 Tamisa Seeko Bandeira Honda
Tamisa Seeko Bandeira Honda John Ku
John Ku Hans-Joachim Anders
Hans-Joachim Anders
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 July 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1214289
This article is part of the Research Topic Community Series in NLRP3 Activation and Regulation in Innate Immune Responses: Volume II View all 9 articles
The NLRP3 inflammasome transforms a wide variety of infectious and non-infectious danger signals that activate pro-inflammatory caspases, which promote the secretion of IL-1β and IL-18, and pyroptosis, a pro-inflammatory form of cell necrosis. Most published evidence documents the presence and importance of the NLRP3 inflammasome in monocytes, macrophages, and neutrophils during host defense and sterile forms of inflammation. In contrast, in numerous unbiased data sets, NLRP3 inflammasome-related transcripts are absent in non-immune cells. However, an increasing number of studies report the presence and functionality of the NLRP3 inflammasome in almost every cell type. Here, we take a closer look at the reported cell type-specific expression of the NLRP3 inflammasome components, review the reported inflammasome-dependent and -independent functions, and discuss possible explanations for this discrepancy.
The nucleotide-binding oligomerization domain (NOD) - and leucine-rich repeat (LRR)-containing receptor pyrin domain-containing (NLRP)-3 have inflammasome-dependent and -independent functions. The NLRP3 inflammasome is a cytosolic pattern recognition platform that integrates infectious and non-infectious danger signals into the secretion of IL-1β and IL-18. The NLRP3 inflammasome is formed by the assembly of NLRP3, the adaptor molecule ASC, and caspase-1, which results in caspase-1 activation, which cleaves pro-IL-1β and pro-IL-18 into their active forms (1). IL-1β and IL-18 can be released actively through gasdermin pores in the plasma membrane or passively during pyroptosis, a type of inflammatory cell death (1, 2). The activation of inflammasomes must be tightly regulated so as to prevent both systemic and tissue inflammation. For example, cryopyrin-associated periodic syndrome (CAPS) results from dysregulated NLRP3 activity due to gain-of-function mutations in NLRP3, which leads to abnormal activation of the NLRP3 inflammasome (3, 4). Overproduction of IL-1β has harmful effects in CAPS patients such as hearing loss, urticaria-like rash, joint pain, and inflammation of the eyes, bones, and central nervous system (5, 6). IL-1 antagonists reverse systemic and local inflammation and protect organs in CAPS patients (7–9). Mice carrying the same gain-of-function mutations in the Nlrp3 gene develop systemic inflammation accompanied by growth retardation and early mortality (10). Conversely, mice lacking Nlrp3, or molecules involved in inflammasome assembly, document the deleterious effect of NLRP3 inflammasome activation in sterile inflammation models (11–14).
The vast majority of data on the NLRP3 inflammasome comes from cells of the myeloid lineage. Indeed, single-cell transcriptome data sets consistently report the absence of NLRP3, ASC, caspase-1, and IL-1β transcripts from cell types other than myeloid cells (https://www.proteinatlas.org/). These data are consistent with the absence of IL-1β protein in parenchymal tissues. Cell type-specific modes of immune activation are an important element in limiting potentially harmful inflammation, cytokine storms, and immunopathology. Nevertheless, a surprising number of studies report NLRP3 expression and activity, and even IL-1β secretion, by human and mouse epithelial cells and other parenchymal tissue cells (Table 1). This is not supported by many unbiased expression data sets.
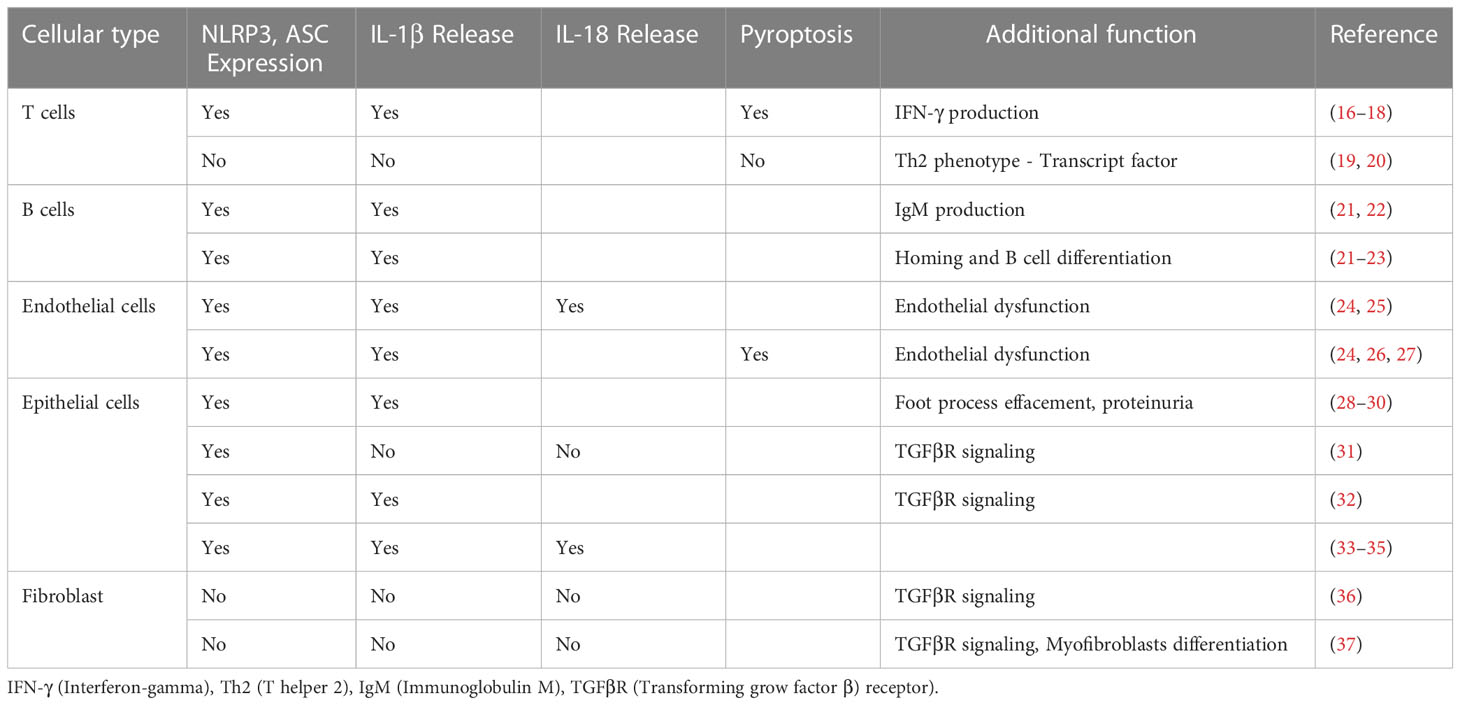
Table 1 Some experimental studies reporting expression and function of the NLRP3 inflammasome in non-myeloid cells.
Here we review the published data on the inflammasome-dependent and inflammasome-independent roles of NLRP3 to better understand the function of NLRP3 in tissue-specific danger signaling and beyond the secretion of IL-1β (Figure 1). Furthermore, we call for a critical discussion of the discrepancy between the apparent absence of transcripts of NLRP3 inflammasome components and of many cell types that are reported to carry out NLRP3 inflammasome functions. In addition, we discuss the experimental tools used in this context.
