 Caitlyn Guldenpfennig
Caitlyn Guldenpfennig Emma Teixeiro
Emma Teixeiro Mark Daniels
Mark Daniels
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 July 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1214095
This article is part of the Research Topic Molecular Regulation of the NFkB Response in Hematopoietic Cells View all 4 articles
NF-κB signaling is essential to an effective innate and adaptive immune response. Many immune-specific functional and developmental outcomes depend in large on NF-κB. The formidable task of sorting out the mechanisms behind the regulation and outcome of NF-κB signaling remains an important area of immunology research. Here we briefly discuss the role of NF-κB in regulating cell fate decisions at various times in the path of B cell development, activation, and the generation of long-term humoral immunity.
In B cells, NF-κB activation was first discovered in the search for transcription factors that regulate immunoglobulin gene recombination (1, 2). Classically, NF-κB is sequestered in its inactive form in the cytoplasm in a complex with inhibitory IκB proteins. There are two pathways of NFκB signaling, namely the canonical and non-canonical pathways (Figure 1). These contain unique members of the NFκB family members whose signals are initiated by the activation of specific receptor proximal signals that lead to the stimulation of their target NFκB complex and the translocation to the nucleus to regulate gene transcription. In B cells, the canonical pathway is activated by B cell antigen receptors (BCR), TNFa, IL-1, lipopolysaccharide (LPS), and others. It involves the phosphorylation and degradation of IκB proteins by the IκB kinases a and b (IKKa and IKKb). This releases and activates NF-κB heterodimers consisting of p50 (NFκB1) associated with REL-A (p65) or REL-B, which translocate to the nucleus and regulate the expression of target genes (3, 4). The noncanonical pathway is activated when stimuli such as B cell-activating factor (BAFF), proliferation-inducing ligand (APRIL), or CD40 ligand bind to receptors such as the BAFF receptor (BAFF-R), B cell maturation antigen (BCMA), transmembrane activator and calcium-modulating cyclophilin ligand interacting protein (TACI), or CD40, to activate IKKa. This leads to phosphorylation and proteolytic processing of the C-terminal ankyrin domain of p100 and results in the release of p52 (NFκB2)-REL-B heterodimers to activate gene expression (5, 6).
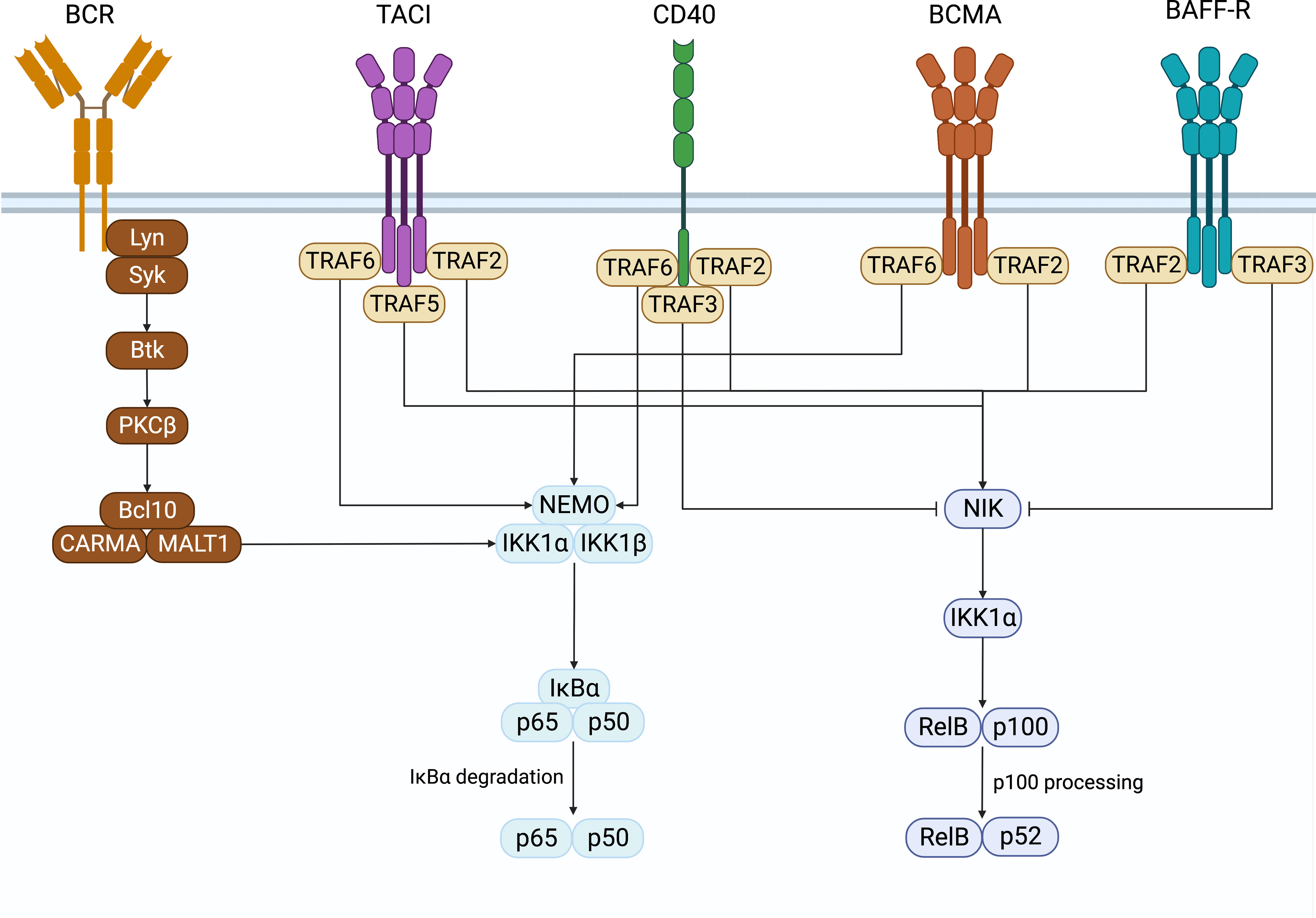
Figure 1 NF-κB signaling. Surface receptors expressed on B cells that induce the activation of the canonical and non-canonical NF-κB signaling pathways. Created with BioRender.com.
The genes induced and signaling pathways influenced by NF-κB play a fundamental role in B cells’ development, differentiation, and survival. It is important to understand the mechanisms that govern target-gene selection by NF-κB, the crosstalk between multiple receptors that can activate NF-κB and the crosstalk between the canonical and non-canonical pathways. NF-κB signaling can also intersect other signaling pathways to influence immune cell fate. Additionally, the outcome of NF-κB activation and the receptors that lead to its activation depends on the developmental or differentiation state and environment of the given B cell. Much of what we know about the mechanisms that regulate NF-κB signaling comes from murine models with engineered deficiencies in the signaling pathway and from human common variable immunodeficiency (CVID) patients with genetic defects and polymorphisms that disrupt NF-κB signaling (Figure 2). Advancing our understanding of these phenomena will aid in our ability to improve vaccine design and address health issues initiated by dysregulation of signaling and mutations in members of the NF-kB signaling pathway, including immunodeficiencies, B cell leukemia, and lymphoma, and autoimmunity, as reviewed in (7–11). This review summarizes how the different NF-κB signaling pathways and their cooperation with signals from co-receptors, B cell receptors, cytokines, inflammation, and T cell help contribute to a healthy and effective B cell compartment.
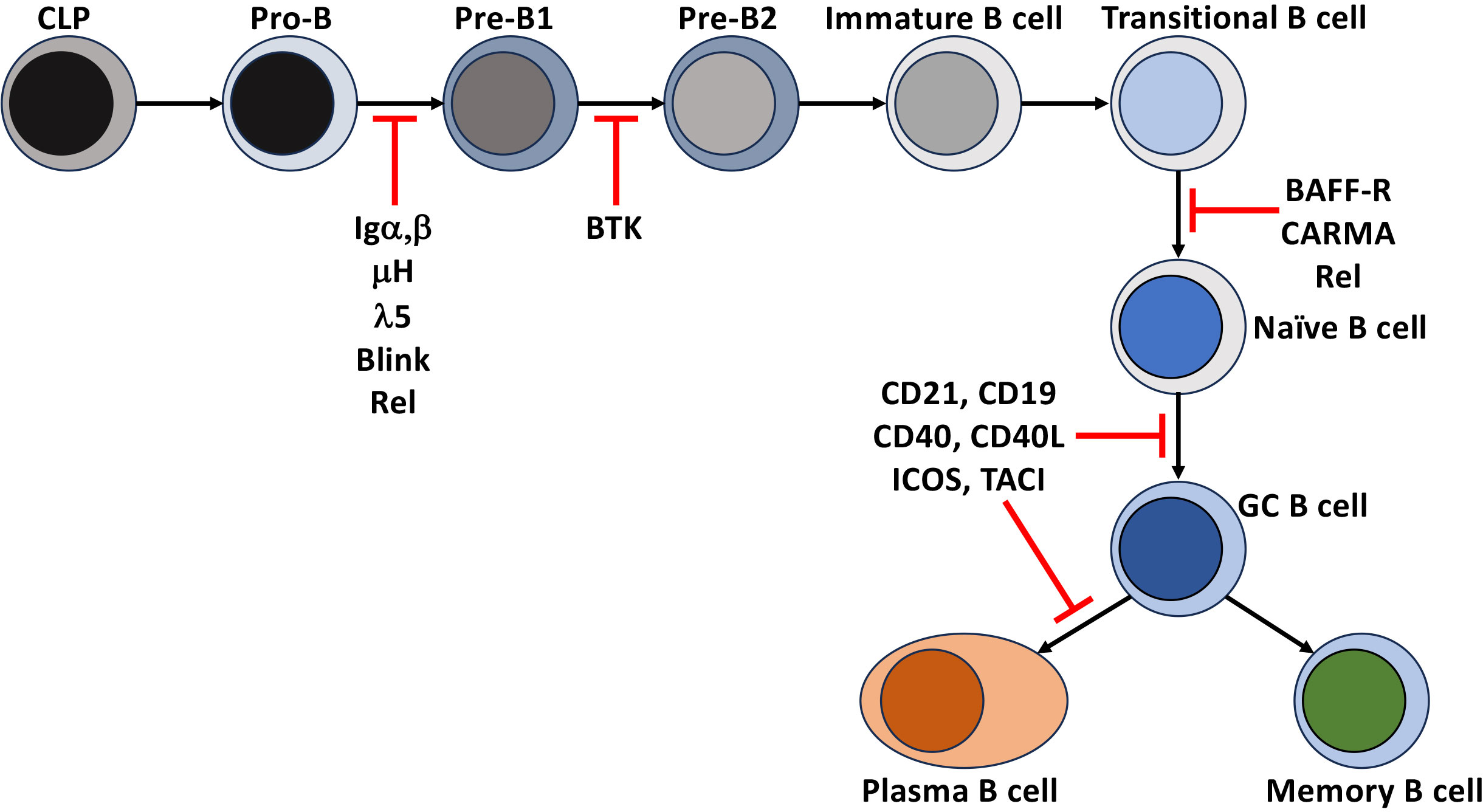
Figure 2 Disruption of NF-κB leads to immunodeficiencies. Points in B cell development and differentiation where mutations, polymorphisms, and deletions of receptors and intermediates of the NF-κB signaling that lead to a block in the development and differentiation of human B cells.
