 Chenxiao Zheng1
Chenxiao Zheng1 Lilu Shu
Lilu Shu Wenjun Liu
Wenjun Liu Peter Wang
Peter Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 June 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1217466
This article is part of the Research TopicEpigenetic, Metabolic, and Transcriptional Regulation of Immune Cell Plasticity and Functions in Cancer and Non-Cancer DiseasesView all 11 articles
Osteoarthritis is non-inflammatory degenerative joint arthritis, which exacerbates disability in elder persons. The molecular mechanisms of osteoarthritis are elusive. Ubiquitination, one type of post-translational modifications, has been demonstrated to accelerate or ameliorate the development and progression of osteoarthritis via targeting specific proteins for ubiquitination and determining protein stability and localization. Ubiquitination process can be reversed by a class of deubiquitinases via deubiquitination. In this review, we summarize the current knowledge regarding the multifaceted role of E3 ubiquitin ligases in the pathogenesis of osteoarthritis. We also describe the molecular insight of deubiquitinases into osteoarthritis processes. Moreover, we highlight the multiple compounds that target E3 ubiquitin ligases or deubiquitinases to influence osteoarthritis progression. We discuss the challenge and future perspectives via modulation of E3 ubiquitin ligases and deubiquitinases expression for enhancement of the therapeutic efficacy in osteoarthritis patients. We conclude that modulating ubiquitination and deubiquitination could alleviate the osteoarthritis pathogenesis to achieve the better treatment outcomes in osteoarthritis patients.
Osteoarthritis (OA) is one common type of non-inflammatory degenerative arthritis, which is a joint disease and influences older persons for walk, leading to disability in elder persons (1, 2). Pain is the primary symptom in osteoarthritis patients. The features of osteoarthritis are the destructions of cartilage extracellular matrix (ECM), loss of mobility, joint dysfunction and synovial inflammation (3, 4). Aging and injure are two common reasons for the development of osteoarthritis. The exact mechanisms of osteoarthritis are not fully understood, but it is thought to involve a complex interplay of genetic, environmental, and biomechanical factors (5–9). The treatments often include pain control and reduction of inflammation (10). It is of great importance to discover the mechanisms of osteoarthritis and develop the novel therapeutic strategies for osteoarthritis (11, 12).
In general, protein is synthesized from the genetic code in DNA and translated into a polypeptide chain. Protein could be modified via the chemical changes, which is named as post-translational modification (PTM) (13, 14). PTMs are found to modulate protein functions via an effect on various protein behavior, such as activity, localization, stability and interactions with other proteins (15). Many common PTM types include methylation (addition of a methyl group), acetylation (addition of an acetyl group), ubiquitination (attachment of a small ubiquitin protein), sumoylation (attachment of the small protein SUMO), glycosylation (addition of sugar molecules) and phosphorylation (addition of a phosphate group), which affect localization, stability, interaction, folding, and gene expression (16–19). PTMs are critical in governing some biological processes, such as cell signaling pathways, cell metabolism, differentiation, metastasis, cell cycle and proliferation (20).
Ubiquitination is one type of PTMs, which involves the covalent attachment of ubiquitin to a target substrate. Ubiquitination affects substrate protein functions via regulation of localization and stability (21). The ubiquitination process is performed by E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase. E1 enzymes activate ubiquitin and transfer it to E2 enzyme. Then, E2 enzymes interact with E3 enzymes, leading to the transfer of ubiquitin from the E2 enzymes to the specific target proteins (22, 23). The ubiquitinated proteins can be recognized and degraded by the proteasome machinery (24, 25). Hence, the E3 ligases determine the specificity of the ubiquitination reaction due to recognizing and interacting with specific substrates (Figure 1). Several types of E3 ligases are RING (really interesting new gene) ligases, HECT (homologous to E6-AP carboxyl terminus) ligases, RBR (RING-between-RING) ligases, U-box ligases and PHD-finger ligases (26, 27). Ubiquitination can be reversed by deubiquitination, which is a process of cleaving ubiquitin from target proteins (28) (Figure 1). Deubiquitination is carried out by a class of deubiquitinases (DUBs), including ovarian tumor proteases (OTUs), ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), and Josephin domain-containing proteins (29). Among these DUBs, USPs are the largest family to cleave ubiquitin from substrates. Dysregulation of USPs have been implicated in various diseases, such as neurodegeneration, inflammation and cancer (30–32). It is clear that the balance between ubiquitination and deubiquitination is pivotal for maintaining proper protein levels and their functions (33).
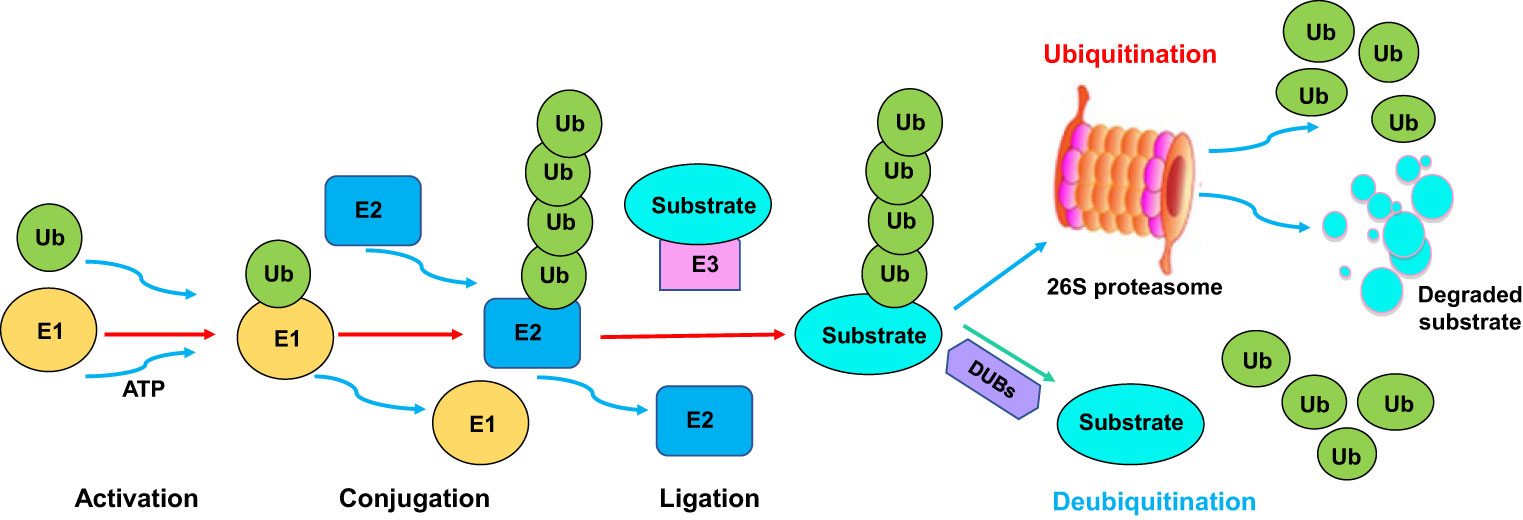
Figure 1 The ubiquitination and deubiquitination processes. The ubiquitination process is performed by E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase. E1 enzymes activate ubiquitin and transfer it to E2 enzyme. Then, E2 enzymes interact with E3 enzymes, leading to the transfer of ubiquitin from the E2 enzymes to the specific target proteins. The ubiquitinated proteins can be recognized and degraded by the proteasome. Ubiquitination can be reversed by deubiquitination, which is a process of cleaving ubiquitin from target proteins. Deubiquitination is carried out by a class of deubiquitinases (DUBs).
In recent years, ubiquitination and deubiquitination have been found to play a potential role in the various pathologies, including osteoarthritis (34). In this review, we will describe the role of ubiquitination in the development and progression of osteoarthritis. Moreover, we discuss the function of deubiquitination in regulating osteoarthritis development. Furthermore, we describe the compounds that target ubiquitination and deubiquitination to influence the osteoarthritis progression. Lastly, we provide the challenge and future perspectives for targeting ubiquitination and deubiquitination to treat with osteoarthritis patients.
NEDD4 E3 ubiquitin ligases are a family of enzymes that share a C2 domain at N-terminal and HECT domain at C-terminal, which regulate protein degradation, membrane trafficking and signaling transduction (35). This family includes several members, such as NEDD4, NEDD4-2, WWP1, WWP2, ITCH, Smurf1, Smurf2, NEDL1 and NEDL2. NEDD4 family has been involved in regulation of various diseases, including cancer, inflammation, and osteoarthritis (36–38). In the following paragraphs, we will discuss the mechanisms of NEDD4 E3 ubiquitin ligases in osteoarthritis initiation and progression (Table 1).

Table 1 E3 ubiquitin ligases regulate osteoarthritis development and progression.
WWP1 (WW domain-containing protein 1), an E3 ubiquitin ligase, plays an essential role in regulating protein degradation, cellular signal transduction, and gene expression. WWP1 targets proteins and make them for degradation by the proteasome (52). Recent studies have implied that WWP1 may involve in the development and progression of osteoarthritis. One bioinformatics analysis using cartilage samples showed that several genes are potential biomarker of osteoarthritis, such as WWP1, MDM2, OAS2, MSH2, UBE2E3, BCL2, RB1, TYMS and EGFR (39). In addition, miR-5692, miR-548e-5p and miR-3613-3p were associated with the abovementioned genes. It is required to determine whether WWP1 could be a useful biomarker of osteoarthritis (39).
WWP2 (WW domain-containing protein 2) belongs to the NEDD4 family of E3 ubiquitin ligases. WWP2 contains several domains, such as a C2 domain, a HECT domain, and multiple WW domains. These domains are responsible for transferring the ubiquitin to target proteins (53). WWP2 controls the protein stability and biological functions via providing ubiquitin molecules to targets, resulting in proteasomal degradation or modulation of protein activity or changing protein subcellular localization (54). Due to that WWP2 is involved in a wide range of biological functions, dysregulation of WWP2 leads to various disease states, such as neurodegeneration, cancer, inflammation and viral infections (55, 56). WWP2 was involved in chondrogenesis and osteoarthritis by regulating cartilage-specific transcription factors (57). WWP2 overexpression led to an inhibition of COL2A1, STC2, ACAN, GDF10 and GJA1, and an upregulation of EPAS1 expression using 3D chondrocyte pellet culture system. In addition, miR-140 overexpression caused an increased WWP2 and WDR1 in chondrocytes (58).
Yang et al. reported that miR-140 co-expressed with WWP2-C isoform, which can be accelerated by Sox9. Silencing of miR-140 reduced chondrogenic proliferation via targeting Sp1 (59). NFAT3 (nuclear factor of activated T-cells protein 3) and TGF-β/SMAD3 (mothers against decapentaplegic homolog 3) controlled miR-140 expression in osteoarthritis (60). Depletion of NFAT5 inhibited the expression of miR-140 and WWP2. NFAT3 and SMAD3 can bind to miR-140, but depletion of NFAT3 reduced miR-140 expression without altering WWP2 (60). Mice without WWP2 and mice with inactivation type of WWP2 E3 ligase (WWP2-C838A) had spontaneous and induced osteoarthritis. WWP2 regulated the expression of Runx2 via poly-ubiquitination and degradation and reduced Adamts5, contributing to cartilage homeostasis. WWP2 mRNA injection attenuated the severity of osteoarthritis in mouse joints (40). One methylation quantitative trait loci (mQTLs) analysis showed that WWP2 was linked to osteoarthritis genetic risk (61). In a word, WWP2 is involved in modulation of osteoarthritis.
