 Jeba Atkia Maisha
Jeba Atkia Maisha Hani S. El-Gabalawy
Hani S. El-Gabalawy Liam J. O’Neil
Liam J. O’Neil- Manitoba Centre for Proteomics and Systems Biology, Department of Internal Medicine, University of Manitoba, Winnipeg, MB, Canada
Rheumatoid Arthritis (RA) is a common autoimmune disease that targets the synovial joints leading to arthritis. Although the etiology of RA remains largely unknown, it is clear that numerous modifiable risk factors confer increased risk to developing RA. Of these risk factors, cigarette smoking, nutrition, obesity, occupational exposures and periodontal disease all incrementally increase RA risk. However, the precise immunological mechanisms by which these risk factors lead to RA are not well understood. Basic and translational studies have provided key insights into the relationship between inflammation, antibody production and the influence in other key cellular events such as T cell polarization in RA risk. Improving our general understanding of the mechanisms which lead to RA will help identify targets for prevention trials, which are underway in at-risk populations. Herein, we review the modifiable risk factors that are linked to RA development and describe immune mechanisms that may be involved. We highlight the few studies that have sought to understand if modification of these risk factors reduces RA risk. Finally, we speculate that modification of risk factors may be an appealing avenue for prevention for some at-risk individuals, specifically those who prefer lifestyle interventions due to safety and economic reasons.
Introduction
Rheumatoid Arthritis (RA) is a common autoimmune disease that primarily targets the synovial joints. At the onset of RA, patients present to clinical care with painful and swollen joints, with symptoms typically in the hands and feet (1). Untreated RA leads to the development of bone erosions, joint damage and functional disability. Furthermore, RA leads to considerable systemic inflammation which is associated with premature cardiovascular disease and early mortality. Fortunately, modern advances in therapeutics, specifically the development of biologic medications, has improved outcomes for many patients with RA. In spite of this, RA is considered a life-long disease without a cure, and although medications are effective, they are costly and have the potential to cause side effects. Further, many patients do not respond to RA therapy, leaving them with chronic pain and poor quality of life (2). Although our understanding of the pathogenesis of RA has improved dramatically over the last number of years, the precise etiology of RA remains elusive.
RA risk factors and progression from a pre-clinical state
It is well understood that the progression into RA occurs in multiple stages, typically over many years. The key genetic risk factor, the shared epitope (SE), is an HLA-DRB1 genetic risk locus (3, 4) that is strongly associated with the development of RA, specifically anti-citrullinated protein antibody (ACPA) positive RA. Studies suggest that the SE binds to citrullinated peptides more efficiently than non-SE HLA-DRB1 variants (5). Other genetic risk single nucleotide polymorphisms (SNPs) have been described, such as PTPN22 and PADI4, pointing towards the involvement of adaptive immune cell signalling (6) and pathogenic citrullination (7) as key steps towards RA development. By in large, the risk imparted by the SE explains the majority (~ 60%) of the genetic risk for RA. RA is also a sex-biased disease, with females being affected more than males at a ratio of 3:1 (8). Being a first-degree relative (FDR) of an RA patient also increases the risk of RA, likely due to both shared genetics and common environmental exposures amongst family members (9, 10).
Ultimately, the combination of these risk factors leads to the development of autoimmunity, marked by autoantibodies which are typically directed towards modified proteins, with citrulline being the best-characterized (3, 9–12). Antigenic targets also include other post-translational modifications such as homocitrulline, the by-product of a process called carbamylation (13), acetylated proteins (14) and malondialdehyde (MDA)/MDA-acetaldehyde (MAA) adducts (15). Rheumatoid Factor (RF), antibodies directed against the Fc portion of IgG, may also be detected at this time. Over time, often years, the immune response matures, with increasing levels of detectable ACPA and accumulation of antibody reactivity to a broader set of proteins (epitope spreading). ACPA has also been shown to undergo variable domain (Fab) N-linked glycosylation, which is closely linked to somatic hypermutation, a process by which the specificity of immunoglobulin is enhanced by T-cell driven maturation of B-cells. Importantly, the glycosylation of ACPA occurs prior to the development of RA, and is seemingly a strong predictor for imminent onset of arthritis (16, 17). Once the ACPA response matures, RA is considered imminent (3), however it is not known what event(s) ultimately triggers the development of inflammatory arthritis from a state of autoimmunity (18).
Aside from non-modifiable risk factors for RA development, much attention has shifted towards modifiable risk factors, specifically lifestyle changes that may reduce the risk of incident RA. This is of specific importance to ACPA+ at-risk individuals where the risk of developing RA may be as high as 45% (10). Herein, we review the modifiable risk factors that are associated with the development of RA and postulate the immunological mechanisms by which this risk is imparted. The focus of the review is on risk factors that are potentially associated with RA risk, namely inhaled exposures, diet and the microbiome. Finally, we review evidence linking modification of these factors with a reduction in RA risk, setting the stage for future RA prevention trials in high-risk populations.
Inhaled exposures
Cigarette smoking
Cigarette smoking, a widely prevalent habit across the world, has been extensively studied for its impact on human health, specifically respiratory and cardiovascular diseases (19). Of all modifiable risk factors, cigarette smoking is the strongest and best studied contributor of RA risk (20, 21). Multiple case-control studies (22–24) and retrospective cohort studies (25–27) have linked cigarette smoking, both duration and intensity (28) (typically measured by pack-years) with enhanced RA risk (Table 1). For example, a meta-analysis of 10 studies showed that in heavy smokers, the risk of developing RA was 2-fold higher compared to never smokers, with evidence of a dose-response based on pack-year exposure. The risk with smoking appears to be much stronger for the development of seropositive RA, rather than seronegative (26, 29), including both RF and ACPA positive RA (30, 36–38). Interestingly, there is an important gene-environment interaction between smoking and the shared epitope risk alleles (21, 30, 39). For example, in the EIRA cohort in Sweden, the odds ratio (OR) of developing seropositive RA in smokers with the SE was substantially higher (SE and Smoking OR 10.0) than the odds for individuals with only 1 (SE alone OR 4.8, Smoking alone OR 1.9) of the 2 risk factors (36). Smoking may also interact with other RA risk allele’s including PADI4, which also displays a sex bias toward enhancing RA risk in men (33). Importantly, smoking enhances the risk of developing RA in seropositive at-risk prospective cohorts (34, 35), although this association is somewhat controversial as it has not been replicated in other prospective cohorts (3, 9, 40), possibly due to limited sample size. Interestingly, passive smoking through parental exposure has been shown to be independently associated with RA risk (41), perhaps suggesting that RA risk may never normalize even with complete cessation. Smoking cessation in established RA is associated with reduced disease activity and cardiovascular disease risk (42), further strengthening the association between smoking and RA pathogenesis.
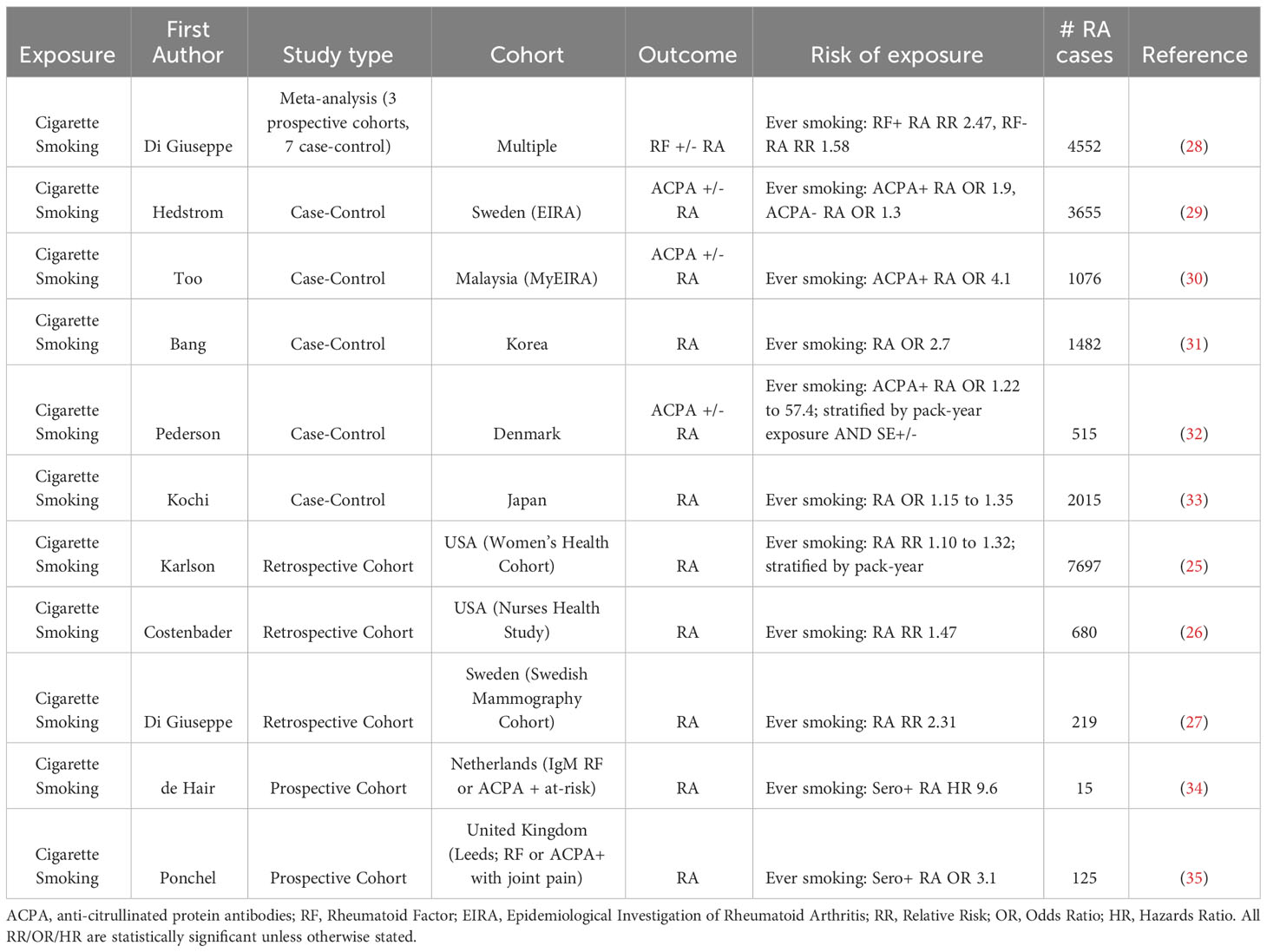
Table 1 Selected studies showing an association between cigarette smoking and Rheumatoid Arthritis (RA) risk.
Cigarette smoke influences RA autoantibody development
The mechanisms by which smoking influences RA development remain unclear. Smoking is strongly associated with the development of RA-specific autoantibodies in unaffected individuals, suggesting a role in breaching immune tolerance. Interestingly, the association between smoking and seropositivity suggests that there may be an influence on the development of both RF and ACPA (43). With respects to RF development, mice exposed to chronic cigarette smoke preferentially develop RF, rather than ACPA. Furthermore, humans with cigarette smoke related lung disease develop RF, as opposed to ACPA (44). In patients with RA, smoking is associated with RF seropositivity, specifically IgA and IgM isotypes (45). In the EIRA cohort, the strength of association between smoking and RA was highest for ACPA+/RF+ (OR 2.0), followed by ACPA-/RF+ (OR 1.6). A positive but nonsignificant association with RF-/ACPA+ RA was observed (OR 1.2), again pointing towards the importance of understanding the relationship between RF, ACPA and smoke exposure (36).
In asymptomatic FDR (First-Degree Relatives) of RA patients and non-RA individuals with joint pain, the association between smoking and ACPA is actually stronger than the association between HLA-SE and ACPA (46). Interestingly, in individuals who are seropositive with joint pain, the effect of smoking on inflammatory arthritis onset is less clear, with HLA-SE potentially playing a more dominant role at this stage (46). Smoking may influence the development of autoantibodies at the mucosal surface (47) through the enhanced production of Peptidyl Arginine Deiminase (PAD) (48), the human enzyme responsible for citrullinating proteins, including known RA autoantigens such as vimentin (49). Enhanced citrullination in the lungs (21, 48) may lead to aberrant autoantibody responses and the development of pulmonary ACPA+ B cell (50). Spontaneous NET formation, a potential source of PAD, is detectable in lungs of individuals at-risk to develop RA which is also associated with sputum IgA ACPA (51) (mucosal ACPA). NETs themselves are decorated with citrullinated proteins (52) and may be responsible for autoreactivity to citrullinated histones in RA patients (53). Some mouse models of RA have corroborated the effects of cigarette smoke exposure on inflammatory arthritis (54) and ACPA development (55). It should be noted however cigarette smoke has been shown to reduce CIA arthritis in some studies (56), a finding that is likely due to the timing of smoke exposure (57).