The development and survival of immature B cells rely largely on NF-κB signals downstream of the BCR, BAFF, and APRIL receptors. During B cell development, the pre-BCR and BCR activate the canonical NF-κB pathway to regulate central tolerance (positive and negative selection), survival, and differentiation. BAFF and APRIL are also essential for B cell development. They differentially bind to three receptors expressed on the surface of B cells. BAFF binds the B cell activating factor receptor (BAFF-R) (12), BCMA, and TACI. In contrast, APRIL binds to only BCMA and TACI. These receptors are part of the TNF receptor superfamily that can activate the non-canonical NF-κB pathway. In this section, we will briefly discuss the roles of NF-κB signaling in immature B cells’ selection, survival, and maturation.
Tonic pre-BCR and BCR signaling (signals that occur independently of receptor engagement) in developing B cells support positive selection and developmental progression through the BCR proximal signaling-based activation of a phosphoinositide 3-kinase (PI3K)/AKT and PKCβ/NF-κB. In the pro-B cell, IL-7 receptor signaling induces Rag1/2 dependent recombination of the Igh loci and expression of the transmembrane form of the μ-Heavy chain (μIg) and the surrogate light chain (Λ5 and VpreB) that, together with Igα, Igβ, make up the pre-BCR and, in addition to serving as a developmental biomarker, provides signals necessary for the transition to the pre-B stage of early B cell development. The contribution of NF-κB to this process was determined when the loss of signals through the pre-BCR by deletion of Igα (CD79a -/-) in pro-B cells or in models deficient in Rel induces the death of pre-B cells developing in the bone marrow. Overexpression of Bcl-2 in both models rescued the defect, indicating that the activation of NF-κB induces Blc-xL and Bcl-2 to promote their survival (13, 14). Similar blocks in B cell development were observed in human common variable immune deficiency (CVID) patients unable to express components of the pre-BCR or components of the BCR signaling pathway, BLINK or BTK. However, these defects led to more severe immunodeficiencies in humans than in mice (15–17).
Signaling by the unligated surface expression of the pre-BCR (and later in development by the BCR) activates receptor proximal signals, which in turn leads to the activation PI3K and PLCγ2/PKCβ, that activate AKT and canonical NF-κB respectively, and aid in the developmental progression of the B cell precursor (18, 19). During positive selection, AKT phosphorylation of forkhead box protein O1 (FOXO1) blocks FOXO1 nuclear localization, stopping the RAG1/2 expression. This increases MYC expression and activity, which induces several rounds of proliferation and cooperation with AKT to support survival, likely through negative regulation of the expression of the pro-apoptotic molecule Bim. This, along with the NF-κB-based induction of Bcl-xL and Bcl-2, supports the survival of developing B cells. As the proliferation of pre-B cells ends, cessation of AKT activity ends the restriction on FOXO1 activity (20). As a result, IL-7 signaling is reduced. The continued NF-κB-based induction of IRF4, along with the expression of PU.1, leads to the downregulation of the surrogate light chain and induction of the rearrangement of the IgL (light chain gene). The expression of IgL that successfully pairs with the previously generated μIgH results in the surface expression of the BCR (20–22). Continued induction of tonic signaling through the newly formed BCR continues to support the completion of positive selection leading to developmental progression and export from the bone marrow as newly formed transitional stage immature B cells (23–25) that continue to be dependent on tonic BCR signaling and NF-κB for their survival and next stage of development.
On the other hand, to avoid the development of autoreactive B cells, BCR ligation of self-antigens during the early stages of B cell development in the bone marrow promotes negative selection through NF-κB-induced receptor editing of the self-reactive BCR or apoptosis (19). Here the developmental stage-specific bi-phasic or bi-modal signaling of NF-κB becomes evident (22). Interestingly, increased signaling through BCR ligation activates the NF-κB signaling pathway and appears to coordinate both IRF4 and IL-7 pathways to enable the synergistic induction of light-chain recombination. The engaged BCR signals through NF-κB to increase IRF4 levels, thereby inducing light chain recombination. IRF4 targets the immunoglobulin 3’Ekappa and E-lambda enhancers to make the kappa and lambda more accessible to RAG1/2. In addition, reduced activation of IL-7 signaling activates the E-kappa promoter through E2A. IRF-4 also induces the expression of chemokine receptor CXCR4 to promote the migration of pre-B cells away from IL-7-expressing stromal cells, ensuring the reduced availability of IL-7 (20). NF-κB signaling promotes the survival of these cells through the induction of the PIM2 and increased expression of Bcl-xL and Bcl-2 reviewed in (19). Allelic exclusion of IgH during this process is critical to blocking the ability of B cells to express more than one BCR. Interestingly, B cells deficient in PLCγ1,2 (upstream of NF-κB) cannot execute the process of allelic exclusion (26, 27). However, whether NF-κB directly regulates this process or not is unclear (28). Here again, once a B cell that was directed to undergo receptor editing successfully expresses a new BCR that does not recognize self, the selection process continues and leads to export from the bone marrow as newly formed transitional stage B cells.
Once these immature B cells leave the bone marrow and enter the secondary lymphatics, even though the rules change slightly, they continue an NF-κB-dependent maturation and lineage commitment to become follicular or marginal zone B cells. These newly formed, immature B cells can be divided into two subsets. They enter the spleen and lymph nodes as transitional stage 1 (T1) B cells and subsequently progress to transitional stage 2 (T2) and T3. Splenic T1 B cells remain targets of negative selection. Unlike their bone marrow precursors, BCR engagement of self leads to apoptosis (29). This ensures that B cells expressing BCRs that recognize self-antigens, either not present in the bone marrow or that are only present in the periphery, are deleted before they become competent to express effector functions. This is supported by studies in mice (29) and humans (30–34) that demonstrated the negative selection of high-affinity self-reactive BCR, and a subsequent narrowing of BCR repertoire diversity in naïve B cell populations when compared to their transitional B cell counterparts. T1 B cells require activation of NF-κB to survive and mature to the T2 stage of development. This was demonstrated by showing that deletion of Rel and RelA blocks peripheral B cell development at the T1 stage (35). The Rel deficient T1 B cells are susceptible to death due to defects in the expression of survival factors Bcl-2 and A1 (36). Human patients with a homozygous deletion of CARMA (also known as CARD11) (Figures 1, 2) abrogated BCR-based activation of the canonical NF-κB pathway while CD40 signaling remained intact. Additionally, BAFF-R expression was also inhibited in these cells, and they were blocked at the transitional stage of development (37). Importantly, BAFF signaling through BAFF-R is required to induce the maturation from T1 to T2. The engagement of BAFF-R activates the non-canonical NF-κB signaling pathway and is essential for this process. In BAFF-deficient mice, like the Rel-deficient animals, all mature B cells are also lost after the T1 stage of maturation (38). This was also seen in BAFF-R signaling mutants and BAFF-R deficient mice and humans (39–41). The loss of BAFF-dependent NF-κB signaling resulted in a decrease in the levels of Bcl-2/Bcl-xL (42) and increased apoptosis of B cells in the periphery. Constitutive expression of Bcl-2 overcame this phenotype to suggest that the loss of mature B cells was due to a loss of non-canonical NF-κB signaling through the BAFF-R (38, 39, 42, 43). These findings were extended and determined that canonical NF-κB signaling was also induced by BAFF early. Thus, the upregulation of Bcl-xL promoted the survival of T1 B cells until BAFF-R-dependent non-canonical (NF-κB2) signaling could take over to promote their maturation and survival (40, 44). While the survival signal provided by BAFF through BAFF-R was thought to be required for all mature B cells, loss of IKKα activity downstream of NIK surprisingly had no impact on mature B cell survival if this activity was lost after maturation had occurred (45). In fact, naïve human follicular and marginal zone B cells are highly dependent on BAFF for survival as a genetic deficiency or pharmacologic inhibition of BAFF depletes these naïve B cell pools. In contrast, memory B cells (MBC) are less dependent on BAFF for survival as in patients treated with the therapeutic anti-BAFF monoclonal antibody, belimumab, the MBC pool appears unaffected (46, 47). This implies that fully mature resting B cells may not rely heavily on BAFF-R-mediated NF-κB signals through the NIK-IKKα axis but may still depend on canonical NF-κB and other signaling pathways downstream of the BAFF-R to survive in the periphery. Thus, BCR-dependent canonical and non-canonical NF-κB activity through BAFF are both necessary for generating a complete and functional repertoire of peripheral mature naïve B cells.
The BAFF-R/BCR axis also controls the lineage commitment to the follicular or marginal zone B cell fate (48). The most abundant population of mature B cells in mice and humans are follicular B cells. These are recirculating cells that home mainly to B cell follicles and thus are well suited to perform their function in T cell-dependent immune responses (48). Marginal zone B cells (MZB) are innate-like antibody producers located at the interface between the white pulp of the spleen and the circulation and rapidly respond to blood-borne antigens. They have been reported to be able to self-renew and survive for very long periods of time (49). Determining the follicular versus marginal zone B cell fate depends on integrating NF-κB signals generated by the BCR and BAFF-R and crosstalk/integration of signals from Notch2 (8, 48, 50). MZBs develop in mice and humans in the presence of weak BCR signals, canonical NF-κB, and Notch2 (8, 48, 51, 52). As mentioned above, BAFF-R deficiency leads to a total loss of mature B cells. Interestingly, the expression of constitutively active IKKβ (a component of the canonical NF-kB pathway) can rescue the MZB development (53, 54). Since this does not occur in BAFF-R/CD19 double knockout mice, this indicates there is cooperativity between AKT and NF-κB in the commitment to the MZB lineage (48–52).
