 Jianshu Zhu
Jianshu Zhu Jiawei Fan
Jiawei Fan Yuanliang Xia
Yuanliang Xia Hengyi Wang
Hengyi Wang Yuehong Li1
Yuehong Li1 Changfeng Fu
Changfeng Fu- 1Department of Spine Surgery, The First Hospital of Jilin University, Changchun, China
- 2Department of Gastroenterology, The First Hospital of Jilin University, Changchun, China
Macrophages are a heterogeneous cell type with high plasticity, exhibiting unique activation characteristics that modulate the progression and resolution of diseases, serving as a key mediator in maintaining tissue homeostasis. Macrophages display a variety of activation states in response to stimuli in the local environment, with their subpopulations and biological functions being dependent on the local microenvironment. Resident tissue macrophages exhibit distinct transcriptional profiles and functions, all of which are essential for maintaining internal homeostasis. Dysfunctional macrophage subpopulations, or an imbalance in the M1/M2 subpopulation ratio, contribute to the pathogenesis of diseases. In skeletal muscle disorders, immune and inflammatory damage, as well as fibrosis induced by macrophages, are prominent pathological features. Therefore, targeting macrophages is of great significance for maintaining tissue homeostasis and treating skeletal muscle disorders. In this review, we discuss the receptor-ligand interactions regulating macrophages and identify potential targets for inhibiting collateral damage and fibrosis in skeletal muscle disorders. Furthermore, we explore strategies for modulating macrophages to maintain tissue homeostasis.
1 Introduction
The hallmark features of musculoskeletal disorders include persistent pain, tissue damage, and limited mobility (1, 2). Therefore, suppressing chronic inflammation and immune responses that cause collateral tissue damage in skeletal muscle disorders, as well as fibrosis resulting from the progression to the terminal stages of the disease, are important therapeutic targets for treating musculoskeletal disorders (3–5).
Macrophages comprise an incredibly diverse and heterogeneous group of cells (6). As their microenvironment constantly changes, macrophages are subject to various regulatory mechanisms that modulate their functional states to new set points in response to tissue alterations or environmental challenges (7, 8). Macrophages of different subpopulations and functional states play crucial roles in the pathogenesis and recovery of various musculoskeletal disorders (9). However, current treatments for musculoskeletal disorders do not always effectively restore the function of affected tissues, and existing therapeutic approaches lack specificity, necessitating the development of personalized, targeted treatment methods (10). The emergence of personalized treatment strategies in cancer therapy, such as immune checkpoint therapy (e.g., PD-1, PD-L1, and CTLA-4 inhibitors) and adoptive T cell therapy (e.g., CAR-T cells), provides inspiration for personalized treatment of musculoskeletal disorders (11, 12). The remarkable clinical success of immune checkpoint therapy and adoptive T cell therapy, as well as the improved understanding of immune cell biological functions, has greatly spurred interest in the field of targeted immunotherapy for musculoskeletal disorders (13–15).
Macrophages are central pathophysiological links in many disease states, such as the chronic inflammation and immune responses caused by their persistent activation, leading to the continuous progression of conditions like Osteoarthritis (OA), Rheumatoid arthritis (RA), and Systemic lupus erythematosus(SLE) (16, 17). Moreover, some studies have reported that macrophages in Systemic Sclerosis (SSc) can transform into myofibroblasts, playing an important role in the terminal stages of musculoskeletal disorders (18). In this review, we discuss recently discovered targets and mechanisms involving macrophage receptor-ligand interactions that activate or inhibit collateral tissue damage and fibrosis, as well as some ongoing clinical studies targeting macrophages for the treatment of musculoskeletal disorders. We aim to identify potential therapeutic targets for suppressing macrophage-induced collateral tissue damage and fibrosis.
2 Roles of macrophage subpopulations in immune-inflammatory injury and pathological fibrosis
Macrophages play a crucial role in the initiation and resolution phases of inflammation, immune response, and pathological fibrosis in musculoskeletal system diseases (Figure 1) (19, 20). Monocyte-derived macrophages can differentiate into various macrophage phenotypes upon recruitment to tissues (21, 22). Changes in macrophage subpopulations and functions during inflammation and immune response are continuous, but corresponding surface markers are lacking (23). Therefore, in diseases of the musculoskeletal system, it is common to classify the course of the disease into different stages, namely, the initial stage of the acute phase, the inflammatory phase, the progressive phase of the subacute period, and the degenerative stage of the chronic period (24, 25). At different stages of the disease, musculoskeletal diseases exhibit distinct clinical and pathophysiological features (26). For instance, during the acute phase, which is often accompanied by severe inflammatory responses, pain, and functional impairment, symptoms usually manifest as pain, redness, increased heat, and restricted function. At this stage, macrophages promote inflammatory responses by secreting inflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). However, the progressive, subacute, and degenerative stages often accompany the ongoing progression of the disease and regressive changes, mainly manifesting as persistent chronic inflammation and tissue fibrosis (27, 28). Researchers typically classify macrophages into M1 pro-inflammatory and M2 anti-inflammatory macrophages based on their phenotypic and functional characteristics.
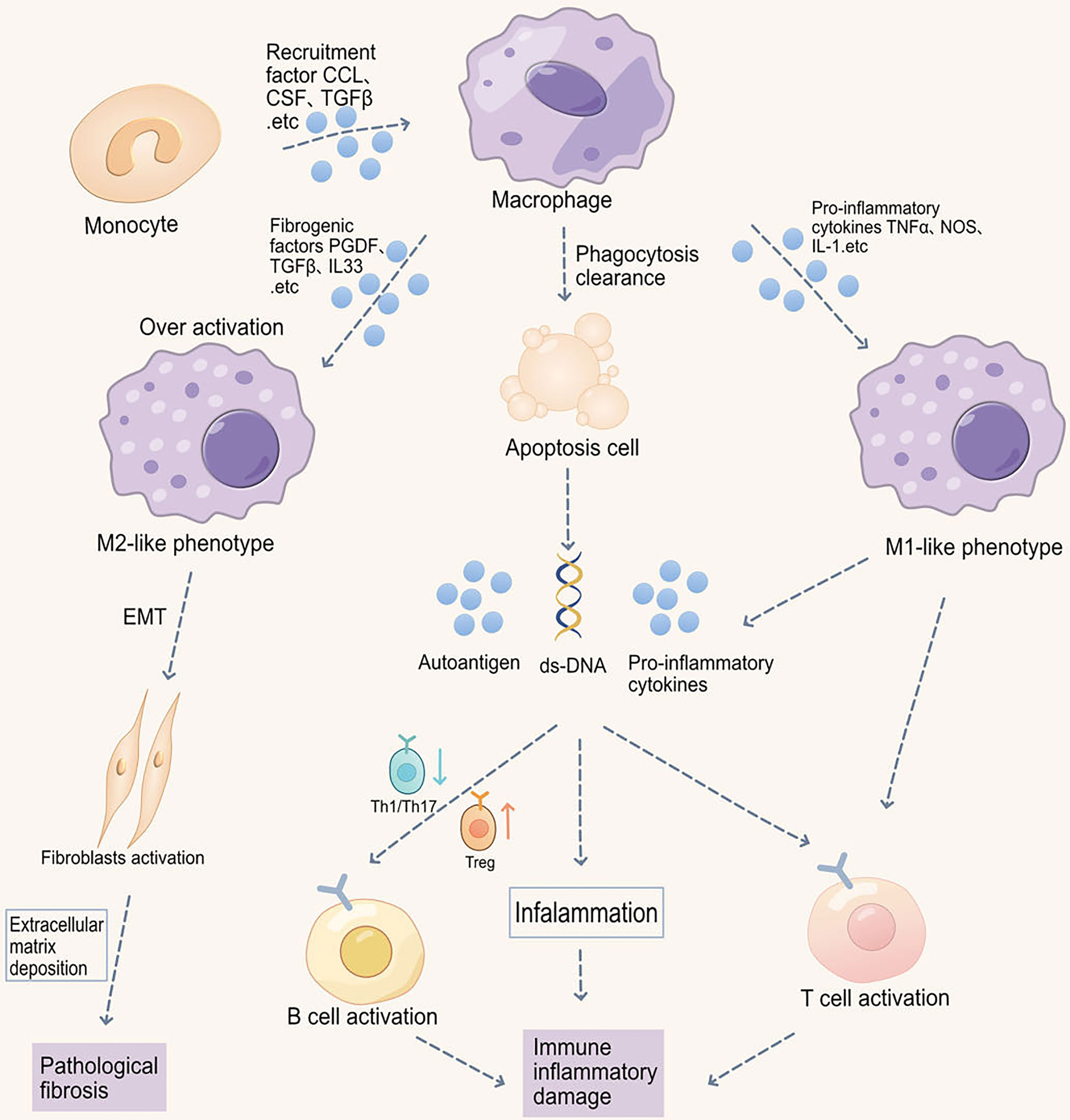
Figure 1 Macrophage-induced immune-inflammatory injury and pathological fibrosis in musculoskeletal disorders. Macrophage phagocytic function is impaired in musculoskeletal disorders, thereby inhibiting the clearance of apoptotic cells. Increased apoptotic cells promote the production of autoantigens and antibodies, exacerbating inflammation. Moreover, macrophages promote the migration and abnormal activation of T cells, including increased Th1/Th17 differentiation and downregulated Treg differentiation, ultimately leading to B cell abnormal activation. Imbalance in M1/M2 macrophage ratio also participates in autoimmunity. Abnormal M1 macrophage activation promotes the production of pro-inflammatory cytokines, such as IL-6, iNOS, TNF-α, and IL-1β, thereby promoting inflammation in target organs. Reduced M2 polarization impairs the production of anti-inflammatory cytokines and immune tolerance. Additionally, M2 macrophage receptor-ligand interactions can also cause epithelial-to-mesenchymal transition (EMT) and fibrosis in autoimmune diseases (e.g., SSc).
2.1 Abnormal function of M1 macrophages and their impact on immune inflammatory injury
Abnormal phagocytosis of M1 macrophages in autoimmune diseases may lead to imbalanced inflammation and immune responses (29). In various autoimmune diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and multiple sclerosis (MS), activation of M1 macrophages and sustained pro-inflammatory responses may exacerbate disease progression (30).
2.1.1 Abnormal phagocytic function of M1 macrophages and immune inflammatory injury
Efficient phagocytosis by macrophages limits the release of intracellular PAMPs driving inflammation, thereby maintaining immune homeostasis (31, 32). In musculoskeletal diseases such as RA and SLE. The expression of inhibitory receptors, such as TIM-3, PD-1, CD32b, and CD200R, can suppress the activation and phagocytic function of macrophages by binding to corresponding ligands (33, 34). As a result, the phagocytic capacity of macrophages against pathogens and cell debris is inhibited [28;29]. Additionally, cytokines like IL-4, IL-10, and TGF-β, along with metabolic substances such as lipopolysaccharide (LPS) and high-density lipoprotein (HDL), can induce macrophage polarization towards M2 phenotype, and suppress macrophage activation and phagocytic function by binding to specific receptors (35–37). Furthermore, macrophage phagocytic capacity for pathogens and cellular debris is inhibited (38, 39). Impaired macrophage phagocytosis promotes the accumulation of uncleared apoptotic or necroptotic cells in autoimmune diseases (40). Increased apoptotic cells promote the production of autoantigens and antibodies, further exacerbating inflammation (41). M1 macrophages produce large amounts of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), which intensify inflammation and cause tissue damage, playing a key role in chronic inflammation in RA, where their release of inflammatory cytokines leads to joint damage and disease worsening (42).
2.1.2 Abnormal aggregation of M1 macrophages and disruption of immune tolerance
Abnormal accumulation of M1 macrophages may lead to the breakdown of immune tolerance, exacerbating the body’s attack on its own tissues (43, 44). The aberrant accumulation of M1 macrophages can regulate immune responses and tissue damage by affecting the activation and function of T cells (45, 46). In certain autoimmune musculoskeletal diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and multiple sclerosis (MS), the activation and accumulation of M1 macrophages lead to excessive Th1 and Th17 responses, thereby exacerbating tissue inflammation and damage (47–49). M1 macrophages produce large amounts of pro-inflammatory cytokines (such as TNF-α, IL-1β, and IL-6), causing massive release of PAMPs, thereby intensifying inflammation and tissue damage (50, 51). M1 macrophages can stimulate cell apoptosis, increase vascular permeability, and recruit more immune cells by producing inflammatory chemokines (such as CCL-2, CCL-3, and CXCL-10), creating a vicious cycle that further exacerbates the course of autoimmune diseases (29, 52, 53). Therefore, interventions targeting the recruitment and abnormal accumulation of M1 macrophages may have potential value in the treatment of musculoskeletal diseases such as RA, SLE, and MS (17, 54). Furthermore, in some fibrosis-related musculoskeletal diseases such as scleroderma (SSc), the abnormal polarization of M2 macrophages leads to an overactive TGF response, thereby promoting pathological tissue fibrosis (55).