The effects of cigarette smoking on innate and adaptive immunity
Cigarette smoking is a proinflammatory process, with multiple effects on key immune cells both systemically and at the mucosal surface. Cigarette smoke contains thousands of chemicals including immunomodulators such as nicotine, carbon monoxide, acrolein and oxygen free radicals (58). At the mucosal surfaces, cigarette smoke actives local epithelial cells to produce pro-inflammatory cytokines (59), which enhances immune cell recruitment to illicit an inflammatory response. Here, cigarette smoke may act through pulmonary dendritic cells to enhance T-cell polarization towards Th1 and Th17 CD4 T-cells (60) (Figure 1). Furthermore, there is a reduction in regulatory T-cells in the lungs of smokers, although this is not observed systemically (61). In-vitro cigarette smoke extract leads to enhanced production of chemokines by monocytes which influences the recruitment of neutrophils (62). This effect is also observed in pulmonary macrophages (alveolar macrophages (63)). Neutrophil chemotaxis is impaired by cigarette smoke extract (64, 65), while their propensity to form NETs appears to be enhanced both in-vitro and at the pulmonary surfaces (66). Systemically, cigarette smoking skews the immune system towards a proinflammatory response. Multiple studies have suggested that smoking influences helper T – cell polarization, and in smokers systemic skewing towards Th1 and Th17 CD4 T-cells has been described (67). Interestingly, studies on the immunomodulatory properties of nicotine have pointed to a primarily immunosuppressive effect, predominantly through nicotinic acetylcholine receptors (68). Specifically, nicotine induces T-cell anergy in mice (69) and attenuates collagen induced arthritis (70). Nonetheless, at least one study has pointed to a role for nicotine in worsening murine arthritis, primarily through the formation of NETs (71).
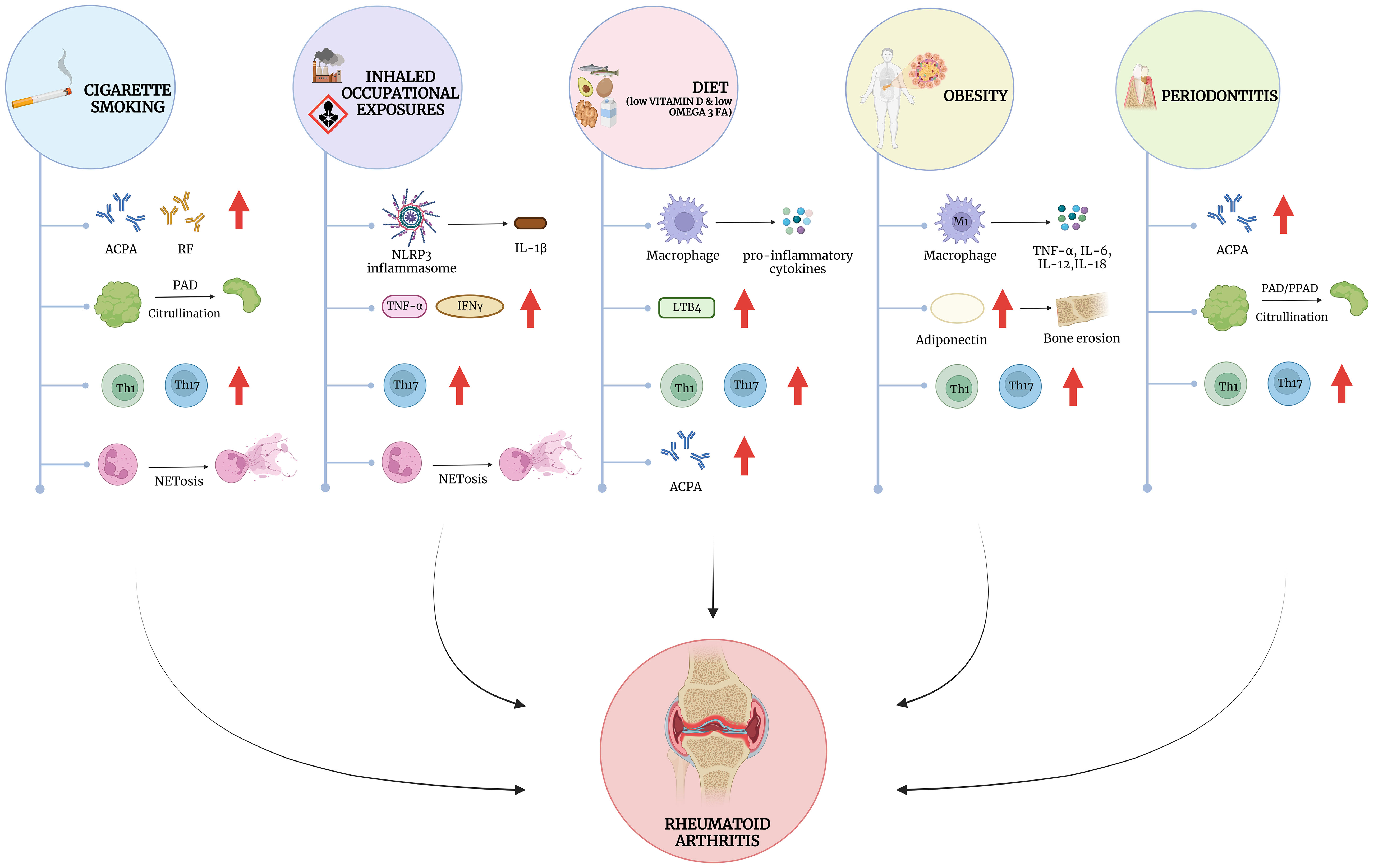
Figure 1 Modifiable risk factors for RA and their impact on autoimmunity and inflammation. Mechanistically, the influence of external risk factors converges on a number of immune cell dysfunction, including the stimulation of the innate and adaptive immune systems. Cigarette smoke has been shown to influence the development of autoantibodies such as anti-citrullinated protein antibodies (ACPA) and Rheumatoid Factor (RF), particularly in the lungs, where it also leads to enhanced citrullination possibly through the activation of neutrophils to form neutrophil extracellular traps (NETosis). Occupational exposures, such as silica dust and textile dust, drives NLRP3 inflammasome activation leading to the release of IL1β, and other cytokines such as TNF-α and IFN-γ. Similar to cigarette smoking, silica may also influence NET formation and the release of citrullinated proteins. Dietary factors such as low vitamin D and Omega-3 fatty acid intake leads to the release of pro-inflammatory cytokines, and eicosanoids such as Leukotriene B4 (LTB4). Obesity has overlapping effects on inflammatory cytokine release with other dietary factors but is also associated with the release of key adipokines such as Adiponectin, which may influence the development of erosive arthritis in RA. Periodontitis shares features with cigarette smoke and occupational exposures, mediating similar processes but in the oral mucosa rather than the lung. Interestingly many exposures strongly influence T-cell polarization, favoring an increase in Th1 and Th17 helper T-cells, which play a crucial role in RA pathogenesis. Created with BioRender.com.
Inhaled occupational exposures
While cigarette smoke remains the most extensively studied pulmonary exposure associated with RA development, there is also evidence linking RA to other inhaled exposures, particularly those encountered in occupational settings. Case-control studies have shown that silica exposure increases the odds of developing both seronegative and seropositive RA (72–74) (Table 2). Furthermore, other inhalants such as pesticides, solvents and other farming related exposures also appear to increase RA risk (88). Air pollution has also been linked to a variety of autoimmune diseases, including RA (78). Textile dust exposure is also linked to RA development, and alike cigarette smoking, displays an important gene-environment interaction with the SE (76). Interestingly, there may be cumulative or synergistic effects of inhalants, with the number of different exposures increasing RA risk in a stepwise fashion. Furthermore, the addition of cigarette smoking and genetic risk to occupational exposures increases the risk of developing seropositive RA dramatically (86, 89). Of the best studied immunopathologic mechanisms, silica dust is not only associated with RA, but there is also clear synergism between silica and cigarette smoke exposure in RA risk (74). Silica is found in nature as quartz, and exposure most often occurs in occupational activities such as mining, drilling and sand blasting. Pathologic exposure to silica occurs primarily though inhalation, where it induces pulmonary fibrosis as a result of inflammation (90–92). Inhaled silica is engulfed by alveolar macrophages, which leads to the production of reactive oxygen species (ROS) (93). Engulfment also leads to rupture of cytosolic lysosomes, which in turn activates the inflammasome and the secretion of IL-1β (94). TNF-α and IFN-γ have been shown to play essential roles in the both the acute and chronic inflammatory responses observed in pulmonary silicosis (95). An exaggerated Th17 response (Figure 1), driven primarily by IL-17A production is partially responsible for acute, but not chronic inflammation mediated by silica (96). Pulmonary silica also leads to the recruitment of neutrophils, and the crystals themselves can induce the production of NETs (97). However, silica does not appear to aggravate murine arthritis, and is not associated with an increased production of PAD enzymes in this murine model (98). Regardless, the combination of cellular death, inflammation and activation of lymphocytes may contribute mechanistically to loss of immune tolerance, particularly in genetically susceptible individuals.
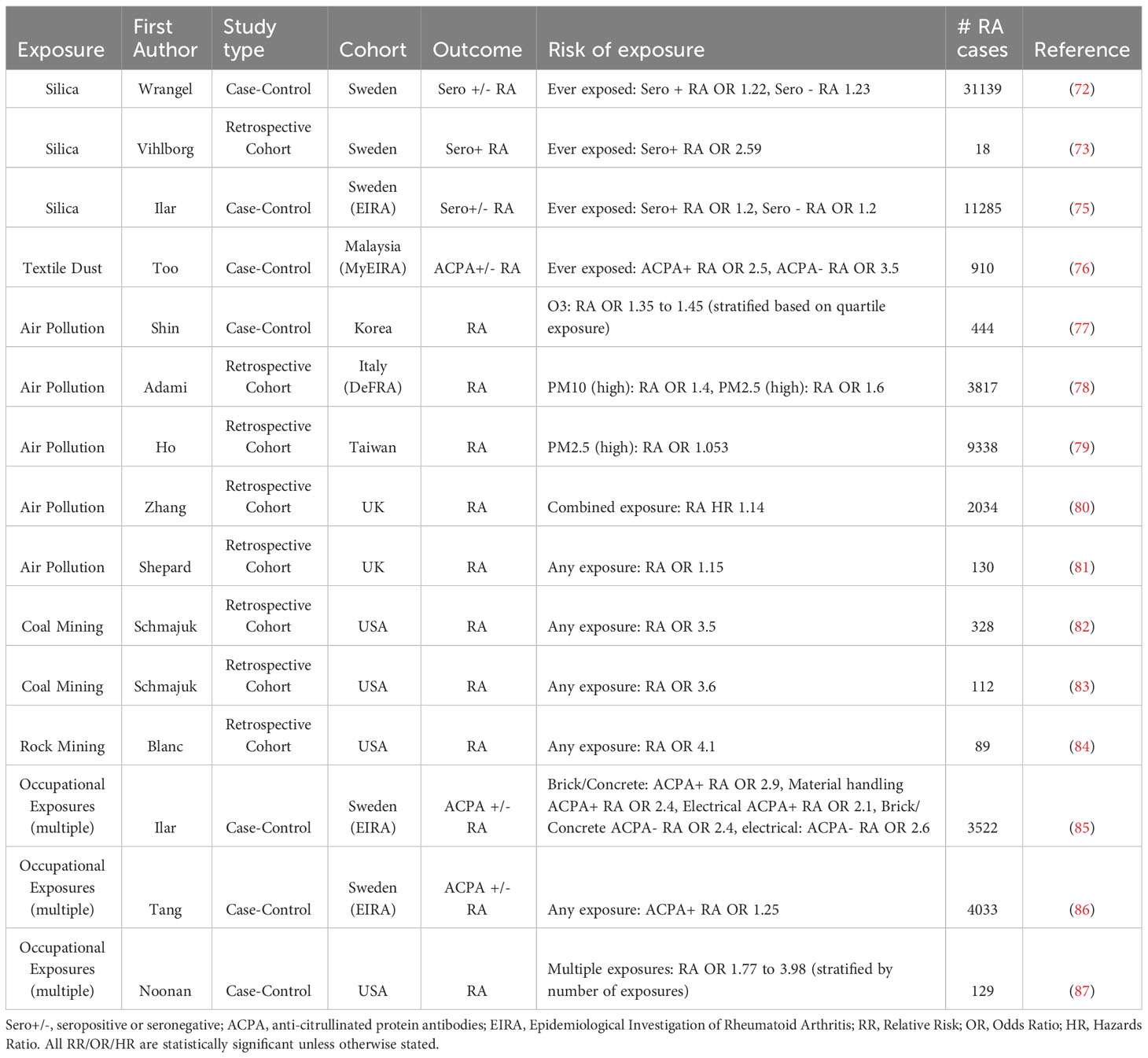
Table 2 Selected studies showing an association between occupational exposures and Rheumatoid Arthritis (RA) risk.