Follicular type II B cell lineage fate occurs in the presence of tonic B cell receptor signaling, mediated by Igα/Igβ (55), and BAFF-R signals, including non-canonical NF-κB. Importantly, tonic BCR signaling increases the expression of p100 as a crucial signaling intermediate downstream of BAFF-R, providing a mechanism for this cell fate (56). On the other hand, the follicular type I B cell lineage arises from strong BCR signals and canonical NF-κB. Here PI3K and BTK function downstream of the BCR to facilitate the activation of canonical NF-κB, necessary for the survival of developing follicular type I B cells (57–59). Despite the wealth of data on the BCR/BAFF developmental axis, the lack of clarity in some experimental models and how the balance between the signals from BCR, BAFF-R, CD19, BAFF levels, or other receptors leads to cell fate outcome requires a more detailed examination of the process.
In an immune response, follicular B cells, primed by their cognate antigen and TLR-based inflammatory cues, become activated and traffic to the T cell zone, where they interact with CD4 + follicular helper T cells (TFH) (60). Newly activated B cells receive help from T cells expressing CD40L and cytokines and, in conjunction with antigen-dependent BCR signals, inflammation, and TLR (61), some differentiate into short-lived antibody-secreting cells in the follicles or pre-germinal center (GC)/GC-independent memory B cells. B cells that do not receive help from TFH die. Others acquire the gene and protein expression profile that enables their entry into the germinal center (62–64).
Within a week of antigen exposure, GCs develop in the center of B cell follicles in the spleen and lymph node (65). As the GC matures, it polarizes into a dark zone (DZ), proximal to the T cell zone, and a light zone (LZ), proximal to the lymph node capsule or the marginal zone of the spleen, extensively reviewed in (66, 67). The DZ consists almost entirely of GC B cells and a network of reticular cells and appears “dark” by light microscopy. By contrast, the LZ, which also gets its name from its “lighter” appearance, contains a network of follicular dendritic cells (FDC), CD4+ TFH, and GC B cells. The DZ functions as the location where selected GC B cells proliferate and undergo somatic hypermutation (SHM) with the goal of increasing BCR affinity for their cognate antigen. After SHM, the GC B cells are directed to traffic to the LZ where their newly modified BCR are selected on antigen bound to FDC and interactions with their cognate TFH. The positive selection signals from this interaction lead to the induction of proliferation and return to the DZ to start the process over. The selection of GC B cells, the cycling between the DZ and LZ, and the decision to leave the GC as memory B cells or long-lived plasma cells require or are significantly influenced by NF-κB (62, 66–69) and are discussed briefly below.
Murine and human GC-B cells receive help from TFH in the form of cytokines (IL-4, IL-21, IL-10), BAFF, APRIL, and CD40L that, together, enable B cells to activate their class-switching machinery and undertake the process of somatic hypermutation required for developing BCR with higher affinity for antigen and high-affinity memory B cells or long-lived high-affinity antibody-producing plasma cells (PC) (63, 70–73). CSR and SHM require the expression of activation-induced cytidine deaminase (AID). DNA methylation and gene expression data from human tonsillar GC B cells, identified genes of the NF-κB signaling pathways that were de-methylated and highly expressed, including those targeted by AID (74). The CSR machinery that controls specific antibody classes is induced by transcription factors downstream of specific inflammatory cytokine receptor signals. For example, on activated B cells, signaling through the IFNγ receptor activates STAT1 to induce T-bet and promote class switching to IgG2a. Similarly, TGFβ signaling through SMAD and RUNX promotes class switching to IgA and so on (75–77). BAFF and APRIL contribute to the CSR due to TACI-induced activation of the canonical NF-κB pathway through MyD88 to induce AID expression (78, 79). Furthermore, the transcriptional regulator BATF, downstream of BCR and CD40-induced NF-kB (80), is also required in B cells to generate germline switch transcripts and to promote AID expression.
BAFF-R is important for maintaining the GC reaction (18). However, the BAFF-R-dependent mechanism and signaling pathways required for this maintenance of the GC have not been fully resolved. B cell-intrinsic non-canonical NF-κB signaling is required for GC formation (49), which may be initiated through the BAFF-R. Still, while BAFF and APRIL are common ligands that induce non-canonical signaling, the induction of NF-κB2 could also be coming from other factors/receptors. TACI has been demonstrated to negatively regulate the germinal center reaction in part by activating cIAP to ubiquitinate NIK for degradation leading to the inhibition of BAFF-R mediated non-canonical NF-κB survival in the germinal center (81, 82). TACI also induces BLIMP-1 expression to inhibit the GC reaction and drive the generation of long-lived plasma cells. Thus, the canonical and non-canonical NF-κB pathways through BAFF-R and TACI are important for regulating the B cell antibody response.
BAFF and APRIL also have key roles in memory B cells and long-lived plasma cell generation and survival. Early studies showed that BAFF and APRIL were not required for memory B cell survival (83, 84). In contrast, more recently, it has been shown that memory B cells need BAFF and BAFF-R to survive in the periphery in mice and in a subset of human memory B cells (85–87). Additionally, Muller-Winkler et al. determined that the NF-κB signals downstream of the BAFF-R are differentially necessary for memory B cells (87). IgM+ memory B cells required both IKKα and IKKβ for optimal survival, while IgG+ memory B cells only relied on IKKβ. Long-lived plasma cells also need BAFF/APRIL signaling to survive, and the signals are thought to occur primarily through BCMA in mice and humans (83, 88, 89). This may be due to IKKα specific signaling, as loss of the signaling through the NIK-IKKα axis led to decreased long-lived plasma cells (90). BCMA also drives memory B cell differentiation toward the plasma cell fate (89, 91, 92). Although, whether this is directly through NF-κB or through the upregulation of BLIMP-1 remains to be determined. BAFF-R deficient CVID patients have severely reduced serum IgM and IgG levels and impaired T cell-independent responses against pneumococcal polysaccharides. However, like BAFF deficient mice, IgA levels and gut IgA+ plasma cell numbers in these patients were normal, reviewed in (16, 89). Deletion of TACI or APRIL in mice and human patients expressing mutant TACI have significantly reduced IgA levels (93–96). Low IgA and IgG antibody titers in TACI-deficient patients suggest its importance for CSR in human B cells. It has also been suggested that decreased numbers of BAFF-binding receptors caused by the absence of TACI give rise to more circulating BAFF binding to BAFF-R, thus supporting B-cell survival (97). High levels of BAFF in mice and humans increase the numbers of mature B cells and may be the mechanism that leads to progressive autoimmune disease in these B cell-deficient patients (89, 98).
CD40 is a costimulatory molecule expressed on B cells and other antigen-presenting cells and contributes to B cell proliferation, antibody class-switching, and rescues B cells from activation-induced apoptosis (99–102). CD40 has been shown to be capable of activating both the canonical and non-canonical NF-κB pathways by interacting with TRAFs (103–105). TRAF2, TRAF3, and TRAF5 have all been shown to associate with CD40 and are important for activating the CD40-induced non-canonical NF-κB signaling (105). CD40’s interaction with either TRAF2 or TRAF3 is also important for inducing canonical NF-κB signaling, while interacting with both is required to induce non-canonical NF-κB (100). Additionally, TRAF6 associates with CD40 to initiate the canonical NF-κB signaling pathway (105).
Like BAFF-R, CD40 plays an important role in B cell antibody class switching and is able to induce class switching alone or in combination with cytokines and the BCR (99). CD40’s ability to activate NF-κB signaling is essential to this process. Jabara et al. demonstrated that mice with B cells expressing CD40 proteins unable to interact with TRAF2 or TRAF3 alone had decreased serum IgG and IgE and smaller germinal center reactions in response to T cell-dependent antigens (100). Furthermore, B cells expressing CD40, unable to bind to both TRAF2 and TRAF3, had further reduced serum IgG and IgE and undetectable germinal center reactions. Ultimately, B cells expressing a mutant form of CD40 that could not signal through TRAF2 and TRAF3 could still induce the canonical NF-κB pathway but not the non-canonical NF-κB pathway. In contrast, CD40 double mutant proteins could not induce either (100). These data demonstrated that CD40’s ability to interact with both TRAF2 and TRAF3 and induction of both the canonical and non-canonical NF-κB signaling is important for CD40-mediated antibody class switching. A second study also demonstrated the reliance of CD40 on canonical NF-κB to induce class switching by inducing AID expression (102). This was shown when B cells deficient in p50 or c-Rel could not induce AID expression in response to CD40, while by contrast, p52 deficient cells could, although to a lesser extent than the wild-type B cells. Thus, both the canonical and non-canonical NF-κB pathways downstream of CD40 are required for CD40-mediated AID expression and antibody class switching.
CD40-mediated NF-κB signaling also regulates other aspects of the B cell response, including the germinal center reaction, B cell trafficking, and memory B cell differentiation. The NF-κB p50/p65 heterodimer binds to the IRF4 promoter and induces its expression downstream of CD40 activation. IRF4 expression can negatively regulate the germinal center reaction by binding to the Bcl-6 promotor and downregulating its expression (106). Thus, CD40 can negatively regulate the germinal center reaction and promote B cell exit from the germinal center reaction as post-GC memory B cells. Additionally, CD40-induced non-canonical NF-κB signaling is important for CXCR5 expression (107), which helps localize newly activated B cells to the follicles to traffic toward and receive help from TFH cells (108). CXCR5 and CXCR4, dependent at least in part on NF-κB signaling, participate in the cycling of GC B cells between the LZ and DZ reviewed in (62, 64, 67). Interestingly, GC in patients with genetic defects in CD40 are poorly organized or nonexistent (109, 110). Finally, CD40 and its induction of NF-κB signaling regulate the differentiation of CD80+PD-L2+ vs. CD80-PD-L2- and CD80-PD-L2+ memory B cells. These two subsets have been shown to be functionally distinct, where CD80+PD-L2+ memory B cells primarily differentiate into plasma cells upon restimulation, whereas CD80-PD-L2- memory B cells primarily generate new germinal center reactions (111). Interestingly, strong CD40 leads to higher expression of CD80 by inducing IRF4 and BAFT expression through NF-κB signaling, which then forms a heterodimer able to bind to the CD80 promotor and induce its expression (80). Thus, CD40-mediated NF-κB signaling is essential for inducing and regulating efficient B cell primary and secondary B cell responses.