Smad ubiquitin regulatory factor 1 (Smurf1) and Smurf2 belong to the family of NEDD4 ubiquitin ligase. Both Smurf1 and Smurf2 are involved in regulation of protein levels in the ubiquitin-dependent manner, which control the cell cycle progression, cell growth and differentiation (62). Although Smurf1 and Smurf2 share many functional similarities, they have distinct roles in governing various cellular processes to maintain cellular homeostasis. Dysregulation of Smurf1 and Smurf2 has been linked to cancer, inflammation and developmental disorders (63–65). One group used chondrogenic progenitor cells (CPCs) and meniscus progenitor cells (MPCs) and found that both Smurf1 and Smurf2 existed in articular cartilage and meniscus. An increased expression of Smurf1 alleviated the levels of TGFBR1, RUNX2 and SOX9, while upregulation of Smurf2 inhibited the levels of RUNX2 and GFFBR1, but not SOX9 levels. Depletion of Smurf2 increased the levels of SOX9 and RUNX2 in CPCs, but not in MPCs. Inhibition of Smurf1 did not affect the levels of RUNX2, SOX9 and TGFBR1 (41). Smurf1 protein was upregulated more in mice with Smurf2 depletion than wildtype mice upon TFG-β3 stimulation (66). Smurf2 has been observed to regulate osteoarthritis via targeting β-catenin in cartilage (67). Smurf2 was strongly expressed in human osteoarthritis compared with nonarthritic cartilage. Mice with Smurf2 overexpression displayed less articular cartilage area, osteophytes, clefting, subchondral sclerosis, and fibrillation. These mice exhibited the high expression of MMP-13 and type X collagen in articular cartilage. Moreover, Smurf2-transgenic mice had a decreased TGF-β pathway and an increased degradation of pSmad3 (68). Mice with Smurf2 deficiency exhibited higher expression of SOX, Col2 and Acan compared with wildtype mice (66). Chondrocytes with Smad3 deficiency accelerated differentiation and activated BMP signaling pathway, but failed to increase the expression of Smurf2, Sno, TGFRII in chondrocytes (69).
Increased expression of Smurf2 in cells of the chondrogenic lineage blocked differentiation of chondrocytes and enhanced maturation as well as promoted osteoblast differentiation. This phenotype could be due to upregulation of beta-catenin in the chondrocytes after Smurf overexpression, indicating that Smurf2 might be responsible for the development of osteoarthritis (70). One study showed that Smurf2 mediated degeneration of articular cartilage via promoting degradation of GSK-3beta and induction of beta-catenin in chondrocytes (71). LncRNA TM1-3P targeted miR-144-3p and ONECUT2 (one cut homeobox 2), governed cell apoptosis, inflammation and proliferation in osteoarthritis (72). LncRNA-CRNDE depletion elevated the ubiquitination of SIRT1 induced by SMURF2, resulting in reduction of SIRT1 stability. Overexpression of lncRNA-CRNDE stimulated the interaction of collagen 2 promoter and SOX9 (42). Circ_0116061 regulated the expression of miR-200b-3p and SMURF2, leading to modulation of cell apoptosis, proliferation, and inflammation in osteoarthritis chondrocytes (73). Lead, an environmental toxin, impaired TGF-β signaling pathway via upregulation of Smurf2 and downregulation of pSmad2 and pSmad3, leading to osteoarthritis in articular chondrocytes (74).
ITCH (also known as AIP4), a member of NEDD4 family of E3 ubiquitin ligases. It is pivotal to regulate signaling pathways and immune system via degradation of different proteins (75–77). One group reported that mice with ITCH deficiency in macrophages exhibited more severe joint damage compared with wildtype mice after post-traumatic osteoarthritis surgery. Mice with ITCH deficiency displayed promotion of the inflammatory macrophage infiltration in the synovium (78). ITCH protein levels were decreased during post-traumatic osteoarthritis. Macrophages with ITCH depletion were stimulated with IL-1β and exhibited pro-inflammatory phenotype. Hence, ITCH attenuated the progression of post-traumatic osteoarthritis via suppression of macrophage polarization (78). Another group identified that ITCH alleviated LPS-mediated chondrocyte injury through regulating JAG1 ubiquitination in osteoarthritis (43). ITCH expression was decreased in osteoarthritis samples compared with normal cartilaginous samples, whereas JAG1 expression was elevated in osteoarthritis specimens. Upregulation of ITCH or depletion of JAG1 induced proliferation, attenuated inflammation and ECM degradation and blocked apoptosis in LPS-mediated chondrocytes (43). Specifically, ITCH interacted with JAG1 via the WW-PPXY motif and caused JAG1 degradation via K48-linked ubiquitination, leading to inactivation of Notch1 pathway. Upregulation of JAG1 restrained the ITCH-mediated protection of chondrocyte damage that was induced by LPS. In conclusion, LPS-mediated chondrocyte injury and osteoarthritis-mediated articular cartilage damage were mitigated by ITCH in part via inhibition of Notch1 activation by enhancement of JAG1 ubiquitination and degradation (43).
The F-box protein is one of the subunits of the Skp1-Cullin1-F-box (SCF) complex, which acts as the substrate recognition component (79). The F-box protein contained a conserved F-box motif, which binds to a scaffold protein Skp1 to provide the link for the F-box protein to the rest of the SCF complex. Cullin1 interacts with Skp1 and F-box protein to form a core structure of the SCF complex. It is important to note that the variable region of F-box proteins determines its specificity for various substrates, leading to the ubiquitination of specific target proteins and degradation by the proteasome (80). F-box proteins regulate cell growth, apoptosis, differentiation, autophagy, cell cycle and metastasis. Abnormal expression of F-box proteins could participate in the development of cancer, inflammation and neurodegenerative disorders (81–83). For example, FBXO45 targeted GGNBP2 for ubiquitination and degradation and enhanced progression of esophageal squamous cell carcinoma (84). In the next paragraphs, we will mention the function of numerous F-box proteins in governing osteoarthritis initiation and progression (Table 1).
FBXW7 (F-box and WD repeat domain-containing 7) has been well studied in regulation of various cellular processes. Numerous targets of FBXW7 have been identified, including c-Myc, c-Jun, Notch, cyclin E and Mcl-1 (85). Trough targeting these proteins, FBXW7 controls cell proliferation, survival, cell cycle, differentiation and apoptosis. Mutations in FBXW7 were reported in various human malignancies. In tumorigenesis, FBXW7 has been revealed to be a tumor suppressive protein (86, 87). In recent years, FBXW7 was characterized to take part in osteoarthritis development. Mechanical overloading led to acceleration of senescence in chondrocyte and in mouse articular cartilage, which accompanied with downregulation of FBXW7 in chondrocytes and mouse cartilage (44). In line with this finding, FBXW7 was decreased in cartilage tissues in ageing mice, osteoarthritis mice, osteoarthritis patients. Depletion of FBXW7 in chondrocytes enhanced cartilage catabolism and caused chondrocyte senescence via promotion of p16, p21 and Colx and reduction of Col2a1 and ACAN, contributing to the exacerbation of osteoarthritis (44). On the contrary, overexpression of FBXW7 by intra-articular injection of adenovirus restrained osteoarthritis in mice. Further experiments indicated that mechanical overloading reduced the transcription of FBXW7 and alleviated FBXW7-induced degradation of MKK7, leading to activation of JNK signaling pathway (44).
Circular RNA VMA21 (circVMA21) expression was impeded by IL-1β in chondrocytes and C28/I2 cells. Ectopic expression of circVMA21 elevated the expression of Bcl-2 and reduced Bax and C-caspase-3 in C28/I2 cells after IL-1β stimulation, resulting in viability promotion and apoptosis attenuation (88). In addition, upregulation of circVMA21 reduced the MMP1 and MMP13 expressions, elevated COL2A1 expression and attenuated the production of NO, PGE2, TNF-α and IL-1β. Mechanistically, circVMA21 sponged miR-495-3p and consequently upregulated the expression of FBXW7, a target of miR-495-3p in chondrocytes (88). Zhu et al. reported that FBXW7 participated in IL-1β-involved chondrocytes degeneration via modulation of HIF-1α and VEGF pathways (89). In particular, lower expression of FBXW7 and higher expression of VEGF and HIF-1α were observed in osteoarthritis cartilage and IL-1β-mediated degenerated chondrocytes. Inhibition of HIF-1α led to reduction of VEGF levels, leading to upregulation of aggrecan, SOX9 and collagen II, and downregulation of collagen I and Runx-2 expression. Therefore, FBXW7 impeded HIF-1α/VEGF pathway and displayed a protective function in IL-1β-mediated chondrocyte degeneration via altering the expression of collagen I, collagen II, SOX9, aggrecan and Runx-2 (89).
FBXO3 has been found to stimulate cytokine secretion via controlling the degradation of Fbxl2 and subsequent upregulation of TNFR-associated factor (TRAF) proteins (90). FBXO3 was also reported to potentiated vascular inflammation and atherosclerosis (91). One study used rat fibroblast-like synoviocytes (FLSs) to construct knee osteoarthritis cell model. This cell model exhibited elevation of IL-18, IL-1β, apoptosis rate, and increased expression of pyroptosis-associated proteins. The abovementioned indicators were downregulated by up-modulation of miR-219a-5p, while downregulation of miR-219a-5p exhibited the opposed results (45). FBXO3 expression was inhibited by miR-219a-5p, and depletion of FBXO3 repressed the expression of IL-18, IL-1β and pyroptosis-related proteins. FBXO3 counteracted the effects of miR-215a-5p-inhibited IL-18, IL-1β and pyroptosis-related proteins. Additionally, miR-219a-5p alleviated knee joint injury and elevated step size of rat with knee osteoarthritis partly via inhibition of FBXO3 and inactivation of NLRP3 (45).
Wang et al. explored the effects of FBXO6 on the osteoarthritis pathogenesis. The human osteoarthritis samples and several mouse osteoarthritis models were used to detect the expression of FBXO6. This study showed that FBXO6 was downregulated in the cartilage from aged mouse samples, spontaneous osteoarthritis samples, ACLT (anterior cruciate ligament transaction)-induced osteoarthritis samples (46). Osteoarthritis process was accelerated in mice with cartilage conditional knockout or global knockout of FBXO6. FBXO6 reduced the MMP14 protein levels via ubiquitination and degradation, contributing to inactivation of MMP13 proteolytic ability. Notably, TGF-β-SMAD2/3 axis can upregulate the expression of FBXO6. Overall, upregulation of FBXO6 attenuated post-injury osteoarthritis development, suggesting that induction of FBXO6 in cartilage could be a useful strategy for treating osteoarthritis (46).
Lin et al. evaluated the effects of FBXO21 in osteoarthritis degeneration and its underlying molecular mechanism. They reported that osteoarthritis patient cartilages had higher expression of FBXO21. Ageing rats and monosodium iodoacetate-driven osteoarthritis rats exhibited an increased expression of FBXO21 in cartilages (47). FBXO21 expression was elevated in chondrocytes after treatment with lipopolysaccharide, IL-1β and TNF-α. Additionally, downregulation of FBXO21 alleviated osteoarthritis-associated cartilage degeneration, which accompanied with promotion of autophagy and anabolism, reduction of apoptosis as well as catabolism. On the contrary, overexpression of FBXO21 accelerated cartilage degeneration. Furthermore, FBXO21 performed their effects on cartilage degeneration via suppression of autophagy by phosphorylating ERK in chondrocytes (47). Strikingly, JUNB directly targeted the promoter of FBXO21 and upregulated the expression of FBXO21, thus contributing to cartilage degeneration in chondrocytes and SW1353 cells. In a word, JUNB/FBXO21/ERK pathway governs cartilage degeneration via blockade of autophagy in osteoarthritis, indicating that downregulation of FBXO21 might be a novel approach for osteoarthritis treatment (47). Jia et al. reported that circRNA-MSR inhibited miR-761 expression and consequently upregulated the expression of FBXO21. FBXO21 and circRNA-MSR expression levels were increased in osteoarthritis, while miR-761 expression levels were decreased in osteoarthritis (92). Depletion of circRNA-MSR facilitated the autophagy of LPS-stimulated cells. Upregulation of FBXO21 abolished the induced autophagy by miR-761 overexpression in chondrocytes. Hence, FBXO21 plays a potential role in osteoarthritis development (92).