Dietary Exposures
Dietary patterns
Over the past few decades, the drastic transformation in dietary habits, particularly the rise of the Western diet, has emerged as a major factor influencing human health and contributing to the escalating burden of chronic diseases worldwide. Diet is considered an important lifestyle risk factor for the development of RA (99). Although an accurate assessment of dietary patterns remains a challenge due to potential confounders and recall bias (100–102), several key studies suggest that dietary factors are implicit in the risk of developing RA (Table 3). Studies of diet and RA risk can be split into those analyzing global diet effects (ie. low inflammatory diet, Mediterranean diet, etc.) or the impact specific food groups/nutrients. Unhealthy diets have been linked to multiple autoimmune diseases (112) including RA, multiple sclerosis and inflammatory bowel disease. In RA, unhealthy eating patterns, measured using the Alternative Healthy Eating Index was associated with incident RA cases in the Nurses’ health study (113) even after adjusting for body mass index (BMI). The effects of the Mediterranean diet, a diet that aims to enrich in mono-unsaturated fatty acids, vegetables, fruits and whole grains, is associated with several important cardiovascular benefits (114), has displayed mixed results in protecting against RA development. For example, a large prospective cohort study suggested that adherence to the Mediterranean diet is protective of incident RA in ever smokers (104) after adjusting for BMI. Interestingly, no protective effects were found in non-smokers. In the EIRA cohort, adherence to the Mediterranean diet was also protective for RA, after adjusting for BMI and physical activity (103). Conversely, there are several studies of similar design that have shown no association between the Mediterranean diet and RA risk (103, 115). However, in established RA, randomized controlled trials (RCTs) of Mediterranean diet have shown a reduction in joint disease activity and improvement of function compared to controls diets (116, 117). Diets that are thought to promote inflammation such as a low fibre diet (118), high carbohydrate diet (115), high meat intake (119) have not confirmed any clear association with RA.

Table 3 Selected studies showing an association between dietary exposures and Rheumatoid Arthritis (RA) risk.
Fish and omega-3 fatty acids
While dietary patterns in general have been associated with the development of RA, the role of fish intake, with specific focus on polyunsaturated fats has been a primary focus in RA risk. Fatty fish is rich in long chain omega-3 fatty acids (eicosapentaenoic acid or EPA, and docosahexaenoic acid or DHA) and is the primary dietary source of these fatty acids. Other sources include krill, algae and nuts (120). Fish intake in particular has been linked to protection from RA development (Table 3). Outside of the studies that investigated the impact of the Mediterranean diet (104) (linked with increased fish intake), a large registry study from Denmark suggested that high fish oil intake reduces the risk of incident RA (121). Further, high omega-3 and fish intake protected against RA development in a cohort of Swedish women (107). Furthermore, data from the Nurses’ Health Study suggested the impact of cigarette smoking on RA development was attenuated by fish consumption (105). Fish oil supplements have also shown efficacy in prospective trials of patients with established RA, which demonstrates their anti-inflammatory potential (122, 123).
Fatty acid metabolism and inflammation
Following ingestion, omega-3 fatty acids are stored in the membrane phospholipids and are thought to exert their predominant anti-inflammatory effect following metabolism into eicosanoids. Eicosanoids are a family of molecules, all roughly 20 carbon units in length, called oxylipins which are predominantly made up of several subfamilies including prostaglandins, thromboxanes and leukotrienes. In contrast, omega-6 fatty acids, derived from animal fats, plant oils and cereals are thought to exert pro-inflammatory effects. The ratio of omega-6 to omega-3 is thought to shift the relative contribution of derivative fatty acids towards or away from inflammation. The effect of EPA/DHA on specific inflammatory cells has been studied both in-vitro and in-vivo (124). In macrophages, EPA/DHA reduces the secretion of pro-inflammatory cytokines after simulation (125, 126), supresses the inflammasome (127) and increases phagocytic capacity (128). Neutrophils exposed to omega-3 FAs similarly display reduced migration capacity (129), enhanced phagocytosis (130), yet enhanced production of reactive oxygen species (131). Interestingly, RA patients supplemented with omega-3 FA displayed reduced production of neutrophil derived Leukotriene B4, a chemotactic leukotriene (132). Omega-3 metabolites, primarily resolvins, reduce cytokine production in activated CD8+ T-cells, T helper 1 (Th1) cells and Th17 cells, and skews T-cell differentiation away from Th1/Th17 (133) (Figure 1), and perhaps towards an increase in Treg cells (134). In asthmatic children omega-3 supplementation reduced IL-17A, a key Th17 cytokine (135). Omega-3 supplementation also reduces the severity of murine arthritis, and this may be in part mediated by effects on T-cell differentiation (134). The effect of omega-3 supplementation on B-cell activation remains less clear. DHA/EPA reduces B-cell activation in human cells treated ex-vivo (136). Conversely, in murine models, enhanced IgM production via B-cell proliferation was observed after EPA/DHA treatment (137, 138). Taken together, the impact of omega-3 supplementation appears to impact both innate and adaptive immune responses, and while further study is required, the majority of evidence suggests a prominent anti-inflammatory effect on a wide range of key processes.
Vitamin D
Due to its immunomodulatory properties, Vitamin D deficiency has been linked to the development of several autoimmune disease, including RA (Table 3). In a large cohort study of older women in the USA, Vitamin D supplementation was shown to be associated with a reduced risk of developing RA (108). In a case-control analysis of the Nurses’ Health Study, vitamin-D levels were lower in the months-years prior to RA development (109). A meta-analysis that included over 200,000 participants suggested that high vitamin D intake reduced the risk of developing RA by 24% (110). We have previously shown that Vitamin D levels are significantly lower in ACPA positive first-degree relatives of RA patients (FDR) suggesting a potential role in breaking immune tolerance (139), although similar results were not observed in a USA cohort (140). A vitamin D genetic risk score analysis suggests that specific SNPs related to Vitamin D may be protective for the development of RA-associated antibodies in FDR (141). Several small studies have also suggested efficacy of Vitamin D supplementation in individuals with established RA (142).
Vitamin D metabolism and influence on the immune system
In order to exert effects physiologically, 1,25-dihydroxyvitamin D (1,25(OH)2D), the active metabolite of Vitamin D, must be synthesized (143). Both Vitamin D2 (ergocalciferol, derived from food/supplements) and Vitamin D3 can undergo conversion by the liver (25(OH)D) and kidneys (1,25(OH)2D). Vitamin D3 is produced in the skin following UVB exposure, typically from sunlight. Vitamin D deficiency is common, particularly in parts of the world where seasonality has a major impact on sun-exposure. Importantly, the immunomodulatory effects of activated Vitamin D(1,25(OH)2D), requires binding to its cognate receptor, Vitamin D Receptor (VDR) which is located in the nucleus where it enacts primarily on the transcription of various genes (144). Vitamin D binding protein (DBP), the major plasma carrier for vitamin D metabolites, plays a crucial role in transporting metabolites to target tissues (145). Importantly, polymorphisms of DBP have been associated with a number of human diseases, including diabetes (146), cancer (147) and RA (146). As such, an important limitation of most of the large studies associating vitamin D status and RA is that they do not take these polymorphisms into account.
Aside from its well-recognized impact on bone homeostasis, vitamin D plays a role in immune homeostasis. In macrophages, activated vitamin D stimulates the production of cathelicidin, an antimicrobial peptide (LL37). Macrophages themselves can hydroxylate 25(OH)D to the active form where local production may allow for a more potent immune response (148). In neutrophils, vitamin D has been shown to enhance bacterial killing, but reduce the production of inflammatory cytokines, thus a dampening of excessive inflammation may be an important effect (149). Upon stimulation, T-cells upregulate VDR allowing vitamin D to supress proliferation and shift T-cell differentiation away from Th17 (150) and Th1 (151) types, favoring the formation of regulatory T-cells which has been shown to influence the development of murine autoimmune diabetes (152) (Figure 1). Vitamin D also plays a crucial role in transitioning Th1 cells to an immunosuppressive, IL-10 producing subset (153). Unlike omega-3 FAs, the immunosuppressive effects of vitamin D in B-cells are more apparent. Activated vitamin D reduces the formation of immunoglobulin producing plasma cells following activation (154), along with class-switch memory B-cells by inducing apoptosis (155).
Obesity
High body mass index (BMI) may also play a role in increasing the risk of RA development. For example, in a case-control study of incident RA, obesity (both BMI and abdominal waist circumference) was associated with an increased risk of RA (156). Several other studies of similar design have also implicated obesity in RA risk (157–159), although studies exist which do not show an association (160) or even a protective role for obesity (161). Interestingly, the risk of obesity and developing RA may be higher in women compared to men (162). Prospective studies of at-risk individuals have also linked obesity with the development of RA, specifically in individuals considered at-risk based on the presence of RA autoantibodies (34), although this association was not found in our study of First Nation FDR (3). Intriguingly, the impact of obesity on RA risk appears to be modified only marginally by physical activity, which is thought to be protective of future RA (163). This points to the complexity of the obesity syndrome, which similar to RA, is an interaction between genetics and environmental factors.
Despite the complexity of obesity, it remains well understood that excessive fat can lead to inflammation through various mechanisms. In obesity, macrophages are skewed towards the pro-inflammatory (M1-like) cells, and away from anti-inflammatory (M2-like) macrophages (150). This plays a key role in perpetuating the inflammatory response. M1-like macrophages display increased secretion of cytokines such as TNF-α, IL-6, IL-12, IL-18, and ROS which are all involved in enhanced inflammatory responses (164–167). These macrophages recruit CD4+ T-cells that eventually differentiate into several effector cells, namely Th1, Th2, Th17 and Treg cells (168, 169). Th2 and Treg cells counter obesity-associated inflammation; However, in obesity, these two types of effector cells decrease in number. As a result, the inflammation-promoting cells Th1 and Th17 constitute the majority of the cell population (Figure 1). Moreover, adipokines secreted by the adipose tissue are strongly correlated with inflammation (170). Studies have shown that Leptin is increased in early RA (171) and is related to increased ROS production (172). A meta-analysis revealed that Adiponectin is significantly higher in RA patients (9) and may contribute to the bone erosions observed in RA (10) via induction of a pro-inflammatory state in osteoblasts and osteoclasts (11). Other adipokines, such as chemerin, resistin, lipocalin 2 have been associated with clinical outcomes in individuals with established RA (173–177).
The microbiome
Gut microbiome
The human microbiome is a vast and intricate ecosystem of microbes residing in our bodies that plays a fundamental role in shaping our health (178). Studies implicate that the gut microbiome have gained ample interest in nearly all fields of medicine. The microbiome has a diverse and still emerging role in digestion and homeostasis of the epithelial layer/mucosal permeability. Further, the influence of diet, medications and other exposures play a crucial role in the overall function and diversity of the microbiota. Dysbiosis is clearly evident in patients with established RA and appears to correlate with disease activity and treatment response (179). Particular interest has been paid to Prevotella copri (180), which is enriched in RA patients and exacerbates murine arthritis (181). Although studies in pre-clinical RA are lacking, a strain of Subdoligranulum has been shown to cross react with IgG and IgA from individuals who are at-risk to develop RA (182), although notably, these were not ACPAs. Fascinatingly, when the bacterium is transferred to germ-free mice, it leads to spontaneous development of inflammatory arthritis. The influence of the gut microbiota on inflammatory processes is complex, yet it is clear that the formation of short chain fatty acids (SCFA) from the processing of fiber plays an important role. SCFA are typically thought to be anti-inflammatory, predominantly though the production of regulatory T-cells (183), but also due to altered permeability of the gastrointestinal tract (184). In ACPA+ at-risk individuals with musculoskeletal symptoms, serum SCFAs were higher in those who did not progress, compared to those that developed RA (185). Components of bacteria can trigger toll-like receptors (TLRs), which propagate both innate and adaptive immune responses (186) in resident leukocytes and epithelial cells. Much attention has been paid to segmented filamentous bacteria (SFB), which induces the production and activation of Th17 cells (187) and follicular helper T-cells (188) in the intestine, which influences murine arthritis disease activity (189). Further studies of the role of the microbiome and the risk of RA, with a specific focus on at-risk individuals are required.
Periodontal disease
Periodontal disease is an oral inflammatory condition by which resident bacteria mediate a cascade of inflammatory responses, ultimately leading to local tissue damage. Periodontitis is a polymicrobial disease whereby overgrowth of oral bacteria interacts with local immune cells leading to inflammation. Similar to RA, inflammatory lesions can lead to radiographic bone loss and irreversible damage (190). To date, the connection between periodontal disease and the development of RA has been shown in a variety of epidemiological studies (191–194). Specifically, a meta-analysis of over 150,000 individuals showed a modest association between RA and periodontitis (195). This association is strongest for ACPA positive RA, and interestingly, RA patients with periodontitis have been shown to have higher concentrations of serum ACPA compared to those without periodontitis (192). Similar to cigarette smoking, there is an association between periodontal disease and the SE that appears to have a strong gene-environment interaction, including leading to more severe/destructive RA (196, 197). Interestingly, treatment of periodontitis in RA patients is associated with improved arthritis disease activity (198, 199). It should be noted however that the co-occurrence of periodontitis and RA does not imply a causative relationship between the two diseases, as there is limited evidence that active periodontitis leads to the development of RA.