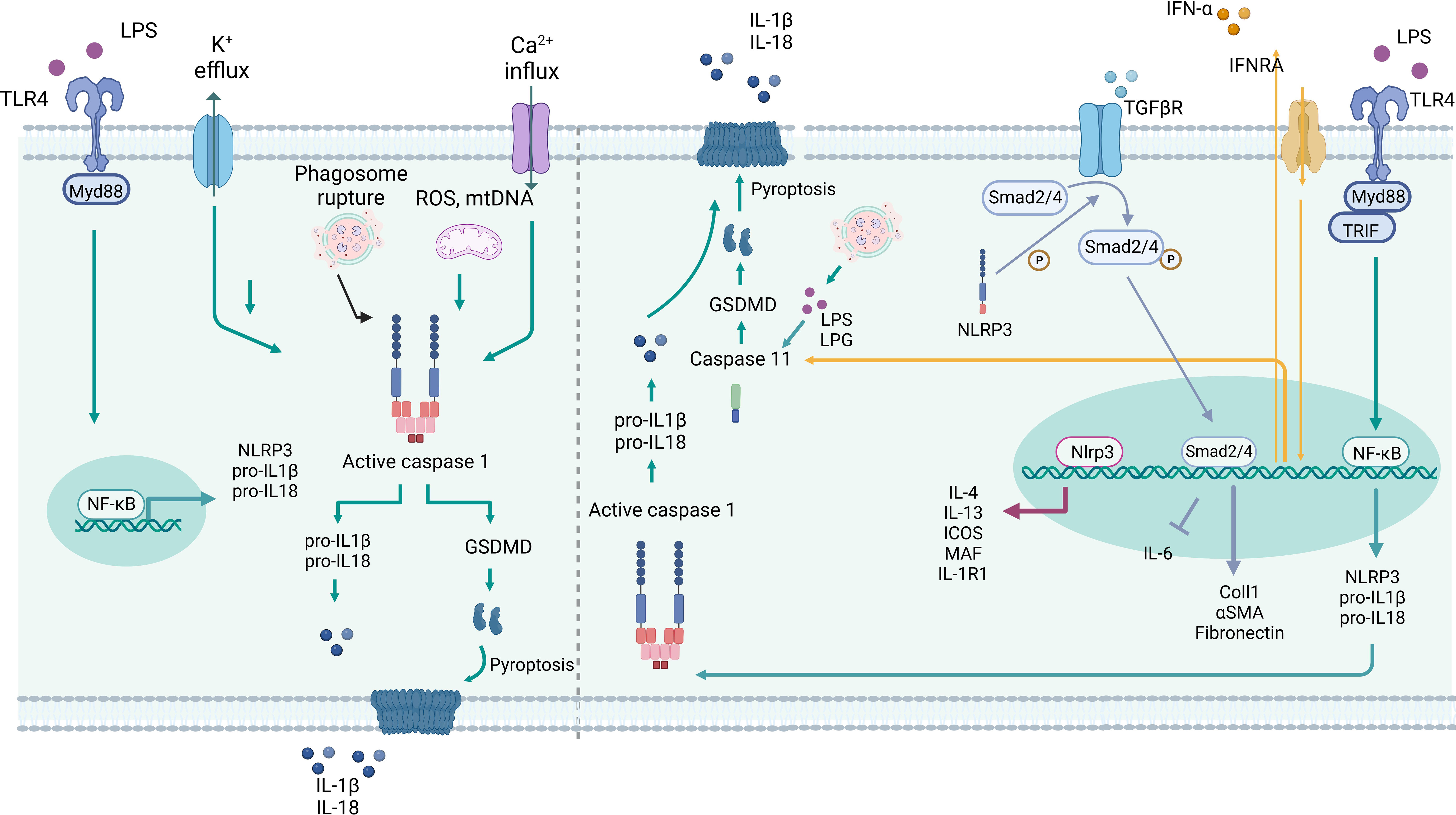
Figure 1 Inflammasome-dependent and –independent roles of NLRP3 in myeloid cells. Left: Canonical activation of NLRP3 starts with TLR activation, mediate by Myd88 and NF-κB, which triggers the expression of NLRP3, pro-IL-1β and -IL-18. Several stimuli can trigger the inflammasome assembly, such as K+ efflux, Ca+2 influx, crystals, phagosome rupture, mtROS and mtDNA. The conjunction of NLRP3, ASC and Caspase 1 lead to Caspase 1 activation and proteolytic cleavage of pro-IL-1β and -IL-18, as well GSDMD resulting in IL-1β and -IL-18 secretion and/or pyroptosis. Right: Non-canonical NLRP3 activation is triggered by TLR activation, via Myd88 and TRIF, or type I interferon receptor (IFRA), which lead to NF-κB activation and induction of NLRP3, pro-IL-1β and -IL-18 expression. The type I interferon triggers caspase-11, and caspase 1 expression. LPS and LPG can bind and directly activate caspase 11, which cleaves GSDMD in GSDMD-N driving pyroptosis. Nlrp3 can act as a transcription factor in T cells by binding directly to the Il4, Il13, Icos, Maf, Il1r1 promoter region. Nlrp3 can induce phosphorylation of Smad2/4 in the TGFβR pathway, thereby regulating the transcription of target genes. Created with BioRender.com.
NLRP3 inflammasome activation is preceded by the canonical and non-canonical pathways. Both pathways initiate the induction of inflammasome components, and their substrates, following cytokine or Toll-like receptor (TLR) activation. This increases the transcription of pro-IL-1β, pro-IL-18, and other NLRP3-related molecules via the translocation of NF-κB into the nucleus. In the canonical pathway, danger signals promote the oligomerization of NLRP3, the adaptor protein ASC, and pro-caspase-1. Once activated, caspase-1 cleaves pro-IL-1β and pro-IL-18 at aspartate residues to generate the effector forms of these cytokines (15). Furthermore, caspase-1 activation leads to the cleavage of gasdermin D (GSDMD), which can disrupt inner membrane lipids (phosphatidylserine and phosphatidylinositol) and cell osmolality, triggering pyroptosis and releasing alarmins such as IL-1α, IL-1β, and IL-18 (15).
Non-canonical NLRP3 inflammasome activation involves activation of caspases 4 and 5 in humans and caspase-11 in mice (15). Prior to this, NF-κB translocates to the nucleus, where it increases the transcription of interferon regulatory factors (IRF) 3 and 7, and induces the expression of IFN-α/β (Figure 2). These IFNs trigger the expression of caspase-11, which cannot cleave pro-IL-1β to IL-1β, but through the induction of caspase-1, the active form of IL-1β can be generated, i.e., the non-canonical pathway to induce IL-1β (38). In addition, caspase-11 can be activated by physically binding to LPS (39) or lipophosphoglycan (LPG) (40). Once activated, caspase-11 can also induce pyroptosis by cleaving GSDMD. The main role of the non-canonical NLRP3 is to protect against Gram-negative bacteria that evade the phagosome and invade the cytosol (41). In this way, Cheng et al. showed that LPS can activate lung endothelial cells towards pyroptosis, which is abolished in Casp11-deficient endothelial cells (42).