Multiple mathematical and experimental models that analyze the LZ to DC cycle indicate that 10%–30% of B cells that traffic into the LZ are selected to re-enter the DZ. The remainder either die by apoptosis or exit the GC as memory B cells or PC (67, 68, 112). T cell help drives cycle reentry and GC selection. In addition, the nature of the B cell-TFH cell interactions regulates the number of divisions and the cell cycle speed of selected B cells in a manner proportional to the strength of that interaction (69, 113, 114). Here we will discuss the contribution of NF-κB signaling to this process.
Recruitment and retention of B cells in the GC require them to upregulate the zinc finger transcription factor B cell lymphoma 6 (BCL-6). This is mediated by antigen-dependent BCR signals and engagement of CD40 by TFH expressing CD40L and cytokine (115, 116). TFH cell expression of IL-4 and IL-21 acts directly on B cells to promote the expression of BCL-6 (117, 118). BCL-6 is a transcriptional repressor that blocks the upregulation of migratory cell receptors S1PR1 and Gpr183. BCL-6 also upregulates the expression of S1PR2, which acts as a retention signal to maintain B cells in the GC (116). BCL-6 regulates the expression of a vast network of genes controlling cellular processes, including the DNA damage response, apoptosis, BCR, and CD40 signaling, inhibition of plasma cell differentiation, and T cell/B cell interactions (62, 64). NF-κB dependent induction of IRF4 is necessary for the initiation, but not the maintenance, of the GC response. Interestingly, transient expression of IRF4 in B cells induces the expression of BCL-6, AID, and POU2AF1 and promotes GC development (119). IRF4 expression is rapidly induced by BCR stimulation and CD40 through the activation of NF-κB (36). However, sustained IRF4 expression is sufficient to directly repress BCL-6 expression and promote plasma cell differentiation, which may reflect the kinetic and dose-dependent function of IRF4 in GC-B cells (36, 37). In-depth genetic analyses indicated that IRF4/PU.1 or BATF bound to Ets or AP-1 motifs to control transcription factors PAX5, BCL-6, and BACH2, which contribute to retention in the GC through negative regulation PC differentiation (120). Conversely, higher concentrations and sustained expression of IRF4 promoted the generation of plasma cells while antagonizing the GC fate (119). The sustained expression of IRF4 upregulates BLIMP1 and downstream target XBP1, transcription factors that direct GC B cells to become long-lived plasma cells (121). Mechanistically, higher concentrations of IRF4 shifted binding to genetic interferon response sequences to induce genes that inhibit the GC program. Expression of Blimp1 terminates the expression of BCL-6 and BACH2, responsible for the maintenance of the GC phenotype, to drive cells to become PC (120, 122–125). Conditional deletion of IRF-4 in GC B cells blocked the generation of post-GC plasma cells, as IRF-4 deficient memory B cells were unable to upregulate BLIMP1 and to differentiate into plasma cells (121). The NF-kB subunits c-Rel and RelA had different roles in this process. Signaling through c-Rel was critical for maintaining newly formed GC, while RelA was dispensable for this process. Importantly, RelA was able to directly induce BLIMP1 independently of IRF-4 and is, therefore, essential for PC development (126). Together, these data indicate that the NF-kB signaling is important for determining the GC and PC fate through the regulation of the IRF-4/Bcl-6/BLIMP1 axis (120, 125, 127). Understanding what signals induce the activation of RelA in vivo may provide valuable clues to the cellular processes involved in PC differentiation.
To achieve a repertoire of B cells with a higher affinity to antigen, one would expect there to be a pronounced difference in how naïve and GC B cells sense or respond to antigens. One difference maps to the type of synapse GC B cells form when engaging antigen (128, 129). Naïve B cells form a classical ‘bullseye’ synapse, directing antigen internalization to a central synaptic structure. By contrast, GC B cells form small foci of synapses and internalized antigens in peripheral synapses (129). Additionally, low-affinity antigens triggered continuous engagement and disengagement of membrane-bound antigens, whereas high-affinity antigens induced stable synapse formation. The mechanical forces needed to pull antigens from the surface of FDC also provide a way to sense antigen affinity. Cytoskeletal contractility disrupted low-affinity and individual BCR-antigen interactions and promoted the internalization of high-affinity, multivalent BCR (130). Here then, these interactions increase the activation of BCR signals, likely increasing the dose of antigen processed and presented to TFH, providing an indirect mechanism by which TFH can sense antigen affinity. These data pose potential mechanical and biochemical mechanisms for differentiating high-affinity BCR.
Considering the NF-κB signals are activated by crosstalk between GC B cells, environmental cues and TFH connects NF-κB to GC selection decisions. A consequence of the interactions between GC B cells and T cells important for the selection decision is the induction of feed-forward or positive feedback loops in both cells. ICOSL on B cells promotes upregulation of CD40L on T cells, which in turn further upregulates more ICOSL expression on the B cell. This interaction promotes the GC B cell TFH interactions described as ‘‘entanglement’’ to promote the differentiation into high-affinity plasma cells (131). ICOS-deficient CVID patients lack isotype-switched B cells and there is a paucity of GC in spleens and lymph nodes (132). Using intravital microscopy, the duration of GC B cell interactions with TFH was also greatly increased by specific high-affinity antigen delivery to GC B cells (113, 114). ICAM has also been implicated in these increased cell-cell interactions. This also increased the T cell production of B cell helper cytokines IL-4 and IL-21. In addition, T cells expressed more BAFF and increased the survival of B cells that have acquired high-affinity mutations (133).
Another change between naïve and GC B cells in the response to an antigen is a qualitative rewiring and quantitative dampening of BCR signaling in GC B cells (128, 129). Although antigen stimulation of BCR can trigger proximal signaling in GC B cells, overall NF-kB signaling is reduced. A critical step of BCR-based NF-kB activation appears to require additional input from TFH cells (129). In fact, BCR signals in mice and humans are dampened 100-fold in GC B cells, and CD40 signaling is rewired in these cells (63, 128, 134). Using a combination of genetic ablation, specific inhibitors, and in vivo delivery, GC stimuli on ex vivo GC B cells and GC B cells in vivo found that CD40 induced signals via NF-kB but not PI3K. At the same time, BCR signals do not activate NF-kB, while BCR-dependent activation of AKT remained intact. In GC B cells, both CD40 and BCR signals are required to induce c-Myc, which is essential for their selection, survival, and proliferation in the GC. This is markedly different from naïve B cells in which either receptor can signal through both PI3K/AKT/mTOR (135) and NF-kB and to c-Myc. The phosphatase PTEN has been implicated as part of the mechanism responsible for the dampening of BCR signaling in GC B cells (136). More recent work using inhibitors of AKT indicated that differential activation of AKT by GC B cell-specific expression of PDK1 and PTEN led to enhanced negative regulation of BCR signaling through CSK, SHP-1, and HPK1 (136). Altogether, these highlight the essential role TFH play in the activation of NF-kB to enhance the successful selection of high-affinity B cells in the GC reaction (67–69, 113). When considered together, these results, along with computational modeling, support a kinetic control model of the PC vs. GC B cell fate determination (62, 63, 123). However, the precise thresholds, cross talk, and synergy between inputs such as BCR affinity, antigen dose, the magnitude and nature of T cell help, and contribution from FDC remain undefined. Furthermore, quantifying these on a per B cell basis under physiological conditions in vivo has not yet been worked out.
The purpose of B cell memory is to provide enhanced antibody responses and immune protection against repeat exposure to homologous or heterologous infection. The B cell memory compartment is comprised of highly diverse subsets of B cells with unique trafficking patterns, longevity, cell surface phenotype, and functions (63, 137). The signals that drive B cell memory vs. the generation of long-lived plasma cells are not worked out and are highly controversial at best. The ability of B cells to differentiate into memory B cells versus PCs changes over the course of an immune response. Memory B cell generation is favored in the pre-GC and early GC periods, and long-lived PC differentiation becomes more pronounced later in the immune response in mice and humans (138–140). Some data from these experiments suggest that lower affinity BCR favors the generation of memory over PC, especially early in the immune response. In addition, a single B cell can differentiate into each of the diverse memory subsets depending on several factors (139, 140). Considering the role of NF-kB in the regulation of the IRF-4/Bcl-6/BLIMP1 axis (120, 125, 127), it is likely to be at least part of the determination of these decisions. TLR signals provides a significant contribution to NF-kB signaling. Thus, as inflammation fades over time, the selection of GC B cells into the PC fate is more likely to be dependent on signals through the BCR, CD40, TFH, cytokines, and BAFF. Importantly, IL-21 has an essential role in conjunction with those signals in determining the nature of the human B cell immune response and programming of B cell memory (72, 85). Other inflammatory and environmental cues also lead to differences in memory subtypes such as the connection between inflammation and CSR INFg and IgG2 or TGFb and IgA or IL-21 and INFg, as discussed earlier (75–77). Another example when human B cells with BCR that bind TLR-7 or 9 ligands, signals from BAFF and concomitant IL-21 or IFNγ promotes T-bet+ B cell development (141–143).
Because the affinity of GC B cells for antigen also increases over time and BCR signaling is rewired (63, 128, 134), the temporal and antigen affinity models may encompass developmental and environmental changes along the course of the immune response that define new thresholds of signals that promote the long-lived PC vs memory B cell fate. Precisely defining this moving target in vivo is a difficult task. Despite this, recent studies provided additional mechanistic detail concerning the selection of GC B cells into the plasma cell lineage, showing that plasma cell lineage commitment was favored by BCL-6lowCD69hi GC B cells that expressed the transcription factor IRF4. The generation of BCL-6lowCD69hi GC B cells, which also expressed high levels of ICAM1 and SLAM, was regulated by T cell-mediated signaling through CD40, suggesting that TFH cell–GC B cell interactions were key to the generation of GC B cells that were prone to differentiate into long-lived plasma cells (144).