P. gingivalis is associated with citrullination and autoantibody responses in RA
The best characterized oral bacteria studied in RA risk is Porphyromonas gingivalis (P. gingivalis). Of considerable interest, P. gingivalis expresses endogenous PAD (PPAD), which can induce citrullination sites of local proteins that are key RA autoantigens (200). Unlike human PAD, PPAD preferentially citrullinates terminal arginine (201) after the digestion of proteins by another enzyme called gingipain (202). Modification of PPAD reduces collagen antibody-induced murine arthritis severity and ACPA levels (203). RA patients have been shown to develop antibodies targeting P. gingivalis (204), and this is observed in the pre-clinical stage of the disease (205). Monoclonal IgG from gingival B-cells cross react with citrullinated peptides derived from human and P. gingivalis proteins, suggesting the potential for molecular mimicry (206). Pre-existing periodontitis has been shown to exacerbate the collagen antibody-induced arthritis mouse (207) and the CIA model (208). In the latter, P. gingivalis was associated with serum cytokines most suggestive of an enhanced Th17/Th1 response (Figure 1). Activation of the innate and adaptive immune responses in periodontitis is crucial. Gingival tissues affected by such inflammation display B and CD4 T-cell infiltration (209), with evidence pointing to Th17 differentiation (210). Neutrophils are recruited to the site of inflammation by IL-8, which expectedly yields the release of destructive, granule proteins, the generation of ROS and the formation of NETs (also yielding enhanced citrullination) (211). A number of other oral microbes, aside from P. gingivalis, have been linked to RA development including Provatella and Veillonella (212, 213). Recently, evidence of oral microbes traversing into the blood of RA patients which can expose citrullinated antigens to ACPA B – cell and promote inflammation by activating inflammatory monocytes (214).
Modification of risk factors to prevent or delay RA onset
Although the studies reviewed above link RA development to several important modifiable risk factors, it remains much less clear if modification of these factors meaningfully reduces RA risk, and at what stage of pre-RA this might be most efficacious. Smoking cessation is not only modifiable, but can be aided with medications, cognitive behaviour therapy (CBT) and nicotine replacement. Further, it is associated with other important health benefits such as reduced cardiovascular disease (215) and cancer (216). The only evidence linking smoking cessation and reduced RA risk comes from observational data. For example, in the Nurses’ Health Study, individuals who quit smoking had a 40% reduction in incident seropositive RA. Interestingly, the effects of smoking on RA risk were still detectable 30 years after quitting, compared to life-long nonsmokers (37). A similar finding was observed in a Swedish cohort study, showing a 30% reduction of incident RA following smoking cessation (27). Many clinicians are likely to recommend smoking cessation to RA patients and at-risk individuals, possibly due to the extended health benefits outside of RA risk. However, it remains unclear if smoking cessation following the development of ACPA has any meaningful impact on RA development.
To our knowledge, there are no observational studies linking changes in diet or nutrition and subsequent risk of incident RA. However, in the VITAL study, a trial of Vitamin D and Omega-3 fatty acid supplementation (217) a prespecified subgroup analysis found that the combination of supplements displayed a reduction in incident RA after 5 years of follow-up (111). It’s important to note that because this trial was originally designed for cardiovascular and oncologic outcomes, thus the population was older and likely had average to below average risk of developing RA. Whether Vitamin D or Omega-3 supplementation can be used to prevent incident RA in high-risk groups, such as ACPA positive individuals remains unclear.
RA prevention has recently moved into the forefront of pre-RA research, and a number of prospective clinical trials are complete or ongoing. Most of these trials have deployed re-purposed RA medications, and so far, none have successfully prevented RA, although Rituximab (218), Abatacept (219, 220) and Methotrexate (221) delayed RA onset by several months. It is notable that recruitment to RA prevention trials remains a challenge, and thus choosing the right intervention (222) is crucial to ensure study feasibility which is in line with participants’ views and priorities (223). There are likely many individuals at increased risk of RA who would not accept targeted, potentially toxic immunosuppressive medications to mitigate their risk. There appears to be a willingness to accept lifestyle interventions in ACPA/RF+ individuals with arthralgia (224) and FDR of RA patients (225, 226) to lower the risk of RA, which for individuals with established RA can also help improve disease activity and progression, as outlined by EULAR (European League against Rheumatism) recommendations from 2021 (227). Hence, there remains an unfilled gap in RA prevention care, which is using nutritional, dietary, and lifestyle interventions to evaluate their protective effects in high-risk individuals, either alone or in combination with more targeted drug therapies.
Further points to consider
When evaluating studies linking modifiable exposures to the risk of RA, it is crucial to consider the strength and levels of evidence supporting the associations. Several limitations apply to retrospective and case-control studies. For instance, even large retrospective population-based studies may lack the power to detect differences due to RA’s relatively low prevalence (~1%). Additionally, how a diagnosis of RA is being made (ACR/EULAR criteria (228), billing codes, self-report) should also be considered and appraised. Case-control studies can be affected by recall bias, specifically with respect to lifestyle exposures. Although prospective cohort studies of at-risk individuals are less common and more difficult to recruit for, they may serve to enrich in the outcome of interest (incident RA), where the prevalence is much higher. However, these cohorts are often smaller, and thus may also suffer from reduced power to detect an association between an exposure and the outcome. Irrespective of the study design, unmeasured or measured confounding factors should be strongly considered as they can influence the interpretation of lifestyle and modifiable exposure studies. For example, many lifestyle factors are interrelated, such as diet and exercise, smoking and alcohol use, and are often connected to socioeconomic status, which itself has been associated with an increased risk of RA (229).
The association between several exposures, including periodontitis, cigarette smoking, occupational exposures all appear to be enriched in seropositive RA, with mixed findings for the strength of association with seronegative RA (Tables 1, 2). Regarding dietary associations, most studies have not stratified based on serological status, but one study found the Mediterranean diet to be protective for seropositive RA (103), and another showed that fish exposure was protective for seronegative RA (105) (Table 3). One consideration is the recent discovery of other post-translational modifications which act as autoantigens in RA, such as homocitrulline, acetyl, and MDA/MAA, suggest a potential overestimation or misclassification of seropositive and seronegative disease (230) in many of these associative studies. It is also intriguing that many of these exposures converge on a similar pathway, with a dominant role for mucosal immunity specifically in the mouth, lung and gut. This notion is reinforced by the finding that secretory IgA may play an important role in the generation of pathogenic autoimmunity in RA (231). It remains clear that a deeper understanding of mucosal immunology will be crucial for identifying the origins of RA, particularly seropositive disease.
Concluding remarks
In conclusion, prospective and retrospective cohort studies have provided key insights into the role of modifiable risk factors and RA risk. Cigarette smoking and diet appear to have substantial impact on incident RA and may be the most practical lifestyle interventions to prevent RA in the future. Further studies are required to understand how these exposures modulate the immune system to promote inflammation, reduce immune tolerance and trigger clinical arthritis in at-risk individuals. Ultimately a combination of prospective clinical trials, longitudinal cohort studies and translational science will help disentangle the key events that drive the development of RA in those at-risk.
Author contributions
JAM: Conceptualized and wrote the manuscript. Performed literature review. HE-G: Edited manuscript, provided expert advice. LO’N: Edited and conceptualized the manuscript. Performed literature review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gravallese EM, Firestein GS. Rheumatoid arthritis - common origins, divergent mechanisms. N Engl J Med (2023) 388:529–42. doi: 10.1056/NEJMra2103726
2. Watanabe R, Okano T, Gon T, Yoshida N, Fukumoto K, Yamada S, et al. Difficult-to-treat rheumatoid arthritis: Current concept and unsolved problems. Front Med (Lausanne) (2022) 9:1049875. doi: 10.3389/fmed.2022.1049875
3. Tanner S, Dufault B, Smolik I, Osorio J, Lemaire I, Lasbleiz S, et al. A prospective study of the development of inflammatory arthritis in the family members of indigenous north American people with rheumatoid arthritis. Arthritis Rheumatol (2019) 71:1494–503. doi: 10.1002/art.40880
4. Du Montcel ST, Michou L, Petit-Teixeira E, Meng X, Anaparti V, Hitchon C, et al. New classification of HLA-DRB1 alleles supports the shared epitope hypothesis of rheumatoid arthritis susceptibility. Arthritis Rheum (2005) 52:1063–8. doi: 10.1002/art.20989
5. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med (2013) 210:2569–82. doi: 10.1084/jem.20131241
6. Saevarsdottir S, Stefansdottir L, Sulem P, Thorleifsson G, Ferkingstad E, Rutsdottir G, et al. Multiomics analysis of rheumatoid arthritis yields sequence variants that have large effects on risk of the seropositive subset. Ann Rheum Dis (2022) 81:1085–95. doi: 10.1136/annrheumdis-2021-221754
7. Massarenti L, Enevold C, Damgaard D, Odum N, Garred P, Frisch M, et al. PADI4 polymorphisms confer risk of anti-CCP-positive rheumatoid arthritis in synergy with HLA-DRB1*04 and smoking. Front Immunol (2021) 12:707690. doi: 10.3389/fimmu.2021.707690
8. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: A review. JAMA (2018) 320:1360–72. doi: 10.1001/jama.2018.13103
9. Bemis EA, Demoruelle MK, Seifert JA, Polinski KJ, Weisman MH, Buckner JH, et al. Factors associated with progression to inflammatory arthritis in first-degree relatives of individuals with RA following autoantibody positive screening in a non-clinical setting. Ann Rheum Dis (2021) 80:154–61. doi: 10.1136/annrheumdis-2020-217066
10. Garcia-Montoya L, Nam JL, Duquenne L, Villota-Eraso C, Di Matteo A, Hartley C, et al. Prioritising referrals of individuals at-risk of RA: guidance based on results of a 10-year national primary care observational study. Arthritis Res Ther (2022) 24:26. doi: 10.1186/s13075-022-02717-w
11. Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, De Vries N, van der Horst-Bruinsma IE, et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis (2010) 69:490–4. doi: 10.1136/ard.2008.105759
12. Di Matteo A, Mankia K, Duquenne L, Mahler M, Corscadden D, Mbara K, et al. Third-generation anti-cyclic citrullinated peptide antibodies improve prediction of clinical arthritis in individuals at risk of rheumatoid arthritis. Arthritis Rheumatol (2020) 72:1820–8. doi: 10.1002/art.41402
13. O'Neil LJ, Oliveira CB, Wang X, Navarrete M, Barrera-Vargas A, Merayo-Chalico J, et al. Neutrophil extracellular trap-associated carbamylation and histones trigger osteoclast formation in rheumatoid arthritis. Ann Rheum Dis (2023) 82:630–8. doi: 10.1136/ard-2022-223568
14. Volkov M, Kampstra ASB, van Schie KA, Kawakami A, Tamai M, Kawashiri S, et al. Evolution of anti-modified protein antibody responses can be driven by consecutive exposure to different post-translational modifications. Arthritis Res Ther (2021) 23:298. doi: 10.1186/s13075-021-02687-5
15. Thiele GM, Duryee MJ, Anderson DR, Klassen LW, Mohring SM, Young KA, et al. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol (2015) 67:645–55. doi: 10.1002/art.38969
16. Rombouts Y, Willemze A, van Beers JJ, Shi J, Kerkman PF, Van Toorn L, et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis (2016) 75:578–85. doi: 10.1136/annrheumdis-2014-206598
17. Kissel T, Hafkenscheid L, Wesemael TJ, Tamai M, Kawashiri SY, Kawakami A, et al. IgG anti-citrullinated protein antibody variable domain glycosylation increases before the onset of rheumatoid arthritis and stabilizes thereafter: A cross-sectional study encompassing ~1,500 samples. Arthritis Rheumatol (2022) 74:1147–58. doi: 10.1002/art.42098
18. Wiens D, Smolik I, Meng X, Anaparti V, El-Gabalawy HS, O'Neil LJ. Functional disability to evaluate the risk of arthritis in first-degree relatives of patients with rheumatoid arthritis. J Rheumatol (2022) 49:244–50. doi: 10.3899/jrheum.210614
19. Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF. Systemic effects of smoking. Chest (2007) 131:1557–66. doi: 10.1378/chest.06-2179
20. Hahn J, Malspeis S, Choi MY, Kallberg H, Bengtsson C, Grunewald J, et al. Association of healthy lifestyle behaviors and the risk of developing rheumatoid arthritis among women. Arthritis Care Res (Hoboken) (2023) 75:272–6. doi: 10.1002/acr.24862
21. Klareskog L, Stolt P, Lundberg K, Stevens E, Karlson EW, Lu B, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum (2006) 54:38–46. doi: 10.1002/art.21575
22. Hutchinson D, Shepstone L, Moots R, Lear JT, Lynch MP. Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann Rheum Dis (2001) 60:223–7. doi: 10.1136/ard.60.3.223
23. Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis (2003) 62:835–41. doi: 10.1136/ard.62.9.835
24. Voigt LF, Koepsell TD, Nelson JL, Dugowson CE, Daling JR. Smoking, obesity, alcohol consumption, and the risk of rheumatoid arthritis. Epidemiology (1994) 5:525–32.
