 Jane Alexandra Shaw
Jane Alexandra Shaw Stephanus T. Malherbe
Stephanus T. Malherbe Gerhard Walzl
Gerhard Walzl Nelita du Plessis
Nelita du Plessis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol., 20 July 2023
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1222911
This article is part of the Research TopicThe Covid-19 and TB Syndemic: Differences and SimilaritiesView all 11 articles
Epidemiologic data show that both current and previous tuberculosis (TB) increase the risk of in-hospital mortality from coronavirus disease-2019 (COVID-19), and there is a similar trend for poor outcomes from Mycobacterium tuberculosis (Mtb) infection after recent SARS-CoV-2. A shared dysregulation of immunity explains the dual risk posed by co-infection, but the specific mechanisms are being explored. While initial attention focused on T cell immunity, more comprehensive analyses revealed a dysfunctional innate immune response in COVID-19, characterized by reduced numbers of dendritic cells, NK cells and a redistribution of mononuclear phagocytes towards intermediate myeloid subsets. During hyper- or chronic inflammatory processes, activation signals from molecules such as growth factors and alarmins lead to the expansion of an immature population of myeloid cells called myeloid-deprived suppressor cells (MDSC). These cells enter a state of pathological activation, lose their ability to rapidly clear pathogens, and instead become broadly immunosuppressive. MDSC are enriched in the peripheral blood of patients with severe COVID-19; associated with mortality; and with higher levels of inflammatory cytokines. In TB, MDSC have been implicated in loss of control of Mtb in the granuloma and ineffective innate and T cell immunity to the pathogen. Considering that innate immune sensing serves as first line of both anti-bacterial and anti-viral defence mechanisms, we propose MDSC as a crucial mechanism for the adverse clinical trajectories of TB-COVID-19 coinfection.
There is now ample evidence that regions with a high prevalence of tuberculosis (TB) disease and latent TB infection (LTBI, where an asymptomatic person has a positive interferon-γ release assay or skin test), also have a high prevalence of recent SARS-CoV-2 infection (1–4). As a result, acute or chronic coinfection, or acute sequential infection with Mycobacterium tuberculosis (Mtb) and SARS-CoV-2 has become inevitable.
We know from epidemiologic reports during the pandemic that coinfection with Mtb and SARS-CoV-2 worsens patient outcomes. Both current active TB disease (defined as culture, molecular test, or other Mtb test positive, with symptoms or imaging changes that justify the initiation of full TB treatment) and previous TB, increase the risk of in-hospital mortality from coronavirus disease-2019 (COVID-19), and the case fatality rate for coinfection is higher than for COVID-19 alone (5, 6). The lymphopaenia which characterizes COVID-19 is exaggerated in coinfection, and markers of inflammation such as D-dimer and ferritin are increased over and above the COVID-19 levels (7, 8). Transcriptomics and RNAseq data from whole blood, peripheral blood mononuclear cells (PBMC) and bronchoalveolar lavage fluid (BALF) of patients with COVID-19, and those with TB across the clinical spectrum, has shown that there is similarity in the immunopathogenesis of the two diseases through commonly enriched genes in 12-gene disease-exacerbation hot spots (9). Moreover, the inflammatory milieu from Mtb-infected human macrophages increased SARS-CoV-2 infection in vitro, which correlated with IFNA1, IFNB1, IFNG, TNF, and other inflammatory gene induction. Interestingly, in Mtb-infected mice, superinfection with SARS-CoV-2 resulted in increased Mtb dissemination, but a lower SARS-CoV-2 viral load in the tissues (10). In a different murine study, the protective effect of pre-existing Mtb infection on the pathological consequences of SARS-CoV-2 occurred without adversely affecting TB outcomes (11). This collection of findings shows that Mtb infection increases the risk of severe COVID-19 in humans, and suggests the possibility that SARS-CoV-2 coinfection may also trigger the progression of subclinical TB to TB disease.
Patients with active TB disease and SARS-CoV-2 coinfection have lower numbers of Mtb-specific T cells (12). They also produce less IFN-γ and other proinflammatory cytokines, chemokines and growth factors on SARS-CoV-2 stimulation; produce less interferon-y (IFN-y) and several other cytokines on Mtb stimulation (though to a lesser extent than the reduction on SARS-CoV-2 stimulation); and have different overall cytokine signatures compared to infection with each pathogen alone (12–14). One possible mechanism underlying this observed immune suppression in the presence of chronic stimulation by Mtb or SARS-CoV-2 antigens, is the presence of suppressive myeloid cells such as myeloid-derived suppressor cells (MDSC). MDSC are known to inhibit many immune pathways, particularly T cell responses. They have now been both directly and indirectly implicated in the pathogenesis of both COVID-19 and TB (15–17). However, their role in coinfection is yet to be explored. In this article we will introduce the reader to MDSC, briefly review the evidence for their involvement in TB disease and COVID-19, and then discuss the potential role of these cells in determining the outcome of Mtb/SARS-CoV-2 coinfection.
Suppressive myeloid cells are considered critical in immune regulation and tolerance, maintaining the delicate balance between healing and harm during the immune response. They limit excessive inflammation and prevent immune-mediated tissue damage in the early response to a tissue insult, promote immune tolerance during tissue repair or pregnancy, and augment protective anti-pathogen responses in acute infection (18–22). However, in pathological conditions such as chronic inflammation, cancer, or extensive tissue trauma, the scales tip toward more harm than help. The function of MDSC in the pathophysiology of cancer is well described (23–25), but in chronic infection and respiratory disease is still in the early stages of investigation. Our understanding of their role in these conditions is hampered by a few ongoing issues. Firstly, the cell type terminology is not globally accepted. Secondly, the nature of MDSC remains poorly defined, partly because these cells likely differentiate into suppressive macrophage subsets upon entering tissue sites. However, most agree that MDSC are cells of myeloid origin which acquire a state of pathological (or alternative) activation in response to the prolonged weak pro-inflammatory signals that are present in chronic infection or cancer (25–27). As a result, they lose their ability to rapidly and effectively clear pathogens, instead becoming immunosuppressive by inhibiting natural killer (NK) cell, B cell, and T cell responses, amongst others.
MDSC are generally divided into two main subtypes (Figure 1). They are named for their cell lineage of origin as polymorphonuclear or granulocytic MDSC (PMN-MDSC) and monocytic MDSC (M-MDSC). A third group which comprises only a small proportion of the total MDSC population is known as ‘early MDSC’, and consists of potent immunosuppressive myeloid progenitors (28). Other subtypes such as eosinophilic MDSC have been proposed, but are not well characterized as yet. In circulation, both PMN- and M-MDSC have a short lifespan of a few days, though the latter survive longer in vitro (29) and the half-life may well be prolonged in inflammatory states (30). Their continuous recruitment to tissues is what results in long term effects (31). In tumors, M-MDSC rapidly differentiate into tumor-associated macrophages (TAMs) which are associated with tumor progression, and inflammatory dendritic cells (32, 33). Tumor associated neutrophils (TANs) are a heterogeneous population of cells which, in mice, includes both neutrophils with anti-tumor (N1) and suppressive/pro-tumor (N2) properties, the latter sharing some cell surface markers and biochemical properties with PMN-MDSC (34, 35).