The E3 ubiquitin ligase HECTD1 (HECT domain E3 ubiquitin protein ligase 1) has been proposed to control autophagy and osteoarthritis pathogenesis via degradation of Rubicon (48). The expression of HECTD1 was remarkably reduced in patients with osteoarthritis compared with healthy cartilage tissues. Surgery- and aging-triggered osteoarthritis pathogenesis was largely enhanced in cartilage with conditional depletion of HECTD1. Consistently, upregulation of HECTD1 alleviated pathogenesis of osteoarthritis in mouse joints. HECTD1 can interact with Rubicon at K534 and cause ubiquitination and proteasomal degradation of Rubicon, contributing to attenuation of stress-mediated chondrocyte death and osteoarthritis progression (48). Casitas B-lineage lymphoma b (Cbl-b) E3 ubiquitin ligase has been demonstrated to regulate cancer immunotherapy (93). One group identified that Cbl-b targeted Tropomyosin-related kinase A (TrkA) for ubiquitination and degradation (94). It has been known that TrkA is a one of nerve growth factors, which participates in the osteoarthritis pain (95). Knee osteoarthritis that was induced by DMM (destabilization of the medial meniscus) exhibited the dissociation of TrkA and Cbl-b E3 ligase in dorsal root ganglion (DRG) neurons, leading to impaired Cbl-b-mediated degradation of TrkA, thereby contributing to TrkA-dependent pain sensitization (49). In line with this result, overexpression of Cbl-b reduced the levels of TrkA protein and attenuated heat hyperalgesia and mechanical allodynia in DRG neurons of mice with osteoarthritis (49).
Ubiquitin-fold modifier 1-specific ligase 1 (UFL1) E3 ligase had a lower expression level in articular tissues of osteoarthritis. IL-1β treatment alleviated the expression of UFL1 in chondrocytes. Increased expression of UFL1 promoted viability of IL-1β-mediated chondrocytes, which was accompanied with inhibition of NO and PGE2 production and suppression of iNOS and COX-2 expression levels (50). IL-6 and TNF-α levels were elevated after IL-1β induction, which was abrogated by UFL1. Moreover, UFL1 reduced the production of ADAMTS-4, ADAMTS-5, MMP-3 and MMP-13 and inactivated the NF-κB signaling pathway in IL-1β-mediated chondrocytes (50). 3-Hydroxy-3-methylglutaryl reductase degradation (HRD1) E3 ubiquitin ligase exhibits a key role in endoplasmic reticulum (ER)-associated degradation (ERAD) via regulating unfolded protein response (UPR) to maintain cellular proteostasis after stress stimulation (96). HRD1 deficient cells had the upregulation of OS9 expression, which was required for degradation of ERAD substrates. HRD1 levels were inversely associated with OS9 expression in clinical synovium tissues of osteoarthritis and rheumatoid arthritis patients (51).
USPs act as deubiquitinating enzymes, which function to remove Ub from Ub conjugates and control the ubiquitination and degradation of proteins. It has been known that USP play a vital role in bone and bone-related diseases (34). For example, one study identified that a total of 463 ubiquitinated peptides were associated with injured articular cartilage tissues after mechanical injury. These ubiquitinated proteins participated in endoplasmic reticulum (ER)-related protein degradation, such as USP5, YOD1 deubiquitinase, BRCA1/BRCA2-containing complex subunit 3 (BRCC3) E3 ligase, Ataxin 3 (ATXN3) deubiquitinating enzyme. The ER stress regulators were also altered in injured articular cartilage, including ubiquilin 1, RAD23 and VCP/p97 (97). ER stress markers were also elevated after cartilage injury. This work suggested that activation of some DUBs and ER stress regulators involved in regulation of cartilage tissue injury and osteoarthritis (97). Herein, we describe the molecular insight of USPs into osteoarthritis processes (Table 2).
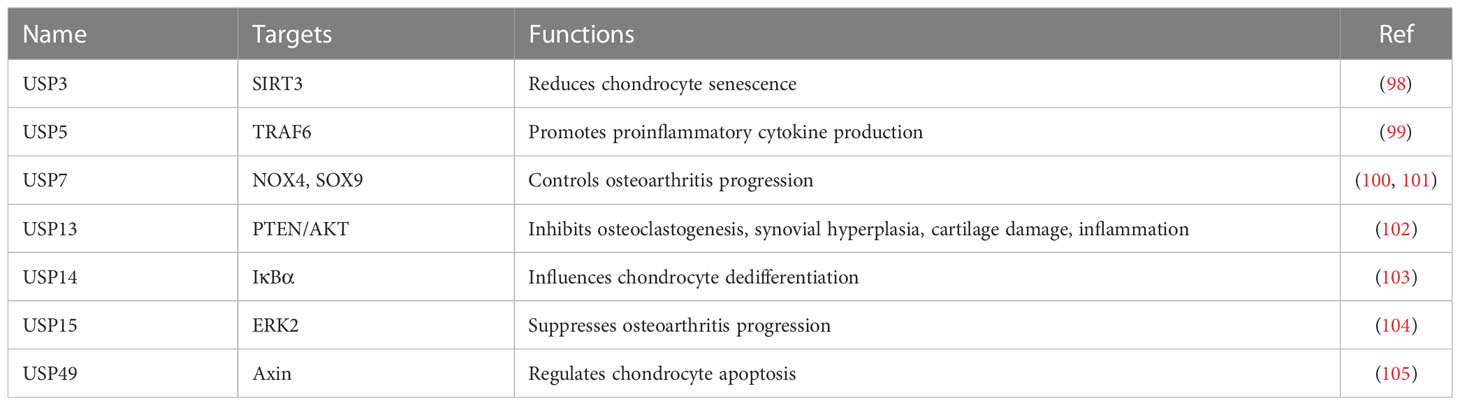
Table 2 USP regulate osteoarthritis progression.
Ubiquitin-specific protease 3 (USP3) has been reported to inhibit type I interferon signaling via deubiquitination of RIG-I-like receptors and antiviral immunity (106). USP3 was found to be downregulated in osteoarthrosis. USP3 upregulation attenuated IL-1β-mediated apoptosis of chondrocytes and inactivation of NF-κB pathway (107). TRAF6 is an adaptor protein for NF-κB pathway and involves in immune response and inflammation. IL-1β elevated the ubiquitination of TRAF6, which can be abrogated by overexpression of USP3 in chondrocytes (107). Increased USP3 expression recued IL-1β-induced cell senescence in rat primary chondrocytes (98). USP3 maintained the protein levels of SIRT3 via inhibition of SIRT3 ubiquitination. Consistently, suppression of SIRT3 attenuated the function of USP3 upregulation in cell senescence in rat chondrocytes (98). Similarly, upregulation of SIRT3 abolished USP3-depletion-mediated ROS production and cell senescence. Mechanistically, upregulation of SIRT3 reduced chondrocyte senescence in part via FOXO3 deacetylation (98).
Evidence has suggested that USP5 is critical involved in inflammation development and processes. For example, USP5 regulated neuropathic and inflammatory pain via promotion of CaV3.2 channel activity (108). Impairment of USP5 and Cav3.2 interaction attenuated inflammatory and neuropathic pain (109). A cell-permeant peptide targeting the cUBP domain of USP5 abolished neuropathic pain and inflammatory (110). IL-1β was an essential mediator to regulate the interaction between Cav3.2 and USP5 in the pain process (111). Impairing the binding between USP5 and Cav3.2 protected mechanical hypersensitivity in female mice with peripheral inflammation (112). USP5 was reported to be related with proinflammatory function in RA-FLS (rheumatoid arthritis-fibroblast-like synoviocytes (99). It has been known that RA is a common chronic autoimmune inflammatory disease. USP5 expression was increased in RA-FLS, while the expression of USP5 was decreased in OA-FLS. USP5 expression was increased after IL-1β stimulation in a time-dependent way (99). Overexpression of USP5 aggravated activation of NF-κB pathway and promoted proinflammatory cytokine production. In addition, depletion of USP5 reduced the cytokines release and inactivated NF-κB activation. USP5 can bind with TRAF6 and stabilize TRAF6 via deubiquitination. USP5 overexpression controls inflammatory processes via maintenance of TRAF6 stability in RA-FLS (99).
One study revealed that hydrogen peroxide (H2O2) induced the expression of USP7 and increased ROS levels and reduced proliferation in rat chondrocytes. Depletion of USP7 abolished H2O2-mediated pyroptosis and ROS induction and inactivated NLRP3 inflammasome activation in rat chondrocytes (100). Consistently, upregulation of USP7 enhanced pyroptosis, IL-1β and IL-18 levels, MMP-1, MMP13, and NLRP3 inflammasome activation via upregulation of ROS levels in rat chondrocytes. Moreover, USP7 interacted with NOX4 and promoted its ubiquitination in rat chondrocytes (100). In addition, USP7 and NOX4 were highly expressed in osteoarthritis patients. Suppression of NOX4 abrogated the function of USP7-mediated pyroptosis, ROS induction and NLRP3 activation in rat chondrocytes. P22077, one USP7 inhibitor, repressed osteoarthritis process in mice with monosodium iodoacetate (MIA) injection. Hence, USP7 regulated NOX4/ROS/NLPR3 pathway to contribute to osteoarthritis progression (100). Ubiquitination of LKB1 increased activation of AMPK pathway and repressed NLRP3 inflammasome response and blocked chondrocyre pyroptosis in osteoarthritis (113).
USP7 expression was decreased in the knee joint cartilage of mice with osteoarthritis. Silencing of USP7 by siRNAs or its inhibitors recued cell proliferation and accelerated apoptosis in chondrocyte. Depletion of USP7 reduced inflammatory response during inflammation process (114). Inhibition of USP7 by its inhibitors promoted cartilage destruction in osteoarthritis mice via activation of the BiP-eIF2α-ATF4-CHOP pathway in ERS and promotion of NF-κB/p65 pathway. Several compounds, including QNZ and 4-PBA, and CHOP siRNAs decreased USP7 expression and contributed to inhibition of chondrocyte proliferation and induction of apoptosis and reduction of inflammatory response after TNF-α stimulation (114). One group found that ADMA promoted SOX9 destabilization, which was mediated by DDAH1 in osteoarthritis. DDAH1 expression was decreased, while DDAH1 was increased in osteoarthritis patients (101). Because DDAH1 was an ADMA hydrolase, mice with global or chondrocyte-conditional knockdown of DDAH1 displayed a rapid development of osteoarthritis. ADMA promoted osteoarthritis progression through induction of degeneration and senescence and disruption of ECM deposition in chondrocytes. ADMA interacted with SOX9 and USP7, protecting the SOX9 deubiquitination by USP7 and resulting in promotion of SOX9 degradation. Hence, upregulation of DDAH1 to inhibit ADMA levels and regulate USP7-mediated SOX9 deubiquitination could be a useful strategy for the treatment of osteoarthritis (101).