2.2 Abnormal immunoregulatory function of macrophages and their impact on immune tolerance
Macrophages, vital components of the immune system, can activate T cells and regulate their function (56). However, an abnormal M1/M2 macrophage ratio may disrupt the immune balance, leading to overactivation or suppression of the immune system and potentially causing disruption of Immune Tolerance (57, 58).
2.2.1 Abnormal antigen-presenting function of macrophages and disruption of immune tolerance
Macrophages are vital antigen-presenting cells (APCs) that can activate T cells by expressing major histocompatibility complex (MHC) molecules and presenting antigen fragments to T cell receptors (TCRs) (56). Macrophages can also regulate T cell polarization and function by expressing co-stimulatory molecules like CD80/CD86 and CD40, and by secreting cytokines such as IL-12 and IL-23 (56, 59). Macrophages can differentiate into different subtypes based on various stimulating factors and microenvironment conditions. M1 macrophages exhibit pro-inflammatory and immune-activating functions, promoting Th1 and Th17 cell differentiation and activation through the production of pro-inflammatory cytokines like IL-12 and IL-23 (60, 61). Conversely, M2 macrophages demonstrate anti-inflammatory and immunoregulatory functions, augmenting the function of regulatory B cells and regulatory T cells (Trg), and inhibiting Th1 and Th17 cell proliferation and differentiation through the production of anti-inflammatory cytokines like IL-10 and TGF-β (62). Moreover, studies have shown that the small protein RELMα, secreted by M2 macrophages, plays a crucial role and mechanism in IL-4-induced inflammatory responses. A deficiency in RELMα leads to a significant reduction in the number of FoxP3+ regulatory T cells. Macrophages expressing RELMα can directly promote the proliferation of regulatory T cells, thus limiting type 2 inflammatory responses (63).
2.2.2 Abnormal immunoregulatory function of macrophages and disruption of immune tolerance
Abnormal innate immune response is an important cause of the collapse of autoimmune tolerance and macrophages are crucial components of the innate immune system (64). M2 macrophages possess strong anti-inflammatory and immune tolerance properties (6, 44). M2a, M2b, and M2c macrophages are anti-inflammatory intermediate macrophage subpopulations with immunoregulatory functions, mainly regulating inflammation and immune responses and participating in tissue repair and regeneration through the production of factors such as TGF-β, IL-6, and IL-10 (8, 53). M2a macrophages can enhance immune tolerance by secreting anti-inflammatory cytokines such as IL-10 and TGF-β, which induce T cells to polarize toward Th2 and inhibit T cell immune responses (65, 66). M2b macrophages are capable of secreting high levels of IL-10 and TGF-β and low levels of IL-12, which strongly regulate immunity and have anti-inflammatory effects. They inhibit the activation and differentiation of T cells such as Th1 and Th17 and NK cells, thereby reducing the risk of diseases such as autoimmune myositis (65, 67). Moreover, IgG4 can induce the transformation of M2a macrophages to an M2b-like phenotype by cross-linking the FcγRIIb receptor, thereby enhancing their ability to inhibit T cells (68). M2c macrophages can also induce immune tolerance by expressing inhibitory ligands such as PD-L1 to inhibit the activation and proliferation of T cells (68).Therefore, abnormal M1/M2 macrophage ratios may disrupt immune balance, leading to excessive activation or suppression of the immune system and, consequently, musculoskeletal diseases (17, 54). Macrophage migration and abnormal activation are related to T cell activation (53), including M1 macrophages producing pro-inflammatory cytokines like IL-12 and IL-23, which promote Th1 and Th17 cell differentiation (6). Conversely, M2 macrophages produce anti-inflammatory cytokines such as IL-10 and TGF-β, significantly enhancing the regulatory function of regulatory B cells, increasing Treg cell generation, and limiting T cell proliferation and differentiation into Th1 and Th17 cells (57, 58).
2.3 Abnormal function of M2 macrophages and their impact on tissue fibrosis
M2 macrophages are a macrophage subpopulation with tissue repair functions (44, 69, 70). If the factors causing tissue injury are not resolved, tissue inflammation induces macrophage polarization towards M2 type through IL-4 and IL-13-triggered signaling pathways such as STAT6 (8).
2.3.1 Abnormal function of different M2 macrophage subsets and their impact on tissue fibrosis
M2a macrophages have the ability to inhibit inflammatory responses, promote tissue repair, and fibrosis. However, if persistently overactivated, they could lead to excessive tissue reconstruction and scar formation, resulting in pathological fibrosis.M2a macrophages express factors like TGF-β1, PDGF, and matrix metalloproteinases (71). These factors can promote the activation of myofibroblasts and the deposition of extracellular matrix, leading to fibrosis (71). In another study, it was found that M2a macrophages release exosomes containing factors such as TGF-β1, PDGF, and matrix metalloproteinases during pathological fibrosis (72). These factors can regulate the activation of myofibroblasts and deposition of extracellular matrix components, resulting in tissue fibrosis, promoting smooth muscle cell migration and adhesion, and causing vascular remodeling and pathological fibrosis (72–74). Moreover, research has found that by inhibiting histone deacetylase (HDAC) with Trichostatin A (TSA), the expression of pro-inflammatory and pro-fibrotic molecules in M2a macrophages can be reduced. This process also inhibits the activation of myofibroblasts, alleviating pathological fibrosis (75). It has been observed that M2b macrophages can mitigate tissue fibrosis by significantly inhibiting the proliferation, migration, and differentiation into myofibroblasts (MFs) of cardiac fibroblasts (CFs) through the suppression of the mitogen-activated protein kinase (MAPK) signaling pathway. Furthermore, they reduce the expression of fibrosis-related proteins such as collagen protein I (COL-1) and α-smooth muscle actin (α-SMA). This suggests that M2b macrophages may be utilized in protective treatments against pathological fibrosis (76, 77). M2c macrophages are generally considered to be macrophages with anti-inflammatory and tissue repair functions. However, if overactivated after an injury, M2c macrophages could promote excessive scar formation and fibrosis. They can lead to pathological fibrosis by enhancing the epithelial-mesenchymal transition of interstitial cells (78). Furthermore, by significantly reducing the M2c subgroup of M2 type macrophages through targeting M2 macrophages, pulmonary fibrosis can be effectively improved (79). Therefore, different subgroups of M2 type macrophages have dual immunomodulatory functions in musculoskeletal diseases. They can both inhibit autoimmune responses and promote tissue repair, but may also cause excessive fibrosis, atrophy, and disruption of immune tolerance.
3 Potential targets of macrophage receptor-ligand interactions
Macrophages are highly plastic and heterogeneous cells that utilize various surface receptors and secreted molecules to monitor and respond to environmental changes (51). Studies on receptor-ligand interactions between macrophages and other components of the immune microenvironment have identified key interactions that regulate macrophage function and abundance, maintaining tissue homeostasis and suppressing autoimmune inflammation and fibrosis (29, 80). Therefore, we have outlined a series of macrophage receptor-ligand interactions with therapeutic potential as potential treatment targets (Figure 2), along with their signaling pathways, biological benefits, and preclinical/clinical trials (Table 1).
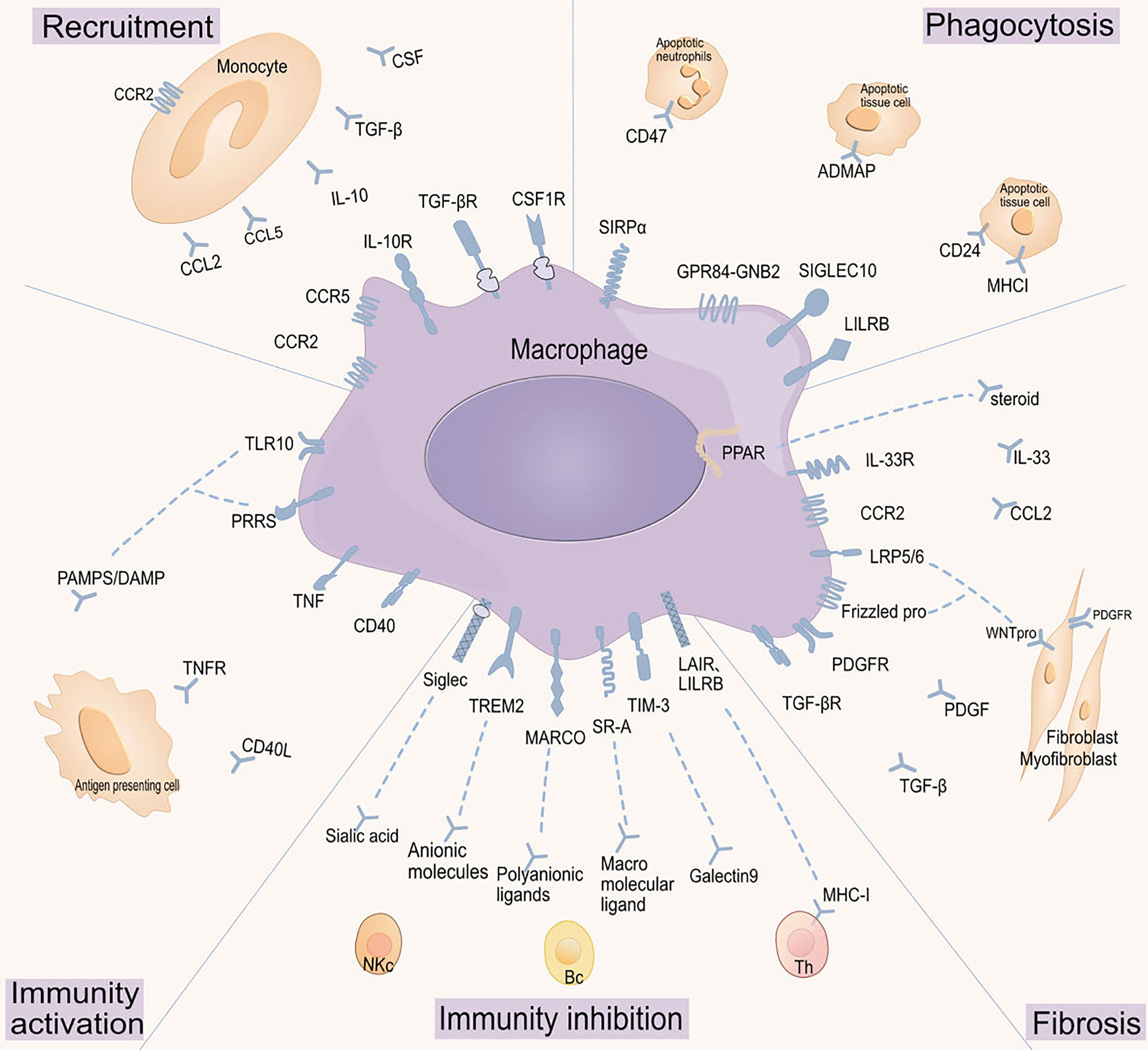
Figure 2 Targeting macrophage receptor-ligand interactions. Macrophage functions are regulated by various receptor-ligand interactions, which have been grouped according to their roles in macrophages: regulating macrophage cell recruitment, modulating phagocytic activity, activating macrophage immune functions, inhibiting macrophage immune functions, and regulating macrophage fibrotic activity. CCR2: C-C chemokine receptor type 2;CCL: Chemokine (C-C motif) ligand;IL-10R: Interleukin-10 receptor;IL-10: Interleukin-10;TGF-βR: Transforming growth factor-beta receptor;TGF-β: Transforming growth factor-beta;CSF1R: Colony-stimulating factor 1 receptor;CSF: Colony-stimulating factor;SIRP α: Signal regulatory protein α;CD47: Cluster of differentiation 47;GPR84-GNB2: G protein-coupled receptor 84 - G protein subunit beta 2;ADMAP: Adhesion and degranulation promoting adapter protein;SIGLEC10: Sialic acid-binding immunoglobulin-like lectin 10;CD24: Cluster of differentiation 24;LILRB: Leukocyte immunoglobulin-like receptor, subfamily B;MHCI: Major histocompatibility complex class I;PPAR: Peroxisome proliferator-activated receptor;IL-33R: Interleukin-33 receptor;IL-33: Interleukin-33;LRP5/6: Low-density lipoprotein receptor-related protein 5/6;WNTpro: Wnt protein;Frizzled pro: Frizzled protein;PDGFR: Platelet-derived growth factor receptor;PDGF: Platelet-derived growth factor;Sialec: Sialic acid-binding immunoglobulin-type lectin;TREM2: Triggering receptor expressed on myeloid cells 2;MARCO: Macrophage receptor with collagenous structure;SR-A: Scavenger receptor class A;TIM-3: T cell immunoglobulin and mucin-domain containing-3;LAIR: Leukocyte-associated immunoglobulin-like receptor;LILRB: Leukocyte immunoglobulin-like receptor, subfamily B;TNF: Tumor necrosis factor;PRRS: Porcine reproductive and respiratory syndrome;TLR10: Toll-like receptor 10;PAMPS/DAMP: Pathogen-associated molecular patterns/Damage-associated molecular patterns.
