 Yong Fu
Yong Fu Renhong Tang1,3
Renhong Tang1,3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 July 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1218082
Cytokines are pivotal mediators of cell communication in the tumor microenvironment. Multiple cytokines are involved in the host antitumor response, but the production and function of these cytokines are usually dysregulated during malignant tumor progression. Considering their clinical potential and the early successful use of cytokines in cancer immunotherapy, such as interferon alpha-2b (IFNα-2b; IntronA®) and IL-2 (Proleukin®), cytokine-based therapeutics have been extensively evaluated in many follow-up clinical trials. Following these initial breakthroughs, however, clinical translation of these natural messenger molecules has been greatly limited owing to their high-degree pleiotropic features and complex biological properties in many cell types. These characteristics, coupled with poor pharmacokinetics (a short half-life), have hampered the delivery of cytokines via systemic administration, particularly because of severe dose-limiting toxicities. New engineering approaches have been developed to widen the therapeutic window, prolong pharmacokinetic effects, enhance tumor targeting and reduce adverse effects, thereby improving therapeutic efficacy. In this review, we focus on the recent progress and competitive landscape in cytokine engineering strategies and preclinical/clinical therapeutics for cancer. In addition, aiming to promote engineered cytokine-based cancer immunotherapy, we present a profound discussion about the feasibility of recently developed methods in clinical medicine translation.
Cytokines constitute a class of immunoregulatory proteins (generally with a molecular weight lower than 30 kDa) that exert biological functions via the extracellular domains of cell membrane receptors and play imperative roles in maintaining physiological homeostasis and modulating pathophysiological processes (1, 2). The main classes of cytokines engaged in cellular communication include interleukins (ILs), interferons (IFNs), certain members of the tumor necrosis factor (TNF) superfamily, and other effector molecules (3). Dysregulated cytokine production in the tumor microenvironment (TME) is involved in all stages of carcinogenesis as well as in responses to therapy, suggesting that cytokines are crucial in cancer initiation, progression and elimination (4). Thus, cytokine-based therapeutics may be promising candidates for cancer immunotherapy, adding to the list of therapies that include radiotherapy (RT)/chemotherapy and immune checkpoint blockade therapy for cancer (5).
In 1986, interferon α-2b (IFNα-2b) was approved by the U.S. Food and Drug Administration (FDA) for the treatment of hairy cell leukemia (6). As the first biotherapeutic agent authorized, IFN-α provides a mechanistic blueprint for the development of many other cytokines into therapeutics. Subsequently, high-dose IL-2 therapy was authorized by the FDA in 1992 and 1998 for the treatment of metastatic renal cell carcinoma (RCC) and metastatic melanoma, respectively (7). Recently, various cytokines have been investigated in human clinical trials (8). However, the utilization of cytokines more broadly as effective therapeutic agents has been restricted due to their poor drug-like capabilities, extensive systemic effects and pleiotropy, which result in high toxicity and limited effectiveness (9). Indeed, a representative cytokine candidate from Nektar Therapeutics and Bristol Myers Squibb (BMS), polyethylene glycol (PEG)ylated IL-2 (Bempegaldesleukin), recently failed in the first Phase III study, delaying the hoped-for therapeutic resurrection of a pleiotropic cytokine (10, 11).
Why do cytokine-based monotherapies largely fail? The following reasons may explain the problems with native cytokines: short half-life in the circulatory system, low biodistribution, pathway redundancy of the target, a narrow therapeutic window, prohibitive toxicities (especially vascular leakage syndrome, central nervous system toxicity and cardiotoxicity), immunosuppressive effects of cytokines, and context-dependent and pleiotropic qualities (3). With a deeper understanding of target immunobiology and through rapid development of engineering biotechnology, preclinical and clinical investigations of cytokine therapeutics for cancer have markedly expanded, which has greatly facilitated the clinical translation of cytokine therapy. Novel approaches based on cytokine engineering are rapidly developing to localize cytokine activity to cells or tissues of interest and to initiate cytokine activity on demand (12). Notably, on July 28, 2022, ImmunityBio reported that the FDA has accepted a biologics license application (BLA) for the IL-15 agonist N-803 (Anktiva™) for the treatment of non-muscle-invasive bladder cancer (NMIBC) carcinoma in situ patients who had not responded to bacillus Calmette-Guerin (BCG) (13–15). The promising clinical data and progress of IL-15 agonists will undoubtedly reinvigorate the field of cytokine engineering for use in cancer therapy. In this review, we mainly concentrate on how engineering strategies and clinical advancements are being exploited for producing cytokines for clinical applications, including the common cytokine receptor γ chain (γc; also designated CD132 or IL-2Rγ) cytokine family (IL-2, IL-15, IL-7 and IL-21), IL-10, IL-12, IL-18, interferons (IFNs) and tumor necrosis factor-α (TNF-α).
As a crucial subset of cell signaling molecules, cytokines with pleiotropy and complex biology can produce serious adverse effects when administered systemically by inducing overactivation of the immune system (16). Recently, locus mutation has been the foundational engineering strategy to improve cytokine affinity or enhance binding selectivity, which in turn activates cells of interest and related signaling pathways (Figures 1A, B).
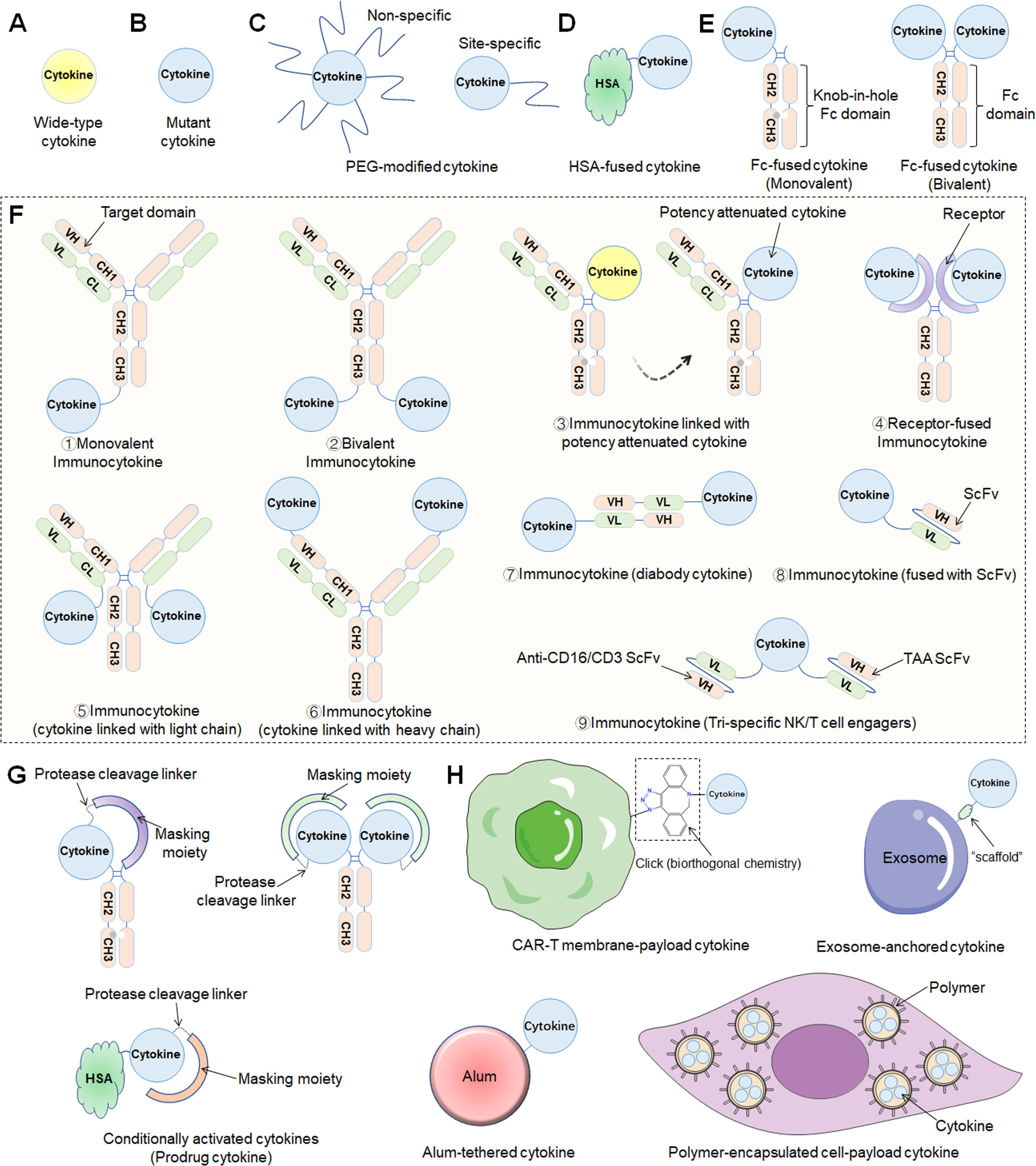
Figure 1 Different engineering approaches to increase the drug-like properties of cytokine-based therapeutics. (A) Wild-type cytokine. (B) Mutant cytokine. (C) PEG modification methods including nonspecific or site-specific chemical conjugation. (D) Fusion to the N- and/or C-terminus of HSA. (E) Fc-fusion strategies. Monovalent (“knob-in-hole” technology) or bivalent cytokine fused to the N-terminus of the IgG Fc-domain. (F) Immunocytokines consist of an antibody fused to a cytokine in various formats (1. Monovalent immunocytokine; 2. Bivalent immunocytokine; 3. immunocytokine with potency attenuated cytokine; 4. Receptor-fused immunocytokine; 5. cytokine linked with light chain; 6. cytokine linked with heavy chain; 7. diabody cytokine-based immunocytokine; 8. immunocytokine (fused with ScFv); 9. Tri-specific NK/T-cell engagers). (G) Prodrug cytokines using conditional activation approaches. (H) Other approaches, including CAR-T-cell membrane-payload cytokines, exosome-anchored cytokines, alum-tethered cytokines, polymer-encapsulated cell-payload cytokines, etc..
Cytokine-mediated therapy is subject to poor pharmacokinetic (PK) properties because the small size of the proteins promotes fast vascular extravasation and renal excretion, ultimately leading to a short half-life (8). Thus, through traditional intravenous administration, high-dose and frequent dosing regimens usually contribute to serious toxic side effects due to the narrow therapeutic window. As a result, multiple innovative strategies have been established to overcome delivery difficulties, such as the coupling of cytokines to high molecular weight carrier proteins such as human serum albumin (HSA), fusion with a fragment crystallizable region (Fc) of an immunoglobulin (IgG), or conjugation with polymer——PEG (Figures 1C, E) (17, 18). Recently, the aforementioned engineering techniques were extensively utilized in preclinical drug development, and multiple drug candidates have thus been developed. Many researchers named these drugs “supercytokines”, which include “superkine” and “superagonist”.
Cytokine targeting is an urgent problem to be solved because without proper targeting, sufficient therapeutic concentrations cannot be realized in tumors. The category of cytokines developed for targeting are antibody cytokine conjugates termed “immunocytokines” (or “fusokines”), which combine the targeting characteristic of antibodies with the ability of cytokines to trigger a local immune response while also tuning the PKs and pharmacodynamics (PDs) of the biologically active molecule (Figure 1F). Notably, a diabody is a novel antibody developed using recombinant DNA technology and consists of two variable heavy light (VH) and two variable light (VL) domains. Each VL domain in a single-chain variable fragment (scFv) is connected with a VH domain via a short linker, resulting in a diabody with two antigen-binding sites pointing in opposite directions (19). In addition, with the development of advanced protein engineering platform technologies, bispecific T/NK cell engagers fused with functional cytokines such as IL-2 and IL-15 have emerged, called trispecific T/NK cell engagers. Recently, diabody-fused cytokines (referred to as “diabody cytokines”) and trispecific T/NK cell engagers have been evaluated in clinical studies, and herein, we classify them as immunocytokines.
Interestingly, other recent approaches to conditionally activate cytokines depending on the TME, referred to herein as “prodrug cytokines”, might reduce systemic adverse events while sustaining antitumor capabilities and allow for enhanced safety and PKs/PDs (Figure 1G).
In general, the most recent cytokine engineering strategies can be broadly classified into the following categories: locus mutation, HSA-cytokine fusion, constant antibody fragment (Fc)-cytokine fusion, polymer conjugation, and immunocytokine and prodrug cytokine production (Figure 1). In fact, most cytokines used in the clinic are the products of multiple engineering strategies that further improve their efficacy and reduce the number and severity of adverse effects.
One of the first engineering strategies used to translate proteins into drugs was the mutation or modification of protein sequences to improve their biological manufacturability (12). More recently, versions of mutated cytokines have been designed to lessen the pleiotropic effects of cytokines. As a consequence, these mutations are employed to either increase or decrease binding affinity to cell receptors, leading to a more selective effect of the engineered cytokine.
The short half-life of cytokines significantly limits their clinical application. The neonatal Fc receptor for IgG (FcRn), which recycles IgG and prolongs its half-life in the circulatory system, contributes to effective humoral immunity (20). HSA is recycled through FcRn, which further extends the circulatory half-life of fusion cytokines by reducing the renal clearance rate and salvaging via FcRn (21). Albumin fusion proteins can be relatively easy to synthesize on a large scale, providing more cost-effective methods for enhancing cytokine PKs.
An Fc-based fusion protein consists of an immunoglobulin Fc domain that is covalently linked to another protein and is designed to alter the PKs of the active molecule (22). Mechanistically similar to HSA-fused proteins, the Fc domain significantly enhances the plasma half-life of the fusion protein, which improves therapeutic effects due to its interaction with the salvaging FcRn, as well as slower kidney excretion of larger molecules (20). Additionally, the Fc domain engages with Fcγ receptors (FcγR) and complement, which may also result in Fc-mediated various biological effects, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (23).
PEG is an amphiphilic nonionic synthetic polymer that readily conjugates cytokines to alter their PKs/PDs (24). Interestingly, in contrast to other strategies used for half-life extension, PEG fusion (PEGylation) is a materials-based approach. Currently, PEGylation can be used to achieve site-specific modification, and it can specifically bind an N-terminal α-amino group of proteins or peptides, which makes PEGylated fusion proteins profitable to manufacture at an industrial level (25).
A crucial safety aspect of cytokine therapy is systemic immune activation mediated by diverse receptor expression profiles, resulting in not only on-target but also off-tumor activity. Cytokines can be fused with targeting or functional proteins to modulate their localization or add new functionalities. Accordingly, structurally different formats of immunocytokines have emerged through the fusion of cytokines with antibodies, thereby locating effector molecules in the TEM and expanding the therapeutic window (26).
Strategies to conditionally activate the cytokine signaling pathway in the TME might further reduce systemic effects while preserving antitumor function and thus increase safety and PKs/PDs. Recently, leveraging tumor-related proteases or the acidic microenvironment for selective activation of cytokines has emerged as a promising strategy to increase safety and efficiency by limiting peripheral activity and localizing cytokine signaling to the TME. Protease-activatable “masked” cytokines have been illustrated and prepared with various masking domains, including anti-cytokine antibodies, antibody fragments, peptides, and native cytokine receptors (27). Selective activation in the TME depends on the linker design and expression of tumor-specific or abundant proteases, such as matrix metalloproteinase and cathepsins (28). Generally, prodrug cytokines consist mainly of the following four parts (29): (i) a cytokine that mediates antitumor response in the TME; (ii) a masking moiety, also referred to as a blocking moiety, which can be any group that physically prevents cytokines from binding and/or activating receptors in or surrounding nontumor tissues vivo until it dissociates and is activated in TME; (iii) “half-life extension elements”, which are usually a part of a chimeric polypeptide that improves PKs by changing the size, shape, charge, or the biodistribution, metabolism and renal clearance of the drug; and (iv) a linker, which is an important component of the prodrug cytokine, by providing flexibility between polypeptides so that the blocking element can suppress the activity of the cytokine polypeptide.
Other engineering approaches have been extensively investigated, including sustained-release cytokine microspheres, microencapsulated engineered cells and exosome-loaded or aluminum hydroxide (alum)-anchored cytokine therapeutics (Figure 1H).
This section focuses on current breakthroughs in the biology of the abovementioned cytokines (IL-2, IL-15, IL-7, IL-21, IL-10, IL-12, IL-18, IFNs and TNF-α), as well as engineering approaches and competitive landscapes aimed at resurrecting cytokine immunotherapy and promoting their clinical translation for the treatment of cancer.
IL-2, which is typically secreted by activated CD4+ T lymphocytes, is a prototypically pleiotropic cytokine that promotes T-cell proliferation, survival and differentiation; regulates natural killer (NK) cell natural lethality; and participates in antibody production on B cells (30). The IL-2 receptor (IL-2R) is a heterotrimeric complex with three subunits: IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132) chains (31). Among these chains, IL-2Rβ and IL-2Rγ (IL-2Rβγ) constitute an intermediate-affinity dimeric receptor (Kd~10-9 M), while IL-2Rα, IL-2Rβ and IL-2Rγ (IL-2Rαβγ) constitute a high-affinity trimeric receptor (Kd~10-11 M) (32). Dimeric intermediate-affinity IL-2R (IL-2Rβγ) is expressed primarily on CD4+ and CD8+ memory T lymphocytes and CD56dim NK cells; trimeric high-affinity IL-2R (IL-2Rαβγ) is expressed mainly on regulatory T (Treg) cells, activated CD4+ and CD8+ effector T lymphocytes, some NK cells, NK T cells, and group 2 innate lymphoid cells (ILC2s) (32). Once IL-2 specifically binds to IL-2R, its configuration changes, initiating the JAK1-JAK3-STAT5, PI3K-AKT-mTOR, and MAPK signaling pathways, inhibiting apoptosis, inducing cell proliferation and differentiation, and producing a variety of biological effects (33). Proleukin® (aldesleukin) was the first recombinant IL-2 immunotherapeutic agent to be developed, jointly by Novartis and Clinigen, but its clinical application is greatly limited due to its short half-life, narrow therapeutic window, profoundly toxic side effects, and stimulation of Treg cell activation (34) (Figure 2A).
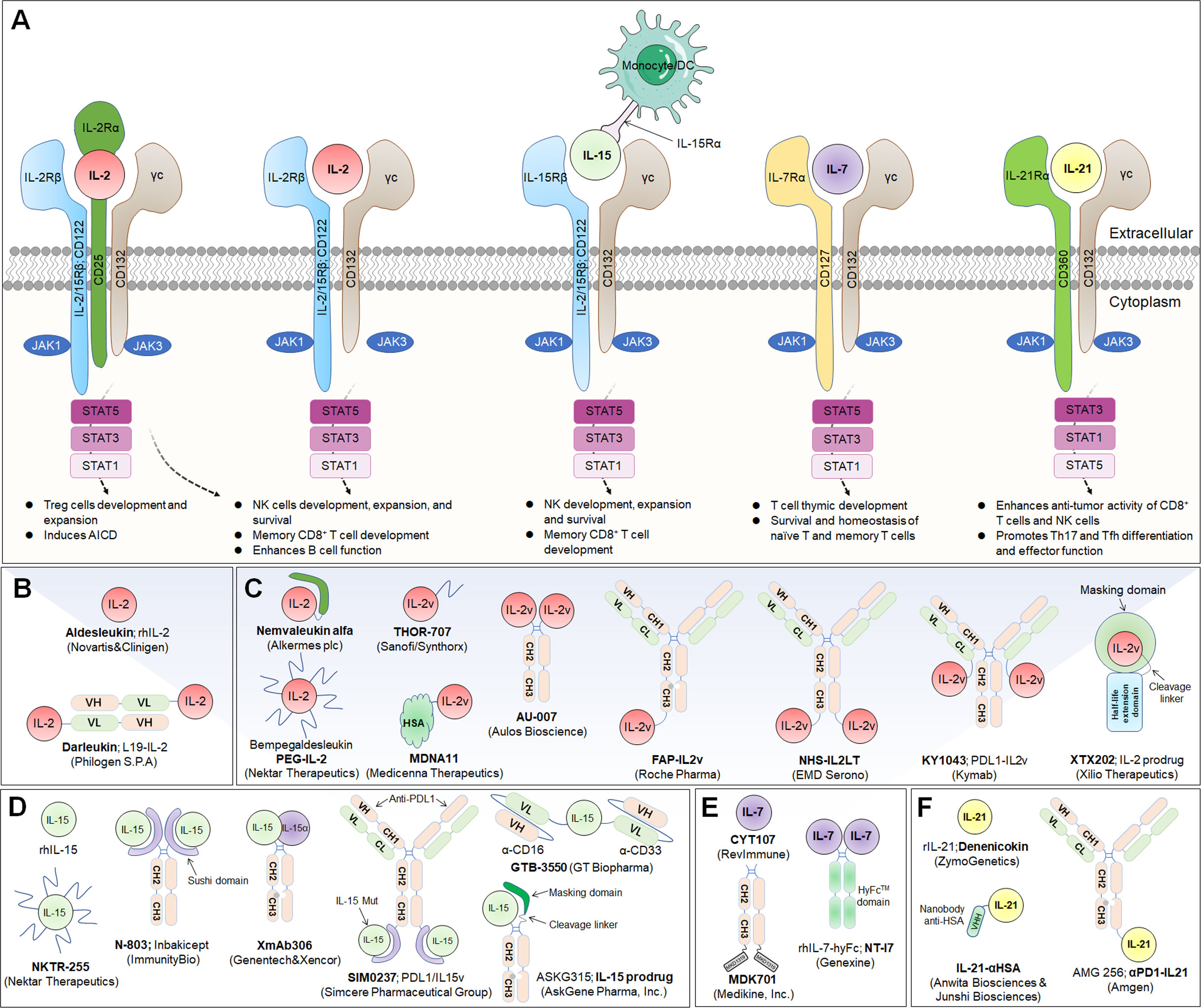
Figure 2 Common γ-chain cytokine (IL-2, IL-15, IL-7 and IL-21) signaling pathways, downstream effector molecules, and representative therapeutic candidates. (A) Common γ-chain cytokine (IL-2, IL-15, IL-7 and IL-21) signaling pathways and downstream effects. IL-2 and IL-15 are the only two of these cytokines to have three receptor chains. IL-2 exerts its biological function through high-affinity (IL-2Rαβγ) and intermediate-affinity (IL-2Rβγ) receptors. Engagement of IL-2 with IL-2Rαβγ strongly activates lymphocytes, driving beneficial effects on NK and T cells for development, increased effector function and enhanced B-cell function. The production of autocrine IL-2 can also lead to activation-induced cell death (AICD). Additionally, IL-2Rα is dramatically increased on Treg cells, so IL-2 is uniquely able to promote the expansion and development of immunosuppressive lymphocytes. IL-15 predominantly enhances the effector functions of NK and T lymphocytes without activating Treg cells. IL-7 primarily potentiates T-cell homeostasis, survival and thymic development, including both native and memory phenotypes. IL-21 dominantly promotes Th17 and Tfh differentiation and effector function and enhances the antitumor activity of CD8+ T and NK cells. (B, C) Representative therapeutic candidates of engineered IL-2 and IL-2 variants. (D-F) Representative therapeutic candidates of engineered IL-15, IL-7 and IL-21.