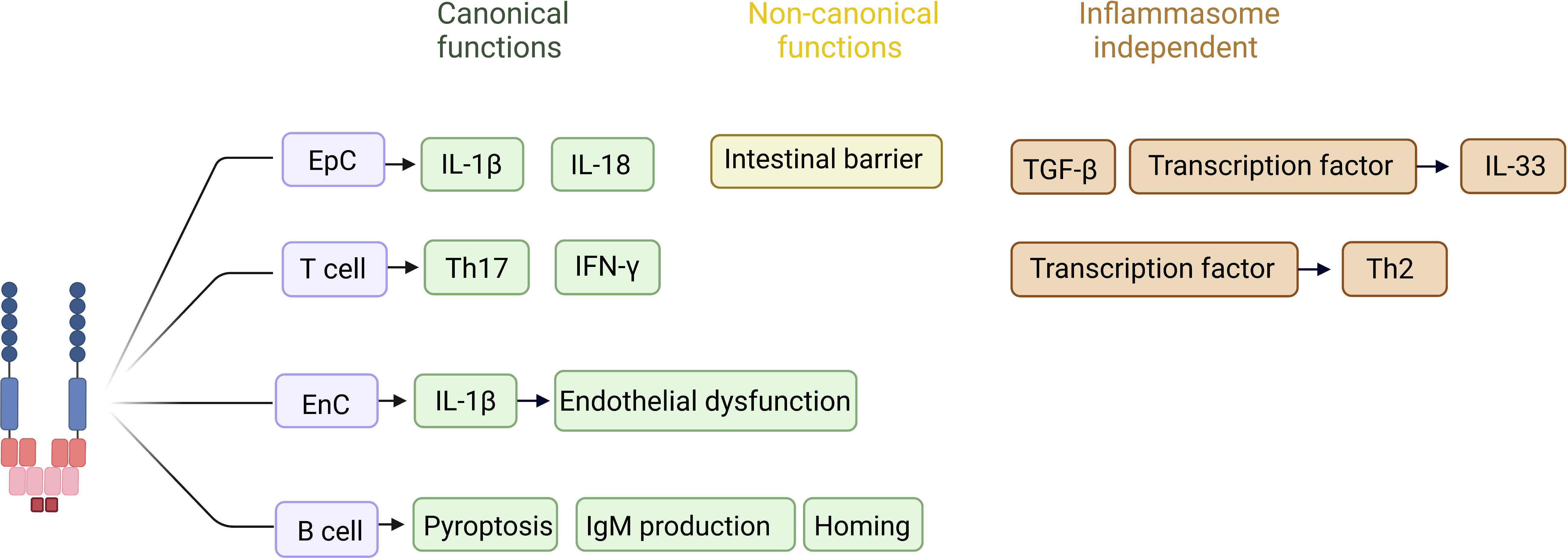
Figure 2 Reported inflammasome-dependent and -independent functions of NLRP3 in non-immune cells. In T cells, canonical activation of NLRP3 has been described to favor differentiation of naive T cells into Th17 cells, as well as to increase IFN-γ production in the face of viral infection. In an inflammasome independent way, the NLRP3 acts as a transcription factor in T cells, favoring a Th2 profile. In epithelial cells, canonical activation of NLRP3 is related to the production and release of IL-1β and IL-18 during inflammatory processes, whereas the non-canonical pathway favors the maintenance of the intestinal epithelial barrier. In epithelial cells the inflammasome independent function is related with TGF-β pathway activation and fibrosis, as well acting as a transcription factor controlling the expression of IL-33. The role of NLRP3 in endothelial cells has been pointed out during inflammatory processes, in which IL-1 β production favors endothelial dysfunction and increased expression of selectins, acting on the recruitment of inflammatory cells to the injured site. In B cells NLRP3 stimulates the IgM production, as well the expression of CXCR4 and CCR7, homing related chemokines. *EpC (epithelial cell). *EnC (endothelial cell). Created with BioRender.com.
An inflammasome-independent function of NLRP3 is the promotion of fibrosis through regulation of the TGF-β signaling pathway (31, 32, 37, 43). The absence of Nlrp3 in epithelial cells, but not caspase 1, IL-1β, or IL-18, reduced their ability to produce αSma and Mmp9, and decreased epithelial-mesenchymal transition when stimulated with TGF-β (31). Numerous studies demonstrate that Nlrp3 regulates TGF-β through the phosphorylation of Smad2/3 (31, 37, 44–46). Furthermore, NLRP3 acts as a transcription factor in the nucleus of T cells, promoting Th2 polarization through its ability to bind directly to DNA, specifically in the promoter regions of Il-4, Il-13, Il1r1, Icos, and Maf (19). Similarly, Nlrp3 acts as a transcription factor and regulates the expression of IL-33 in epithelial cells (47). In addition, Nlrp3 deficiency or inhibition prevented the production of IL-33 in a model of atopic dermatitis (47). We will discuss in more detail the functions of NLRP3 that are not related to inflammasome activation in specific cell types.
Monocytes and macrophages are mononuclear phagocytes that play a key role in innate immunity and that have distinct roles in tissue homeostasis and immune response (48). Both cell types can recognize pathogen- and damage-associated molecular patterns that trigger inflammation and immunity. Monocytes are circulating cells that differentiate into tissue macrophages upon transmigration into injured tissues, whereas tissue macrophages recognize, phagocytose, and degrade pathogens or cellular debris, contributing to tissue repair and fibrosis (49).
In mononuclear phagocytes, a broad spectrum of exogenous stimuli ranging from crystalline microparticles to viral proteins (SARS-CoV viroporin, hepatitis C virus core protein, and influenza virus M2) can activate the NLRP3 inflammasome (50–52). In addition, NLRP3 acts as a sensor of host-derived danger signals and can detect changes in membrane lipids, ion efflux, mitochondrial dysfunction, and the production of reactive oxygen species (ROS) (53, 54). The ability to secrete large amounts of IL-1β and ROS contributes to the defense against pathogens, but the mechanism by which these effector molecules control pathogen load differs from one type of microorganism to the next (55–57) (57–59). The mechanism by which macrophages resist intracellular protozoan infection depends on IL-1β production via activation of the NLRP3 inflammasome. Binding to IL-1R and activation of Myd88 signaling trigger NO production (55, 57). The dependence of ROS on NLRP3 inflammasome activation has also been shown in a model of the influenza virus, where treatment of infected macrophages with N-acetyl-L-cysteine (NAc) reduced inflammasome-dependent IL-1β production and, consequently, impaired pathogen control (58).
NLRP3 activation is associated with a change in macrophage phenotype, a mechanism independent of IL-1β (36, 60–62). Indeed, the lack of silencing of NLRP3 reduces the number of anti-inflammatory macrophages and/or their ability to secrete anti-inflammatory cytokines, such as IL-10 and IL-4 (36, 61). However, NLRP3 inhibition favors macrophage infiltration and/or macrophage reprogramming to a pro-inflammatory phenotype in models of chronic kidney disease (CKD) and pancreatic cancer (62–64) .
The fact that macrophages do not require a priming step to assemble the components of the NLRP3 inflammasome highlights the importance of regulatory mechanisms to avoid chronic and systemic inflammation (65). Macrophage migration inhibitory factor (MIF) regulates NLRP3 inflammasome assembly and activation via the inhibition of ASC speck formation and caspase-1 cleavage, rather than by interfering with NF-κB activation (66). Another mechanism of regulation of NLRP3 in macrophages is the methylation of Cpt1a, a gene involved in ROS and energy production via fatty acid oxidation and oxidative phosphorylation (67).
Dendritic cells are professional antigen-presenting cells due to their ability to induce activation and differentiation of naive T cells (68). DCs upregulate inflammasome components upon a priming signal via NF-κB-dependent transcription (69, 70).
In DCs, the activation of the NLRP3 inflammasome can lead to pyroptosis or hyperactivation (71). During hyperactivation, DCs produce high amounts of IL-1β while remaining viable and increase their migratory ability to lymph nodes (72, 73). Furthermore, it was observed that through IL-1β production, NLRP3 induces a lowered tolerogenic profile in DCs (74). In addition to its role in the activation of DCs, IL-1β secretion is essential for the priming of naive T cells. DCs lacking Casp1 fail to activate CD8+ T cells (75).
In different pathological models, activation of the NLRP3 inflammasome in DCs can have deleterious effects. Mice lacking Nlrp3 or by gene inhibition, have been shown to favor DCs toward a tolerogenic phenotype, leading to a better disease outcome (74, 76–78). This is in surprising contrast to the role of NLRP3 in a model of lupus-like systemic autoimmunity, where the absence of Nlrp3 and Casp1 accelerated the systemic autoimmune process; NLRP3 is needed to maintain the immunosuppressive effect of TGFβR signaling that counterbalances adaptive immunity (45). In this disease context, NLRP3 contributes to the generation of suppressor DCs (79). Indeed, NLRP3 -/- and Casp1 -/- DCs show less Smad2/4 phosphorylation, which is required in the TGFβR signaling pathway to suppress the expression of pro-inflammatory cytokines such as IL-6 (44, 45).
Neutrophils are the most abundant granulocyte in blood circulation and are the fastest line of defense against bacterial and fungal infections (80). When confronted with a microorganism, neutrophils can release antimicrobial proteins from their granules, ROS from their cytosol, and extracellular traps (NETs) to entrap pathogens (81). The same effector mechanisms of neutrophils are involved in sterile inflammation (82).
Knowledge about NLRP3 and neutrophils is not as well established as knowledge about macrophages and dendritic cells. Neutrophils possess the machinery required for IL-1β production, in an NLRP3 inflammasome-dependent manner in response to Staphylococcus aureus infection (83, 84). Furthermore, neutrophils carrying gain-of-function mutations in Nlrp3 have been observed to increase cytoplasmic granule exocytosis as well as NETosis capacity (85, 86).