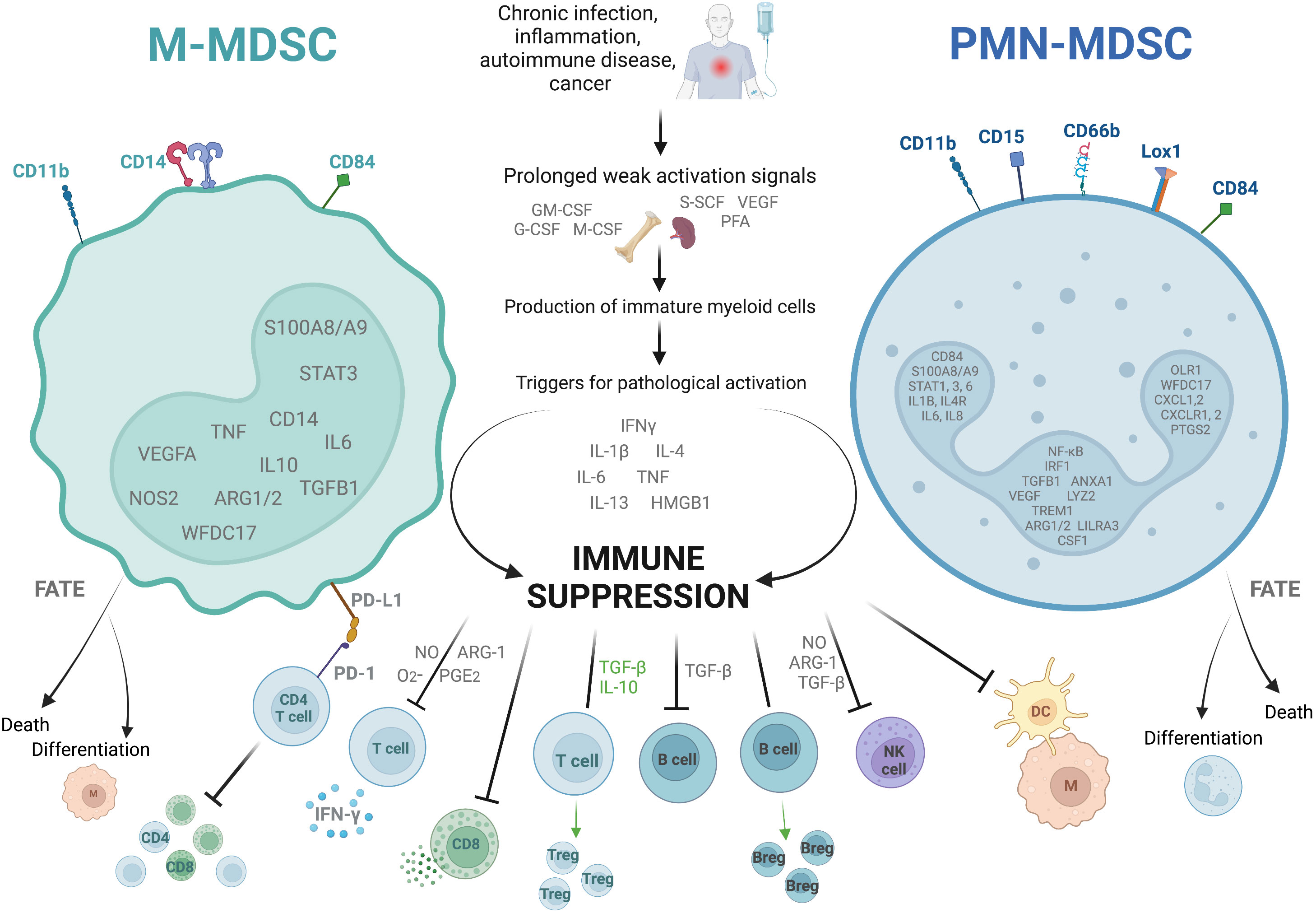
Figure 1 Characteristics of the two dominant subtypes of myeloid derived suppressor cells. The figure shows the two main types of myeloid derived suppressor cells (MDSC), monocytic (M)- and polymorphonuclear (PMN)-MDSC. They are identified as cells in the low density portion of a Ficoll gradient with a low expression of HLA-DR, as well as specific combinations of cell surface markers, and the upregulation of specific genes (shown inside the nucleus). They arise in situations of prolonged weak activation signals, by which they are pathologically activated to become immunosuppressive. They exert their immunosuppressive effects through several different mechanisms including suppression of CD4+ and CD8+ T cell differentiation, cytokine release, and cytotoxic degranulation; promotion of T regulatory cell (Treg) and B regulatory cell (Breg) differentiation and function; and inhibition of B cell, natural killer cell (NK), and antigen presenting cell (M, macrophage; DC, dendritic cell) functions. They are short lived, either dying after a few days in circulation or differentiating into suppressive macrophages or suppressive neutrophils. ARG-1, Arginase-1; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HMGB-1, high mobility box group-1; IFNγ, interferon-γ; IL, interleukin; Lox-1, lectin- type oxidized LDL receptor 1; M-CSF, macrophage colony- stimulating factor; NO, nitric oxide; PD-1, programmed cell death protein-1; PD-L1, programmed death ligand-1; PFA, polyunsaturated fatty acids; PG-E2, prostaglandin-E2; SCF, stem cell factor; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor. Created with BioRender.com.
Identifying these cells is complex, and not always consistent between studies. Cell surface markers which identify MDSC differ between mice and humans. For the purpose of this review we will focus on human-relevant markers only. PMN-MDSC are identified as CD11b+CD14-CD15+/CD66b+ cells in the low density Ficoll gradient fraction of PBMC. Other marker combinations have been proposed which do not need a Ficoll gradient, such as CD15+/CD66b+CD14-LOX1+ and CD15+/CD66b+CD14-CD84+. All human MDSC are HLA-DRlo/-. M-MDSC are identified as CD11b+CD14+CD15-HLA-DRlo/- cells in the low density Ficoll fraction of PBMC, or alternatively CD14+/CD66b-CXCR1+ or CD14+/CD66b-CD84+ (28, 31, 36). Early MDSC are identified as Lin-HLA-DR-CD33+/hi (where Lin is CD3, CD14, CD15, CD19 and CD56) (25, 28). Some of the newer markers such as LOX-1 have yet to be validated in infection-induced MDSC.
Classical neutrophils and monocytes are activated by pathogen- and damage-associated molecular patterns (such as lipopolysaccharide and heat shock proteins respectively) binding to pattern recognition receptors such as Toll-like receptors (TLRs). This interaction triggers the innate immune mechanisms for the rapid clearance of microbes and infected cells, such as phagocytosis, the respiratory burst, the production of proinflammatory cytokines and upregulation of co-stimulatory molecules and MHC-II (25). In contrast, MDSC are the result of prolonged but weak stimulation with growth factors, cytokines and other stimuli (such as GM-CSF, VEGF, NLRP3, and the S100A8/A9 alarmins), and activation is triggered by further stimulation with inflammatory cytokines and pathogen- or damage-associated molecular patterns including iIFN-γ, interleukin (IL)-1β, IL-4, IL-6, IL-13, tumor necrosis factor (TNF), high mobility group box (HMGB)-1 (37–41). The transcription factor STAT3 is also invariably upregulated in MDSC, as the main regulator of genes controlling the expansion of MDSC (31, 42).
Overall, the most important characteristic of these cells which distinguishes them from classical neutrophils and monocytes is their ability to inhibit immune responses, specifically T cell activation and function (28, 31). They use multiple mechanisms to achieve their suppressive effect. Depletion of L-arginine from the microenvironment through upregulation of arginase-1 (ARG1) and inducible nitric oxide synthase (iNOS), inhibits the key human T cell receptor (TCR) signaling molecule CD3ζ in vitro (43, 44). Nitric oxide from the iNOS also interferes with JAK/STAT signaling in the T cells of mice in vitro (45, 46). In addition to this, increased reactive oxygen species (ROS) in murine immature myeloid cells and human low density granulocytes reduces the expression of CD3ζ on T cells, and reactive nitrogen species (RNS) block T cell activation ex vivo by nitrating the TCR and CD8 molecule (47–49). MDSC of both humans and mice express programmed death-ligand-1 (PD-L1) which causes T cell dysfunction, exhaustion and IL-10 secretion when it interacts with programmed death protein-1 (PD-1) on the T cell surface ex vivo (50–52). Lastly, in murine models MDSC exert their immunosuppressive effects by the secretion of TGF-β and IL-10, which directly suppress T cells, induce differentiation into T regs, and suppress macrophage IL-12 production. Expression of membrane-bound TGF-β also suppresses NK cells (53–55).
Several host-directed therapies which target MDSC have shown efficacy in cancer therapy, through several mechanisms. MDSC expansion and recruitment can be inhibited with, for example, 5-Fluorouracil or tyrosine kinase inhibitors (56, 57). MDSC function can be inhibited through, for example, phosphodiesterase-5 inhibitors or PD-L1 inhibitors (58, 59). Lastly, agents such as All-trans retinoic acid (ATRA) promote the differentiation of MDSC into mature leukocytes or tumor-specific cells (60). Several of these MDSC targeting agents are in the experimental stages of investigation for use in TB (61, 62). Tasquinimod causes exhaustion of MDSC, and has been shown to enhance mycobacterial clearance in mice (63). Both sildenafil and ATRA initially showed promise as TB host directed therapies, but recent data using human MDSC have been disappointing (64, 65). Many of these MDSC-targeting therapies have been identified as possible treatments for COVID-19, but little data is available on their efficacy (66). The studies in cancer and TB demonstrate the complexity of MDSC. It is likely that particular MDSC subsets predominate in particular conditions, and that they employ different suppressive mechanisms in different disease microenvironments (64). This means that even if an agent is effective in treating one condition, it may not be equally effective in treating a second condition, even if the two conditions have a similar immunopathogenesis. This must be kept in mind for SARS-CoV-2 infection as well.
Classically activated myeloid cells are the initial effectors of antimycobacterial responses. They sense Mtb through multiple PRRs, phagocytose the bacteria, contain them, limit replication, kill them, release cytokines and chemokines, and activate T cells which in turn increase the activation state of the myeloid cells to enable then to kill Mtb more effectively. However, while alternatively activated myeloid cell subsets – initially labelled natural suppressor cells but later renamed MDSC – also phagocytose Mtb, they have less effective mycobactericidal activity, low expression of MHC class II, secrete immune mediators which suppress T cell responses, and promote lung damage (17, 62, 67, 68).
Mtb contains structural moieties such as glycolipids, that are known to induce MDSC generation (69). MDSC have been detected in the blood of BCG vaccinated mice, where they were found to reduce T cell priming through an IL-1R-dependent pathway (70). Where mice are in the advanced clinical stages of TB disease, MDSC accumulate in the lungs, bone marrow, spleen, and blood, and suppress T cell proliferation and IFN-γ production in vitro through NO-dependent mechanisms (71). In humans, MDSC are enriched in the peripheral blood, bronchoalveolar lavage fluid (BALF), and pleural fluid of patients with active pulmonary and pleural TB disease, to levels and phenotype comparable to lung cancer (17, 72). The predominant subset seems to depend on the anatomical site, as PMN-MDSC were preferentially found in BALF, but M-MDSC were the main subset in the pleural fluid (17, 72). Moreover, after successful TB treatment, not only do peripheral blood levels of MDSC decline (particularly PMN-MDSC), but the MDSC express more maturation surface markers (17, 72). During active TB disease, MDSC inhibit T cell proliferation (possibly through a NO-dependent mechanism), suppress CD4+ T-cell production of IL-2, TNF-α, IFN-γ in vitro (17). MDSC also inhibit IL-10 production by CD4+ T cells, thereby inhibiting the regulation of IL-2, TNF and IFNs, and perhaps demonstrating the global shutdown of T cell activation regardless of which cytokines they produce. Furthermore, MDSC suppress CD8+ T cell production of TNF-α, IL-2, IFN-γ, and IL-10 in active TB disease, and subvert effector T-cell-mediated containment of Mtb in monocyte-derived macrophages (17, 72, 73). MDSC impair T cell-mediated killing of Mtb infected cells through down-regulation of Th1 cytokines (17, 70). In addition, CD8+ T cell mediated killing of infected cells using cytotoxic granules such as perforin and granulysin, is critical for Mtb control (74). Through skewing of the immune response toward a regulatory phenotype, MDSC likely suppress granule-associated effector molecules, and thereby impair killing of infected cells (74–76).