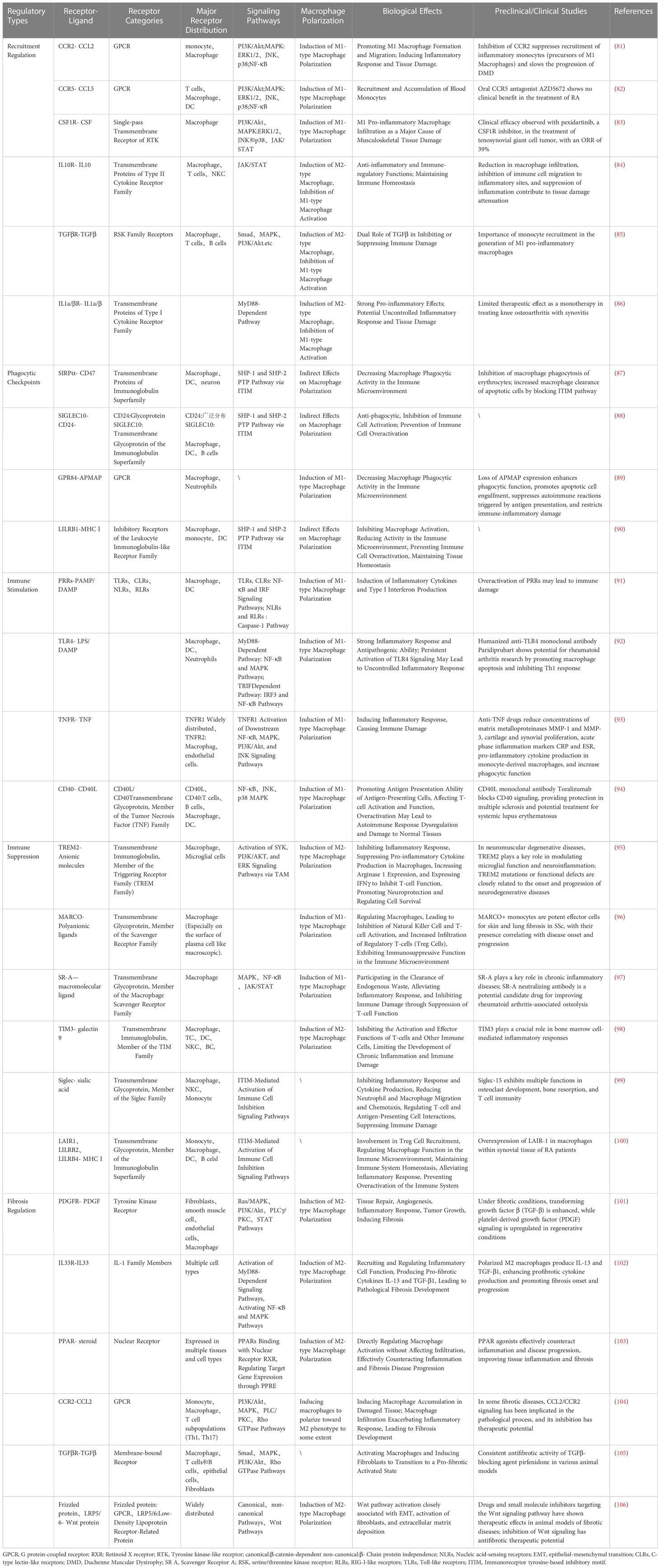
Table 1 Macrophage receptor-ligand interaction signaling pathways, biological benefits, and preclinical/clinical trials.
3.1 Recruitment and aggregation: regulation of macrophage cell abundance
Recruitment and accumulation of macrophages are related to the prognosis and treatment effects of musculoskeletal disorders. The accumulation of macrophages in blood vessels and interstitial tissue is a significant feature of acute and chronic inflammatory musculoskeletal disorders (9). Macrophages that accumulate in damaged musculoskeletal tissues are primarily derived from bone marrow monocytes (107), and their local proliferation is a characteristic of inflammatory damage (108).
Various chemokines (such as CCL-2, CCL-5, CXCL-9, CXCL-10) and cytokines (such as IL-4, IL-10, IL-13, IL-1, TGF-β) play roles in the recruitment and polarization of macrophages (44, 109–111). Chemokine axes like CCL-2-CXCR-2, CCL-5-CCR-5, and CSF-CSF-1R, and cytokine interactions like IL-10-IL-10R, TGF-β-TGF-βR play key roles in the recruitment and function of suppressive macrophages (110, 112–114). These studies demonstrate the importance of monocyte recruitment for the generation of M1 pro-inflammatory macrophages (31–33) and establish that infiltration of M1 pro-inflammatory macrophages is a major cause of tissue damage in musculoskeletal disorders (115–117) and establish that infiltration of M1 pro-inflammatory macrophages is a major cause of tissue damage in musculoskeletal disorders (34–36). In preclinical studies, targeting these pathways has led to a significant reduction in the recruitment and accumulation of blood monocytes (85, 118, 119). In preclinical studies, targeting these pathways has led to a significant reduction in the recruitment and accumulation of blood monocytes (120, 121). The reduction of macrophage infiltration, inhibition of immune cell arrival at the site of inflammation, and suppression of inflammatory responses can mitigate tissue damage (51, 81, 84). Although there are clear reasons to target CCR-5, clinical studies on patients with active rheumatoid arthritis (RA) have shown that oral CCR-5 antagonist AZD5672 provides no clinical benefits, suggesting that the use of CCR-5 antagonists alone is unlikely to be a viable treatment strategy for RA (82).
Research shows that M1 macrophages play a key role in the development of Duchenne muscular dystrophy (DMD). Inhibiting CCR-2 suppresses the recruitment of inflammatory monocytes (precursors of M1 macrophages) and slows the progression of DMD (83). In addition, patients with tenosynovial giant cell tumors caused by genetic translocation-induced CSF1 overexpression have shown clinical efficacy when treated with the CSF-1R inhibitor pexidartinib, with an ORR of 39% (86). However, the use of IL-1a/β inhibitors as monotherapy for treating knee osteoarthritis with synovitis has limited therapeutic effects (122, 123). The suboptimal therapeutic effect of macrophage chemokine blockade might be due to the heterogeneity of macrophage populations in the immune microenvironment and the differential effects of these targeting strategies (51, 124).
Furthermore, the activation of the complement cascade can drive the recruitment of monocytes to damaged tissues, resulting in the deposition of complement component C3b and the local release of effective chemotactic molecules C3a and C5a (125). The complement system plays a role in promoting macrophage recruitment in chronic inflammatory demyelinating polyneuropathy (CIDP), generating a pro-inflammatory environment and mediating demyelination (126–128).
3.2 Phagocytic Checkpoint Receptor-Ligand Interactions
Macrophages’ phagocytosis and clearance of apoptotic cells are essential for suppressing autoimmune diseases (129). In musculoskeletal disorders such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), the expression of phagocytosis-related receptors on macrophages (e.g., Fc receptors and complement receptors) may change, thereby affecting their phagocytic capacity (130, 131).
Anti-phagocytic signals in tissue cells, such as CD47 and CD24, interact with signal regulatory protein-alpha (SIRP-α) and sialic acid-binding immunoglobulin-like lectin 10 (SIGLEC10), both of which are highly expressed on monocytes and macrophages, leading to the inhibition of macrophages’ phagocytic function towards apoptotic cells (88, 132). For example, CD47 acts as a self-marker on red blood cells, interacting with SIRPα on macrophages to inhibit the phagocytosis of red blood cells (87).
Although immune checkpoint therapy is primarily used for cancer treatment, its application in autoimmune diseases has been explored in recent years (133–135). Therefore, CD47-SIRPα axis-targeted therapies are currently being investigated in various clinical trials, with blockade of this pathway enhancing macrophages’ ability to phagocytose and clear apoptotic cells (136, 137). Studies using mouse models of autoimmune diseases have found that CD47-SIRPα and CD24-SIGLEC10 polymorphisms affect macrophages’ phagocytosis of apoptotic cells, suggesting that modulating the CD24-SIGLEC10 axis may have potential value in various musculoskeletal disorders (87, 88). Although there are not yet many clinical trial reports on CD24-SIGLEC10-targeted therapies for autoimmune diseases, existing research provides a foundation for further exploration in this field (88, 138). In the future, more studies and clinical trials may focus on the application of CD47-SIRPα and CD24-SIGLEC10 axes in autoimmune diseases.
Despite the promise of CD47 and SIRPα as clinical targets, the widespread expression of CD47 has led to different off-target effects and responses, posing challenges for the development of clinical treatments targeting these molecules (139, 140). Recent research has identified additional phagocytic checkpoints that alter macrophages’ phagocytic function. One such checkpoint is the G-protein coupled receptor GPR84 and its signaling partner GNB2, which interact with the anti-phagocytic factor APMAP expressed on tissue cells, resulting in enhanced phagocytosis in APMAP-deficient cells (89, 141, 142). When APMAP is knocked out, the level of CD47 on the cell surface decreases, making it easier for macrophages to recognize and engulf them (89, 143). Adipocyte plasma membrane-associated protein (APMAP) is ubiquitously expressed in all cell lines and various types of musculoskeletal disorders (89), APMAP deficiency synergizes with CD47-blocking monoclonal antibodies to enhance phagocytic function, promotes the engulfment of apoptotic cells, suppress antigen presentation-induced autoimmune responses, limit immune-inflammatory damage, and contribute to tissue homeostasis maintenance (144–146). Another phagocytic checkpoint is the LILRB1 on the surface of macrophages, which is an inhibitory immunoglobulin-like receptor. It can bind to MHC class I molecules expressed on tissue cells, reducing macrophages’ phagocytic activity in the immune microenvironment (90, 147, 148). Thus, these newly discovered receptor-ligand interactions may become important therapeutic targets in the future.
3.3 Immunoregulation
Similar to the modulation of T cells through the activation and inhibition of checkpoint receptors, the immunostimulatory and immunosuppressive roles of macrophages are also regulated by various modulatory molecules (30, 149, 150). Macrophages express multiple receptors that interact with various ligands on different cells in the immune microenvironment, which have been shown to reduce the extent and duration of inflammatory responses and, in some cases, contribute to the resolution of fibrosis (16, 84). In this section, we discuss the newly discovered macrophage activation and inhibitory receptors that may play important roles in limiting immune damage in musculoskeletal disorders.
3.3.1 Immune stimulatory receptor-ligand interactions
The activation of M1 macrophages is mainly stimulated by pathogen-associated molecular patterns (PAMPs) and cytokines produced by Th1 cells, such as interferon-γ (IFN-γ). Type I and II interferon responses mediate immune damage responses through intrinsic cellular cytotoxicity and immune activation (70, 151, 152). Pattern recognition receptors (PRRs) are primarily expressed on antigen-presenting cells (APCs), including macrophages and dendritic cells (153, 154). The interaction between PRRs and PAMPs/DAMPs activates macrophages and dendritic cells, leading to the expression of pro-inflammatory cytokines and other immunoregulatory molecules and enhancing their immunostimulatory effects (154–156).
Co-stimulatory molecules of the tumor necrosis factor (TNF) receptor superfamily play an essential role in initiating T cell responses in dendritic cells (DCs) (157–159). Pro-inflammatory cytokines can be induced in macrophages by stimulating Toll-like receptor 7 (TLR7) and TLR9 with CL097 or the interferon gene stimulator (STING) (160, 161). Preclinical studies have shown that activation of PRRs can cause immune damage (91, 162), suggesting that these PRRs are essential targets for immunotherapy (91, 155).TLR agonists are critical targets for immunotherapy because they bridge the gap between the innate and adaptive immune systems (92, 163). Currently, ligands for different members of the TLR family are being studied as potential therapeutic agents, both as monotherapies and in combination with other immunotherapies. Paridiprubart (NI-0101) is a humanized anti-TLR4 monoclonal antibody (93). Paridiprubart has potential in rheumatoid arthritis research by promoting macrophage apoptosis and inhibiting Th1 responses to reduce macrophage accumulation (93). Adalimumab, an anti-TNF biologic antirheumatic drug, has been shown in vitro to block the interaction of TNF with p55 and p75 cell surface TNF receptors, reduce the concentrations of matrix metalloproteinase MMP-1 and MMP-3, reduce cartilage and synovial proliferation, and decrease the concentrations of acute-phase inflammatory reactants (CRP and ESR), reduce the production of pro-inflammatory cytokines by monocyte-derived macrophages and increase phagocytosis (164–166), all of which alleviate the inflammatory response and limit immune damage.
