447
40. Lochner M, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med (2008) 205:1381–93. doi: 10.1084/jem.20080034
41. Choi G, Park YJ, Cho M, Moon H, Kim D, Kang CY, et al. A critical role for Th17 cell-derived TGF-beta1 in regulating the stability and pathogenicity of autoimmune Th17 cells. Exp Mol Med (2021) 53:993–1004. doi: 10.1038/s12276-021-00632-9
42. Wu B, Zhang S, Guo Z, Bi Y, Zhou M, Li P, et al. The TGF-beta superfamily cytokine Activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation. Immunity (2021) 54:308–323.e306. doi: 10.1016/j.immuni.2020.12.010
43. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol (2014) 14:585–600. doi: 10.1038/nri3707
44. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
45. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
46. Hwang ES. Transcriptional regulation of T helper 17 cell differentiation. Yonsei Med J (2010) 51:484–91. doi: 10.3349/ymj.2010.51.4.484
47. Hall JA, Pokrovskii M, Kroehling L, Kim BR, Kim SY, Wu L, et al. Transcription factor RORalpha enforces stability of the Th17 cell effector program by binding to a Rorc cis-regulatory element. Immunity (2022) 55:2027–2043.e2029. doi: 10.1016/j.immuni.2022.09.013
48. Martinez GJ, Dong C. BATF: bringing (in) another Th17-regulating factor. J Mol Cell Biol (2009) 1:66–8. doi: 10.1093/jmcb/mjp016
49. Pham D, Silberger DJ, Nguyen KN, Gao M, Weaver CT, Hatton RD. Batf stabilizes Th17 cell development via impaired Stat5 recruitment of Ets1-Runx1 complexes. EMBO J (2023) 42:e109803. doi: 10.15252/embj.2021109803
50. Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature (2009) 460:405–9. doi: 10.1038/nature08114
51. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol (2007) 8:958–66. doi: 10.1038/ni1500
52. Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, et al. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity (2008) 29:899–911. doi: 10.1016/j.immuni.2008.10.011
53. de Lima KA, Donate PB, Talbot J, Davoli-Ferreira M, Peres RS, Cunha TM, et al. TGFbeta1 signaling sustains aryl hydrocarbon receptor (AHR) expression and restrains the pathogenic potential of T(H)17 cells by an AHR-independent mechanism. Cell Death Dis (2018) 9:1130. doi: 10.1038/s41419-018-1107-7
54. Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC. Ets-1 is a negative regulator of Th17 differentiation. J Exp Med (2007) 204:2825–35. doi: 10.1084/jem.20070994
55. Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, et al. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood (2006) 108:1595–601. doi: 10.1182/blood-2006-04-015016
56. Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, et al. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J Exp Med (2009) 206:329–41. doi: 10.1084/jem.20081666
57. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453:236–40. doi: 10.1038/nature06878
58. Lim HW, Lee J, Hillsamer P, Kim CH. Human Th17 cells share major trafficking receptors with both polarized effector T cells and FOXP3+ regulatory T cells. J Immunol (2008) 180:122–9. doi: 10.4049/jimmunol.180.1.122
59. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204:2803–12. doi: 10.1084/jem.20071397
60. Kara EE, McKenzie DR, Bastow CR, Gregor CE, Fenix KA, Ogunniyi AD, et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat Commun (2015) 6:8644. doi: 10.1038/ncomms9644
61. Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity (2014) 40:355–66. doi: 10.1016/j.immuni.2014.01.002
62. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood (2010) 115:335–43. doi: 10.1182/blood-2009-04-216085
63. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34:149–62. doi: 10.1016/j.immuni.2011.02.012
64. Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, et al. The cytokine IL-17A limits th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity (2020) 53:384–397 e385. doi: 10.1016/j.immuni.2020.06.022
65. Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflammation (2015) 2015:568543. doi: 10.1155/2015/568543
66. Long D, Chen Y, Wu H, Zhao M, Lu Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun (2019) 99:1–14. doi: 10.1016/j.jaut.2019.01.013
67. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and th17 cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
68. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448:480–3. doi: 10.1038/nature05969
69. Perez LG, Kempski J, McGee HM, Pelzcar P, Agalioti T, Giannou A, et al. TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat Commun (2020) 11:2608. doi: 10.1038/s41467-020-16363-w
70. Wagner A, Wang C, Fessler J, DeTomaso D, Avila-Pacheco J, Kaminski J, et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell (2021) 184:4168–4185.e4121. doi: 10.1016/j.cell.2021.05.045
71. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. CD5L/AIM regulates lipid biosynthesis and restrains th17 cell pathogenicity. Cell (2015) 163:1413–27. doi: 10.1016/j.cell.2015.10.068
72. Camporeale A, Poli V. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci (Landmark Ed) (2012) 17:2306–26. doi: 10.2741/4054
73. Poholek CH, Raphael I, Wu D, Revu S, Rittenhouse N, Uche UU, et al. Noncanonical STAT3 activity sustains pathogenic Th17 proliferation and cytokine response to antigen. J Exp Med (2020) 217(10):e20191761. doi: 10.1084/jem.20191761
74. Castro G, Liu X, Ngo K, De Leon-Tabaldo A, Zhao S, Luna-Roman R, et al. RORgammat and RORalpha signature genes in human Th17 cells. PloS One (2017) 12:e0181868. doi: 10.1371/journal.pone.0181868
75. Wang Q, Wang Y, Liu Q, Chu Y, Mi R, Jiang F, et al. MALT1 regulates Th2 and Th17 differentiation via NF-kappaB and JNK pathways, as well as correlates with disease activity and treatment outcome in rheumatoid arthritis. Front Immunol (2022) 13:913830. doi: 10.3389/fimmu.2022.913830
76. Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature (2010) 464:1381–5. doi: 10.1038/nature08922
77. Shimizu M, Kondo Y, Tanimura R, Furuyama K, Yokosawa M, Asashima H, et al. T-bet represses collagen-induced arthritis by suppressing Th17 lineage commitment through inhibition of RORgammat expression and function. Sci Rep (2021) 11:17357. doi: 10.1038/s41598-021-96699-5
78. Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, et al. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J Hepatol (2012) 57:1044–51. doi: 10.1016/j.jhep.2012.07.008
79. Hou L, Yuki K. CCR6 and CXCR6 identify the th17 cells with cytotoxicity in experimental autoimmune encephalomyelitis. Front Immunol (2022) 13:819224. doi: 10.3389/fimmu.2022.819224
80. Zhao Z, Wang Y, Gao Y, Ju Y, Zhao Y, Wu Z, et al. The PRAK-NRF2 axis promotes the differentiation of Th17 cells by mediating the redox homeostasis and glycolysis. Proc Natl Acad Sci U.S.A. (2023) 120:e2212613120. doi: 10.1073/pnas.2212613120
81. Lu Y, Li Y, Liu Q, Tian N, Du P, Zhu F, et al. MondoA-thioredoxin-interacting protein axis maintains regulatory T-cell identity and function in colorectal cancer microenvironment. Gastroenterology (2021) 161:575–591.e516. doi: 10.1053/j.gastro.2021.04.041
82. Hong HS, Mbah NE, Shan M, Loesel K, Lin L, Sajjakulnukit P, et al. OXPHOS promotes apoptotic resistance and cellular persistence in T(H)17 cells in the periphery and tumor microenvironment. Sci Immunol (2022) 7:eabm8182. doi: 10.1126/sciimmunol.abm8182
83. Wyatt MM, Huff LW, Nelson MH, Neal LR, Medvec AR, Rangel Rivera GO, et al. Augmenting TCR signal strength and ICOS costimulation results in metabolically fit and therapeutically potent human CAR Th17 cells. Mol Ther (2023) 31(7):2120–2131. doi: 10.1016/j.ymthe.2023.04.010
84. Ge D, Dauchy RT, Liu S, Zhang Q, Mao L, Dauchy EM, et al. Insulin and IGF1 enhance IL-17-induced chemokine expression through a GSK3B-dependent mechanism: a new target for melatonin’s anti-inflammatory action. J Pineal Res (2013) 55:377–87. doi: 10.1111/jpi.12084
