 Olalekan Chris Akinsulie1*
Olalekan Chris Akinsulie1* Sammuel Shahzad1
Sammuel Shahzad1 Seto Charles Ogunleye2
Seto Charles Ogunleye2 Ifeoluwa Peace Oladapo1
Ifeoluwa Peace Oladapo1 Melina Joshi3
Melina Joshi3 Charles Egede Ugwu4
Charles Egede Ugwu4 Joy Olaoluwa Gbadegoye5
Joy Olaoluwa Gbadegoye5 Fasilat Oluwakemi Hassan5
Fasilat Oluwakemi Hassan5 Richard Adeleke6
Richard Adeleke6 Qudus Afolabi Akande7
Qudus Afolabi Akande7 Ridwan Olamilekan Adesola8
Ridwan Olamilekan Adesola8- 1Department of Veterinary Microbiology and Pathology, College of Veterinary Medicine, Washington State University, Pullman, WA, United States
- 2College of Veterinary Medicine, Mississippi State University, Starkville, MS, United States
- 3Center for Molecular Dynamics Nepal, Kathmandu, Nepal
- 4Paul G. Allen School for Global Health, Washington State University, Pullman, WA, United States
- 5Department of Physiology, University of Tennessee Health Science Center, Memphis, TN, United States
- 6College of Veterinary Medicine, Cornell University, Ithaca, NY, United States
- 7Department of Biological Sciences, University of Notre Dame, Notre Dame, IN, United States
- 8Faculty of Veterinary Medicine, University of Ibadan, Ibadan, Oyo State, Nigeria
There are overwhelming reports on the promotional effect of hypoxia on the malignant behavior of various forms of cancer cells. This has been proposed and tested exhaustively in the light of cancer immunotherapy. However, there could be more interesting functions of a hypoxic cellular micro-environment than malignancy. There is a highly intricate crosstalk between hypoxia inducible factor (HIF), a transcriptional factor produced during hypoxia, and nuclear factor kappa B (NF‐κB) which has been well characterized in various immune cell types. This important crosstalk shares common activating and inhibitory stimuli, regulators, and molecular targets. Impaired hydroxylase activity contributes to the activation of HIFs. Inflammatory ligands activate NF-κB activity, which leads to the expression of inflammatory and anti-apoptotic genes. The eventual sequelae of the interaction between these two molecular players in immune cells, either bolstering or abrogating functions, is largely cell-type dependent. Importantly, this holds promise for interesting therapeutic interventions against several infectious diseases, as some HIF agonists have helped prevent immune‐related diseases. Hypoxia and inflammation are common features of infectious diseases. Here, we highlighted the role of this crosstalk in the light of functional immunity against infection and inflammation, with special focus on various innate and adaptive immune cells. Particularly, we discussed the bidirectional effects of this crosstalk in the regulation of immune responses by monocytes/macrophages, dendritic cells, neutrophils, B cells, and T cells. We believe an advanced understanding of the interplay between HIFs and NF-kB could reveal novel therapeutic targets for various infectious diseases with limited treatment options.
Introduction
Mammalian cells generally adapt to hypoxia (low oxygen tension) by activating a major heterodimeric transcription factor known as hypoxia inducible factor (HIF)-1 (1). HIF-1 is composed of the two main subunits; HIF-1α and HIF-1β. HIF-1α levels are modulated by changes in oxygen partial pressure (pO2) within the cellular micro-environment. On the other hand, HIF-1β are constitutively expressed regardless of changes in the extracellular flux (2). HIF-1 regulates the robust expression of hypoxia-inducible genes (HIGs) downstream which are mainly involved in glycolysis (mediated by hexokinase), angiogenesis (mediated by vascular endothelial growth factor), erythropoiesis (mediated by erythropoietin), and cellular proliferation (mediated by adrenomedullin) (3). Mechanistically, HIF-1 activation requires the enzymatic inhibition of the prolyl hydroxylase domain-containing proteins (PHDs), a process which occurs strictly during hypoxia. In a normoxic (normal/optimum oxygen tension) situation, PHDs-mediated hydroxylation of the prolyl residues of the HIF-1α subunit ensures its binding to the von-Hippel-Lindau (VHL) protein, the recognition component of an E3 ubiquitin-protein ligase, leading to an eventual proteasomal degradation (2). However, hypoxia creates an inhibitory effect on PHD-hydroxylation thereby mediating the stabilization of HIF-1α, subsequent binding of the HIF-1 heterodimer to the promoter regions of HIGs resulting in the induction of those genes (4) (Figure 1A). Accumulating evidence from literature strongly suggest that HIF-1α plays a key role in the immune response to infection and inflammatory process in mammalian hosts (5–7). Essentially, HIF-1α is critical for the bactericidal activities of phagocytes like macrophages and neutrophils, and vital for inhibiting the systemic dissemination of certain bacteria (8). The role of nuclear factor kappa B (NF-kB) signaling pathway in infectious disease development and control has been well characterized (9, 10). Here, we discussed the interaction between HIF-1α and NF-kB, and their crosstalk within vital components of the innate immune system, highlighting the importance of this crosstalk as a plausible therapeutic target for infectious diseases.
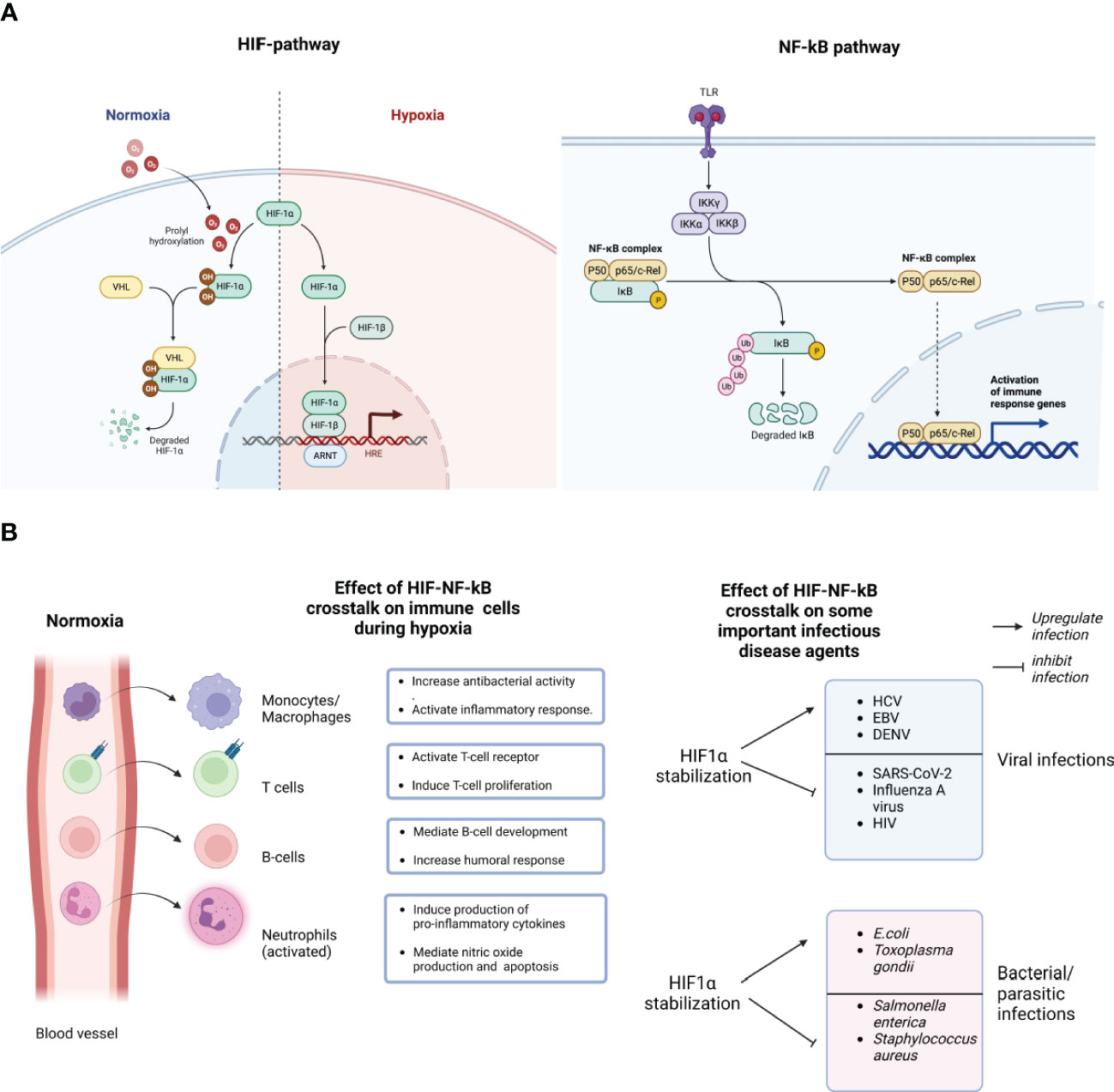
Figure 1 Schematic representation of the molecular crosstalk between the HIF-1a and NF-kB pathways. During normoxia (normal/optimum oxygen tension), prolyl hydroxylase domain (PHDs)-mediated hydroxylation of the prolyl residues of the HIF-1a subunit enables its binding to the von-Hippei-Lindau (VHL) protein, the recognition component of an E3 ubiquitin-protein ligase, resulting in proteasomal degradation. However, during hypoxia, HIF-1α activation requires the enzymatic inhibition of the PHDs-containing proteins. Hypoxia creates an inhibitory effect on PHD-hydroxylation, mediating the stabilization of HIF-1α, subsequent binding of the HIF-1 heterodimer to the promoter regions of HIGs resulting in the hypoxia inducible genes (A). The stabilization of HIF-1α stimulates NF-kB activation by negatively modulating the catalytic activity of the inhibitor of nuclear factor Kappa-b (IKKb). NF-kB has several sub-units like P50, P65 and c-Rel which can be activated by many stimuli, including bacterial lipopolysaccharide (LPS), viral pathogens, cytokines or growth factors, leading to the activation of Toll like receptors (TLR) (A). HIF-1α stabilization enhances the expression of NF-kB-regulated inflammatory cytokines from immune cells like macrophages, neutrophils, T and B cells, after lipopolysaccharides (LPS) stimulation, mediating NF-kB activation of those cells. As shown here, most bone marrow-derived phagocytic cells, essentially granulocytes like neutrophils, monocytes (macrophages in tissues), during peripheral circulation of oxygenated blood in blood vessels, sustain their bactericidal and pro-inflammatory abilities in a "naive state". However, following extravasation and upon invading an infected tissues, they encounter a robust decline in oxygen gradients, and a consequent bolstered activation of their bactericidal and pro-inflammatory abilities ("on-state") (B). Consequently, HIF-1α stabilization could either inhibit the infection and replication of some pathogens while upregulating others. Created with BioRender.com.