To achieve the beneficial activation of cytotoxic T lymphocytes to enhance cancer immunotherapy, a variety of engineering strategies have concentrated on altering the binding affinity of IL-2 to distinct receptor subunits (Table 1; Figures 2B, C). Subsequently, multiple new-generation IL-2 variants have been developed. In 2000, Bayer designed Bay50-4798, which selectively binds IL-2Raβγ, thereby reducing IL-2Rβγ+ NK cell-mediated toxicity, but the ultimate clinical benefit was not better than that of traditional IL-2 (35). In two ground-breaking studies published in 2005 and 2012, the prestigious scientist K. Christopher Garcia and colleagues analyzed the structure of the IL-2 quaternary complex with its different IL-2 receptors (36) and elucidated the underlying molecular mechanism by which IL-2 sensitizes T cells (37), which facilitated the use of biased IL-2 variants in clinical trials.
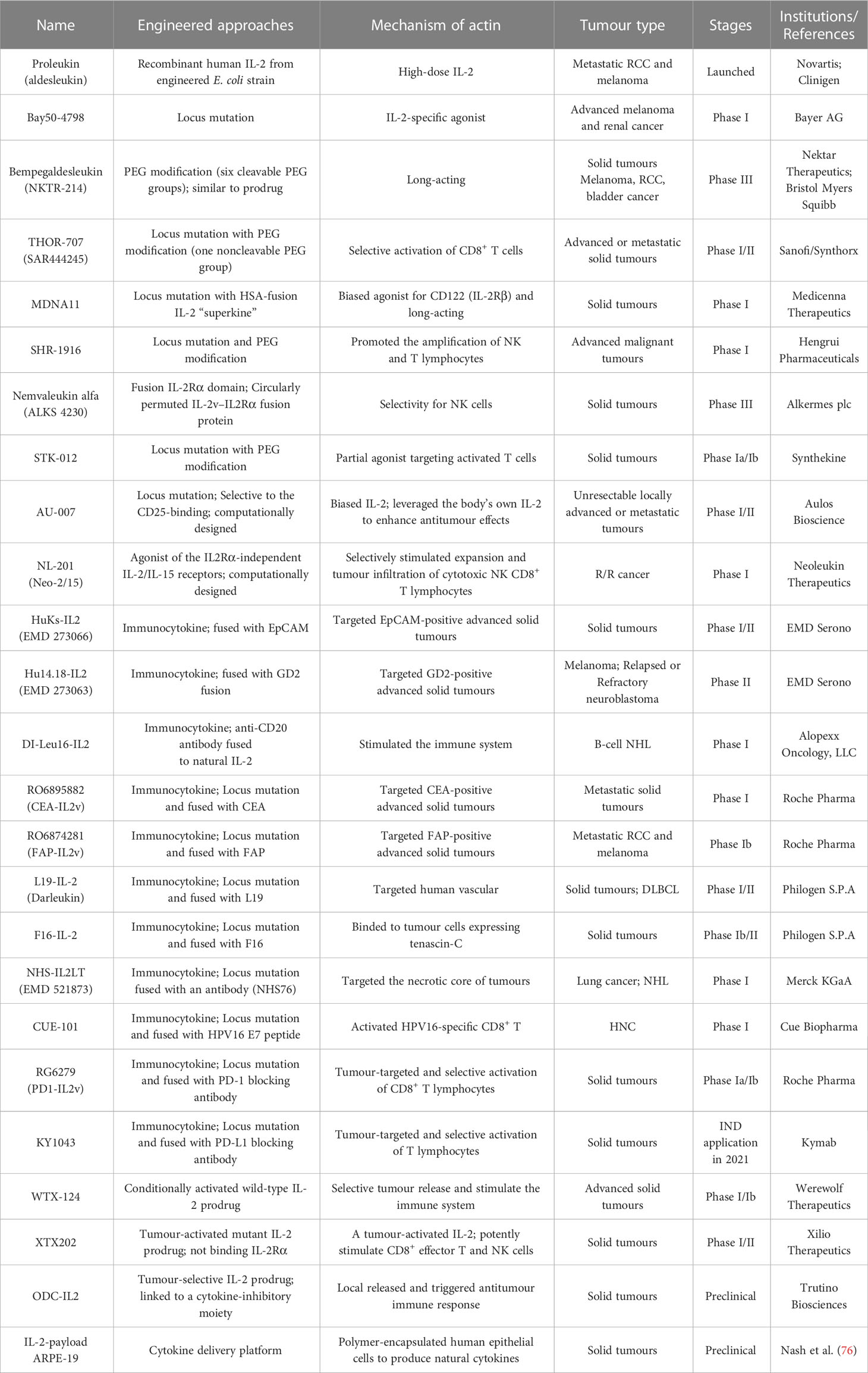
Table 1 Characteristics of engineered IL-2 variants.
NKTR-214 (bempegaldesleukin) is a representative PEGylated IL-2 with six slowly released functional groups that systematically deliver and preferentially bind to the IL-2Rβγ subunit but not the IL-2Rα subunit, thus promoting the effect of IL-2 on effector NK and T lymphocytes but not Treg cells (38). This mechanism of action resulted in selective expansion of effector T lymphocytes and inhibition of Treg cell proliferation in preclinical animal models (39). By modifying aldesleukin with PEG, the resulting compound NKTR-214 showed an effective reduction in severe toxic and other side effects and prolonged cancer onset compared to unmodified aldesleukin. However, on March 14, 2022, based on data from the Phase III PIVOT IO-001 study (NCT03635983), BMS and Nektar Therapeutics disclosed that anti-PD-1 antibody (Opdivo®) in combination with Bempegaldesleukin in the treatment of metastatic melanoma did not show key differences in the primary endpoints of progression-free survival (PFS) and overall response rate (ORR) compared with those of Opdivo® monotherapy (40). Additionally, according to the findings from PIVOT IO-001, the companies decided to terminate enrolment and unblind the ongoing Phase III PIVOT-12 study for patients with adjuvant melanoma, which was designed to evaluate differences in the dual therapy of Bempegaldesleukin combined with Opdivo® versus Opdivo® monotherapy in participants with high risk for recurrence after resection of melanoma (NCT04410445) (41).
THOR-707 (SAR444245) is a novel recombinant human IL-2 (rhIL-2) molecule that is irreversibly bound to a PEG moiety via click chemistry to block α-chain binding while maintaining appropriate affinity for IL-2Rβ/γ subunits (42). In preclinical studies, THOR-707, administered either as a single agent or in combination with an anti-PD-1 antibody, showed clear antitumor benefits without inducing severe side effects (43). A HAMMER Phase I/II trial documented that THOR-707 had good tolerability and showed antitumor effects in cancer patients, including those who were previously treated with checkpoint inhibitors. Combination therapy with pembrolizumab and cetuximab leveraged the effects of SAR44245 on CD8+ T and NK cells (NCT04009681) (44).
MDNA11, designed by Medicenna Therapeutics, is an IL-2-based “Superkine” fused with human recombinant albumin designed to increase the cytokine half-life and minimize dosing requirements without sacrificing efficacy or safety (45). Notably, this next-generation IL-2 drug preferentially binds CD122 (and shows no CD25 affinity) on immune cells and acts as a powerful switch for activating and proliferating tumor-killing effector NK and T lymphocytes to fight cancer (46). A Phase I/II ABILITY trial is ongoing to evaluate the safety, PKs/PDs, and antitumor properties of various dosages of MDNA11 in participants with advanced solid tumors (NCT05086692).
SHR-1916 is a new PEG-modified and site-mutated IL-2 molecule independently developed by Hengrui Medicine. It can activate the JAK1-JAK3-STAT5 signaling pathway, boost the proliferation of NK and CD8+ T lymphocytes, and exert antitumor effects (47). A Phase I clinical study of SHR-1916 is under underway based on clinical approval to assess its tolerability, safety, PK characteristics and preliminary efficacy in individuals with advanced malignant tumors (NCT04842630) (47).
Another clinically investigated IL-2-related candidate agent is ALKS 4230 (Nemvaleukin alfa, from Alkermes plc), which is a fusion protein composed of a circularly permuted IL-2 with an extracellular IL-2Rα domain that was designed for selective activation of effector lymphocytes with intermediate affinity for IL-2R (48, 49). Currently, a Phase III study (ARTISTRY-7) of ALKS 4230 combined with pembrolizumab for participants with platinum-unresponsive epithelial ovarian cancer is ongoing (NCT05092360) (50).
AU-007, designed by Aulos Bioscience, is a computationally developed, human IgG1 monoclonal antibody with LALA mutations in the Fc domain and is highly selective for the CD25-binding portion of IL-2 (51). Interestingly, AU-007 shows a different mechanism of action than other IL-2 therapies being developed, as it leverages the body’s own IL-2 to increase its innate antitumor immune effects. This increased effect was achieved by preventing IL-2 secreted from effector T cells from binding to trimeric receptors on Treg cells while still allowing IL-2 to bind to and induce the expansion of the effector T cells (52). A Phase I/II dose-escalation and expansion trial of AU-007 in participants with unresectable locally advanced or metastatic cancer is ongoing (NCT05267626) (52).
According to the report published in Nature magazine in January 2019, NL-201 (Neoleukin-2/15; Neo-2/15), developed by Neoleukin Therapeutics, is the first computationally designed de novo CD25-independent IL-2/IL-15 receptor agonist that specifically boosts the proliferation and tumor infiltration of NK and CD8+ T lymphocytes, thus improving antitumor performance (53). A first-in-human (FIH) Phase I trial of NL-201 as a monotherapy and combination therapy with pembrolizumab for participants with advanced cancer is ongoing (NCT04659629) (54).
To further enhance the transmission of IL-2 to tumor lesions and improve its therapeutic effect, several groups have combined IL-2/IL-2R mutations with antibody-mediated tumor-targeted delivery strategies (Table 1; Figures 2B, C). IL-2 fused with tumor-associated antigen (TAA)-targeted antibodies has been exploited, and these drugs can be classified into two main categories: cell-surface antigens, including epithelial cell adhesion molecule (EpCAM), ganglioside D2 (GD2), CD20, and carcinoembryonic antigen (CEA), and tumor extracellular matrix (ECM) targets, such as angiogenesis-related extra domain A/B (EDA/EDB) of fibronectin, tenascin-C, and collagen.
Humanized KS-IL-2 (HuKs-IL2; EMD 273066) is an immunocytokine with specific affinity for EpCAM fused at the Fc end to two IL-2 molecules (55), and it has been investigated in combination with cyclophosphamide in a Phase I/II trial of participants with recurrent EpCAM+ ovarian cancer, prostate cancer, colorectal cancer (CRC) or non-small cell lung cancers (NSCLC) (NCT00132522) (56). Using the same molecular construction strategy, EMD Serono developed Hu14.18-IL2 (EMD 273063), which is composed of anti-GD2 antibody 14.18 and IL-2 molecule, and Phase I/II studies were performed to determine its effect on patients with recurrent melanoma (NCT00590824) (57) or with relapsed/refractory (R/R) neuroblastoma (NCT01334515) (58). Based on compelling preclinical data, the maximal tolerated dose (MTD) of Hu14.18-IL2 was established to be 12 mg/m2/day (59).
In a Phase I dose escalation study, subcutaneous administration of DI-Leu16-IL2 (anti-CD20 antibody fused to wild-type IL-2) resulted in complete response (CR) in multiple cancer patients with minimal or no adverse events (60). CEA-IL2v (RG7813; RO6895882; cergutuzumab amunaleukin) and FAP-IL2v (RG7461; RO6874281; simlukafusp alfa) are CEA/fibroblast activating protein (FAP)-targeted immunocytokines designed by Roche Pharma with a novel monomeric IL-2 variant (IL2v) that completely lacks CD25 binding capacity (61). The IL2v component is engineered by structurally changing critical residues in the IL-2Rα interface (F42A, Y45A, and L72G) to prevent binding to IL-2Rα while preserving affinity for IL-2Rβγ (62, 63). Moreover, in multiple clinical Phase I trials, CEA-IL2v has been assessed for use against advanced and/or metastatic solid tumors (NCT02004106) (64). In parallel, FAP-IL2v has been investigated for use against metastatic melanoma (NCT03875079) and metastatic RCC (NCT03063762) and with trastuzumab or cetuximab (NCT02627274) or atezolizumab (NCT03386721) for use against metastatic pancreatic ductal adenocarcinoma (NCT03193190).
Darleukin (L19-IL2), developed in collaboration with academia and Philogen S.p.A., is a human vasculature-targeting monoclonal antibody (L19) fused to IL-2, and L19 targets the EDB region, which is contained in the ECM-associated fibronectin (B-FN) isoform (65, 66). A Phase I/II trial of L19-IL2 combined with rituximab against R/R diffuse large B-cell lymphoma (DLBCL) is ongoing (NCT02957019). Meanwhile, Philogen S.p.A. developed an F16-IL-2 immunocytokine in which the human monoclonal antibody fragment F16 scFv against EDA of tenascin-C was fused by linking to rhIL-2, and it showed potential immunostimulatory and antineoplastic activities (67).
Another clinical exploration of IL-2-based immunocytokines is NHS-IL2LT (EMD 521873; Selectikine; LT indicates low toxicity), developed by Merck KGaA, which contains an IL-2v (D20T) fused with an antibody (NHS76) that targets the necrotic core of tumors (68). NHS76 shows high affinity for both double- and single-stranded DNA (dsDNA/ssDNA), thus targeting exposed DNA and directing the cytokine to regions of tumor necrosis and apoptosis (69). To date, NHS-IL2LT has shown the lowest toxicity among all IL-2 variants in clinical trials (70).
To solve the problem of drug resistance to immune checkpoint monoclonal antibodies and further strengthen synergistic effects and targeting precision, immune cytokines based on immune checkpoint monoclonal antibodies have been developed. PD1-IL2v (RG6279; RO7284755, from Roche) is an antibody protein fusion that blocks PD-1 and is combined with an engineered IL-2v that induces immune-modulating activity (71). KY1043 (from Kymab; acquired by Sanofi in 2021) is a differentiated immunocytokine consisting of a PD-L1 antibody fused to an IL-2v (retaining the IL-2Rα affinity) designed to enhance antitumor immune responses by blocking the interaction between PD-L1 and PD-1 and activating antigen-experience T lymphocytes while reducing IL-2 activity to improve tolerability (72).
Tumor-selective IL-2 prodrugs have also recently attracted considerable interest from different research groups for the purpose of increasing intratumoral IL-2 abundance and reducing toxic side effects caused by systemic administration via injection (Table 1; Figure 2C). Its activity is based on a protease-cleaved substrate, which is sensitive and specific to proteases and preferentially overexpressed in tumors. WTX-124 is a novel systemically administered, tumor-selective IL-2 prodrug candidate consisting of a wild-type IL-2 cytokine tethered to an inactivation domain to prevent activation in nontarget tissue, a tumor protease-specific linker to allow activation in the TME and a half-life extension element to improve tumor exposure (73). At present, WTX-124 exhibits favorable antitumor abilities with beneficial PKs and tolerability profiles in preclinical experiments. In July 2022, Werewolf Therapeutics started enrolling patients in a FIH Phase I, multicenter study of WTX-124 administered as a single agent and in combination with pembrolizumab to participants with advanced solid tumors (NCT05479812).
XTX202 is a conditionally activated, βγ-chain biased (non-α-chain), engineered IL-2 molecule that is masked with a protein domain to limit binding activity until it is cleaved by TME-specific proteases. In preclinical studies, XTX202 was activated in a protease-dependent manner, inhibited tumor growth and had favorable tolerability (74). Xilio Therapeutics recently sponsored a Phase I/II clinical study for assessing XTX202 in the treatment of solid tumors (NCT05052268) (75).
Several other novel strategies have been used to engineer IL-2 prodrugs. For example, Nash et al. designed a clinically applicable cytokine delivery platform consisting of polymer-encapsulated human retinal pigmented epithelial cells, called ARPE-19, that produces natural cytokines, such as IL-2, and it significantly eliminated peritoneal tumors in ovarian and colorectal mouse models (76).
Interleukin-15 (IL-15) is a well-documented potent immunoregulator that is typically produced by monocytes and macrophages and can stimulate the proliferation of immune cells such as NK cells, CD8+ T lymphocytes, and γδ T cells (77). The biological functions of IL-15 are similar to those of IL-2, and their receptors share the same β chain and γ chain, while they also carry their own distinct α-receptors, IL-15Rα (CD215) and IL-2Rα (CD25), respectively (78). Independent of the IL-15Rβ and γ chains, IL-15Rα is normally expressed by antigen-presenting cells (79). It specifically binds to IL-15, forms an immunological synapse with the IL-15Rβγ chain on peripheral effector NK cells and T lymphocytes, and activates the JAK-STAT signaling pathway in NK cells and T cells (80). Due to its powerful effects on cytolytic NK and T cells without causing the expansion of suppressive Treg cells, IL-15 has recently emerged as an alternative to IL-2 in cancer therapy (81) (Figure 2A). Structurally, IL-15Rα comprises a Sushi domain (amino acids 1-65) that interacts with IL-15 and plays an essential role in the biological function of IL-15 (82). However, similar to other cytokines, because of a “cytokine sink”, native IL-15 is characterized by a short half-life and low monomeric bioactivity (83). Moreover, low systemic exposure and poor bioefficacy are associated with dose-dependent systemic toxicity. Therefore, the development of IL-15-based drugs has focused on optimizing their pharmaceutical properties by increasing their biological activity and half-life.
Currently, several IL-15-based superagonists with different structures are in clinical development (Table 2; Figure 2D). For example, N-803 (formerly ALT-803; Anktiva®), an IL-15 superagonist, consists of an IL-15 mutant (IL-15N72D) bound to the IL-15Rα/IgG1 Fc fusion protein and directly and specifically boosts the proliferation and activation of NK and CD8+ T lymphocytes through βγ receptor binding without activating Treg cells (84). Compared to native IL-15, N-803 shows better PK properties, a longer lymphoid tissue retention time, and higher antitumor activity in vivo. In a recent QUILT-3.032 Phase II/III trial (NCT03022825) (13), a total of 160 individuals with BCG-unresponsive NMIBC were enrolled, and the overall survival (OS) rate for bladder cancer was 99% at two years. For CIS patients, the complete response (CR) rate was 71%, with a 24.1-month median response duration, and the papillary disease-free survival (DFS) rate was 53% at 18 months. At the 2-year follow-up, more than 90% of patients avoided cystectomy. N-803 in combination with BCG outperformed other current intravesical and systemic treatments for BCG-refractory NMIBC in terms of efficacy and safety. Based on the aforementioned study, the FDA accepted the biologics BLA for N-803 in July 2022 for the treatment of NMIBC carcinoma in situ patients who had not responded to BCG. However, in May 2023, the FDA has delayed this approval decision due to deficiencies identified during a pre-licensing inspection of the manufacturer’s third-party contracting company.