USP13 has been known to participate in the pathogenesis of infection, cancers, inflammation and neurodegenerative diseases (115). For example, USP13 inhibited lung inflammation via stabilization of the anti-inflammatory receptor IL-1R8/single immunoglobin interleukin-1-related receptor (Sigirr) (116). Inhibition of miR-19a-3p reduced sepsis-mediated lung injury through upregulation of USP13 expression (117). USP13 impaired the symptoms of lipopolysaccharides-mediated sepsis via deubiquitination of IRAK4 (118). USP13 inhibited sepsis-induced inflammation and cardiomyocyte oxidative stress via induction of nuclear factor erythroid 2-related factor 2 (Nrf2) (119). One work showed that USP13 regulated PTEN to ameliorate osteoarthritis via reducing oxidative stress, regulating apoptosis and inflammation (102). Specifically, USP13 expression was increased in synovial specimens from RA patients. Interestingly, USP13 expression was decreased in human-FLSs after stimulation by LPS, TNF-α and IL-1β. Upregulation of USP13 repressed inflammatory response in H-FLSs upon TNF-α or IL-1β challenge due to the improvement of PTEN and reduction of AKT phosphorylation as well as inactivation of NF-κB pathway (102). USP13 performed the protective functions due to the upregulation of Nrf-2 and downregulation of caspase-3. Mechanistically, USP13 bound with PTEN and regulated AKT activation. Moreover, upregulation of USP13 suppressed the expression of osteoclast-related genes and repressed osteoclastogenesis. In collagen-mediated arthritis (CIA) mice, USP13 attenuated synovial hyperplasia, cartilage damage and inflammation and bone loss (102).
USP14 has been known to regulate protein degradation and associate with carcinogenesis, neurodegenerative diseases and viral infections (120, 121). Li et al. reported that USP14 exacerbated activation of NF-κB signaling pathway and accelerated IL-1β-induced chondrocyte dedifferentiation via modulation of IκBα degradation (103). USP14 was highly elevated in osteoarthritis articular cartilage and IL-1β-induced chondrocytes. ACHP, an inhibitor of IKK-β, inactivated NF-κB pathway and abrogated USP14 upregulation. In turn, USP14 enhanced IκBα deubiquitination and promoted NF-κB activation. Suppression of NF-κB abolished USP14-mediated dedifferentiation effect of IL-1β on chondrocytes (103). Hence, USP14 might be a useful target for osteoarthritis intervention.
USP15 has been uncovered to participate in several cellular processes and tumorigenesis as well as other noncancer diseases (122). USP15 plays the multifaceted roles in various diseases via regulating diverse signaling pathways by deubiquitination of target proteins (123). In addition, USP15 has been implied to involve in immune and inflammatory processes by regulation of several pathways, including TLR signaling, NF-κB, RIG-1 pathway, TGF-β, and p53 pathways (124). USP15 was found to stimulate TGF-β/SMAD2 signaling. Moreover, USP15 was required for ERK2 to affect TGF-β/SMAD2 signaling and to control the cartilage phenotype. Mechanistically, USP15 interacted with ERK2 and form a complex to govern the ubiquitination of ERK2. USP15 elevated the levels of phosphor-ERK1/2, but not ERK2 stability, to activate the TGF-β/SMAD2 signaling pathway. Overall, USP15 suppressed osteoarthritis progression via targeting ERK and TGF-β/SMAD2 signaling (104).
Ubiquitin-specific protease 49 (USP49) has been reported to govern the tumorigenesis. For instance, Fbxo45 promoted cell proliferation and invasion via induction of USP49 for ubiquitination and degradation (125). USP49 targets Yes-associated protein 1 (YAP1) for maintaining the stability of YAP1 in gastric cancer, leading to induction of cell proliferation, metastasis, chemoresistance (126). USP49 controlled the oncogenic ability of glucocorticoid receptor beta in glioblastoma cells (127). USP49 was reported to regulate rat chondrocyte apoptosis via governing Axin deubiquitination (105). In osteoarthritis patients, the expression of USP49 was lower compared with normal healthy persons. IL-1β stimulation on primary rat chondrocytes caused the downregulation of USP49 expression. Consistently, overexpression of USP49 reduced chondrocyte apoptotic death that was induced by IL-1β stimulation via enhancement of Axin deubiquitination, leading to upregulation of Axin protein levels. The increased Axin protein led to β-catenin degradation and suppression of Wnt/β-catenin pathway (105).
Numerous compounds have been discovered to target ubiquitination and regulate osteoarthritis (Table 3). Digoxin has been approved by FDA to treat several medical conditions, such as heart failure, atrial fibrillation, supraventricular tachycardia, and cardiomyopathy (133, 134). One study showed the effects of digoxin on the inflammatory microenvironment and chondrogenesis in osteoarthritis (128). Digoxin was found to alleviate osteoarthritis synovitis via suppression of the M1-like polarization of synovial macrophages in patients with osteoarthritis. The similar results were found in collagenase-mediated osteoarthritis mice (128). By analysis of exosomes produced by macrophages and M1-like macrophages after digoxin treatments, this study found that digoxin governed inflammatory microenvironment of osteoarthritis and enhanced chondrogenesis via inhibition of the synovial macrophage M1-like polarization and exosomal miR-146b-5p, USP3 and SOX5 pathways in osteoarthritis (128).

Table 3 Compounds target ubiquitination in osteoarthritis.
It has been known that [6]-gingerol protected against osteoarthritis via regulation of a number of factors, such as Nrf2 and GSTA4-4 (135, 136). One group showed that [6]-gingerol, an anti-osteoarthritis compound, increased the protein levels of USP49 and inhibited the activation of the Wnt/β-catenin signaling pathway in primary rat chondrocytes (105). Resveratrol was reported to inhibit IL-1β-mediated apoptosis via regulation of caspase-3 activation and PARP cleavage, and reduce inflammatory signaling via inhibition of the degradation of IκBα in articular chondrocytes, suggesting that resveratrol might be a natural compound for treating osteoarthritis (129). ALLN (N-Ac-Leu-norleucinal), the proteasome inhibitor, was also found to repress the degradation of IκBα in chondrocytes and decreased chondrocyte apoptosis (129). 5-azacytidine promoted maturation via modulation of TGF-β (transforming growth factor-beta) and BMP (bone morphogenetic protein) pathways in articular chondrocytes (130). 5-azacytidine increased the expression of Smurf2 and decreased the protein levels of Smad2 and Smad3, but elevated the expression of Smad1 and Smad5 as well as inactivation of TGF-β in articular chondrocytes (130).
Spermidine, a natural inducer of autophagy, performs antiaging, anti-inflammation and antitumor activities (137). Spermidine exhibits anti-inflammatory effects on osteoarthritis via attenuation of synovitis, cartilage degeneration and osteophyte formation (131). Spermidine facilitated RIP1 deubiquitination by CYLD to block TNF-α-mediated NF-κB signaling pathway in osteoarthritis (131). Alpinetin, a natural flavonoid compound, displayed antitumor, anti-inflammation and antibacterial ability (138). Alpinetin attenuated the TNF-α-mediated upregulation of MMP-13 and ADAMTS-5 and downregulation of COL2A1 expression. Alpinetin also inhibited p65 nuclear translocation and inactivated IκB phosphorylation and regulated ERK phosphorylation. In DMM-induced rats, alpinetin abolished cartilage matrix degradation (132).
In conclusion, ubiquitination and deubiquitination play a potential role in osteoarthritis progression via regulating protein degradation (Figure 2). Although E3 ubiquitin ligases and DUBs have demonstrated to modulate osteoarthritis processes, several issues should be mentioned. In some studies, the researchers reported the role of ubiquitination in osteoarthritis pathogenesis. However, these studies did not point out the E3 ubiquitin ligases or deubiquitinases. For example, RUNX2 stability was regulated by lncRNAs LINC02035 and LOC100130207, leading to hypertrophic changes in human chondrocytes (139). Depletion of these two lncRNAs facilitated ubiquitin-involved proteasomal degradation of RUNX2 and attenuated hypertrophic differentiation of chondrocytes. Overexpression of LINC02035 and LOC100130207 promoted the stability of RUNX2 protein and induced hypertrophic changes. Therefore, LINC02035 and LOC100130207 might be potential targets for blocking osteoarthritis development via delaying chondrocyte hypertrophy (139).
Figure 2 E3 ubiquitin ligases and DUBs in osteoarthritis progression. E3 ubiquitin ligases and DUBs regulate protein ubiquitination and degradation to participate in osteoarthritis development and progression.
Besides lncRNAs, circRNAs are also involved in osteoarthritis progression (140, 141). Circ_DHRS3 sponged miR-183-5p and elevated the expression of GREM1, resulting in influencing IL-1β-induced chondrocyte proliferation, apoptosis and ECM degradation (142). Circ-9119 intercepted the miR-26a/PTEN axis and prevented chondrocyte apoptosis after IL-1β treatment (143). Circ_0134111 impaired the interaction between miR-224-5p and CCL1 and enhanced osteoarthritis progression (144). CircSERPINE2 protected IL-1β−induced apoptosis and ECM degradation via modulation of miR-495/TGFBR2 pathway in chondrocytes (145). CircFNDC3B regulated miR-525-5p and HO-1 pathways and subsequently governed osteoarthritis and oxidative stress (146).
In addition, m6A (N6-methyladenosine) modification has been known to regulate osteoarthritis (147–149). METTL3, an enzyme promoted the m6A formation on the mRNA, modulated inflammatory response and apoptosis and increased osteoarthritis development (150). METTL3 affected ECM degradation and governed the inflammatory response, leading to osteoarthritis progression (151). METTL3 reduced the chondrocyte autophagy and apoptosis via regulation of Bcl-2 stability by YTHDF1-involved m6A modification (152). METTL3 induced ATG7 m6A modification and enhanced cellular senescence and osteoarthritis progression via targeting autophagy and GATA4 pathways (153). METTL3 stimulated LINC00680 expression and facilitated osteoarthritis via SIRT1 m6A modification (154). AC008 was increased after m6A modification, leading to accelerating osteoarthritis progression via modulation of miR-328-3p and AQP1/ANKH pathways (155). One study implicated that m6A (N6-methyladenosine)-modified circRNA RERE regulated osteoarthritis via control of β-catenin ubiquitination and degradation (156).
One group found that knockdown of FBXO32 did not alter cartilage destruction in mouse osteoarthritis, although FBXO32 was elevated in osteoarthritis cartilage, suggesting that FBXO32 was not necessary for cartilage destruction in mouse osteoarthritis (157). Besides osteoarthritis, E3 ubiquitin ligases have been revealed to regulate rheumatoid arthritis pathogenesis. For example, E3 ubiquitin ligase TRIM32 (tripartite motif protein 32) overexpression accelerated the production of pro-inflammatory cytokines in FLS of patients with rheumatoid arthritis (158). The expression of TRIM32 was higher in FLS of rheumatoid arthritis patients than that in FLS of osteoarthritis patients. TRIM32 activated NF-κB pathway and bound to TRAF2 to induce the K63-linked polyubiquitination of TRAF2 in rheumatoid arthritis FLS (158). CUL1 affected rheumatoid arthritis via alteration of lymphocyte signal transduction (159). E3 ubiquitin ligase STUB1/CHIP regulated the ubiquitination of aryl hydrocarbon receptor and led to the imbalance between Treg and Th17 in rheumatoid arthritis (160).
Like ubiquitination, other PTMs, such as acetylation, methylation, phosphorylation and SUMOylation, have also participated in osteoarthritis progression. Aging increased superoxide dismutase 2 acetylation, which was dependent on Sirtuin 3 in cartilage, leading to osteoarthritis progression (161). GDF11 (growth differentiation factor 11) suppressed the abnormal adipogenesis of chondrocytes and reduced TMJ condylar cartilage degeneration via modulation of PPARγ SUMOylation in cartilage of osteoarthritis (162). Desumoylation of aggrecan and collagen II by SENP2 triggered osteoarthritis after IL-1β treatment (163). SIRT1 maintained chondrocyte ECM upon activation of SOX9 via deacetylation of FOXO4 (164). Acetylation attenuated SOX9 nuclear accumulation and transactivation of ACAN gene in chondrocytes (161). Protein arginine methyltransferase PRMT1 regulated AKT/FOXO1 pathway and enhanced ECM degradation and apoptosis of chondrocytes in joint osteoarthritis (165). The activity of PRMT1 was elevated and controlled osteoarthritis via induction of DHX9 arginine methylation and activation of NF-κB signaling pathway (166). Suppression of PRMT5 reduced MAPK and NF-κB signaling pathway and blocked cartilage degradation in osteoarthritis (167).