25. Karlson EW, Lee IM, Cook NR, Manson JE, Buring JE, Hennekens CH. A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum (1999) 42:910–7. doi: 10.1002/1529-0131(199905)42:5<910::AID-ANR9>3.0.CO;2-D
26. Costenbader KH, Feskanich D, Mandl LA, Karlson EW. Smoking intensity, duration, and cessation, and the risk of rheumatoid arthritis in women. Am J Med (2006) 119:503 e1–9. doi: 10.1016/j.amjmed.2005.09.053
27. Di Giuseppe D, Orsini N, Alfredsson L, Askling J, Wolk A. Cigarette smoking and smoking cessation in relation to risk of rheumatoid arthritis in women. Arthritis Res Ther (2013) 15:R56. doi: 10.1186/ar4218
28. Di Giuseppe D, Discacciati A, Orsini N, Wolk A. Cigarette smoking and risk of rheumatoid arthritis: a dose-response meta-analysis. Arthritis Res Ther (2014) 16:R61. doi: 10.1186/ar4498
29. Hedstrom AK, Stawiarz L, Klareskog L, Alfredsson L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur J Epidemiol (2018) 33:415–23. doi: 10.1007/s10654-018-0360-5
30. Too CL, Yahya A, Murad S, Dhaliwal JS, Larsson PT, Muhamad NA, et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: results from the Malaysian epidemiological investigation of rheumatoid arthritis (MyEIRA). Arthritis Res Ther (2012) 14:R89. doi: 10.1186/ar3813
31. Bang SY, Lee KH, Cho SK, Lee HS, Lee KW, Bae SC. Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum (2010) 62:369–77.
32. Pedersen M, Jacobsen S, Garred P, Madsen HO, Klarlund M, Svejgaard A, et al. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: a nationwide case-control study in Denmark. Arthritis Rheum (2007) 56:1446–53. doi: 10.1002/art.22597
33. Kochi Y, Thabet MM, Suzuki A, Okada Y, Daha NA, Toes RE, et al. PADI4 polymorphism predisposes male smokers to rheumatoid arthritis. Ann Rheum Dis (2011) 70:512–5. doi: 10.1136/ard.2010.130526
34. de Hair MJ, Landewe RB, van de Sande MG, Van Schaardenburg D, Van Baarsen LG, Gerlag DM, et al. Smoking and overweight determine the likelihood of developing rheumatoid arthritis. Ann Rheum Dis (2013) 72:1654–8. doi: 10.1136/annrheumdis-2012-202254
35. Ponchel F, Duquenne L, Xie X, Corscadden D, Shuweihdi F, Mankia K, et al. Added value of multiple autoantibody testing for predicting progression to inflammatory arthritis in at-risk individuals. RMD Open (2022) 8. doi: 10.1136/rmdopen-2022-002512
36. Hedstrom AK, Ronnelid J, Klareskog L, Alfredsson L. Complex relationships of smoking, HLA-DRB1 genes, and serologic profiles in patients with early rheumatoid arthritis: update from a swedish population-based case-control study. Arthritis Rheumatol (2019) 71:1504–11. doi: 10.1002/art.40852
37. Liu X, Tedeschi SK, Barbhaiya M, Leatherwood CL, Speyer CB, Lu B, et al. Impact and timing of smoking cessation on reducing risk of rheumatoid arthritis among women in the nurses' health studies. Arthritis Care Res (Hoboken) (2019) 71:914–24. doi: 10.1002/acr.23837
38. Svard A, Skogh T, Alfredsson L, Ilar A, Klareskog L, Bengtsson C, et al. Associations with smoking and shared epitope differ between IgA- and IgG-class antibodies to cyclic citrullinated peptides in early rheumatoid arthritis. Arthritis Rheumatol (2015) 67:2032–7. doi: 10.1002/art.39170
39. Hensvold AH, Magnusson PK, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis (2015) 74:375–80. doi: 10.1136/annrheumdis-2013-203947
40. Eloff E, Martinsson K, Ziegelasch M, Cedergren J, Reckner A, Skogh T, et al. Autoantibodies are major predictors of arthritis development in patients with anti-citrullinated protein antibodies and musculoskeletal pain. Scand J Rheumatol (2021) 50:189–97. doi: 10.1080/03009742.2020.1818820
41. Yoshida K, Wang J, Malspeis S, Marchand N, Lu B, Prisco LC, et al. Passive smoking throughout the life course and the risk of incident rheumatoid arthritis in adulthood among women. Arthritis Rheumatol (2021) 73:2219–28. doi: 10.1002/art.41939
42. Roelsgaard IK, Ikdahl E, Rollefstad S, Wibetoe G, Esbensen BA, Kitas GD, et al. Smoking cessation is associated with lower disease activity and predicts cardiovascular risk reduction in rheumatoid arthritis patients. Rheumatol (Oxford) (2020) 59:1997–2004. doi: 10.1093/rheumatology/kez557
43. van Wesemael TJ, Ajeganova S, Humphreys J, Terao C, Muhammad A, Symmons DP, et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: a multicenter cohort study. Arthritis Res Ther (2016) 18:285. doi: 10.1186/s13075-016-1177-9
44. Newkirk MM, Mitchell S, Procino M, Li Z, Cosio M, Mazur W, et al. Chronic smoke exposure induces rheumatoid factor and anti-heat shock protein 70 autoantibodies in susceptible mice and humans with lung disease. Eur J Immunol (2012) 42:1051–61. doi: 10.1002/eji.201141856
45. Masdottir B, Jonsson T, Manfredsdottir V, Vikingsson A, Brekkan A, Valdimarsson H. Smoking, rheumatoid factor isotypes and severity of rheumatoid arthritis. Rheumatol (Oxford) (2000) 39:1202–5. doi: 10.1093/rheumatology/39.11.1202
46. Wouters F, Maurits MP, van Boheemen L, Verstappen M, Mankia K, Matthijssen XME, et al. Determining in which pre-arthritis stage HLA-shared epitope alleles and smoking exert their effect on the development of rheumatoid arthritis. Ann Rheum Dis (2022) 81:48–55. doi: 10.1136/annrheumdis-2021-220546
47. Demoruelle MK, Weisman MH, Simonian PL, Lynch DA, Sachs PB, Pedraza IF, et al. Brief report: airways abnorMalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum (2012) 64:1756–61. doi: 10.1002/art.34344
48. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
49. Lugli EB, Correia RE, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther (2015) 17:9. doi: 10.1186/s13075-015-0520-x
50. Joshua V, Loberg Haarhaus M, Hensvold A, Wahamaa H, Gerstner C, Hansson M, et al. Rheumatoid arthritis specific autoimmunity in the lung before and at the onset of disease. Arthritis Rheumatol (2023). doi: 10.1002/art.42549
51. Okamoto Y, Devoe S, Seto N, Minarchick V, Wilson T, Rothfuss HM, et al. Association of sputum neutrophil extracellular trap subsets with igA anti-citrullinated protein antibodies in subjects at risk for rheumatoid arthritis. Arthritis Rheumatol (2022) 74:38–48. doi: 10.1002/art.41948
52. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
53. Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis (2014) 73:1414–22. doi: 10.1136/annrheumdis-2012-202765
54. Okamoto S, Adachi M, Chujo S, Yamada K, Akita K, Itoh S, et al. Etiological role of cigarette smoking in rheumatoid arthritis: Nasal exposure to cigarette smoke condensate extracts augments the development of collagen-induced arthritis in mice. Biochem Biophys Res Commun (2011) 404:1088–92. doi: 10.1016/j.bbrc.2010.12.118
55. Kang J, Jeong SH, Lee K, Park N, Jung H, Lee K, et al. Exacerbation of symptomatic arthritis by cigarette smoke in experimental arthritis. PloS One (2020) 15:e0230719. doi: 10.1371/journal.pone.0230719
56. Lindblad SS, Mydel P, Jonsson IM, Senior RM, Tarkowski A, Bokarewa M. Smoking and nicotine exposure delay development of collagen-induced arthritis in mice. Arthritis Res Ther (2009) 11:R88. doi: 10.1186/ar2728
57. Yu H, Yang YH, Rajaiah R, Moudgil KD. Nicotine-induced differential modulation of autoimmune arthritis in the Lewis rat involves changes in interleukin-17 and anti-cyclic citrullinated peptide antibodies. Arthritis Rheum (2011) 63:981–91. doi: 10.1002/art.30219
58. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res (2012) 91:142–9. doi: 10.1177/0022034511421200
59. Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med (2002) 166:849–54. doi: 10.1164/rccm.200202-097OC
60. Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med (2009) 1:4ra10.
61. Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med (2007) 13:567–9. doi: 10.1038/nm1583
62. Walters MJ, Paul-Clark MJ, McMaster SK, Ito K, Adcock IM, Mitchell JA. Cigarette smoke activates human monocytes by an oxidant-AP-1 signaling pathway: implications for steroid resistance. Mol Pharmacol (2005) 68:1343–53. doi: 10.1124/mol.105.012591
63. Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol (2002) 2:372–7. doi: 10.1038/nri803
64. White PC, Hirschfeld J, Milward MR, Cooper PR, Wright HJ, Matthews JB, et al. Cigarette smoke modifies neutrophil chemotaxis, neutrophil extracellular trap formation and inflammatory response-related gene expression. J Periodontal Res (2018) 53:525–35. doi: 10.1111/jre.12542
65. Bokaba RP, Anderson R, Theron AJ, Tintinger GR. Cigarette smoke condensate attenuates phorbol ester-mediated neutrophil extracellular trap formation. Afr Health Sci (2017) 17:896–904. doi: 10.4314/ahs.v17i3.33
66. Zou Y, Chen X, He B, Xiao J, Yu Q, Xie B, et al. Neutrophil extracellular traps induced by cigarette smoke contribute to airway inflammation in mice. Exp Cell Res (2020) 389:111888. doi: 10.1016/j.yexcr.2020.111888
67. Vargas-Rojas MI, Ramirez-Venegas A, Limon-Camacho L, Ochoa L, Hernandez-Zenteno R, Sansores RH. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Med (2011) 105:1648–54. doi: 10.1016/j.rmed.2011.05.017
68. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature (2003) 421:384–8. doi: 10.1038/nature01339
69. Geng Y, Savage SM, Razani-Boroujerdi S, Sopori ML. Effects of nicotine on the immune response. II. chronic nicotine treatment induces T cell anergy. J Immunol (1996) 156:2384–90. doi: 10.4049/jimmunol.156.7.2384
70. Wu S, Luo H, Xiao X, Zhang H, Li T, Zuo X. Attenuation of collagen induced arthritis via suppression on Th17 response by activating cholinergic anti-inflammatory pathway with nicotine. Eur J Pharmacol (2014) 735:97–104. doi: 10.1016/j.ejphar.2014.04.019
71. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatol (Oxford) (2017) 56:644–53.