Some of the most compelling evidence for MDSC’s role in TB comes from granuloma research. In mouse models, MDSC accumulate at the edges of necrotic granulomas in the lung parenchyma of infected Mtb-susceptible mice, and this finding has been associated with TB disease progression and uncontrolled bacterial replication (62, 68). Conversely, mice that are Mtb-resistant, with no necrotic granulomas, have very low levels of MDSC in their lungs (68, 77). Suppressive neutrophils which exhibit immunoregulatory functions resembling MDSC subsets have also been identified in TB granulomas from non-human primates (NHPs) (78). Another recent report in NHPs suggested that PMN-MDSC in the periphery of TB granulomas may restrict T cell access to the granuloma core and Mtb-infected cells (79). In an in vitro human granuloma model, ex vivo generated human M-MDSC promoted mycobacterial replication, changing the structure of the granuloma and adversely affecting bacterial containment (80). In lymph node granulomas from TB/HIV coinfected people and TB-only controls, MDSC expressing Arg-1 were highly expressed in TB/HIV granulomas (81), and the proportion of CD15+ MDSC correlated with plasma HIV viral load and Mtb antigen load in tissue, but was negatively correlated with peripheral CD4+ T cell numbers. In the same study, PMN-MDSC were also elevated in blood samples from TB/HIV coinfected patients (81). Recently, multiplexed ion beam imaging by time of flight (MIBI-TOF) was used to generate a comprehensive spatial map of 19 cell subsets across 8 spatial microenvironments within TB granulomas from multiple human tissues, including the lung (82). The myeloid core of the granulomas was characterized by expression of the tolerogenic proteins IDO1 and PD-L1, which was highest in CD11b+CD11c+ macrophages (identical to immunosuppressive TAMs). This expression was also associated with downregulation of HLA-DR in the ‘intermediate monocyte’ subset. These data support the existing evidence for a highly localized, myeloid-mediated immune suppression in the granuloma (82). These findings also seem to support the hypothesis that MDSC which enter a granuloma differentiate into suppressive macrophages which are permissive to Mtb growth, similar to their activity in solid tumors (74, 83).
The role of MDSC in events early in the TB disease spectrum is less clear. Recent attempts to define more clearly the spectrum and pathogenesis of TB before clinical disease highlight the gaps in knowledge of factors promoting progression from infection to disease or to cure (84). Household contacts and people with presumed LTBI have far lower median frequencies of MDSC in peripheral blood than those with active TB disease, comparable to healthy donors (72, 73, 85). Higher frequencies of peripheral blood M-MDSC have been correlated with more severe TB disease (based on time to positivity of Mtb culture, cavitary disease on chest radiography, symptoms score, ESR and monocyte/lymphocyte ratio), but higher frequencies of PMN-MDSC have been associated with a lower radiological TB severity score (85, 86). This suggests that when considering the early TB spectrum, which includes such entities as subclinical TB – where there are no symptoms but may be radiographic changes, and which may or may not progress to active TB disease – the function of the subsets of MDSC need to be examined separately.
As with TB and other viral infections, myeloid cells are the first responders to infection with SARS-CoV-2. After navigating the upper airways, the virus is taken up by alveolar macrophages, without active viral replication, which become activated, and are subsequently responsible for the proinflammatory anti-viral immune response (87). Other innate cells are recruited to the site of infection, and in most people the result is mild disease with eventual eradication of the virus. Yet, a proportion of people infected with SARS-CoV-2 will suffer a marked dysregulation of the innate immune response, especially the myeloid cell compartment, as a result of emergency myelopoiesis (88). This dysregulated state is characterized by the emergence of immature neutrophils and monocytes with suppressive features, including MDSC, which have been directly implicated in the pathogenesis of this dysregulated response, as well as shown to be predictive of severe or fatal disease (88–91).
As we have come to expect from the pandemic literature, there is an abundance of evidence on the topic of MDSC in COVID-19. Several studies in humans have now provided direct evidence of high peripheral blood frequencies of both subsets of MDSC in COVID-19, across all levels of COVID-19 severity but particularly in severe disease, fatal disease, and acute respiratory distress syndrome (ARDS) (91–101). Again, the different roles of the MDSC subsets are evident. Early M-MDSC frequency predicted subsequent COVID-19 severity and mortality, but transient early expansion of PMN-MDSC was associated with survivors of severe COVID-19 (91, 99, 102–104). Studies using single cell RNA sequencing (scRNAseq) in combination with flow cytometry, CyTOF and other assays, have found that populations of immature neutrophils with features of PMN-MDSC and, to a lesser extent, monocytes with features of M-MDSC, emerge in the blood and BALF of patients with severe COVID-19. These cells differentiate them from patients with mild COVID-19, in whom frequencies are still higher than healthy donors (15, 88, 105–107). M-MDSC are not increased in airway samples from nasopharyngeal and endotracheal aspirates, but large numbers of CD66b+ cells with a high expression of intracytoplasmic Arg1, in line with PMN-MDSC, were found in lung tissue from patients who died of COVID-19 (91, 97).
The immunosuppressive abilities of these SARS-CoV-2-induced MDSC have been demonstrated. Both PMN-MDSC and M-MDSC from the peripheral blood of patients with moderate and severe COVID-19 inhibit T cell proliferation and IFN-γ production in vitro (91, 94–96, 108). In bacterial sepsis, MDSC expansion, IFN-γ production, and TNF-α production reduced over time from admission, but in COVID-19 these responses accelerated over time, despite initial lesser physiological derangement (109). The presence of PMN-MDSC increased the expression of Arg1 and iNOS mRNA compared to PMN-MDSC-depleted PBMC, and plasma levels of TGF-β directly correlated with PMN-MDSC frequency. Moreover, PMN-MDSC depletion significantly improved the SARS-CoV-2 specific T cell response of PBMC (95). Similarly, PD-L1, ILT-3 and IDO-1-expressing M-MDSC were the dominant producers of IL-10 and IL-6 in severe COVID-19 patients, and this correlated with increased inflammatory markers, as well as accumulation of regulatory T and B cells (110). Cocultures with M-MDSC had high levels of Arg1, the suppressive effect of M-MDSC on T cell proliferation was reduced by the addition of L-Arginine, and plasma levels of Arg-1 and IL-6 were elevated in COVID-19 patients, which increased with increasing severity of disease (91). Several other papers have also reported elevated levels of Arg1, with low plasma levels of L-Arginine in association with MDSC in COVID-19, which may not only have implications for immune function but also for increased platelet aggregation (97, 111–113).
Another consideration is the influence of MDSC on the genesis of lung fibrosis in COVID-19 patients. MDSC can transdifferentiate into extracellular matrix (collagen type I)-producing fibrocytes, which interact with activated T-cells, resulting in the production of IDO and leading to Treg expansion (114). Murine models have suggested that MDSC promote lung fibrogenesis by inhibiting collagen degradation through TGF-β production (115). Elevated serum levels of TGF-β correlated with lung fibrosis in a cohort of severe COVID-19 patients (108).This study also reported increased serum levels of TGF-β and of MDSC in COVID-19 patients, as well as a significant correlation between COVID-19 severity and serum TGF-β levels; and showed that isolated M-MDSC from these patients produced higher levels of intracellular TGF-β than non-M-MDSC (108).
These effects do not appear to be short-lived. Elevations in PMN-MDSC persist from hospital admission to convalescence, and longer than three months after acute COVID-19 – across all severities, but at higher levels in those with severe compared to mild disease (94, 116, 117). Elevated levels of circulating M-MDSC were found up to seven months after moderate to severe COVID-19, along with elevated levels of the immune checkpoint marker CD86 (characteristic of ongoing immune activation and chronic inflammation) (118). In another report, MDSC numbers had normalized by three months after acute COVID-19, but the immune dysfunction persisted (119). When monocytes from COVID-19 patients at three months after hospital discharge were stimulated with LPS and R848, both TNF and IL-6 production was impaired, and levels of other cytokines in plasma were also lower, in both moderate and severe COVID-19 (119). At five months after SARS-CoV-2 infection, levels of M-MDSC remained elevated compared to healthy controls, and continued to suppress SARS-CoV-2-specific T cell cytokine production through arginase, ROS and TGF-β dependent pathways (120). These data suggest a long-lasting impairment in the immune response after COVID-19, attributable to the suppressive effects of MDSC.
The evidence for MDSC involvement in COVID-19 and TB are summarized in Table 1.