MHCII and co-stimulatory molecules expressed on macrophages (such as CD40, CD80, and CD86) promote T cell activation (92, 167). CD40L-CD40 binding can activate dendritic cells (DCs), and activated DCs promote T cell differentiation and trigger effective CTL responses by enhancing the expression of B7 molecules and the secretion of cytokines such as interleukin-12 (IL-12) (168, 169). Various STING and CD40 agonists are also being tested in clinical trials, either as monotherapies or combination therapies. Bleselumab (ASKP 1240) is a human anti-CD40 monoclonal antibody (mAb) that binds human CD40 with high affinity and inhibits immune responses by blocking the interaction between CD40 and its ligand CD40L (170, 171). Ruplizumab (BG 9588) is a humanized monoclonal anti-CD40L (TNF Receptor) IgG1 antibody with potential for use in systemic lupus erythematosus research (94). Toralizumab (IDEC-131) is a humanized monoclonal antibody (mAb) targeting CD40L (CD154) that specifically binds to human CD40L on T cells, thereby blocking CD40 signaling. Toralizumab, as an immunosuppressive agent, has been shown to be safe and effective in multiple sclerosis research (172), and also has potential for use in active systemic lupus erythematosus (SLE) research. Therefore, it is necessary to better understand the mechanistic basis of their modes of action to optimize their immunotherapeutic efficacy. When developing therapeutic strategies targeting these receptors, factors such as the duration of receptor-ligand interactions, the type of exposed cells, and the nature of the inflammatory environment in the immune microenvironment should be carefully considered.
3.3.2 Immune inhibitory receptor-ligand interactions
Inhibitory receptors on macrophages suppress the activation of pro-inflammatory myeloid cells, skewing their function towards an immunosuppressive phenotype. M2 macrophages primarily create an anti-inflammatory environment in the immune milieu by producing anti-inflammatory cytokines, such as IL-10 and TGF-β, which help maintain tissue homeostasis (173, 174). Their activation is mainly regulated by cytokines produced by Th2 cells, such as IL-4 and IL-13 (51, 175). In this section, we highlight several novel inhibitory receptors with promising therapeutic potential.
PRR-dependent immune injury responses are mainly driven by interferons. However, other studies have shown that interferons can also exert immunosuppressive effects (176–178). Chronic interferon signaling can increase the expression of immune checkpoint ligands such as PDL1 and PDL2 and immunosuppressive molecules, thereby limiting immune injury (179, 180). Scavenger receptors are widely expressed on immune cells, particularly macrophages, and exert immunosuppressive effects through phagocytosis and regulation of inflammatory responses (181). In the immune microenvironment, TREM2-mediated signaling pathways in macrophages suppress the production of pro-inflammatory cytokines, increase the expression of arginase-1, and express IFNγ to inhibit T cell function (182–184). In neuromuscular system neurodegenerative diseases, the TREM2 receptor on macrophages plays a crucial role in regulating microglial function and neuroinflammation. Mutations or functional defects in TREM2 are closely related to the onset and progression of neurodegenerative diseases (95, 185). MARCO interacts with multiple anionic ligands, including nucleic acids, anionic proteins, and lipids, modulating macrophages, inhibiting the activation of natural killer cells and T cells, and increasing the infiltration of regulatory T cells (Tregs), indicating its suppressive function in the immune microenvironment (96, 186). Moreover, research shows that the MARCO+ macrophage subpopulation is associated with driving the onset and progression of diffuse cutaneous systemic sclerosis (SSc), and MARCO+ monocyte-derived macrophages are potent effector cells causing tissue fibrosis (96, 187). SR-A (Scavenger Receptor-A) participates in the clearance of endogenous waste and alleviates inflammatory responses by inhibiting signaling pathways such as NF-κB and MAPK, and suppresses immune injury by inhibiting T cell function. Thus, targeting these scavenger receptors may be a promising approach (97, 188, 189). Blocking SR-A with an anti-SR-A neutralizing antibody may offer a hopeful treatment strategy for bone destruction in RA (97). In addition, some receptors of the immunoglobulin family have been found to promote inhibitory functions. For example, TIM-3 (T cell immunoglobulin and mucin domain-containing protein 3) is mainly expressed on T cells (CD4+ Th1, CD8+ subsets), macrophages, and dendritic cells (190). When TIM-3 binds to its ligand galectin-9, it inhibits the activation and effector functions of T cells and other immune cells, thereby exerting immunosuppressive effects (191, 192). In addition to its inhibitory effects on Th1 cells, recent compelling experiments have emphasized the indispensable role of TIM-3 in bone marrow cell-mediated inflammatory responses (98). The Siglec family (Sialic Acid-Binding Immunoglobulin-like Lectins), such as Siglec-9, upon interaction with ligands, suppresses inflammatory responses and cytokine production, reduces the migration and chemotaxis of neutrophils and macrophages, and regulates the interaction between T cells and dendritic cells, further inhibiting immune responses (193–195). Siglec-15 is an immune receptor that plays multiple roles in osteoclast development, bone resorption, and macrophage-mediated T cell immune responses, serving as a potential target for the treatment of osteoporosis (99, 196). Additionally, LILRB2 and LILRB4, along with the related receptor Leukocyte-Associated Immunoglobulin-like Receptor 1 (LAIR1), are involved in the recruitment of Treg cells and regulation of macrophage function in the immune microenvironment (197, 198). LAIR-1 is highly expressed in CD14(+) mononuclear cells and local CD68(+) macrophages in the synovial tissue of RA patients. Upon TNF-α stimulation, LAIR-4 expression in helper T cells (Th)1 and Th1 CD2(+) T cells from healthy donors is reduced. These results suggest that LAIR-1 exerts distinct functions on T cells and mononuclear cells/macrophages and indicates that LAIR-1 may be a novel therapeutic target for RA (100).
The aforementioned preclinical studies demonstrate that targeting these receptors can reverse the immune damage effects in various musculoskeletal diseases and restore tissue homeostasis, suggesting the potential of these receptors as macrophage-specific targets for monotherapy or in combination with other immunotherapeutic drugs. Consequently, various monoclonal antibodies targeting biomolecules produced by macrophages can be used as therapeutic options for RA. Notably, both inhibitory and activating receptors are widely expressed in various immune and non-immune cell subpopulations in the immune microenvironment.
3.4 Fibrosis regulation
Musculoskeletal diseases are characterized by limited activity due to fibrosis, such as systemic sclerosis, which is a musculoskeletal disease characterized by fibrosis of various tissues (199). Multiple studies have linked the fibrotic features of M2 macrophages to the pathogenesis of this disease. Myofibroblasts are generated from various sources, including the epithelial/endothelial-to-mesenchymal (EMT/EndMT) transition process, as well as circulating fibrocyte-like cells derived from bone marrow stem cells (200). Activated M2 macrophages are particularly abundant in the blood and skin of patients with systemic sclerosis and have been shown to be potential major sources of fibrosis. Tissue fibrosis is an aberrant pathological process involving excessive extracellular matrix (ECM) deposition, resulting in impaired tissue structure and function (201). Macrophages play a crucial role in fibrosis, and macrophage receptors and ligands associated with fibrosis are being investigated as potential targets for anti-fibrotic drugs. Although current treatments for fibrotic diseases such as idiopathic fibrosis, systemic sclerosis, and musculoskeletal disease fibrosis typically target inflammatory responses, increasing evidence suggests that mechanisms driving fibrosis differ from those regulating inflammation (202).
Platelet-derived growth factor (PDGF) is a critical pro-fibrotic factor that binds to the PDGF receptor on the surface of macrophages, activating fibroblast proliferation and migration, thereby increasing ECM synthesis (203). Platelet-derived growth factors (PDGFs) occupy a central role in SSc-related fibrosis and represent potential molecular targets for systemic sclerosis (SSc) (199, 204). Receptor/ligand analysis of macrophage-mesenchymal progenitor cell (MPC) cross-talk reveals that under fibrotic conditions, transforming growth factor-β (TGF-β) is enhanced, while platelet-derived growth factor (PDGF) signaling is enhanced under regenerative conditions (101) providing targets for fibrosis treatment. Interleukin-13 (IL-13) is a Th2 cell factor that can activate the JAK-STAT pathway by acting on the IL-13 receptor on the surface of macrophages, promoting M2 macrophage polarization and leading to the development of pathological fibrosis (205, 206). IL-33 is a novel pro-fibrotic cytokine that signals through ST2, recruiting and directing inflammatory cell function and enhancing pro-fibrotic cytokine production through polarization of M2 macrophages in an ST2- and macrophage-dependent manner, generating IL-13 and TGF-β1, thereby promoting the onset and progression of fibrosis (102).
Peroxisome proliferator-activated receptors (PPARs) are important regulators of metabolism and inflammation (207, 208). PPARs can directly modulate macrophage activation without affecting infiltration, effectively counteracting inflammation and fibrosis progression (209). The pan-PPAR agonist lanifibranor has been shown to effectively improve tissue inflammation and fibrosis (103). C-C motif chemokine ligand 2 (CCL-2) binds to C-C motif chemokine receptor 2 (CCR2), promoting macrophage aggregation at damaged tissues. Macrophage infiltration exacerbates the inflammatory response, further promoting fibrosis development. Studies have shown that chemokine receptors CCR-2 and CX3CR1 regulate skin fibrosis in cytokine-induced systemic sclerosis mouse models, suggesting that blocking C-C motif chemokine ligand (CCL-24) or CCL-2 to inhibit monocyte recruitment may be an attractive new therapy to limit SSc fibrosis manifestations (104).
Transforming growth factor-β (TGF-β) is the most important cytokine in the pro-fibrotic process (210, 211). Macrophages express TGF-β receptors on their surface, which, upon binding to TGF-β, can activate macrophages and induce fibroblast transformation into a pro-fibrotic activated state. TGF-β signaling is considered a key pathway in almost all types of fibrosis (212). CCL-2/CCR-2 interactions also induce fibrosis via the TGF-β pathway (104). Since the approval of pirfenidone, targeting TGF-β signaling has been anticipated as an effective treatment for fibrosis (105, 212). The Wnt ligand Wnt3a enhances IL-4 or TGF-β1-induced M2 macrophage polarization. Wnt/β-catenin signaling works in conjunction with TGF-β signaling during fibrosis; TGF-β signaling can induce the expression of Wnt/β-catenin superfamily members and vice versa (106, 213). Drugs and small molecule inhibitors targeting the Wnt signaling pathway have demonstrated some efficacy in animal models of fibrotic diseases (106). Thus, inhibiting the Wnt signaling pathway may have therapeutic potential for anti-fibrotic treatment. These receptor-ligand interactions affecting fibrosis may become a suitable and promising therapeutic strategy in the future. As for the Wnt/β-catenin signaling pathway, some small molecule inhibitors have been developed, among which MSAB is an effective and selective inhibitor of Wnt/β-catenin signal transduction. MSAB binds with β-catenin, promoting its degradation and specifically downregulating Wnt/β-catenin target genes (214). TGFβ1-IN-1 (Compound 42) is an effective orally active inhibitor of TGF-β1. TGFβ1-IN-1 inhibits the upregulation of fibrosis markers (α-SMA and fibronectin) induced by TGF-β1, making it suitable for fibrotic disease research. In vivo studies have shown that TGFβ1-IN-1 inhibits TGF-β1-induced tissue damage and fibrosis, suppresses the activation of epithelial-mesenchymal transition (EMT), and improves the immune microenvironment of tissues (215). In clinical settings, the effects of these inhibitors vary across different diseases and conditions. Some early studies indicate that inhibitors of the Wnt/β-catenin and TGF-β signaling pathways, such as SSc, have shown some efficacy in certain musculoskeletal diseases (215). However, there have yet to be large-scale, randomized controlled trials to verify the effectiveness and safety of these drugs in a clinical setting.
4 Targeted macrophage therapeutic strategies
Chimeric antigen receptor T (CAR-T) cell therapy has achieved evolutionary success in hematologic malignancies and has expanded its application to solid tumors (216). However, adoptive T-cell therapy (ACT) for autoimmune diseases requires a high degree of specificity to avoid attacking healthy tissue and identifying and targeting pathogenic T-cell subpopulations remains challenging (217). Furthermore, in musculoskeletal diseases, the therapeutic goal is to restore immune system balance rather than simply enhancing or suppressing immune responses.
We have summarized the key approaches for utilizing genetically engineered macrophages in the treatment of musculoskeletal disorders (Figure 3). Genetically engineered macrophages (GEMs) may minimize the impact on the immune microenvironment and improve both innate and adaptive immune responses, making them suitable for treating musculoskeletal diseases such as rheumatoid arthritis and systemic lupus erythematosus (218, 219). Lentiviral expression systems have been validated for generating transduced monocytes and monocyte-derived macrophages, and transgene expression has been demonstrated to be stable over several weeks to months in vitro and in mouse xenograft models of GBM (220). GEMs employ the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system for gene editing to correct gene mutations and improve disease, such as by expressing TGF-βR or IL-10R to modulate immune cell activation (221–223). Modulation of immune responses is also possible by using the CRISPR system to knock out genes that cause aberrant activation of cytotoxic cell functions, including IL-10R and PD-1R (224, 225). These results suggest that GEMs are an ideal approach for manipulating the immune microenvironment and suppressing immune damage. CARs may potentially contribute to the treatment of some common musculoskeletal genetic mutation diseases, such as cystic fibrosis and amyotrophic lateral sclerosis.