85. Zhang Q, Liu S, Parajuli KR, Zhang W, Zhang K, Mo Z, et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene (2017) 36:687–99. doi: 10.1038/onc.2016.240
86. Cantor H, Shinohara ML. Regulation of T-helper-cell lineage development by osteopontin: the inside story. Nat Rev Immunol (2009) 9:137–41. doi: 10.1038/nri2460
87. Pawlik A, Anisiewicz A, Filip-Psurska B, Klopotowska D, Maciejewska M, Mazur A, et al. Divergent effect of tacalcitol (PRI-2191) on th17 cells in 4T1 tumor bearing young and old ovariectomized mice. Aging Dis (2020) 11:241–53. doi: 10.14336/AD.2019.0618
88. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen T-LM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature (2018) 565:101–5. doi: 10.1038/s41586-018-0806-7
89. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD(+)Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab (2018) 27:85–100.e108. doi: 10.1016/j.cmet.2017.10.006
90. Chatterjee S, Thyagarajan K, Kesarwani P, Song JH, Soloshchenko M, Fu J, et al. Reducing CD73 expression by IL1beta-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res (2014) 74:6048–59. doi: 10.1158/0008-5472.CAN-14-1450
91. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood (2009) 113:1250–6. doi: 10.1182/blood-2008-04-146480
92. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254:1643–7. doi: 10.1126/science.1840703
93. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol (2012) 23 Suppl 8:viii6–9. doi: 10.1093/annonc/mds256
94. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
95. Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell (2019) 179:1177–1190 e1113. doi: 10.1016/j.cell.2019.10.029
96. Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang CH. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci U.S.A. (2013) 110:2270–5. doi: 10.1073/pnas.1206048110
97. Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, et al. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology (2018) 155:33–37.e36. doi: 10.1053/j.gastro.2018.04.001
98. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature (2012) 491:254–8. doi: 10.1038/nature11465
99. Dmitrieva-Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, Thovarai V, et al. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity (2019) 50:166–180.e167. doi: 10.1016/j.immuni.2018.11.015
100. Xing C, Wang M, Ajibade AA, Tan P, Fu C, Chen L, et al. Microbiota regulate innate immune signaling and protective immunity against cancer. Cell Host Microbe (2021) 29:959–974.e957. doi: 10.1016/j.chom.2021.03.016
101. Cao Y, Wang Z, Yan Y, Ji L, He J, Xuan B, et al. Enterotoxigenic bacteroidesfragilis promotes intestinal inflammation and Malignancy by inhibiting exosome-packaged miR-149-3p. Gastroenterology (2021) 161:1552–1566.e1512. doi: 10.1053/j.gastro.2021.08.003
102. Wallace K, El Nahas GJ, Bookhout C, Thaxton JE, Lewin DN, Nikolaishvili-Feinberg N, et al. Immune responses vary in preinvasive colorectal lesions by tumor location and histology. Cancer Prev Res (Phila) (2021) 14:885–92. doi: 10.1158/1940-6207.CAPR-20-0592
103. Pinchuk IV, Morris KT, Nofchissey RA, Earley RB, Wu JY, Ma TY, et al. Stromal cells induce Th17 during Helicobacter pylori infection and in the gastric tumor microenvironment. PloS One (2013) 8:e53798. doi: 10.1371/journal.pone.0053798
104. Avalos-Navarro G, Munoz-Valle JF, Daneri-Navarro A, Quintero-Ramos A, Franco-Topete RA, Moran-Mendoza AJ, et al. Circulating soluble levels of MIF in women with breast cancer in the molecular subtypes: relationship with Th17 cytokine profile. Clin Exp Med (2019) 19:385–91. doi: 10.1007/s10238-019-00559-6
105. Voigt C, May P, Gottschlich A, Markota A, Wenk D, Gerlach I, et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci U.S.A. (2017) 114:12994–9. doi: 10.1073/pnas.1705165114
106. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med (2009) 206:1457–64. doi: 10.1084/jem.20090207
107. Xu ZS, Zhang HX, Li WW, Ran Y, Liu TT, Xiong MG, et al. FAM64A positively regulates STAT3 activity to promote Th17 differentiation and colitis-associated carcinogenesis. Proc Natl Acad Sci U.S.A. (2019) 116:10447–52. doi: 10.1073/pnas.1814336116
108. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, et al. beta-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med (2014) 6:225ra228. doi: 10.1126/scitranslmed.3007607
109. Straus DS. TNFalpha and IL-17 cooperatively stimulate glucose metabolism and growth factor production in human colorectal cancer cells. Mol Cancer (2013) 12:78. doi: 10.1186/1476-4598-12-78
110. Wang Y, Du X, Wei J, Long L, Tan H, Guy C, et al. LKB1 orchestrates dendritic cell metabolic quiescence and anti-tumor immunity. Cell Res (2019) 29:391–405. doi: 10.1038/s41422-019-0157-4
111. Peng DH, Rodriguez BL, Diao L, Gaudreau PO, Padhye A, Konen JM, et al. Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers. Nat Commun (2021) 12:2606. doi: 10.1038/s41467-021-22875-w
112. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood (2008) 112:362–73. doi: 10.1182/blood-2007-11-120998
113. Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood (2009) 114:1141–9. doi: 10.1182/blood-2009-03-208249
114. Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med (2011) 3(104):104ra100. doi: 10.1126/scitranslmed.3002949
115. Knochelmann HM, Dwyer CJ, Smith AS, Bowers JS, Wyatt MM, Nelson MH, et al. IL6 fuels durable memory for th17 cell-mediated responses to tumors. Cancer Res (2020) 80:3920–32. doi: 10.1158/0008-5472.CAN-19-3685
116. Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol (2010) 184:1630–41. doi: 10.4049/jimmunol.0902813
117. Yi F-S, Zhai K, Shi H-Z. Helper T cells in Malignant pleural effusion. Cancer Lett (2021) 500:21–8. doi: 10.1016/j.canlet.2020.12.016
118. Yan J, Yu W, Lu C, Liu C, Wang G, Jiang L, et al. High ORAI3 expression correlates with good prognosis in human muscle-invasive bladder cancer. Gene (2022) 808:145994. doi: 10.1016/j.gene.2021.145994
119. Kirshberg S, Izhar U, Amir G, Demma J, Vernea F, Beider K, et al. Involvement of CCR6/CCL20/IL-17 axis in NSCLC disease progression. PloS One (2011) 6:e24856. doi: 10.1371/journal.pone.0024856
120. Li H, Liu H, Fu H, Li J, Xu L, Wang G, et al. Peritumoral tertiary lymphoid structures correlate with protective immunity and improved prognosis in patients with hepatocellular carcinoma. Front Immunol (2021) 12:648812. doi: 10.3389/fimmu.2021.648812
121. Melcher C, Yu J, Duong VHH, Westphal K, Helmi Siasi Farimany N, Shaverskyi A, et al. B cell-mediated regulatory mechanisms control tumor-promoting intestinal inflammation. Cell Rep (2022) 40:111051. doi: 10.1016/j.celrep.2022.111051
122. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of TH cells: A balancing act for cancer immunotherapy. Front Immunol (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
123. Cerboni S, Gehrmann U, Preite S, Mitra S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology (2021) 163:3–18. doi: 10.1111/imm.13280
124. Graf T, Enver T. Forcing cells to change lineages. Nature (2009) 462:587–94. doi: 10.1038/nature08533
125. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature (2015) 523:221–5. doi: 10.1038/nature14452
126. Zhang S. The role of transforming growth factor beta in T helper 17 differentiation. Immunology (2018) 155:24–35. doi: 10.1111/imm.12938
127. Downs-Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, Odunsi K, et al. Suppressive IL-17A(+)Foxp3(+) and ex-Th17 IL-17A(neg)Foxp3(+) T(reg) cells are a source of tumour-associated T(reg) cells. Nat Commun (2017) 8:14649. doi: 10.1038/ncomms14649