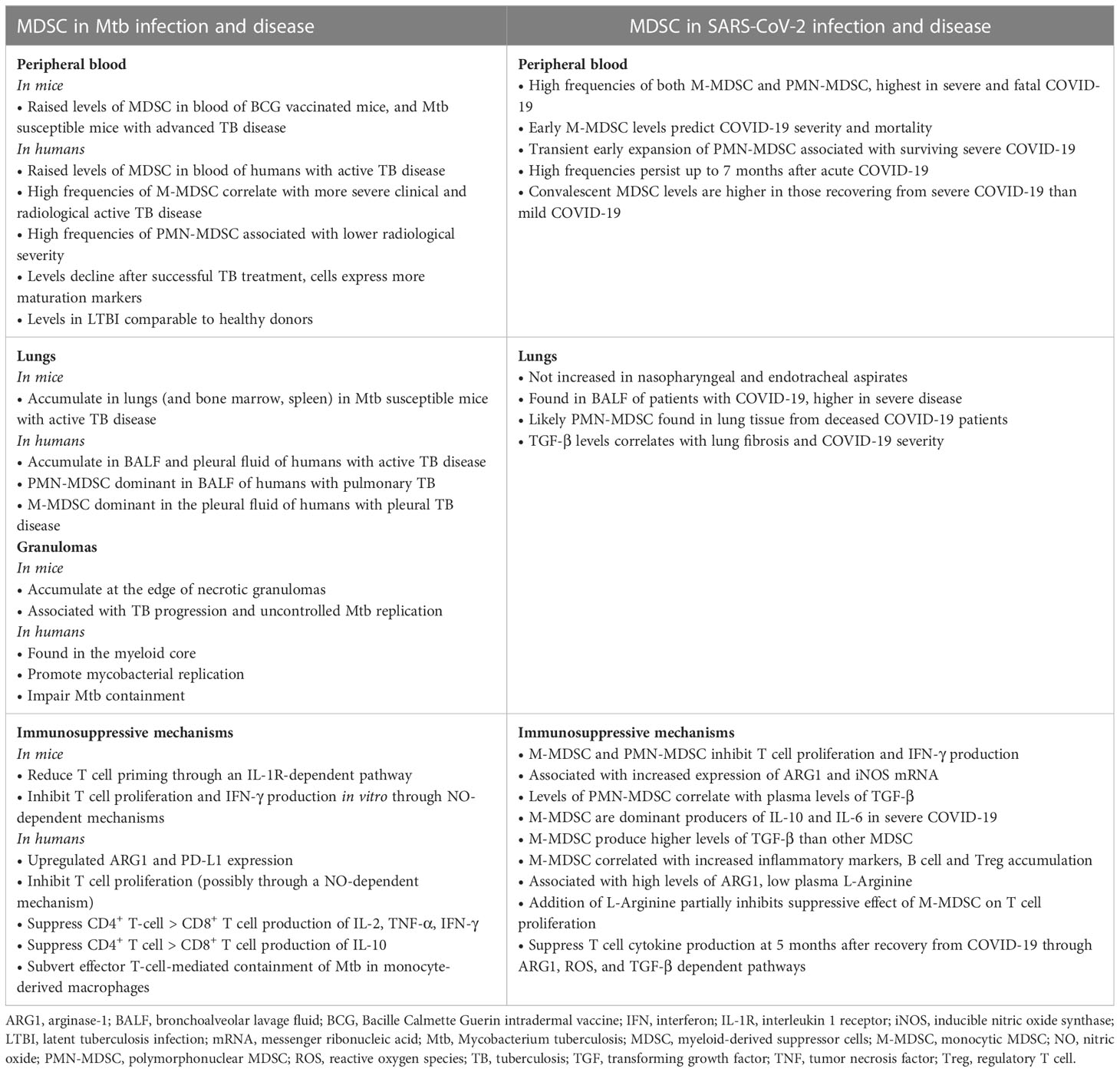
Table 1 Evidence for the role of myeloid derived suppressor cells in the pathogenesis of Tuberculosis compared to COVID-19.
It is thought that up to a quarter of the world’s population have been latently infected with Mtb, with at most 15% progressing to active disease in their lifetime, often with a long latency period between initial infection and active TB disease (121). This means that a coinfection sequence of pre-existing LTBI followed by SARS-CoV-2 infection is a highly likely scenario. However, the fact that many patients present with a long history of symptoms and cavitary lung lesions at TB diagnosis, implying chronicity of disease, suggests that a scenario of active TB disease followed by SARS-CoV-2 infection is also likely to be a common occurrence (Figure 2).
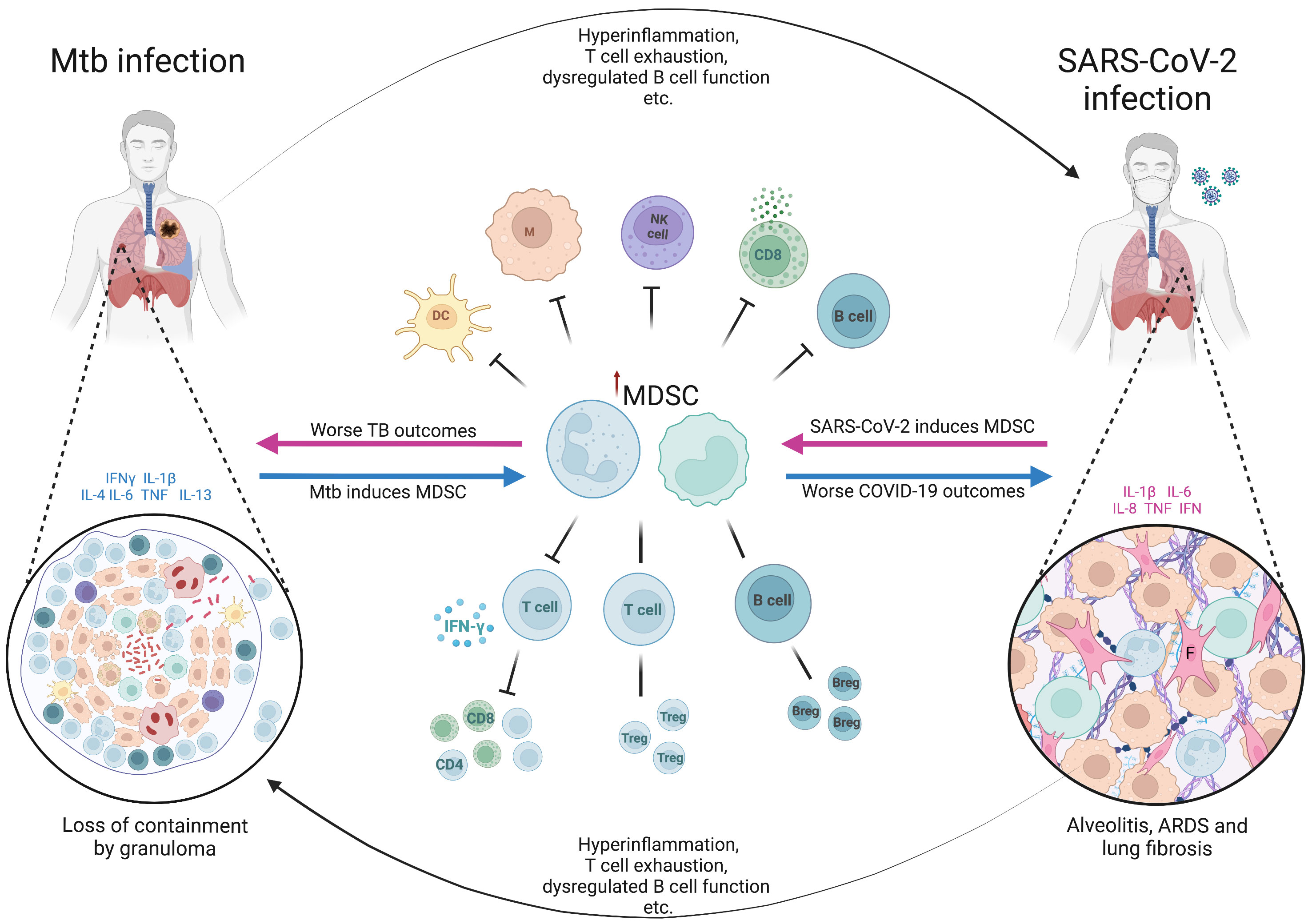
Figure 2 The hypothetical role of myeloid derived suppressor cells in Mtb/SARS-CoV-2 coinfection. The figure shows the bidirectional effects of myeloid derived suppressor cells (MDSC) on the clinical outcomes in patients coinfected with Mycobacterium tuberculosis (Mtb) and SARS-CoV-2. In latent infection with Mtb or active TB disease, the presence of cytokines such as IFN-γ, IL-1β, IL-4, IL-6, TNF and IL-13, triggers the pathological activation of MDSC (blue arrows), which become immune suppressive. The effects of MDSC, along with other effects of Mtb infection and disease (hyperinflammation, T cell exhaustion etc.), predispose to worse clinical outcomes from SARS-CoV-2 infection including acute respiratory distress syndrome (ARDS) and lung fibrosis. MDSC accumulate in the lungs of severe SARS-CoV-2 cases (shown in the cut-out on the right), further exacerbating the immune suppression and profibrotic effects. Similarly, in current or recent SARS-CoV-2 infection, there is induction of proinflammatory cytokines, often pathologically high (termed hyperinflammation), along with other aspects of SARS-CoV-2-related immune dysfunction and dysregulation, and pathological activation of MDSC (pink arrows). These factors combined result in worse clinical outcomes from active tuberculosis disease, as well as the potential loss of containment of Mtb within granulomas in latent infection. Suppressive neutrophils and macrophages within the myeloid core of the granuloma (shown in the cut-out on the left) further contribute to the loss of Mtb control. Breg, regulatory B cell; COVID-19, coronavirus disease-2019; DC, dendritic cell; F, fibroblast; IFN, interferon; IL, interleukin; M, macrophage; NK, natural killer cell; TB, tuberculosis; Treg, regulatory T cell. Created with BioRender.com
The number of Mtb-specific CD4 T cells is lower in coinfected patients who have active TB disease/SARS-CoV-2, than in those with TB alone (12). Also, COVID-19 itself is characterized by reduced T cell numbers and T cell exhaustion (12, 122). Therefore, SARS-CoV-2 infection could hypothetically trigger progression from LTBI to active TB disease. As we have shown in the sections above, MDSC may be responsible for the lower T cell numbers in COVID-19, and for reduced expression of key cytokines required to maintain the immune response to Mtb.