HIF-1α and NF-kB molecular crosstalk: broad effect on the immune system
The hypoxic response is functionally inter-connected with innate and adaptive immunity (11, 12). Interestingly, the interaction between these stress responses is essentially mediated by a molecular loop regulated by two important transcription factors, HIF-1α and NF-kB (13). Specifically, the stabilization of HIF-1α stimulates NF-kB activation by negatively modulating the catalytic activity of the inhibitor of nuclear factor Kappa-B (IKKb) (14). The hypoxia-induced activation of the NF-kB pathway characterized a while ago (15), shows a direct link between changes in the phosphorylation pattern of I kappa B alpha (IkB-alpha) with NF-kB activation. Following immunoprecipitation of IkB-alpha at different levels of hypoxic exposure, the finding showed an upregulated tyrosine phosphorylation (15). Consequently, the modulation in the phosphorylation of IkB-alpha prevents its degradation and ensures NF-kB binding (15). The ripple effect of this interaction can be seen in various immune cells. For example, bacteria-infected macrophages show a dysfunctional HIF-1α expression following the deletion of the IKKb-encoding gene (14). Also, the role of HIF-1α in enhancing the expression of NF-kB-regulated inflammatory cytokines from macrophages after lipopolysaccharides (LPS) stimulation, and mediating NF-kB activation in anoxic neutrophils has been thoroughly studied (16, 17). Briefly put, most bone marrow-derived phagocytic cells, essentially granulocytes like neutrophils, eosinophils, monocytes (macrophages in tissues), during peripheral circulation in oxygenated blood, sustain their bactericidal and pro-inflammatory abilities in a “naive state”. However, upon invading an infected tissue, they encounter a robust decline in oxygen gradients, and a consequent bolstered activation of their bactericidal and pro-inflammatory abilities (Figure 1B) (5). Generally, HIF activity is involved in a series of events promoting the release of proinflammatory cytokines and antimicrobial peptides, enhancing phagocytosis, sustaining phagocytes’ half-life by inhibiting apoptosis, and activating nitric oxide (NO) production (18). NO generally interferes with HIF degradation, causing an amplification loop for phagocyte activation, and the attendant immune response downstream (19). Interestingly, the relatively well-characterized ability of NF-kB to crosstalk with HIF in vivo and in vitro under inflammatory conditions is still majorly attributed to HIF-1α (5, 6). Very little attention has been given to HIF-1β and HIF-2α subunits in the context of inflammation and infection. Essentially, because both subunits have connections with NF-kB, this could be a premise for future studies. For example, studies evaluating the functional relatedness of HIF‐2α, HIF-1β and HIF‐1α, their roles in the regulation of infection and inflammatory process, and the involvement of other co-stimulatory factors, like pro-inflammatory cytokines, would bolster current molecular understanding of these transcriptional molecules.
HIF-1α and NF-kB crosstalk: effect on the innate and adaptive immune cells
Monocytes and macrophages
Macrophages are very important hematopoietic cells derived from circulating monocytes upon migration into tissues and becoming differentiated. These myeloid cells play a vital role both in innate and adaptive immune response (20). They initiate inflammatory responses, phagocytose, and destroy pathogens, and ensure lymphocytes recruitment. They also have the intrinsic ability to show extensive plasticity by undergoing two different types of activation: the classical M1 profile induced by Toll-like receptor (TLR) ligands, inducing a characteristic proinflammatory activity; and the alternative M2 profile, induced by specific interleukins like interleukin (IL)-4 and IL-13), which are largely involved in anti-inflammatory activities (21, 22). During an infection or inflammatory process, a typical host response is monocytosis; infiltration and accumulation of these myeloid cells into the infection and inflammatory site (23). Hypoxia represents an important factor modulating monocytopoiesis, specifically, the maturation from monocytes to macrophages (24). HIF-1α and TLRs bidirectionally interact on a plethora of biologically relevant levels (5). While HIF-1α regulates the surface expression of various TLRs (e.g., TLR-2, -4, -6, and -9), intracellular signal transduction of several TLRs as aforelisted is often mediated by HIF-1α. NF-kB is a major downstream player of this pathway (5). NF-kB remains a major control factor of the mammalian immune system and has been exhaustively reported as being strongly induced by LPS (25). Interestingly, LPS-induced HIF-1α activation is dependent on NF-kB in both human and murine monocytes (26, 27). Functionally, it’s been shown that in response to hypoxia, monocytes switch to a glycolytic metabolism thereby blocking their migration mediated by the chemotactic gradient (28). Gene expression modulations, such as the upregulation of essential molecules needed for macrophage survival, such as glucose transporter 1 (GLUT1) and their association with hypoxia has been well studied as well (29). Conversely, in both human and murine macrophages, hypoxia can initiate gene expression in a HIF-independent manner (14), however, not only through the upregulation of NF-kB, but also activation of transcription factor 4 (ATF4), and early growth response-1 (Egr-1) (30). The strict importance of HIF-1α for TLR activation could provide a plausible molecular explanation for the functional importance of HIF-1α in many pathogens’ defense.
Neutrophils
The interaction between NF-kB and neutrophils in a hypoxic cellular milieu is critical in influencing immunological responses and inflammatory processes (5). NF-κB regulates immune-related gene expression, and neutrophils, as major actors in innate immunity, are known for their quick reaction to infections and tissue injury (31). Neutrophils have a short lifespan and are highly motile cells, largely depending on glycolysis as their major energy source (32). As they migrate from the circulation to sites of inflammation, they are required to adapt to and function within lower oxygen tensions because the physiological oxygen gradient is often greatly exaggerated in disease situations. Hypoxia bolsters the association of HIF-1α with the p65 component of NF-kB in neutrophils, hence increasing NF-κB-dependent gene expression (33). Furthermore, the generation of reactive oxygen species (ROS) influences NF-κB activation in neutrophils under hypoxia (34). Hypoxia causes neutrophils to produce more ROS, which then activates NF-κB (34). A study on cancer cells revealed a positive feedback loop between ROS and NF-kB, demonstrating that hypoxia-induced ROS generation activates NF-κB in neutrophils, resulting in the overexpression of pro-inflammatory genes (35). Notably, the connection between NF-κB and neutrophils in a hypoxic milieu is bidirectional since NF-κB activation can impact neutrophil activities in a reciprocal manner (35). Neutrophil migration, survival, and cytokine production have all been demonstrated to be influenced by NF-kB signaling (36). Also, NF-κB activation in neutrophils under hypoxia improves survival by upregulating anti-apoptotic genes (37). HIF-1α-induced activation of NF-κB in neutrophils can also influence the production of cytokines and chemokines, which are important mediators of inflammatory reactions (35).