128. Thibaudin M, Chaix M, Boidot R, Vegran F, Derangere V, Limagne E, et al. Human ectonucleotidase-expressing CD25(high) Th17 cells accumulate in breast cancer tumors and exert immunosuppressive functions. Oncoimmunology (2016) 5:e1055444. doi: 10.1080/2162402X.2015.1055444
129. Roy D, Bose S, Pati S, Guin A, Banerjee K, Saha S, et al. GFI1/HDAC1-axis differentially regulates immunosuppressive CD73 in human tumor-associated FOXP3(+) Th17 and inflammation-linked Th17 cells. Eur J Immunol (2021) 51:1206–17. doi: 10.1002/eji.202048892
130. Soheilifar MH, Vaseghi H, Seif F, Ariana M, Ghorbanifar S, Habibi N, et al. Concomitant overexpression of mir-182-5p and mir-182-3p raises the possibility of IL-17-producing Treg formation in breast cancer by targeting CD3d, ITK, FOXO1, and NFATs: A meta-analysis and experimental study. Cancer Sci (2021) 112:589–603. doi: 10.1111/cas.14764
131. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity (2012) 36:362–73. doi: 10.1016/j.immuni.2011.12.019
132. Xia C, Yin S, To KKW, Fu L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer (2023) 22:44. doi: 10.1186/s12943-023-01733-x
133. Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell (2018) 34:757–774.e757. doi: 10.1016/j.ccell.2018.10.006
134. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35:972–85. doi: 10.1016/j.immuni.2011.09.019
135. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125:222–230.e221-224. doi: 10.1016/j.jaci.2009.10.012
136. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol (2014) 15:856–65. doi: 10.1038/ni.2947
137. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
138. Salazar Y, Zheng X, Brunn D, Raifer H, Picard F, Zhang Y, et al. Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J Clin Invest (2020) 130:3560–75. doi: 10.1172/JCI124037
139. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med (2013) 19:1114–23. doi: 10.1038/nm.3291
140. Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood (2003) 101:2620–7. doi: 10.1182/blood-2002-05-1461
141. Wang D, Yu W, Lian J, Wu Q, Liu S, Yang L, et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J Hematol Oncol (2020) 13(1):68. doi: 10.1186/s13045-020-00897-z
142. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther (2021) 6:263. doi: 10.1038/s41392-021-00658-5
143. Li K, Tandurella JA, Gai J, Zhu Q, Lim SJ, Thomas DL 2nd, et al. Multi-omic analyses of changes in the tumor microenvironment of pancreatic adenocarcinoma following neoadjuvant treatment with anti-PD-1 therapy. Cancer Cell (2022) 40:1374–1391 e1377. doi: 10.1016/j.ccell.2022.10.001
144. Bellone M, Brevi A, Huber S. Microbiota-propelled T helper 17 cells in inflammatory diseases and cancer. Microbiol Mol Biol Rev (2020) 84(2):e00064-19. doi: 10.1128/MMBR.00064-19
145. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood (2013) 121:2402–14. doi: 10.1182/blood-2012-09-378653
146. Liu HP, Cao AT, Feng T, Li Q, Zhang W, Yao S, et al. TGF-beta converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol (2015) 45:1010–8. doi: 10.1002/eji.201444726
147. Obermajer N, Dahlke MH. (Compl)Ex-Th17-Treg cell inter-relationship. Oncoimmunology (2016) 5:e1040217. doi: 10.1080/2162402X.2015.1040217
Keywords: Th17 cells, tumor microenvironment, anti-/pro-tumor, molecular regulation, transdifferentiation
Citation: Xing J, Man C, Liu Y, Zhang Z and Peng H (2023) Factors impacting the benefits and pathogenicity of Th17 cells in the tumor microenvironment. Front. Immunol. 14:1224269. doi: 10.3389/fimmu.2023.1224269
Received: 17 May 2023; Accepted: 07 August 2023;
Published: 23 August 2023.
Edited by:
Jiajia Zhou, University of Michigan, United StatesReviewed by:
Frederique Vegran, INSERM U1231 Lipides, Nutrition, Cancer (LNC), FranceCopyright © 2023 Xing, Man, Liu, Zhang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiyong Peng, cGVuZ2h1aXlvbmczMzgxNUAxNjMuY29t; Yingzhao Liu, empsaXV5aW5nemhhb0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.