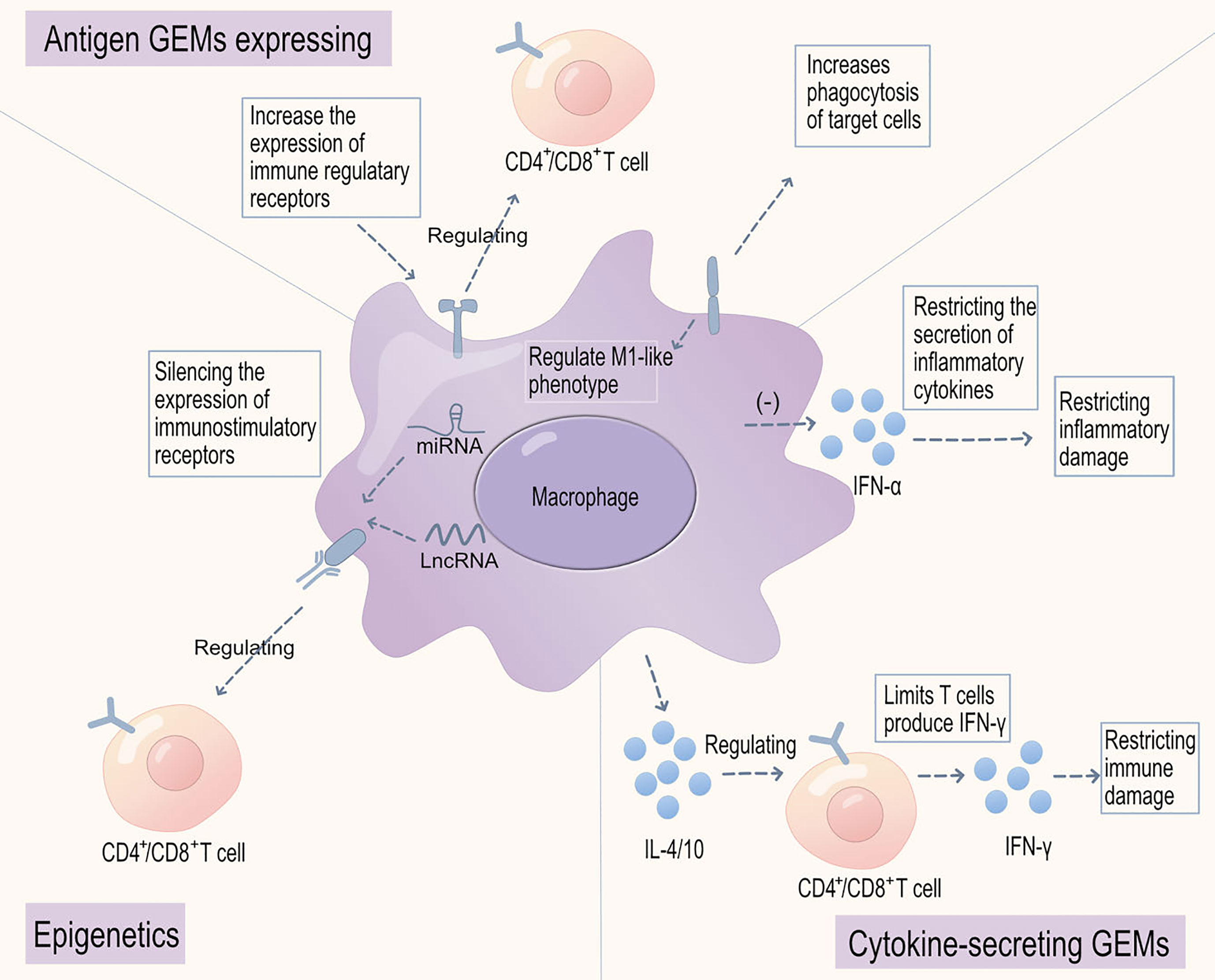
Figure 3 Genetically engineered macrophages for the treatment of musculoskeletal disorders. There are three major categories of genetically engineered macrophages (GEMs): firstly, those with engineered cell surface molecules, including increased expression of membrane regulatory receptors and MOTO-CAR with TGF-βR or IL-10R receptor signaling domains, to modulate immune cell activation, reduce immune-inflammatory injury, or enhance macrophage phagocytic checkpoints for apoptotic cell clearance, and reduce autoantigen presentation and immune-inflammatory injury; secondly, those engineered to secrete inflammatory mediators and cytokines, such as increasing the secretion of immunoregulatory cytokines IL-4/10 to enhance immune cell regulation and limit interferon-α (IFNα) secretion, restricting immune-inflammatory injury; finally, those that can silence the expression of immune-stimulating receptors through epigenetics, alleviating the stimulatory effect on activated immune cells and reducing immune-inflammatory injury.
Furthermore, the use of epigenetic RNA interference (such as miRNA or lncRNA) can specifically reduce the expression of particular receptors (226). This approach is achieved by degrading the target gene’s miRNA, thereby affecting receptor protein synthesis. The exploration and pathway analysis of microRNAs (miRNAs) have paved the way for discovering potential therapeutic targets (226, 227). miRNAs are small non-coding oligonucleotides characterized by their role in gene regulation, transcription, and immune modulation mechanisms (228). Research on lncRNAs has underscored their importance as both immune markers of active disease progression and immune modulators of innate processes such as apoptosis and autophagy (229). Epigenetic silencing of miRNAs remodels macrophages through receptor expression or paracrine secretion of cytokines, such as macrophage migration inhibitory factor (230).
In addition to engineering macrophages to express cell surface receptors, research has also focused on modulating the expression of ligand cytokines that regulate the immune microenvironment and immune cell activation (231, 232). Genetically engineered macrophages secreting anti-inflammatory cytokines, such as interleukin-4 (IL-4) and interleukin-10 (IL-10), effectively reach the primary site, significantly alleviate inflammatory responses, and protect tissue cells from LPS-induced functional impairment, suggesting that these engineered macrophages may have inhibitory effects on inflammatory damage (233, 234). Furthermore, in preclinical models, macrophages carrying engineered particles containing interferon-γ (referred to as “backpacks”) exhibit enhanced phagocytic activity in the immune microenvironment, polarizing macrophages towards a pro-inflammatory phenotype, resulting in sustained phagocytic function enhancement and reduced self-antigen presentation (235, 236).
Concerning these strategies, future research may continue to explore how to apply these methods in clinical settings, such as using macrophage-based therapies in musculoskeletal diseases or other inflammatory diseases. In addition, further research is needed on how to precisely manipulate the polarization and function of macrophages in vivo, such as by developing new drugs or cell therapies. Overall, the field of genetically engineered macrophages has opened an exciting avenue for targeting the immune damage microenvironment in musculoskeletal diseases. However, the heterogeneity and plasticity of macrophage subpopulations need to be carefully analyzed in clinical and preclinical models to optimize specific clinical responses.
5 Conclusion and perspectives
In this review, we have focused on the various functions of macrophage receptor-ligand interactions, which serve as potential targets for immune-related musculoskeletal disease therapies. However, the development of such therapies is challenged by increasing complexity on several levels.
First, all these receptor-ligand interactions are intricately interconnected. On the one hand, macrophage receptors, upon stimulation by ligands such as cytokines or other molecules, activate or inhibit a series of signaling pathways, thereby influencing macrophage polarization and function. On the other hand, the polarization state and function of macrophages can also affect the expression of their surface receptors, which in turn further influences receptor-ligand interactions (237).
For instance, receptor-ligand interactions regulate multiple aspects of macrophage polarization and function, including aggregation, phagocytosis, and macrophage-derived products that, in turn, regulate receptor-ligand interactions (51, 73). A second, largely unexplored area is the determination of specific macrophage phenotypes in particular etiologies or pathological processes by selectively altering macrophage phenotype and function (29, 70). Lastly, the persistence of macrophage-directed therapy effects is a prominent challenge that will impact whether macrophage-targeted therapies can serve as standalone treatments or only in conjunction with other forms of therapeutic targets (238). Crosstalk among these receptors in the immune microenvironment must be considered to enhance efficacy and minimize off-target toxicological effects. Additionally, due to the marked diversity and expression of human and murine myeloid cell subpopulations, the relevance of inhibiting receptors in human myeloid cell subpopulations requires careful evaluation.
Macrophage-targeted therapy has several undeniable advantages in the treatment of musculoskeletal diseases: first, macrophages can suppress excessive immune responses and restore immune system balance in the treatment of musculoskeletal diseases through the release of anti-inflammatory cytokines and promotion of regulatory T-cell proliferation, among other pathways (44, 239). Second, macrophages exhibit high plasticity and can differentiate into subtypes with distinct functions and phenotypes in response to environmental signals and stimuli. By modulating macrophages ex vivo, they can be directed to differentiate into immune-regulatory phenotypes, ultimately improving musculoskeletal disease conditions (8). Lastly, macrophage therapy allows for personalized treatment plans for each patient. Ex vivo modulation and genetic engineering of macrophages can provide patient-specific treatment strategies to address different types of musculoskeletal diseases, enhancing specificity for disease treatment while minimizing the impact on healthy tissues (240, 241).
In conclusion, the field of immune therapy for musculoskeletal diseases has already provided benefits to many patients, and these studies will enable the next wave of musculoskeletal disease immunotherapies targeting macrophage subpopulations to further enhance immune responses and clinical outcomes. The beneficial effects of adoptive polarized macrophage transfer therapy are currently being evaluated, with significant improvements observed in several different animal models. Another promising strategy is the treatment of autoimmune diseases with miRNA-based epigenetic therapies. The high plasticity of macrophages allows them to alter their effector functions, and therefore, they can potentially be manipulated to inhibit chronic inflammatory immune damage and fibrotic processes.
Author contributions
JZ wrote the initial manuscript. CF and JZ contributed new ideas. JF and HW created the figures. JF and ZF created Table 1. YX, YL, ZF, and HW revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82071391), the Science and Technology Development Program of Jilin Province (Grant No. 20200404182YY).
Acknowledgments
We would like to express our appreciation to everyone involved in drafting and preparing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Burmester GR, Bijlsma JWJ, Cutolo M, McInnes IB. Managing rheumatic and musculoskeletal diseases - past, present and future. Nat Rev Rheumatol (2017) 13:443–8. doi: 10.1038/nrrheum.2017.95
2. Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benkő S, Szűcs G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol (2021) 17:585–95. doi: 10.1038/s41584-021-00652-9
3. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum (2012) 64:1697–707. doi: 10.1002/art.34453