72. Wrangel O, Graff P, Bryngelsson IL, Fornander L, Wiebert P, Vihlborg P. Silica dust exposure increases risk for rheumatoid arthritis: A Swedish national registry case-control study. J Occup Environ Med (2021) 63:951–5. doi: 10.1097/JOM.0000000000002281
73. Vihlborg P, Bryngelsson IL, Andersson L, Graff P. Risk of sarcoidosis and seropositive rheumatoid arthritis from occupational silica exposure in Swedish iron foundries: a retrospective cohort study. BMJ Open (2017) 7:e016839. doi: 10.1136/bmjopen-2017-016839
74. Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis (2010) 69:1072–6. doi: 10.1136/ard.2009.114694
75. Ilar A, Klareskog L, Saevarsdottir S, Wiebert P, Askling J, Gustavsson P, et al. Occupational exposure to asbestos and silica and risk of developing rheumatoid arthritis: findings from a Swedish population-based case-control study. RMD Open (2019) 5:e000978. doi: 10.1136/rmdopen-2019-000978
76. Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Ann Rheum Dis (2016) 75:997–1002. doi: 10.1136/annrheumdis-2015-208278
77. Shin J, Lee J, Lee J, Ha EH. Association between exposure to ambient air pollution and rheumatoid arthritis in adults. Int J Environ Res Public Health (2019) 16. doi: 10.3390/ijerph16071227
78. Adami G, Pontalti M, Cattani G, Rossini M, Viapiana O, Orsolini G, et al. Association between long-term exposure to air pollution and immune-mediated diseases: a population-based cohort study. RMD Open (2022) 8. doi: 10.1136/rmdopen-2021-002055
79. Ho WC, Chou LW, Wang RY, Doan TN, Yu HL, Chou TH, et al. Association between exposure to ambient air pollution and the risk of rheumatoid arthritis in Taiwan: A population-based retrospective cohort study. Int J Environ Res Public Health (2022) 19. doi: 10.3390/ijerph19127006
80. Zhang J, Fang XY, Wu J, Fan YG, Leng RX, Liu B, et al. Association of combined exposure to ambient air pollutants, genetic risk, and incident rheumatoid arthritis: A prospective cohort study in the UK biobank. Environ Health Perspect (2023) 131:37008. doi: 10.1289/EHP10710
81. Shepherd A, Mullins JT. Arthritis diagnosis and early-life exposure to air pollution. Environ Pollut (2019) 253:1030–7. doi: 10.1016/j.envpol.2019.07.054
82. Schmajuk G, Trupin L, Yelin EH, Blanc PD. Dusty trades and associated rheumatoid arthritis in a population-based study in the coal mining counties of Appalachia. Occup Environ Med (2022) 79:308–14. doi: 10.1136/oemed-2021-107899
83. Schmajuk G, Trupin L, Yelin E, Blanc PD. Prevalence of arthritis and rheumatoid arthritis in coal mining counties of the united states. Arthritis Care Res (Hoboken) (2019) 71:1209–15. doi: 10.1002/acr.23874
84. Blanc PD, Trupin L, Yelin EH, Schmajuk G. Assessment of risk of rheumatoid arthritis among underground hard rock and other mining industry workers in Colorado, new Mexico, and Utah. JAMA Netw Open (2022) 5:e2236738. doi: 10.1001/jamanetworkopen.2022.36738
85. Ilar A, Alfredsson L, Wiebert P, Klareskog L, Bengtsson C. Occupation and risk of developing rheumatoid arthritis: results from a population-based case-control study. Arthritis Care Res (Hoboken) (2018) 70:499–509. doi: 10.1002/acr.23321
86. Tang B, Liu Q, Ilar A, Wiebert P, Hagg S, Padyukov L, et al. Occupational inhalable agents constitute major risk factors for rheumatoid arthritis, particularly in the context of genetic predisposition and smoking. Ann Rheum Dis (2023) 82:316–23. doi: 10.1136/ard-2022-223134
87. Noonan CW, Pfau JC, Larson TC, Spence MR. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ Health Perspect (2006) 114:1243–7. doi: 10.1289/ehp.9203
88. Prisco LC, Martin LW, Sparks JA. Inhalants other than personal cigarette smoking and risk for developing rheumatoid arthritis. Curr Opin Rheumatol (2020) 32:279–88. doi: 10.1097/BOR.0000000000000705
89. Kronzer VL, Sparks JA. Occupational inhalants, genetics and the respiratory mucosal paradigm for ACPA-positive rheumatoid arthritis. Ann Rheum Dis (2023) 82:303–5. doi: 10.1136/ard-2022-223286
90. Callis AH, Sohnle PG, Mandel GS, Wiessner J, Mandel NS. Kinetics of inflammatory and fibrotic pulmonary changes in a murine model of silicosis. J Lab Clin Med (1985) 105:547–53.
91. Krefft S, Wolff J, Rose C. Silicosis: an update and guide for clinicians. Clin Chest Med (2020) 41:709–22. doi: 10.1016/j.ccm.2020.08.012
92. Adamcakova J, Mokra D. New insights into pathomechanisms and treatment possibilities for lung silicosis. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22084162
93. Castranova V. Signaling pathways controlling the production of inflammatory mediators in response to crystalline silica exposure: role of reactive oxygen/nitrogen species. Free Radic Biol Med (2004) 37:916–25. doi: 10.1016/j.freeradbiomed.2004.05.032
94. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U.S.A. (2008) 105:9035–40.
95. Davis GS, Holmes CE, Pfeiffer LM, Hemenway DR. Lymphocytes, lymphokines, and silicosis. J Environ Pathol Toxicol Oncol (2001) 20(Suppl 1):53–65. doi: 10.1615/JEnvironPatholToxicolOncol.v20.iSuppl.1.50
96. Lo Re S, Dumoutier L, Couillin I, Van Vyve C, Yakoub Y, Uwambayinema F, et al. IL-17A-producing gammadelta T and Th17 lymphocytes mediate lung inflammation but not fibrosis in experimental silicosis. J Immunol (2010) 184:6367–77. doi: 10.4049/jimmunol.0900459
97. Rizzi M, Carniato F, Tonello S, Migliario M, Invernizzi M, Rocchetti V, et al. Charged molecular silica trigger in vitro NETosis in human granulocytes via both oxidative and autophagic pathways. Eur Rev Med Pharmacol Sci (2018) 22:7058–68.
98. Engelmann R, Muller-Hilke B. Experimental silicosis does not aggravate collagen-induced arthritis in mice. J Negat Results BioMed (2017) 16:5. doi: 10.1186/s12952-017-0071-6
99. Backlund R, Drake I, Bergstrom U, Compagno M, Sonestedt E, Turesson C. Diet and the risk of rheumatoid arthritis - a systematic literature review. Semin Arthritis Rheum (2023) 58:152118. doi: 10.1016/j.semarthrit.2022.152118
100. Jackson KA, Byrne NM, Magarey AM, Hills AP. Minimizing random error in dietary intakes assessed by 24-h recall, in overweight and obese adults. Eur J Clin Nutr (2008) 62:537–43. doi: 10.1038/sj.ejcn.1602740
101. Livingstone MB. Assessment of food intakes: are we measuring what people eat? Br J BioMed Sci (1995) 52:58–67.
102. Salvador Castell G, Serra-Majem L, Ribas-Barba L. What and how much do we eat? 24-hour dietary recall method. Nutr Hosp (2015) 31(Suppl 3):46–8.
103. Johansson K, Askling J, Alfredsson L, Di Giuseppe D, Group ES. Mediterranean Diet and risk of rheumatoid arthritis: a population-based case-control study. Arthritis Res Ther (2018) 20:175. doi: 10.1186/s13075-018-1680-2
104. Nguyen Y, Salliot C, Gelot A, Gambaretti J, Mariette X, Boutron-Ruault MC, et al. Mediterranean Diet and risk of rheumatoid arthritis: findings from the french E3N-EPIC cohort study. Arthritis Rheumatol (2021) 73:69–77. doi: 10.1002/art.41487
105. Sparks JA, O'Reilly EJ, Barbhaiya M, Tedeschi SK, Malspeis S, Lu B, et al. Association of fish intake and smoking with risk of rheumatoid arthritis and age of onset: a prospective cohort study. BMC Musculoskelet Disord (2019) 20:2. doi: 10.1186/s12891-018-2381-3
106. Shapiro JA, Koepsell TD, Voigt LF, Dugowson CE, Kestin M, Nelson JL. Diet and rheumatoid arthritis in women: a possible protective effect of fish consumption. Epidemiology (1996) 7:256–63. doi: 10.1097/00001648-199605000-00007
107. Di Giuseppe D, Wallin A, Bottai M, Askling J, Wolk A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis (2014) 73:1949–53. doi: 10.1136/annrheumdis-2013-203338
108. Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG, et al. Vitamin d intake is inversely associated with rheumatoid arthritis: results from the Iowa women's health study. Arthritis Rheum (2004) 50:72–7. doi: 10.1002/art.11434
109. Hiraki LT, Arkema EV, Cui J, Malspeis S, Costenbader KH, Karlson EW. Circulating 25-hydroxyvitamin d level and risk of developing rheumatoid arthritis. Rheumatol (Oxford) (2014) 53:2243–8. doi: 10.1093/rheumatology/keu276
110. Song GG, Bae SC, Lee YH. Association between vitamin d intake and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol (2012) 31:1733–9. doi: 10.1007/s10067-012-2080-7
111. Hahn J, Cook NR, Alexander EK, Friedman S, Walter J, Bubes V, et al. Vitamin d and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial. BMJ (2022) 376:e066452. doi: 10.1136/bmj-2021-066452
112. Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of "Western diet" in inflammatory autoimmune diseases. Curr Allergy Asthma Rep (2014) 14:404. doi: 10.1007/s11882-013-0404-6
113. Hu Y, Sparks JA, Malspeis S, Costenbader KH, Hu FB, Karlson EW, et al. Long-term dietary quality and risk of developing rheumatoid arthritis in women. Ann Rheum Dis (2017) 76:1357–64. doi: 10.1136/annrheumdis-2016-210431
114. Dinu M, Pagliai G, Casini A, Sofi F. Mediterranean Diet and multiple health outcomes: an umbrella review of meta-analyses of observational studies and randomised trials. Eur J Clin Nutr (2018) 72:30–43. doi: 10.1038/ejcn.2017.58
115. Sundstrom B, Johansson I, Rantapaa-Dahlqvist S. Diet and alcohol as risk factors for rheumatoid arthritis: a nested case-control study. Rheumatol Int (2015) 35:533–9. doi: 10.1007/s00296-014-3185-x
116. Skoldstam L, Hagfors L, Johansson G. An experimental study of a Mediterranean diet intervention for patients with rheumatoid arthritis. Ann Rheum Dis (2003) 62:208–14. doi: 10.1136/ard.62.3.208
117. Garcia-Morales JM, Lozada-Mellado M, Hinojosa-Azaola A, Llorente L, Ogata-Medel M, Pineda-Juarez JA, et al. Effect of a dynamic exercise program in combination with mediterranean diet on quality of life in women with rheumatoid arthritis. J Clin Rheumatol (2020) 26:S116–S22. doi: 10.1097/RHU.0000000000001064
118. Rubin KH, Rasmussen NF, Petersen I, Kopp TI, Stenager E, Magyari M, et al. Intake of dietary fibre, red and processed meat and risk of late-onset chronic inflammatory diseases: A prospective Danish study on the "diet, cancer and health" cohort. Int J Med Sci (2020) 17:2487–95. doi: 10.7150/ijms.49314
119. Sundstrom B, Ljung L, Di Giuseppe D. Consumption of meat and dairy products is not associated with the risk for rheumatoid arthritis among women: A population-based cohort study. Nutrients (2019) 11. doi: 10.3390/nu11112825
120. Gammone MA, Riccioni G, Parrinello G, D'Orazio N. Omega-3 polyunsaturated fatty acids: benefits and endpoints in sport. Nutrients (2018) 11. doi: 10.3390/nu11010046
121. Pedersen M, Stripp C, Klarlund M, Olsen SF, Tjonneland AM, Frisch M. Diet and risk of rheumatoid arthritis in a prospective cohort. J Rheumatol (2005) 32:1249–52.
122. Proudman SM, James MJ, Spargo LD, Metcalf RG, Sullivan TR, Rischmueller M, et al. Fish oil in recent onset rheumatoid arthritis: a randomised, double-blind controlled trial within algorithm-based drug use. Ann Rheum Dis (2015) 74:89–95. doi: 10.1136/annrheumdis-2013-204145
123. Miles EA, Calder PC. Influence of marine n-3 polyunsaturated fatty acids on immune function and a systematic review of their effects on clinical outcomes in rheumatoid arthritis. Br J Nutr (2012) 107(Suppl 2):S171–84. doi: 10.1017/S0007114512001560
124. Gutierrez S, Svahn SL, Johansson ME. Effects of omega-3 fatty acids on immune cells. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20205028
125. Takashima A, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Nishimoto S, et al. Combination of n-3 polyunsaturated fatty acids reduces atherogenesis in apolipoprotein e-deficient mice by inhibiting macrophage activation. Atherosclerosis (2016) 254:142–50. doi: 10.1016/j.atherosclerosis.2016.10.002
126. Allam-Ndoul B, Guenard F, Barbier O, Vohl MC. A study of the differential effects of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) on gene expression profiles of stimulated thp-1 macrophages. Nutrients (2017) 9. doi: 10.3390/nu9050424
127. Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity (2013) 38:1154–63. doi: 10.1016/j.immuni.2013.05.015
128. Chang HY, Lee HN, Kim W, Surh YJ. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor gamma activation. Life Sci (2015) 120:39–47. doi: 10.1016/j.lfs.2014.10.014
129. Jones CN, Dalli J, Dimisko L, Wong E, Serhan CN, Irimia D. Microfluidic chambers for monitoring leukocyte trafficking and humanized nano-proresolving medicines interactions. Proc Natl Acad Sci U.S.A. (2012) 109:20560–5.