In neutrophils, NLRP3 positively regulates their recruitment to an inflammatory site through IL-1β production. Once in the absence of Nlrp3, neutrophils secrete lower amounts of IL-1β, which in turn decreases the activation of endothelial cells and the expression of P-selectin, which is necessary for neutrophil rolling and transmigration (87).
In a model of Streptococcus pneumonieae infection, tumor necrosis factor-α (TNF-α) and pneumolysin toxin can trigger IL-1β production through NLRP3 inflammasome activation. IL-1β production is required to activate γδT17 cells and, consequently, IL-17 production, which supports infection control (88). On the other hand, inhibition of NLRP3 and caspase-1 increased ROS production by neutrophils and their ability to control the levels of uropathogenic Escherichia coli (89).
The mechanism connecting the NLRP3 inflammasome to NET extrusion or even NETosis is caspase-1-independent. Chen et al. showed in a PAMP-rich milieu, that neutrophils produce IL-1β, but do not undergo caspase-1-dependent pyroptosis, due to the inability of caspase-1 to cleave the GSDMD pore-forming p30 fragment (90, 91). On the other hand, caspase-11 is more efficient at cleaving GSDMD in neutrophils. The combination of high caspase-11 expression and cleaved GSDMD is necessary for neutrophil plasma membrane rupture and NET extrusion, which acts as a defense mechanism against cytoplasmic microorganisms (90, 92). The requirement for plasma membrane rupture for NET release is still under debate, as it has recently been shown that NETs can be released from viable neutrophils that have not undergone pyroptosis (93). This distinct resistance to pyroptosis may be explained by the ability of neutrophils to resist mitochondrial depolarization, which is a trigger for NLRP3 inflammasome activation (91).
In a model of hepatic ischemia/reperfusion, the absence of Nlrp3 was associated with fewer neutrophil infiltrates, but not in Asc -/- or Casp1-/-mice, which indicates an inflammasome-independent role of Nlrp3 in neutrophils. Mechanistically, Nlrp3-deficient neutrophils are less responsive to chemokines that guide their migration (94).
The role of NLRP3 in eosinophils has not been well characterized. In a helminth infection model, Alhallaf et al. observed that the absence of NLRP3 increased the number of eosinophils in mesenteric lymph nodes (95). This phenomenon was also found in allergic mice and could be explained by a microenvironment with high levels of IL-33, IL-13, and IL-5 (96).
Mast cells are granulocytes found in connective tissues throughout the body. Their localization around blood vessels enables them to regulate vasodilation and vascular homeostasis by secreting angiogenic or vasoactive factors, including vascular endothelial growth factor (VEGF), TNFα, and histamine (97).
Like the other granulocytes, mast cells have functional inflammasome components to activate caspase-1 and induce the secretion of IL-1β. In the skin of CAPS patients, mast cells are the major producers of IL-1β; cell-producers in the skin of CAPS patients are mast cells (98). The ability of mast cells to produce IL-1β has also been demonstrated in a model of endometriosis, in which estrogen-stimulated mast cells can produce IL-1β via K+ efflux. Furthermore, it has been observed that the NLRP3 promoter region contains estrogen responsive elements (99). The anti-allergic drug Tranilast can directly inhibit the NLRP3 inflammasome assembly and reduce IL-1β and IL-6 production (100).
Lymphocytes are produced in the bone marrow and mostly patrol and reside in lymphoid tissues, where they can undergo maturation and clonal expansion upon presentation of their cognate antigens as part of antigen-specific adaptive immune responses (16). These lymphocyte lineage cells can be divided into T and B cells, which are part of the adaptive immune system, while NK cells and other innate lymphocytes support innate immunity (16).
According to RNAseq analysis provided by the Human Protein Atlas, T cells express the machinery necessary to assemble the NLRP3 inflammasome (NLRP3, CASP1, and PYCARD), while B cells express only CASP1 and PYCARD.
Studies in mice carrying an NLRP3 gain-of-function mutation have shown that hyperactivation of the NLRP3 inflammasome induces differentiation of naive T cells into Th17 cells, in a mechanism supported by high levels of IL-1β (17, 101). The role of NLRP3 in regulating T cell phenotype has been described in HIV patients and models of autoimmune disease (18, 20). In addition, autocrine canonical activation of the NLRP3 inflammasome in CD4+ T cells is necessary to enhance IFN-γ production during viral infection (102). During intracellular microorganism infection, the NLRP3 inflammasome-caspase-1 axis induces T cell pyroptosis, which is considered to be the major mechanism associated with T cell depletion (20, 21, 23).
In a mechanism independent of IL-1β generation, NLRP3 can regulate T cell subsets by promoting a Th2-profile (22). Due to its nuclear localization in Th2 cells, NLRP3 may act as a transcription factor capable of binding to promoter regions of genes involved in the Th2 signature, such as IL-4, IL-13, IL1R1, ICOS, and MAF (19, 103).
B cells are key regulators of the adaptive humoral immune system and are responsible for the production of immunoglobulins directed against pathogen-related antigens (104). Unlike T cells, which clearly express the NLRP3 inflammasome, RNAseq analysis indicates that B cells express low amounts of NLRP3, although they express ASC and CASP1. B cells are capable of canonically activating the NLRP3 inflammasome and driving IL-1β production upon stimulation with β-glucan, a fungal antigen, or even with B cell activating factor (BAFF) (105, 106).
Although the role of NLRP3 in B cells is not yet fully understood, it is believed to be associated with the maintenance of an inflammatory environment that favors the development, homing, and retention of B cells in lymphoid organs. When stimulated with BAFF (one of the major pro-survival factors involved in B cell homeostasis), human B cells assemble the NLRP3-caspase-1 complex, triggering IL-1β production (107). When activated in the absence of Nlrp3, B cells produce lower IgM titers (106). The absence of Nlrp3 was associated with a lower expression of markers of B cell homing and differentiation, such as CXCR4 and CCR7 (105, 106). However, it has not yet been proven that this function is associated with the activation of the NLRP3 inflammasome.
Endothelial cells form the inner lining of the heart, blood, and lymphatic vessels. In addition to their barrier function, endothelial cells help maintain the immune privilege of certain tissues. Under homeostatic conditions, endothelial cells produce antiplatelet and anticoagulant molecules to prevent platelet aggregation and fibrin formation.
During endothelial dysfunction, endothelial cells express adhesion molecules and chemokines that allow for the recruitment and transmigration of immune cells to the inflamed sites and the upregulation of procoagulant mediators, such as tissue factor and von Willebrand factor (87, 108). Chronic inflammation is considered one of the main causes of endothelial dysfunction (24). IL-1β and ROS, among other inflammatory mediators, can activate endothelial cells and contribute to their dysfunction and are produced by circulating immune cells, e.g., neutrophils (25, 87). The Human Protein Atlas reports transcript expression under basal conditions in endothelial cells from different organs that do not appear to express relevant levels of NLRP3 mRNA, although they do express ASC and CASP1.
Diabetes, obesity, atherosclerosis, and stroke are triggers that can activate endothelial cells to increase NLRP3 expression, which may contribute to endothelial dysfunction (26, 27, 109). DAMPs (HMGB1 and cold-inducible RNA-binding protein) increase the expression of the NLRP3 inflammasome in endothelial cells (110–112). However, activation of the NLRP3 inflammasome generates high concentrations of IL-1β, which contributes to endothelial injury (111). The cleavage of GSDMD and induction of pyroptosis, by both canonical and non-canonical pathways, have been observed in endothelial cells under pathological conditions (28, 110, 112). Some approved drugs, such as statins, hypoglycemic agents, and anti-inflammatory drugs reduce endothelial injury by inhibiting NLRP3 inflammasome activation (27, 29, 33, 109).