The levels of IFN-γ produced by whole blood on stimulation by the SARS-CoV-2 spike protein-derived peptide CD4S were lower in patients with active TB disease/COVID-19 coinfection than in those with LTBI/COVID-19 and COVID-19 alone (13). In a more comprehensive analysis by the same group, an immune signature consisting of TNF-α, macrophage inflammatory protein-1β and IL-9 associated with active TB disease/COVID-19 compared to COVID-19 alone, and another signature including TNF-α, IL-1β, IL-17A, IL-5, fibroblast growth factor-basic, and GM-CSF, associated with active TB disease/COVID-19 compared to TB alone (14). In addition to this they confirmed the reduced SARS-CoV-2 specific response in coinfected patients for IFN-γ, as well as IL-10, IP-10, and other key cytokines (14). This suggests the possibility that the pre-existing immune milieu of active TB disease impaired the critical T cell response to SARS-CoV-2 infection. Hypothetically, MDSC induced by Mtb infection may be responsible for this, through the expression of PD-L1 which directly inhibits further T cell proliferation and induces both dysregulation and an exhausted immune profile in T cells. If this is the case, then the clinical implication may be a higher viral load, more severe virus-related lung damage, and an increased immune dysregulation which results in more severe clinical manifestations.
The impact of MDSC on initial or recent SARS-CoV-2 infection with secondary Mtb infection must also be considered. As described in detail above, on infection with SARS-CoV-2, emergency myelopoiesis is stimulated and MDSC released into the peripheral blood, accumulating in the lungs in severe COVID-19. Those who die from COVID-19 will most likely have early and persistently elevated M-MDSC, whereas survivors are more likely to have an early peak of PMN-MDSC which reduces as they improve. Nonetheless, the immune suppression mediated by these cells persists for many months irrespective of severity. In theory, Mtb infection during the acute phase of SARS-CoV-2 illness or in the months thereafter, may therefore have a higher likelihood of a poor outcome, as the key T cell responses to Mtb are impaired. This is supported by reports of deficient IFN-γ release assay (IGRA) responses to Mtb antigens (and mitogen) in patients with severe COVID-19, which suggest a generalized unresponsiveness of T cells to all antigens, and specifically to Mtb (123, 124). Production of IFN-γ, IP-10, and IL-1β, amongst others, in response to Mtb-antigen stimulation was impaired in the whole blood from coinfected active TB disease/COVID-19 patients (14). On the other hand, some data implies that the Mtb-antigen cytokine responses are augmented by recent SARS-CoV-2 infection, and immunosuppressive responses are reduced (125). The latter study examined the baseline, Mtb antigen-, and mitogen-stimulated levels of key cytokines and chemokines in elderly patients with positive and negative SARS-CoV-2 serology, both with and without LTBI (based on IGRA). They found higher baseline/unstimulated and Mtb antigen-stimulated levels of IFN-γ, IL-2, TNF-α, and others, in patients with LTBI who were seropositive for SARS-CoV-2 compared to those with LTBI who were seronegative for SARS-CoV-2. Moreover, the levels of immunosuppressive IL-10 were lower in LTBI/seropositive SARS-CoV-2 individuals. There were no differences in response to mitogen between groups in this study, and the LTBI negative control group did not show any enhanced cytokine response to Mtb antigen (125). These data show that if SARS-CoV-2-induced MDSC are the mechanism underlying the poor response of T cells to Mtb antigen, then their effects are likely Mtb antigen-specific, rather than part of a non-antigen-specific response. These observations also suggest that SARS-CoV-2-induced MDSC may have a different effect on the outcome of Mtb infection, in different COVID-19 severities.
Whilst it is not direct evidence for the effect of MDSC, the fact that Mtb-specific T cell numbers are reduced in SARS-CoV-2 coinfection suggests that if MDSC are involved, then it is in an antigen-specific manner (12). In other words, MDSC induced by SARS-CoV-2 may well suppress Mtb-specific T cell responses, as well as suppressing global T cell proliferation and cytokine production in an antigen non-specific way. Similarly, evidence that there is a reduced T cell response to stimulation with SARS-CoV-2 antigen in patients with preexisting Mtb infection might imply that Mtb-induced MDSC also suppress SARS-CoV-2 T cells in both an antigen specific and non-specific way (14). This is not unknown, as MDSC induced by HIV infection also suppress T cell function in vitro by both antigen-specific and non-specific mechanisms (126). However, this antigen-specific effect may be limited to CD8 + T cells because of the low expression of MHC Class II by MDSC – a theory supported by the inhibitory effects of tumor-associated MDSC on CD8+ T cells demonstrated in human cells in vitro and mouse models, which can be reversed by anti-MDSC host directed therapies (127, 128).
Whether any of the MDSC-targeting host directed therapies with efficacy in TB will also prove effective in Mtb/SARS-CoV-2 co-infection remains to be seen. Because of the high prevalence of both infections, it is likely that by default coinfected participants will be included in any human trials in the future. An agent such as Imatinib (a tyrosine kinase inhibitor which targets the ABL kinase domain) which has experimental evidence in both diseases – despite disappointing clinical disease outcomes in COVID-19 – is an attractive option for future investigation in coinfection (129–132).
Lastly, we must consider if preexisting Mtb infection (LTBI or active TB disease) with MDSC induction might affect the efficacy of a subsequent SARS-CoV-2 vaccination, and if recent COVID-19 might adversely affect a subsequent Mtb vaccine response. MDSC impair both T and B cell responses, even inducing regulatory and suppressive B cells in the tumor microenvironment (133). Theoretically, the presence of MDSC from Mtb or SARS-CoV-2 infection might impair adaptive and cell-mediated responses to a vaccine administered in their presence, resulting in reduced immune memory to the vaccine (133). Adults with TB have lower levels of anti-SARS-CoV-2 antibodies after three doses of inactivated vaccine (134). Assuming antibody levels are a correlate of protection, this would mean that a SARS-CoV-2 vaccinated person with preexisting Mtb infection would lose the vaccine-mediated protection for severe COVID-19, and lead to worse clinical outcomes. MDSC are known to be part of the response to Bacillus Calmette-Guérin vaccination (BCG), possibly contributing to incomplete protection against Mtb by restraining T cell priming (70). BCG has immunomodulatory effects on myeloid cells, including epigenetic reprogramming, which affect these cells’ ability to respond to pathogens (including Mtb) (135, 136). A person receiving BCG or another Mtb vaccine candidate in a clinical trial may not develop the desired immune response if they have had recent COVID-19, and importantly, the SARS-CoV-2-induced MDSCs continue to exert their effects months after the acute infection. This would need to be adjusted for in the analysis.
Overall, there is evidence supporting a potential role for MDSC in determining the outcomes of Mtb/SARS-CoV-2 coinfection, in all disease severities and iterations of the coinfection sequence. More evidence is needed to find out if MDSC impact the outcome of SARS-CoV-2 infection in early TB or LTBI; if there are any vaccine-relevant interactions; and if a host directed therapy aimed at modulating the effect of MDSC might improve the outcome of active TB disease/COVID-19 coinfection.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
NdP, GW, SM and JS conceived of the article, JS wrote the first draft of the manuscript and all authors edited and approved the final version.
NP received funding from the NIAID (12446SUB). STM received funding from the EDCTP2 program supported by the European Union (grant number CDF1576). GW received funding from the South African National Research Foundation (SANRF; SARChI TB Biomarkers #86535), the South African Medical Research Council (SAMRC), the Bill and Melinda Gates Foundation (INV-023825), and the NIH (DAA9-20-669180-1). JS is funded by the SAMRC through its Division of Research Capacity Development under the Bongani Mayosi National Health Scholars Programme from funding received from the Public Health Enhancement Fund/South African National Department of Health. The content hereof is the sole responsibility of the authors and does not necessarily represent the official views of any of the funders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nasimiyu C, Ngere I, Dawa J, Amoth P, Oluga O, Ngunu C, et al. Near-Complete SARS-CoV-2 Seroprevalence among Rural and Urban Kenyans despite Significant Vaccine Hesitancy and Refusal. Vaccines (Basel). (2022) 11(1):68. doi: 10.3390/vaccines11010068
2. Madhi SA, Kwatra G, Myers JE, Jassat W, Dhar N, Mukendi CK, et al. Population immunity and covid-19 severity with omicron variant in South Africa. N Engl J Med (2022) 386(14):1314–26. doi: 10.1056/NEJMoa2119658
3. Jahan N, Brahma A, Kumar MS, Bagepally BS, Ponnaiah M, Bhatnagar T, et al. Seroprevalence of IgG antibodies against SARS-CoV-2 in India, March 2020 to August 2021: a systematic review and meta-analysis. Int J Infect Dis (2022) 116:59–67. doi: 10.1016/j.ijid.2021.12.353
4. World Health Organization. Global Tuberculosis Report 2022 (2022). Geneva. Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (Accessed 20 Jan 2023).