Dendritic cells
Dendritic cells (DCs) are vital antigen presenting cells (APCs) between the innate and adaptive immune system (38). Hypoxia and inflammation contribute significantly towards DC differentiation, migration, and survival, essentially mediated by HIF-1α (38). An important activity of DCs is migration to inflammatory sites, often characterized by low oxygen levels. Under this condition, there is an upregulation of HIF-1α levels, and immature myeloid DCs may undergo cell death due to hypoxia (39). However, DC maturation induced by LPS provides protection against hypoxia-induced cell death. This protective activity is mediated by the P13K/AKT pathway, a key intracellular signaling pathway involved in cell growth, motility, survival metabolism, and angiogenesis (39). Importantly, LPS stimulation in a hypoxic environment leads to higher levels of IL-6 and TNF compared to levels produced in normoxia (40). For the induction of TNF in hypoxic conditions, there is a need for HIF-dependent upregulation of MAP3K8 (MAPK kinase kinase 8), an upstream player in the p38/MAPK pathway (39). Conversely, murine experiments with HIF-1α-deficient DCs have shown a decrease in the expressions of IL-12p70, IL-10, TNF, IL-1b, and IL-23 following LPS stimulation in hypoxic bone marrow-derived DCs, strongly suggesting that the induction of these cytokines during hypoxia is independent of HIF-1α in mice (38). HIF-1α has been shown to be vital for DC migration, and the activation of the chemokine receptor CCR7, resulting in an increased glycolysis in humans (39). The elevated glycolysis is required for the rapid, yet transient, migration of DCs to draining lymph nodes, needed for the initiation of protective immune responses and the maintaining homeostasis.
T- cells
T cells are central players in adaptive immunity (41). In T cell development, NF-kB is a vital factor in the early antigen-independent phase of thymocyte differentiation and the final antigen-dependent lineage commitment and post-selection maturation (42). HIF-1α stabilization in T cells has been shown to occur in response to hypoxia and is essential for maintaining T cell homeostasis and functionality (43). Moreover, HIF-1α stabilization in T cells has been reported to bolster their survival and proliferation, as well as promote the production of effector cytokines, such as interferon-gamma (IFN-γ) and interleukin-17 (IL-17) (43). Furthermore, the interaction between HIFs and T cells in a hypoxic microenvironment influences T cell differentiation (44). HIF-1α has also been implicated in regulating T cell lineage commitment and polarization (44). Specifically, HIF-1α has been shown to promote the differentiation of T helper 17 (Th17) cells, a subset of CD4+ T cells that play a critical role in inflammatory responses and autoimmune diseases (45). The study by Dang et al. demonstrated that HIF-1α directly interacts with the transcription factor RORγt, a master regulator of Th17 cell differentiation, thereby promoting Th17 cell differentiation under hypoxic conditions (46). Moreover, HIF-1α and NF-kB molecular crosstalk can also influence regulatory T cell (Treg) function in a hypoxic microenvironment (47). Tregs are crucial for maintaining immune tolerance and suppressing excessive immune responses. HIF-1α has been found to modulate Treg stability and suppressive capacity (47). This crosstalk promotes the stability and immunosuppressive function of Tregs by upregulating the expression of Foxp3, a key transcription factor involved in Treg development and function (48). Considering the numerous shared activators, inhibitors, and molecular targets between HIF-1α and NF-kB, a deeper insight into their crosstalk in T-cell development and function would greatly advance efforts toward discovering novel targets for T-cell-mediated therapeutic development.
B cells
B cells are essential components of the adaptive immune system responsible for antibody-mediated immune responses (49). Currently, there is a dearth of information on the effect of the HIF-NF-kB crosstalk in B cells. However, in a hypoxic cellular microenvironment, the interaction between HIF-1 and B cells significantly modulates B cell development, activation, and antibody production (13). Understanding the interplay between HIF-1 and NF-kB in B cells in a hypoxic setting could provide valuable insights into the molecular mechanisms governing B cell responses and immune regulation. An interesting aspect of the interaction between HIF-1 and B cells in a hypoxic microenvironment is the effect of HIF-1 on B cell development and differentiation (12). Hypoxia has been implicated in regulating immunoglobulin production (50). Specifically, HIF-1α has been shown to promote the generation of B cell precursors and enhance their maturation into antibody-secreting plasma cells (50). Also, the molecular interaction between HIF-1α and B cells has been connected to immunoglobulin class switching (51). Moreover, HIF-1α has been shown to regulate the expression of key molecules involved in B cell activation, such as CD40 and CD86 (52).
HIF-NF-kB crosstalk in inflammatory conditions
The physiological process of inflammation is intricate and involves the activation of multiple coordinated signaling pathways in response to stress (53, 54). Typically, the response entails the expression of small peptides (e.g., cytokines), glycoproteins (e.g., the cluster of differentiation (CD)], and transcription factors, including NF-κB, which contribute to both anti-inflammatory and pro-inflammatory mediators. NF-κB is recognized as the primary pro-inflammatory family of transcription factors and has been identified as a direct modulator of HIF expression in the context of inflammation and hypoxia (53, 54). Generally, chronic inflammatory diseases are characterized by a significant interplay between HIF and NF-κB signaling pathways (53). Hypoxia-induced activation of HIF-dependent genes, along with impaired hydroxylase activity, contribute to the activation of HIF. Inflammatory ligands activate NF-κB activity, which leads to the expression of inflammatory and anti-apoptotic genes (53). For example, mice that overexpress HIF-1α in keratinocytes show enhanced NF-κB activity and elevated expression of pro-inflammatory genes resulting in heightened sensitivity to inflammatory stimuli (55). In contrast, mice lacking HIF-1α in neutrophils revealed the involvement of HIF-dependent NF-κB signaling in the regulation of neutrophil survival. The presence of common target genes and the physical interactions between HIF subunits and NF-κB remains the major reason contributing to their crosstalk in inflammatory settings (4). Studies on several inflammatory conditions, such as rheumatoid arthritis, inflammatory bowel disease, colorectal cancer, asthma, and many others, show the presence of potential points and the similarity in the cellular microenvironment in those conditions, which underscore the importance of the crosstalk between HIF and NF-κB (55–57).
HIF-NF-kB crosstalk in infectious disease conditions
There is growing evidence that the crosstalk between these two pathways may be critical in the pathogenesis of a range of infectious diseases, including viral, bacterial, and parasitic infections (58). Figure 1B provides a short list of viral, bacterial and parasitic infectious diseases that are impacted by hypoxia and consequently could be modulated, either inhibition or upregulation, by the molecular crosstalk between HIFs and NF-kB. Essentially, as described above, the hypoxic response is crucial for the optimum activities of tissue macrophages and infiltrating neutrophils that encounter low oxygen pressure in infected tissues. Also, HIF-1α has been proposed to bolster the expression of inflammatory cytokines that are known to be regulated by NF-κB, particularly in LPS-stimulated macrophages (59). It has been suggested that the activation of HIF-1α during Infection is particularly attributed to common mechanisms associated with the Infection, such as a hypoxic environment caused by bacterial oxygen consumption (59). The role of hypoxia on infectious agents is bidirectional. Several studies have shown the activation of HIF-1α in response to infections caused by many pathogens either upregulate or downregulates their replication in host cells (60, 61). On the other hand, it has been suggested that the presence of bacteria can also stabilize HIF-1α in both immune cells and epithelial cells (60). Upon stabilization and activation, HIF-1α initiates the release of anti-microbial peptides like cathelicidins and granule proteases, which facilitate the production of TNF- α and nitric oxide (62). Considering a highly debilitating infectious disease, tuberculosis caused by Mycobacterium tuberculosis (Mtb), NF-κB is involved in the transcriptional activation of HIF-1 (61). Studies have observed a higher expression of HIF-1α and related immune responses in Mtb infection both in vivo and in vitro (63, 64). When murine macrophages are infected with Mtb, inducible NO synthase (NOS2/iNOS) increases nitric oxide (NO), leading to the inhibition of PHDs and creating a positive feedback loop that results in sustained high levels of HIF-1α and increased macrophage activation (61). Through the regulation of HIF-1α activation in macrophages during microbial infections, which can result in decreased oxygen levels, NF-κB has the ability to boost glycolytic energy metabolism, the production of angiogenic factors, and the expression of pro-inflammatory cytokines, chemokines, and anti-microbial peptides. As a result, the ability of NF-κB to enhance the expression of HIF-1α broadens its regulatory capabilities, leading to the more effective execution of the host-defense response. Furthermore, modulations in oxygen tension and attendant HIF-1α signaling may also play a vital role in viral tropism and pathogenesis (65, 66). Therefore, pharmaceutical agents that modulate the HIFs pathway could provide novel treatment options for infections and associated pathological conditions. However, advanced investigations are required to decipher the complex interactions between different pathogen-specific genes and the crosstalk between HIFs and NF-kB. One aspect of advanced investigations involves unraveling the intricate interplay between HIFs and pathogen-specific genes. Different pathogens have evolved diverse mechanisms to interact with host cells and manipulate cellular processes. Understanding how HIFs modulate the expression of pathogen-specific genes is crucial for deciphering the underlying molecular mechanisms of pathogenesis and identifying potential therapeutic targets. This requires comprehensive studies using advanced genomics, and transcriptomics approaches to characterize the transcriptional changes induced by HIFs during Infection (66, 67).