4. Woolf AD, Pfleger B. Burden of major musculoskeletal conditions. Bull World Health Organ (2003) 81:646–56.
5. Munir H, McGettrick HM. Mesenchymal stem cell therapy for autoimmune disease: risks and rewards. Stem Cells Dev (2015) 24:2091–100. doi: 10.1089/scd.2015.0008
6. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi: 10.1038/nri2448
7. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14:392–404. doi: 10.1038/nri3671
8. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
9. Tidball JG. Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol (2017) 17:165–78. doi: 10.1038/nri.2016.150
10. Castillero E, Nieto-Bona MP, Fernández-Galaz C, Martín AI, López-Menduiña M, Granado M, et al. Fenofibrate, a PPAR{alpha} agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am J Physiol Endocrinol Metab (2011) 300:E790–9. doi: 10.1152/ajpendo.00590.2010
11. Benveniste O, Stenzel W, Allenbach Y. Advances in serological diagnostics of inflammatory myopathies. Curr Opin Neurol (2016) 29:662–73. doi: 10.1097/WCO.0000000000000376
12. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget (2017) 8:38022–43. doi: 10.18632/oncotarget.16723
13. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
14. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-Small-Cell lung cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
15. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
16. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
17. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
18. Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev (2008) 22:1747–52. doi: 10.1101/gad.465908
19. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol (2016) 16:378–91. doi: 10.1038/nri.2016.49
20. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity (2014) 41:116–26. doi: 10.1016/j.immuni.2014.05.018
21. Luddy KA, Robertson-Tessi M, Tafreshi NK, Soliman H, Morse DL. The role of toll-like receptors in colorectal cancer progression: evidence for epithelial to leucocytic transition. Front Immunol (2014) 5:429. doi: 10.3389/fimmu.2014.00429
22. Kugelberg E. Neutrophils: bugging transplantation. Nat Rev Immunol (2014) 14:430–1. doi: 10.1038/nri3703
23. Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, et al. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity (2014) 40:865–79. doi: 10.1016/j.immuni.2014.03.014
24. Murphey MD, Kransdorf MJ. Staging and classification of primary musculoskeletal bone and soft-tissue tumors according to the 2020 WHO update, from the AJR special series on cancer staging. AJR Am J Roentgenol (2021) 217:1038–52. doi: 10.2214/AJR.21.25658
25. Ofluoglu O, Boriani S, Gasbarrini A, De Iure F, Donthineni R. Diagnosis and planning in the management of musculoskeletal tumors: surgical perspective. Semin Intervent Radiol (2010) 27:185–90. doi: 10.1055/s-0030-1253516
26. Tucker-Bartley A, Lemme J, Gomez-Morad A, Shah N, Veliu M, Birklein F, et al. Pain phenotypes in rare musculoskeletal and neuromuscular diseases. Neurosci Biobehav Rev (2021) 124:267–90. doi: 10.1016/j.neubiorev.2021.02.009
27. Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL, et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res (2017) 5:16044. doi: 10.1038/boneres.2016.44
28. Elma Ö., Yilmaz ST, Deliens T, Clarys P, Nijs J, Coppieters I, et al. Chronic musculoskeletal pain and nutrition: where are we and where are we heading? Pm r (2020) 12:1268–78. doi: 10.1002/pmrj.12346
29. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi: 10.1038/nri3073
30. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
31. Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol (2012) 12:492–502. doi: 10.1038/nri3244
32. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm (2015) 2015:816460. doi: 10.1155/2015/816460
33. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol (2019) 20:1425–34. doi: 10.1038/s41590-019-0512-0
34. Shi X, Li CW, Tan LC, Wen SS, Liao T, Zhang Y, et al. Immune Co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: a Large cohort study. J Clin Endocrinol Metab (2021) 106:120–32. doi: 10.1210/clinem/dgaa701
35. Sun X, Gao J, Meng X, Lu X, Zhang L, Chen R. Polarized macrophages in periodontitis: characteristics, function, and molecular signaling. Front Immunol (2021) 12:763334. doi: 10.3389/fimmu.2021.763334
36. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol (2018) 330:27–42. doi: 10.1016/j.cellimm.2018.01.020
37. Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol (2022) 19:384–408. doi: 10.1038/s41423-021-00791-9
38. Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med (2012) 18:871–82. doi: 10.1038/nm.2752
39. Kennedy A, Fearon U, Veale DJ, Godson C. Macrophages in synovial inflammation. Front Immunol (2011) 2:52. doi: 10.3389/fimmu.2011.00052
40. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi: 10.1038/nrrheum.2010.46
41. Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell (2010) 140:619–30. doi: 10.1016/j.cell.2010.02.014
42. Joosten LA, Abdollahi-Roodsaz S, Dinarello CA, O'Neill L, Netea MG. Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol (2016) 12:344–57. doi: 10.1038/nrrheum.2016.61
43. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
44. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
45. Du T, Yang CL, Ge MR, Liu Y, Zhang P, Li H, et al. M1 macrophage derived exosomes aggravate experimental autoimmune neuritis via modulating Th1 response. Front Immunol (2020) 11:1603. doi: 10.3389/fimmu.2020.01603
46. Paul S, Chhatar S, Mishra A, Lal G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J Immunother Cancer (2019) 7:208. doi: 10.1186/s40425-019-0697-7
47. Kim SJ, Chang HJ, Volin MV, Umar S, Van Raemdonck K, Chevalier A, et al. Macrophages are the primary effector cells in IL-7-induced arthritis. Cell Mol Immunol (2020) 17:728–40. doi: 10.1038/s41423-019-0235-z
48. Mizutani S, Nishio J, Kondo K, Motomura K, Yamada Z, Masuoka S, et al. Treatment with an anti-CX3CL1 antibody suppresses M1 macrophage infiltration in interstitial lung disease in SKG mice. Pharm (Basel) (2021) 14(5):474. doi: 10.3390/ph14050474
49. Ahamada MM, Jia Y, Wu X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front Immunol (2021) 12:734008. doi: 10.3389/fimmu.2021.734008
50. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity (2006) 25:383–92. doi: 10.1016/j.immuni.2006.08.010
51. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
52. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci (2014) 10:520–9. doi: 10.7150/ijbs.8879
53. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
54. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15:545–58. doi: 10.1038/nri3871
55. Hu M, Yao Z, Xu L, Peng M, Deng G, Liu L, et al. M2 macrophage polarization in systemic sclerosis fibrosis: pathogenic mechanisms and therapeutic effects. Heliyon (2023) 9:e16206. doi: 10.1016/j.heliyon.2023.e16206
56. Guerriero JL. Macrophages: their untold story in T cell activation and function. Int Rev Cell Mol Biol (2019) 342:73–93. doi: 10.1016/bs.ircmb.2018.07.001
57. Hu WT, Howell JC, Ozturk T, Gangishetti U, Kollhoff AL, Hatcher-Martin JM, et al. CSF cytokines in aging, multiple sclerosis, and dementia. Front Immunol (2019) 10:480. doi: 10.3389/fimmu.2019.00480
58. Carvajal Alegria G, Cornec D, Saraux A, Devauchelle-Pensec V, Jamin C, Hillion S, et al. Abatacept promotes regulatory b cell functions, enhancing their ability to reduce the Th1 response in rheumatoid arthritis patients through the production of IL-10 and TGF-β. J Immunol (2021) 207:470–82. doi: 10.4049/jimmunol.2000455
59. Dikiy S, Rudensky AY. Principles of regulatory T cell function. Immunity (2023) 56:240–55. doi: 10.1016/j.immuni.2023.01.004
60. Yang X, Zhou F, Yuan P, Dou G, Liu X, Liu S, et al. T Cell-depleting nanoparticles ameliorate bone loss by reducing activated T cells and regulating the Treg/Th17 balance. Bioact Mater (2021) 6:3150–63. doi: 10.1016/j.bioactmat.2021.02.034
61. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity (2018) 49:666–677.e6. doi: 10.1016/j.immuni.2018.07.015
62. Li N, Gao Z, Zhao L, Du B, Ma B, Nian H, et al. MSC-derived small extracellular vesicles attenuate autoimmune dacryoadenitis by promoting M2 macrophage polarization and inducing tregs via miR-100-5p. Front Immunol (2022) 13:888949. doi: 10.3389/fimmu.2022.888949
63. Li J, Kim SY, Lainez NM, Coss D, Nair MG. Macrophage-regulatory T cell interactions promote type 2 immune homeostasis through resistin-like molecule α. Front Immunol (2021) 12:710406. doi: 10.3389/fimmu.2021.710406
64. Schett G, Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun (2018) 9:3261. doi: 10.1038/s41467-018-05800-6
65. Zhang Q, Sioud M. Tumor-associated macrophage subsets: shaping polarization and targeting. Int J Mol Sci (2023) 24(8):7493. doi: 10.3390/ijms24087493
66. Goetz C, Hammerbeck C, Boss K, Peterson R, Bonnevier J. Phenotyping of M1 and M2a macrophages and differential expression of ACE-2 on monocytes by flow cytometry: impact of cell culture conditions and sample processing. Methods Mol Biol (2023) 2593:197–212. doi: 10.1007/978-1-0716-2811-9_12
67. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106:345–58. doi: 10.1002/JLB.3RU1018-378RR
68. Chen S, Saeed A, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther (2023) 8:207. doi: 10.1038/s41392-023-01452-1
69. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
70. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
71. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
72. Niu C, Wang X, Zhao M, Cai T, Liu P, Li J, et al. Macrophage foam cell-derived extracellular vesicles promote vascular smooth muscle cell migration and adhesion. J Am Heart Assoc (2016) 5(10):e004099. doi: 10.1161/JAHA.116.004099
73. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229:176–85. doi: 10.1002/path.4133
74. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
75. Tseng WC, Tsai MT, Chen NJ, Tarng DC. Trichostatin a alleviates renal interstitial fibrosis through modulation of the M2 macrophage subpopulation. Int J Mol Sci (2020) 21:123–30. doi: 10.3390/ijms21175966
76. Yue Y, Huang S, Wang L, Wu Z, Liang M, Li H, et al. M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ J (2020) 84:626–35. doi: 10.1253/circj.CJ-19-0959
77. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
78. Tian L, Yu Q, Liu D, Chen Z, Zhang Y, Lu J, et al. Epithelial-mesenchymal transition of peritoneal mesothelial cells is enhanced by M2c macrophage polarization. Immunol Invest (2022) 51:301–15. doi: 10.1080/08820139.2020.1828911
79. Choi SM, Mo Y, Bang JY, Ko YG, Ahn YH, Kim HY, et al. Classical monocyte-derived macrophages as therapeutic targets of umbilical cord mesenchymal stem cells: comparison of intratracheal and intravenous administration in a mouse model of pulmonary fibrosis. Respir Res (2023) 24:68. doi: 10.1186/s12931-023-02357-x
80. Brown JM, Recht L, Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res (2017) 23:3241–50. doi: 10.1158/1078-0432.CCR-16-3122
81. Morioka S, Maueröder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
82. Zoshima T, Baba T, Tanabe Y, Ishida Y, Nakatani K, Nagata M, et al. CCR2- and CCR5-mediated macrophage infiltration contributes to glomerular endocapillary hypercellularity in antibody-induced lupus nephritis. Rheumatol (Oxford) (2022) 61:3033–48. doi: 10.1093/rheumatology/keab825
83. Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, et al. Inflammatory monocytes promote progression of duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med (2014) 6:1476–92. doi: 10.15252/emmm.201403967
84. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
85. Liu F, Qiu H, Xue M, Zhang S, Zhang X, Xu J, et al. MSC-secreted TGF-β regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res Ther (2019) 10:345. doi: 10.1186/s13287-019-1447-y
86. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay JY, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet (2019) 394:478–87. doi: 10.1016/S0140-6736(19)30764-0
87. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (2000) 288:2051–4. doi: 10.1126/science.288.5473.2051
88. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
89. Kamber RA, Nishiga Y, Morton B, Banuelos AM, Barkal AA, Vences-Catalán F, et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature (2021) 597:549–54. doi: 10.1038/s41586-021-03879-4
90. Kang X, Kim J, Deng M, John S, Chen H, Wu G, et al. Inhibitory leukocyte immunoglobulin-like receptors: immune checkpoint proteins and tumor sustaining factors. Cell Cycle (2016) 15:25–40. doi: 10.1080/15384101.2015.1121324
91. Zhang Y, Liang C. Innate recognition of microbial-derived signals in immunity and inflammation. Sci China Life Sci (2016) 59:1210–7. doi: 10.1007/s11427-016-0325-6
92. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov (2020) 19:860–83. doi: 10.1038/s41573-020-0081-9
93. Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf K, Reijers J, et al. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin Pharmacol Ther (2017) 101:200–8. doi: 10.1002/cpt.522
94. Chamberlain C, Colman PJ, Ranger AM, Burkly LC, Johnston GI, Otoul C, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis (2017) 76:1837–44. doi: 10.1136/annrheumdis-2017-211388