Tissue-resident memory B cells in humans and mice have been found in non-lymphoid tissues that are phenotypically and functionally distinct from their classical lymphoid-associated counterparts (145–147). Tissue-resident memory B cells (BRM) perform protective functions in peripheral tissues dependent on their ability to engage antigen at the site of infection, secrete cytokines, and differentiate into plasma cells (147). The tissues BRM are directed to enter depend in part on the pathogen and site of infection. Much less is known about the mechanism of their generation and maintenance (147). A role for NF-κB in regulating this process is suggested by a recent model of influenza infection. Here CD40L blockade through the first 2 weeks blocked HA and NP-specific B cell response in both the LNs and the lung. CD40 blockade between day 10 and day 20 or between day 20 and day 30 allowed BRM to appear in the lung. Importantly, when CD40L was blocked between days 30 and 40, all GC B cells were lost, while flu-specific BRM cells in the lung were maintained. Since BRM generation also depended on the presence of their cognate antigen in the lung, it suggests that cooperation between CD40 and BCR (NF-kB/AKT axis)? regulates their generation. Interestingly this parallels our recent work in T cells, where temporal regulation of NF-kB controls both the generation and development of tissue-resident T cells (148).
MZ B cells are innate-like B cells specialized to mount rapid T-independent, but also T-dependent responses against blood-borne pathogens and are a major source of IgM antibodies in humans (49). Whilst there are similarities to mouse and human MZB development (52), their phenotype as memory/PC are different. Human MZB cells express hypermutated IgM and are circulatory, while their murine counterparts express unmutated IgM and remain in the spleen (149, 150). Interestingly, the MZ B cells in children less than 2 years of age express low levels of AID, while AID was not detected in MZ B cells from adults (151–153). In fact, children show a higher clonal diversity in the MZ subset than in class-switched B cells (149). Furthermore, MZB with mutated IgM found in patients with genetic defects in CD40 suggest they can arise independent of a GC reaction (109, 110). Whether this is due to the differences in the physiology of mouse and splenic marginal zones, environmental cues, NF-κB activity, or other factors is not clear.
Our reductionist methodology and ability to design well-controlled experimental approaches have provided us with a solid picture of the function of NF-κB in humoral immunity. Other mechanistic information has come from exhaustive studies of CVID patients with various genetic deletions, mutations, and polymorphisms in NF-κB. These works have been used to develop FDA-approved treatments for inflammatory diseases and multiple cancers (154–158). However, the precise quantification of the dose of antigen or cytokine a B cell is exposed to, how many and which cell-cell encounters occur within a given time, or what is the variability in paths a cell can follow to arrive at a given cell fate in vivo is not able to be determined by these methods. Thus, some of the fine details in human and mouse B cell development and differentiation remain unknown. The progressive development of new ‘multi-omics’ approaches enables the potential to provide these data.
Phenotype tracking human and mouse B cells through development and differentiation has provided a consistent and high-definition classification of B cell subsets during B cell development and differentiation into MBC and PC. In addition, they have corroborated checkpoints in development in mice with those across a population of healthy human donors. RNA velocity analyses have begun to precisely map out the quantity, quality and kinetics of gene expression over the course of early development and differentiation, defining pathways of development. Lineage tracing and single-cell analyses across a diverse population have been used for the discovery of new B cell subsets and their plasticity. In addition, aligning surface phenotypes with genetic and signaling profiles provide starting points to extend our understanding of NF-κB signaling in B cell extensively reviewed in (159–164). These new methodologies can lead to our ability to fill gaps in our understanding of NF-κB. For example, how can we improve vaccine response using our understanding of the interplay between inflammation and long-term immunity to improve vaccine design? Is a given B cell memory subset better at providing immune protection against a pathogen? When is the best time in an immune response to manipulate NF-κB activity to direct tissue-resident B cells to (or away from) an organ of interest such as the lung to increase protection and safety of vaccines against respiratory infections?
When working in harmony, the pathways regulating NF-κB described above lead to the generation and maintenance of a complete repertoire of the diverse subsets of healthy B cells. However, many of these pathways are susceptible to dysregulation, leading to aberrant NF-κB signaling and lymphoid tumorigenesis. Many B cell leukemias and lymphomas, including Hodgkin lymphoma, activated B-cell-like diffuse large B-cell lymphoma (ABC-DLBCL), lymphomas of the mucosa-associated lymphoid tissue (MALT), and chronic lymphocytic leukemia (CLL) are associated with constitutively active NF-κB signaling which contributes to these cancer’s proliferation and survival (165–168). While we will not cover the role of NF-κB in B cell malignancies in depth (comprehensively reviewed in (169, 170)), we will summarize a few key points at which NF-κB may be dysregulated to drive B cell transformation and other tumors.
In healthy B cells, MyD88 can coordinate the activation of B cells downstream of TLRs and induce AID expression by activating the NF-κB pathway, as described earlier. However, mutations in MyD88, most notably the L265P mutation, lead to the constitutive activation of NF-κB and are commonly found in ABC-DLCBL and can also be found in MALT lymphomas and CLL (171–174). This mutation allows poly-ubiquitination by an E3 ubiquitin ligase RNF138 which leads to activation of MyD88L265P and drives activation of the NF-κB and B cells transformation (175). Ubiquitination of MyD88L265P is negatively regulated by the ubiquitin editing protein A20-mediated downregulation of RNF138 and, as such, knock-down of A20 in this model enhanced the efficiency of transformation. These suggest two complementary mechanisms by which the dysregulation of NF-κB signaling is caused by MyD88 mutations. A20 down-regulates canonical NF-κB signaling mediated by multiple receptors through multiple mechanisms (176, 177). Interestingly, inactivation or deletion of A20 is also commonly linked to B cell malignancies on its own, including ABC-DLBL.
Signals from both the pre-BCR and BCR can activate NF-κB. Signals downstream of the pre-BCR and BCR signaling are commonly dysregulated in B cell malignancies, where enhanced activation or disrupted negative regulation of components of the BCR pathway leads to the constitutive activation of NF-κB (178). The chronic activation of the BCR by either self or non-self-antigens has been shown to lead to the development of lymphoid cancers (179, 180). Mutations within the CD79B and CD79A co-receptors are common and maintain this activation (181). Mutation of CD79B can increase the surface expression of the BCR and limit its internalization to maintain chronic signaling (182). As described above, engagement of the BCR leads to the activation of BTK, which in turn activates canonical NF-κB signaling. Cancers that are dependent on mutations in CD79 are sensitive to Bruton tyrosine kinase (BTK) inhibitors and, thus, downstream inhibition of NF-κB (183). A20 down-regulates canonical NF-κB signaling mediated by multiple receptors through multiple mechanisms (176, 177). Interestingly, the inactivation or deletion of A20 enhances the transformation of these cells (178, 184, 185).
Finally, the mutation of proteins within the CARMA/MALT1/BCL10 (CBM) complex also leads to constitutive NF-κB signaling and oncogenesis. In healthy B cells, the CBM complex is activated downstream of the BCR through the phosphorylation of CARMA. This phosphorylation induces a conformational change which then allows it to associate with BCL10 and MALT1. The active CBM complex will then recruit the IKK and TAK1, which will then be able to phosphorylate IKKβ and induce NF-κB signaling. Gain of function mutations within any of the three proteins that make up the CBM complex can result in the constitutive activation of NF-κB and drive B cell oncogenesis. Again, multiple leukemias and lymphomas depend on the crosstalk between AKT and NF-κB (178, 184, 186). This suggests the potential inclusion of other scaffolds, such as POSH (plenty of SH3 domains), that are implicated in crosstalk between these and other signaling pathways important to the survival of other cancers and for lymphocyte survival and differentiation (187–191).
Stepping briefly outside of the hematopoietic system, considering the general function and the genes targeted by the NF-κB signaling pathway, it has the potential to play a role in multiple tumor types. Mutations in NF-κB genes are rare in solid tumors. However, activation and nuclear translocation of RelA are found in many tumors and associated with tumor progression. In these tumors, aberrant activation of signaling molecules upstream of NF-κB, such as mutant or oncogene-driven RAS, EGFR, PGF, and HER2, likely induce increased NF-kB signaling, reviewed in (192–194). NF-kB then, in turn, upregulates genes that control cell survival, proliferation, metastasis, angiogenesis, and others. For example, BCR-ABL signaling in AML, CML, and B-ALL activates NF-κB, and tumorigenesis driven by BCR-ABL-expressing cells was blocked upon inhibition of NF-κB (195). In addition, NF-kB’s ability to induce the expression of AID and APOBEC was demonstrated as a mechanism for increased mutagenesis in cervical cancer (196). High levels of c-Rel have been found in non-small cell lung carcinoma (197), breast cancer (198), and squamous cell carcinomas of the head and neck (199). Defective IkBα was found in several solid tumors such as breast, colon, ovarian, pancreatic, bladder, prostate carcinomas, and melanoma (193). Taken together, the data from solid tumors and hematological malignancies provide the information that can be used to develop was to inhibit NF-κB to enhance current tumor treatment protocols.
NF-κB signaling plays a major role in the development of a healthy B cell immune response. It has unique functions depending on the developmental/differentiation state of the cell and cooperation with multiple other signaling pathways responsible for cell fate decisions. In addition, the timing and kinetics of the induction of NF-κB signaling (transient vs. sustained) greatly influence the outcome of its activation. The cooperation between different receptors able to activate canonical and non-canonical NF-κB aids in the diversity of function for this signaling pathway. Much is known about this amazing pathway; much more is still to be known. In knowing this, we can address ways to enhance immune protection and vaccination as well as improve treatment protocols when NF-κB signaling gets out of control.
MD: Headed the teamwriting and editing. ET: Contributed to writing and editing. CG: Contributed to writing and editing. All authors contributed to the article and approved the submitted version.