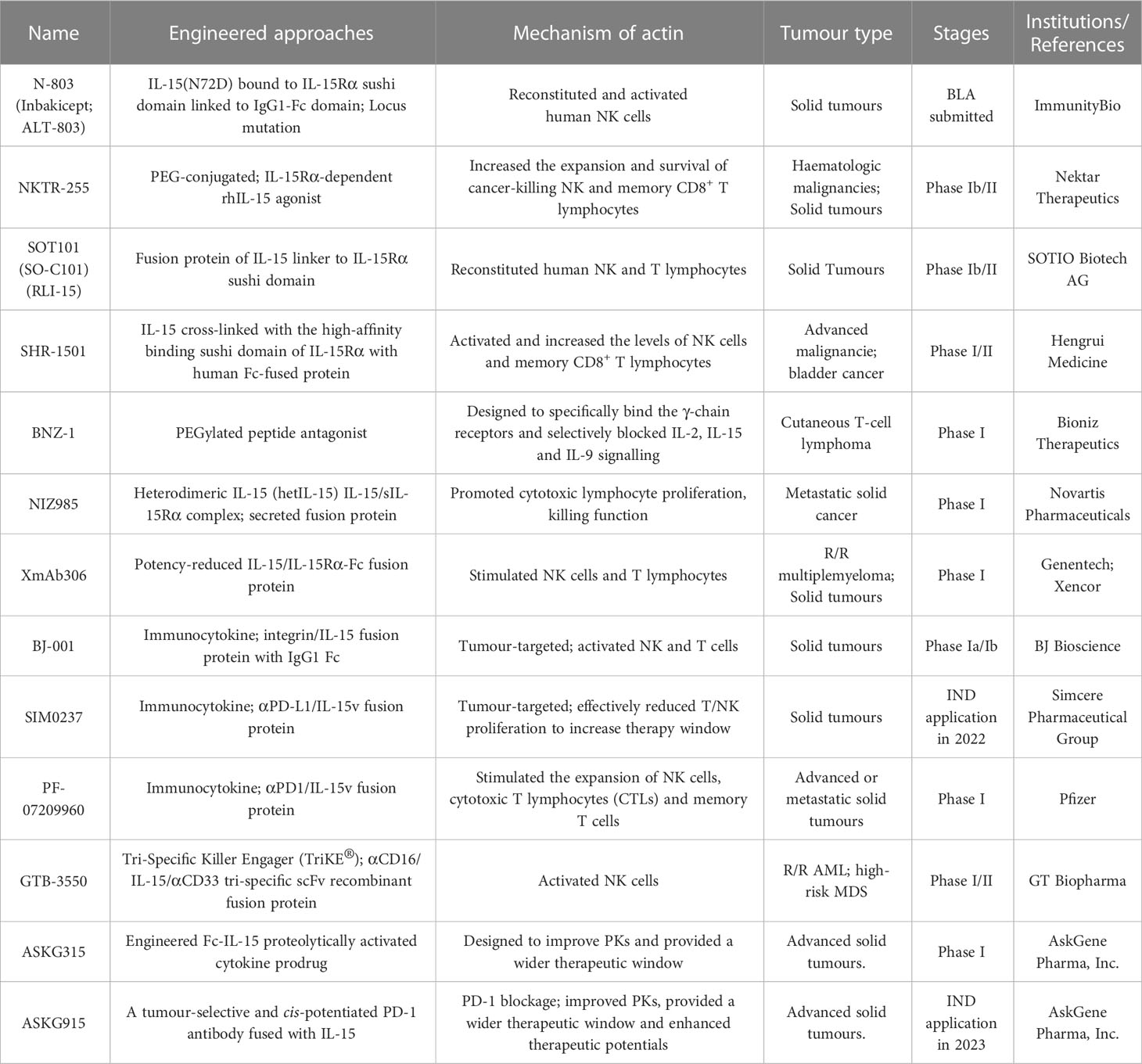
Table 2 Characteristics of engineered IL-15 variants.
NKTR-255 is an experimental PEG-conjugated IL-15R agonist aimed at increasing the intrinsic anticancer capacity of the immune system by promoting the survival and expansion of tumor-killing NK cells as well as memory CD8+ T lymphocytes (85, 86). NKTR-255 engages the complete IL-15 receptor complex (IL‐15Rα/IL‐15Rβγ) to improve the formation of long-term immunological memory, perhaps leading to persistent antitumor effects (87). NKTR-255 was developed to overcome the challenges of natural IL-15, which must be administered in high dosages due to quick clearance, limiting its effectiveness because of toxicity and inconvenience of use. NKTR-255 is now being studied in a Phase I trial for participants with R/R non-Hodgkin’s lymphoma (NHL) and multiple myeloma (MM) (NCT04136756) (88), and a Phase Ib/II trial is ongoing for evaluating NKTR-255 therapeutic effects in combination with ERBITUX® for R/R colorectal cancer (CRC) and squamous cell carcinoma of the head and neck (SCCHN) (NCT04616196) (89).
SOT101 (also known as RLI-15 and formerly as SO-C101) is an IL-15 superagonist fused to the IL-15Rα chain and designed to selectively bind only to cytotoxic NK and T lymphocytes but not to other cell types to minimize adverse events when administered subcutaneously. In addition, the PKs of SOT101 enable it to ideally stimulate NK and T lymphocytes through pulses in cytokine concentration not tonic stimulation (90). In a Phase I/Ib trial, the AURELIO-03 trial, SOT101 demonstrated a favorable safety profile and encouraging efficacy both when given as a monotherapy and in combination with KEYTRUDA® (pembrolizumab) (NCT04234113) (91). Subsequently, a Phase II AURELIO-04 trial of SOT101 in combination with pembrolizumab (NCT05256381) is ongoing to assess its efficacy and safety for patients with selected advanced/refractory solid tumors (90).
BNZ-1 is a PEGylated peptide antagonist designed to specifically bind γ-chain receptors and selectively block IL-2, IL-15 and IL-9 signaling (92). Preliminary Phase I data clarified that BNZ-1 was well tolerated and inhibited IL-2 and IL-15 activity, leading to clinical improvement in participants with cutaneous T-cell lymphoma (CTCL) by rejuvenating anti-lymphoma immunity and reducing inflammatory responses (NCT03239392) (93).
NIZ985 is a recombinant heterodimer of two polypeptide chains—physiologically active IL-15 and IL-15Rα—which together are named heterodimeric IL-15 (hetIL-15) (94). In preclinical studies, NIZ985 promoted cytotoxic NK and T lymphocyte expansion, tumor-killing function, and tumor infiltration, leading to significant anticancer ability. A recent FIH Phase I study was performed to assess the safety, PKs, and immune efficacy of NIZ985 in participants with metastatic or unresectable solid tumors (NCT02452268) (95). Subcutaneous NIZ985 three times a week was well tolerated in patients with advanced solid tumors and triggered a favorable immune response paralleling those identified in preclinical observations, manifesting as the induction of IFN-γ and amplification of cytotoxic lymphocytes.
XmAb306 (XmAb24306; RO7310729, from Genentech, a member of the Roche Group) is a potency-reduced IL-15/IL-15Rα fusion protein with an XmAb® bispecific Fc domain (96). Currently, two Phase I clinical trials to evaluate XmAb24306 as a monotherapy and in combination with atezolizumab or daratumumab in participants with locally advanced/metastatic solid tumors (NCT05243342) or R/R M/M (NCT04250155) are ongoing. No dose-limiting toxicities (DLTs) or severe drug-related side effects have been identified to date.
IL-15-based immunocytokines are also being developed in the clinic to further improve targeting (Table 2; Figure 2D). Recently, the Simcere Pharmaceutical Group developed a PD-L1/IL-15v bifunctional immunocytokine (SIM0237) that has best-in-class potential with improved tumor control and low safety risk. It blocks PD-1/PD-L1 signaling and delivers IL-15 directly to the TME for full activation of CD8+ T lymphocytes. The IL-15 pharmacological profile was fine-tuned through protein engineering. According to preclinical data, SIM0237 effectively reduced T/NK proliferation to increase the therapy window in vitro and significantly inhibited tumor growth in an NCI-H292-hPBMC xenograft model in vivo. In October 2022, the Simcere Pharmaceutical Group submitted an Investigational New Drug Application (IND) application for SIM0237 to the FDA.
PF-07209960 is a fusion protein composed of PD-1 fused to an IL-15 mutant designed to demonstrate potential immune checkpoint inhibition, immunomodulation, and antitumor activity. PF-07209960 is produced by Pfizer, which is currently enrolling participants for a Phase I clinical trial (NCT04628780) (97).
GTB 3550 (OXIS 3550) is a single-chain, triple compound-specific scFv recombinant fusion protein (Tri-specific NK cell engagers; TriKE®) composed of anti-CD16 and anti-CD33 antibodies and an IL-15 variant developed by GT Biopharma (owner of Oxis Biotech) for the treatment of patients with R/R acute myelogenous leukemia (AML) and high-risk myelodysplastic syndrome (MDS) (98–101). The candidate was also intended to be investigated in CD33+ hematopoietic malignancies in addition to MDS and AML. The drug was developed based on the hypothesis that GTB-3550 TriKE® induces NK cell function by targeting malignant cells as well as CD33+ myeloid-derived suppressor cells (MDSCs). Because CD16 is the most potent activating receptor on NK cells, GTB-3550 may elicit a targeted anti-CD33+ tumor response. Interim results from the current Phase I/II clinical trial showed that patients treated with GTB-3550 presented with up to a 63.7% downregulation in bone marrow blast levels, suggesting a clinical benefit (NCT03214666). Due to the discovery of second-generation camelid single-domain antibody technology, which presents several advantages over traditional IgG monoclonal antibodies, GTB-3550 development was halted, and a related clinical study was terminated. In September 2021, GT Biopharma announced the advancement of the second-generation camelid-derived nanobody TriKE agent product GTB-3650 into IND-enabling studies.
IL-15-based prodrug candidates have also attracted the attention of scientists (Table 2; Figure 2D). ASKG315 (ASKG215β) is an engineered proteolytically activated Fc-IL-15 cytokine prodrug designed to show better PKs and provide a wide therapeutic window. It was independently developed by AskGene Pharma, an overseas subsidiary of Jiangsu Aosaikang Pharmaceutical Co., Ltd (102). To fully harness the therapeutic potential of cytokine molecules, AskGene created a novel cytokine prodrug platform (SmartKine®), wherein cytokine molecules avoid the cytokine sink when systematically injected and are activated at the site of the lesion. The IL-15-based prodrug candidate ASKG315 has shown sustained PKs from 7 to 15 days in animal models. In the third quarter of 2022, two Phase I clinical trials were established to assess the safety, tolerability, PKs and PDs of ASKG315 as monotherapy and in combination with pembrolizumab in individuals with advanced solid tumors (NCT05554666; NCT05509985). In addition, AskGene Pharma designed ASKG915, a tumor-selective and cis-potentiated anti-PD-1 antibody-IL15 bispecific therapeutic, and ASKG915 was licensed to an IND in the first quarter of 2023 (103).
In a preclinical study, Zhao et al. developed an IL-15 prodrug masked by biocompatible polymers through chemical linkers. It responds to a TME with tumor-specific high reducing potential and acidic pH value, which are features aimed at reducing systemic exposure and thus minimizing toxicity (104).
IL-7 is a fundamental lymphopoietic cytokine that is predominantly expressed in non-hematopoietic cells, including stromal cells, epithelial cells, endothelial cells, fibroblasts, smooth muscle cells and keratinocytes, and has been proven to be essential for the development and survival of T and B lymphocytes (105). Moreover, IL-7 deficiency leads to severely impaired immune cell development (106). IL-7 receptor (IL-7R) is a heterodimeric complex consisting of IL-7Rα (also known as CD127) and the common cytokine receptor γ-chain, which is shared with the receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 (107). Whereas γc is expressed in most hematopoietic cells, IL-7Rα is almost exclusively expressed in cells of the lymphoid lineage, including B-cell progenitors (but not mature B cells), ILCs, naïve T cells and central memory T cells (107). Mechanistically, IL-7 crosslinks the extracellular domains of IL-7Rα and γc and promotes IL-7Rα and γc heterodimerization, leading to activation of JAK1 and JAK3, which ultimately activates major downstream signaling molecules, including STAT5 and PI3K (108) (Figure 2A).
As a multifunctional cytokine, IL‐7 can restore T‐cell numbers under lymphopenic conditions by boosting homeostatic cell proliferation (109). Early animal experiments consistently showed that IL-7 administered pharmacologically induced T lymphocyte proliferation while inducing little toxicity, implying that IL-7 may have a therapeutic benefit in humans. CYT107 is a glycosylated recombinant human IL-7 (rhIL-7) developed by RevImmune (formerly Cytheris) that is well tolerated and expands the lymphocyte pool in humans after allogeneic stem cell transplantation (Figure 2E) (110). In a randomized placebo-controlled Phase IIa trial (NCT01362107), CYT107 significantly increased the number of CD4+ and CD8+ T lymphocytes in twenty lymphopenic metastatic breast cancer patients when administered before chemotherapy (110). Due to the small sample size, a larger clinical trial is required to manifest its impact on clinical outcomes.
Because of its low structural stability and low productivity, the commercial use of IL-7 as a therapeutic agent has been hindered by many intrinsic technical challenges. Recently, NT-I7 (also named HyLeukin-7™; TJ107; rhIL-7-hyFc; GX-I7; and Efineptakin alfa), developed by Genexine (originating from a NeoImmuneTech-related company), which relied on its own hyFc® technology platform, is a long-acting cytokine consisting of rhIL-7 fused to a hybrid Fc (hyFc) region composed of the hinge-CH2 region of immunoglobulin D (IgD) and the CH2-CH3 region of immunoglobulin G4 (IgG4) (Figure 2E) (111, 112). NT-I7 was engineered to serve as a stable, long-acting factor for T-cell production, maturation, amplification, trafficking, functioning and survival. Moreover, it was designed to minimize the loss of the bioactivity of drug candidates and prevent ADCC- or CDC-mediated cell killing effects. Notably, NT-I7 enhances T-cell-mediated antitumor immunity through multiple mechanisms, including expansion of the TCR repertoire; sensitization of T cells to weaker antigens; boosting naïve T cells, stem cell-like memory T cells (TSCM) and memory T cells; promotion of memory T-cell differentiation; enhancement of T-cell trafficking and infiltration into tumors; augmentation of the antigen-specific T-cell response; antagonization of T-cell exhaustion and other inhibitory networks; and stimulation of the proinflammatory cytokine milieu in the TME (113).
At present, NT-I7 is the only clinical-stage long-acting rhIL-7 being investigated for oncological and immunological indications, in which it promotes T lymphocyte proliferation and enhances T-cell functionality. Previous data from a Phase I study with 30 healthy volunteers (NCT02860715) showed that NT-I7 dramatically boosted the absolute number of lymphocytes, especially T lymphocytes, and was well tolerated (114); another Phase Ib trial with 21 patients with advanced cancer (NCT03478995) showed that the drug was also well tolerated, and no DLT or cytokine release syndrome (CRS) was evident, and a dose-dependent increase in the number of lymphocytes and T lymphocytes (except Treg cells) was identified. Recently, Campian et al. demonstrated that NT-I7 mitigated RT-related lymphopenia, increased cytotoxic CD8+ T lymphocyte numbers throughout the body and in the tumor, and prolonged the survival of orthotopic glioma-bearing mice (111). NT-I7 is currently being evaluated for the treatment of high-grade glioblastoma in an ongoing Phase I/II trial (NCT03687957) (111). Additionally, the results from the trial confirmed that NT-I7 promoted TME inflammation by significantly upregulating the CD8+ T-cell to Treg cell ratio and enhanced the antitumor response by increasing the infiltration of IFN-γ-expressing T lymphocytes into the tumor. In other clinical trials, NT-I7 has exhibited favorable PKs/PDs and safety profiles when used both as a single agent and in combination with other antitumor treatments. Correspondingly, several Genexine-sponsored clinical studies of NT-I7 in combination with immune checkpoint monoclonal antibodies (namely pembrolizumab and atezolizumab) in the treatment of solid tumors including triple-negative breast cancer (TNBC), Merkel cell carcinoma (MCC), cutaneous squamous cell carcinoma (SCC) and melanoma are ongoing (NCT03752723; NCT03901573).
MDK1319 is a small IL-7R peptidyl agonist developed by Medikine, Inc., and is fused to an IgG Fc domain to generate MDK701 to improve in vivo performance (Figure 2E) (115). Intravenous injection of MDK-701 into cynomolgus macaques (single dose at 1 mg/kg) exhibited a circulating terminal half-life of approximately 32 hours and induced peripheral lymphocyte profiles similar to those observed after IL-7 treatment, followed by sustained elevation of peripheral lymphocyte levels, which remained higher than the baseline for 29 days with no side effects observed (115).
IL-21 is a pleiotropic cytokine that promotes the activation, expansion, and survival of tumor-specific CD8+ cytotoxic T cells while also enhancing B-cell proliferation and antibody production (116, 117). In addition, IL-21 enhances the antineoplastic activity of CD8+ T cells and NK cells (118, 119). IL-21 is primarily produced by CD4+ T and NKT cells, and the IL-21 receptor (IL-21R) is expressed on many hematopoietic cells, including CD4+ T follicular helper (Tfh), Treg, and T helper 17 (Th17) cells, naïve and memory CD8+ T cells, and B, NK, and dendritic cells (DCs) (116). IL-21R is a heterodimer consisting of a common γ-chain subunit and a specific subunit (CD360). IL-21/IL-21R engagement triggers a cascade of events that includes activation of the tyrosine kinases JAK1 and JAK3, followed by activation and phosphorylation of the transcription factors STAT3, STAT1 and STAT5 (120) (Figure 2A).
Early preclinical studies leveraged the overexpression of IL-21 directly in transplanted tumors or systemic administration of IL-21 plasmid DNA by hydrodynamic-mediated methods, and both of these delivery methods significantly reduced tumor growth in an NK cell- and T-cell-dependent manner (119, 121). Moreover, treatment of cynomolgus monkeys with recombinant IL-21 (rIL-21; denenicokin) developed by ZymoGenetics (acquired by Bristol-Myers Squibb) was associated with a dose-dependent increase in soluble CD25 (sCD25), an important marker of T-cell and NK-cell activation (Figure 2F) (122). A Phase I dose-elevation trial was performed, and the toxicity profile, immunomodulatory properties, and PKs of rIL-21 in patients with metastatic melanoma and RCC were recorded (122). Subsequently, in a Phase IIa clinical study, treatment with rIL-21 (30 μg/kg per dose) led to 1 CR and 1 partial response (PR) in 24 patients with metastatic melanoma. Further biomarker analyses demonstrated that rIL-21 treatment led to increased frequencies and selective activation of NK and CD8+ T lymphocytes, as indicated by upregulated expression of CD25, IFN-γ, perforin and granzyme B (123).
Due to the modest efficacy and short half-life of IL-21, its widespread therapeutic utility as a monotherapy has been limited. Recently, scientists from Anwita Biosciences and Shanghai Junshi Biosciences jointly engineered an IL-21-based fusion protein (IL-21-αHSA) in which a nanobody targeting HSA was fused to the C-terminus of rhIL-21 (Figure 2F) (124). αHSA nanobodies exhibited extensive species cross-reactivity and bound to an HSA epitope that does not overlap with FcRn-binding sites, prolonging the IL-21 half-life. Notably, in two syngeneic animal models, IL-21-αHSA demonstrated improved antitumor effects and exhibited synergistic antitumor benefits when combined with PD-1 and T-cell immunoreceptor with Ig and ITIM domain (TIGIT) blockers (124).
In a comparable preclinical study, Amgen Inc. developed a highly attenuated variant of IL-21 (R9E:R76A) and fused it to an anti-PD-1 antibody, and it provided protection against a humanized mouse tumor model that was resistant to anti-PD-1 monotherapy (125). Fusion of engineered IL-21 variants to anti-PD-1 antibodies can improve the drug-like characteristics of IL-21 cytokines, thereby increasing serum cytokine half-life and allowing for less frequent dosing. Bifunctional fusion proteins can block PD-1/PD-L1 interactions while delivering the IL-21 cytokine to PD-1-positive T cells. Furthermore, Amgen Inc. sponsored a FIH Phase I trial to assess the safety, tolerability, and estimated dosing of the abovementioned agent (also known as AMG 256; a chimaera consisting of anti-human PD-1 variable region, a mouse IgG1 constant region and human IL-21 variant R9E:R76A monomer fused to the C-terminus) and utilized it as a single agent in patients with advanced solid tumors (NCT04362748) (Figure 2F) (126).
Interleukin-12 (IL-12) is a powerful proinflammatory cytokine that has piqued the interest of researchers seeking to develop long-term cancer immunotherapy. Due to its prominent proinflammatory and immunomodulatory capabilities, IL-12 can convert immunologically suppressed “cold” tumors into inflamed “hot” tumors (127). Structurally, IL-12 is a typical heterodimeric cytokine consisting of two subunits——an α subunit (IL-12p35) and a β subunit (IL-12p40), which are linked by disulfide bonds (128). Biologically active IL-12 is produced primarily by activated inflammatory cells, including monocytes, macrophages, DCs, and other antigen-presenting cells. IL-12 activates the immune system by binding to its receptor (IL-12R), which consists of IL-12Rβ1 and IL-12Rβ2 and is predominantly expressed by activated T cells, NK cells, and DCs (128). Engagement of IL-12 with its heterodimeric receptors induces phosphorylation of JAK2 and TYK2, followed by STAT4 phosphorylation, dimerization, nuclear translocation, and IFN-γ secretion (129). Functionally speaking, IL-12 induces the differentiation of Th1 cells; increases the activation and cytotoxicity of NK and T lymphocytes; inhibits or reprograms immunosuppressive cells, such as MDSCs and tumor-associated macrophages (TAMs); upregulates the expression of MHC-I and MHC-II on tumor cells to enhance recognition and lysis; suppresses IgE production but enhances IgG production by B cells; and shows anti-angiogenic effects (130) (Figure 3A).