There are cross-talks between several PTMs. For instance, SIRT1 restoration regulated PTEN-involved EGFR ubiquitination and accelerated chondrocyte autophagy in osteoarthritis (168). SIRT1 depletion diminished the acetylation of PTEN and then elevated the expression of PTEN. Inactivation of PTEN alleviated EGFR ubiquitination and aggravated EGFR expression via destabilization of the EGFR-Cbl complex, contributing to suppression of extracellular matrix degradation in cartilage and promotion of autophagy in chondrocyte (168). In addition, it is necessary to describe that ubiquitin conjugating enzymes involved in osteoarthritis pathogenesis (169, 170). For example, miR-101a-3p increased apoptosis of chondrocytes via targeting FZD4 and ubiquitin-conjugating enzyme 2D1 (UBE2D1), leading to involvement of pathogenesis of temporomandibular joint osteoarthritis (169). Ubiquitin conjugating enzyme E2 M (UBE2M) induced apoptosis of chondrocyte via upregulation of Wnt/β-catenin signaling in osteoarthritis (170). Without a doubt, it is pivotal to further explore the mechanisms of ubiquitination and deubiquitination in involvement of osteoarthritis development.
CZ searched the literature and drafted the original manuscript. JC, YW, XW, and YL made the figures and tables and edited the manuscript. LS and WL corrected and edited the manuscript. PW conceived and supervised this study and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part supported by GuangDong Basic and Applied Basic Research Foundation (2022B1515230008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DDAH1, dimethylarginine dimethylaminohydrolase-1; ADMA, metabolite asymmetric dimethylarginine; ECM, extracellular matrix; ERS, endoplasmic reticulum stress; FOXO3, forkhead box O3; H2O2, hydrogen peroxide; HIF-1α, hypoxia-inducible factor-1α; IL-1β, interleukin-1β; ITCH, itchy E3 ubiquitin protein ligase; LPS, lipopolysaccharide; MMP-1, matrix metalloproteinases; NEDL1, neural precursor cell expressed, developmentally down-regulated protein 1; NEDD4, neural precursor cell expressed developmentally down-regulated 4; NF-κB, nuclear factor κB NLRP3, Nod-like receptor protein 3; NOX4, NADPH oxidase 4; OA, osteoarthritis; ROS, reactive oxygen species; SMURF1, SMAD ubiquitination regulatory factor 1; TRAF6, tumor necrosis factor-receptor-associated factor 6; TNF-α, tumor necrosis factor alpha; USP7, ubiquitin specific protease 7; VEGF, vascular endothelial growth factor; WWP1, WW domain-containing protein 1.
1. Bernabei I, So A, Busso N, Nasi S. Cartilage calcification in osteoarthritis: mechanisms and clinical relevance. Nat Rev Rheumatol (2023) 19(1):10–27. doi: 10.1038/s41584-022-00875-4
2. Sanchez-Lopez E, Coras R, Torres A, Lane NE, Guma M. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol (2022) 18(5):258–75. doi: 10.1038/s41584-022-00749-9
3. Atukorala I, Hunter DJ. A review of quality-of-life in elderly osteoarthritis. Expert Rev Pharmacoecon Outcomes Res (2023) 23(4):365–81. doi: 10.1080/14737167.2023.2181791
4. Jansen MP, Mastbergen SC. Joint distraction for osteoarthritis: clinical evidence and molecular mechanisms. Nat Rev Rheumatol (2022) 18(1):35–46. doi: 10.1038/s41584-021-00695-y
5. Zhang XA, Kong H. Mechanism of HIFs in osteoarthritis. Front Immunol (2023) 14:1168799. doi: 10.3389/fimmu.2023.1168799
6. Rosado SE. Osteoarthritis affects us too: an expert panel survey of factors important for younger adult wellbeing. Soc Work Health Care (2023) 62(2-4):73–92. doi: 10.1080/00981389.2023.2191654
7. Liu S, Pan Y, Li T, Zou M, Liu W, Li Q, et al. The role of regulated programmed cell death in osteoarthritis: from pathogenesis to therapy. Int J Mol Sci (2023) 24(6). doi: 10.3390/ijms24065364
8. Kim M, Rubab A, Chan WCW, Chan D. Osteoarthritis year in review: genetics, genomics and epigenetics. Osteoarthritis Cartilage (2023). doi: 10.1016/j.joca.2023.03.003
9. Rice SJ, Beier F, Young DA, Loughlin J. Interplay between genetics and epigenetics in osteoarthritis. Nat Rev Rheumatol (2020) 16(5):268–81. doi: 10.1038/s41584-020-0407-3
10. Foster NE, Eriksson L, Deveza L, Hall M. Osteoarthritis year in review 2022: epidemiology & therapy. Osteoarthritis Cartilage (2023). doi: 10.1016/j.joca.2023.03.008
11. Hadzic E, Beier F. Emerging therapeutic targets for osteoarthritis. Expert Opin Ther Targets (2023) 27(2):111–20. doi: 10.1080/14728222.2023.2185133
12. Lv Z, Cai X, Bian Y, Wei Z, Zhu W, Zhao X, et al. Advances in mesenchymal stem cell therapy for osteoarthritis: from preclinical and clinical perspectives. Bioengineering (Basel) (2023) 10(2). doi: 10.3390/bioengineering10020195
13. Chen J, Tsai YH. Applications of genetic code expansion in studying protein post-translational modification. J Mol Biol (2022) 434(8):167424. doi: 10.1016/j.jmb.2021.167424
14. Singh HR, Ostwal YB. Post-translational modification, phase separation, and robust gene transcription. Trends Genet (2019) 35(2):89–92. doi: 10.1016/j.tig.2018.11.002
15. Mowen KA, David M. Unconventional post-translational modifications in immunological signaling. Nat Immunol (2014) 15(6):512–20. doi: 10.1038/ni.2873
16. Bhat KP, Umit Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discovery (2021) 20(4):265–86. doi: 10.1038/s41573-020-00108-x
17. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol (2019) 20(3):156–74. doi: 10.1038/s41580-018-0081-3
18. Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol (2018) 19(1):59–70. doi: 10.1038/nrm.2017.83
19. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol (2017) 18(7):452–65. doi: 10.1038/nrm.2017.22
20. Chen T, Liu J, Li S, Wang P, Shang G. The role of protein arginine n-methyltransferases in inflammation. Semin Cell Dev Biol (2022). doi: 10.1016/j.semcdb.2022.08.005
21. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
22. Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol (2005) 7(8):742–9. doi: 10.1038/ncb0805-742
23. Liu J, Chen T, Li S, Liu W, Wang P, Shang G. Targeting matrix metalloproteinases by E3 ubiquitin ligases as a way to regulate the tumor microenvironment for cancer therapy. Semin Cancer Biol (2022) 86(Pt 2):259–68. doi: 10.1016/j.semcancer.2022.06.004
24. Chen X, Ma J, Wang ZW, Wang Z. The E3 ubiquitin ligases regulate inflammation in cardiovascular diseases. Semin Cell Dev Biol (2023). doi: 10.1016/j.semcdb.2023.02.008
25. Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol (2014) 21(4):301–7. doi: 10.1038/nsmb.2780
26. Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Canc (2021) 21(10):638–54. doi: 10.1038/s41568-021-00365-x
27. Dikic I, Schulman BA. An expanded lexicon for the ubiquitin code. Nat Rev Mol Cell Biol (2023) 24(4):273–87. doi: 10.1038/s41580-022-00543-1
28. Estavoyer B, Messmer C, Echbicheb M, Rudd CE, Milot E, Affar EB. Mechanisms orchestrating the enzymatic activity and cellular functions of deubiquitinases. J Biol Chem (2022) 298(8):102198. doi: 10.1016/j.jbc.2022.102198
29. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discovery (2018) 17(1):57–78. doi: 10.1038/nrd.2017.152
30. Parihar N, Bhatt LK. Deubiquitylating enzymes: potential target in autoimmune diseases. Inflammopharmacology (2021) 29(6):1683–99. doi: 10.1007/s10787-021-00890-z
31. Do HA, Baek KH. Cellular functions regulated by deubiquitinating enzymes in neurodegenerative diseases. Ageing Res Rev (2021) 69:101367. doi: 10.1016/j.arr.2021.101367
32. Liu J, Cheng Y, Zheng M, Yuan B, Wang Z, Li X, et al. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduct Target Ther (2021) 6(1):28. doi: 10.1038/s41392-020-00418-x
33. Snyder NA, Silva GM. Deubiquitinating enzymes (DUBs): regulation, homeostasis, and oxidative stress response. J Biol Chem (2021) 297(3):101077. doi: 10.1016/j.jbc.2021.101077
34. Luo W, Zhang G, Wang Z, Wu Y, Xiong Y. Ubiquitin-specific proteases: vital regulatory molecules in bone and bone-related diseases. Int Immunopharmacol (2023) 118:110075. doi: 10.1016/j.intimp.2023.110075
35. Wang ZW, Hu X, Ye M, Lin M, Chu M, Shen X. NEDD4 E3 ligase: functions and mechanism in human cancer. Semin Cancer Biol (2020) 67(Pt 2):92–101. doi: 10.1016/j.semcancer.2020.03.006
36. Zhang R, Shi S. The role of NEDD4 related HECT-type E3 ubiquitin ligases in defective autophagy in cancer cells: molecular mechanisms and therapeutic perspectives. Mol Med (2023) 29(1):34. doi: 10.1186/s10020-023-00628-3
37. Jayaprakash S, Hegde M, BharathwajChetty B, Girisa S, Alqahtani MS, Abbas M, et al. Unraveling the potential role of NEDD4-like E3 ligases in cancer. Int J Mol Sci (2022) 23(20). doi: 10.3390/ijms232012380
38. Xu K, Chu Y, Liu Q, Fan W, He H, Huang F. NEDD4 E3 ligases: functions and mechanisms in bone and tooth. Int J Mol Sci (2022) 23(17). doi: 10.3390/ijms23179937
39. Wang B, Zhong JL, Jiang N, Shang J, Wu B, Chen YF, et al. Exploring the mystery of osteoarthritis using bioinformatics analysis of cartilage tissue. Comb Chem High Throughput Screen (2022) 25(1):53–63. doi: 10.2174/1386207323666201207100905
40. Mokuda S, Nakamichi R, Matsuzaki T, Ito Y, Sato T, Miyata K, et al. Wwp2 maintains cartilage homeostasis through regulation of Adamts5. Nat Commun (2019) 10(1):2429. doi: 10.1038/s41467-019-10177-1
41. Schminke B, Kauffmann P, Schubert A, Altherr M, Gelis T, Miosge N. SMURF1 and SMURF2 in progenitor cells from articular cartilage and meniscus during late-stage osteoarthritis. Cartilage. (2021) 13(2_suppl):117S–28S. doi: 10.1177/1947603520967069