130. Svahn SL, Ulleryd MA, Grahnemo L, Stahlman M, Boren J, Nilsson S, et al. Dietary omega-3 fatty acids increase survival and decrease bacterial load in mice subjected to staphylococcus aureus-induced sepsis. Infect Immun (2016) 84:1205–13. doi: 10.1128/IAI.01391-15
131. Paschoal VA, Vinolo MA, Crisma AR, Magdalon J, Curi R. Eicosapentaenoic (EPA) and docosahexaenoic (DHA) acid differentially modulate rat neutrophil function in vitro. Lipids (2013) 48:93–103. doi: 10.1007/s11745-012-3726-6
132. Kremer JM, Lawrence DA, Jubiz W, Digiacomo R, Rynes R, Bartholomew LE, et al. Dietary fish oil and olive oil supplementation in patients with rheumatoid arthritis. Clin immunologic effects. Arthritis Rheum (1990) 33:810–20. doi: 10.1002/art.1780330607
133. Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med (2016) 8:353ra111. doi: 10.1126/scitranslmed.aaf7483
134. Kim JY, Lim K, Kim KH, Kim JH, Choi JS, Shim SC. N-3 polyunsaturated fatty acids restore Th17 and treg balance in collagen antibody-induced arthritis. PloS One (2018) 13:e0194331. doi: 10.1371/journal.pone.0194331
135. Farjadian S, Moghtaderi M, Kalani M, Gholami T, Hosseini Teshnizi S. Effects of omega-3 fatty acids on serum levels of T-helper cytokines in children with asthma. Cytokine (2016) 85:61–6. doi: 10.1016/j.cyto.2016.06.002
136. Weise C, Hilt K, Milovanovic M, Ernst D, Ruhl R, Worm M. Inhibition of IgE production by docosahexaenoic acid is mediated by direct interference with STAT6 and NFkappaB pathway in human b cells. J Nutr Biochem (2011) 22:269–75. doi: 10.1016/j.jnutbio.2010.02.004
137. Ramon S, Gao F, Serhan CN, Phipps RP. Specialized proresolving mediators enhance human b cell differentiation to antibody-secreting cells. J Immunol (2012) 189:1036–42. doi: 10.4049/jimmunol.1103483
138. Teague H, Harris M, Fenton J, Lallemand P, Shewchuk BM, Shaikh SR. Eicosapentaenoic and docosahexaenoic acid ethyl esters differentially enhance b-cell activity in murine obesity. J Lipid Res (2014) 55:1420–33. doi: 10.1194/jlr.M049809
139. Anaparti V, Meng X, Hemshekhar M, Smolik I, Mookherjee N, El-Gabalawy H. Circulating levels of free 25(OH)D increase at the onset of rheumatoid arthritis. PloS One (2019) 14:e0219109. doi: 10.1371/journal.pone.0219109
140. Feser M, Derber LA, Deane KD, Lezotte DC, Weisman MH, Buckner JH, et al. Plasma 25,OH vitamin d concentrations are not associated with rheumatoid arthritis (RA)-related autoantibodies in individuals at elevated risk for RA. J Rheumatol (2009) 36:943–6. doi: 10.3899/jrheum.080764
141. Vanderlinden LA, Bemis EA, Seifert J, Guthridge JM, Young KA, Demoruelle MK, et al. Relationship between a vitamin d genetic risk score and autoantibodies among first-degree relatives of probands with rheumatoid arthritis and systemic lupus erythematosus. Front Immunol (2022) 13:881332. doi: 10.3389/fimmu.2022.881332
142. Guan Y, Hao Y, Guan Y, Bu H, Wang H. The effect of vitamin d supplementation on rheumatoid arthritis patients: A systematic review and meta-analysis. Front Med (Lausanne) (2020) 7:596007. doi: 10.3389/fmed.2020.596007
143. Charoenngam N, Holick MF. Immunologic effects of vitamin d on human health and disease. Nutrients (2020) 12. doi: 10.3390/nu12072097
144. Haussler MR, Haussler CA, Jurutka PW, Thompson PD, Hsieh JC, Remus LS, et al. The vitamin d hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol (1997) 154 Suppl:S57–73.
145. Rozmus D, Ciesielska A, Plominski J, Grzybowski R, Fiedorowicz E, Kordulewska N, et al. Vitamin d binding protein (VDBP) and its gene polymorphisms-the risk of malignant tumors and other diseases. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21217822
146. Yan X, Zhao Y, Pan J, Fang K, Wang Y, Li Z, et al. Vitamin d-binding protein (group-specific component) has decreased expression in rheumatoid arthritis. Clin Exp Rheumatol (2012) 30:525–33.
147. Yin J, Liu H, Yi X, Wu W, Amos CI, Fang S, et al. Genetic variants in the vitamin d pathway genes VDBP and RXRA modulate cutaneous melanoma disease-specific survival. Pigment Cell Melanoma Res (2016) 29:176–85. doi: 10.1111/pcmr.12437
148. Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, et al. Vitamin d-directed rheostatic regulation of monocyte antibacterial responses. J Immunol (2009) 182:4289–95. doi: 10.4049/jimmunol.0803736
149. Subramanian K, Bergman P, Henriques-Normark B. Vitamin d promotes pneumococcal killing and modulates inflammatory responses in primary human neutrophils. J Innate Immun (2017) 9:375–86. doi: 10.1159/000455969
150. Tang J, Zhou R, Luger D, Zhu W, Silver PB, Grajewski RS, et al. Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J Immunol (2009) 182:4624–32. doi: 10.4049/jimmunol.0801543
151. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A. 1alpha,25-dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol (2001) 167:4974–80. doi: 10.4049/jimmunol.167.9.4974
152. Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes (2002) 51:1367–74. doi: 10.2337/diabetes.51.5.1367
153. Chauss D, Freiwald T, McGregor R, Yan B, Wang L, Nova-Lamperti E, et al. Autocrine vitamin d signaling switches off pro-inflammatory programs of T(H)1 cells. Nat Immunol (2022) 23:62–74. doi: 10.1038/s41590-021-01080-3
154. Lemire JM, Adams JS, Sakai R, Jordan SC. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J Clin Invest (1984) 74:657–61. doi: 10.1172/JCI111465
155. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human b cell differentiation. J Immunol (2007) 179:1634–47. doi: 10.4049/jimmunol.179.3.1634
156. Ljung L, Rantapaa-Dahlqvist S. Abdominal obesity, gender and the risk of rheumatoid arthritis - a nested case-control study. Arthritis Res Ther (2016) 18:277. doi: 10.1186/s13075-016-1171-2
157. Hahn J, Malspeis S, Choi MY, Stevens E, Karlson EW, Lu B, et al. Association of healthy lifestyle behaviors and the risk of developing rheumatoid arthritis among women. Arthritis Care Res (Hoboken) (2022). doi: 10.1002/acr.24862
158. Lu B, Hiraki LT, Sparks JA, Malspeis S, Chen CY, Awosogba JA, et al. Being overweight or obese and risk of developing rheumatoid arthritis among women: a prospective cohort study. Ann Rheum Dis (2014) 73:1914–22. doi: 10.1136/annrheumdis-2014-205459
159. Qin B, Yang M, Fu H, Ma N, Wei T, Tang Q, et al. Body mass index and the risk of rheumatoid arthritis: a systematic review and dose-response meta-analysis. Arthritis Res Ther (2015) 17:86. doi: 10.1186/s13075-015-0601-x
160. Wesley A, Bengtsson C, Elkan AC, Klareskog L, Alfredsson L, Wedren S, et al. Association between body mass index and anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis: results from a population-based case-control study. Arthritis Care Res (Hoboken) (2013) 65:107–12. doi: 10.1002/acr.21749
161. Turesson C, Bergstrom U, Pikwer M, Nilsson JA, Jacobsson LT. A high body mass index is associated with reduced risk of rheumatoid arthritis in men, but not in women. Rheumatol (Oxford) (2016) 55:307–14. doi: 10.1093/rheumatology/kev313
162. Linauskas A, Overvad K, Symmons D, Johansen MB, Stengaard-Pedersen K, de Thurah A. Body fat percentage, waist circumference, and obesity as risk factors for rheumatoid arthritis: A Danish cohort study. Arthritis Care Res (Hoboken) (2019) 71:777–86. doi: 10.1002/acr.23694
163. Liu X, Tedeschi SK, Lu B, Zaccardelli A, Speyer CB, Costenbader KH, et al. Long-term physical activity and subsequent risk for rheumatoid arthritis among women: A prospective cohort study. Arthritis Rheumatol (2019) 71:1460–71. doi: 10.1002/art.40899
164. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi: 10.1172/JCI29881
165. Mantovani A, Sozzani S, Locati M, Schioppa T, Saccani A, Allavena P, et al. Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp (2004) 256:137–45; discussion 46-8, 259-69. doi: 10.1002/0470856734.ch10
166. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett (2008) 267:204–15. doi: 10.1016/j.canlet.2008.03.028
167. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi: 10.1016/j.semcancer.2008.03.004
168. Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol (2019) 10:1587. doi: 10.3389/fimmu.2019.01587
169. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PloS One (2011) 6:e16376. doi: 10.1371/journal.pone.0016376
170. Gonzalez-Rodriguez M, Ruiz-Fernandez C, Cordero-Barreal A, Ait Eldjoudi D, Pino J, Farrag Y, et al. Adipokines as targets in musculoskeletal immune and inflammatory diseases. Drug Discovery Today (2022) 27:103352. doi: 10.1016/j.drudis.2022.103352
171. Rodriguez J, Lafaurie GI, Bautista-Molano W, Chila-Moreno L, Bello-Gualtero JM, Romero-Sanchez C. Adipokines and periodontal markers as risk indicators of early rheumatoid arthritis: a cross-sectional study. Clin Oral Investig (2021) 25:1685–95. doi: 10.1007/s00784-020-03469-0
172. Sun X, Wei J, Tang Y, Wang B, Zhang Y, Shi L, et al. Leptin-induced migration and angiogenesis in rheumatoid arthritis is mediated by reactive oxygen species. FEBS Open Bio (2017) 7:1899–908. doi: 10.1002/2211-5463.12326
173. Gonzalez-Ponce F, Gamez-Nava JI, Perez-Guerrero EE, Saldana-Cruz AM, Vazquez-Villegas ML, Ponce-Guarneros JM, et al. Serum chemerin levels: A potential biomarker of joint inflammation in women with rheumatoid arthritis. PloS One (2021) 16:e0255854. doi: 10.1371/journal.pone.0255854
174. Vazquez-Villegas ML, Gamez-Nava JI, Saldana-Cruz AM, Celis A, Sanchez-Rodriguez EN, Perez-Guerrero EE, et al. Functional disability is related to serum chemerin levels in rheumatoid arthritis. Sci Rep (2021) 11:8360. doi: 10.1038/s41598-021-87235-6
175. Sato H, Muraoka S, Kusunoki N, Masuoka S, Yamada S, et al. Resistin upregulates chemokine production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res Ther (2017) 19:263. doi: 10.1186/s13075-017-1472-0
176. Vuolteenaho K, Tuure L, Nieminen R, Celis A, Sanchez-Rodriguez EN, Perez-Guerrero EE, et al. Pretreatment resistin levels are associated with erosive disease in early rheumatoid arthritis treated with disease-modifying anti-rheumatic drugs and infliximab. Scand J Rheumatol (2022) 51:180–5. doi: 10.1080/03009742.2021.1929456
177. Gulkesen A, Akgol G, Poyraz AK, Aydin S, Denk A, Yildirim T, et al. Lipocalin 2 as a clinical significance in rheumatoid arthritis. Cent Eur J Immunol (2017) 42:269–73. doi: 10.5114/ceji.2017.70969
178. Ogunrinola GA, Oyewale JO, Oshamika OO, Olasehinde GI. The human microbiome and its impacts on health. Int J Microbiol (2020) 2020:8045646. doi: 10.1155/2020/8045646
179. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly norMalized after treatment. Nat Med (2015) 21:895–905. doi: 10.1038/nm.3914
180. Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal prevotella copri correlates with enhanced susceptibility to arthritis. Elife (2013) 2:e01202. doi: 10.7554/eLife.01202
181. Jiang L, Shang M, Yu S, Liu Y, Zhang H, Zhou Y, et al. A high-fiber diet synergizes with prevotella copri and exacerbates rheumatoid arthritis. Cell Mol Immunol (2022) 19:1414–24. doi: 10.1038/s41423-022-00934-6
182. Chriswell ME, Lefferts AR, Clay MR, Hsu AR, Seifert J, Feser ML, et al. Clonal IgA and IgG autoantibodies from individuals at risk for rheumatoid arthritis identify an arthritogenic strain of subdoligranulum. Sci Transl Med (2022) 14:eabn5166. doi: 10.1126/scitranslmed.abn5166
183. Arpaia N, Campbell C, Fan X, Dikiy S, van der 0 J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
184. Horta-Baas G, Romero-Figueroa MDS, Montiel-Jarquin AJ, Pizano-Zarate ML, Garcia-Mena J, Ramirez-Duran N. Intestinal dysbiosis and rheumatoid arthritis: A link between gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res (2017) 2017:4835189. doi: 10.1155/2017/4835189