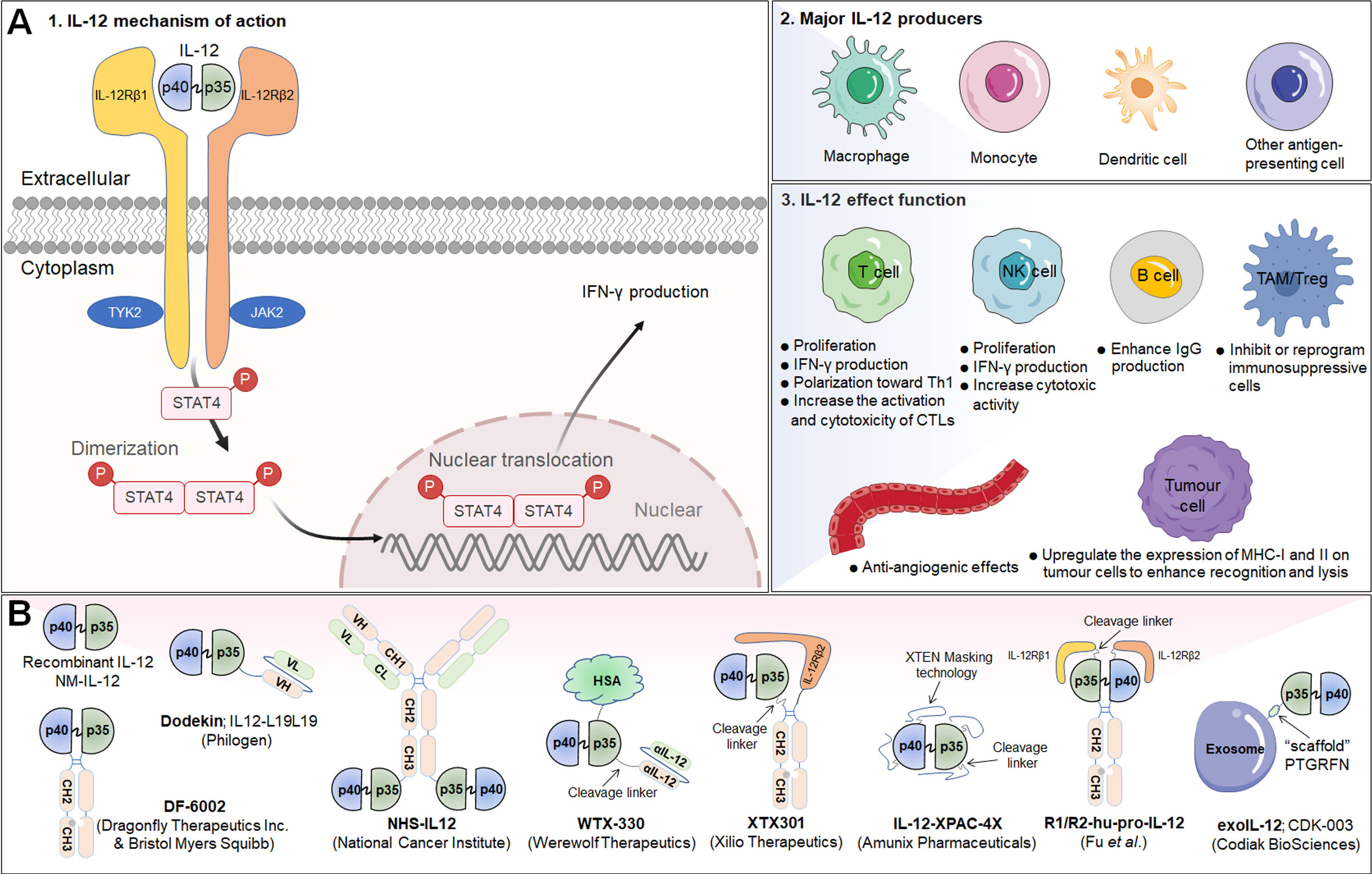
Figure 3 IL-12 signaling pathway, downstream effector molecules, major IL-12 producers, IL-12 effect function, and representative therapeutic candidates. (A-1) IL-12 is a heterodimeric cytokine consisting of IL-12p35 and IL-12p40, which bind to IL-12Rβ2 and IL-12Rβ1, respectively. IL-12 interacts with IL-12R to induce Jak2 and Tyk2 phosphorylation, followed by STAT4 phosphorylation, dimerization and nuclear translocation, ultimately promoting IFN-γ gene transcription and IFN-γ secretion. (A-2) Biologically active IL-12 is produced mainly by activated inflammatory cells including monocytes, macrophages, DCs, and other antigen-presenting cells. (A-3) Biological effects of IL-12 on different cells. (B) Representative therapeutic candidates for engineered IL-12.
The overwhelming majority of the evidence has indicated that IL-12 activates the innate and adaptive immune systems and exerts excellent therapeutic effects by shrinking syngeneic tumors. However, the systematic administration of recombinant IL-12 (rIL-12) in clinical trials has been associated with serious immune-related side effects and a narrow therapeutic window, limiting the response rates, and no IL-12-based therapeutic drug has been approved to date. Moreover, natural IL-12 may not specifically accumulate in the TME, limiting its efficacy and potentially causing the aforementioned immune-associated adverse events. Thus, modification of IL-12 or its combination with other targeted agents may increase its efficacy and overcome safety issues (Table 3; Figure 3B).
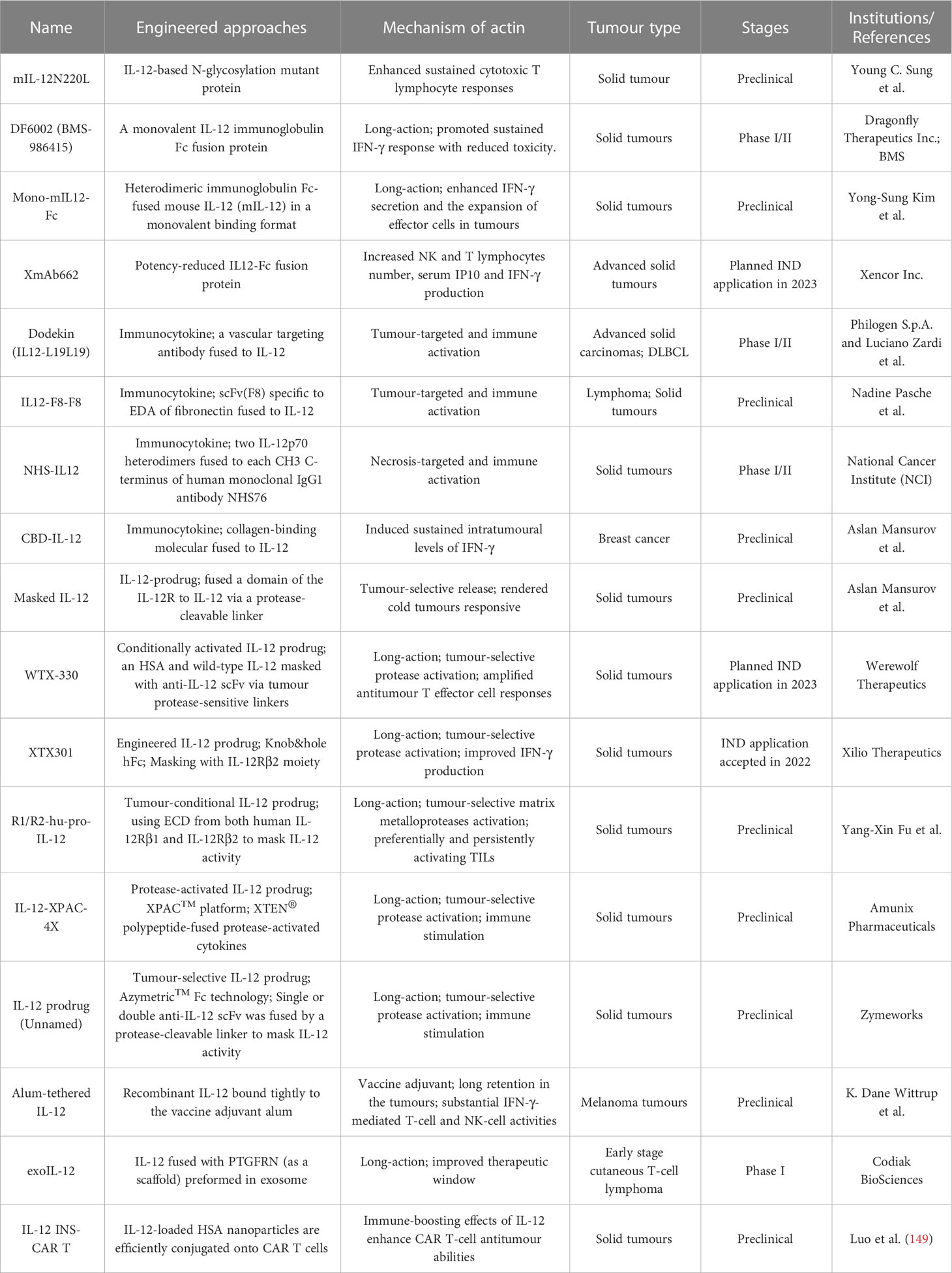
Table 3 Characteristics of engineered IL-12 variants.
In a prior report, Young C. Sung and team engineered an IL-12-based N-glycosylation mutant (mIL-12N220L) that increases durable cytotoxic T-cell immune responses and induces more effective protection against tumor challenge than wild-type IL-12. Thus, this mutant IL-12 was employed to develop DNA vaccines that can be used as an adjuvant for the production of long-term memory T-cell responses (131).
To further improve the half-life of IL-12 in vivo and broaden the therapeutic window, several fusion proteins of IL-12 and immunoglobulin Fc with comparable structures were applied for clinical development. For example, a preclinical research team from Ajou University School of Medicine, developed heterodimeric immunoglobulin Fc-fused mouse IL-12 (mIL-12) in a monovalent-binding format (Mono-mIL12-Fc) to produce long-acting mIL-12 in the natural heterodimeric form. Mono-mIL12-Fc displayed a much longer plasma half-life than recombinant mIL-12 and can be administered systemically twice weekly, which eliminated established tumors in syngeneic mouse models without inducing noticeable toxicity. More importantly, mono-mIL12-Fc elicited weak IL-12 signaling, favoring the generation of functional and protective memory CD8+ T lymphocytes (132).
Subsequently, Dragonfly Therapeutics Inc. and BMS jointly developed DF-6002 (BMS-986415), a monovalent IL-12-immunoglobulin Fc fusion protein, as an experimental immunotherapy for the treatment of advanced solid tumors in adult patients. A Phase I/II trial established to evaluate DF6002 administered alone and in combination with nivolumab in participants with locally advanced or metastatic solid tumors is ongoing (NCT04423029) (133).
Another clinically investigated potency-reduced IL-12-Fc fusion protein is XmAb662, which is aimed at increasing tumor immunogenicity. XmAb662 is being further studied and, to date, has demonstrated significant antitumor activity in vivo, and has shown the ability to increase the numbers of NK cells and T lymphocytes and the levels of interferon gamma-induced protein 10 (IP-10) and IFN-γ in serum. Moreover, these effects were enhanced when XmAb662 was combined with an anti-PD-1 antibody. In 2023, Xencor Inc., a biopharmaceutical company that develops clinical-stage drugs, plans to submit an IND application for XmAb662 and initiate a Phase I trial for participants with advanced solid tumors.
Considering that immune cells in the tumor and surrounding draining lymph nodes are ideal targets for IL-12 immunotherapy, there have been various initiatives to develop tumor-targeted IL-12 therapeutics (Table 3; Figure 3B). In an earlier study, Verena Gafner et al. developed three structurally similar IL-12-based fusion proteins (IL12-scFv(L19)-FLAG) based on the sequential fusion of a single-chain IL-12 derivative to an anti-EDB antibody fragment scFv (scFv(L19)) (134). One of three candidate compounds, in which p40 and p35 formed a covalent heterodimer and each subunit was fused to a molecule of scFv(L19) (known as p40-scFv(L19)/scFv(L19)-p35), demonstrated excellent tumor-targeting efficacy, as evaluated via biodistribution analysis, and potent antitumor activity in animal models of teratocarcinoma, showing superior performance compared to that of the other two IL12-scFv(L19)-FLAG candidates (134). Based on the aforementioned study, Philogen developed Dodekin (IL12-L19L19), a fully human immunostimulatory candidate consisting of a vasculature-targeting antibody fused to IL-12, a tumor-targeted product that selectively localizes to the site of tumor lesions while safeguarding healthy tissues (135). Considering encouraging preclinical data, Philogen has initiated a Phase I clinical study to assess the safety and early signs of efficacy of Dodekin in patients with advanced solid carcinomas and DLBCL that have progressed after immune-checkpoint blockade therapy (NCT04471987).
NHS-IL12 is another necrosis factor-targeting immunocytokine consisting of two IL-12p70 heterodimers in which each CH3 C-terminus is fused to the anti-human monoclonal IgG1 antibody NHS76, capitalizing on the specific affinity of NHS76 antibody for ssDNA and dsDNA, which are frequently exposed in the necrotic portions of tumors (136). Because human IL-12 does not cross-react with murine IL-12R, a chimeric surrogate immunocytokine, NHS-muIL12, was constructed and tested in preclinical experiments with immunocompetent mice. Systemic administration of the murine analogue NHS-muIL12 delayed the growth of MC38-CEA+ colorectal carcinomas in carcinoembryonic antigen transgenic (CEA.Tg) mice (137). Currently, NHS-IL12 is being investigated in Phase I/II clinical trials as a single agent (NCT01417546) or in combination with M7824 (an anti-PD-L1/TGFβ trap fusion protein) (NCT04303117) in solid tumors. Interim data showed that NHS-IL12 had good tolerability with an MTD of 16.8 µg/kg, with 9% of cancer patients experiencing disease stability (138).
Recently, Aslan Mansurov and his coworkers created a novel fusion protein by fusing collagen-binding molecule to IL-12, designated CBD-IL-12, which can target tumor lesions and accumulate in the tumor stroma, where collagen is exposed due to a disorder in the tumor vascular system. In vivo data illustrated that intravenous administration of CBD-IL-12 stimulated sustained IFN-γ levels within tumors, significantly reduced IFN-γ levels in the peripheral circulatory system, attenuated organ damage and exhibited excellent anticancer activity, triggering complete remission of checkpoint inhibitor-resistant breast tumors (139).
Notably, there are several structurally distinct IL-12 prodrugs in clinical development designed to further improve the safety of treatment (Table 3; Figure 3B). WTX-330 is a systemically administered, selectively activated IL-12 prodrug consisting of an HSA to extend its half-life and wild-type IL-12 masked with a high-affinity anti-IL-12 scFv that are connected via tumor protease-sensitive linkers (140, 141). WTX-330 was developed to include a high-affinity blockade of IL-12/IL-12R engagement systemically via the circulatory system and in nontumor tissues to prolong the half-life of IL-12 for optimal tumor exposure and tumor-selective protease activation through a proprietary mechanism. In preclinical investigations, WTX-330 exhibited prominent antitumor potential, as well as a favorable PK and tolerability profile. Robust antitumor activity was mediated through stimulation of innate and adaptive antitumor responses, including DC maturation and cross-presentation of antigens, Th1 cell differentiation, and expansion of antitumor effector T lymphocyte responses. Werewolf Therapeutics filed an IND for WTX-330 in the third quarter of 2022.
XTX301 is a protein-engineered IL-12 prodrug that is masked with a protein domain to block binding activity until IL-12 is cleaved by TME-related proteases. To assess tumor-selective activity, antitumor potency and peripheral toxicities, a murine surrogate (mXTX301) was constructed. Preclinical results showed that intravenous administration of mXTX301 at a single dose of 0.5 mg/kg inhibited tumor growth by up to 90% in MC38 (inflamed) and B16F10 (noninflamed) syngeneic mouse models. Moreover, mXTX301 induced an approximately 3-fold increase in the IFN-γ levels within the tumor compared to the vehicle control and an approximately 150-fold reduction in peripheral IFN-γ levels compared to mXTX300 (the unmasked control). More importantly, XTX301 was well tolerated and activated in a protease-dependent manner. Xilio Therapeutics is progressing XTX301 through IND-enabling studies and plans to investigate XTX301 as a single agent and combination therapy for the treatment of solid tumors (142).
Recently, in a preclinical study, Professor Yang-Xin Fu and team generated a conditional IL-12 prodrug (Pro-IL-12; R1/R2-hu-pro-IL-12) composed of IL-12 masked by IL-12 extracellular receptor-binding domains consisting of both human IL12Rβ1 and IL12Rβ2 that can be released by cleavage of an MMP linker, which is preferentially activated inside the TME and shows limited systemic toxicity. The results showed that the activity of this pro-IL-12 shielded IL-12 compound in vitro, showed reduced systemic toxicity in vivo, and effectively controlled both primary and metastatic tumor growth in MC38, B16-F10, and 4T1 mouse tumor models. Importantly, Pro-IL-12 used in combination with an anti-PD-L1 antibody and a tyrosine kinase inhibitor (TKI) induced robust tumor regression in a TUBO model, a representative model of cold tumors, and an MC38 model (143).
Notably, Amunix Pharmaceuticals (acquired by Sanofi in 2021) recently applied its proprietary masking technology (the XPAC™ platform; XTEN® polypeptide-fused protease-activated cytokines) to its first masked protease-activated cytokine platform, IL-12-XPAC-4X (29). Concurrently, Zymeworks engineered another novel IL-12-prodrug (unnamed to date) in which single-chain IL-12 was fused to one C-terminus of Zymeworks’ Azymetric™ Fc heterodimer (144). An anti-IL-12 scFv or anti-p35 scFv was fused to the other Fc heterodimer C-terminus via a protease-cleavable linker to block IL-12 activity. Leveraging linkers engineered to be cleaved by highly active intratumoral proteases, blocking antibodies are released selectively within the TME, hence boosting intratumoral IL-12 activity. Comprehensive published data on these two IL-12-based prodrugs are currently unavailable.
Alum has been safely used in humans for approximately 100 years and has been approved by the FDA as a vaccine adjuvant (145). Alum adjuvants consist of micrometer-sized (1-10 mm) aggregates of rod-shaped aluminum oxyhydroxide nanocrystals, which form a physical deposit at injection sites in tissue that persists for weeks. Interestingly, researchers from MIT constructed an engineered alum-tethered IL-12 in which recombinant IL-12 was bound tightly to the vaccine adjuvant alum via ligand exchange between hydroxyls on the surface of alum and phosphoserine residues attached to the cytokine by an alum-binding peptide. Specifically, the in vivo results demonstrated that in a murine melanoma model, the intratumoral infusion of alum-tethered IL-12 exhibited retention for weeks in the tumors along and induced minimal side effects while inducing substantial IFN-γ-mediated NK and T lymphocyte activity, thus increasing tumor antigen accumulation in draining lymph nodes and triggering strong tumor-specific T-cell priming (146).
Exosomes are natural, cell-derived vesicles that compose a nonimmunogenic delivery system for a variety of therapeutic payloads (147). Codiak BioSciences recently generated a novel engineered IL-12-based exosome therapeutic candidate (exoIL-12; CDK-003) by displaying functional IL-12 on the exosome surface by fusing the abundant exosomal surface protein PTGFRN as a “scaffold” (148). ExoIL-12 was designed to address the limitations of undesired systemic exposure and unpredictable pharmacology. In preclinical studies, exoIL-12 demonstrated longer tumor retention and better antitumor effects than wild-type IL-12 following intratumoral injection, thereby creating a therapeutic window for this powerful cytokine (148). Codiak BioSciences has initiated a Phase I/IIa clinical study for exoIL-12 with healthy volunteers (subcutaneous administration) and patients with early stage cutaneous T-cell lymphoma (intralesional administration) (NCT05156229).
Chimeric antigen receptor T (CAR T) cell immunotherapy has shown significant success in the treatment of hematological tumors, but its therapeutic benefits are greatly limited in solid tumors due to the lack of activated T lymphocytes effectively infiltrating the immunosuppressed TME. Recently, Luo et al. designed an IL-12-based nanostimulant (INS; as a nanochaperone)-functionalized CAR T-cell (INS-CAR T) biohybrid, in which IL-12-loaded HSA nanoparticles were efficiently conjugated with CAR T cells via biorthogonal chemistry without affecting the antitumor potential of these engineered cells. IL-12 is reactively released from INS-CAR T-cell biohybrids after the number of tumor antigen-triggered thiol groups is increased. Finally, the immune-boosting effects of the IL-12 nanochaperone greatly enhanced CAR T-cell antitumor activity, markedly eradicated solid tumors and reduced undesired adverse events (149).
Interleukin (IL)-18, formerly known as interferon gamma-inducing factor (IGIF), belongs to the IL-1 cytokine family and mediates inflammation downstream of activated NLRP3 and NLRP1 inflammasomes. IL-18 is abundant in macrophages, DCs and epithelial cells in the form of an inactive precursor (pro-IL-18) (150). When inflammation is triggered, pro-IL-18 is cleaved by caspase-1, a reconstituted NLRP3 inflammasome component, becoming active IL-18 and is rapidly released (151). Mechanistically, IL-18 drives the MyD88/NF-κB and MAPK signaling pathways through heterodimerization of its receptor subunits IL-18Rα (IL18R1; CD218a) and IL-18Rβ (IL18RAP; CD218b) (152). IL-18R has been shown to be expressed on normal peripheral blood lymphocytes (153). Increasing evidence indicates that IL-18 not only strongly induces the production of cytokines such as IFN-γ and TNF-α from immune cells but also increases the expression of FasL and enhances the toxicity of NK cells (154) (Figure 4).