5. Jassat W, Cohen C, Tempia S, Masha M, Goldstein S, Kufa T, et al. Risk factors for COVID-19-related in-hospital mortality in a high HIV and tuberculosis prevalence setting in South Africa: a cohort study. Lancet HIV. (2021) 8(9):e554–67. doi: 10.1016/S2352-3018(21)00151-X
6. Sy KTL, Haw NJL, Uy J. Previous and active tuberculosis increases risk of death and prolongs recovery in patients with COVID-19. Infect Dis (London England) (2020) 52(12):902–7. doi: 10.1080/23744235.2020.1806353
7. Wang Y, Chen Y, Gu L, Lou L, Zhang J, Zhang K. The clinical characteristics and risk factors for severe COVID-19 in patients with COVID-19 and tuberculosis coinfection. Front Microbiol (2022) 13:1061879. doi: 10.3389/fmicb.2022.1061879
8. du Bruyn E, Stek C, Daroowala R, Said-Hartley Q, Hsiao M, Schafer G, et al. Effects of tuberculosis and/or HIV-1 infection on COVID-19 presentation and immune response in Africa. Nat Commun (2023) 14(1):188. doi: 10.1038/s41467-022-35689-1
9. Sheerin D, Abhimanyu, Peton N, Vo W, Allison CC, Wang X, et al. Immunopathogenic overlap between COVID-19 and tuberculosis identified from transcriptomic meta-analysis and human macrophage infection. iScience (2022) 25(6):104464. doi: 10.1016/j.isci.2022.104464
10. Hildebrand RE, Chandrasekar SS, Riel M, Touray BJB, Aschenbroich SA, Talaat AM. Superinfection with SARS-CoV-2 Has Deleterious Effects on Mycobacterium bovis BCG Immunity and Promotes Dissemination of Mycobacterium tuberculosis. Microbiol Spectr. (2022) 10(5):e0307522. doi: 10.1128/spectrum.03075-22
11. Rosas Mejia O, Gloag ES, Li J, Ruane-Foster M, Claeys TA, Farkas D, et al. Mice infected with Mycobacterium tuberculosis are resistant to acute disease caused by secondary infection with SARS-CoV-2. PloS Pathog (2022) 18(3):e1010093. doi: 10.1371/journal.ppat.1010093
12. Riou C, du Bruyn E, Stek C, Daroowala R, Goliath RT, Abrahams F, et al. Relationship of SARS-CoV-2–specific CD4 response to COVID-19 severity and impact of HIV-1 and tuberculosis coinfection. J Clin Invest. (2021) 131(12):e149125. doi: 10.1172/JCI149125
13. Petrone L, Petruccioli E, Vanini V, Cuzzi G, Gualano G, Vittozzi P, et al. Coinfection of tuberculosis and COVID-19 limits the ability to in vitro respond to SARS-CoV-2. Int J Infect Dis (2021) 113 Suppl 1:S82–7. doi: 10.1016/j.ijid.2021.02.090
14. Najafi-Fard S, Aiello A, Navarra A, Cuzzi G, Vanini V, Migliori GB, et al. Characterization of the immune impairment of tuberculosis and COVID-19 coinfected patients. Int J Infect Dis (2023) 130 Suppl 1:S34–42. doi: 10.1016/j.ijid.2023.03.021
15. Silvin A, Chapuis N, Dunsmore G, Goubet A-G, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell (2020) 182(6):1401–1418.e18. doi: 10.1016/j.cell.2020.08.002
16. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
17. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med (2013) 188(6):724–32. doi: 10.1164/rccm.201302-0249OC
18. Ou L, Shi Y, Dong W, Liu C, Schmidt TJ, Nagarkatti P, et al. Kruppel-like factor KLF4 facilitates cutaneous wound healing by promoting fibrocyte generation from myeloid-derived suppressor cells. J Invest Dermatol (2015) 135(5):1425–34. doi: 10.1038/jid.2015.3
19. Mahdipour E, Charnock JC, Mace KA. Hoxa3 promotes the differentiation of hematopoietic progenitor cells into proangiogenic Gr-1+CD11b+ myeloid cells. Blood (2011) 117(3):815–26. doi: 10.1182/blood-2009-12-259549
20. Köstlin N, Kugel H, Spring B, Leiber A, Marmé A, Henes M, et al. Granulocytic myeloid derived suppressor cells expand in human pregnancy and modulate T-cell responses. Eur J Immunol (2014) 44(9):2582–91. doi: 10.1002/eji.201344200
21. Kang X, Zhang X, Liu Z, Xu H, Wang T, He L, et al. Granulocytic myeloid-derived suppressor cells maintain feto-maternal tolerance by inducing Foxp3 expression in CD4+CD25–T cells by activation of the TGF-β/β-catenin pathway. Mol Hum Reprod (2016) 22(7):499–511. doi: 10.1093/molehr/gaw026
22. Poe SL, Arora M, Oriss TB, Yarlagadda M, Isse K, Khare A, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol (2013) 6(1):189–99. doi: 10.1038/mi.2012.62
23. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: A meta-analysis. PloS One (2016) 11(10):e0164514. doi: 10.1371/journal.pone.0164514
24. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
25. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
26. Landskron G, de la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi: 10.1155/2014/149185
27. Youn J-I, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 91(1):167–81. doi: 10.1189/jlb.0311177
28. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
29. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol (2018) 200(2):422–31. doi: 10.4049/jimmunol.1701019
30. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res (2007) 67(20):10019–26. doi: 10.1158/0008-5472.CAN-07-2354
31. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
32. Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
33. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344(6186):921–5. doi: 10.1126/science.1252510
34. Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PloS One (2014) 9(6):e98259. doi: 10.1371/journal.pone.0098259
35. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
36. Condamine T, Dominguez GA, Youn J-I, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
37. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92(11):4150–66. doi: 10.1182/blood.V92.11.4150
38. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res (2004) 64(17):6337–43. doi: 10.1158/0008-5472.CAN-04-0757
39. Wu C-T, Hsieh C-C, Lin C-C, Chen W-C, Hong J-H, Chen M-F. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J Mol Med (Berl). (2012) 90(11):1343–55. doi: 10.1007/s00109-012-0916-x
40. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. (2006) 116(10):2777–90. doi: 10.1172/JCI28828
41. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity (2013) 38(3):541–54. doi: 10.1016/j.immuni.2013.02.007
42. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res (2005) 65(20):9525–35. doi: 10.1158/0008-5472.CAN-05-0529
43. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
44. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol (2003) 171(3):1232–9. doi: 10.4049/jimmunol.171.3.1232
45. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol (1998) 160(12):5729–34. doi: 10.4049/jimmunol.160.12.5729
46. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
47. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172(2):989–99. doi: 10.4049/jimmunol.172.2.989
48. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi: 10.1038/nm1609
49. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res (2001) 61(12):4756–60.