Furthermore, investigating the crosstalk between HIFs and NF-kB is of paramount importance. Both HIFs and NF-kB are central players in inflammation and immune responses, and their interactions can significantly influence the outcome of infectious diseases. Deciphering the molecular mechanisms underlying their cross-regulation is essential to uncover the complex dynamics of host-pathogen interactions. Advanced investigations, including molecular and cellular studies, can shed light on the reciprocal regulation between HIFs and NF-kB, enabling the development of therapeutic strategies that target both pathways synergistically (68, 69). In addition, advanced investigations should explore the functional consequences of modulating HIFs on host immune responses. HIFs can modulate immune cell function, cytokine production, and anti-microbial responses, thereby influencing the outcome of infections. Elucidating the impact of HIFs modulation on immune cell behavior, including phagocytosis, antigen presentation, and T cell responses, is critical for understanding the immunological consequences of targeting the HIFs pathway. This can be achieved through advanced immunological techniques, such as flow cytometry, functional assays, and in vivo models (70, 71). Moreover, advanced investigations should incorporate systems biology approaches to gain a comprehensive understanding of the complex molecular networks involved in infectious diseases. Integrating multi-omics data, including genomics, transcriptomics, proteomics, and metabolomics, can provide a holistic view of the host-pathogen interactions including signaling cascades for different pathogens. These research along with computational analyses can unravel the intricate molecular pathways and identify key nodes or modules that can be targeted for therapeutic intervention (72, 73).
Concluding remarks and perspectives: HIF-NF-kB crosstalk as potential therapeutic targets
Developing pharmaceutical HIF-1α agonists to inhibit the microbial activity of pathogens in different cell types could be an exciting strategy for supplementary therapy against complex infections caused by antibiotic-resistant pathogens or treating infections in patients with compromised host immunity. As aforementioned, HIF-1α is increasingly being investigated as a potential target in the fight against bacterial, viral and parasitic infections, particularly multidrug-resistant ones. Modulating HIF-1α as a pharmacologic intervention for treating chronic inflammatory disorders or improving innate immune function has been exhaustively proposed (58). Moreover, inhibiting HIF-1α activity may be a promising therapeutic approach for sepsis triggered by LPS (74). To develop effective therapeutic strategies for infectious and inflammatory diseases, a solid grasp and understanding of the underlying biology is required (75). First, there is a need to identify novel genes and targets specific to various pathogens which can be modulated directly or indirectly by HIF-1α inhibition. Table 1A provides a list of genes that have been tested for various pathogens in the context of HIF-NF-kB crosstalk and therapeutic development. Moreover, in general terms, comprehensive research efforts are required to uncover specific molecules, receptors, and signaling pathways that play crucial roles in the establishment of Infection and evasion of host immune responses. Identifying these novel targets would enable the design of more precise and targeted therapeutic interventions. Second, an important point for consideration in therapeutic design is usually having a grasp of the dynamics of the pathogen’s variability. Infectious agents exhibit genetic and phenotypic variability, which can influence their virulence, drug resistance, and ability to evade host immune responses (75). Therefore, investigating the molecular mechanisms underlying pathogen variability and its impact on disease progression and therapeutic outcomes could be germane in this context. This includes studying genetic diversity, mutations, and genetic exchanges among pathogens, as well as characterizing their phenotypic changes and adaptation to different host environments (either in normoxia and hypoxic cellular environment). Additionally, as mentioned above, the integration of multi-omics approaches is necessary to gain a comprehensive understanding of the molecular pathways involved in infectious diseases. Utilizing genomics, transcriptomics, proteomics, and metabolomics can provide insights into the complex interactions between host and pathogen (75). Data garnered from these advanced approaches, together with computational modeling and network analysis could provide a robust understanding of the effect of the HIF-NF-kB crosstalk on infectious disease processes. This integrative approach is vital for obtaining a holistic view of the underlying biology and identifying novel therapeutic targets. Lastly, it is crucial to validate findings from preclinical studies and translate them into clinical applications. While in vitro and animal studies provide valuable insights, it is essential to validate the efficacy and safety of potential therapeutic strategies in human subjects. Table 1B shows a list of current HIF-1α inhibitory or upregulating molecules and drugs currently under clinical trials. These studies would ideally involve conducting well-designed clinical trials to assess the therapeutic potential of identified targets. For this, physiologically relevant disease models can be employed to answer the needed research questions. In this regard, novel animal models such as the zebrafish, in combination with traditional cell lines and murine models, could help to elucidate the extent of the effects of HIF modulation on cellular physiology (157). We strongly believe targeting HIF-NF-kB crosstalk holds promise for therapeutic interventions for various microbial infections. Studies employing genetic and chemical inhibition of the PHD proteins have demonstrated therapeutic benefits in various disease models, such as colitis, where inflammation and NF-kB activity are significant contributors (158). Therefore, it would be intriguing to investigate the potential therapeutic impact of HIF activators and inhibitors on other infectious agents. More research efforts are needed to test the efficacy of available HIF modulators in different disease models, which could provide valuable insights into the various forms of HIF-NF-kB crosstalk that occur in different cell types in the context of individual pathogen. Collectively, a progressive understanding of the interplay between HIF-1 and NF-κB pathways through hydroxylation (159) can lead to the development of novel therapies for infectious and inflammatory disorders.
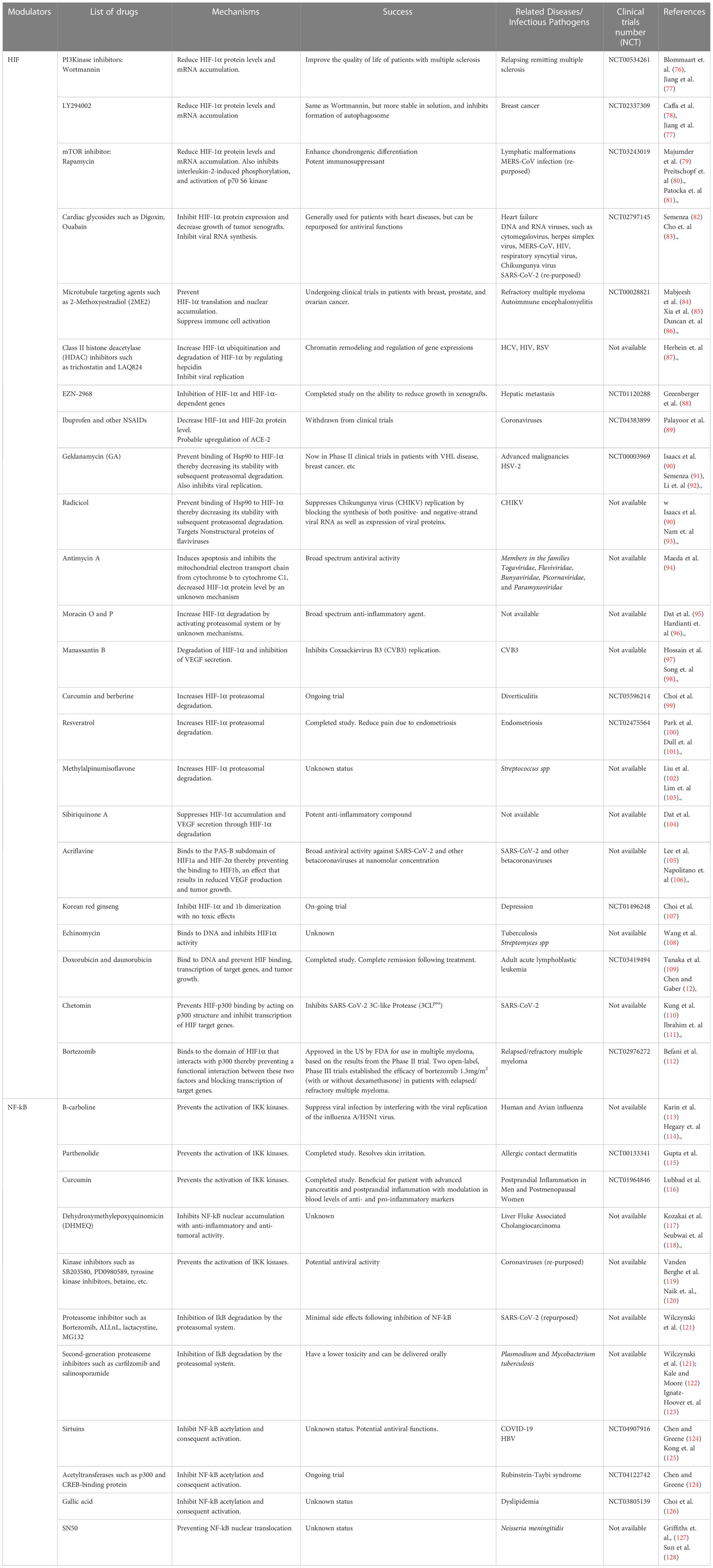
Table 1A Table showing the effects of hypoxia on genes of various infectious agents.