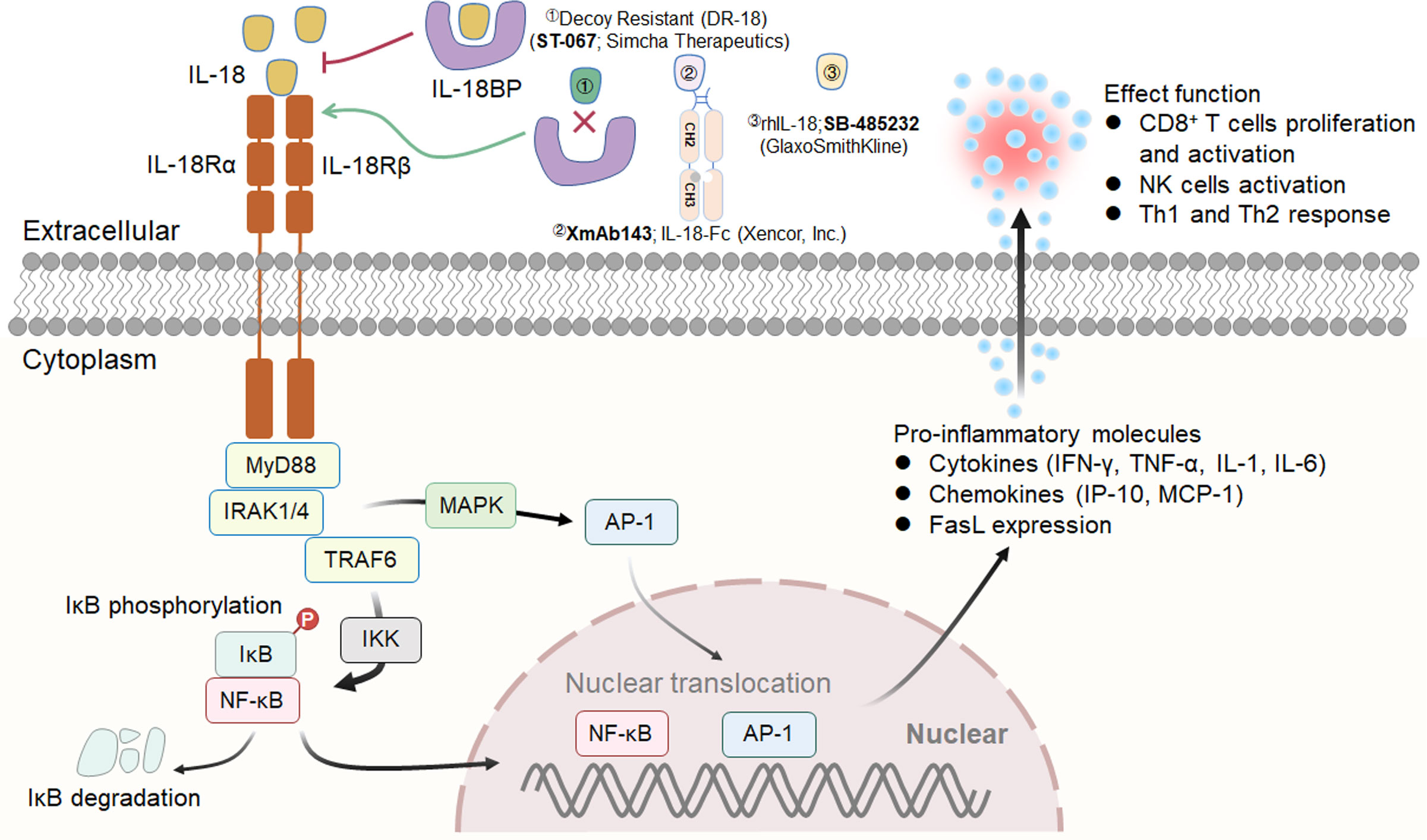
Figure 4 IL-18 signaling pathway, downstream effector molecules and representative therapeutic candidates. IL-18 drives the MyD88/NF-κB and MAPK signaling pathways through heterodimerization of its receptor subunits IL-18Rα and IL-18Rβ. Three representative therapeutic candidates of engineered IL-18 are shown (above).
In a preclinical animal study, Ma et al. investigated the effect of recombinant mouse IL-18 (provided by GlaxoSmithKline) on the peritoneal dissemination of CT-26 cells or tail vein infusion of metastatic B16/F10 cells in combination with an anti-PD-L1 antibody and/or anti-CTLA-4 antibody (155). The experimental data illuminated that IL-18 boosted the therapeutic efficacy of immunological checkpoint blockade by mediating the accumulation of premature NK cells and memory-type CD8+ T lymphocytes while suppressing Treg cells (155). Thus, IL-18 in combination with immune checkpoint inhibitors may be a promising strategy for developing next-generation cancer immunotherapy.
An early Phase I, dose-escalation trial documented the toxicity, PKs, and biological activities of recombinant human IL-18 (rhIL-18; SB-485232, developed by GlaxoSmithKline) in participants with advanced solid tumors and lymphomas, and the data demonstrated that rhIL-18 administration was safe and that the agent was well tolerated (Figure 4) (156). Subsequently, a Phase II trial was planned to assess the antitumor effect of three dose groups of rhIL-18 (0.01, 0.1, and 1.0 mg/kg/day) delivered as monotherapy in patients with previously untreated metastatic melanoma (157). Unfortunately, rhIL-18 failed to show efficacy in clinical studies, and none of the three doses investigated were recommended for Phase III valuation.
In a recent study, Professor Aaron M. Ring and colleagues revealed that IL-18-binding protein (IL-18BP), a secreted high-affinity (Kd<1 nM) IL-18 decoy receptor, is often upregulated in a variety of human and murine tumors and is a barrier to the antitumor activity of IL-18 (158). Subsequently, these authors engineered a variant of decoy-resistant IL-18 (DR-18) that does not bind the decoy receptor IL-18BP and retains the ability to bind tumor-infiltrating lymphocytes. Surprisingly, injecting DR-18 into tumor-bearing animals triggered the selective proliferation of stem-like TCF1+CD8+ T lymphocytes in tumors, as well as preferential differentiation towards polyfunctional T lymphocytes rather than an exhaustion phenotype. Additionally, DR-18 stimulated the expansion of NK cells in MHC class I-deficient tumors, suggesting a potential treatment option for immune checkpoint blockade therapy-unresponsive tumors. Simcha Therapeutics is currently sponsoring a Phase I/II clinical study (NCT04787042) designed to evaluate the safety of DR-18 (ST-067) as a single agent administered subcutaneously weekly for a variety of cancer indications, including melanoma, RCC, TNBC and NSCLC (Figure 4). Additional results of the Phase II trial, which is expected to be completed in 2023, will include PKs/PDs, clinical activity, and safety data (159).
Another IL-18 fusion protein investigated is XmAb143 developed by Xencor, which is an engineered IL18 heterodimer fc fusion protein with improved stability, reduced potency, and insensitivity to IL18BP (Figure 4). In a preclinical study, XmAb143 (IL18-Fc) promoted NK and T lymphocyte expansion and IFN-γ secretion in huPBMC-engrafted NSG mice (GvHD model) and demonstrated tumor growth inhibition, T-cell proliferation and activation in CD34+ humanized mice (160).
IL-10 is a soluble homodimer cytokine that is generally considered an immunosuppressive mediator due to its prominent anti-inflammatory effects, which ensure host protection from excessive responses to pathogens and microorganisms (161, 162). Genetic ablation of Il10 demonstrated the critical role of Il10 in regulating inflammation, as IL-10-deficient mice spontaneously developed colitis (163). IL-10 can be produced by most T cells (including Treg cells), DCs, macrophages, epithelial cells and tumor cells (164). Mechanistically, IL-10 exerts its effects after binding to a surface IL-10 receptor (IL-10R), which is a tetramer composed of two α subunits (IL-10Rα) and two β subunits (IL-10Rβ); the α subunit is expressed on hematopoietic cells, while the β subunit is expressed ubiquitously (165). IL-10 has a strong affinity for IL-10Rα but a substantially weaker affinity for IL-10Rβ (166). IL-10 induces the dimerization of IL-10Rα and IL-10Rβ, resulting in the phosphorylation and activation of the JAK1/TYK2/STAT3/STAT1 signaling pathway and the induction of IL-10-related gene expression, including c-myc, bcl-2, and bcl-xL (167) (Figure 5A).
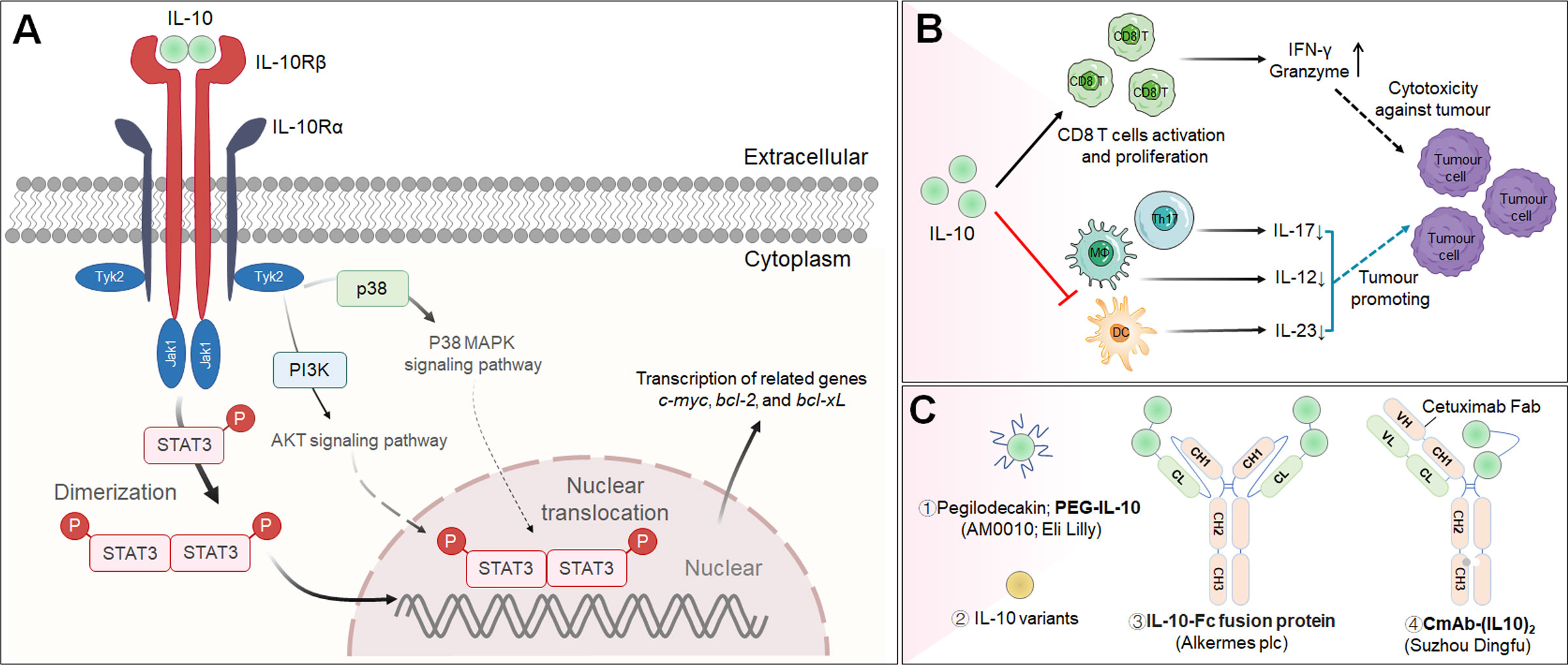
Figure 5 The cascade of the IL-10 signaling pathway and its downstream effector molecules and representative therapeutic candidates. (A) IL-10 signaling pathway and downstream cellular effects. (B) Immunopromoting and immunosuppresive mechanisms of IL-10 in cancer immunotherapy. (C) Representative therapeutic candidates of engineered IL-10.
Recently, increasing evidence has proven that IL-10 induces antitumor activity increasing CD8+ T lymphocyte activation in immunologically “cold” cancers (168, 169). For example, Mumm et al. suggested that IL-10 participates in effective antitumor immune surveillance through several basic mechanisms, including by increasing the count of intratumoral tumor-specific cytotoxic CD8+ T lymphocytes, promoting the secretion of the cytokine IFN-γ from Th1 cells and granzyme from CD8+ T lymphocytes, improving the activity antigen presentation machinery and thus inhibiting tumor growth (Figure 5B) (170). Although the infusion of IL-10 has been identified as a potential strategy for cancer immunotherapy, its long-term therapeutic efficacy has been hampered partially because it is rapidly cleared from the circulatory system.
Pegilodecakin (PEGylated IL-10; PEG-IL-10; AM0010) is an IL-10-based antineoplastic agent that stimulates systemic immune responses and enhances intratumoral CD8+ T lymphocyte invigoration in cancer patients, according to early preclinical studies (Figure 5C) (171). Subsequently, a Phase I clinical study elucidated that pegilodecakin exhibits an acceptable toxicity profile and shows promising antitumor effects in the treatment of participants with advanced solid tumors (NCT02009449) (172). Despite these encouraging preclinical and Phase I clinical results, pegilodecakin in combination with FOLFOX chemotherapy (folinic acid, 5-fluorouracil and oxaliplatin) for metastatic pancreatic cancer failed to prolong OS in a Phase III clinical study (NCT02923921) (173). In addition, two Phase II clinical trials of pegilodecakin combined with immune checkpoint monoclonal antibodies (pembrolizumab or nivolumab) for metastatic NSCLC failed to meet primary endpoint criteria (NCT03382899 and NCT03382912) (174). Because the rationale for using pegilodecakin is based on strong scientific evidence, the reasons for its clinical failure require more research, including studies on the dose, activity, and concentration that is trafficked to tumors. Indeed, an early preclinical study revealed that untargeted IL-10 treatment poses substantial risks for generating toxicity by inducing CD8+ T lymphocyte infiltration in healthy tissue, possibly due to off-tumor delivery of PEG-IL-10 (170).
Interestingly, Gorby et al. developed multiple IL-10 variants with different affinities for IL-10Rβ that can recruit IL-10Rβ more efficiently than the wild type to activate cellular surface signaling complexes and trigger STAT1 and STAT3 activation at a greater rate in monocytes and CD8+ T lymphocytes, potentially opening up new avenues for IL-10-based antitumor immunotherapy (Figure 5C). Furthermore, this study shed light on how IL-10R complex stability fine-tunes IL-10 biology and suggested new possibilities for reviving failed IL-10 therapies (175).
T-cell exhaustion is a crucial obstacle to effective cancer therapy. Guo et al. engineered an IL-10-Fc fusion protein with a longer half-life than previous IL-10-based drugs that directly and potently increased the proliferation and effector function of terminally exhausted tumor-infiltrating CD8+ T cells by enhancing oxidative phosphorylation, a process that was triggered independent of exhausted progenitor T cells (Figure 5C). When combined with adoptive T-cell transfer immunotherapy, IL-10-Fc was discovered to be a safe and highly effective metabolic intervention to eradicate well-established solid tumors and provide long-lasting treatment for most treated mice. These results testified that metabolic reprogramming through activation of mitochondrial pyruvate carrier-dependent oxidative phosphorylation can revive terminally exhausted T lymphocytes and improve the responsiveness to cancer treatment (176).
More recently, Professor Fu and team developed a cetuximab-based IL-10 immunocytokine (CmAb-(IL10)2) for EGFR-targeted delivery of IL-10 to tumor locations in conjunction with prolonged PKs and reduced toxicity via IL-10 regulation of IFN-γ production in DCs and enhanced CD8+ T lymphocyte-dependent antitumor responsiveness (Figure 5C). The fusion protein showed great effectiveness when administered alone or combined with anti-PD-L1/CTLA-4 antibody. Structurally, CmAb-(IL10)2 is a bispecific Fc fusion protein that links the Fab fragment of cetuximab and an IL-10 dimer, respectively (177). At present, the National Medical Products Administration (NMPA) of China accepted Suzhou Dingfu Target Biotechnology’s clinical application of an anti-EGFR antibody/IL-10 fusion protein in 2020.
Interferons (IFNs) constitute a vast family of α-helical cytokines that were first discovered because of their powerful antiviral properties. IFNs are now acknowledged for their capacity to have a wide range of cellular effects, from direct cytotoxicity to immunological modulation. According to an IFN sequence identity and engagement with distinct cell surface receptors, three major types of IFNs have been classified: Type-I IFNs (IFN-α, IFN-β, IFN-ϵ, IFN-κ and IFN-ω), Type-II IFN (IFN-γ), and Type-III IFN (IFN-λ), and their receptors are IFNαR1/IFNαR2, IFNγR1/IFNγR2, IFNλR1/IL1-Rβ, respectively (178). IFNs interact with their cognate receptors to initiate signal transduction, mainly through the JAK/STAT pathway, followed by the phosphorylation and nuclear translocation of STAT1 and STAT2, and to activate a large panel of interferon-stimulated genes (ISGs) that regulate innate and adaptive immune responses as well as antiviral and antitumor immunity (179) (Figure 6A).
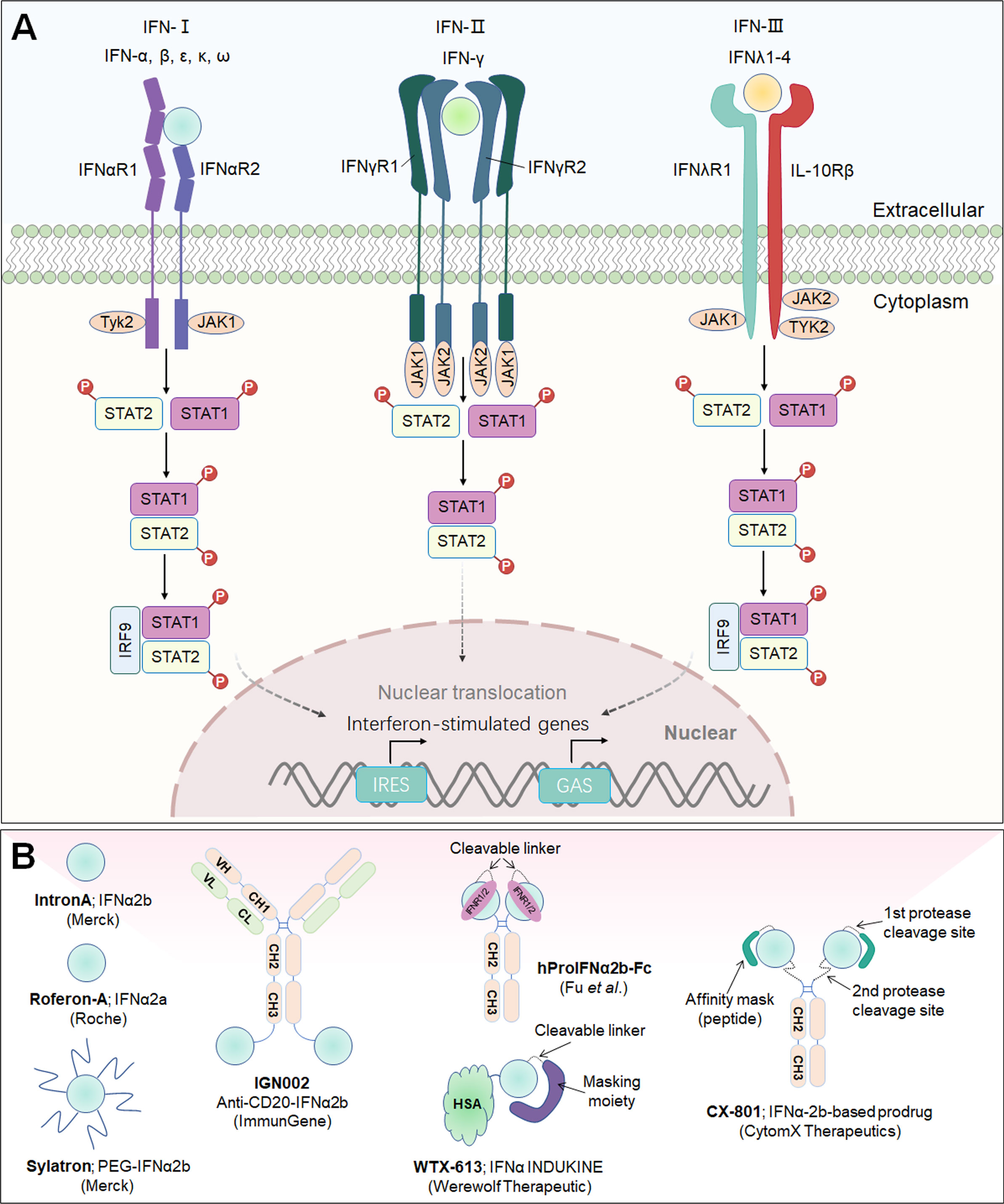
Figure 6 Interferon signaling pathway and downstream effector molecules and representative therapeutic candidates. (A) Type-I, type-II and type-III interferons, their receptors and signaling pathways. Engagement of IFNs with their corresponding receptors triggers signal transduction, mainly through the JAK/STAT pathway, followed by the phosphorylation and nuclear translocation of STAT1 and STAT2 and the activation of a large panel of ISGs. (B) Representative therapeutic candidates of engineered IFNs.
Interferons have been approved anticancer therapies, but their use has been limited by narrow therapeutic windows. Several IFNα2 alleles have been described. The best characterized are IFNα2a and IFNα2b, both of which have been marketed for clinical use as Roferon A and Intron A, respectively. Intron-A® (recombinant interferon alfa-2b from Merck) was the first agent approved for adjuvant therapy for melanoma patients with a high risk for recurrence after surgical resection (Figure 6B) (180). Roferon-A® (recombinant interferon alfa-2a from Roche) is authorized for the treatment of chronic hepatitis C and hairy cell leukemia in patients aged 18 years and older (Figure 6B) (181).
Sylatron™ (an agent launched by the Schering Corporation, a subsidiary of Merck & Co., Inc.) is a covalent coupling of recombinant IFNα-2b with monomethoxy PEG that is approved for the adjuvant therapy of melanoma patients with microscopic or gross nodal involvement within 84 days after surgical resection, including complete lymph node dissection (Figure 6B) (182).
IGN002 is a novel recombinant immunocytokine composed of an anti-CD20 antibody (rituximab) fused to human IFNα-2b via a peptide linker developed by ImmunGene (Figure 6B). A Phase I study of IGN002 is currently underway to evaluate its efficacy in patients with refractory NHL (NCT02519270) (183).