42. Shi C, Zheng W, Wang J. lncRNA-CRNDE regulates BMSC chondrogenic differentiation and promotes cartilage repair in osteoarthritis through SIRT1/SOX9. Mol Cell Biochem (2021) 476(4):1881–90. doi: 10.1007/s11010-020-04047-4
43. Qi L, Wang M, He J, Jia B, Ren J, Zheng S. E3 ubiquitin ligase ITCH improves LPS-induced chondrocyte injury by mediating JAG1 ubiquitination in osteoarthritis. Chem Biol Interact (2022) 360:109921. doi: 10.1016/j.cbi.2022.109921
44. Zhang H, Shao Y, Yao Z, Liu L, Zhang H, Yin J, et al. Mechanical overloading promotes chondrocyte senescence and osteoarthritis development through downregulating FBXW7. Ann Rheum Dis (2022) 81(5):676–86. doi: 10.1136/annrheumdis-2021-221513
45. Wang Q, Huang PY, Wu JG, Zhang TQ, Li LF, Huang LD, et al. miR-219a-5p inhibits the pyroptosis in knee osteoarthritis by inactivating the NLRP3 signaling via targeting FBXO3. Environ Toxicol (2022) 37(11):2673–82. doi: 10.1002/tox.23627
46. Wang G, Chen S, Xie Z, Shen S, Xu W, Chen W, et al. TGFbeta attenuates cartilage extracellular matrix degradation via enhancing FBXO6-mediated MMP14 ubiquitination. Ann Rheum Dis (2020) 79(8):1111–20. doi: 10.1136/annrheumdis-2019-216911
47. Lin Z, Miao J, Zhang T, He M, Wang Z, Feng X, et al. JUNB-FBXO21-ERK axis promotes cartilage degeneration in osteoarthritis by inhibiting autophagy. Aging Cell (2021) 20(2):e13306. doi: 10.1111/acel.13306
48. Liao S, Zheng Q, Shen H, Yang G, Xu Y, Zhang X, et al. HECTD1-mediated ubiquitination and degradation of Rubicon regulates autophagy and osteoarthritis pathogenesis. Arthritis Rheumatol (2023) 75(3):387–400. doi: 10.1002/art.42369
49. Chen HK, Li YZ, Ge AN, Zhu YB, Wu SJ, Bai X, et al. Cbl-b modulated TrkA ubiquitination and function in the dorsal root ganglion of mice. Eur J Pharmacol (2022) 921:174876. doi: 10.1016/j.ejphar.2022.174876
50. Yang G, Wang Y, Chen Y, Huang R. UFL1 attenuates IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes. Int Immunopharmacol (2020) 81:106278. doi: 10.1016/j.intimp.2020.106278
51. Ye Y, Baek SH, Ye Y, Zhang T. Proteomic characterization of endogenous substrates of mammalian ubiquitin ligase Hrd1. Cell Biosci (2018) 8:46. doi: 10.1186/s13578-018-0245-z
52. Hu X, Yu J, Lin Z, Feng R, Wang ZW, Chen G. The emerging role of WWP1 in cancer development and progression. Cell Death Discovery (2021) 7(1):163. doi: 10.1038/s41420-021-00532-x
53. Chen W, Jiang X, Luo Z. WWP2: a multifunctional ubiquitin ligase gene. Pathol Oncol Res (2014) 20(4):799–803. doi: 10.1007/s12253-014-9838-y
54. Zhang R, Zhang J, Luo W, Luo Z, Shi S. WWP2 is one promising novel oncogene. Pathol Oncol Res (2019) 25(2):443–6. doi: 10.1007/s12253-018-0506-5
55. Lu X, Xu H, Xu J, Lu S, You S, Huang X, et al. The regulatory roles of the E3 ubiquitin ligase NEDD4 family in DNA damage response. Front Physiol (2022) 13:968927. doi: 10.3389/fphys.2022.968927
56. Haouari S, Vourc'h P, Jeanne M, Marouillat S, Veyrat-Durebex C, Lanznaster D, et al. The roles of NEDD4 subfamily of HECT E3 ubiquitin ligases in neurodevelopment and neurodegeneration. Int J Mol Sci (2022) 23(7). doi: 10.3390/ijms23073882
57. Chantry A. WWP2 ubiquitin ligase and its isoforms: new biological insight and promising disease targets. Cell Cycle (2011) 10(15):2437–9. doi: 10.4161/cc.10.15.16874
58. Tuerlings M, Janssen GMC, Boone I, van Hoolwerff M, Rodriguez Ruiz A, Houtman E, et al. WWP2 confers risk to osteoarthritis by affecting cartilage matrix deposition via hypoxia associated genes. Osteoarthritis Cartilage (2023) 31(1):39–48. doi: 10.1016/j.joca.2022.09.009
59. Yang J, Qin S, Yi C, Ma G, Zhu H, Zhou W, et al. MiR-140 is co-expressed with Wwp2-c transcript and activated by Sox9 to target Sp1 in maintaining the chondrocyte proliferation. FEBS Lett (2011) 585(19):2992–7. doi: 10.1016/j.febslet.2011.08.013
60. Tardif G, Pelletier JP, Fahmi H, Hum D, Zhang Y, Kapoor M, et al. NFAT3 and TGF-beta/SMAD3 regulate the expression of miR-140 in osteoarthritis. Arthritis Res Ther (2013) 15(6):R197. doi: 10.1186/ar4387
61. Rice SJ, Cheung K, Reynard LN, Loughlin J. Discovery and analysis of methylation quantitative trait loci (mQTLs) mapping to novel osteoarthritis genetic risk signals. Osteoarthritis Cartilage (2019) 27(10):1545–56. doi: 10.1016/j.joca.2019.05.017
62. Koganti P, Levy-Cohen G, Blank M. Smurfs in protein homeostasis, signaling, and cancer. Front Oncol (2018) 8:295. doi: 10.3389/fonc.2018.00295
63. Xia Q, Li Y, Han D, Dong L. SMURF1, a promoter of tumor cell progression? Cancer Gene Ther (2021) 28(6):551–65. doi: 10.1038/s41417-020-00255-8
64. David D, Nair SA, Pillai MR. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim Biophys Acta (2013) 1835(1):119–28. doi: 10.1016/j.bbcan.2012.11.003
65. Fu L, Cui CP, Zhang X, Zhang L. The functions and regulation of smurfs in cancers. Semin Cancer Biol (2020) 67(Pt 2):102–16. doi: 10.1016/j.semcancer.2019.12.023
66. Huang H, Veien ES, Zhang H, Ayers DC, Song J. Skeletal characterization of Smurf2-deficient mice and in vitro analysis of Smurf2-deficient chondrocytes. PloS One (2016) 11(1):e0148088. doi: 10.1371/journal.pone.0148088
67. Wu Q, Zhu M, Rosier RN, Zuscik MJ, O'Keefe RJ, Chen D. Beta-catenin, cartilage, and osteoarthritis. Ann N Y Acad Sci (2010) 1192(1):344–50. doi: 10.1111/j.1749-6632.2009.05212.x
68. Wu Q, Kim KO, Sampson ER, Chen D, Awad H, O'Brien T, et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheumatol (2008) 58(10):3132–44. doi: 10.1002/art.23946
69. Li TF, Darowish M, Zuscik MJ, Chen D, Schwarz EM, Rosier RN, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res (2006) 21(1):4–16. doi: 10.1359/JBMR.050911
70. Wu Q, Chen D, Zuscik MJ, O'Keefe RJ, Rosier RN. Overexpression of Smurf2 stimulates endochondral ossification through upregulation of beta-catenin. J Bone Miner Res (2008) 23(4):552–63. doi: 10.1359/jbmr.071115
71. Wu Q, Huang JH, Sampson ER, Kim KO, Zuscik MJ, O'Keefe RJ, et al. Smurf2 induces degradation of GSK-3beta and upregulates beta-catenin in chondrocytes: a potential mechanism for Smurf2-induced degeneration of articular cartilage. Exp Cell Res (2009) 315(14):2386–98. doi: 10.1016/j.yexcr.2009.05.019
72. Yi Y, Yang N, Yang Z, Tao X, Li Y. LncRNA TM1-3P regulates proliferation, apoptosis and inflammation of fibroblasts in osteoarthritis through miR-144-3p/ONECUT2 axis. Orthop Surg (2022) 14(11):3078–91. doi: 10.1111/os.13530
73. Zheng W, Hou G, Li Y. Circ_0116061 regulated the proliferation, apoptosis, and inflammation of osteoarthritis chondrocytes through regulating the miR-200b-3p/SMURF2 axis. J Orthop Surg Res (2021) 16(1):253. doi: 10.1186/s13018-021-02391-9
74. Holz JD, Beier E, Sheu TJ, Ubayawardena R, Wang M, Sampson ER, et al. Lead induces an osteoarthritis-like phenotype in articular chondrocytes through disruption of TGF-beta signaling. J Orthop Res (2012) 30(11):1760–6. doi: 10.1002/jor.22117
75. Yin Q, Wyatt CJ, Han T, Smalley KSM, Wan L. ITCH as a potential therapeutic target in human cancers. Semin Cancer Biol (2020) 67(Pt 2):117–30. doi: 10.1016/j.semcancer.2020.03.003
76. Moser EK, Oliver PM. Regulation of autoimmune disease by the E3 ubiquitin ligase itch. Cell Immunol (2019) 340:103916. doi: 10.1016/j.cellimm.2019.04.004
77. Aki D, Zhang W, Liu YC. The E3 ligase itch in immune regulation and beyond. Immunol Rev (2015) 266(1):6–26. doi: 10.1111/imr.12301
78. Lin X, Wang W, McDavid A, Xu H, Boyce BF, Xing L. The E3 ubiquitin ligase itch limits the progression of post-traumatic osteoarthritis in mice by inhibiting macrophage polarization. Osteoarthritis Cartilage (2021) 29(8):1225–36. doi: 10.1016/j.joca.2021.04.009
79. Wang Z, Liu P, Inuzuka H, Wei W. Roles of f-box proteins in cancer. Nat Rev Canc (2014) 14(4):233–47. doi: 10.1038/nrc3700
80. Tekcham DS, Chen D, Liu Y, Ling T, Zhang Y, Chen H, et al. F-box proteins and cancer: an update from functional and regulatory mechanism to therapeutic clinical prospects. Theranostics (2020) 10(9):4150–67. doi: 10.7150/thno.42735
81. Randle SJ, Laman H. F-box protein interactions with the hallmark pathways in cancer. Semin Cancer Biol (2016) 36:3–17. doi: 10.1016/j.semcancer.2015.09.013
82. Yumimoto K, Yamauchi Y, Nakayama KI. F-box proteins and cancer. Stem Cell Res Ther (2020) 12(5). doi: 10.3390/cancers12051249
83. Song Y, Lin M, Liu Y, Wang ZW, Zhu X. Emerging role of f-box proteins in the regulation of epithelial-mesenchymal transition and stem cells in human cancers. Stem Cell Res Ther (2019) 10(1):124. doi: 10.1186/s13287-019-1222-0
84. Wang Q, Wu L, Cao R, Gao J, Chai D, Qin Y, et al. Fbxo45 promotes the malignant development of esophageal squamous cell carcinoma by targeting GGNBP2 for ubiquitination and degradation. Oncogene (2022) 41(43):4795–807. doi: 10.1038/s41388-022-02468-7
85. Yumimoto K, Nakayama KI. Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol (2020) 67(Pt 2):1–15. doi: 10.1016/j.semcancer.2020.02.017
86. Shen W, Zhou Q, Peng C, Li J, Yuan Q, Zhu H, et al. FBXW7 and the hallmarks of cancer: underlying mechanisms and prospective strategies. Front Oncol (2022) 12:880077. doi: 10.3389/fonc.2022.880077
87. Fan J, Bellon M, Ju M, Zhao L, Wei M, Fu L, et al. Clinical significance of FBXW7 loss of function in human cancers. Mol Cancer (2022) 21(1):87. doi: 10.1186/s12943-022-01548-2
88. Li Z, Meng D, Liu Y, Bi F, Tian K, Xu J, et al. Circular RNA VMA21 ameliorates IL-1beta-engendered chondrocyte injury through the miR-495-3p/FBWX7 signaling axis. Clin Immunol (2022) 238:108995. doi: 10.1016/j.clim.2022.108995