Epithelial cells cover body surfaces and cavities and provide barrier function and transepithelial exchange of fluids, ions, metabolites, and other nanomolecules as part of absorption, excretion, and filtration. The Human Protein Atlas reports that under healthy conditions, epithelial cells from the kidneys, lung, skin, intestine, and other organs lack NLRP3 expression, which for methodological reasons does not exclude low-level transcripts. NLRP3 inflammasome-related transcripts (ASC and CASP1) are present in goblet cells, are poorly expressed in keratinocytes, and are not detectable in lung or kidney epithelial cells. However, NLRP3 induction under stress conditions has been demonstrated (30, 34, 35, 113).
Although kidney epithelial cells do not appear to express much of NLRP3 mRNA under healthy conditions, kidney biopsies from patients with lupus nephritis showed an increase in NLRP3 expression in podocytes (34). The increase in NLRP3 was accompanied by higher expression of CASP1 and ILBβ and correlated positively with higher levels of proteinuria (114). In a similar manner, pharmacological inhibition of Nlrp3 improved kidney function in a model of APOL1-associated podocytopathy through the reduction of Il1b and IL-6 (35). Podocyte-specific depletion of NLRP3 demonstrated a positive role for the NLRP3 inflammasome in protecting podocytes from glomerular pathology in a mouse model of diabetic kidney disease. Evidence also points to NLRP3 exerting a non-canonical effect on podocytes, as the absence of caspase-1 and IL-1β was only partially protective (115).
Beyond the classical pro-inflammatory roles of NLRP3, in vitro studies with primary tubular epithelial cells (TECs) isolated from Nlrp3-deficient mice showed that NLRP3 regulates the TGF-β pathway in an inflammasome-independent manner. Using NLRP3-/- TECs, Wang et al. observed a reduction of EMT markers, such as TGFβ-1, MMP-9, and ACTA2 after stimulation with TGF-β1 (31). A similar study in lung epithelial cells showed that inhibition of NLRP3 prevented EMT by upregulating CHD1, while downregulating TGFβ-1, and ACTA2, preventing morphological changes towards a fusiform shape (32). In contrast, anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3 inflammasome-mediated activation of caspase-1 (116).
Keratinocytes are specialized epithelial cells of the epidermis that constitutively express pro-IL-1β, as a defense mechanism against foreign antigens found in the skin. However, under homeostatic conditions, keratinocytes cannot release the active form of IL-1β (117).
Skin inflammation caused by UVB irradiation induces caspase-1 activation and IL-1β processing by increasing cytoplasmic Ca2 (113, 118). Viral dsDNA and self-DNA, released from cells damaged by mechanical insults, UVB inflammation, and/or skin diseases such as psoriasis, can lead to increased NLRP3 activation (119, 120) In a psoriasis model, it was observed that keratinocytes stimulated by IL-17 and IL-22 upregulated Il1β, via ROS-induced NLRP3 activation (121). The use of drugs that inhibit NLRP3 and/or caspase-1 activation, such as metformin and ginsenoside Rg1, has shown a positive effect in the treatment of psoriasis-like lesions by inhibiting Il1b and keratinocyte proliferation (122, 123).
Recently, NLRP3 has been shown to localize to the keratinocyte nucleus, act as a transcription factor, and regulate IL-33 expression. Lack of NLRP3 has been shown to reduce IL-33 mRNA and protein levels, thereby improving the lesions associated with topical dermatitis (47).
Intestinal epithelial cells have pleiotropic functions ranging from hormone-like secretion to regulation of the intestinal microbiota and the host immune system. Maintenance of the epithelial barrier in the intestine is essential to prevent microorganism translocation.
In a model of Citrobacter rodentium infection, it was observed that mice lacking NLRP3 and Casp1 were more susceptible to bacterial penetration into the intestinal crypts (124). In a model of dextran sodium sulfate colitis, it was observed that Il1β played a role in intestinal epithelial repair and epithelial barrier formation. Il1β deficiency reduced the proliferation of intestinal epithelial cells and the expression of tight junction proteins while impairing intestinal permeability (125). Similarly, DNA sequencing analysis showed that mutations in the downstream regulatory region of NLRP3 are associated with lower expression of Il1β and consequently increased susceptibility to Crohn’s disease (126). Taken together, these data show that activation of the NLRP3 inflammasome in intestinal epithelial cells plays an important physiological role in maintaining the intestinal barrier and limiting pathogen colonization.
Non-canonical activation of NLRP3 via activation of caspase-11 positively regulates the integrity of the intestinal epithelium by stimulating intestinal epithelial cell proliferation. In a model of colitis, it was observed that the absence of Casp11 impaired IL-18 production, increasing susceptibility to colitis (127, 128). Although the function of cytokines related to the activation of the NLRP3 inflammasome in the protection of the intestinal barrier is recognized, its origin seems to be unclear, since it has been observed that in the absence of caspase-11, there is a compensation of caspase-1 expression and the production of IL-1β (129), which has a protective role in intestinal epithelial cells (125).
Hepatocytes are the main parenchymal cells in the liver involved in detoxification, lipid metabolism, albumin synthesis, and the secretion of coagulation factors. The Human Protein Atlas shows that under homeostatic conditions, hepatocytes do not express NLRP3 and ASC, although they express low levels of CASP1. Some studies show that diabetes, liver inflammation, and steatosis induce the expression of Nlrp3, Casp1, and Il1b in the liver tissue, including immune and non-immune cells (46, 124, 125). Activation of the NLRP3 inflammasome by ROS induces the release of IL-1β and IL-18 by hepatocytes, which then undergo pyroptosis (130). Hepatocytes carrying gain-of-function mutations were sufficient to drive spontaneous collagen deposition in the liver (131). Conversely, hepatocytes with Nlrp3-deficiency and/or pharmacological inhibition of Nlrp3 ameliorate liver inflammation by reducing IL-1β, IL-6, and TNF-α production; they also ameliorate liver fibrosis by reducing Col1a and aSma. Together, this information shows that canonical inflammasome activation in hepatocytes is sufficient to propagate liver injury and fibrosis.
There is a discrepancy between transcriptional data obtained from RNAseq analysis and functional studies of the NLRP3 inflammasome in non-immune cells. One of the possible explanations is the low sensitivity of single-cell RNA sequencing analysis, which may miss low levels of transcripts under homeostatic conditions. However, different RNA sequencing data sets and data available on the Human Protein Atlas from certain disease conditions, such as diabetes, ischemia-reperfusion, or tissue remodeling, still do not report NLRP3 inflammasome-related transcripts in epithelial cells (GSE131882, GSE119531, and GSE206084). The most common form of functional evidence is provided by in vitro studies employing immortalized cell lines that may have undergone somatic mutations and phenotypic changes over time. In this regard, primary cell cultures would seem to provide more solid evidence. However, such primary isolates are frequently contaminated by tissue-resident immune cells, leading to erroneous conclusions. The use of non-specific antibodies in immunohistochemistry and Western blotting may also be one of the reasons for this discrepancy (132). In fact, less than half of the routinely used antibodies bind to their specific targets (133). Therefore, the specificity of the NLRP3 antibody should be questioned, especially when used as a negative control, when documenting tissue samples as NLRP3-deficient.
The use of constitutive NLRP3 knockout animals has greatly contributed to several discoveries about the canonical role of the NLRP3 inflammasome, mainly in immune cells. However, such animals cannot determine the cell type-specific effects of the NLRP3 inflammasome. Similarly, NLRP3 inhibitors or whole-body knockout mice cannot attribute an improvement of the condition to the sole and exclusive inhibition of the inflammasome in parenchymal cells without considering the participation of immune cells (27, 29, 109, 122). Thus, studies concluding on NLRP3 inflammasome activity in non-immune cells have to be carefully evaluated for methodological consistency and possible involvement of myeloid cells.