95. Silvin A, Uderhardt S, Piot C, Da Mesquita S, Yang K, Geirsdottir L, et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity (2022) 55:1448–1465.e6. doi: 10.1016/j.immuni.2022.07.004
96. Xue D, Tabib T, Morse C, Yang Y, Domsic RT, Khanna D, et al. Expansion of fcγ receptor IIIa-positive macrophages, ficolin 1-positive monocyte-derived dendritic cells, and plasmacytoid dendritic cells associated with severe skin disease in systemic sclerosis. Arthritis Rheumatol (2022) 74:329–41. doi: 10.1002/art.41813
97. Xie Y, Jiang X, Wang P, Zheng X, Song J, Bai M, et al. SR-a neutralizing antibody: potential drug candidate for ameliorating osteoclastogenesis in rheumatoid arthritis. Clin Exp Immunol (2022) 207:297–306. doi: 10.1093/cei/uxac010
98. Lu C, Chen H, Wang C, Yang F, Li J, Liu H, et al. An emerging role of TIM3 expression on T cells in chronic kidney inflammation. Front Immunol (2021) 12:798683. doi: 10.3389/fimmu.2021.798683
99. Kameda Y, Takahata M, Mikuni S, Shimizu T, Hamano H, Angata T, et al. Siglec-15 is a potential therapeutic target for postmenopausal osteoporosis. Bone (2015) 71:217–26. doi: 10.1016/j.bone.2014.10.027
100. Zhang Y, Lv K, Zhang CM, Jin BQ, Zhuang R, Ding Y. The role of LAIR-1 (CD305) in T cells and monocytes/macrophages in patients with rheumatoid arthritis. Cell Immunol (2014) 287:46–52. doi: 10.1016/j.cellimm.2013.12.005
101. Ridderstad A, Abedi-Valugerdi M, Möller E. Cytokines in rheumatoid arthritis. Ann Med (1991) 23:219–23. doi: 10.3109/07853899109148051
102. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol (2014) 134:1422–1432.e11. doi: 10.1016/j.jaci.2014.05.011
103. Boyer-Diaz Z, Aristu-Zabalza P, Andrés-Rozas M, Robert C, Ortega-Ribera M, Fernández-Iglesias A, et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol (2021) 74:1188–99. doi: 10.1016/j.jhep.2020.11.045
104. Arai M, Ikawa Y, Chujo S, Hamaguchi Y, Ishida W, Shirasaki F, et al. Chemokine receptors CCR2 and CX3CR1 regulate skin fibrosis in the mouse model of cytokine-induced systemic sclerosis. J Dermatol Sci (2013) 69:250–8. doi: 10.1016/j.jdermsci.2012.10.010
105. Birnhuber A, Jandl K, Biasin V, Fließer E, Valzano F, Marsh LM, et al. Pirfenidone exacerbates Th2-driven vasculopathy in a mouse model of systemic sclerosis-associated interstitial lung disease. Eur Respir J (2022) 60(4). doi: 10.1183/13993003.02347-2021
106. Lv Q, Wang J, Xu C, Huang X, Ruan Z, Dai Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways. Mol Med (2020) 26:49. doi: 10.1186/s10020-020-00173-3
107. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med (2007) 204:1057–69. doi: 10.1084/jem.20070075
108. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (2011) 332:1284–8. doi: 10.1126/science.1204351
109. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
110. Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F, et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis (2017) 76:914–22. doi: 10.1136/annrheumdis-2016-210426
111. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer (2020) 19:41. doi: 10.1186/s12943-020-01165-x
112. Li M, Sun X, Zhao J, Xia L, Li J, Xu M, et al. CCL5 deficiency promotes liver repair by improving inflammation resolution and liver regeneration through M2 macrophage polarization. Cell Mol Immunol (2020) 17:753–64. doi: 10.1038/s41423-019-0279-0
113. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ (2020) 27:1765–81. doi: 10.1038/s41418-019-0460-0
114. Qiu Y, Cao Y, Tu G, Li J, Su Y, Fang F, et al. Myeloid-derived suppressor cells alleviate renal fibrosis progression via regulation of CCL5-CCR5 axis. Front Immunol (2021) 12:698894. doi: 10.3389/fimmu.2021.698894
115. Chávez-Galán L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol (2015) 6:263. doi: 10.3389/fimmu.2015.00263
116. Li H, Jiang T, Li MQ, Zheng XL, Zhao GJ. Transcriptional regulation of macrophages polarization by MicroRNAs. Front Immunol (2018) 9:1175. doi: 10.3389/fimmu.2018.01175
117. Yao Y, Xu XH, Jin L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol (2019) 10:792. doi: 10.3389/fimmu.2019.00792
118. Nakkala JR, Duan Y, Ding J, Muhammad W, Zhang D, Mao Z, et al. Macrophage membrane-functionalized nanofibrous mats and their immunomodulatory effects on macrophage polarization. Acta Biomater (2022) 141:24–38. doi: 10.1016/j.actbio.2021.12.026
119. Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol (2010) 298:R1173–87. doi: 10.1152/ajpregu.00735.2009
120. Dou A, Fang J. Heterogeneous myeloid cells in tumors. Cancers (Basel) (2021) 13(15):3772. doi: 10.3390/cancers13153772
121. Kumari N, Choi SH. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin Cancer Res (2022) 41:68. doi: 10.1186/s13046-022-02272-x
122. Fleischmann RM, Bliddal H, Blanco FJ, Schnitzer TJ, Peterfy C, Chen S, et al. A phase II trial of lutikizumab, an anti-Interleukin-1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol (2019) 71:1056–69. doi: 10.1002/art.40840
123. Kloppenburg M, Peterfy C, Haugen IK, Kroon F, Chen S, Wang L, et al. Placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1α and anti-interleukin-1β dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann Rheum Dis (2019) 78:413–20. doi: 10.1136/annrheumdis-2018-213336
124. Wang WM, Zhao ZL, Ma SR, Yu GT, Liu B, Zhang L, et al. Epidermal growth factor receptor inhibition reduces angiogenesis via hypoxia-inducible factor-1α and Notch1 in head neck squamous cell carcinoma. PloS One (2015) 10:e0119723. doi: 10.1371/journal.pone.0119723
125. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi: 10.1038/ni.1923
126. Querol LA, Hartung HP, Lewis RA, van Doorn PA, Hammond TR, Atassi N, et al. The role of the complement system in chronic inflammatory demyelinating polyneuropathy: implications for complement-targeted therapies. Neurotherapeutics (2022) 19:864–73. doi: 10.1007/s13311-022-01221-y
127. Sommer C, Koch S, Lammens M, Gabreels-Festen A, Stoll G, Toyka KV. Macrophage clustering as a diagnostic marker in sural nerve biopsies of patients with CIDP. Neurology (2005) 65:1924–9. doi: 10.1212/01.wnl.0000188879.19900.b7
128. Vallat JM, Mathis S, Vegezzi E, Richard L, Duchesne M, Gallouedec G, et al. Antibody- and macrophage-mediated segmental demyelination in chronic inflammatory demyelinating polyneuropathy: clinical, electrophysiological, immunological and pathological correlates. Eur J Neurol (2020) 27:692–701. doi: 10.1111/ene.14133
129. Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity (2011) 35:445–55. doi: 10.1016/j.immuni.2011.09.004
130. Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, et al. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ (2001) 8:327–34. doi: 10.1038/sj.cdd.4400826
131. Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, et al. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest (2009) 119:20–32. doi: 10.1172/JCI36226
132. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol (2018) 19:76–84. doi: 10.1038/s41590-017-0004-z
133. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci USA (2016) 113:E2383–92. doi: 10.1073/pnas.1603892113
134. Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res (2013) 1:320–31. doi: 10.1158/2326-6066.CIR-13-0086
135. Weiss R, Bitton A, Ben Shimon M, Elhaik Goldman S, Nahary L, Cooper I, et al. Annexin A2, autoimmunity, anxiety and depression. J Autoimmun (2016) 73:92–9. doi: 10.1016/j.jaut.2016.06.011
136. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536:86–90. doi: 10.1038/nature18935
137. Yurdagul A Jr., Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med (2017) 4:86. doi: 10.3389/fcvm.2017.00086
138. Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer (2017) 76:100–9. doi: 10.1016/j.ejca.2017.02.013
139. Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol Rev (2017) 276:145–64. doi: 10.1111/imr.12527
140. Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U.S.A. (2016) 113:E2646–54. doi: 10.1073/pnas.1604268113
141. Gaidarov I, Anthony T, Gatlin J, Chen X, Mills D, Solomon M, et al. Embelin and its derivatives unravel the signaling, proinflammatory and antiatherogenic properties of GPR84 receptor. Pharmacol Res (2018) 131:185–98. doi: 10.1016/j.phrs.2018.02.021
142. Recio C, Lucy D, Purvis GSD, Iveson P, Zeboudj L, Iqbal AJ, et al. Activation of the immune-metabolic receptor GPR84 enhances inflammation and phagocytosis in macrophages. Front Immunol (2018) 9:1419. doi: 10.3389/fimmu.2018.01419
143. Wang M, Zhang X, Zhang S, Liu Z. Zebrafish fatty acids receptor Gpr84 enhances macrophage phagocytosis. Fish Shellfish Immunol (2019) 84:1098–9. doi: 10.1016/j.fsi.2018.11.023
144. Sheng M, Lin Y, Xu D, Tian Y, Zhan Y, Li C, et al. CD47-mediated Hedgehog/SMO/GLI1 signaling promotes mesenchymal stem cell immunomodulation in mouse liver inflammation. Hepatology (2021) 74:1560–77. doi: 10.1002/hep.31831
145. Theruvath J, Menard M, Smith BAH, Linde MH, Coles GL, Dalton GN, et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat Med (2022) 28:333–44. doi: 10.1038/s41591-021-01625-x
146. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater (2020) 32:e2002054. doi: 10.1002/adma.202002054
147. Belkin D, Torkar M, Chang C, Barten R, Tolaini M, Haude A, et al. Killer cell ig-like receptor and leukocyte ig-like receptor transgenic mice exhibit tissue- and cell-specific transgene expression. J Immunol (2003) 171:3056–63. doi: 10.4049/jimmunol.171.6.3056
148. Shi B, Chu J, Huang T, Wang X, Li Q, Gao Q, et al. The scavenger receptor MARCO expressed by tumor-associated macrophages are highly associated with poor pancreatic cancer prognosis. Front Oncol (2021) 11:771488. doi: 10.3389/fonc.2021.771488
149. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol (2022) 19:37–50. doi: 10.1038/s41571-021-00552-7
150. Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci (2015) 72:4111–26. doi: 10.1007/s00018-015-1995-y
151. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol (2022) 19:402–21. doi: 10.1038/s41571-022-00620-6
152. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin d in pyroptosis. Acta Pharm Sin B (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
153. Andersen MN, Al-Karradi SN, Kragstrup TW, Hokland M. Elimination of erroneous results in flow cytometry caused by antibody binding to fc receptors on human monocytes and macrophages. Cytometry A (2016) 89:1001–9. doi: 10.1002/cyto.a.22995
154. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
155. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
156. Celhar T, Magalhães R, Fairhurst AM. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res (2012) 53:58–77. doi: 10.1007/s12026-012-8270-1
157. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol Rev (2019) 99:115–60. doi: 10.1152/physrev.00045.2017
158. Tafalla C, Granja AG. Novel insights on the regulation of b cell functionality by members of the tumor necrosis factor superfamily in jawed fish. Front Immunol (2018) 9:1285. doi: 10.3389/fimmu.2018.01285
159. Sonar S, Lal G. Role of tumor necrosis factor superfamily in neuroinflammation and autoimmunity. Front Immunol (2015) 6:364. doi: 10.3389/fimmu.2015.00364
160. Petricevic B, Wessner B, Sachet M, Vrbanec D, Spittler A, Bergmann M. CL097, a TLR7/8 ligand, inhibits TLR-4–dependent activation of IRAK-m and BCL-3 expression. Shock (2009) 32:484–90. doi: 10.1097/SHK.0b013e3181a5ac8a
161. Pietrobon AJ, Yoshikawa FSY, Oliveira LM, Pereira NZ, Matozo T, de Alencar BC, et al. Antiviral response induced by toll-like receptor (TLR) 7/TLR8 activation inhibits human immunodeficiency virus type 1 infection in cord blood macrophages. J Infect Dis (2022) 225:510–9. doi: 10.1093/infdis/jiab389
162. Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol (2014) 25:1387–400. doi: 10.1681/ASN.2014010117
163. Stuart RW, Racke MK. Targeting T cell costimulation in autoimmune disease. Expert Opin Ther Targets (2002) 6:275–89. doi: 10.1517/14728222.6.3.275
164. Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol (2013) 304:G970–9. doi: 10.1152/ajpgi.00183.2012
165. Hatterer E, Shang L, Simonet P, Herren S, Daubeuf B, Teixeira S, et al. A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res Ther (2016) 18:224. doi: 10.1186/s13075-016-1128-5
166. Monnet E, Choy EH, McInnes I, Kobakhidze T, de Graaf K, Jacqmin P, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis (2020) 79:316–23. doi: 10.1136/annrheumdis-2019-216487
167. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and treg homeostasis in autoimmunity and tumor immunity. J Autoimmun (2018) 95:77–99. doi: 10.1016/j.jaut.2018.08.007
168. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity (2018) 49:819–828.e6. doi: 10.1016/j.immuni.2018.09.008
169. de Silva S, Fromm G, Shuptrine CW, Johannes K, Patel A, Yoo KJ, et al. CD40 enhances type I interferon responses downstream of CD47 blockade, bridging innate and adaptive immunity. Cancer Immunol Res (2020) 8:230–45. doi: 10.1158/2326-6066.CIR-19-0493
170. Harland RC, Klintmalm G, Jensik S, Yang H, Bromberg J, Holman J, et al. Efficacy and safety of bleselumab in kidney transplant recipients: a phase 2, randomized, open-label, noninferiority study. Am J Transplant (2020) 20:159–71. doi: 10.1111/ajt.15591