Table 1B Table showing a list of HIF-NF-kB modulators highlighting their mechanism of action, accompany Clinical trial numbers (NTC), and related pathogen/diseases.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
Conceptualization: OCA. Figure and Table: OCA, SS, SCO, RA, ROA. Article draft and review: All authors contributed to the writing and review of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through HIFs and beyond. Nat Rev Mol Cell Biol (2020) 21(5):268. doi: 10.1038/s41580-020-0227-y
2. Ziello JE, Jovin IS, Huang Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med (2007) 80(2):51–60.
3. Vadlapatla RK, Vadlapudi AD, Mitra AK. Hypoxia-inducible factor-1 (HIF-1): a potential target for intervention in ocular neovascular diseases. Curr Drug Targets (2013) 14(8):919. doi: 10.2174/13894501113149990015
4. Wu L, He Y, Zhu L. Possible role of PHD inhibitors as hypoxia-mimicking agents in the maintenance of neural stem cells’ self-renewal properties. Front Cell Dev Biol (2018) 6:169. doi: 10.3389/fcell.2018.00169
5. McGettrick AF, O’Neill LA. The role of HIF in immunity and inflammation. Cell Metab (2020) 32(4):524–36. doi: 10.1016/j.cmet.2020.08.002
6. Tian M, Liu W, Li X, Zhao P, Shereen MA, Zhu C, et al. HIF-1α promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct Targeted Ther (2021) 6(1):1–13. doi: 10.1038/s41392-021-00726-w
7. Lei X, Teng W, Fan Y, Zhu Y, Yao L, Li Y, et al. The protective effects of HIF-1α activation on sepsis induced intestinal mucosal barrier injury in rats model of sepsis. PloS One (2022) 17(5):e0268445. doi: 10.1371/journal.pone.0268445
8. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest (2005) 115(7):1806–15. doi: 10.1172/JCI23865
9. Gudowska-Sawczuk M, Mroczko B. The role of nuclear factor kappa b (NF-κB) in development and treatment of COVID-19: review. Int J Mol Sci (2022) 23(9). doi: 10.3390/ijms23095283
10. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Targeted Ther (2020) 5(1):1–23. doi: 10.1038/s41392-020-00312-6
11. Harris AJ, Thompson AR, Whyte MK, Walmsley SR. HIF-mediated innate immune responses: cell signaling and therapeutic implications. Hypoxia (2013) 2:47–58. doi: 10.2147/HP.S50269
12. Chen Y, Gaber T. Hypoxia/HIF modulates immune responses. Biomedicines (2021) 9(3):260. doi: 10.3390/biomedicines9030260
13. Zhang J, Wu X, Ma J, Long K, Sun J, Li M, et al. Hypoxia and hypoxia-inducible factor signals regulate the development, metabolism, and function of b cells. Front Immunol (2022) 13:967576. doi: 10.3389/fimmu.2022.967576
14. Bandarra D, Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J (2016) 283(3):413–24. doi: 10.1111/febs.13578
15. Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappa b through the phosphorylation of I kappa b alpha on tyrosine residues. Cancer Res (1994) 54(6):1425–30.
16. Scholz CC, Cavadas MA, Tambuwala MM, Hams E, Rodríguez J, Kriegsheim AV, et al. Regulation of IL-1β–induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci (2013) 110(46):18490–5. doi: 10.1073/pnas.1309718110
17. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD-L1 expression by NF-κB in cancer. Front Immunol (2020) 11:584626. doi: 10.3389/fimmu.2020.584626
18. Kumar R, Singh P, Kolloli A, Shi L, Bushkin Y, Tyagi S, et al. Immunometabolism of phagocytes during mycobacterium tuberculosis infection. Front Mol Biosci (2019) 6:105. doi: 10.3389/fmolb.2019.00105
19. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol (2009) 9(9):609. doi: 10.1038/nri2607
20. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci (2018) 19(1). doi: 10.3390/ijms19010092
21. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci (2018) 19(6). doi: 10.3390/ijms19061801
22. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado D, Popovich PG, Partida-Sanchez S. Novel markers to delineate murine M1 and M2 macrophages. PloS One (2015) 10(12):e0145342. doi: 10.1371/journal.pone.0145342
23. Shi C. & pamer, e. g. monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762. doi: 10.1038/nri3070
24. Strehl C, Fangradt M, Fearon U, Gaber T, Buttgereit F, Veale DJ. Hypoxia: how does the monocyte-macrophage system respond to changes in oxygen availability? J Leukocyte Biol (2014) 95(2):233–41. doi: 10.1189/jlb.1212627
25. Liu T, Zhang L, Joo D, Sun C. NF-κB signaling in inflammation. Signal Transduct Targeted Ther (2016) 2:17023. doi: 10.1038/sigtrans.2017.23
26. Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem J (2006) 396(Pt 3):517–27. doi: 10.1042/BJ20051839
27. Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr topics Microbiol Immunol (2009) 345:105. doi: 10.1007/82_2010_74
28. Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation – a potential mechanism. Eur J Immunol (2001) 31(2):480–9. doi: 10.1002/1521-4141(200102)31:2<480::AID-IMMU480>3.0.CO;2-L
29. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun (2016) 7(1):1–10. doi: 10.1038/ncomms11635
30. Pang Z, Xu Y, Zhu Q. Early growth response 1 suppresses macrophage phagocytosis by inhibiting NRF2 activation through upregulation of autophagy during pseudomonas aeruginosa infection. Front Cell Infection Microbiol (2022) 11:773665. doi: 10.3389/fcimb.2021.773665
31. Selders GS, Fetz AE, Radic MZ, Bowlin GL. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regenerative Biomater (2017) 4(1):55–68. doi: 10.1093/rb/rbw041
32. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int J Mol Sci (2020) 21(1). doi: 10.3390/ijms21010287
33. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J Experimental Med (2005) 201(1):105–15. doi: 10.1084/jem.20040624
34. LU J, LIU J, LI A. Roles of neutrophil reactive oxygen species (ROS) generation in organ function impairment in sepsis. J Zhejiang Univ Sc B (2022) 23(6):437–50. doi: 10.1631/jzus.B2101075
35. Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res (2014) 2(9):823. doi: 10.1158/2326-6066.CIR-14-0112
36. Lodge KM, Cowburn AS, Li W, Condliffe AM. The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci (2020) 21(4). doi: 10.3390/ijms21041183
37. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol (2019) 10:85. doi: 10.3389/fimmu.2019.00085
38. Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol (2012) 42(5):1226–36. doi: 10.1002/eji.201142053
39. Paardekooper LM, Bendix MB, Ottria A, Radstake DJ, Marut W. Hypoxia potentiates monocyte-derived dendritic cells for release of tumor necrosis factor α via MAP3K8. Biosci Rep (2018) 38(6). doi: 10.1042/BSR20182019
40. Krzywinska E, Stockmann C. Hypoxia, metabolism and immune cell function. Biomedicines (2018) 6(2):56. doi: 10.3390/biomedicines6020056
41. DeMaio A, Mehrotra S, Sambamurti K, Husain S. The role of the adaptive immune system and T cell dysfunction in neurodegenerative diseases. J Neuroinflamm (2022) 19:251. doi: 10.1186/s12974-022-02605-9
42. Xing Y, Wang X, Jameson SC, Hogquist KA. Late stages of T cell maturation in the thymus involve NF-κB and tonic type I interferon signaling. Nat Immunol (2016) 17(5):565. doi: 10.1038/ni.3419
43. Tao H, Barbi J, Pan F. Hypoxia-inducible factors in T lymphocyte differentiation and function. a review in the theme: cellular responses to hypoxia. Am J Physiol - Cell Physiol (2015) 309(9):C580. doi: 10.1152/ajpcell.00204.2015
44. Palazon A, Goldrath A, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41(4):518. doi: 10.1016/j.immuni.2014.09.008
45. Arias C, Sepúlveda P, Castillo RL, Salazar LA. Relationship between hypoxic and immune pathways activation in the progression of neuroinflammation: role of HIF-1α and Th17 cells. Int J Mol Sci (2023) 24(4). doi: 10.3390/ijms24043073
46. Dang EV, Barbi J, Yang Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772. doi: 10.1016/j.cell.2011.07.033
47. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol (2022) 15:104. doi: 10.1186/s13045-022-01322-3
48. Barbi J, Pardoll DM, Pan F. Ubiquitin-dependent regulation of Foxp3 and treg function. Immunol Rev (2015) 266(1):27. doi: 10.1111/imr.12312
49. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2018) 14(Suppl 2):49. doi: 10.1186/s13223-018-0278-1
50. Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal center hypoxia and regulation of antibody qualities by a hypoxia response system. Nature (2016) 537(7619):234. doi: 10.1038/nature19334
51. Abbott RK, Thayer M, Labuda J, Silva M, Philbrook P, Cain DW, et al. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J Immunol (Baltimore Md. 1950) (2016) 197(10):4014. doi: 10.4049/jimmunol.1601401
52. Hazrati A, Soudi S, Malekpour K, Mahmoudi M, Rahimi A, Hashemi SM, et al. Immune cells-derived exosomes function as a double-edged sword: role in disease progression and their therapeutic applications. Biomark Res (2022) 10:30. doi: 10.1186/s40364-022-00374-4
53. Batie M, Rocha S. Hypoxia and inflammation in cancer, focus on HIF and NF-κB. Biomedicines (2017) 5(2). doi: 10.3390/biomedicines5020021
54. Scortegagna M, Cataisson C, Martin RJ, Hicklin DJ, Schreiber RD, Yuspa SH, et al. HIF-1α regulates epithelial inflammation by cell autonomous NF-κB activation and paracrine stromal remodeling. Blood (2008) 111(7):3343–54. doi: 10.1182/blood-2007-10-115758
55. BIDDLESTONE J, BANDARRA D, ROCHA S. The role of hypoxia in inflammatory disease (Review). Int J Mol Med (2015) 35(4):859–69. doi: 10.3892/ijmm.2015.2079
56. Pham K, Parikh K, Heinrich EC. Hypoxia and inflammation: insights from high-altitude physiology. Front Physiol (2021) 12:676782. doi: 10.3389/fphys.2021.676782
57. Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med (2019) 25(1):33–46. doi: 10.1016/j.molmed.2018.10.006
58. Santos SAD, Andrade DR,Júnior. HIF-1alpha and infectious diseases: a new frontier for the development of new therapies. Rev do Instituto Medicina Trop Sao Paulo (2017) 59:e92. doi: 10.1590/S1678-9946201759092
59. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
60. Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A, et al. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PloS One (2010) 5(7):e11576. doi: 10.1371/journal.pone.0011576
61. Zenk SF, Hauck S, Mayer D, Grieshober M, Stenger S. Stabilization of hypoxia-inducible factor promotes antimicrobial activity of human macrophages against mycobacterium tuberculosis. Front Immunol (2020) 12:678354. doi: 10.3389/fimmu.2021.678354
62. Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med (2007) 85:1339–46. doi: 10.1007/s00109-007-0282-2
63. Liu R, Muliadi V, Mou W, Li H, Yuan J, Holmberg J, et al. HIF-1 stabilization in T cells hampers the control of mycobacterium tuberculosis infection. Nat Commun (2021) 13. doi: 10.1038/s41467-022-32639-9
64. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α is an essential mediator of IFN-γ dependent immunity to mycobacterium tuberculosis. J Immunol (Baltimore Md. 1950) (2016) 197(4):1287. doi: 10.4049/jimmunol.1600266
65. Liu PJ, Balfe P, McKeating JA, Schilling M. Oxygen sensing and viral replication: implications for tropism and pathogenesis. Viruses (2020) 12(11). doi: 10.3390/v12111213
66. Wing PAC, Prange-Barczynska M, Cross A, Crotta S, Orbegozo Rubio C, Cheng X, et al. Hypoxia inducible factors regulate infectious SARS-CoV-2, epithelial damage and respiratory symptoms in a hamster COVID-19 model. PloS Pathog (2022) 18(9):e1010807. doi: 10.1371/journal.ppat.1010807
67. Norman TM, Lord ND, Paulsson J, Losick R. Stochastic switching of cell fate in microbes. Annu Rev Microbiol (2015) 69:381–403. doi: 10.1146/annurev-micro-091213-112852
68. Le Moan N, Baeten KM, Rafalski VA, Ryu JK, Rios Coronado PE, Bedard C, et al. Hypoxia inducible factor-1α in astrocytes and/or myeloid cells is not required for the development of autoimmune demyelinating disease. eNeuro (2015) 2(2). doi: 10.1523/ENEURO.0050-14.2015a. ENEURO.0050-14.2015.
69. Vaupel P, Schmidberger H, Mayer A. The warburg effect: an essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
70. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1 alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57. doi: 10.1016/s0092-8674(03)00154-5
71. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
72. Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship? Mol Cell Biochem (2010) 336(1-2):25–37. doi: 10.1007/s11010-009-0267-2
73. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol (2019) 20(7):436–50. doi: 10.1038/s41580-019-0123-5
74. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol (Baltimore Md. 1950) (2007) 178(12):7516–9. doi: 10.4049/jimmunol.178.12.7516
75. Cohen J. The immunopathogenesis of sepsis. Nature (2002) 420(6917):885–91. doi: 10.1038/nature01326
76. Dull M, Moga MA, Dimienescu OG, Sechel G, Burtea V, Anastasiu CV. Therapeutic approaches of resveratrol on endometriosis via anti-inflammatory and anti-angiogenic pathways. Molecules (2019) 24(4). doi: 10.3390/molecules24040667
77. Liu Y, Veena CK, Morgan JB, Mohammed KA, Jekabsons MB, Nagle DG, et al. Methylalpinumisoflavone inhibits hypoxia-inducible factor-1 (HIF-1) activation by simultaneously targeting multiple pathways. J Biol Chem (2009) 284(9):5859–68. doi: 10.1074/jbc.M806744200
78. Lim JW, Jo YH, Choi JS, Lee MK, Lee KY, Kang SY. Antibacterial activities of prenylated isoflavones from Maclura tricuspidata against fish pathogenic Streptococcus: their structure-activity relationships and extraction optimization. Molecules (Basel Switzerland) (2021) 26(24):7451. doi: 10.3390/molecules26247451
79. Dat NT, Jin X, Lee JH, Lee D, Hong YS, Lee K, et al. Abietane diterpenes from salvia miltiorrhiza inhibit the activation of hypoxia-inducible factor-1. J Nat Prod (2007) 70(7):1093–7. doi: 10.1021/np060482d
80. Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA (2009) 106(42):17910–5. doi: 10.1073/pnas.0909353106
81. Napolitano V, Dabrowska A, Schorpp K, Mourão A, Barreto-Duran E, Benedyk M, et al. Acriflavine, a clinically approved drug, inhibits SARS-CoV-2 and other betacoronaviruses. Cell Chem Biol (2022) 29(5):774–84. doi: 10.1016/j.chembiol.2021.11.006
82. Choi YJ, Choi H, Cho CH, Park JW. Red ginseng deregulates hypoxia-induced genes by dissociating the HIF-1 dimer. J Nat Med (2011) 65(2):344–52. doi: 10.1007/s11418-010-0504-8
83. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF-1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell (2011) 8(4):399–411. doi: 10.1016/j.stem.2011.02.006
84. Tanaka T, Yamaguchi J, Shoji K, Nangaku M. Anthracycline inhibits recruitment of hypoxia-inducible transcription factors and suppresses tumor cell migration and cardiac angiogenic response in the host. J Biol Chem (2012) 287(42):34866–82. doi: 10.1074/jbc.M112.374587
85. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell (2004) 6(1):33–43. doi: 10.1016/j.ccr.2004.06.009
86. Ibrahim MAA, Abdelrahman AHM, Mohamed DEM, Abdeljawaad KAA, Naeem MA, Gabr GA, et al. Chetomin, a SARS-CoV-2 3C-like protease (3CLpro) inhibitor: in silico screening, enzyme docking, molecular dynamics and pharmacokinetics analysis. Viruses (2023) 15(1). doi: 10.3390/v15010250
87. Befani CD, Vlachostergios PJ, Hatzidaki E, Patrikidou A, Bonanou S, Simos G, et al. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med (Berl) (2012) 90(1):45–54. doi: 10.1007/s00109-011-0805-8
88. Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa b system: a treasure trove for drug development. Nat Rev Drug Discov (2004) 3(1):17–26. doi: 10.1038/nrd1279
89. Hegazy A, Mahmoud SH, Elshaier YA, Shama NM, Nasr NF, Ali MA, et al. Antiviral activities of plant-derived indole and β-carboline alkaloids against human and avian influenza viruses. Sci Rep (2023) 13(1):1–15. doi: 10.1038/s41598-023-27954-0
90. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta (2010) 1799(10-12):775–87. doi: 10.1016/j.bbagrm.2010.05.004
91. Lubbad A, Oriowo MA, Khan I. Curcumin attenuates inflammation through inhibition of TLR-4 receptor in experimental colitis. Mol Cell Biochem (2009) 322(1-2):127–35. doi: 10.1007/s11010-008-9949-4
92. Kozakai N, Kikuchi E, Hasegawa M, Suzuki E, Ide H, Miyajima A, et al. Enhancement of radiosensitivity by a unique novel NF-κB inhibitor, DHMEQ, in prostate cancer. Br J Cancer (2012) 107(4):652–7. doi: 10.1038/bjc.2012.321