The use of cell type-specific knockout mice, e.g., with a floxed NLRP3 motif under the control of cell type-specific expression of Cre recombinase, may help to determine the role of the NLRP3 inflammasome in parenchymal cells. However, such experiments need a series of controls to validate the cell type-specific depletion and exclude Cre leakage or epigenetic silencing of Cre recombinase (134–136). Another approach to evaluating the role of NLRP3 in specific cell types is through tools that induce NLRP3 expression, such as the use of animals with gain-of-function mutations and the transfection of cells with the gene of interest. For example, animals with the human A350V gain-of-function mutation and/or deletion of NLRP3 in podocytes have shown that activation of this inflammasome is sufficient to promote the glomerular damage observed in animals with diabetic kidney disease (115). In a similar manner, the specific deletion of Nlrp3 in epithelial cells demonstrated the importance of this inflammasome in promoting TGF-β signaling (31).
NLRP3 is involved in multiple cellular mechanisms. The canonical activation of NLRP3 is well-known and understood. However, inflammasome-independent functions of NLRP3 may play various roles in different cell types.Here we show that NLRP3 can act to induce tissue fibrosis by enhancing TGF-β receptor signaling and fibrosis-associated markers (31, 32, 44). NLRP3 can also regulate the translocation of intestinal microorganisms and enhance endothelial cell damage by increasing the expression of selectins, which promote neutrophil recruitment and thereby NETosis and granule secretion.
However, many of the cell-specific functions attributed to NLRP3 are inconsistent with the expression of its transcripts in various tissues and/or cell types, such as lung and kidney epithelial cells. It is likely that many of these conflicting results are due to problematic in vitro tools, such as immortalized cell lines of untested nature or, in the case of primary cells, possible immune cell contamination. Thus, the use of tools to delete or induce NLRP3 or its related molecules in specific cell types, e.g., knockout, knockin, should be better explored so that new therapeutic alternatives can be created and even expand beyond merely immune-mediated diseases.
All authors contributed equally to this review. All authors contributed to the article and approved the submitted version.
TH received support from the Deutsche Akademischer Auslandsdienst (57552338). H-JA received support from the Deutsche Forschungsgemeinschaft (AN372/27-1 and 30-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
2. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature (2021) 593:607–11. doi: 10.1038/s41586-021-03478-3
3. Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM, et al. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest (2013) 123:4695–705. doi: 10.1172/jci71543
4. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
5. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J, et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20:319–25. doi: 10.1016/s1074-7613(04)00046-9
6. Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol (2019) 39:277–86. doi: 10.1007/s10875-019-00638-z
7. Leinonen M, Hallén B, Olivecrona H. The IL-1 receptor antagonist anakinra (kineret®) stabilizes the NLRP3 mutation-specific risk for hearing loss in patients with severe cryopyrin-associated periodic syndromes (CAPS). Pediatr Rheumatol (2014) 12:P76. doi: 10.1186/1546-0096-12-S1-P76
8. Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch Dermatol (2006) 142:1591–7. doi: 10.1001/archderm.142.12.1591
9. Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis rheumatism (2008) 58:2443–52. doi: 10.1002/art.23687
10. Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity (2009) 30:875–87. doi: 10.1016/j.immuni.2009.05.005
11. Kim HJ, Lee DW, Ravichandran K, Keys DO, Akcay A, Nguyen Q, et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther (2013) 346:465–72. doi: 10.1124/jpet.113.205732
12. Zhong C, Wang R, Hua M, Zhang C, Han F, Xu M, et al. NLRP3 inflammasome promotes the progression of acute myeloid leukemia via IL-1β pathway. Front Immunol (2021) 12:661939. doi: 10.3389/fimmu.2021.661939
13. Zhang Y, Huang R, Cheng M, Wang L, Chao J, Li J, et al. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome (2019) 7:116. doi: 10.1186/s40168-019-0733-3
14. Wu M, Han W, Song S, Du Y, Liu C, Chen N, et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol (2018) 478:115–25. doi: 10.1016/j.mce.2018.08.002
15. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
16. Abbas AK, Janeway CA Jr. Immunology: improving on nature in the twenty-first century. Cell (2000) 100:129–38. doi: 10.1016/s0092-8674(00)81689-x
17. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med (2012) 209:1595–609. doi: 10.1084/jem.20111453
18. Braga TT, Brandao WN, Azevedo H, Terra FF, Melo ACL, Pereira FV, et al. NLRP3 gain-of-function in CD4(+) T lymphocytes ameliorates experimental autoimmune encephalomyelitis. Clin Sci (Lond) (2019) 133:1901–16. doi: 10.1042/cs20190506
19. Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, Boidot R, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol (2015) 16:859–70. doi: 10.1038/ni.3202
20. Zhang C, Song JW, Huang HH, Fan X, Huang L, Deng JN, et al. NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1-infected patients. J Clin Invest (2021) 131(6). doi: 10.1172/jci138861
21. He X, Aid M, Ventura JD, Borducchi E, Lifton M, Liu J, et al. Rapid loss of CD4 T cells by pyroptosis during acute SIV infection in rhesus macaques. J Virol (2022) 96:e0080822. doi: 10.1128/jvi.00808-22
22. Andersen K, Eltrich N, Lichtnekert J, Anders HJ, Vielhauer V. The NLRP3/ASC inflammasome promotes T-cell-dependent immune complex glomerulonephritis by canonical and noncanonical mechanisms. Kidney Int (2014) 86:965–78. doi: 10.1038/ki.2014.161
23. Linder A, Bauernfried S, Cheng Y, Albanese M, Jung C, Keppler OT, et al. CARD8 inflammasome activation triggers pyroptosis in human T cells. EMBO J (2020) 39:e105071. doi: 10.15252/embj.2020105071
24. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi: 10.1038/nri2171
25. Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol (2017) 37:e108–14. doi: 10.1161/atvbaha.117.309813
26. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis (2015) 24:2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
27. Lv ZH, Phuong TA, Jin SJ, Li XX, Xu M. Protection by simvastatin on hyperglycemia-induced endothelial dysfunction through inhibiting NLRP3 inflammasomes. Oncotarget (2017) 8:91291–305. doi: 10.18632/oncotarget.20443
28. Ito H, Kimura H, Karasawa T, Hisata S, Sadatomo A, Inoue Y, et al. NLRP3 inflammasome activation in lung vascular endothelial cells contributes to intestinal Ischemia/Reperfusion-induced acute lung injury. J Immunol (2020) 205:1393–405. doi: 10.4049/jimmunol.2000217
29. Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang L, et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm Sin B (2019) 9:711–23. doi: 10.1016/j.apsb.2019.02.008
30. Rao X, Zhou D, Deng H, Chen Y, Wang J, Zhou X, et al. Activation of NLRP3 inflammasome in lung epithelial cells triggers radiation-induced lung injury. Respir Res (2023) 24:25. doi: 10.1186/s12931-023-02331-7
31. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol (Baltimore Md.: 1950) (2013) 190:1239–49. doi: 10.4049/jimmunol.1201959
32. Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H, et al. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res (2017) 357:328–34. doi: 10.1016/j.yexcr.2017.05.028
33. Wu LM, Wu SG, Chen F, Wu Q, Wu CM, Kang CM, et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis (2020) 293:26–34. doi: 10.1016/j.atherosclerosis.2019.11.033
34. Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol (2017) 69:1636–46. doi: 10.1002/art.40155
35. Wu J, Raman A, Coffey NJ, Sheng X, Wahba J, Seasock MJ, et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest (2021) 131(20). doi: 10.1172/jci136329
36. Kobayashi M, Usui F, Karasawa T, Kawashima A, Kimura H, Mizushina Y, et al. NLRP3 deficiency reduces macrophage interleukin-10 production and enhances the susceptibility to doxorubicin-induced cardiotoxicity. Sci Rep (2016) 6:26489. doi: 10.1038/srep26489
37. Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR Jr, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem (2014) 289:19571–84. doi: 10.1074/jbc.M114.550624
38. Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem (1996) 271:20580–7. doi: 10.1074/jbc.271.34.20580
39. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
40. de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep (2019) 26:429–437.e425. doi: 10.1016/j.celrep.2018.12.047
41. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
42. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest (2017) 127:4124–35. doi: 10.1172/jci94495
43. Alyaseer AAA, de Lima MHS, Braga TT. The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol (2020) 11:883. doi: 10.3389/fimmu.2020.00883
44. Lech M, Lorenz G, Kulkarni OP, Grosser MO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-β receptor signalling. Ann Rheum Dis (2015) 74:2224–35. doi: 10.1136/annrheumdis-2014-205496
45. Kang H, Seo E, Oh YS, Jun HS. TGF-β activates NLRP3 inflammasome by an autocrine production of TGF-β in LX-2 human hepatic stellate cells. Mol Cell Biochem (2022) 477:1329–38. doi: 10.1007/s11010-022-04369-5
46. Shi C, Wang Q, Rao Z, Shi Y, Wei S, Wang H, et al. Diabetes induces hepatocyte pyroptosis by promoting oxidative stress-mediated NLRP3 inflammasome activation during liver ischaemia and reperfusion injury. Ann Transl Med (2020) 8:739. doi: 10.21037/atm-20-1839
47. Zheng J, Yao L, Zhou Y, Gu X, Wang C, Bao K, et al. A novel function of NLRP3 independent of inflammasome as a key transcription factor of IL-33 in epithelial cells of atopic dermatitis. Cell Death Dis (2021) 12:871. doi: 10.1038/s41419-021-04159-9
48. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
49. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med (1968) 128:415–35. doi: 10.1084/jem.128.3.415
50. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11:404–10. doi: 10.1038/ni.1861
51. Negash AA, Olson RM, Griffin S, Gale M Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PloS Pathog (2019) 15:e1007593. doi: 10.1371/journal.ppat.1007593
52. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol (2019) 10:50. doi: 10.3389/fmicb.2019.00050
53. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
54. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi: 10.1016/j.immuni.2013.08.001
55. Gonçalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, et al. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PloS Negl Trop Dis (2013) 7:e2469. doi: 10.1371/journal.pntd.0002469
56. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature (2022) 606(7914):585–93. doi: 10.1038/s41586-022-04802-1
57. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, et al. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to leishmania. Nat Med (2013) 19:909–15. doi: 10.1038/nm.3221
58. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
59. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206:79–87. doi: 10.1084/jem.20081667
60. Wisitpongpun P, Potup P, Usuwanthim K. Oleamide-mediated polarization of M1 macrophages and IL-1β production by regulating NLRP3-inflammasome activation in primary human monocyte-derived macrophages. Front Immunol (2022) 13:856296. doi: 10.3389/fimmu.2022.856296
61. Liu Y, Gao X, Miao Y, Wang Y, Wang H, Cheng Z, et al. NLRP3 regulates macrophage M2 polarization through up-regulation of IL-4 in asthma. Biochem J (2018) 475:1995–2008. doi: 10.1042/bcj20180086
62. Anders HJ, Suarez-Alvarez B, Grigorescu M, Foresto-Neto O, Steiger S, Desai J, et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int (2018) 93:656–69. doi: 10.1016/j.kint.2017.09.022
63. Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med (2017) 214:1711–24. doi: 10.1084/jem.20161707
64. Anders HJ. Of inflammasomes and alarmins: IL-1β and IL-1α in kidney disease. J Am Soc Nephrol (2016) 27:2564–75. doi: 10.1681/asn.2016020177
65. Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols EM, Davis DM, et al. Priming is dispensable for NLRP3 inflammasome activation in human monocytes In vitro. Front Immunol (2020) 11:565924. doi: 10.3389/fimmu.2020.565924
66. Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun (2018) 9:2223. doi: 10.1038/s41467-018-04581-2
67. Li X, Zhang X, Xia J, Zhang L, Chen B, Lian G, et al. Macrophage HIF-2α suppresses NLRP3 inflammasome activation and alleviates insulin resistance. Cell Rep (2021) 36:109607. doi: 10.1016/j.celrep.2021.109607
68. Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol (2017) 17:30–48. doi: 10.1038/nri.2016.116
69. de Carvalho RVH, Silva ALN, Santos LL, Andrade WA, de Sá KSG, Zamboni DS. Macrophage priming is dispensable for NLRP3 inflammasome activation and restriction of leishmania amazonensis replication. J leukoc Biol (2019) 106:631–40. doi: 10.1002/jlb.Ma1118-471r
70. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
71. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol (2013) 43:3336–42. doi: 10.1002/eji.201243224
72. Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, et al. Inflammasomes within hyperactive murine dendritic cells stimulate long-lived T cell-mediated anti-tumor immunity. Cell Rep (2020) 33:108381. doi: 10.1016/j.celrep.2020.108381
73. Hatscher L, Lehmann CHK, Purbojo A, Onderka C, Liang C, Hartmann A, et al. Select hyperactivating NLRP3 ligands enhance the T(H)1- and T(H)17-inducing potential of human type 2 conventional dendritic cells. Sci Signaling (2021) 14(680). doi: 10.1126/scisignal.abe1757
74. Mak'Anyengo R, Duewell P, Reichl C, Hörth C, Lehr HA, Fischer S, et al. Nlrp3-dependent IL-1β inhibits CD103+ dendritic cell differentiation in the gut. JCI Insight (2018) 3(5). doi: 10.1172/jci.insight.96322
75. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
76. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med (2013) 210:1899–910. doi: 10.1084/jem.20130084
77. Chen S, Smith BA, Iype J, Prestipino A, Pfeifer D, Grundmann S, et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood (2015) 126:103–12. doi: 10.1182/blood-2014-12-617258
78. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab (2017) 25:1294–1304.e1296. doi: 10.1016/j.cmet.2017.04.005
79. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res (2010) 70:10161–9. doi: 10.1158/0008-5472.Can-10-1921
80. Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
81. Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discovery (2020) 19:253–75. doi: 10.1038/s41573-019-0054-z
82. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science (2017) 358:111–6. doi: 10.1126/science.aam9690
83. Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, et al. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol (Baltimore Md.: 1950) (2015) 194:1763–75. doi: 10.4049/jimmunol.1401624
84. Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1β processing in neutrophils. Eur J Immunol (2012) 42:710–5. doi: 10.1002/eji.201141921
85. Johnson JL, Ramadass M, Haimovich A, McGeough MD, Zhang J, Hoffman HM, et al. Increased neutrophil secretion induced by NLRP3 mutation links the inflammasome to azurophilic granule exocytosis. Front Cell Infect Microbiol (2017) 7:507. doi: 10.3389/fcimb.2017.00507
86. Skendros P, Papagoras C, Mitroulis I, Ritis K. Autoinflammation: lessons from the study of familial Mediterranean fever. J Autoimmun (2019) 104:102305. doi: 10.1016/j.jaut.2019.102305
87. Fukui S, Fukui S, Van Bruggen S, Shi L, Sheehy CE, Chu L, et al. NLRP3 inflammasome activation in neutrophils directs early inflammatory response in murine peritonitis. Sci Rep (2022) 12:21313. doi: 10.1038/s41598-022-25176-4
88. Hassane M, Demon D, Soulard D, Fontaine J, Keller LE, Patin EC, et al. Neutrophilic NLRP3 inflammasome-dependent IL-1β secretion regulates the γδT17 cell response in respiratory bacterial infections. Mucosal Immunol (2017) 10:1056–68. doi: 10.1038/mi.2016.113
89. Demirel I, Persson A, Brauner A, Särndahl E, Kruse R, Persson K. Activation of NLRP3 by uropathogenic Escherichia coli is associated with IL-1β release and regulation of antimicrobial properties in human neutrophils. Sci Rep (2020) 10:21837. doi: 10.1038/s41598-020-78651-1
90. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aar6676