171. Okimura K, Maeta K, Kobayashi N, Goto M, Kano N, Ishihara T, et al. Characterization of ASKP1240, a fully human antibody targeting human CD40 with potent immunosuppressive effects. Am J Transplant (2014) 14:1290–9. doi: 10.1111/ajt.12678
172. Fadul CE, Mao-Draayer Y, Ryan KA, Noelle RJ, Wishart HA, Channon JY, et al. Safety and immune effects of blocking CD40 ligand in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2021) 8(6). doi: 10.1212/NXI.0000000000001096
173. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
174. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol (2022) 13:867260. doi: 10.3389/fimmu.2022.867260
175. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
176. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
177. Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res (2011) 17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482
178. Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity (2012) 36:166–74. doi: 10.1016/j.immuni.2012.01.011
179. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
180. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
181. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjärvi S, Oliynyk G, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci USA (2020) 117:32005–16. doi: 10.1073/pnas.2015343117
182. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
183. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest (2021) 131(4). doi: 10.1172/JCI135197
184. Chen S, Peng J, Sherchan P, Ma Y, Xiang S, Yan F, et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J Neuroinflamm (2020) 17:168. doi: 10.1186/s12974-020-01853-x
185. Hwang M, Savarin C, Kim J, Powers J, Towne N, Oh H, et al. Trem2 deficiency impairs recovery and phagocytosis and dysregulates myeloid gene expression during virus-induced demyelination. J Neuroinflamm (2022) 19:267. doi: 10.1186/s12974-022-02629-1
186. La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. MARCO and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res (2021) 81:956–67. doi: 10.1158/0008-5472.CAN-20-1885
187. Xu D, Bhattacharyya S, Wang W, Ifergan I, Wong MAC, Procissi D, et al. PLG nanoparticles target fibroblasts and MARCO+ monocytes to reverse multiorgan fibrosis. JCI Insight (2022) 7(5). doi: 10.1172/jci.insight.151037
188. Mäkinen PI, Lappalainen JP, Heinonen SE, Leppänen P, Lähteenvuo MT, Aarnio JV, et al. Silencing of either SR-a or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res (2010) 88:530–8. doi: 10.1093/cvr/cvq235
189. Yu H, Ha T, Liu L, Wang X, Gao M, Kelley J, et al. Scavenger receptor a (SR-a) is required for LPS-induced TLR4 mediated NF-κB activation in macrophages. Biochim Biophys Acta (2012) 1823:1192–8. doi: 10.1016/j.bbamcr.2012.05.004
190. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol (2011) 32:345–9. doi: 10.1016/j.it.2011.05.003
191. Li ZH, Wang LL, Liu H, Muyayalo KP, Huang XB, Mor G, et al. Galectin-9 alleviates LPS-induced preeclampsia-like impairment in rats via switching decidual macrophage polarization to M2 subtype. Front Immunol (2018) 9:3142. doi: 10.3389/fimmu.2018.03142
192. Tang L, Li G, Zheng Y, Hou C, Gao Y, Hao Y, et al. Tim-3 relieves experimental autoimmune encephalomyelitis by suppressing MHC-II. Front Immunol (2021) 12:770402. doi: 10.3389/fimmu.2021.770402
193. Matsumoto T, Takahashi N, Kojima T, Yoshioka Y, Ishikawa J, Furukawa K, et al. Soluble siglec-9 suppresses arthritis in a collagen-induced arthritis mouse model and inhibits M1 activation of RAW264.7 macrophages. Arthritis Res Ther (2016) 18:133. doi: 10.1186/s13075-016-1035-9
194. von Gunten S, Simon HU. Autophagic-like cell death in neutrophils induced by autoantibodies. Autophagy (2007) 3:67–8. doi: 10.4161/auto.3436
195. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin siglec-9. Nat Immunol (2016) 17:1273–81. doi: 10.1038/ni.3552
196. Kang FB, Chen W, Wang L, Zhang YZ. The diverse functions of siglec-15 in bone remodeling and antitumor responses. Pharmacol Res (2020) 155:104728. doi: 10.1016/j.phrs.2020.104728
197. Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest (2018) 128:5647–62. doi: 10.1172/JCI97570
198. Jiang Z, Qin JJ, Zhang Y, Cheng WL, Ji YX, Gong FH, et al. LILRB4 deficiency aggravates the development of atherosclerosis and plaque instability by increasing the macrophage inflammatory response via NF-κB signaling. Clin Sci (Lond) (2017) 131:2275–88. doi: 10.1042/CS20170198
199. Paolini C, Agarbati S, Benfaremo D, Mozzicafreddo M, Svegliati S, Moroncini G. PDGF/PDGFR: a possible molecular target in scleroderma fibrosis. Int J Mol Sci (2022) 23(7):3904. doi: 10.3390/ijms23073904
200. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol (2008) 214:199–210. doi: 10.1002/path.2277
201. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol (2019) 15:705–30. doi: 10.1038/s41584-019-0322-7
202. Antar SA, Ashour NA, Marawan ME, Al-Karmalawy AA. Fibrosis: types, effects, markers, mechanisms for disease progression, and its relation with oxidative stress, immunity, and inflammation. Int J Mol Sci (2023) 24(4):4004. doi: 10.3390/ijms24044004
203. Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev (2004) 15:255–73. doi: 10.1016/j.cytogfr.2004.03.006
204. Iwayama T, Olson LE. Involvement of PDGF in fibrosis and scleroderma: recent insights from animal models and potential therapeutic opportunities. Curr Rheumatol Rep (2013) 15:304. doi: 10.1007/s11926-012-0304-0
205. Gieseck RL, Wilson 3MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18:62–76. doi: 10.1038/nri.2017.90
206. Melo-Cardenas J, Bezavada L, Crawford JC, Gurbuxani S, Cotton A, Kang G, et al. IL-13/IL-4 signaling contributes to fibrotic progression of the myeloproliferative neoplasms. Blood (2022) 140:2805–17. doi: 10.1182/blood.2022017326
207. Toobian D, Ghosh P, Katkar GD. Parsing the role of PPARs in macrophage processes. Front Immunol (2021) 12:783780. doi: 10.3389/fimmu.2021.783780
208. Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res (2005) 51:85–94. doi: 10.1016/j.phrs.2004.07.012
209. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophagesDifferential effects of selective- and pan-PPAR agonists on experimenta (☆). J Hepatol (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
210. Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J Exp Med (2020) 217:e20190103. doi: 10.1084/jem.20190103
211. Györfi AH, Matei AE, Distler JHW. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol (2018) 68-69:8–27. doi: 10.1016/j.matbio.2017.12.016
212. Li X, Ding Z, Wu Z, Xu Y, Yao H, Lin K. Targeting the TGF-β signaling pathway for fibrosis therapy: a patent review (2015-2020). Expert Opin Ther Pat (2021) 31:723–43. doi: 10.1080/13543776.2021.1896705
213. Guo Y, Xiao L, Sun L, Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol Res (2012) 61:337–46. doi: 10.33549/physiolres.932289
214. Hwang SY, Deng X, Byun S, Lee C, Lee SJ, Suh H, et al. Direct targeting of β-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic wnt/β-catenin signaling. Cell Rep (2016) 16:28–36. doi: 10.1016/j.celrep.2016.05.071
215. Yue L, Xue T, Su X, Liu Z, Liu H, Tan Z, et al. Discovery and evaluation of phenacrylanilide derivatives as novel potential anti-liver fibrosis agents. Eur J Med Chem (2022) 242:114685. doi: 10.1016/j.ejmech.2022.114685
216. Zhang L, Ding J, Li HY, Wang ZH, Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys Acta Rev Cancer (2020) 1874:188441. doi: 10.1016/j.bbcan.2020.188441
217. D'Ippolito E, Schober K, Nauerth M, Busch DH. T Cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer Immunol Immunother (2019) 68:1701–12. doi: 10.1007/s00262-019-02395-9
218. Zeballos CM, Gaj T. Next-generation CRISPR technologies and their applications in gene and cell therapy. Trends Biotechnol (2021) 39:692–705. doi: 10.1016/j.tibtech.2020.10.010
219. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-cas technologies and applications. Nat Rev Mol Cell Biol (2019) 20:490–507. doi: 10.1038/s41580-019-0131-5
220. Hong Y, Casimir M, Houghton BC, Zhang F, Jensen B, Omoyinmi E, et al. Lentiviral mediated ADA2 gene transfer corrects the defects associated with deficiency of adenosine deaminase type 2. Front Immunol (2022) 13:852830. doi: 10.3389/fimmu.2022.852830
221. Zhang L, Li Y, Chen Q, Xia Y, Zheng W, Jiang X. The construction of drug-resistant cancer cell lines by CRISPR/Cas9 system for drug screening. Sci Bull (Beijing) (2018) 63:1411–9. doi: 10.1016/j.scib.2018.09.024
222. Ren J, Yu D, Fu R, An P, Sun R, Wang Z, et al. IL2RG-deficient minipigs generated via CRISPR/Cas9 technology support the growth of human melanoma-derived tumours. Cell Prolif (2020) 53:e12863. doi: 10.1111/cpr.12863
223. Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD-1hiCXCR5- T peripheral helper cells promote b cell responses in lupus via MAF and IL-21. JCI Insight (2019) 4(20). doi: 10.1172/jci.insight.130062
224. Shi H, Guo J, Yu Q, Hou X, Liu L, Gao M, et al. CRISPR/Cas9 based blockade of IL-10 signaling impairs lipid and tissue homeostasis to accelerate atherosclerosis. Front Immunol (2022) 13:999470. doi: 10.3389/fimmu.2022.999470
225. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547:413–8. doi: 10.1038/nature23270
226. Yang B, Huang X, Xu S, Li L, Wu W, Dai Y, et al. Decreased miR-4512 levels in monocytes and macrophages of individuals with systemic lupus erythematosus contribute to innate immune activation and neutrsophil NETosis by targeting TLR4 and CXCL2. Front Immunol (2021) 12:756825. doi: 10.3389/fimmu.2021.756825
227. Luo Z, Qi B, Sun Y, Chen Y, Lin J, Qin H, et al. Engineering bioactive M2 macrophage-polarized, anti-inflammatory, miRNA-based liposomes for functional muscle repair: from exosomal mechanisms to biomaterials. Small (2022) 18:e2201957. doi: 10.1002/smll.202201957
228. Ghafouri-Fard S, Abak A, Tavakkoli Avval S, Shoorei H, Taheri M, Samadian M. The impact of non-coding RNAs on macrophage polarization. BioMed Pharmacother (2021) 142:112112. doi: 10.1016/j.biopha.2021.112112
229. Ahmad I, Valverde A, Naqvi RA, Naqvi AR. Long non-coding RNAs RN7SK and GAS5 regulate macrophage polarization and innate immune responses. Front Immunol (2020) 11:604981. doi: 10.3389/fimmu.2020.604981
230. Zhao J, Li H, Zhao S, Wang E, Zhu J, Feng D, et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol Cancer (2021) 20:46. doi: 10.1186/s12943-021-01343-5
231. Cannac M, Nikolic J, Benaroch P. Cancer immunotherapies based on genetically engineered macrophages. Cancer Immunol Res (2022) 10:1156–66. doi: 10.1158/2326-6066.CIR-22-0030
232. Moradinasab S, Pourbagheri-Sigaroodi A, Ghaffari SH, Bashash D. Targeting macrophage-mediated tumor cell phagocytosis: an overview of phagocytosis checkpoints blockade, nanomedicine intervention, and engineered CAR-macrophage therapy. Int Immunopharmacol (2022) 103:108499. doi: 10.1016/j.intimp.2021.108499
233. Liu H, He Y, Lu C, Zhang P, Zhou C, Ni Y, et al. Efficacy of pulmonary transplantation of engineered macrophages secreting IL-4 on acute lung injury in C57BL/6J mice. Cell Death Dis (2019) 10:664. doi: 10.1038/s41419-019-1900-y
234. Lee SW, Park HJ, Jeon J, Park YH, Kim TC, Jeon SH, et al. Chromatin regulator SRG3 overexpression protects against LPS/D-GalN-Induced sepsis by increasing IL10-producing macrophages and decreasing IFNγ-producing NK cells in the liver. Int J Mol Sci (2021) 22(6):3043. doi: 10.3390/IJMS22063043
235. Mucci A, Varesio L, Neglia R, Colombari B, Pastorino S, Blasi E. Antifungal activity of macrophages engineered to produce IFNgamma: inducibility by picolinic acid. Med Microbiol Immunol (2003) 192:71–8. doi: 10.1007/s00430-002-0118-1
236. Wu M, Hussain S, He YH, Pasula R, Smith PA, Martin WJ. 2nd, genetically engineered macrophages expressing IFN-gamma restore alveolar immune function in scid mice. Proc Natl Acad Sci USA (2001) 98:14589–94. doi: 10.1073/pnas.251451498
237. Xiao L, Zhou Y, Friis T, Beagley K, Xiao Y. S1P-S1PR1 signaling: the "Sphinx" in osteoimmunology. Front Immunol (2019) 10:1409. doi: 10.3389/fimmu.2019.01409
238. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
239. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol (2017) 15:53. doi: 10.1186/s12915-017-0392-4
240. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
Keywords: macrophages, targeted therapy, immune damage, musculoskeletal diseases, inflammatory
Citation: Zhu J, Fan J, Xia Y, Wang H, Li Y, Feng Z and Fu C (2023) Potential therapeutic targets of macrophages in inhibiting immune damage and fibrotic processes in musculoskeletal diseases. Front. Immunol. 14:1219487. doi: 10.3389/fimmu.2023.1219487
Received: 09 May 2023; Accepted: 04 July 2023;
Published: 20 July 2023.
Edited by:
Lan Xiao, Queensland University of Technology, AustraliaReviewed by:
Donglin Sun, Guangzhou Medical University, ChinaGuangchuan Wang, Jinzhou Medical University, China
Copyright © 2023 Zhu, Fan, Xia, Wang, Li, Feng and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changfeng Fu, ZnVjZkBqbHUuZWR1LmNu