50. Sun Z, Fourcade J, Pagliano O, Chauvin J-M, Sander C, Kirkwood JM, et al. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res (2015) 75(8):1635–44. doi: 10.1158/0008-5472.CAN-14-3016
51. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology (2016) 5(12):e1247135. doi: 10.1080/2162402X.2016.1247135
52. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am J Respir Crit Care Med (2018) 198(6):777–87. doi: 10.1164/rccm.201708-1707OC
53. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
54. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
55. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179(2):977–83. doi: 10.4049/jimmunol.179.2.977
56. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi: 10.1158/0008-5472.CAN-09-3690
57. du Plessis N, Kotze LA, Leukes V, Walzl G. Translational potential of therapeutics targeting regulatory myeloid cells in tuberculosis. Front Cell Infect Microbiol (2018) 8:332. doi: 10.3389/fcimb.2018.00332
58. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203(12):2691–702. doi: 10.1084/jem.20061104
59. Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology (2017) 6(4):e1294299. doi: 10.1080/2162402X.2017.1294299
60. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
61. Singh V, Brecik M, Mukherjee R, Evans JC, Svetlíková Z, Blaško J, et al. The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem Biol (2015) 22(1):63–75. doi: 10.1016/j.chembiol.2014.11.006
62. Knaul JK, Jörg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190(9):1053–66. doi: 10.1164/rccm.201405-0828OC
63. Gupta S, Krug S, Pokkali S, Leanderson T, Isaacs JT, Srikrishna G, et al. Pharmacologic exhaustion of suppressor cells with tasquinimod enhances bacterial clearance during tuberculosis. Am J Respir Crit Care Med (2019) 199(3):386–9. doi: 10.1164/rccm.201805-0820LE
64. Leukes VN, Dorhoi A, Malherbe ST, Maasdorp E, Khoury J, McAnda S, et al. Targeting of myeloid-derived suppressor cells by all-trans retinoic acid as host-directed therapy for human tuberculosis. Cell Immunol (2021) 364:104359. doi: 10.1016/j.cellimm.2021.104359
65. Leukes VN, Malherbe ST, Hiemstra A, Kotze LA, Roos K, Keyser A, et al. Sildenafil, a type-5 phosphodiesterase inhibitor, fails to reverse myeloid-derived suppressor cell-mediated T cell suppression in cells isolated from tuberculosis patients. Front Immunol (2022) 13:883886. doi: 10.3389/fimmu.2022.883886
66. Park S-J, Nam D-E, Seong HC, Hahn YS. New discovery of myeloid-derived suppressor cell’s tale on viral infection and COVID-19. Front Immunol (2022) 13:842535. doi: 10.3389/fimmu.2022.842535
67. Dorhoi A, Kaufmann SHE. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol (2015) 45(8):2191–202. doi: 10.1002/eji.201545493
68. Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. Gr1intCD11b+ Myeloid-derived suppressor cells in mycobacterium tuberculosis infection. PloS One (2013) 8(11):e80669. doi: 10.1371/journal.pone.0080669
69. Wang Z, Jiang J, Li Z, Zhang J, Wang H, Qin Z. A myeloid cell population induced by Freund adjuvant suppresses T-cell-mediated antitumor immunity. J Immunother. (2010) 33(2):167–77. doi: 10.1097/CJI.0b013e3181bed2ba
70. Martino A, Badell E, Abadie V, Balloy V, Chignard M, Mistou M-Y, et al. Mycobacterium bovis bacillus Calmette-Guérin vaccination mobilizes innate myeloid-derived suppressor cells restraining in vivo T cell priming via IL-1R-dependent nitric oxide production. J Immunol (2010) 184(4):2038–47. doi: 10.4049/jimmunol.0903348
71. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol (2014) 192(10):4718–27. doi: 10.4049/jimmunol.1301365
72. El Daker S, Sacchi A, Tempestilli M, Carducci C, Goletti D, Vanini V, et al. Granulocytic myeloid derived suppressor cells expansion during active pulmonary tuberculosis is associated with high nitric oxide plasma level. PloS One (2015) 10(4):e0123772. doi: 10.1371/journal.pone.0123772
73. Davids M, Pooran A, Smith L, Tomasicchio M, Dheda K. The frequency and effect of granulocytic myeloid-derived suppressor cells on mycobacterial survival in patients with tuberculosis: A preliminary report. Front Immunol (2021) 12:676679. doi: 10.3389/fimmu.2021.676679
74. Ashenafi S, Brighenti S. Reinventing the human tuberculosis (TB) granuloma: Learning from the cancer field. Front Immunol (2022) 13:1059725. doi: 10.3389/fimmu.2022.1059725
75. Jiang H, Gong H, Zhang Q, Gu J, Liang L, Zhang J. Decreased expression of perforin in CD8(+) T lymphocytes in patients with Mycobacterium tuberculosis infection and its potential value as a marker for efficacy of treatment. J Thorac Dis (2017) 9(5):1353–60. doi: 10.21037/jtd.2017.05.74
76. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol (2012) 3:51. doi: 10.3389/fimmu.2012.00051
77. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188(9):1137–46. doi: 10.1164/rccm.201304-0803OC
78. Gideon HP, Phuah J, Junecko BA, Mattila JT. Neutrophils express pro- and anti-inflammatory cytokines in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Mucosal Immunol (2019) 12(6):1370–81. doi: 10.1038/s41385-019-0195-8
79. Singh B, Singh DK, Ganatra SR, Escobedo RA, Khader S, Schlesinger LS, et al. Myeloid-derived suppressor cells mediate T cell dysfunction in nonhuman primate TB granulomas. MBio (2021) 12(6):e0318921. doi: 10.1128/mbio.03189-21
80. Agrawal N, Streata I, Pei G, Weiner J, Kotze L, Bandermann S, et al. Human Monocytic Suppressive Cells Promote Replication of Mycobacterium tuberculosis and Alter Stability of in vitro Generated Granulomas. Front Immunol (2018) 9:2417. doi: 10.3389/fimmu.2018.02417
81. Ashenafi S, Muvva JR, Mily A, Snäll J, Zewdie M, Chanyalew M, et al. Immunosuppressive features of the microenvironment in lymph nodes granulomas from tuberculosis and HIV-co-infected patients. Am J Pathol (2022) 192(4):653–70. doi: 10.1016/j.ajpath.2021.12.013
82. McCaffrey EF, Donato M, Keren L, Chen Z, Delmastro A, Fitzpatrick MB, et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat Immunol (2022) 23(2):318–29. doi: 10.1038/s41590-021-01121-x
83. Dorhoi A, Kotzé LA, Berzofsky JA, Sui Y, Gabrilovich DI, Garg A, et al. Therapies for tuberculosis and AIDS: myeloid-derived suppressor cells in focus. J Clin Invest. (2020) 130(6):2789–99. doi: 10.1172/JCI136288
84. Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, et al. Incipient and subclinical tuberculosis: a clinical review of early stages and progression of infection. Clin Microbiol Rev (2018) 31(4):e00021–18. doi: 10.1128/CMR.00021-18
85. Grassi G, Vanini V, De Santis F, Romagnoli A, Aiello A, Casetti R, et al. PMN-MDSC frequency discriminates active versus latent tuberculosis and could play a role in counteracting the immune-mediated lung damage in active disease. Front Immunol (2021) 12:594376. doi: 10.3389/fimmu.2021.594376
86. Jøntvedt Jørgensen M, Jenum S, Tonby K, Mortensen R, Walzl G, Du Plessis N, et al. Monocytic myeloid-derived suppressor cells reflect tuberculosis severity and are influenced by cyclooxygenase-2 inhibitors. J Leukoc Biol (2021) 110(1):177–86. doi: 10.1002/JLB.4A0720-409RR
87. Hönzke K, Obermayer B, Mache C, Fathykova D, Kessler M, Dökel S, et al. Human lungs show limited permissiveness for SARS-CoV-2 due to scarce ACE2 levels but virus-induced expansion of inflammatory macrophages. Eur Respir J (2022) 60(6):2102725. doi: 10.1183/13993003.02725-2021
88. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell (2020) 182(6):1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
89. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med (2020) 26(7):1070–6. doi: 10.1038/s41591-020-0944-y
90. Laing AG, Lorenc A, del Molino del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med (2020) 26(10):1623–35. doi: 10.1038/s41591-020-1038-6
91. Falck-Jones S, Vangeti S, Yu M, Falck-Jones R, Cagigi A, Badolati I, et al. Functional monocytic myeloid-derived suppressor cells increase in blood but not airways and predict COVID-19 severity. J Clin Invest. (2021) 131(6):e144734. doi: 10.1172/JCI144734
92. Bordoni V, Sacchi A, Cimini E, Notari S, Grassi G, Tartaglia E, et al. An inflammatory profile correlates with decreased frequency of cytotoxic cells in coronavirus disease 2019. Clin Infect Dis (2020) 71(16):2272–5. doi: 10.1093/cid/ciaa577
93. Coudereau R, Waeckel L, Cour M, Rimmele T, Pescarmona R, Fabri A, et al. Emergence of immunosuppressive LOX-1+ PMN-MDSC in septic shock and severe COVID-19 patients with acute respiratory distress syndrome. J Leukoc Biol (2022) 111(2):489–96. doi: 10.1002/JLB.4COVBCR0321-129R
94. Agrati C, Sacchi A, Bordoni V, Cimini E, Notari S, Grassi G, et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ (2020) 27(11):3196–207. doi: 10.1038/s41418-020-0572-6
95. Sacchi A, Grassi G, Bordoni V, Lorenzini P, Cimini E, Casetti R, et al. Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis (2020) 11(10):921. doi: 10.1038/s41419-020-03125-1
96. Cabrera LE, Pekkarinen PT, Alander M, Nowlan KHA, Nguyen NA, Jokiranta S, et al. Characterization of low-density granulocytes in COVID-19. PloS Pathog (2021) 17(7):e1009721. doi: 10.1371/journal.ppat.1009721
97. Dean MJ, Ochoa JB, Sanchez-Pino MD, Zabaleta J, Garai J, Del Valle L, et al. Severe COVID-19 is characterized by an impaired type I interferon response and elevated levels of arginase producing granulocytic myeloid derived suppressor cells. Front Immunol (2021) 12:695972. doi: 10.3389/fimmu.2021.695972