CX-801, designed by CytomX Therapeutics, is a wholly owned IFNα-2b-based prodrug based on Probody™ technology (Figure 6B). According to reports of preclinical results, CX-801 revealed a broad therapeutic scope with an improved tolerability profile when compared to unmasked IFN without compromised antitumor effects (184, 185). CX-801 has a broad range of potential applications in both traditionally immuno-oncology-sensitive (hot) and immuno-oncology-insensitive (cold) tumors. An IND submission is planned for 2023.
WTX-613 (licensed by Jazz Pharmaceuticals) is a systemically administered, selectively activated IFNα molecule based on INDUKINE™ approaches and is being developed to lessen the serious toxic side effects associated with IFNα monotherapy while maximizing clinical benefit when used as a single agent or in combination with checkpoint antibodies in patients with refractory and/or immunologically unsensitive tumors (Figure 6B) (186). This drug candidate was designed to induce an efficient blockade of IFNα/IFNR engagement in the circulatory system and nontumor tissues, extending the half-life for optimal tumor exposure and proprietary tumor-selective protease activation. In experimental animal models, WTX-613 exhibited potent antitumor efficacy mediated through the induction of a type I interferon immune response with favorable PKs and tolerability profiles (186, 187).
Recently, Professor Yang-Xin Fu and team engineered a receptor-masked IFNα-2b prodrug (hProIFNα2b-Fc; ProIFN) in which components are connected by a linker that can be cleaved by tumor-related proteases (Figure 6B). hProIFNα2b-Fc showed an extended serum half-life, reduced systemic toxicity, and a long-term tumor-targeting ability compared to the unmasked form. Strikingly, ProIFN-challenged mice exhibited enhanced DC cross-priming and significantly boosted CD8+ T lymphocyte infiltration and effector function within the TME. ProIFN also demonstrated synergistic effects with checkpoint blockade in established tumors, as well as radiotherapy efficacy in both primary and metastatic tumors, along with superior long-term PKs with minimal toxicity in nonhuman primates (188).
Notably, the abundant expression of the IFN-γ receptor on almost all tissues limits the use of IFN-γ as a therapeutic approach. While current evidence has clarified that it is possible to leverage the immunostimulatory ability of IFN-γ, early clinical data showed inconsistent outcomes with this cytokine, which could be partially attributed to immune checkpoint receptor upregulation (i.e., PD-L1) antagonizing the immunostimulatory effects. As a consequence, IFN-γ is not approved by the FDA for the treatment of cancers thus far, although it remains a promising candidate in the modern era of immunotherapy, in which the use of checkpoint blockades is increasing.
Tumor necrosis factor alpha (TNF-α; also called TNF or TNFSF2) is a homotrimeric proinflammatory cytokine in the TNF superfamily and a multifunctional molecule involved in regulating important biological processes, including the induction of apoptotic cell death and inflammatory immune responses and the inhibition of tumorigenesis and viral replication (189). It is predominantly synthesized by activated monocytes/macrophages as a 26 kDa transmembrane protein that can be enzymatically cleaved to produce a 17 kDa soluble TNF form (189). Notably, TNF-α is unique in that it activates biological effects at two distinct receptors—TNF receptor type 1 (TNFR1; CD120a; TNFR60) and TNF receptor type 2 (TNFR2; CD120b; TNFR80) (190). Recent evidence suggests that TNFR1 is commonly expressed on almost all cells, whereas TNFR2 expression is limited largely to Treg cells (191). Additionally, cross-linking of soluble TNF-α with TNFR1 primarily triggers proinflammatory pathways that activate NF-κB, MAPK and antiapoptotic response factors and subsequently produce other proinflammatory cytokines, such as IL-1, IL-6 and GM-CSF. Nevertheless, transmembrane TNF binding to TNFR2 typically activates different signaling pathways than those activated by TNFR1, initiating immune modulation and tissue regeneration (192) (Figure 7A).
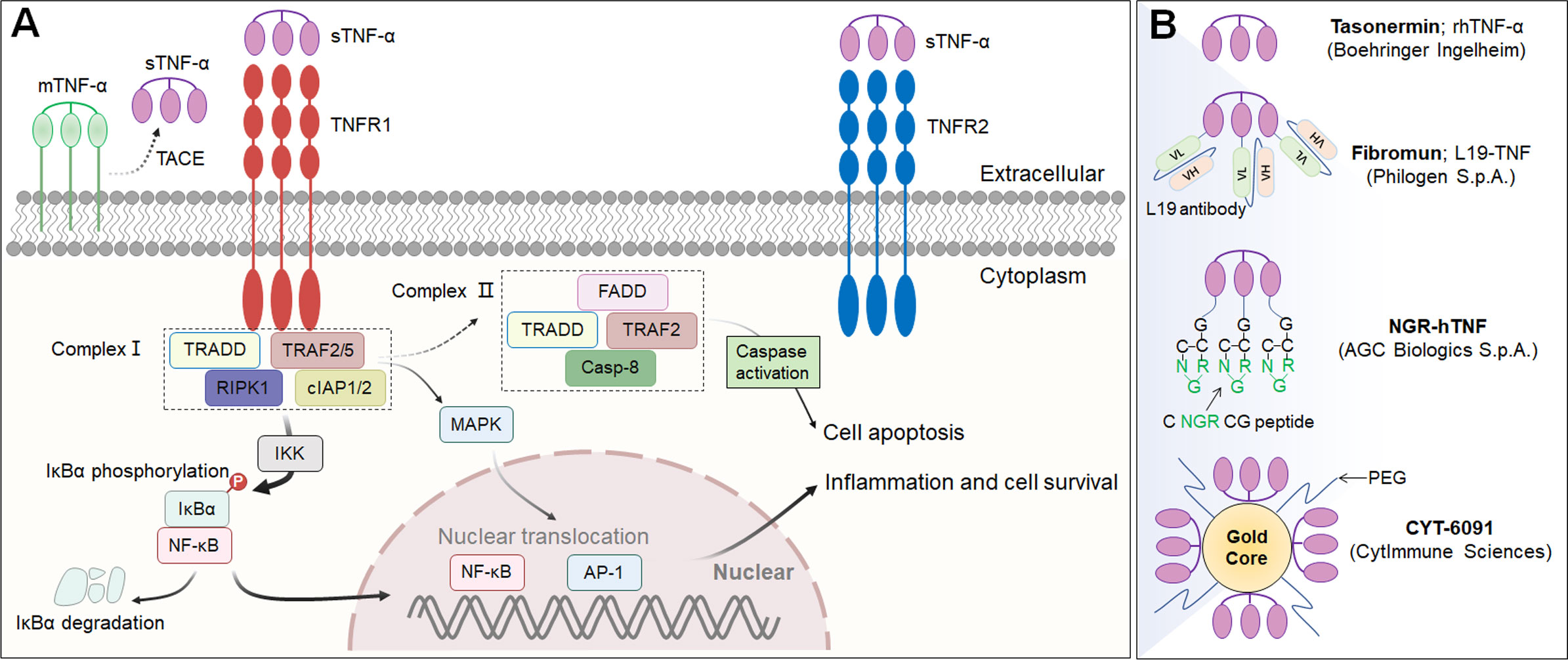
Figure 7 The cascade of the TNF-α signaling pathway and downstream effector molecules and representative therapeutic candidates. (A) TNF-α signaling pathway and downstream effector molecules. Cross-linking of soluble TNF-α with TNFR1 primarily triggers proinflammatory pathways that activate NF-κB, MAPK and antiapoptotic response factors, followed by the production of other proinflammatory cytokines. (B) Representative therapeutic candidates for engineered TNF-α. TACE: Tumor necrosis factor (TNF)-alpha converting enzyme; mTNF-α: transmembrane TNF-α; sTNF-α: soluble TNF-α.
TNF-α is considered to be an effective tumoricidal cytokine because of its capacity to directly induce apoptosis and stimulate the inflammatory response at tumor sites. More importantly, recombinant TNF-α (rTNF-α) can rapidly destroy tumor blood vessels, eliminate hypertension inside tumors, disrupt tumor tissue growth, and activate CD8+ T cells. However, systemic administration of a clinically relevant dose of rTNF-α, that is, a dose greater than 300 μg/m2 has been limited due to substantial toxic side effects (193). Hence, the early clinical applications of rTNF-α have been restricted to localized regional uses.
Tasonermin (Beromun®; developed by Boehringer Ingelheim), an agent approved for use by the European Medicine Agency (EMA) in 1999, is a human recombinant form of soluble TNF-α produced in Escherichia coli cell culture that can kill cancer cells directly, destroy the blood vessels that supply tumors with nutrients and oxygen, and activate the immune system to fight tumor vascular structure (Figure 7A). Tasonermin is available for the treatment of patients with soft tissue sarcoma in combination with melphalan and is administered via isolated limb perfusion (ILP) to minimize systemic exposure (194). The recommended dose is 3 mg in the arm or 4 mg in the leg administered over 90 minutes.
Recombinant human TNF-α (rhTNF-α) and thiolated PEG were concurrently bound to the surface of 27 nanometer colloidal gold particles to generate CYT-6091 (Aurimune™), a revolutionary tumor-targeting nanomedicine (Figure 7B) (195). Preclinical studies have shown that CYT-6091 was enriched in tumors 6 hours after intravenous injection due to enhanced permeability and retention (EPR) effects inside the tumor, and a negligible amount of CYT-6091 was taken up by the liver or spleen (196). In a Phase I clinical study, Cytimmune Sciences disclosed the ability of CYT-6091 to deliver 4-fold the MTD of native TNF-α with no dose limiting toxicities (NCT00356980; NCT00436410). Furthermore, a clinical trial agreement has been signed with the National Cancer Institute (NCI) for establishing a Phase II trial to assess CYT-6091 effectiveness in combination with nab-paclitaxel (Abraxane) in participants with late stage endocrine tumors of the pancreas and thyroid.
Concurrently, a number of TNF-α fusion proteins have been created to guide TNF-α towards tumor tissue by incorporating targeting domains that recognize tumor or tumor-related stromal cell markers. Fibromun (L19-TNF) is a fully human immunomodulatory agent consisting of a human anti-L19 antibody in the scFv format linked to the proinflammatory cytokine TNF-α, which yields a stable noncovalent trimetric L19-TNF immunocytokine (Figure 7B) (197). Based on encouraging preclinical data, L19-TNF is being studied in numerous Phase I/II clinical trials for the treatment of advanced or metastatic soft tissue sarcoma in combination with doxorubicin (NCT04650984; NCT04733183) and for the treatment of glioblastoma in combination with lomustine (NCT04573192) or temozolomide chemoradiotherapy (NCT04443010) (198). In addition, several Phase II studies on the intratumoral administration of L19-IL2 plus L19-TNF in patients with metastatic melanoma (NCT02076633) or nonmelanoma skin cancer (NCT04362722; NCT05329792) are ongoing (199).
Another TNF-α fusion protein under clinical investigation is NGR-hTNF, in which homotrimeric TNF-α is fused to the C-terminus of a cyclic CNGRCG peptide, which targets a CD13 (aminopeptidase N) isoform expressed on angiogenic blood vessels (Figure 7B) (200). Recent preclinical results suggested that CNGRC binding to CD13 not only determined the homing specificity of NGR-hTNF to tumor vessels but also reduced the activation of prosurvival pathways and increased the activation of caspases (200). NGR-hTNF is in an ongoing Phase III clinical study for the treatment of advanced malignant pleural mesothelioma (NCT01098266), but recent data have suggested that the study did not meet primary endpoints because patients whose disease progressed rapidly after first-line therapy had a poor prognosis (201). Additionally, NGR-hTNF is also being investigated alone or in combination with chemotherapy in five other randomized Phase II studies for the treatment of participants with ovarian cancer (NCT03804866; NCT01358071), CRC (NCT00483080), hepatocellular carcinoma (NCT00484211), small cell lung cancer (NCT00483509; NCT00994097), and metastatic adult soft tissue sarcoma (NCT00484341).
Although their bioactivity is increased upon targeting, most TNF-α-based immunocytokines induce a notable risk of adverse side effects because of the potential off-target effects of the active TNF-α effector moiety. Accordingly, to minimize the risk of off-target effects and improve the safety of highly bioactive and potentially toxic TNF-α fusion proteins, researchers have sought to develop TNF-α-based prodrugs. For example, Klaus Pfizenmaier and team from the University of Stuttgart developed a TNF-α-based prodrug fusion protein composed of a FAP antibody scFv fragment specific to a target antigen, a TNF-α homotrimeric module, a protease-sensitive linker and a masking fragment from the TNFR1-driven extracellular domain (ECD) (202). To overcome the known heterogeneity of protease expression, the authors constructed two effective protease-sensitive linkers, matrix metalloproteinase-2 (MMP-2) and urokinase-type plasminogen activator (uPA), both of which exhibited effective proteolysis with the respective recombinant protease and led to greatly increased target antigen-independent TNF-α bioactivity (203).
Cancer immunotherapy, represented by immune checkpoint monoclonal antibodies, provides favorable tools for reinvigorating effector immune cells, but the vast majority of cancer patients (more than 80%) do not benefit from these immunotherapies due to limited responsiveness and primary and acquired drug resistance. Strikingly, cytokines can overcome the related resistance mechanisms because of their potential to expand and reactivate effector T and NK cells and to enhance lymphocyte infiltration, in addition to their persistence in the TME (8). However, as a class of complex immune mediators, the use of cytokines as therapeutic agents faces huge challenges, including the difficulty of large-scale manufacturing, which requires increased understanding of cytokine biology and the use of modern biotechnology to develop their antitumor activity while minimizing toxicity. Large industrial companies, risk-taking startups, and academic laboratories have all extensively invested in basic research aimed at engineering and manufacturing cytokine drugs, which has led to a significant resurgence in ongoing clinical trials, and the results are eagerly anticipated.
For future cytokine-based drug development, multiple crucial challenges should be considered: (i) overcoming the naturally quick clearance and poor exposure of endogenous cytokines, (ii) concentrating the delivery of cytokines to diseased tissues or particular cell types to prevent systemic toxicity, and (iii) developing combination regimens to achieve synergistic effects with immune checkpoint blockade antibodies, CAR-T therapy, or gene therapy. To date, many strategies have been employed to meet clinical needs, including half-life extension, antigen targeting, conditional activation, potency manipulation and local delivery. These emerging strategies allow fine-tuning of cytokine signaling pathways and a tailored cellular response. Accordingly, based on current clinical data and progress of immunocytokine-based therapeutics, the IL-15 agonist N-803 (Anktiva™) is currently the only one that submitted a BLA application to the FDA for NMIBC and is more likely to be successful in this indication or beyond in terms of clinical efficacy and might also work in other formats e.g., with antibody conjugation. Therefore, IL-15-related immunocytokine therapeutics would be an important direction for future industrial development suitable for clinical use, although it now faces some challenges related to chemical, manufacturing and control (CMC) development and would more likely to be addressed in the near future. The key to using cytokines as therapeutic agents is to improve therapeutic windows either by reducing potency applying mutational design by protein engineering or by tissue/tumor targeted delivery or the combination of the two strategies. The other direction might be tissue/tumor-specific activation of cytokines by applying prodrug strategies that render tumor-specific cleavage of masking peptide, scFv, or natural binding receptors conjugated to the binding regions of immunocytokines. Thus, potency-reduced cytokines conjugated with antibodies for tumor targeting or cytokine-based prodrug strategies may more likely lead to the development of new immunotherapeutics suitable for future clinical use. Additionally, immunogenicity is an intractable problem for all mutant and conjugated nonendogenous proteins, and methods to reduce immunogenicity also need to be considered in future developments.
Most importantly, a deeper understanding of the biology and intricate cellular processes of cytokines in the TME, determined using approaches such as cryo-electron microscope techniques, single-cell RNA sequencing, transcriptome analysis, and preclinical mechanistic studies, will help shed light on the currently unknown biological activity of cytokines. Concurrently, molecular designs based on contemporary protein engineering technologies to fine-tune cytokine biology, deconvolute the pleiotropism of cytokines, enhance cytokine drug-like properties, and drive selective proinflammatory effects will further facilitate the clinical translation of cytokine-based therapeutics to enhance cancer immunotherapy.
YF designed and wrote the manuscript. XZ gave the original idea and design. XZ and RT confirmed the final release.