93. Seubwai W, Wongkham C, Puapairoj A, Khuntikeo N, Pugkhem A, Hahnvajanawong C, et al. Aberrant expression of NF-κB in liver fluke associated cholangiocarcinoma: implications for targeted therapy. PloS One (2014) 9(8):e106056. doi: 10.1371/journal.pone.0106056
94. Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem (1998) 273(6):3285–90. doi: 10.1074/jbc.273.6.3285
95. Wilczynski J, Duechler M, Czyz M. Targeting NF-κB and HIF-1 pathways for the treatment of cancer: part I. Arch Immunol Ther Exp (Warsz) (2011) 59(4):289–99. doi: 10.1007/s00005-011-0131-4
96. Kale AJ, Moore BS. Molecular mechanisms of acquired proteasome inhibitor resistance. J Med Chem (2012) 55(23):10317–27. doi: 10.1021/jm300434z
97. Ignatz-Hoover JJ, Murphy EV, Driscoll JJ. Targeting proteasomes in cancer and infectious disease: a parallel strategy to treat malignancies and microbes. Front Cell Infection Microbiol (2021) 12:925804. doi: 10.3389/fcimb.2022.925804
98. Legendre C, Reen FJ, Mooij MJ, McGlacken GP, Adams C, O'Gara F. Pseudomonas aeruginosa alkyl quinolones repress hypoxia-inducible factor 1 (HIF-1) signaling through HIF-1α degradation. Infect Immun (2012) 80(11):3985–92. doi: 10.1128/IAI.00554-12
99. Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol (2004) 5(5):392–401. doi: 10.1038/nrm1368
100. Kong F, Li Q, Zhang F, Li X, You H, Pan X, et al. Sirtuins as potential therapeutic targets for hepatitis b virus infection. Front Med (2021) 8:751516. doi: 10.3389/fmed.2021.751516
101. Choi KC, Lee YH, Jung MG, Kwon SH, Kim MJ, Jun WJ, et al. Gallic Acid suppresses lipopolysaccharide-induced nuclear factor-kappaB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol Cancer Res (2009) 7(12):2011–21. doi: 10.1158/1541-7786.MCR-09-0239
102. Storey J, Gobbetti T, Olzinski A, Berridge BR. A structured approach to optimizing animal model selection for human translation: the animal model quality assessment. ILAR J (2020) 62(1-2):66–76. doi: 10.1093/ilar/ilac004
103. Tambuwala MM, Manresa MC, Cummins EP, Aversa V, Coulter IS, Taylor CT. Targeted delivery of the hydroxylase inhibitor DMOG provides enhanced efficacy with reduced systemic exposure in a murine model of colitis. J Controlled release Off J Controlled Release Soc (2015) 217:221–7. doi: 10.1016/j.jconrel.2015.09.022
104. Masson N, Ratcliffe PJ. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J Cell Sci (2003) 116(Pt 15):3041–9. doi: 10.1242/jcs.00655
105. Kaihami GH, Breda LCD, de Almeida JRF, de Oliveira Pereira T, Nicastro GG, Boechat AL, et al. The atypical response regulator AtvR is a new player in pseudomonas aeruginosa response to hypoxia and virulence. Infect Immun (2017) 85(8):e00207–17. doi: 10.1128/IAI.00207-17
106. Crocker AW, Harty CE, Hammond JH, Willger SD, Salazar P, Botelho NJ, et al. Pseudomonas aeruginosa ethanol oxidation by AdhA in low-oxygen environments. J Bacteriol (2019) 201(23). doi: 10.1128/JB.00393-19
107. Schaible B, Crifo B, Schaffer K, Taylor CT. The putative bacterial oxygen sensor pseudomonas prolyl hydroxylase (PPHD) suppresses antibiotic resistance and pathogenicity in pseudomonas aeruginosa. J Biol Chem (2020) 295(5):1195–201. doi: 10.1074/jbc.RA119.010033
108. Hayek I, Fischer F, Schulze-Luehrmann J, Dettmer K, Sobotta K, Schatz V, et al. Limitation of TCA cycle intermediates represents an oxygen-independent nutritional antibacterial effector mechanism of macrophages. Cell Rep (2019) 26(13):3502–3510.e6. doi: 10.1016/j.celrep.2019.02.103
109. Riess T, Andersson GE, Lupas A, Schaller M, Schäfer A, Kyme P, et al. Bartonella adhesin a mediates a proangiogenic host cell response. J Exp Med (2004) 200(10):1267–78. doi: 10.1084/jem.20040500
110. Pasipoularides A, Murgo JP, Miller JW, Craig WE. Nonobstructive left ventricular ejection pressure gradients in man. Circ Res (1987) 61(2):220–7. doi: 10.1161/01.res.61.2.220
111. Chen T, He L, Deng W, Xie J. The mycobacterium DosR regulon structure and diversity revealed by comparative genomic analysis. J Cell Biochem (2013) 114(1):1–6. doi: 10.1002/jcb.24302
112. Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, et al. The mycobacterium tuberculosis regulatory network and hypoxia. Nature (2013) 499(7457):178–83. doi: 10.1038/nature12337
113. McGillivray A, Golden NA, Kaushal D. The mycobacterium tuberculosis clp gene regulator is required for in vitro reactivation from hypoxia-induced dormancy. J Biol Chem (2015) 290(4):2351–67. doi: 10.1074/jbc.M114.615534
114. Eoh H, Wang Z, Layre E, Rath P, Morris R, Moody DB, et al. Metabolic anticipation in mycobacterium tuberculosis. Nat Microbiol (2017) 2:17084. doi: 10.1038/nmicrobiol.2017.84
115. Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol (2020) 17(6):613–20. doi: 10.1038/s41423-020-0400-4
116. Gutierrez-Rodarte M, Kolappan S, Burrell BA, Craig L. The Vibrio cholerae minor pilin TcpB mediates uptake of the cholera toxin phage CTXφ. J Biol Chem (2019) 294(43):15698–710. doi: 10.1074/jbc.RA119.009980
117. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology (2014) 146(6):1489–99. doi: 10.1053/j.gastro.2014.02.009
118. Kinkel TL, Roux CM, Dunman PM, Fang FC. The staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. mBio (2013) 4(6):e00696–13. doi: 10.1128/mBio.00696-13
119. Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, et al. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem (2003) 278(33):31277–85. doi: 10.1074/jbc.M300763200
120. Frakolaki E, Kaimou P, Moraiti M, Kalliampakou KI, Karampetsou K, Dotsika E, et al. The role of tissue oxygen tension in dengue virus replication. Cells (2018) 7(12). doi: 10.3390/cells7120241
121. Charles S, Ammosova T, Cardenas J, Foster A, Rotimi J, Jerebtsova M, et al. Regulation of HIV-1 transcription at 3% versus 21% oxygen concentration. J Cell Physiol (2009) 221(2):469. doi: 10.1002/jcp.21882
122. Deshmane SL, Amini S, Sen S, Khalili K, Sawaya BE. Regulation of the HIV-1 promoter by HIF-1α and vpr proteins. Virol J (2011) 8:477. doi: 10.1186/1743-422X-8-477
123. Connor JH, Naczki C, Koumenis C, Lyles DS. Replication and cytopathic effect of oncolytic vesicular stomatitis virus in hypoxic tumor cells In vitro and In vivo. J Virol (2004) 78(17):8960–70. doi: 10.1128/JVI.78.17.8960-8970.2004
124. Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, et al. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol (2009) 297(4):L631–40. doi: 10.1152/ajplung.90415.2008
125. Jiang JH, Wang N, Li A, Liao WT, Pan ZG, Mai SJ, et al. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. J Clin Virol Off Publ Pan Am Soc Clin Virol (2006) 37(2):98–103. doi: 10.1016/j.jcv.2006.06.013
126. Aghi MK, Liu TC, Rabkin S, Martuza RL. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol Ther J Am Soc Gene Ther (2009) 17(1):51–6. doi: 10.1038/mt.2008.232
127. Shen BH, Hermiston TW. Effect of hypoxia on Ad5 infection, transgene expression and replication. Gene Ther (2005) 12(11):902–10. doi: 10.1038/sj.gt.3302448
128. Singh AK, Mukhopadhyay C, Biswas S, Singh VK, Mukhopadhyay CK. Intracellular pathogen leishmania donovani activates hypoxia inducible factor-1 by dual mechanism for survival advantage within macrophage. PloS One (2012) 7(6):e38489. doi: 10.1371/journal.pone.0038489
129. Saraiva FMS, Cosentino-Gomes D, Inacio JDF, Almeida-Amaral EE, Louzada-Neto O, Rossini A, et al. Hypoxia effects on trypanosoma cruzi epimastigotes proliferation, differentiation, and energy metabolism. Pathogens (2022) 11(8):897. doi: 10.3390/pathogens11080897
130. Wiley M, Sweeney KR, Chan DA, Brown KM, McMurtrey C, Howard EW, et al. Toxoplasma gondii activates hypoxia-inducible factor (HIF) by stabilizing the HIF-1α subunit via type I activin-like receptor kinase receptor signaling. J Biol Chem (2010) 285(35):26852–60. doi: 10.1074/jbc.M110.147041
131. Ng S, March S, Galstian A, Hanson K, Carvalho T, Mota MM, et al. Hypoxia promotes liver-stage malaria infection in primary human hepatocytes in vitro. Dis Models Mech (2014) 7(2):215–24. doi: 10.1242/dmm.013490
132. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem (1997) 243(1-2):240–6. doi: 10.1111/j.1432-1033.1997.0240a.x
133. Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ (2001) 12(7):363–9.
134. Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature (2020) 583(7817):620–4. doi: 10.1038/s41586-020-2502-7
135. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, et al. mTOR inhibition reverses akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med (2004) 10(6):594–601. doi: 10.1038/nm1052
136. Preitschopf A, Schörghofer D, Kinslechner K, Schütz B, Zwickl H, Rosner M, et al. Rapamycin-induced hypoxia inducible factor 2A is essential for chondrogenic differentiation of amniotic fluid stem cells. Stem Cells Trans Med (2016) 5(5):580–90. doi: 10.5966/sctm.2015-0262
137. Patocka J, Kuca K, Oleksak P, Nepovimova E, Valis M, Novotny M, et al. Rapamycin: drug repurposing in SARS-CoV-2 infection. Pharmaceuticals (2021) 14(3). doi: 10.3390/ph14030217
138. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33(4):207–14. doi: 10.1074/jbc.271.51.32529
139. Cho J, Lee YJ, Kim JH, Kim SI, Kim SS, Choi BS, et al. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci Rep (2020) 10(1):16200. doi: 10.1038/s41598-020-72879-7
140. Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell (2003) 3(4):363–75. doi: 10.1016/s1535-6108(03)00077-1
141. Xia Y, Choi HK, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem (2012) 49:24–40. doi: 10.1016/j.ejmech.2012.01.033
142. Duncan GS, Brenner D, Tusche MW, Brüstle A, Knobbe CB, Elia AJ, et al. 2-methoxyestradiol inhibits experimental autoimmune encephalomyelitis through suppression of immune cell activation. Proc Natl Acad Sci United States America (2012) 109(51):21034–9. doi: 10.1073/pnas.1215558110
143. Herbein G, Wendling D. Histone deacetylases in viral infections. Clin Epigenet (2010) 1(1-2):13–24. doi: 10.1007/s13148-010-0003-5
144. Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther (2008) 7(11):3598–608. doi: 10.1158/1535-7163.MCT-08-0510
145. Palayoor ST, Tofilon PJ, Coleman CN. Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1α and HIF-2α in prostate cancer cells. Clin Cancer Res (2003) 9(8):3150–7.
146. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von hippel lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem (2002) 277(33):29936–44. doi: 10.1074/jbc.M204733200
147. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148(3):399–408. doi: 10.1016/j.cell.2012.01.021
148. Li Y, Lu Q, Wang H, Tao P, Jiang J. Geldanamycin, a ligand of heat shock protein 90, inhibits herpes simplex virus type 2 replication both in vitro and in vivo. J Antibiotics (2012) 65(10):509–12. doi: 10.1038/ja.2012.67
149. Nam S, Ga YJ, Lee Y, Hwang Y, Jung E, Shin JS, et al. Radicicol inhibits chikungunya virus replication by targeting nonstructural protein 2. Antimicrobial Agents Chemother (2021) 65(7). doi: 10.1128/AAC.00135-21
150. Maeda M, Hasebe Y, Egawa K, Shibanuma M, Nose K. Inhibition of angiogenesis and HIF-1alpha activity by antimycin A1. Biol Pharm Bull (2006) 29(7):1344–8. doi: 10.1248/bpb.29.1344
151. Dat NT, Jin X, Lee K, Hong YS, Kim YH, Lee JJ. Hypoxia-inducible factor-1 inhibitory benzofurans and chalcone-derived diels-alder adducts from morus species. J Nat Prod (2009) 72(1):39–43. doi: 10.1021/np800491u
152. Hardianti B, Umeyama L, Li F, Yokoyama S, Hayakawa Y. Anti_inflammatory compounds moracin O and p from morus alba linn. (Sohakuhi) target the NF_κB pathway. Mol Med Rep (2020) 22(6):5385–91. doi: 10.3892/mmr.2020.11615
153. Hossain CF, Kim YP, Baerson SR, Zhang L, Bruick RK, Mohammed KA, et al. Saururus cernuus lignans–potent small molecule inhibitors of hypoxia-inducible factor-1. Biochem Biophys Res Commun (2005) 333(3):1026–33. doi: 10.1016/j.bbrc.2005.05.191
154. Song J, Ahn J, Kim S, Cho S, Hong E, Kwon B, et al. Manassantin b shows antiviral activity against coxsackievirus B3 infection by activation of the STING/TBK-1/IRF3 signalling pathway. Sci Rep (2019) 9(1):1–10. doi: 10.1038/s41598-019-45868-8
155. Choi H, Chun YS, Kim SW, Kim MS, Park JW. Curcumin inhibits hypoxia-inducible factor-1 by degrading aryl hydrocarbon receptor nuclear translocator: a mechanism of tumor growth inhibition. Mol Pharmacol (2006) 70(5):1664–71. doi: 10.1124/mol.106.025817
156. Park SY, Jeong KJ, Lee J, Yoon DS, Choi WS, Kim YK, et al. Hypoxia enhances LPA-induced HIF-1alpha and VEGF expression: their inhibition by resveratrol. Cancer Lett (2007) 258(1):63–9. doi: 10.1016/j.canlet.2007.08.011
157. Sun YX, Dai DK, Liu R, Wang T, Luo CL, Bao HJ,, et al Therapeutic effect of SN50, an inhibitor of nuclear factor-kB, in treatment of TBI in mice. Neurol Sci (2013) 34(3):345–55. doi: 10.1007/s10072-012-1007-z
158. Naik RR, Shakya AK, Aladwan SM, El-Tanani M. (2021). Kinase inhibitors as potential therapeutic agents in the treatment of COVID-19. Front Pharmacol 13. doi: 10.3389/fphar.2022.806568.
Keywords: hypoxia, HIFs, NF-kB, immune cells, infectious diseases, inflammation
Citation: Akinsulie OC, Shahzad S, Ogunleye SC, Oladapo IP, Joshi M, Ugwu CE, Gbadegoye JO, Hassan FO, Adeleke R, Afolabi Akande Q and Adesola RO (2023) Crosstalk between hypoxic cellular micro-environment and the immune system: a potential therapeutic target for infectious diseases. Front. Immunol. 14:1224102. doi: 10.3389/fimmu.2023.1224102
Received: 17 May 2023; Accepted: 26 June 2023;
Published: 31 July 2023.
Edited by:
Jean-louis Mege, Aix-Marseille Université, FranceReviewed by:
Farokh Jal Dotiwala, Ocugen, Inc., United StatesBardin Nathalie, Aix Marseille Université, France
Copyright © 2023 Akinsulie, Shahzad, Ogunleye, Oladapo, Joshi, Ugwu, Gbadegoye, Hassan, Adeleke, Afolabi Akande and Adesola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olalekan Chris Akinsulie, b2xhbGVrYW4uYWtpbnN1bGllQHdzdS5lZHU=