98. Jiménez-Cortegana C, Liró J, Palazón-Carrión N, Salamanca E, Sojo-Dorado J, de la Cruz-Merino L, et al. Increased blood monocytic myeloid derived suppressor cells but low regulatory T lymphocytes in patients with mild COVID-19. Viral Immunol (2021) 34(9):639–45. doi: 10.1089/vim.2021.0044
99. Thompson EA, Cascino K, Ordonez AA, Zhou W, Vaghasia A, Hamacher-Brady A, et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep (2021) 34(11):108863. doi: 10.1016/j.celrep.2021.108863
100. Rendeiro AF, Casano J, Vorkas CK, Singh H, Morales A, DeSimone RA, et al. Profiling of immune dysfunction in COVID-19 patients allows early prediction of disease progression. Life Sci Alliance. (2021) 4(2):e202000955. doi: 10.26508/lsa.202000955
101. Mortaz E, Movassaghi M, Bassir A, Dezfuli NK, Roofchayee ND, Jamaati H, et al. Evaluation of myeloid-derived suppressor cells in the blood of iranian COVID-19 patients. Iran J Allergy Asthma Immunol (2022) 21(4):467–77. doi: 10.18502/ijaai.v21i4.10294
102. Takano T, Matsumura T, Adachi Y, Terahara K, Moriyama S, Onodera T, et al. Myeloid cell dynamics correlating with clinical outcomes of severe COVID-19 in Japan. Int Immunol (2021) 33(4):241–7. doi: 10.1093/intimm/dxab005
103. Jiménez-Cortegana C, Sánchez-Jiménez F, Pérez-Pérez A, Álvarez N, Sousa A, Cantón-Bulnes L, et al. Low levels of granulocytic myeloid-derived suppressor cells may be a good marker of survival in the follow-up of patients with severe COVID-19. Front Immunol (2022) 12:801410. doi: 10.3389/fimmu.2021.801410
104. Marais C, Claude C, Semaan N, Charbel R, Barreault S, Travert B, et al. Myeloid phenotypes in severe COVID-19 predict secondary infection and mortality: a pilot study. Ann Intensive Care (2021) 11(1):111. doi: 10.1186/s13613-021-00896-4
105. Grassi G, Notari S, Gili S, Bordoni V, Casetti R, Cimini E, et al. Myeloid-derived suppressor cells in COVID-19: the paradox of good. Front Immunol (2022) 13:842949. doi: 10.3389/fimmu.2022.842949
106. Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Hed Myrberg I, Lourda M, et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc Natl Acad Sci (2021) 118(6):e2018587118. doi: 10.1073/pnas.2018587118
107. Xu G, Qi F, Li H, Yang Q, Wang H, Wang X, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discovery (2020) 6(1):73. doi: 10.1038/s41421-020-00225-2
108. Kiaee F, Jamaati H, Shahi H, Roofchayee ND, Varahram M, Folkerts G, et al. Immunophenotype and function of circulating myeloid derived suppressor cells in COVID-19 patients. Sci Rep (2022) 12(1):22570. doi: 10.1038/s41598-022-26943-z
109. Loftus TJ, Ungaro R, Dirain M, Efron PA, Mazer MB, Remy KE, et al. Overlapping but disparate inflammatory and immunosuppressive responses to SARS-coV-2 and bacterial sepsis: an immunological time course analysis. Front Immunol (2021) 12:792448. doi: 10.3389/fimmu.2021.792448
110. Tomić S, Đokić J, Stevanović D, Ilić N, Gruden-Movsesijan A, Dinić M, et al. Reduced expression of autophagy markers and expansion of myeloid-derived suppressor cells correlate with poor T cell response in severe COVID-19 patients. Front Immunol (2021) 12:614599. doi: 10.3389/fimmu.2021.614599
111. Reizine F, Lesouhaitier M, Gregoire M, Pinceaux K, Gacouin A, Maamar A, et al. SARS-coV-2-induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J Clin Immunol (2021) 41(3):515–25. doi: 10.1007/s10875-020-00920-5
112. Sacchi A, Grassi G, Notari S, Gili S, Bordoni V, Tartaglia E, et al. Expansion of myeloid derived suppressor cells contributes to platelet activation by L-arginine deprivation during SARS-coV-2 infection. Cells (2021) 10(8):2111. doi: 10.3390/cells10082111
113. Bost P, De Sanctis F, Canè S, Ugel S, Donadello K, Castellucci M, et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat Commun (2021) 12(1):1428. doi: 10.1038/s41467-021-21702-6
114. Zoso A, Mazza EMC, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur J Immunol (2014) 44(11):3307–19. doi: 10.1002/eji.201444522
115. Lebrun A, Lo Re S, Chantry M, Izquierdo Carerra X, Uwambayinema F, Ricci D, et al. CCR2(+) monocytic myeloid-derived suppressor cells (M-MDSCs) inhibit collagen degradation and promote lung fibrosis by producing transforming growth factor-β1. J Pathol (2017) 243(3):320–30. doi: 10.1002/path.4956
116. Ruenjaiman V, Sodsai P, Kueanjinda P, Bunrasmee W, Klinchanhom S, Reantragoon R, et al. Impact of SARS-CoV-2 infection on the profiles and responses of innate immune cells after recovery. J Microbiol Immunol Infect (2022) 55(6 Pt 1):993–1004. doi: 10.1016/j.jmii.2022.09.001
117. Siemińska I, Węglarczyk K, Surmiak M, Kurowska-Baran D, Sanak M, Siedlar M, et al. Mild and asymptomatic COVID-19 convalescents present long-term endotype of immunosuppression associated with neutrophil subsets possessing regulatory functions. Front Immunol (2021) 12:748097. doi: 10.3389/fimmu.2021.748097
118. Hopkins FR, Govender M, Svanberg C, Nordgren J, Waller H, Nilsdotter-Augustinsson Å, et al. Major alterations to monocyte and dendritic cell subsets lasting more than 6 months after hospitalization for COVID-19. Front Immunol (2023) 13:1082912. doi: 10.3389/fimmu.2022.1082912
119. Schrijver IT, Théroude C, Antonakos N, Regina J, Le Roy D, Bart P-A, et al. COVID-19 rapidly increases MDSCs and prolongs innate immune dysfunctions. Eur J Immunol (2022) 52(10):1676–9. doi: 10.1002/eji.202249827
120. Beliakova-Bethell N, Maruthai K, Xu R, Salvador LCM, Garg A. Monocytic-myeloid derived suppressor cells suppress T-cell responses in recovered SARS CoV2-infected individuals. Front Immunol (2022) 13:894543. doi: 10.3389/fimmu.2022.894543
121. Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA (1999) 282(7):677–86. doi: 10.1001/jama.282.7.677
122. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
123. Ward JD, Cornaby C, Schmitz JL. Indeterminate quantiFERON gold plus results reveal deficient interferon gamma responses in severely ill COVID-19 patients. J Clin Microbiol (2021) 59(10):e0081121. doi: 10.1128/JCM.00811-21
124. Gupta A, Sural S, Gupta A, Rousa S, Koner BC, Bhalotra A, et al. Positive QuantiFERON test and the severity of COVID-19 disease: A prospective study. Indian J Tuberc. (2021) 68(4):474–80. doi: 10.1016/j.ijtb.2020.12.013
125. Rajamanickam A, Pavan Kumar N, Chandrasekaran P, Nancy A, Bhavani PK, Selvaraj N, et al. Effect of SARS-CoV-2 seropositivity on antigen – specific cytokine and chemokine responses in latent tuberculosis. Cytokine (2022) 150:155785. doi: 10.1016/j.cyto.2021.155785
126. Qin A, Cai W, Pan T, Wu K, Yang Q, Wang N, et al. Expansion of monocytic myeloid-derived suppressor cells dampens T cell function in HIV-1-seropositive individuals. J Virol (2013) 87(3):1477–90. doi: 10.1128/JVI.01759-12
127. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother cancer. (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
128. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia (2013) 15(12):1400–9. doi: 10.1593/neo.131748
129. Cleverley TL, Peddineni S, Guarner J, Cingolani F, Garcia PK, Koehler H, et al. The host-directed therapeutic imatinib mesylate accelerates immune responses to Mycobacterium marinum infection and limits pathology associated with granulomas. PloS Pathog (2023) 19(5):e1011387. doi: 10.1371/journal.ppat.1011387
130. Napier RJ, Rafi W, Cheruvu M, Powell KR, Zaunbrecher MA, Bornmann W, et al. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe (2011) 10(5):475–85. doi: 10.1016/j.chom.2011.09.010
131. Xia L, Yuan L-Z, Hu Y-H, Liu J-Y, Hu G-S, Qi R-Y, et al. A SARS-CoV-2-specific CAR-T-cell model identifies felodipine, fasudil, imatinib, and caspofungin as potential treatments for lethal COVID-19. Cell Mol Immunol (2023) 20(4):351–64. doi: 10.1038/s41423-023-00985-3
132. Aman J, Duijvelaar E, Botros L, Kianzad A, Schippers JR, Smeele PJ, et al. Imatinib in patients with severe COVID-19: a randomised, double-blind, placebo-controlled, clinical trial. Lancet Respir Med (2021) 9(9):957–68. doi: 10.1016/S2213-2600(21)00237-X
133. Shen M, Wang J, Yu W, Zhang C, Liu M, Wang K, et al. A novel MDSC-induced PD-1(-)PD-L1(+) B-cell subset in breast tumor microenvironment possesses immuno-suppressive properties. Oncoimmunology (2018) 7(4):e1413520. doi: 10.1080/2162402X.2017.1413520
134. Zhan H, Yang L, Liu Y, Li H, Li X, Wang H, et al. Acceptance, safety and immunogenicity of a booster dose of inactivated SARS-CoV-2 vaccine in adults with tuberculosis. J Infect (2023) 86(5):e138–41. doi: 10.1016/j.jinf.2023.01.030
135. Verma D, Parasa VR, Raffetseder J, Martis M, Mehta RB, Netea M, et al. Anti-mycobacterial activity correlates with altered DNA methylation pattern in immune cells from BCG-vaccinated subjects. Sci Rep (2017) 7(1):12305. doi: 10.1038/s41598-017-12110-2
Keywords: myeloid derived suppressor cells, tuberculosis, SARS-CoV-2, COVID-19, coinfection
Citation: Shaw JA, Malherbe ST, Walzl G and du Plessis N (2023) Suppressive myeloid cells in SARS-CoV-2 and Mycobacterium tuberculosis co-infection. Front. Immunol. 14:1222911. doi: 10.3389/fimmu.2023.1222911
Received: 15 May 2023; Accepted: 06 July 2023;
Published: 20 July 2023.
Edited by:
Stefan H.E. Kaufmann, Max Planck Institute for Infection Biology, GermanyReviewed by:
Björn Corleis, Friedrich-Loeffler-Institute, GermanyCopyright © 2023 Shaw, Malherbe, Walzl and du Plessis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane Alexandra Shaw, amFuZXNoYXdAc3VuLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.