Authors YF, RT and XZ were employed by the company Jiangsu Simcere Pharmaceutical Co., Ltd. Author RT was employed by the company Simcere Zaiming Pharmaceutical Co., Ltd.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lin JX, Leonard WJ. Fine-tuning cytokine signals. Annu Rev Immunol (2019) 37:295–324. doi: 10.1146/annurev-immunol-042718-041447
2. Briukhovetska D, Dorr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer (2021) 21(8):481–99. doi: 10.1038/s41568-021-00363-z
3. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol (2022) 19(4):237–53. doi: 10.1038/s41571-021-00588-9
4. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer (2004) 4(1):11–22. doi: 10.1038/nrc1252
5. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol (2018) 10(12):a028472. doi: 10.1101/cshperspect.a028472
6. Wang YS, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Adv Drug Deliv Rev (2002) 54(4):547–70. doi: 10.1016/S0169-409X(02)00027-3
7. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
8. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer (2019) 120(1):6–15. doi: 10.1038/s41416-018-0328-y
9. Zidek Z, Anzenbacher P, Kmonickova E. Current status and challenges of cytokine pharmacology. Br J Pharmacol (2009) 157(3):342–61. doi: 10.1111/j.1476-5381.2009.00206.x
10. Bempeg failure unlikely to affect other IL2 drugs. Cancer Discov (2022) 12(7):1604–5. doi: 10.1158/2159-8290.CD-NB2022-0036
11. Mullard A. Engineered IL-2 cytokine takes pivotal immuno-oncology blow. Nat Rev Drug Discov (2022) 21(5):327. doi: 10.1038/d41573-022-00069-3
12. Pires IS, Hammond PT, Irvine DJ. Engineering strategies for immunomodulatory cytokine therapies - challenges and clinical progress. Adv Ther (Weinh) (2021) 4(8):2100035. doi: 10.1002/adtp.202100035
13. Chamie K, Chang SS, Gonzalgo M, Kramolowsky EV, Sexton WJ, Bhar P, et al. Final clinical results of pivotal trial of IL-15RαFc superagonist n-803 with BCG in BCG-unresponsive CIS and papillary nonmuscle-invasive bladder cancer (NMIBC). J Clin Oncol (2022) 40(16_suppl):4508–8. doi: 10.1200/JCO.2022.40.16_suppl.4508
14. Chang SS, Chamie K, Gonzalgo ML, Kramolowsky EV, Sexton WJ, Reddy SK, et al. Positive efficacy and safety phase 3 results in both CIS and papillary cohorts BCG-unresponsive nonmuscle invasive bladder cancer (NMIBC) after IL-15RαFc superagonist n-803 (Anktiva) and BCG infusion. J Clin Oncol (2022) 40(6_suppl):431–1. doi: 10.1200/JCO.2022.40.6_suppl.431
15. Zhang P, Ding Y. Novel therapeutic strategies for BCG unresponsive non-muscle invasive bladder cancer. Ann Urol Oncol (2022) 5(2):59–67. doi: 10.32948/auo.2022.11.27
16. McFarlane A, Pohler E, Moraga I. Molecular and cellular factors determining the functional pleiotropy of cytokines. FEBS J (2022). doi: 10.1111/febs.16420
17. Holder PG, Lim SA, Huang CS, Sharma P, Dagdas YS, Bulutoglu B, et al. Engineering interferons and interleukins for cancer immunotherapy. Adv Drug Deliv Rev (2022) 182:114112. doi: 10.1016/j.addr.2022.114112
18. Zheng X, Wu Y, Bi J, Huang Y, Cheng Y, Li Y, et al. The use of supercytokines, immunocytokines, engager cytokines, and other synthetic cytokines in immunotherapy. Cell Mol Immunol (2022) 19(2):192–209. doi: 10.1038/s41423-021-00786-6
19. Poljak RJ. Production and structure of diabodies. Structure (1994) 2(12):1121–3. doi: 10.1016/S0969-2126(94)00113-8
20. Kuo TT, Aveson VG. Neonatal fc receptor and IgG-based therapeutics. MAbs (2011) 3(5):422–30. doi: 10.4161/mabs.3.5.16983
21. Sockolosky JT, Szoka FC. The neonatal fc receptor, FcRn, as a target for drug delivery and therapy. Adv Drug Deliv Rev (2015) 91:109–24. doi: 10.1016/j.addr.2015.02.005
22. Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med (2012) 4(10):1015–28. doi: 10.1002/emmm.201201379
23. Bournazos S, Gupta A, Ravetch JV. The role of IgG fc receptors in antibody-dependent enhancement. Nat Rev Immunol (2020) 20(10):633–43. doi: 10.1038/s41577-020-00410-0
24. Eliason JF. Pegylated cytokines: potential application in immunotherapy of cancer. BioDrugs (2001) 15(11):705–11. doi: 10.2165/00063030-200115110-00001
25. Dozier JK, Distefano MD. Site-specific PEGylation of therapeutic proteins. Int J Mol Sci (2015) 16(10):25831–64. doi: 10.3390/ijms161025831
26. Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol (2016) 40:96–102. doi: 10.1016/j.coi.2016.03.006
27. Lucchi R, Bentanachs J, Oller-Salvia B. The masking game: design of activatable antibodies and mimetics for selective therapeutics and cell control. ACS Cent Sci (2021) 7(5):724–38. doi: 10.1021/acscentsci.0c01448
28. Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer (2007) 7(10):800–8. doi: 10.1038/nrc2228
29. Bleuez C, Koch WF, Urbach C, Hollfelder F, Jermutus L. Exploiting protease activation for therapy. Drug Discov Today (2022) 27(6):1743–54. doi: 10.1016/j.drudis.2022.03.011
30. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
31. Cheng G, Yu A, Malek TR. T-Cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev (2011) 241(1):63–76. doi: 10.1111/j.1600-065X.2011.01004.x
32. Hernandez R, Poder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol (2022) 22(10):614–28. doi: 10.1038/s41577-022-00680-w
33. Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol (2004) 4(9):665–74. doi: 10.1038/nri1435
34. Noble S, Goa KL. Aldesleukin (recombinant interleukin-2). BioDrugs (1997) 7(5):394–422. doi: 10.2165/00063030-199707050-00007
35. Matthews L, Chapman S, Ramchandani MS, Lane HC, Davey RT Jr., Sereti I. BAY 50-4798, a novel, high-affinity receptor-specific recombinant interleukin-2 analog, induces dose-dependent increases in CD25 expression and proliferation among unstimulated, human peripheral blood mononuclear cells in vitro. Clin Immunol (2004) 113(3):248–55. doi: 10.1016/j.clim.2004.07.009
36. Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science (2005) 310(5751):1159–63. doi: 10.1126/science.1117893
37. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature (2012) 484(7395):529–33. doi: 10.1038/nature10975
38. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res (2016) 22(3):680–90. doi: 10.1158/1078-0432.CCR-15-1631
39. Doberstein SK. Bempegaldesleukin (NKTR-214): a CD-122-biased IL-2 receptor agonist for cancer immunotherapy. Expert Opin Biol Ther (2019) 19(12):1223–8. doi: 10.1080/14712598.2019.1685489
40. Khushalani NI, Diab A, Ascierto PA, Larkin J, Sandhu S, Sznol M, et al. Bempegaldesleukin plus nivolumab in untreated, unresectable or metastatic melanoma: phase III PIVOT IO 001 study design. Future Oncol (2020) 16(28):2165–75. doi: 10.2217/fon-2020-0351
41. Eggermont AM, Ascierto PA, Khushalani NI, Schadendorf D, Boland G, Weber J, et al. PIVOT-12: a phase III study of adjuvant bempegaldesleukin plus nivolumab in resected stage III/IV melanoma at high risk for recurrence. Future Oncol (2022) 18(8):903–13. doi: 10.2217/fon-2021-1286
42. Ptacin JL, Caffaro CE, Ma L, San Jose Gall KM, Aerni HR, Acuff NV, et al. An engineered IL-2 reprogrammed for anti-tumor therapy using a semi-synthetic organism. Nat Commun (2021) 12(1):4785. doi: 10.1038/s41467-021-24987-9
43. Janku F, Abdul-Karim R, Azad A, Bendell J, Falchook G, Gan HK, et al. Abstract LB041: THOR-707 (SAR444245), a novel not-alpha IL-2 as monotherapy and in combination with pembrolizumab in advanced/metastatic solid tumors: interim results from HAMMER, an open-label, multicenter phase 1/2 study. Cancer Res (2021) 81(13_Supplement):LB041–1. doi: 10.1158/1538-7445.AM2021-LB041
44. Falchook G, Gan H, Fu S, McKean M, Azad A, Sommerhalder D, et al. 481 phase 1/2 study of THOR-707 (SAR444245), a pegylated recombinant non-alpha IL-2, as monotherapy and in combination with pembrolizumab or cetuximab in patients (pts) with advanced solid tumors. J ImmunoTher Cancer (2021) 9(Suppl 2):A511–1. doi: 10.1136/jitc-2021-SITC2021.481
45. Rafei M, To M, Merchant F, Merchant N. In vitro and in vivo characterization of MDNA11: a long-acting “beta-only” IL-2 superkine in syngeneic mice tumor models and nonhuman primates. J Clin Oncol (2020) 38(15_suppl):3036–6. doi: 10.1200/JCO.2020.38.15_suppl.3036
46. Merchant R, Galligan C, Munegowda MA, Pearce LB, Lloyd P, Smith P, et al. Fine-tuned long-acting interleukin-2 superkine potentiates durable immune responses in mice and non-human primate. J Immunother Cancer (2022) 10(1):e003155. doi: 10.1136/jitc-2021-003155
47. Raeber ME, Sahin D, Boyman O. Interleukin-2-based therapies in cancer. Sci Transl Med (2022) 14(670):eabo5409. doi: 10.1126/scitranslmed.abo5409
48. Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J Immunother Cancer (2020) 8(1):e000673. doi: 10.1136/jitc-2020-000673
49. Lopes JE, Sun L, Flick HL, Murphy EA, Losey HC. Pharmacokinetics and pharmacodynamic effects of nemvaleukin Alfa, a selective agonist of the intermediate-affinity IL-2 receptor, in cynomolgus monkeys. J Pharmacol Exp Ther (2021) 379(2):203–10. doi: 10.1124/jpet.121.000612
50. Herzog T, Hays J, Moore K, Konstantinopoulos P, Gilbert L, Monk B, et al. TP025/1399 artistry-7: phase 3 multicenter study of nemvaleukin alfa plus pembrolizumab versus chemotherapy in patients with platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer. Int J Gynecol Cancer (2022) 32(Suppl 3):A235–5. doi: 10.1136/ijgc-2022-igcs.534
51. Amit I, Levin I, Wyant T, Levitin N, Barak R, Ben-Mayor M, et al. 704 the computationally designed human antibody, AU-007, mediates human immune activation by endogenous IL-2, while uniquely breaking the IL-2 auto-inhibitory loop and preventing treg expansion. J ImmunoTher Cancer (2021) 9(Suppl 2):A732–4. doi: 10.1136/jitc-2021-SITC2021.704
52. Vasselli JR, Frentzas S, Weickhardt AJ, de Souza PL, Tang J, Wyant T, et al. (2022). Trial in progress: a phase 1-2, first-in-Human, open label, dose escalation and expansion study of AU-007, a monoclonal antibody that binds to IL-2 and inhibits IL-2Rα binding, in patients with advanced solid tumors. J Clin Oncol (2022) 40(16_suppl):TPS2671–TPS2671. doi: 10.1200/JCO.2022.40.16_suppl.TPS2671
53. Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature (2019) 565(7738):186–91. doi: 10.1038/s41586-018-0830-7
54. Naing A, Callahan M, Costello BA, Curti B, Hall E, Hansen A, et al. Abstract CT250: a first-in-human phase 1 study of NL-201 in patients with relapsed or refractory cancer. Cancer Res (2022) 82(12_Supplement):CT250–0. doi: 10.1158/1538-7445.AM2022-CT250
55. Holden SA, Lan Y, Pardo AM, Wesolowski JS, Gillies SD. Augmentation of antitumor activity of an antibody-interleukin 2 immunocytokine with chemotherapeutic agents. Clin Cancer Res (2001) 7(9):2862–9.
56. Connor JP, Cristea MC, Lewis NL, Lewis LD, Komarnitsky PB, Mattiacci MR, et al. A phase 1b study of humanized KS-interleukin-2 (huKS-IL2) immunocytokine with cyclophosphamide in patients with EpCAM-positive advanced solid tumors. BMC Cancer (2013) 13:20. doi: 10.1186/1471-2407-13-20
57. Yang RK, Kuznetsov IB, Ranheim EA, Wei JS, Sindiri S, Gryder BE, et al. Outcome-related signatures identified by whole transcriptome sequencing of resectable stage III/IV melanoma evaluated after starting Hu14.18-IL2. Clin Cancer Res (2020) 26(13):3296–306. doi: 10.1158/1078-0432.CCR-19-3294
58. Shusterman S, London WB, Hank JA, Parisi MT, Shulkin BL, Servaes S.-E.-N., et al. A feasibility and phase II study of the hu14.18-IL2 immunocytokine in combination with GM-CSF and isotretinoin in patients with recurrent or refractory neuroblastoma: a children’s oncology group study. J Clin Oncol (2015) 33(15_suppl):10017–7. doi: 10.1200/jco.2015.33.15_suppl.10017
59. Osenga K, Hank J, Albertini M, Gan J, Sternberg A, Seeger R, et al. A phase I trial of immunocytokine HU14.18-IL2 in children with recurrent or refractory neuroblastoma and other GD2 positive malignancies: a study of the children's oncology group. J Immunother (2004) 27(6):S55–6. doi: 10.1158/1078-0432.CCR-05-2000
60. Lansigan F, Nakamura R, Quick DP, Vlock D, Raubitschek A, Gillies SD, et al. DI-Leu16-IL2, an anti-CD20-Interleukin-2 immunocytokine, is safe and active in patients with relapsed and refractory b-cell lymphoma: a report of maximum tolerated dose, optimal biologic dose, and recommended phase 2 dose. Blood (2016) 128(22):620–0. doi: 10.1182/blood.V128.22.620.620
61. Nicolini V, Waldhauer I, Freimoser-Grundschober A, Evers S, Saro J, Bacac M, et al. Abstract 2217: combining CEA-IL2v and FAP-IL2v immunocytokines with PD-L1 checkpoint blockade. Cancer Res (2016) 76(14_Supplement):2217–7. doi: 10.1158/1538-7445.AM2016-2217
62. Waldhauer I, Gonzalez-Nicolini V, Freimoser-Grundschober A, Nayak TK, Fahrni L, Hosse RJ, et al. Simlukafusp alfa (FAP-IL2v) immunocytokine is a versatile combination partner for cancer immunotherapy. MAbs (2021) 13(1):1913791. doi: 10.1080/19420862.2021.1913791
63. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology (2017) 6(3):e1277306. doi: 10.1080/2162402X.2016.1277306
64. Schellens JHM, Tabernero J, Lassen UN, Melero I, Homicsko K, Argilés G, et al. CEA-targeted engineered IL2: clinical confirmation of tumor targeting and evidence of intra-tumoral immune activation. J Clin Oncol (2015) 33(15_suppl):3016–6. doi: 10.1200/jco.2015.33.15_suppl.3016
65. Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, et al. Intralesional treatment of stage III metastatic melanoma patients with L19–IL2 results in sustained clinical and systemic immunologic responses. Cancer Immunol Res (2014) 2(7):668–78. doi: 10.1158/2326-6066.CIR-13-0206
66. Carnemolla B, Borsi L, Balza E, Castellani P, Meazza R, Berndt A, et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood (2002) 99(5):1659–65. doi: 10.1182/blood.V99.5.1659
67. Marlind J, Kaspar M, Trachsel E, Sommavilla R, Hindle S, Bacci C, et al. Antibody-mediated delivery of interleukin-2 to the stroma of breast cancer strongly enhances the potency of chemotherapy. Clin Cancer Res (2008) 14(20):6515–24. doi: 10.1158/1078-0432.CCR-07-5041
68. Gillies SD, Lan Y, Hettmann T, Brunkhorst B, Sun Y, Mueller SO, et al. A low-toxicity IL-2-based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clin Cancer Res (2011) 17(11):3673–85. doi: 10.1158/1078-0432.CCR-10-2921
69. Laurent J, Touvrey C, Gillessen S, Joffraud M, Vicari M, Bertrand C, et al. T-Cell activation by treatment of cancer patients with EMD 521873 (Selectikine), an IL-2/anti-DNA fusion protein. J Transl Med (2013) 11:5. doi: 10.1186/1479-5876-11-5
70. Gillessen S, Gnad-Vogt US, Gallerani E, Beck J, Sessa C, Omlin A, et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur J Cancer (2013) 49(1):35–44. doi: 10.1016/j.ejca.2012.07.015
71. Mullard A. Restoring IL-2 to its cancer immunotherapy glory. Nat Rev Drug Discov (2021) 20(3):163–5. doi: 10.1038/d41573-021-00034-6
72. Van Krinks C, Malcolm T, Lecointre M, Brown R, Ali H, Craig H, et al. KY1043, a novel PD-L1 IL-2 immunocytokine directed towards CD25, delivers potent anti-tumour activity in vitro and in vivo. J Immunother Cancer (2019) 4.
73. Nirschl CJ, Brodkin HR, Hicklin DJ, Ismail N, Morris K, Seidel-Dugan C, et al. Discovery of a conditionally activated IL-2 that promotes antitumor immunity and induces tumor regression. Cancer Immunol Res (2022) 10(5):581–96. doi: 10.1158/2326-6066.CIR-21-0831
74. O'Neil J, Guzman W, Yerov O, Johnson P, Fanny M, Greene J, et al. Tumor-selective activity of XTX202, a protein-engineered IL-2, in mice without peripheral toxicities in nonhuman primates. J Clin Oncol (2021) 39(15_suppl):2563–3. doi: 10.1200/JCO.2021.39.15_suppl.2563
75. McKean M, Powderly JD, Duncan M, Norman T, Patel E, O'Neil J, et al. A first-in-human, multicenter, phase 1/2, open-label study of XTX202, a masked and tumor-selective recombinant human interleukin-2 (IL-2) protein, in patients with advanced solid tumors. J Clin Oncol (2022) 40(16_suppl):TPS2697–TPS2697. doi: 10.1200/JCO.2022.40.16_suppl.TPS2671
76. Nash AM, Jarvis MI, Aghlara-Fotovat S, Mukherjee S, Hernandez A, Hecht AD, et al. Clinically translatable cytokine delivery platform for eradication of intraperitoneal tumors. Sci Adv (2022) 8(9):eabm1032. doi: 10.1126/sciadv.abm1032
77. Waldmann TA, Miljkovic MD, Conlon KC. Interleukin-15 (dys)regulation of lymphoid homeostasis: implications for therapy of autoimmunity and cancer. J Exp Med (2020) 217(1):e20191062. doi: 10.1084/jem.20191062
78. Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol (2012) 13(12):1187–95. doi: 10.1038/ni.2449
79. Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci (2012) 33(1):35–41. doi: 10.1016/j.tips.2011.09.004
80. Chirifu M, Hayashi C, Nakamura T, Toma S, Shuto T, Kai H, et al. Crystal structure of the IL-15-IL-15Ralpha complex, a cytokine-receptor unit presented in trans. Nat Immunol (2007) 8(9):1001–7. doi: 10.1038/ni1492
81. Yang Y, Lundqvist A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers (Basel) (2020) 12(12):3586. doi: 10.3390/cancers12123586
82. Wei X, Orchardson M, Gracie JA, Leung BP, Gao B, Guan H, et al. The sushi domain of soluble IL-15 receptor alpha is essential for binding IL-15 and inhibiting inflammatory and allogenic responses in vitro and in vivo. J Immunol (2001) 167(1):277–82. doi: 10.4049/jimmunol.167.1.277
83. Hangasky JA, Waldmann TA, Santi DV. Interleukin 15 pharmacokinetics and consumption by a dynamic cytokine sink. Front Immunol (2020) 11:1813. doi: 10.3389/fimmu.2020.01813
84. Knudson KM, Hodge JW, Schlom J, Gameiro SR. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opin Biol Ther (2020) 20(7):705–9. doi: 10.1080/14712598.2020.1738379
85. Miyazaki T, Maiti M, Hennessy M, Chang T, Kuo P, Addepalli M, et al. NKTR-255, a novel polymer-conjugated rhIL-15 with potent antitumor efficacy. J Immunother Cancer (2021) 9(5):e002024. doi: 10.1136/jitc-2020-002024
86. Robinson TO, Hegde SM, Chang A, Gangadharan A, Rivas S, Madakamutil L, et al. NKTR-255 is a polymer-conjugated IL-15 with unique mechanisms of action on T and natural killer cells. J Clin Invest (2021) 131(19):e144365. doi: 10.1172/JCI144365
87. Fernandez RA, Mayoral JE, Pierre-Louis L, Yao Y, Xu Y, Mu S, et al. Improving NK cell function in multiple myeloma with NKTR-255, a novel polymer-conjugated human IL-15. Blood Adv (2023) 7(1):9–19. doi: 10.1182/bloodadvances.2022007985
88. Shah N, Perales MA, Turtle CJ, Cairo MS, Cowan AJ, Saeed H, et al. Phase I study protocol: NKTR-255 as monotherapy or combined with daratumumab or rituximab in hematologic malignancies. Future Oncol (2021) 17(27):3549–60. doi: 10.2217/fon-2021-0576
89. Altan M, Patnaik A, Barve M, Dunn L, Cobb P, Rosenberg A, et al. 957 NKTR-255+cetuximab in patients with solid tumors: interim safety and efficacy results from the phase 1b dose-escalation study. J ImmunoTher Cancer (2021) 9(Suppl 2):A1007–7. doi: 10.1136/jitc-2021-SITC2021.957
90. Antosova Z, Podzimkova N, Tomala J, Augustynkova K, Sajnerova K, Nedvedova E, et al. SOT101 induces NK cell cytotoxicity and potentiates antibody-dependent cell cytotoxicity and anti-tumor activity. Front Immunol (2022) 13:989895. doi: 10.3389/fimmu.2022.989895
91. Garralda E, Galvao V, Champiat S, LoRusso P, Grell P, Naing A, et al. 484 preliminary efficacy of the IL-15 superagonist SO-C101 in combination with pembrolizumab in patients with advanced/metastatic solid tumors. J ImmunoTher Cancer (2021) 9(Suppl 2):A514–4. doi: 10.1136/jitc-2021-SITC2021.484
92. Wang TT, Yang J, Zhang Y, Zhang M, Dubois S, Conlon KC, et al. IL-2 and IL-15 blockade by BNZ-1, an inhibitor of selective gamma-chain cytokines, decreases leukemic T-cell viability. Leukemia (2019) 33(5):1243–55. doi: 10.1038/s41375-018-0290-y
93. Frohna PA, Ratnayake A, Doerr N, Basheer A, Al-Mawsawi LQ, Kim WJ, et al. Results from a first-in-Human study of BNZ-1, a selective multicytokine inhibitor targeting members of the common gamma (gammac) family of cytokines. J Clin Pharmacol (2020) 60(2):264–73. doi: 10.1002/jcph.1522
94. Kurz E, Hirsch CA, Dalton T, Shadaloey SA, Khodadadi-Jamayran A, Miller G, et al. Exercise-induced engagement of the IL-15/IL-15Ralpha axis promotes anti-tumor immunity in pancreatic cancer. Cancer Cell (2022) 40(7):720–737 e5. doi: 10.1016/j.ccell.2022.05.006
95. Conlon K, Watson DC, Waldmann TA, Valentin A, Bergamaschi C, Felber BK, et al. Phase I study of single agent NIZ985, a recombinant heterodimeric IL-15 agonist, in adult patients with metastatic or unresectable solid tumors. J Immunother Cancer (2021) 9(11):e003388. doi: 10.1136/jitc-2021-003388
96. Miyahira AK, Soule HR. The 28th annual prostate cancer foundation scientific retreat report. Prostate (2022) 82(14):1346–77. doi: 10.1002/pros.24409
97. Roviello G, Gambale E, Giorgione R, Santini D, Stellato M, Fornarini G, et al. Effect of systemic therapies or best supportive care after disease progression to both nivolumab and cabozantinib in metastatic renal cell carcinoma: the meet-uro 19BEYOND study. Cancer Med (2022) 11(16):3084–92. doi: 10.1002/cam4.4681
98. Felices M, Warlick E, Juckett M, Weisdorf D, Vallera D, Miller S, et al. 444 GTB-3550 tri-specific killer engager TriKE™ drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. J ImmunoTher Cancer (2021) 9(Suppl 2):A473–3. doi: 10.1136/jitc-2021-SITC2021.444
99. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, In vivo expansion, and enhanced function. Clin Cancer Res (2016) 22(14):3440–50. doi: 10.1158/1078-0432.CCR-15-2710
100. Sarhan D, Brandt L, Felices M, Guldevall K, Lenvik T, Hinderlie P, et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv (2018) 2(12):1459–69. doi: 10.1182/bloodadvances.2017012369
101. Yun HD, Felices M, Vallera DA, Hinderlie P, Cooley S, Arock M, et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv (2018) 2(13):1580–4. doi: 10.1182/bloodadvances.2018018176
102. Yu C, Shanebeck K, Sun J, Wang C, Yu S, Luiz J, et al. 1101 ASKG315 – an IL-15 prodrug with antibody-like PK, enhanced safety and expanded therapeutic window. J ImmunoTher Cancer (2022) 10(Suppl 2):A1144–4. doi: 10.1136/jitc-2022-SITC2022.1101
103. Shanebeck K, Yu C, Yu S, Sun J, Wang D, Luiz J, et al. 1183 ASKG915 – an anti-PD-1 antibody-IL-15 prodrug fusion molecule with enhanced therapeutic potentials. J ImmunoTher Cancer (2022) 10(Suppl 2):A1227–7.