185. Martinsson K, Durholz K, Schett G, Zaiss MM, Kastbom A. Higher serum levels of short-chain fatty acids are associated with non-progression to arthritis in individuals at increased risk of RA. Ann Rheum Dis (2022) 81:445–7. doi: 10.1136/annrheumdis-2021-221386
186. Walker WA. Initial intestinal colonization in the human infant and immune homeostasis. Ann Nutr Metab (2013) 63(Suppl 2):8–15. doi: 10.1159/000354907
187. Lee N, Kim WU. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med (2017) 49:e340. doi: 10.1038/emm.2017.36
188. Block KE, Zheng Z, Dent AL, Kee BL, Huang H. Gut microbiota regulates K/BxN autoimmune arthritis through follicular helper T but not Th17 cells. J Immunol (2016) 196:1550–7. doi: 10.4049/jimmunol.1501904
189. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32:815–27. doi: 10.1016/j.immuni.2010.06.001
190. Kindstedt E, Johansson L, Palmqvist P, Koskinen Holm C, Kokkonen H, Johansson I, et al. Association between marginal jawbone loss and onset of rheumatoid arthritis and relationship to plasma levels of RANKL. Arthritis Rheumatol (2018) 70:508–15. doi: 10.1002/art.40394
191. Kasser UR, Gleissner C, Dehne F, Michel A, Willershausen-Zonnchen B, Bolten WW. Risk for periodontal disease in patients with longstanding rheumatoid arthritis. Arthritis Rheum (1997) 40:2248–51. doi: 10.1002/art.1780401221
192. Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, et al. Periodontitis and porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol (2014) 66:1090–100. doi: 10.1002/art.38348
193. Mankia K, Cheng Z, Do T, Hunt L, Meade J, Kang J, et al. Prevalence of periodontal disease and periodontopathic bacteria in anti-cyclic citrullinated protein antibody-positive at-risk adults without arthritis. JAMA Netw Open (2019) 2:e195394. doi: 10.1001/jamanetworkopen.2019.5394
194. Cheng Z, Do T, Mankia K, Meade J, Hunt L, Clerehugh V, et al. Dysbiosis in the oral microbiomes of anti-CCP positive individuals at risk of developing rheumatoid arthritis. Ann Rheum Dis (2021) 80:162–8. doi: 10.1136/annrheumdis-2020-216972
195. Fuggle NR, Smith TO, Kaul A, Sofat N. Hand to mouth: A systematic review and meta-analysis of the association between rheumatoid arthritis and periodontitis. Front Immunol (2016) 7:80. doi: 10.3389/fimmu.2016.00080
196. Marotte H, Farge P, Gaudin P, Alexandre C, Mougin B, Miossec P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann Rheum Dis (2006) 65:905–9. doi: 10.1136/ard.2005.036913
197. Kharlamova N, Jiang X, Sherina N, Potempa B, Israelsson L, Quirke AM, et al. Antibodies to porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol (2016) 68:604–13. doi: 10.1002/art.39491
198. Moura MF, Cota LOM, Silva TA, Cortelli SC, Ferreira GA, Lopez MM, et al. Clinical and microbiological effects of non-surgical periodontal treatment in individuals with rheumatoid arthritis: a controlled clinical trial. Odontology (2021) 109:484–93. doi: 10.1007/s10266-020-00566-0
199. Bialowas K, Radwan-Oczko M, Dus-Ilnicka I, Korman L, Swierkot J. Periodontal disease and influence of periodontal treatment on disease activity in patients with rheumatoid arthritis and spondyloarthritis. Rheumatol Int (2020) 40:455–63. doi: 10.1007/s00296-019-04460-z
200. Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, et al. Peptidylarginine deiminase from porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum (2010) 62:2662–72. doi: 10.1002/art.27552
201. Montgomery AB, Kopec J, Shrestha L, Thezenas ML, Burgess-Brown NA, Fischer R, et al. Crystal structure of porphyromonas gingivalis peptidylarginine deiminase: implications for autoimmunity in rheumatoid arthritis. Ann Rheum Dis (2016) 75:1255–61. doi: 10.1136/annrheumdis-2015-207656
202. Cheng Z, Meade J, Mankia K, Emery P, Devine DA. Periodontal disease and periodontal bacteria as triggers for rheumatoid arthritis. Best Pract Res Clin Rheumatol (2017) 31:19–30. doi: 10.1016/j.berh.2017.08.001
203. Gully N, Bright R, Marino V, Marchant C, Cantley M, Haynes D, et al. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PloS One (2014) 9:e100838. doi: 10.1371/journal.pone.0100838
204. Mikuls TR, Payne JB, Reinhardt RA, Thiele GM, Maziarz E, Cannella AC, et al. Antibody responses to porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol (2009) 9:38–42. doi: 10.1016/j.intimp.2008.09.008
205. Johansson L, Sherina N, Kharlamova N, Potempa B, Larsson B, Israelsson L, et al. Concentration of antibodies against porphyromonas gingivalis is increased before the onset of symptoms of rheumatoid arthritis. Arthritis Res Ther (2016) 18:201. doi: 10.1186/s13075-016-1100-4
206. Sherina N, de Vries C, Kharlamova N, Sippl N, Jiang X, Brynedal B, et al. Antibodies to a citrullinated porphyromonas gingivalis epitope are increased in early rheumatoid arthritis, and can be produced by gingival tissue b cells: implications for a bacterial origin in RA etiology. Front Immunol (2022) 13:804822. doi: 10.3389/fimmu.2022.804822
207. Cantley MD, Haynes DR, Marino V, Bartold PM. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J Clin Periodontol (2011) 38:532–41. doi: 10.1111/j.1600-051X.2011.01714.x
208. Marchesan JT, Gerow EA, Schaff R, Taut AD, Shin SY, Sugai J, et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Res Ther (2013) 15:R186. doi: 10.1186/ar4376
209. Campbell L, Millhouse E, Malcolm J, Culshaw S. T Cells, teeth and tissue destruction - what do T cells do in periodontal disease? Mol Oral Microbiol (2016) 31:445–56. doi: 10.1111/omi.12144
210. Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol (2000) 2014:64:57–80.
211. White PC, Chicca IJ, Cooper PR, Milward MR, Chapple IL. Neutrophil extracellular traps in periodontitis: A web of intrigue. J Dent Res (2016) 95:26–34. doi: 10.1177/0022034515609097
212. Kroese JM, Brandt BW, Buijs MJ, Crielaard W, Lobbezoo F, Loos BG, et al. Differences in the oral microbiome in patients with early rheumatoid arthritis and individuals at risk of rheumatoid arthritis compared to healthy individuals. Arthritis Rheumatol (2021) 73:1986–93. doi: 10.1002/art.41780
213. Esberg A, Johansson L, Johansson I, Dahlqvist SR. Oral microbiota identifies patients in early onset rheumatoid arthritis. Microorganisms (2021) 9. doi: 10.3390/microorganisms9081657
214. Brewer RC, Lanz TV, Hale CR, Crielaard W, Lobbezoo F, Loos BG, et al. Oral mucosal breaks trigger anti-citrullinated bacterial and human protein antibody responses in rheumatoid arthritis. Sci Transl Med (2023) 15:eabq8476. doi: 10.1126/scitranslmed.abq8476
215. Duncan MS, Freiberg MS, Greevy RA Jr., Kundu S, Vasan RS, Tindle HA. Association of smoking cessation with subsequent risk of cardiovascular disease. JAMA (2019) 322:642–50. doi: 10.1001/jama.2019.10298
216. Dobson Amato KA, Hyland A, Reed R, Mahoney MC, Marshall J, Giovino G, et al. Tobacco cessation may improve lung cancer patient survival. J Thorac Oncol (2015) 10:1014–9. doi: 10.1097/JTO.0000000000000578
217. Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, et al. Vitamin d supplements and prevention of cancer and cardiovascular disease. N Engl J Med (2019) 380:33–44. doi: 10.1056/NEJMoa1809944
218. Gerlag DM, Safy M, Maijer KI, Tang MW, Tas SW, Starmans-Kool MJF, et al. Effects of b-cell directed therapy on the preclinical stage of rheumatoid arthritis: the PRAIRI study. Ann Rheum Dis (2019) 78:179–85. doi: 10.1136/annrheumdis-2017-212763
219. Cope A, Jasenecova M, Vasconcelos J, Filer A, Raza K, Qureshi S, et al. OP0130 abatacept in individuals at risk of developing rheumatoid arthritis: results from the arthritis prevention in the pre-clinical phase of ra with abatacept (apippra) trial. Ann Rheumatic Dis (2023) 82:86–. doi: 10.1136/annrheumdis-2023-eular.1751
220. Rech J, Kleyer A, Østergaard M, Hagen M, Valor L, Tascilar K, et al. POS0531 abatacept delays the development of ra– clinical results after 18 months from the randomized, placebo-controlled ariaa study in ra-at risk patients. Ann Rheumatic Dis (2022) 81:526–7. doi: 10.1136/annrheumdis-2022-eular.1693
221. Krijbolder DI, Verstappen M, van Dijk BT, Dakkak YJ, Burgers LE, Boer AC, et al. Intervention with methotrexate in patients with arthralgia at risk of rheumatoid arthritis to reduce the development of persistent arthritis and its disease burden (TREAT EARLIER): a randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet (2022) 400:283–94. doi: 10.1016/S0140-6736(22)01193-X
222. O'Neil LJ, Deane KD. Striking a balance in rheumatoid arthritis prevention trials. Nat Rev Rheumatol (2021) 17:385–6. doi: 10.1038/s41584-021-00627-w
223. van Boheemen L, Ter Wee MM, Seppen B, van Schaardenburg D. How to enhance recruitment of individuals at risk of rheumatoid arthritis into trials aimed at prevention: understanding the barriers and facilitators. RMD Open (2021) 7. doi: 10.1136/rmdopen-2021-001592
224. van Boheemen L, Bolt JW, Ter Wee MM, de Jong HM, van de Sande MG, van Schaardenburg D. Patients' and rheumatologists' perceptions on preventive intervention in rheumatoid arthritis and axial spondyloarthritis. Arthritis Res Ther (2020) 22:217. doi: 10.1186/s13075-020-02314-9
225. Simons G, Stack RJ, Stoffer-Marx M, Englbrecht M, Mosor E, Buckley CD, et al. Perceptions of first-degree relatives of patients with rheumatoid arthritis about lifestyle modifications and pharmacological interventions to reduce the risk of rheumatoid arthritis development: a qualitative interview study. BMC Rheumatol (2018) 2:31. doi: 10.1186/s41927-018-0038-3
226. Novotny F, Haeny S, Hudelson P, Escher M, Finckh A. Primary prevention of rheumatoid arthritis: a qualitative study in a high-risk population. Joint Bone Spine (2013) 80:673–4. doi: 10.1016/j.jbspin.2013.05.005
227. Gwinnutt JM, Wieczorek M, Balanescu A, Bischoff-Ferrari HA, Boonen A, Cavalli G, et al. EULAR recommendations regarding lifestyle behaviours and work participation to prevent progression of rheumatic and musculoskeletal diseases. Ann Rheum Dis (2021 2023) 82:48–56. doi: 10.1136/annrheumdis-2021-222020
228. Kay J, Upchurch KS. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatol (Oxford) (2012) 51(Suppl 6):vi5–9. doi: 10.1093/rheumatology/kes279
229. Yang DH, Huang JY, Chiou JY, Wei JC. Analysis of socioeconomic status in the patients with rheumatoid arthritis. Int J Environ Res Public Health (2018) 15. doi: 10.3390/ijerph15061194
230. Reed E, Hedstrom AK, Hansson M, Mathsson-Alm L, Brynedal B, Saevarsdottir S, et al. Presence of autoantibodies in "seronegative" rheumatoid arthritis associates with classical risk factors and high disease activity. Arthritis Res Ther (2020) 22:170. doi: 10.1186/s13075-020-02191-2
Keywords: rheumatoid arthritis, prevention, environment, risk factors, smoking, diet, mucosal
Citation: Maisha JA, El-Gabalawy HS and O’Neil LJ (2023) Modifiable risk factors linked to the development of rheumatoid arthritis: evidence, immunological mechanisms and prevention. Front. Immunol. 14:1221125. doi: 10.3389/fimmu.2023.1221125
Received: 11 May 2023; Accepted: 24 August 2023;
Published: 12 September 2023.
Edited by:
Alf Kastbom, Linköping University, SwedenReviewed by:
Solbritt Rantapää-Dahlqvist, Umeå University, SwedenAnna Svärd, Linköping University, Sweden
Karin Lundberg, Karolinska Institutet (KI), Sweden
Copyright © 2023 Maisha, El-Gabalawy and O’Neil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liam J. O’Neil, TGlhbS5vbmVpbEB1bWFuaXRvYmEuY2E=