89. Zhu WJ, Chang BY, Wang XF, Zang YF, Zheng ZX, Zhao HJ, et al. FBW7 regulates HIF-1alpha/VEGF pathway in the IL-1beta induced chondrocytes degeneration. Eur Rev Med Pharmacol Sci (2020) 24(11):5914–24. doi: 10.26355/eurrev_202006_21484
90. Mallampalli RK, Coon TA, Glasser JR, Wang C, Dunn SR, Weathington NM, et al. Targeting f box protein Fbxo3 to control cytokine-driven inflammation. J Immunol (2013) 191(10):5247–55. doi: 10.4049/jimmunol.1300456
91. Chandra D, Londino J, Alexander S, Bednash JS, Zhang Y, Friedlander RM, et al. The SCFFBXO3 ubiquitin E3 ligase regulates inflammation in atherosclerosis. J Mol Cell Cardiol (2019) 126:50–9. doi: 10.1016/j.yjmcc.2018.11.006
92. Jia Z, Liu J, Wang J. circRNA-MSR regulates the expression of FBXO21 to inhibit chondrocyte autophagy by targeting miR-761 in osteoarthritis. Kaohsiung J Med Sci (2022) 38(12):1168–77. doi: 10.1002/kjm2.12604
93. Augustin RC, Bao R, Luke JJ. Targeting cbl-b in cancer immunotherapy. J Immunother Cancer (2023) 11(2). doi: 10.1136/jitc-2022-006007
94. Emdal KB, Pedersen AK, Bekker-Jensen DB, Tsafou KP, Horn H, Lindner S, et al. Temporal proteomics of NGF-TrkA signaling identifies an inhibitory role for the E3 ligase cbl-b in neuroblastoma cell differentiation. Sci Signal (2015) 8(374):ra40. doi: 10.1126/scisignal.2005769
95. Yu H, Huang T, Lu WW, Tong L, Chen D. Osteoarthritis pain. Int J Mol Sci (2022) 23(9). doi: 10.3390/ijms23094642
96. Karamali N, Ebrahimnezhad S, Khaleghi Moghadam R, Daneshfar N, Rezaiemanesh A. HRD1 in human malignant neoplasms: molecular mechanisms and novel therapeutic strategy for cancer. Life Sci (2022) 301:120620. doi: 10.1016/j.lfs.2022.120620
97. Kaokhum N, Pinto-Fernandez A, Wilkinson M, Kessler BM, Ismail HM. The mechano-ubiquitinome of articular cartilage: differential ubiquitination and activation of a group of ER-associated DUBs and ER stress regulators. Mol Cell Proteomics (2022) 21(12):100419. doi: 10.1016/j.mcpro.2022.100419
98. Zhou Q, Wang W, Wu J, Qiu S, Yuan S, Fu PL, et al. Ubiquitin-specific protease 3 attenuates interleukin-1beta-mediated chondrocyte senescence by deacetylating forkhead box O-3 via sirtuin-3. Bioengineered. (2022) 13(2):2017–27. doi: 10.1080/21655979.2021.2012552
99. Luo XB, Xi JC, Liu Z, Long Y, Li LT, Luo ZP, et al. Proinflammatory effects of ubiquitin-specific protease 5 (USP5) in rheumatoid arthritis fibroblast-like synoviocytes. Mediators Inflamm (2020) 2020:8295149. doi: 10.1155/2020/8295149
100. Liu G, Liu Q, Yan B, Zhu Z, Xu Y. USP7 inhibition alleviates H(2)O(2)-induced injury in chondrocytes via inhibiting NOX4/NLRP3 pathway. Front Pharmacol (2020) 11:617270. doi: 10.3389/fphar.2020.617270
101. Wu Y, Shen S, Chen J, Ni W, Wang Q, Zhou H, et al. Metabolite asymmetric dimethylarginine (ADMA) functions as a destabilization enhancer of SOX9 mediated by DDAH1 in osteoarthritis. Sci Adv (2023) 9(6):eade5584. doi: 10.1126/sciadv.ade5584
102. Huang J, Ye Z, Wang J, Chen Q, Huang D, Liu H. USP13 mediates PTEN to ameliorate osteoarthritis by restraining oxidative stress, apoptosis and inflammation via AKT-dependent manner. BioMed Pharmacother (2021) 133:111089. doi: 10.1016/j.biopha.2020.111089
103. Li M, Zhao J, Jia L. USP14-mediated IkappaBalpha degradation exacerbates NF-kappaB activation and IL-1beta-stimulated chondrocyte dedifferentiation. Life Sci (2019) 218:147–52. doi: 10.1016/j.lfs.2018.12.014
104. Wang W, Zhu Y, Sun Z, Jin C, Wang X. Positive feedback regulation between USP15 and ERK2 inhibits osteoarthritis progression through TGF-beta/SMAD2 signaling. Arthritis Res Ther (2021) 23(1):84. doi: 10.1186/s13075-021-02456-4
105. Yang L, Wang Z, Zou C, Mi Y, Tang H, Wu X. Ubiquitin-specific protease 49 attenuates IL-1beta-induced rat primary chondrocyte apoptosis by facilitating axin deubiquitination and subsequent wnt/beta-catenin signaling cascade inhibition. Mol Cell Biochem (2020) 474(1-2):263–75. doi: 10.1007/s11010-020-03850-3
106. Cui J, Song Y, Li Y, Zhu Q, Tan P, Qin Y, et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-i-like receptors. Cell Res (2014) 24(4):400–16. doi: 10.1038/cr.2013.170
107. Zhou Q, Xiao Z, Zhou R, Zhou Y, Fu P, Li X, et al. Ubiquitin-specific protease 3 targets TRAF6 for deubiquitination and suppresses IL-1beta induced chondrocyte apoptosis. Biochem Biophys Res Commun (2019) 514(2):482–9. doi: 10.1016/j.bbrc.2019.04.163
108. Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. (2014) 83(5):1144–58. doi: 10.1016/j.neuron.2014.07.036
109. Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA, et al. Small organic molecule disruptors of Cav3.2 - USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain (2015) 11:12. doi: 10.1186/s12990-015-0011-8
110. Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain (2016) 12. doi: 10.1177/1744806916642444
111. Stemkowski PL, Garcia-Caballero A, Gadotti VM, M'Dahoma S, Chen L, Souza IA, et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3.2-USP5 interactions in the pain pathway. Mol Pain (2017) 13:1744806917724698. doi: 10.1177/1744806917724698
112. Gadotti VM, Zamponi GW. Disrupting USP5/Cav3.2 interactions protects female mice from mechanical hypersensitivity during peripheral inflammation. Mol Brain (2018) 11(1):60. doi: 10.1186/s13041-018-0405-4
113. Chen Y, Liu Y, Jiang K, Wen Z, Cao X, Wu S. Linear ubiquitination of LKB1 activates AMPK pathway to inhibit NLRP3 inflammasome response and reduce chondrocyte pyroptosis in osteoarthritis. J Orthop Translat (2023) 39:1–11. doi: 10.1016/j.jot.2022.11.002
114. Dong X, Yang C, Luo Y, Dong W, Xu X, Wu Y, et al. USP7 attenuates endoplasmic reticulum stress and NF-kappaB signaling to modulate chondrocyte proliferation, apoptosis, and inflammatory response under inflammation. Oxid Med Cell Longev (2022) 2022:1835900. doi: 10.1155/2022/1835900
115. Wang Q, Sun Z, Xia W, Sun L, Du Y, Zhang Y, et al. Role of USP13 in physiology and diseases. Front Mol Biosci (2022) 9:977122. doi: 10.3389/fmolb.2022.977122
116. Li L, Wei J, Li S, Jacko AM, Weathington NM, Mallampalli RK, et al. The deubiquitinase USP13 stabilizes the anti-inflammatory receptor IL-1R8/Sigirr to suppress lung inflammation. EBioMedicine (2019) 45:553–62. doi: 10.1016/j.ebiom.2019.06.011
117. Ren H, Mu W, Xu Q. miR-19a-3p inhibition alleviates sepsis-induced lung injury via enhancing USP13 expression. Acta Biochim Pol (2021) 68(2):201–6. doi: 10.18388/abp.2020_5505
118. Wang Z, Jiang L, Zhang D, Chen D, Wang L, Xiao D. USP13-mediated IRAK4 deubiquitination disrupts the pathological symptoms of lipopolysaccharides-induced sepsis. Microbes Infect (2021) 23(9-10):104867. doi: 10.1016/j.micinf.2021.104867
119. Wu D, Yuan R, Zhang L, Sun M. USP13 reduces septic mediated cardiomyocyte oxidative stress and inflammation by inducing Nrf2. Allergol Immunopathol (Madr) (2023) 51(2):160–7. doi: 10.15586/aei.v51i1.813
120. Wang F, Ning S, Yu B, Wang Y. USP14: structure, function, and target inhibition. Front Pharmacol (2021) 12:801328. doi: 10.3389/fphar.2021.801328
121. Wang D, Ma H, Zhao Y, Zhao J. Ubiquitin-specific protease 14 is a new therapeutic target for the treatment of diseases. J Cell Physiol (2021) 236(5):3396–405. doi: 10.1002/jcp.30124
122. Li YC, Cai SW, Shu YB, Chen MW, Shi Z. USP15 in cancer and other diseases: from diverse functionsto therapeutic targets. Biomedicines (2022) 10(2):474. doi: 10.3390/biomedicines10020474
123. Das T, Song EJ, Kim EE. The multifaceted roles of USP15 in signal transduction. Int J Mol Sci (2021) 22(9). doi: 10.3390/ijms22094728
124. Georges A, Gros P, Fodil N. USP15: a review of its implication in immune and inflammatory processes and tumor progression. Genes Immun (2021) 22(1):12–23. doi: 10.1038/s41435-021-00125-9
125. Wu L, Yu K, Chen K, Zhu X, Yang Z, Wang Q, et al. Fbxo45 facilitates pancreatic carcinoma progression by targeting USP49 for ubiquitination and degradation. Cell Death Dis (2022) 13(3):231. doi: 10.1038/s41419-022-04675-2
126. Liu Z, Li J, Ding Y, Ma M, Chen J, Lei W, et al. USP49 mediates tumor progression and poor prognosis through a YAP1-dependent feedback loop in gastric cancer. Oncogene (2022) 41(18):2555–70. doi: 10.1038/s41388-022-02267-0
127. Hu Y, Jiang Y, Zhang Z, Wang J, Zhang B, Gong L, et al. Oncogenic activity of glucocorticoid receptor beta is controlled by ubiquitination-dependent interaction with USP49 in glioblastoma cells. Mol Cancer Res (2022) 20(1):92–101. doi: 10.1158/1541-7786.MCR-20-1068
128. Jia H, Duan L, Yu P, Zhou Y, Liu R, Wang H. Digoxin ameliorates joint inflammatory microenvironment by downregulating synovial macrophage M1-like-polarization and its-derived exosomal miR-146b-5p/Usp3&Sox5 axis. Int Immunopharmacol (2022) 111:109135. doi: 10.1016/j.intimp.2022.109135
129. Shakibaei M, Csaki C, Nebrich S, Mobasheri A. Resveratrol suppresses interleukin-1beta-induced inflammatory signaling and apoptosis in human articular chondrocytes: potential for use as a novel nutraceutical for the treatment of osteoarthritis. Biochem Pharmacol (2008) 76(11):1426–39. doi: 10.1016/j.bcp.2008.05.029
130. Zuscik MJ, Baden JF, Wu Q, Sheu TJ, Schwarz EM, Drissi H, et al. 5-azacytidine alters TGF-beta and BMP signaling and induces maturation in articular chondrocytes. J Cell Biochem (2004) 92(2):316–31. doi: 10.1002/jcb.20050
131. Chen Z, Lin CX, Song B, Li CC, Qiu JX, Li SX, et al. Spermidine activates RIP1 deubiquitination to inhibit TNF-alpha-induced NF-kappaB/p65 signaling pathway in osteoarthritis. Cell Death Dis (2020) 11(7):503. doi: 10.1038/s41419-020-2710-y