104. Zhao Y, Xie YQ, Van Herck S, Nassiri S, Gao M, Guo Y, et al. Switchable immune modulator for tumor-specific activation of anticancer immunity. Sci Adv (2021) 7(37):eabg7291. doi: 10.1126/sciadv.abg7291
105. Kang J, Coles M. IL-7: the global builder of the innate lymphoid network and beyond, one niche at a time. Semin Immunol (2012) 24(3):190–7. doi: 10.1016/j.smim.2012.02.003
106. Chen D, Tang TX, Deng H, Yang XP, Tang ZH. Interleukin-7 biology and its effects on immune cells: mediator of generation, differentiation, survival, and homeostasis. Front Immunol (2021) 12:747324. doi: 10.3389/fimmu.2021.747324
107. Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol (2019) 20(12):1584–93. doi: 10.1038/s41590-019-0479-x
108. Palmer MJ, Mahajan VS, Trajman LC, Irvine DJ, Lauffenburger DA, Chen J. Interleukin-7 receptor signaling network: an integrated systems perspective. Cell Mol Immunol (2008) 5(2):79–89. doi: 10.1038/cmi.2008.10
109. Capitini CM, Chisti AA, Mackall CL. Modulating T-cell homeostasis with IL-7: preclinical and clinical studies. J Intern Med (2009) 266(2):141–53. doi: 10.1111/j.1365-2796.2009.02085.x
110. Tredan O, Menetrier-Caux C, Ray-Coquard I, Garin G, Cropet C, Verronese E, et al. ELYPSE-7: a randomized placebo-controlled phase IIa trial with CYT107 exploring the restoration of CD4+ lymphocyte count in lymphopenic metastatic breast cancer patients. Ann Oncol (2015) 26(7):1353–62. doi: 10.1093/annonc/mdv173
111. Campian JL, Ghosh S, Kapoor V, Yan R, Thotala S, Jash A, et al. Long-acting recombinant human interleukin-7, NT-I7, increases cytotoxic CD8 T cells and enhances survival in mouse glioma models. Clin Cancer Res (2022) 28(6):1229–39. doi: 10.1158/1078-0432.CCR-21-0947
112. Kim MY, Jayasinghe R, Devenport JM, Ritchey JK, Rettig MP, O'Neal J, et al. A long-acting interleukin-7, rhIL-7-hyFc, enhances CAR T cell expansion, persistence, and anti-tumor activity. Nat Commun (2022) 13(1):3296. doi: 10.1038/s41467-022-30860-0
113. Kim S, Lee SW, Koh JY, Choi D, Heo M, Chung JY, et al. A single administration of hIL-7-hyFc induces long-lasting T-cell expansion with maintained effector functions. Blood Adv (2022) 6(23):6093–107. doi: 10.1182/bloodadvances.2021006591
114. Lee SW, Choi D, Heo M, Shin EC, Park SH, Kim SJ, et al. hIL-7-hyFc, a long-acting IL-7, increased absolute lymphocyte count in healthy subjects. Clin Transl Sci (2020) 13(6):1161–9. doi: 10.1111/cts.12800
115. Dower W, Bakker A, Cwirla S, Joshi P, Pongtornpipat P, Williams B, et al. 567 MDK1319/MDK-701: a potent fully efficacious peptidyl agonist of IL-7Rαγc, designed with no reference to cytokine or receptor structure and unrelated to IL-7, fused to an fc-domain for PK enhancement. J ImmunoTher Cancer (2020) 8(Suppl 3):A341–2. doi: 10.1136/jitc-2020-SITC2020.0567
116. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discovery (2014) 13(5):379–95. doi: 10.1038/nrd4296
117. Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat Rev Immunol (2005) 5(9):688–98. doi: 10.1038/nri1688
118. Moroz A, Eppolito C, Li Q, Tao J, Clegg CH, Shrikant PA. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol (2004) 173(2):900–9. doi: 10.4049/jimmunol.173.2.900
119. Wang G, Tschoi M, Spolski R, Lou Y, Ozaki K, Feng C, et al. In vivo antitumor activity of interleukin 21 mediated by natural killer cells. Cancer Res (2003) 63(24):9016–22.
120. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21–mediated proliferation. Blood (2007) 109(10):4135–42. doi: 10.1182/blood-2006-10-054973
121. Allard EL, Hardy MP, Leignadier J, Marquis M, Rooney J, Lehoux D, et al. Overexpression of IL-21 promotes massive CD8+ memory T cell accumulation. Eur J Immunol (2007) 37(11):3069–77. doi: 10.1002/eji.200637017
122. Thompson JA, Curti BD, Redman BG, Bhatia S, Weber JS, Agarwala SS, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol (2008) 26(12):2034–9. doi: 10.1200/JCO.2007.14.5193
123. Davis ID, Brady B, Kefford RF, Millward M, Cebon J, Skrumsager BK, et al. Clinical and biological efficacy of recombinant human interleukin-21 in patients with stage IV malignant melanoma without prior treatment: a phase IIa trial. Clin Cancer Res (2009) 15(6):2123–9. doi: 10.1158/1078-0432.CCR-08-2663
124. Liu H, Wang R, An D, Liu H, Ye F, Li B, et al. An engineered IL-21 with half-life extension enhances anti-tumor immunity as a monotherapy or in combination with PD-1 or TIGIT blockade. Int Immunopharmacol (2021) 101(Pt A):108307. doi: 10.1016/j.intimp.2021.108307
125. Shen S, Sckisel G, Sahoo A, Lalani A, Otter DD, Pearson J, et al. Engineered IL-21 cytokine muteins fused to anti-PD-1 antibodies can improve CD8+ T cell function and anti-tumor immunity. Front Immunol (2020) 11:832. doi: 10.3389/fimmu.2020.00832
126. Durm G, Frentzas S, Rasmussen E, Najmi S, Sadraei N. 417 design and rationale of a phase 1 study evaluating AMG 256, a novel, targeted, IL-21 receptor agonist and anti-PD-1 antibody, in patients with advanced solid tumors. J ImmunoTher Cancer (2020) 8(Suppl 3):A254–4. doi: 10.1136/jitc-2020-SITC2020.0417
127. Ge Y, Wang H, Ren J, Liu W, Chen L, Chen H, et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J Immunother Cancer (2020) 8(1):e000710. doi: 10.1136/jitc-2020-000710
128. L. S, He C, Nair L, Yeung J, Egwuagu CE. Interleukin 12 (IL-12) family cytokines: role in immune pathogenesis and treatment of CNS autoimmune disease. Cytokine (2015) 75(2):249–55. doi: 10.1016/j.cyto.2015.01.030
129. Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev (2004) 202:139–56. doi: 10.1111/j.0105-2896.2004.00211.x
130. Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized interleukin-12 for cancer immunotherapy. Front Immunol (2020) 11:575597. doi: 10.3389/fimmu.2020.575597
131. Ha SJ, Chang J, Song MK, Suh YS, Jin HT, Lee CH, et al. Engineering n-glycosylation mutations in IL-12 enhances sustained cytotoxic T lymphocyte responses for DNA immunization. Nat Biotechnol (2002) 20(4):381–6. doi: 10.1038/nbt0402-381
132. Jung K, Ha JH, Kim JE, Kim JA, Kim YJ, Kim CH, et al. Heterodimeric fc-fused IL12 shows potent antitumor activity by generating memory CD8(+) T cells. Oncoimmunology (2018) 7(7):e1438800. doi: 10.1080/2162402X.2018.1438800
133. Gutierrez E, Bigelow M, LaCroix C, Kirby P, Markowitz L, Naill M, et al. Abstract 1714: preclinical characterization of DF6002/BMS-986415, a novel differentiated IL-12 fc-fusion protein with robust antitumor activity as monotherapy or in combination with anti-PD-1. Cancer Res (2021) 81(13_Supplement):1714–4. doi: 10.1158/1538-7445.AM2021-1714
134. Gafner V, Trachsel E, Neri D. An engineered antibody-interleukin-12 fusion protein with enhanced tumor vascular targeting properties. Int J Cancer (2006) 119(9):2205–12. doi: 10.1002/ijc.22101
135. Ongaro T, Matasci M, Cazzamalli S, Gouyou B, De Luca R, Neri D, et al. A novel anti-cancer L19-interleukin-12 fusion protein with an optimized peptide linker efficiently localizes in vivo at the site of tumors. J Biotechnol (2019) 291:17–25. doi: 10.1016/j.jbiotec.2018.12.004
136. Greiner JW, Morillon YM 2nd, Schlom J. NHS-IL12, a tumor-targeting immunocytokine. Immunotargets Ther (2021) 10:155–69. doi: 10.2147/ITT.S306150
137. Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget (2014) 5(7):1869–84. doi: 10.18632/oncotarget.1853
138. Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-Human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res (2019) 25(1):99–109. doi: 10.1158/1078-0432.CCR-18-1512
139. Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat BioMed Eng (2020) 4(5):531–43. doi: 10.1038/s41551-020-0549-2
140. Morris K, Brodkin H, Hicklin D, Ismail N, Nirschl C, Salmeron A, et al. 715 WTX-330 is an IL-12 pro-drug that is conditionally activated within the tumor microenvironment and induces regressions in mouse tumor models. J ImmunoTher Cancer (2021) 9(Suppl 2):A744–4. doi: 10.1136/jitc-2021-SITC2021.715
141. Steiner P, Brodkin H, Hicklin D, Ismail N, Morris K, Nirschl C, et al. Abstract 1716: WTX-330, a conditionally activated IL-12 INDUKINE, releases IL-12 selectively in the tumor microenvironment to activate anti-tumor immune responses and induce regressions in mouse tumor models. Cancer Res (2021) 81(13_Supplement):1716–6. doi: 10.1158/1538-7445.AM2021-1716
142. Patel E, Malkova N, Vu S, O'Donnell R, Fanny M, Guzman W, et al. 719 XTX301, a protein-engineered IL-12, exhibits tumor-selective activity in mice without peripheral toxicities and is well tolerated in non-human primates. J ImmunoTher Cancer (2021) 9(Suppl 2):A748–8. doi: 10.1136/jitc-2021-SITC2021.719
143. Xue D, Moon B, Liao J, Guo J, Zou Z, Han Y, et al. A tumor-specific pro-IL-12 activates preexisting cytotoxic T cells to control established tumors. Sci Immunol (2022) 7(67):eabi6899. doi: 10.1126/sciimmunol.abi6899
144. Bishop JL, Blackler R, Volkers G, Poffenberger M, Yu I, Smith J, et al. Increasing the therapeutic index of IL12 by engineering for tumor specific protease activation. Cancer Res (2021) 81(13_Supplement):1788–8. doi: 10.1158/1538-7445.AM2021-1788
145. Schmidt CS, Morrow WJ, Sheikh NA. Smart adjuvants. Expert Rev Vaccines (2007) 6(3):391–400. doi: 10.1586/14760584.6.3.391
146. Agarwal Y, Milling LE, Chang JYH, Santollani L, Sheen A, Lutz EA, et al. Intratumourally injected alum-tethered cytokines elicit potent and safer local and systemic anticancer immunity. Nat BioMed Eng (2022) 6(2):129–43. doi: 10.1038/s41551-021-00831-9
147. Ke W, Afonin KA. Exosomes as natural delivery carriers for programmable therapeutic nucleic acid nanoparticles (NANPs). Adv Drug Deliv Rev (2021) 176:113835. doi: 10.1016/j.addr.2021.113835
148. Lewis ND, Sia CL, Kirwin K, Haupt S, Mahimkar G, Zi T, et al. Exosome surface display of IL12 results in tumor-retained pharmacology with superior potency and limited systemic exposure compared with recombinant IL12. Mol Cancer Ther (2021) 20(3):523–34. doi: 10.1158/1535-7163.MCT-20-0484
149. Luo Y, Chen Z, Sun M, Li B, Pan F, Ma A, et al. IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials (2022) 281:121341. doi: 10.1016/j.biomaterials.2021.121341
150. Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev (2018) 281(1):138–53. doi: 10.1111/imr.12616
151. Van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol (2011) 32(3):110–6. doi: 10.1016/j.it.2011.01.003
152. Hirooka Y, Nozaki Y. Interleukin-18 in inflammatory kidney disease. Front Med (2021) 8:639103. doi: 10.3389/fmed.2021.639103
153. Nakamura S, Otani T, Okura R, Ijiri Y, Motoda R, Kurimoto M, et al. Expression and responsiveness of human interleukin-18 receptor (IL-18R) on hematopoietic cell lines. Leukemia (2000) 14(6):1052–9. doi: 10.1038/sj.leu.2401789
154. Fabbi M, Carbotti G, Ferrini S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J leukocyte Biol (2015) 97(4):665–75. doi: 10.1189/jlb.5RU0714-360RR
155. Ma Z, Li W, Yoshiya S, Xu Y, Hata M, El-Darawish Y, et al. Augmentation of immune checkpoint cancer immunotherapy with IL18IL18 plus anti-PD-L1 and anti-CTLA-4 antibodies. Clin Cancer Res (2016) 22(12):2969–80. doi: 10.1158/1078-0432.CCR-15-1655
156. Simpkins F, Flores AM, Chu C, Lucci JA, Berek JS, Coukos G, et al. A phase I, dose escalation trial to assess the safety and biological activity of recombinant human interleukin-18 (SB-485232) in combination with pegylated liposomal doxorubicin in platinum-resistant recurrent ovarian cancer. J Clin Oncol (2012) 30(15_suppl):5065. doi: 10.1200/jco.2012.30.15_suppl.5065
157. Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, et al. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer: Interdiscip Int J Am Cancer Soc (2009) 115(4):859–68. doi: 10.1002/cncr.24100
158. Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature (2020) 583(7817):609–14. doi: 10.1038/s41586-020-2422-6
159. Xu X, Dai W, Li C. Interleukins in the treatment of melanoma. Chin Med J (2022) 135(04):393–9. doi: 10.1097/CM9.0000000000001929
160. Nisthal A, Lee S-H, Bonzon C, Love R, Avery K, Rashid R, et al. 1372 XmAb143, an engineered IL18 heterodimeric fc-fusion, features improved stability, reduced potency, and insensitivity to IL18BP. J ImmunoTher Cancer (2022) 10(Suppl 2):A1426. doi: 10.1136/jitc-2022-SITC2022.1372
161. Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev (2008) 226:205–18. doi: 10.1111/j.1600-065X.2008.00706.x
162. Ouyang W, O'Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
163. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun (1998) 66(11):5224–31. doi: 10.1128/IAI.66.11.5224-5231.1998
164. Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10(3):170–81. doi: 10.1038/nri2711
165. Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J (1997) 16(19):5894–903. doi: 10.1093/emboj/16.19.5894
166. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
167. Saraiva M, Vieira P, O'Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020) 217(1):e20190418. doi: 10.1084/jem.20190418
168. Fujii S, Shimizu K, Shimizu T, Lotze MT. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood (2001) 98(7):2143–51. doi: 10.1182/blood.V98.7.2143
169. Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res (2012) 72(14):3570–81. doi: 10.1158/0008-5472.CAN-12-0721
170. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell (2011) 20(6):781–96. doi: 10.1016/j.ccr.2011.11.003
171. Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, et al. PEGylated IL-10 (Pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell (2018) 34(5):775–791 e3. doi: 10.1016/j.ccell.2018.10.007
172. Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol (2016) 34(29):3562–9. doi: 10.1200/JCO.2016.68.1106
173. Hecht JR, Lonardi S, Bendell J, Sim HW, Macarulla T, Lopez CD, et al. Randomized phase III study of FOLFOX alone or with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer that progressed after gemcitabine (SEQUOIA). J Clin Oncol (2021) 39(10):1108–18. doi: 10.1200/JCO.20.02232
174. Spigel D, Jotte R, Nemunaitis J, Shum M, Schneider J, Goldschmidt J, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with pegilodecakin in patients with metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol (2021) 16(2):327–33. doi: 10.1016/j.jtho.2020.10.001
175. Gorby C, Sotolongo Bellon J, Wilmes S, Warda W, Pohler E, Fyfe PK, et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci Signal (2020) 13(649):eabc0653. doi: 10.1126/scisignal.abc0653
176. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol (2021) 22(6):746–56. doi: 10.1038/s41590-021-00940-2
177. Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis. Cancer Cell (2019) 35(6):901–915 e4. doi: 10.1016/j.ccell.2019.05.005
178. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16(3):131–44. doi: 10.1038/nrc.2016.14
179. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol (2017) 25(7):573–84. doi: 10.1016/j.tim.2017.01.001
180. Herndon TM, Demko SG, Jiang X, He K, Gootenberg JE, Cohen MH, et al. US Food and drug administration approval: peginterferon-alfa-2b for the adjuvant treatment of patients with melanoma. oncol (2012) 17(10):1323–8. doi: 10.1634/theoncologist.2012-0123
181. Stuart-Harris RC, Lauchlan R, Day R. The clinical application of the interferons: a review. Med J Aust (1992) 156(12):869–72. doi: 10.5694/j.1326-5377.1992.tb137004.x
182. Patel JN, Walko CM. Sylatron: a pegylated interferon for use in melanoma. Ann Pharmacother (2012) 46(6):830–8. doi: 10.1345/aph.1Q791
183. Young PA, Dang NH, Nastoupil LJ, Minning D, Gresser MJ, Timmerman J. Antibody-interferon-alpha fusion protein (IGN002) for the treatment of b-cell non-Hodgkin lymphomas: a phase 1, first-in-human, dose-escalation trial. J Clin Oncol (2016) 34(15_suppl):TPS3109–TPS3109. doi: 10.1200/JCO.2016.34.15_suppl.TPS3109
184. Howng B, Winter MB, LePage C, Popova I, Krimm M, Vasiljeva O. Novel ex vivo zymography approach for assessment of protease activity in tissues with activatable antibodies. Pharmaceutics (2021) 13(9):1390. doi: 10.3390/pharmaceutics13091390
185. Berezhnoy A, Wang H, Cai N, Assi H, Lapuyade N, Paidhungat M, et al. 706 conditional cytokine therapeutics for tumor-selective biological activity: preclinical characterization of a dual-masked IFN-a2b. J ImmunoTher Cancer (2021) 9(Suppl 2):A735–5. doi: 10.1136/jitc-2021-SITC2021.706
186. Tyagi E, Brodkin H, Canales J, Hicklin D, Ismail N, Morris K, et al. 723 WTX-613, a conditionally activated IFNα INDUKINE™ molecule, induces anti-tumor immune responses resulting in strong tumor growth control in syngeneic mouse tumor models. J ImmunoTher Cancer (2021) 9(Suppl 2):A752–2. doi: 10.1136/jitc-2021-SITC2021.723
187. Steiner P, Brodkin H, Canales J, Hicklin D, Isaacs R, Ismail N, et al. Conditionally activated IL-12 or IFNα indukine™ molecules inhibit syngeneic lymphoma tumor growth in mice, induce anti-tumor immune responses and are tolerated in non-human primates. Blood (2021) 138:2258. doi: 10.1182/blood-2021-152758
188. Cao X, Liang Y, Hu Z, Li H, Yang J, Hsu EJ, et al. Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance. Nat Commun (2021) 12(1):5866. doi: 10.1038/s41467-021-26112-2
189. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12(1):49–62. doi: 10.1038/nrrheum.2015.169
190. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell (1995) 83(5):793–802. doi: 10.1016/0092-8674(95)90192-2
191. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front Immunol (2018) 9:784. doi: 10.3389/fimmu.2018.00784
192. Wajant H, Siegmund D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front Cell Dev Biol (2019) 7:91. doi: 10.3389/fcell.2019.00091
193. Roberts NJ, Zhou S, Diaz LA Jr., Holdhoff M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget (2011) 2(10):739–51. doi: 10.18632/oncotarget.344
194. Hayes AJ, Neuhaus SJ, Clark MA, Thomas JM. Isolated limb perfusion with melphalan and tumor necrosis factor alpha for advanced melanoma and soft-tissue sarcoma. Ann Surg Oncol (2007) 14(1):230–8. doi: 10.1245/s10434-006-9040-x
195. Libutti SK, Paciotti GF, Byrnes AA, Alexander HR Jr., Gannon WE, Walker M, et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res (2010) 16(24):6139–49. doi: 10.1158/1078-0432.CCR-10-0978
196. Goel R, Shah N, Visaria R, Paciotti GF, Bischof JC. Biodistribution of TNF-alpha-coated gold nanoparticles in an in vivo model system. Nanomed (Lond) (2009) 4(4):401–10. doi: 10.2217/nnm.09.21
197. Schliemann C, Hemmerle T, Berdel AF, Angenendt L, Kerkhoff A, Hering JP, et al. Dose escalation and expansion phase I studies with the tumour-targeting antibody-tumour necrosis factor fusion protein L19TNF plus doxorubicin in patients with advanced tumours, including sarcomas. Eur J Cancer (2021) 150:143–54. doi: 10.1016/j.ejca.2021.03.038
198. Dakhel S, Lizak C, Matasci M, Mock J, Villa A, Neri D, et al. An attenuated targeted-TNF localizes to tumors in vivo and regains activity at the site of disease. Int J Mol Sci (2021) 22(18):10020. doi: 10.3390/ijms221810020
199. Danielli R, Patuzzo R, Di Giacomo AM, Gallino G, Maurichi A, Di Florio A, et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: results of a phase II study. Cancer Immunol Immunother (2015) 64:999–1009. doi: 10.1007/s00262-015-1704-6
200. Valentinis B, Porcellini S, Asperti C, Cota M, Zhou D, Di Matteo P, et al. Mechanism of action of the tumor vessel targeting agent NGR-hTNF: role of both NGR peptide and hTNF in cell binding and signaling. Int J Mol Sci (2019) 20(18):4511. doi: 10.3390/ijms20184511
201. Gregorc V, Gaafar RM, Favaretto A, Grossi F, Jassem J, Polychronis A, et al. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol (2018) 19(6):799–811. doi: 10.1016/S1470-2045(18)30193-1
202. Wuest T, Gerlach E, Banerjee D, Gerspach J, Moosmayer D, Pfizenmaier K. TNF-selectokine: a novel prodrug generated for tumor targeting and site-specific activation of tumor necrosis factor. Oncogene (2002) 21(27):4257–65. doi: 10.1038/sj.onc.1205193
Keywords: engineering cytokine, fusion protein, polyethylene glycol, immunocytokine, prodrug cytokine, cancer immunotherapy
Citation: Fu Y, Tang R and Zhao X (2023) Engineering cytokines for cancer immunotherapy: a systematic review. Front. Immunol. 14:1218082. doi: 10.3389/fimmu.2023.1218082
Received: 06 May 2023; Accepted: 19 June 2023;
Published: 06 July 2023.
Edited by:
Soren Skov, University of Copenhagen, DenmarkReviewed by:
Silvano Ferrini, University of Genoa, ItalyCopyright © 2023 Fu, Tang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Zhao, eGlhb2Zlbmcuemhhb0Bjbi5zaW1jZXJlLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.