91. Son S, Yoon SH, Chae BJ, Hwang I, Shim DW, Choe YH, et al. Neutrophils facilitate prolonged inflammasome response in the DAMP-rich inflammatory milieu. Front Immunol (2021) 12:746032. doi: 10.3389/fimmu.2021.746032
92. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
93. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res (2022) 1:732–47. doi: 10.1038/s44161-022-00108-7
94. Inoue Y, Shirasuna K, Kimura H, Usui F, Kawashima A, Karasawa T, et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J Immunol (Baltimore Md.: 1950) (2014) 192:4342–51. doi: 10.4049/jimmunol.1302039
95. Alhallaf R, Agha Z, Miller CM, Robertson AAB, Sotillo J, Croese J, et al. The NLRP3 inflammasome suppresses protective immunity to gastrointestinal helminth infection. Cell Rep (2018) 23:1085–98. doi: 10.1016/j.celrep.2018.03.097
96. Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol (2015) 7:351–65. doi: 10.1093/jmcb/mjv012
97. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol (2015) 6:620. doi: 10.3389/fimmu.2015.00620
98. Nakamura Y, Kambe N, Saito M, Nishikomori R, Kim YG, Murakami M, et al. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med (2009) 206:1037–46. doi: 10.1084/jem.20082179
99. Guo X, Xu X, Li T, Yu Q, Wang J, Chen Y, et al. NLRP3 inflammasome activation of mast cells by estrogen via the nuclear-initiated signaling pathway contributes to the development of endometriosis. Front Immunol (2021) 12:749979. doi: 10.3389/fimmu.2021.749979
100. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10(4). doi: 10.15252/emmm.201708689
101. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity (2009) 30:860–74. doi: 10.1016/j.immuni.2009.04.012
102. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T Helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science (2016) 352:aad1210. doi: 10.1126/science.aad1210
103. Huanosta-Murillo E, Alcántara-Hernández M, Hernández-Rico B, Victoria-Acosta G, Miranda-Cruz P, Domínguez-Gómez MA, et al. NLRP3 regulates IL-4 expression in TOX(+) CD4(+) T cells of cutaneous T cell lymphoma to potentially promote disease progression. Front Immunol (2021) 12:668369. doi: 10.3389/fimmu.2021.668369
104. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol (2020) 20:229–38. doi: 10.1038/s41577-019-0244-2
105. Hsu ML, Zouali M. Inflammasome is a central player in B cell development and homing. Life Sci Alliance (2023) 6. doi: 10.26508/lsa.202201700
106. Ali MF, Dasari H, Van Keulen VP, Carmona EM. Canonical stimulation of the NLRP3 inflammasome by fungal antigens links innate and adaptive B-lymphocyte responses by modulating IL-1β and IgM production. Front Immunol (2017) 8:1504. doi: 10.3389/fimmu.2017.01504
107. Lim KH, Chen LC, Hsu K, Chang CC, Chang CY, Kao CW, et al. BAFF-driven NLRP3 inflammasome activation in B cells. Cell Death Dis (2020) 11:820. doi: 10.1038/s41419-020-03035-2
108. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord (2015) 15:130. doi: 10.1186/s12872-015-0124-z
109. Luo X, Hu Y, He S, Ye Q, Lv Z, Liu J, et al. Dulaglutide inhibits high glucose- induced endothelial dysfunction and NLRP3 inflammasome activation. Arch Biochem Biophys (2019) 671:203–9. doi: 10.1016/j.abb.2019.07.008
110. Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis (2019) 10:778. doi: 10.1038/s41419-019-2021-3
111. Yang WL, Sharma A, Wang Z, Li Z, Fan J, Wang P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep (2016) 6:26571. doi: 10.1038/srep26571
112. Beltrán-García J, Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, et al. Extracellular histones activate endothelial NLRP3 inflammasome and are associated with a severe sepsis phenotype. J Inflammation Res (2022) 15:4217–38. doi: 10.2147/jir.S363693
113. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr biology: CB (2007) 17:1140–5. doi: 10.1016/j.cub.2007.05.074
114. Yan J, Li Y, Yang H, Zhang L, Yang B, Wang M, et al. Interleukin-17A participates in podocyte injury by inducing IL-1β secretion through ROS-NLRP3 inflammasome-caspase-1 pathway. Scand J Immunol (2018) 87:e12645. doi: 10.1111/sji.12645
115. Shahzad K, Fatima S, Khawaja H, Elwakiel A, Gadi I, Ambreen S, et al. Podocyte-specific Nlrp3 inflammasome activation promotes diabetic kidney disease. Kidney Int (2022) 102:766–79. doi: 10.1016/j.kint.2022.06.010
116. Hicks JM. Point-of-care testing: is it a must in pediatrics? Clin Biochem (2011) 44:516–7. doi: 10.1016/j.clinbiochem.2011.03.021
117. Kupper TS, Ballard DW, Chua AO, McGuire JS, Flood PM, Horowitz MC, et al. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J Exp Med (1986) 164:2095–100. doi: 10.1084/jem.164.6.2095
118. Sand J, Haertel E, Biedermann T, Contassot E, Reichmann E, French LE, et al. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis (2018) 9:24. doi: 10.1038/s41419-017-0009-4
119. Hasegawa T, Nakashima M, Suzuki Y. Nuclear DNA damage-triggered NLRP3 inflammasome activation promotes UVB-induced inflammatory responses in human keratinocytes. Biochem Biophys Res Commun (2016) 477:329–35. doi: 10.1016/j.bbrc.2016.06.106
120. Dai X, Tohyama M, Murakami M, Sayama K. Epidermal keratinocytes sense dsRNA via the NLRP3 inflammasome, mediating interleukin (IL)-1β and IL-18 release. Exp Dermatol (2017) 26:904–11. doi: 10.1111/exd.13334
121. Cho KA, Suh JW, Lee KH, Kang JL, Woo SY. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1β by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int Immunol (2012) 24:147–58. doi: 10.1093/intimm/dxr110
122. Tsuji G, Hashimoto-Hachiya A, Yen VH, Takemura M, Yumine A, Furue K, et al. Metformin inhibits IL-1β secretion via impairment of NLRP3 inflammasome in keratinocytes: implications for preventing the development of psoriasis. Cell Death Discovery (2020) 6:11. doi: 10.1038/s41420-020-0245-8
123. Mao J, Ma X, Zhu J, Zhang H. Ginsenoside Rg1 ameliorates psoriasis-like skin lesions by suppressing proliferation and NLRP3 inflammasomes in keratinocytes. J Food Biochem (2022) 46:e14053. doi: 10.1111/jfbc.14053
124. Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol (2014) 7:763–74. doi: 10.1038/mi.2013.94
125. Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut (2014) 63:598–609. doi: 10.1136/gutjnl-2012-303329
126. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41:71–6. doi: 10.1038/ng.285
127. Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol (2015) 194:1252–60. doi: 10.4049/jimmunol.1400501
128. Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, et al. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PloS Pathog (2014) 10:e1004410. doi: 10.1371/journal.ppat.1004410
129. Williams TM, Leeth RA, Rothschild DE, McDaniel DK, Coutermarsh-Ott SL, Simmons AE, et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am J Physiol Gastrointest Liver Physiol (2015) 308:G139–150. doi: 10.1152/ajpgi.00234.2014
130. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
131. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol (2021) 74:156–67. doi: 10.1016/j.jhep.2020.07.041
132. Bradbury A, Plückthun A. Reproducibility: standardize antibodies used in research. Nature (2015) 518:27–9. doi: 10.1038/518027a
133. Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics (2008) 7:2019–27. doi: 10.1074/mcp.R800013-MCP200
134. Song AJ, Palmiter RD. Detecting and avoiding problems when using the Cre-lox system. Trends Genet (2018) 34:333–40. doi: 10.1016/j.tig.2017.12.008
135. Stifter SA, Greter M. STOP floxing around: specificity and leakiness of inducible Cre/loxP systems. Eur J Immunol (2020) 50:338–41. doi: 10.1002/eji.202048546
Keywords: inflammation, infection, regulated necrosis, innate immunity, interleukin
Citation: Honda TSB, Ku J and Anders HJ (2023) Cell type-specific roles of NLRP3, inflammasome-dependent and -independent, in host defense, sterile necroinflammation, tissue repair, and fibrosis. Front. Immunol. 14:1214289. doi: 10.3389/fimmu.2023.1214289
Received: 29 April 2023; Accepted: 27 June 2023;
Published: 25 July 2023.
Edited by:
Chaofeng Han, Second Military Medical University, ChinaReviewed by:
Anna Lisa Giuliani, University of Ferrara, ItalyCopyright © 2023 Honda, Ku and Anders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Joachim Anders, aGphbmRlcnNAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.