132. Gao Y, Wang S, He L, Wang C, Yang L. Alpinetin protects chondrocytes and exhibits anti-inflammatory effects via the NF-kappaB/ERK pathway for alleviating osteoarthritis. Inflammation. (2020) 43(5):1742–50. doi: 10.1007/s10753-020-01248-3
133. Gazzaniga G, Menichelli D, Scaglione F, Farcomeni A, Pani A, Pastori D. Effect of digoxin on all-cause and cardiovascular mortality in patients with atrial fibrillation with and without heart failure: an umbrella review of systematic reviews and 12 meta-analyses. Eur J Clin Pharmacol (2023) 79(4):473–83. doi: 10.1007/s00228-023-03470-y
134. Hashemi-Shahri SH, Aghajanloo A, Ghavami V, Arasteh O, Mohammadpour AH, Reiner Z, et al. Digoxin and outcomes in patients with heart failure and preserved ejection fraction (HFpEF) patients: a systematic review and meta- analysis. Curr Drug Targets (2023) 24(2):191–200. doi: 10.2174/1389450123666220906093058
135. Abusarah J, Benabdoune H, Shi Q, Lussier B, Martel-Pelletier J, Malo M, et al. Elucidating the role of protandim and 6-gingerol in protection against osteoarthritis. J Cell Biochem (2017) 118(5):1003–13. doi: 10.1002/jcb.25659
136. Sharma S, Shukla MK, Sharma KC, Tirath, Kumar L, Anal JMH, et al. Revisiting the therapeutic potential of gingerols against different pharmacological activities. Naunyn Schmiedebergs Arch Pharmacol (2023) 396(4):633–47. doi: 10.1007/s00210-022-02372-7
137. Zou D, Zhao Z, Li L, Min Y, Zhang D, Ji A, et al. A comprehensive review of spermidine: safety, health effects, absorption and metabolism, food materials evaluation, physical and chemical processing, and bioprocessing. Compr Rev Food Sci Food Saf (2022) 21(3):2820–42. doi: 10.1111/1541-4337.12963
138. Zhao G, Tong Y, Luan F, Zhu W, Zhan C, Qin T, et al. Alpinetin: a review of its pharmacology and pharmacokinetics. Front Pharmacol (2022) 13:814370. doi: 10.3389/fphar.2022.814370
139. Yoon DS, Kim EJ, Cho S, Jung S, Lee KM, Park KH, et al. RUNX2 stabilization by long non-coding RNAs contributes to hypertrophic changes in human chondrocytes. Int J Biol Sci (2023) 19(1):13–33. doi: 10.7150/ijbs.74895
140. Xue Q, Huang Y, Chang J, Cheng C, Wang Y, Wang X, et al. CircRNA-mediated ceRNA mechanism in osteoarthritis: special emphasis on circRNAs in exosomes and the crosstalk of circRNAs and RNA methylation. Biochem Pharmacol (2023) 212:115580. doi: 10.1016/j.bcp.2023.115580
141. Li Z, Lu J. CircRNAs in osteoarthritis: research status and prospect. Front Genet (2023) 14:1173812. Cited in: Pubmed. doi: 10.3389/fgene.2023.1173812
142. Jiang R, Gao H, Cong F, Zhang W, Song T, Yu Z. Circ_DHRS3 positively regulates GREM1 expression by competitively targeting miR-183-5p to modulate IL-1beta-administered chondrocyte proliferation, apoptosis and ECM degradation. Int Immunopharmacol (2021) 91:107293. doi: 10.1016/j.intimp.2020.107293
143. Chen C, Yin P, Hu S, Sun X, Li B. Circular RNA-9119 protects IL-1beta-treated chondrocytes from apoptosis in an osteoarthritis cell model by intercepting the microRNA-26a/PTEN axis. Life Sci (2020) 256:117924. doi: 10.1016/j.lfs.2020.117924
144. Liu Y, Zhang Y. Hsa_circ_0134111 promotes osteoarthritis progression by regulating miR-224-5p/CCL1 interaction. Aging (Albany NY) (2021) 13(16):20383–94. doi: 10.18632/aging.203420
145. Zhang Q, Qiao X, Xia W. CircSERPINE2 weakens IL-1beta-caused apoptosis and extracellular matrix degradation of chondrocytes by regulating miR-495/TGFBR2 axis. Biosci Rep (2020) 40(11). doi: 10.1042/BSR20201601
146. Chen Z, Huang Y, Chen Y, Yang X, Zhu J, Xu G, et al. CircFNDC3B regulates osteoarthritis and oxidative stress by targeting miR-525-5p/HO-1 axis. Commun Biol (2023) 6(1):200. doi: 10.1038/s42003-023-04569-9
147. Yu Y, Lu S, Li Y, Xu J. Overview of distinct N6-methyladenosine profiles of messenger RNA in osteoarthritis. Front Genet (2023) 14:1168365. doi: 10.3389/fgene.2023.1168365
148. Zhai G, Xiao L, Jiang C, Yue S, Zhang M, Zheng J, et al. Regulatory role of N6-methyladenosine (m6A) modification in osteoarthritis. Front Cell Dev Biol (2022) 10:946219. doi: 10.3389/fcell.2022.946219
149. Liu H, Zheng YL, Wang XQ. The emerging roles of N(6)-methyladenosine in osteoarthritis. Front Mol Neurosci (2022) 15:1040699. doi: 10.3389/fnmol.2022.1040699
150. Liu Q, Li M, Jiang L, Jiang R, Fu B. METTL3 promotes experimental osteoarthritis development by regulating inflammatory response and apoptosis in chondrocyte. Biochem Biophys Res Commun (2019) 516(1):22–7. doi: 10.1016/j.bbrc.2019.05.168
151. Sang W, Xue S, Jiang Y, Lu H, Zhu L, Wang C, et al. METTL3 involves the progression of osteoarthritis probably by affecting ECM degradation and regulating the inflammatory response. Life Sci (2021) 278:119528. doi: 10.1016/j.lfs.2021.119528
152. He Y, Wang W, Xu X, Yang B, Yu X, Wu Y, et al. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m(6)A modification. Bone (2022) 154:116182. doi: 10.1016/j.bone.2021.116182
153. Chen X, Gong W, Shao X, Shi T, Zhang L, Dong J, et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis (2022) 81(1):87–99. doi: 10.1136/annrheumdis-2021-221091
154. Ren J, Li Y, Wuermanbieke S, Hu S, Huang G. N(6)-methyladenosine (m(6)A) methyltransferase METTL3-mediated LINC00680 accelerates osteoarthritis through m(6)A/SIRT1 manner. Cell Death Discovery (2022) 8(1):240. doi: 10.1038/s41420-022-00890-0
155. Yang J, Zhang M, Yang D, Ma Y, Tang Y, Xing M, et al. m(6)A-mediated upregulation of AC008 promotes osteoarthritis progression through the miR-328-3p−AQP1/ANKH axis. Exp Mol Med (2021) 53(11):1723–34. doi: 10.1038/s12276-021-00696-7
156. Liu Y, Yang Y, Lin Y, Wei B, Hu X, Xu L, et al. N(6) -methyladenosine-modified circRNA RERE modulates osteoarthritis by regulating beta-catenin ubiquitination and degradation. Cell Prolif (2023) 56(1):e13297. doi: 10.1111/cpr.13297
157. Kim HE, Rhee J, Park S, Yang J, Chun JS. Upregulation of atrogin-1/FBXO32 is not necessary for cartilage destruction in mouse models of osteoarthritis. Osteoarthritis Cartilage (2017) 25(3):397–400. doi: 10.1016/j.joca.2016.07.008
158. Liang T, Song M, Xu K, Guo C, Xu H, Zhang H, et al. TRIM32 promotes inflammatory responses in rheumatoid arthritis fibroblast-like synoviocytes. Scand J Immunol (2020) 91(6):e12876. doi: 10.1111/sji.12876
159. Kawaida R, Yamada R, Kobayashi K, Tokuhiro S, Suzuki A, Kochi Y, et al. CUL1, a component of E3 ubiquitin ligase, alters lymphocyte signal transduction with possible effect on rheumatoid arthritis. Genes Immun (2005) 6(3):194–202. doi: 10.1038/sj.gene.6364177
160. Wang W, Xiang T, Yang Y, Wang Z, Xie J. E3 ubiquitin ligases STUB1/CHIP contributes to the Th17/Treg imbalance via the ubiquitination of aryl hydrocarbon receptor in rheumatoid arthritis. Clin Exp Immunol (2022) 209(3):280–90. doi: 10.1093/cei/uxac072
161. Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, et al. Aging promotes sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis Rheumatol (2016) 68(8):1887–98. doi: 10.1002/art.39618
162. Wang H, Shi Y, He F, Ye T, Yu S, Miao H, et al. GDF11 inhibits abnormal adipogenesis of condylar chondrocytes in temporomandibular joint osteoarthritis. Bone Joint Res (2022) 11(7):453–64. doi: 10.1302/2046-3758.117.BJR-2022-0019.R1
163. Zhu B, Cui G, Zhang Q, Cheng X, Tang S. Desumoylation of aggrecan and collagen II facilitates degradation via aggrecanases in IL-1beta-mediated osteoarthritis. J Pain Res (2019) 12:2145–53. doi: 10.2147/JPR.S194306
164. Ma ZX, Xu H, Xiang W, Qi J, Xu YY, Zhao ZG. Deacetylation of FOXO4 by Sirt1 stabilizes chondrocyte extracellular matrix upon activating SOX9. Eur Rev Med Pharmacol Sci (2021) 25(2):626–35. doi: 10.26355/eurrev_202101_24621
165. Shen Q, Xiao Y, Cheng B, Sun Z, Hu Y, Yang H, et al. PRMT1 promotes extracellular matrix degradation and apoptosis of chondrocytes in temporomandibular joint osteoarthritis via the AKT/FOXO1 signaling pathway. Int J Biochem Cell Biol (2021) 141:106112. doi: 10.1016/j.biocel.2021.106112
166. Tang S, Cao Y, Cai Z, Nie X, Ruan J, Zhou Z, et al. The lncRNA PILA promotes NF-kappaB signaling in osteoarthritis by stimulating the activity of the protein arginine methyltransferase PRMT1. Sci Signal (2022) 15(735):eabm6265. doi: 10.1126/scisignal.abm6265
167. Dong Y, Wang P, Yang Y, Huang J, Dai Z, Zheng W, et al. PRMT5 inhibition attenuates cartilage degradation by reducing MAPK and NF-kappaB signaling. Arthritis Res Ther (2020) 22(1):201. doi: 10.1186/s13075-020-02304-x
168. Lu Q, Liu P, Miao Z, Luo D, Li S, Lu M. SIRT1 restoration enhances chondrocyte autophagy in osteoarthritis through PTEN-mediated EGFR ubiquitination. Cell Death Discovery (2022) 8(1):203. doi: 10.1038/s41420-022-00896-8
169. Mao D, Wu M, Wei J, Zhou X, Yang L, Chen F. MicroRNA-101a-3p could be involved in the pathogenesis of temporomandibular joint osteoarthritis by mediating UBE2D1 and FZD4. J Oral Pathol Med (2021) 50(2):236–43. doi: 10.1111/jop.13131
Keywords: ubiquitination, deubiquitination, osteoarthritis, E3 ligase, DUBs
Citation: Zheng C, Chen J, Wu Y, Wang X, Lin Y, Shu L, Liu W and Wang P (2023) Elucidating the role of ubiquitination and deubiquitination in osteoarthritis progression. Front. Immunol. 14:1217466. doi: 10.3389/fimmu.2023.1217466
Received: 05 May 2023; Accepted: 01 June 2023;
Published: 09 June 2023.
Edited by:
Dong Ren, UC Irvine Medical Center, United StatesReviewed by:
Jiateng Zhong, Xinxiang Medical University, ChinaCopyright © 2023 Zheng, Chen, Wu, Wang, Lin, Shu, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Wang, d2FuZ3BldGVyMkBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.