CG: MU Life Sciences Fellowship. MD: NIH-MBARC. National Institutes of Health NIH AI110420-01A1 (ET), NCI CA244314 (ET), NIH IB026560 (MAD), NIH-REACH (MAD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein nf-kappa b by a posttranslational mechanism. Cell (1986) 47(6):921–8. doi: 10.1016/0092-8674(86)90807-x
2. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell (1986) 46(5):705–16. doi: 10.1016/0092-8674(86)90346-6
3. Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J (2004) 23(21):4202–10. doi: 10.1038/sj.emboj.7600391
4. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol (2004) 25(6):280–8. doi: 10.1016/j.it.2004.03.008
5. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev (2004) 18(18):2195–224. doi: 10.1101/gad.1228704
6. Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res (2011) 21(2):223–44. doi: 10.1038/cr.2011.13
7. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol (2011) 12(8):715–23. doi: 10.1038/ni.2060
8. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol (2011) 12(8):695–708. doi: 10.1038/ni.2065
9. Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol (2011) 12(8):709–14. doi: 10.1038/ni.2055
11. Smale ST. Hierarchies of NF-kappaB target-gene regulation. Nat Immunol (2011) 12(8):689–94. doi: 10.1038/ni.2070
12. Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-r, a newly identified TNF receptor that specifically interacts with BAFF. Science (2001) 293(5537):2108–11. doi: 10.1126/science.1061965
13. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol (2010) 2(5):a000182. doi: 10.1101/cshperspect.a000182
14. Gerondakis S, Strasser A. The role of Rel/NF-kappaB transcription factors in b lymphocyte survival. Semin Immunol (2003) 15(3):159–66. doi: 10.1016/s1044-5323(03)00036-8
15. Ghia P, ten Boekel E, Rolink AG, Melchers F. B-cell development: a comparison between mouse and man. Immunol Today (1998) 19(10):480–5. doi: 10.1016/s0167-5699(98)01330-9
16. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi: 10.1016/j.jaci.2013.01.046
17. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in btk-deficient mice. Immunity (1995) 3(3):283–99. doi: 10.1016/1074-7613(95)90114-0
18. Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, et al. Essential role of src-family protein tyrosine kinases in NF-kappaB activation during B cell development. Nat Immunol (2003) 4(3):274–9. doi: 10.1038/ni893
19. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol (2017) 17(5):281–94. doi: 10.1038/nri.2017.19
20. Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity (2008) 28(3):335–45. doi: 10.1016/j.immuni.2007.12.019
21. Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, et al. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol (2008) 9(12):1388–98. doi: 10.1038/ni.1667
22. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol (2001) 19:595–621. doi: 10.1146/annurev.immunol.19.1.595
23. Allman DM, Ferguson SE, Cancro MP. Peripheral B cell maturation. i. immature peripheral B cells in adults are heat-stable antigenhi and exhibit unique signaling characteristics. J Immunol (1992) 149(8):2533–40.
24. Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation. II. heat-stable antigen(hi) splenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J Immunol (1993) 151(9):4431–44.
25. Cancro MP, Allman DM, Hayes CE, Lentz VM, Fields RG, Sah AP, et al. B cell maturation and selection at the marrow-periphery interface. Immunol Res (1998) 17(1-2):3–11. doi: 10.1007/BF02786425
26. Marklund U, Lightfoot K, Cantrell D. Intracellular location and cell context-dependent function of protein kinase d. Immunity (2003) 19(4):491–501. doi: 10.1016/s1074-7613(03)00260-7
27. Wen R, Chen Y, Schuman J, Fu G, Yang S, Zhang W, et al. An important role of phospholipase Cgamma1 in pre-b-cell development and allelic exclusion. EMBO J (2004) 23(20):4007–17. doi: 10.1038/sj.emboj.7600405
28. Vettermann C, Schlissel MS. Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunol Rev (2010) 237(1):22–42. doi: 10.1111/j.1600-065X.2010.00935.x
29. Sandel PC, Monroe JG. Negative selection of immature B cells by receptor editing or deletion is determined by site of antigen encounter. Immunity (1999) 10(3):289–99. doi: 10.1016/s1074-7613(00)80029-1
30. Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. J Immunol (2009) 182(10):5982–93. doi: 10.4049/jimmunol.0801859
31. Vossenkamper A, Lutalo PM, Spencer J. Translational mini-review series on B cell subsets in disease. transitional B cells in systemic lupus erythematosus and sjogren's syndrome: clinical implications and effects of B cell-targeted therapies. Clin Exp Immunol (2012) 167(1):7–14. doi: 10.1111/j.1365-2249.2011.04460.x
32. Vossenkamper A, Spencer J. Transitional B cells: how well are the checkpoints for specificity understood? Arch Immunol Ther Exp (Warsz) (2011) 59(5):379–84. doi: 10.1007/s00005-011-0135-0
33. Wardemann H, Nussenzweig MC. B-cell self-tolerance in humans. Adv Immunol (2007) 95:83–110. doi: 10.1016/S0065-2776(07)95003-8
34. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
35. Grossmann M, O'Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S. The anti-apoptotic activities of rel and RelA required during b-cell maturation involve the regulation of bcl-2 expression. EMBO J (2000) 19(23):6351–60. doi: 10.1093/emboj/19.23.6351
36. Tomayko MM, Punt JA, Bolcavage JM, Levy SL, Allman DM, Cancro MP. Expression of the bcl-2 family member A1 is developmentally regulated in T cells. Int Immunol (1999) 11(11):1753–61. doi: 10.1093/intimm/11.11.1753
37. Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol (2013) 131(2):477–85 e1. doi: 10.1016/j.jaci.2012.11.050
38. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science (2001) 293(5537):2111–4. doi: 10.1126/science.1061964
39. Shulga-Morskaya S, Dobles M, Walsh ME, Ng LG, MacKay F, Rao SP, et al. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol (2004) 173(4):2331–41. doi: 10.4049/jimmunol.173.4.2331
40. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. (2009) 106(33):13945–50. doi: 10.1073/pnas.0903543106
41. Pieper K, Rizzi M, Speletas M, Smulski CR, Sic H, Kraus H, et al. A common single nucleotide polymorphism impairs b-cell activating factor receptor's multimerization, contributing to common variable immunodeficiency. J Allergy Clin Immunol (2014) 133(4):1222–5. doi: 10.1016/j.jaci.2013.11.021
42. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol (2002) 3(10):958–65. doi: 10.1038/ni842
43. Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity (2002) 17(4):515–24. doi: 10.1016/s1074-7613(02)00425-9
44. Hatada EN, Do RK, Orlofsky A, Liou HC, Prystowsky M, MacLennan IC, et al. NF-kappa B1 p50 is required for BLyS attenuation of apoptosis but dispensable for processing of NF-kappa B2 p100 to p52 in quiescent mature B cells. J Immunol (2003) 171(2):761–8. doi: 10.4049/jimmunol.171.2.761
45. Jellusova J, Miletic AV, Cato MH, Lin WW, Hu Y, Bishop GA, et al. Context-specific BAFF-r signaling by the NF-kappaB and PI3K pathways. Cell Rep (2013) 5(4):1022–35. doi: 10.1016/j.celrep.2013.10.022
46. Scholz JL, Crowley JE, Tomayko MM, Steinel N, O'Neill PJ, Quinn WJ 3rd, et al. BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc Natl Acad Sci U S A. (2008) 105(40):15517–22. doi: 10.1073/pnas.0807841105
47. Ramskold D, Parodis I, Lakshmikanth T, Sippl N, Khademi M, Chen Y, et al. B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine (2019) 40:517–27. doi: 10.1016/j.ebiom.2018.12.035
48. Pillai S, Cariappa A. The follicular versus marginal zone b lymphocyte cell fate decision. Nat Rev Immunol (2009) 9(11):767–77. doi: 10.1038/nri2656
49. Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol (2013) 13(2):118–32. doi: 10.1038/nri3383
50. Cancro MP. Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol (2009) 9(9):657–61. doi: 10.1038/nri2621
51. Descatoire M, Weller S, Irtan S, Sarnacki S, Feuillard J, Storck S, et al. Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J Exp Med (2014) 211(5):987–1000. doi: 10.1084/jem.20132203
52. Morgan D, Tergaonkar V. Unraveling B cell trajectories at single cell resolution. Trends Immunol (2022) 43(3):210–29. doi: 10.1016/j.it.2022.01.003
53. Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, et al. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity (2006) 24(6):729–39. doi: 10.1016/j.immuni.2006.04.005
54. Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol (2004) 173(4):2245–52. doi: 10.4049/jimmunol.173.4.2245
55. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature b lymphocytes depends on BCR signaling via the igalpha/beta heterodimer. Cell (2004) 117(6):787–800. doi: 10.1016/j.cell.2004.05.014
56. Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, et al. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol (2008) 9(12):1379–87. doi: 10.1038/ni.1666
57. Inabe K, Miyawaki T, Longnecker R, Matsukura H, Tsukada S, Kurosaki T. Bruton's tyrosine kinase regulates B cell antigen receptor-mediated JNK1 response through Rac1 and phospholipase c-gamma2 activation. FEBS Lett (2002) 514(2-3):260–2. doi: 10.1016/S0014-5793(02)02375-X
58. Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunol (2010) 28:21–55. doi: 10.1146/annurev.immunol.021908.132541
59. Yamazaki T, Kurosaki T. Contribution of BCAP to maintenance of mature B cells through c-rel. Nat Immunol (2003) 4(8):780–6. doi: 10.1038/ni949
60. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
61. Akkaya M, Akkaya B, Kim AS, Miozzo P, Sohn H, Pena M, et al. Toll-like receptor 9 antagonizes antibody affinity maturation. Nat Immunol (2018) 19(3):255–66. doi: 10.1038/s41590-018-0052-z
62. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45(3):471–82. doi: 10.1016/j.immuni.2016.09.001
63. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol (2020) 20(4):229–38. doi: 10.1038/s41577-019-0244-2
64. Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol (2021) 21(4):209–20. doi: 10.1038/s41577-020-00446-2
65. MacLennan IC. Germinal centers. Annu Rev Immunol (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
66. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
67. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408
68. Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner KM. A theory of germinal center B cell selection, division, and exit. Cell Rep (2012) 2(1):162–74. doi: 10.1016/j.celrep.2012.05.010
69. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T Follicular helper cell dynamics in germinal centers. Science (2013) 341(6146):673–7. doi: 10.1126/science.1241680
70. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3(9):822–9. doi: 10.1038/ni829
71. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol (2011) 12(1):24–34. doi: 10.1038/nri3128
72. Karnell JL, Ettinger R. The interplay of IL-21 and BAFF in the formation and maintenance of human B cell memory. Front Immunol (2012) 3:2. doi: 10.3389/fimmu.2012.00002
73. Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10(12):1292–9. doi: 10.1038/ni.1814
74. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA Methyltransferase 1 and DNA methylation patterning contribute to germinal center b-cell differentiation. Blood (2011) 118(13):3559–69. doi: 10.1182/blood-2011-06-357996
75. Mohr E, Cunningham AF, Toellner KM, Bobat S, Coughlan RE, Bird RA, et al. IFN-gamma produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc Natl Acad Sci U S A. (2010) 107(40):17292–7. doi: 10.1073/pnas.1004879107
76. Peng SL, Szabo SJ, Glimcher LH. T-Bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. (2002) 99(8):5545–50. doi: 10.1073/pnas.082114899
77. Watanabe K, Sugai M, Nambu Y, Osato M, Hayashi T, Kawaguchi M, et al. Requirement for runx proteins in IgA class switching acting downstream of TGF-beta 1 and retinoic acid signaling. J Immunol (2010) 184(6):2785–92. doi: 10.4049/jimmunol.0901823
78. He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol (2010) 11(9):836–45. doi: 10.1038/ni.1914
79. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-r mediate isotype switching in B cells. J Exp Med (2005) 201(1):35–9. doi: 10.1084/jem.20032000
80. Koike T, Harada K, Horiuchi S, Kitamura D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. Elife (2019) 8. doi: 10.7554/eLife.44245
81. Kanno Y, Sakurai D, Hase H, Kojima H, Kobata T. TACI induces cIAP1-mediated ubiquitination of NIK by TRAF2 and TANK to limit non-canonical NF-kappaB signaling. J Recept Signal Transduct Res (2010) 30(2):121–32. doi: 10.3109/10799891003634509
82. Tsuji S, Stein L, Kamada N, Nunez G, Bram R, Vallance BA, et al. TACI deficiency enhances antibody avidity and clearance of an intestinal pathogen. J Clin Invest. (2014) 124(11):4857–66. doi: 10.1172/JCI74428
83. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol (2008) 180(6):3655–9. doi: 10.4049/jimmunol.180.6.3655
84. Rauch M, Tussiwand R, Bosco N, Rolink AG. Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PloS One (2009) 4(5):e5456. doi: 10.1371/journal.pone.0005456
85. Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, et al. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol (2007) 178(5):2872–82. doi: 10.4049/jimmunol.178.5.2872
86. Lau AWY, Turner VM, Bourne K, Hermes JR, Chan TD, Brink R. BAFFR controls early memory B cell responses but is dispensable for germinal center function. J Exp Med (2021) 218(2). doi: 10.1084/jem.20191167
87. Muller-Winkler J, Mitter R, Rappe JCF, Vanes L, Schweighoffer E, Mohammadi H, et al. Critical requirement for BCR, BAFF, and BAFFR in memory B cell survival. J Exp Med (2021) 218(2). doi: 10.1084/jem.20191393
88. O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
89. Tangye SG, Bryant VL, Cuss AK, Good KL. BAFF, APRIL and human B cell disorders. Semin Immunol (2006) 18(5):305–17. doi: 10.1016/j.smim.2006.04.004
90. Mills DM, Bonizzi G, Karin M, Rickert RC. Regulation of late B cell differentiation by intrinsic IKKalpha-dependent signals. Proc Natl Acad Sci U S A. (2007) 104(15):6359–64. doi: 10.1073/pnas.0700296104
91. Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. (2003) 112(2):286–97. doi: 10.1172/JCI18025
92. Nakayama Y, Kosek J, Capone L, Hur EM, Schafer PH, Ringheim GE. Aiolos overexpression in systemic lupus erythematosus B cell subtypes and BAFF-induced memory B cell differentiation are reduced by CC-220 modulation of cereblon activity. J Immunol (2017) 199(7):2388–407. doi: 10.4049/jimmunol.1601725
93. von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity (2001) 14(5):573–82. doi: 10.1016/s1074-7613(01)00130-3
94. Castigli E, Scott S, Dedeoglu F, Bryce P, Jabara H, Bhan AK, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A. (2004) 101(11):3903–8. doi: 10.1073/pnas.0307348101
95. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet (2005) 37(8):829–34. doi: 10.1038/ng1601
96. Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet (2005) 37(8):820–8. doi: 10.1038/ng1600
97. Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, et al. Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol (2001) 2(7):638–43. doi: 10.1038/89790
98. Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in b-cell autoimmune disease. Nature (2000) 404(6781):995–9. doi: 10.1038/35010115
99. Jabara HH, Fu SM, Geha RS, Vercelli D. CD40 and IgE: synergism between anti-CD40 monoclonal antibody and interleukin 4 in the induction of IgE synthesis by highly purified human B cells. J Exp Med (1990) 172(6):1861–4. doi: 10.1084/jem.172.6.1861
100. Jabara HH, Weng Y, Sannikova T, Geha RS. TRAF2 and TRAF3 independently mediate ig class switching driven by CD40. Int Immunol (2009) 21(4):477–88. doi: 10.1093/intimm/dxp013
101. Tsubata T, Wu J, Honjo T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature (1993) 364(6438):645–8. doi: 10.1038/364645a0
102. Zarnegar B, He JQ, Oganesyan G, Hoffmann A, Baltimore D, Cheng G. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-kappaB activation pathways. Proc Natl Acad Sci U S A. (2004) 101(21):8108–13. doi: 10.1073/pnas.0402629101
103. Berberich I, Shu GL, Clark EA. Cross-linking CD40 on B cells rapidly activates nuclear factor-kappa b. J Immunol (1994) 153(10):4357–66. doi: 10.4049/jimmunol.153.10.4357
104. Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, et al. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J (2002) 21(20):5375–85. doi: 10.1093/emboj/cdf542
105. Tsukamoto N, Kobayashi N, Azuma S, Yamamoto T, Inoue J. Two differently regulated nuclear factor kappaB activation pathways triggered by the cytoplasmic tail of CD40. Proc Natl Acad Sci U S A. (1999) 96(4):1234–9. doi: 10.1073/pnas.96.4.1234
106. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell (2007) 12(3):280–92. doi: 10.1016/j.ccr.2007.08.011
107. Wei C, Chen Y, Xu L, Yu B, Lu D, Yu Y, et al. CD40 signaling promotes CXCR5 expression in B cells via noncanonical NF-kappaB pathway activation. J Immunol Res (2020) 2020:1859260. doi: 10.1155/2020/1859260
108. Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol (2000) 18:593–620. doi: 10.1146/annurev.immunol.18.1.593
109. Berkowska MA, Driessen GJ, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood (2011) 118(8):2150–8. doi: 10.1182/blood-2011-04-345579
110. Weller S, Faili A, Garcia C, Braun MC, Le Deist FF, de Saint Basile GG, et al. CD40-CD40L independent ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci U S A. (2001) 98(3):1166–70. doi: 10.1073/pnas.98.3.1166
111. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol (2014) 15(7):631–7. doi: 10.1038/ni.2914
112. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi: 10.1016/j.cell.2010.10.032
113. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509(7502):637–40. doi: 10.1038/nature13300
114. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345(6200):1058–62. doi: 10.1126/science.1257861
115. Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J Exp Med (1997) 186(3):439–48. doi: 10.1084/jem.186.3.439
116. Huang C, Gonzalez DG, Cote CM, Jiang Y, Hatzi K, Teater M, et al. The BCL6 RD2 domain governs commitment of activated B cells to form germinal centers. Cell Rep (2014) 8(5):1497–508. doi: 10.1016/j.celrep.2014.07.059
117. Chevrier S, Kratina T, Emslie D, Tarlinton DM, Corcoran LM. IL4 and IL21 cooperate to induce the high Bcl6 protein level required for germinal center formation. Immunol Cell Biol (2017) 95(10):925–32. doi: 10.1038/icb.2017.71
118. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate bcl-6 expression and germinal center responses. J Exp Med (2010) 207(2):353–63. doi: 10.1084/jem.20091738
119. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center b and plasma cell fates by dynamical control of IRF4. Immunity (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
120. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol (2011) 23(5):341–9. doi: 10.1016/j.smim.2011.08.010
121. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol (2006) 7(7):773–82. doi: 10.1038/ni1357
122. Muto A, Ochiai K, Kimura Y, Itoh-Nakadai A, Calame KL, Ikebe D, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J (2010) 29(23):4048–61. doi: 10.1038/emboj.2010.257
123. Sciammas R, Li Y, Warmflash A, Song Y, Dinner AR, Singh H. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol Syst Biol (2011) 7:495. doi: 10.1038/msb.2011.25
124. Turner CA Jr., Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of b lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77(2):297–306. doi: 10.1016/0092-8674(94)90321-2
125. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15(3):160–71. doi: 10.1038/nri3795
126. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med (2014) 211(10):2103–18. doi: 10.1084/jem.20132613
127. Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity (2013) 39(1):136–47. doi: 10.1016/j.immuni.2013.06.011
128. Kwak K, Quizon N, Sohn H, Saniee A, Manzella-Lapeira J, Holla P, et al. Intrinsic properties of human germinal center B cells set antigen affinity thresholds. Sci Immunol (2018) 3(29). doi: 10.1126/sciimmunol.aau6598
129. Nowosad CR, Spillane KM, Tolar P. Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat Immunol (2016) 17(7):870–7. doi: 10.1038/ni.3458
130. Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P. B cells use mechanical energy to discriminate antigen affinities. Science (2013) 340(6140):1587–90. doi: 10.1126/science.1237572
131. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517(7533):214–8. doi: 10.1038/nature13803
132. Tangye SG, Good KL. Human IgM+CD27+ B cells: memory B cells or "memory" B cells? J Immunol (2007) 179(1):13–9. doi: 10.4049/jimmunol.179.1.13
133. Goenka R, Matthews AH, Zhang B, O'Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211(1):45–56. doi: 10.1084/jem.20130505
134. Luo W, Weisel F, Shlomchik MJ. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-myc transcription factor in germinal center B cells. Immunity (2018) 48(2):313–26 e5. doi: 10.1016/j.immuni.2018.01.008
135. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity (2017) 46(6):1045–58 e6. doi: 10.1016/j.immuni.2017.06.005
136. Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science (2012) 336(6085):1178–81. doi: 10.1126/science.1213368
137. Palm AE, Henry C. Remembrance of things past: long-term B cell memory after infection and vaccination. Front Immunol (2019) 10:1787. doi: 10.3389/fimmu.2019.01787
138. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory b and plasma cells. Immunity (2016) 44(1):116–30. doi: 10.1016/j.immuni.2015.12.004
139. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science (2011) 331(6021):1203–7. doi: 10.1126/science.1201730
140. Taylor JJ, Pape KA, Steach HR, Jenkins MK. Humoral immunity. apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science (2015) 347(6223):784–7. doi: 10.1126/science.aaa1342
141. Sindhava VJ, Oropallo MA, Moody K, Naradikian M, Higdon LE, Zhou L, et al. A TLR9-dependent checkpoint governs B cell responses to DNA-containing antigens. J Clin Invest. (2017) 127(5):1651–63. doi: 10.1172/JCI89931
142. Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, et al. The transcription factor T-bet resolves memory B cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity (2020) 52(5):842–55 e6. doi: 10.1016/j.immuni.2020.03.020
143. Knox JJ, Myles A, Cancro MP. T-Bet(+) memory B cells: generation, function, and fate. Immunol Rev (2019) 288(1):149–60. doi: 10.1111/imr.12736
144. Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, et al. T Follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity (2018) 48(4):702–15 e4. doi: 10.1016/j.immuni.2018.03.027
145. Chen C, Laidlaw BJ. Development and function of tissue-resident memory B cells. Adv Immunol (2022) 155:1–38. doi: 10.1016/bs.ai.2022.08.001
146. Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol (2019) 20(1):97–108. doi: 10.1038/s41590-018-0260-6
147. Allie SR, Randall TD. Resident memory B cells. Viral Immunol (2020) 33(4):282–93. doi: 10.1089/vim.2019.0141
148. Pritzl CJ, Luera D, Knudson KM, Quaney MJ, Daniels MA, Teixeiro E. IKK2/NFkB signaling controls lung resident CD8 T cell memory during influenza infection. bioRxiv (2022) 2022:2. doi: 10.1101/2022.02.15.480615
149. Weill JC, Weller S, Reynaud CA. Human marginal zone B cells. Annu Rev Immunol (2009) 27:267–85. doi: 10.1146/annurev.immunol.021908.132607
150. Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, et al. Human blood IgM "memory" B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood (2004) 104(12):3647–54. doi: 10.1182/blood-2004-01-0346
151. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell (2000) 102(5):565–75. doi: 10.1016/s0092-8674(00)00079-9
152. Weller S, Mamani-Matsuda M, Picard C, Cordier C, Lecoeuche D, Gauthier F, et al. Somatic diversification in the absence of antigen-driven responses is the hallmark of the IgM+ IgD+ CD27+ B cell repertoire in infants. J Exp Med (2008) 205(6):1331–42. doi: 10.1084/jem.20071555
153. Willenbrock K, Jungnickel B, Hansmann ML, Kuppers R. Human splenic marginal zone B cells lack expression of activation-induced cytidine deaminase. Eur J Immunol (2005) 35(10):3002–7. doi: 10.1002/eji.200535134
154. Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem (2007) 14(3):367–76. doi: 10.2174/092986707779941113
155. Ouk S, Liou ML, Liou HC. Direct Rel/NF-kappaB inhibitors: structural basis for mechanism of action. Future Med Chem (2009) 1(9):1683–707. doi: 10.4155/fmc.09.96
156. Rasmi RR, Sakthivel KM, Guruvayoorappan C. NF-kappaB inhibitors in treatment and prevention of lung cancer. BioMed Pharmacother. (2020) 130:110569. doi: 10.1016/j.biopha.2020.110569
157. Vrabel D, Pour L, Sevcikova S. The impact of NF-kappaB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev (2019) 34:56–66. doi: 10.1016/j.blre.2018.11.003
158. Zhou J, Ching YQ, Chng WJ. Aberrant nuclear factor-kappa b activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target. Oncotarget (2015) 6(8):5490–500. doi: 10.18632/oncotarget.3545
159. Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature (2018) 564(7735):219–24. doi: 10.1038/s41586-018-0744-4
160. Del Pino-Molina L, Lopez-Granados E, Lecrevisse Q, Torres Canizales J, Perez-Andres M, Blanco E, et al. Dissection of the pre-germinal center b-cell maturation pathway in common variable immunodeficiency based on standardized flow cytometric EuroFlow tools. Front Immunol (2020) 11:603972. doi: 10.3389/fimmu.2020.603972
161. Fei L, Chen H, Ma L, EW, Wang R, Fang X, et al. Systematic identification of cell-fate regulatory programs using a single-cell atlas of mouse development. Nat Genet (2022) 54(7):1051–61. doi: 10.1038/s41588-022-01118-8
162. van der Burg M, Kalina T, Perez-Andres M, Vlkova M, Lopez-Granados E, Blanco E, et al. The EuroFlow PID orientation tube for flow cytometric diagnostic screening of primary immunodeficiencies of the lymphoid system. Front Immunol (2019) 10:246. doi: 10.3389/fimmu.2019.00246
163. Wagner DE, Klein AM. Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet (2020) 21(7):410–27. doi: 10.1038/s41576-020-0223-2
164. Wentink MWJ, Kalina T, Perez-Andres M, Del Pino Molina L, IJ H, Kavelaars FG, et al. Delineating human B cell precursor development with genetically identified PID cases as a model. Front Immunol (2019) 10:2680. doi: 10.3389/fimmu.2019.02680
165. Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, et al. High-level nuclear NF-kappa b and Oct-2 is a common feature of cultured Hodgkin/Reed-sternberg cells. Blood (1996) 87(10):4340–7. doi: 10.1182/blood.V87.10.4340.bloodjournal87104340
166. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med (2001) 194(12):1861–74. doi: 10.1084/jem.194.12.1861
167. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes b-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
168. Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science (2011) 331(6016):468–72. doi: 10.1126/science.1198946
169. Grondona P, Bucher P, Schulze-Osthoff K, Hailfinger S, Schmitt A. NF-kappaB activation in lymphoid malignancies: genetics, signaling, and targeted therapy. Biomedicines (2018) 6(2). doi: 10.3390/biomedicines6020038
170. Pasqualucci L, Klein U. NF-kappaB mutations in germinal center b-cell lymphomas: relation to NF-kappaB function in normal B cells. Biomedicines (2022) 10(10). doi: 10.3390/biomedicines10102450
171. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470(7332):115–9. doi: 10.1038/nature09671
172. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475(7354):101–5. doi: 10.1038/nature10113
173. Troen G, Warsame A, Delabie J. CD79B and MYD88 mutations in splenic marginal zone lymphoma. ISRN Oncol (2013) 2013:252318. doi: 10.1155/2013/252318
174. Yan Q, Huang Y, Watkins AJ, Kocialkowski S, Zeng N, Hamoudi RA, et al. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica (2012) 97(4):595–8. doi: 10.3324/haematol.2011.054080
175. Yu X, Li W, Deng Q, Liu H, Wang X, Hu H, et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-kappaB activation and lymphomagenesis. Blood (2021) 137(12):1615–27. doi: 10.1182/blood.2020004918
176. Pujari R, Hunte R, Khan WN, Shembade N. A20-mediated negative regulation of canonical NF-kappaB signaling pathway. Immunol Res (2013) 57(1-3):166–71. doi: 10.1007/s12026-013-8463-2
177. Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol (2012) 9(2):123–30. doi: 10.1038/cmi.2011.59
178. Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol (2013) 13(8):578–91. doi: 10.1038/nri3487
179. Suarez F, Lortholary O, Hermine O, Lecuit M. Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood (2006) 107(8):3034–44. doi: 10.1182/blood-2005-09-3679
180. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires b-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A. (2015) 112(44):13447–54. doi: 10.1073/pnas.1514944112
181. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active b-cell-receptor signalling in diffuse large b-cell lymphoma. Nature (2010) 463(7277):88–92. doi: 10.1038/nature08638
182. Busman-Sahay K, Drake L, Sitaram A, Marks M, Drake JR. Cis and trans regulatory mechanisms control AP2-mediated B cell receptor endocytosis via select tyrosine-based motifs. PloS One (2013) 8(1):e54938. doi: 10.1371/journal.pone.0054938
183. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med (2015) 21(8):922–6. doi: 10.1038/nm.3884
184. Young RM, Shaffer AL 3rd, Phelan JD, Staudt LM. B-cell receptor signaling in diffuse large b-cell lymphoma. Semin Hematol (2015) 52(2):77–85. doi: 10.1053/j.seminhematol.2015.01.008
185. Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug discovery. (2013) 12(3):229–43. doi: 10.1038/nrd3937
186. Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol (2010) 2(6):a000109. doi: 10.1101/cshperspect.a000109
187. Cunningham CA, Knudson KM, Peng BJ, Teixeiro E, Daniels MA. The POSH/JIP-1 scaffold network regulates TCR-mediated JNK1 signals and effector function in CD8 T cells. Eur J Immunol (2013) 43:12. doi: 10.1002/eji.201343635
188. Figueroa C, Tarras S, Taylor J, Vojtek AB. Akt2 negatively regulates assembly of the POSH-MLK-JNK signaling complex. J Biol Chem (2003) 278(48):47922–7. doi: 10.1074/jbc.M307357200
189. Kim G-H, Park E, Kong Y-Y, Han J-K. Novel function of POSH, a JNK scaffold, as an E3 ubiquitin ligase for the hrs stability on early endosomes. Cell Signal (2006) 18(4):553–63. doi: 10.1016/j.cellsig.2005.05.026
190. Lyons TR, Thorburn J, Ryan PW, Thorburn A, Anderson SM, Kassenbrock CK. Regulation of the pro-apoptotic scaffolding protein POSH by akt. J Biol Chem (2007) 282(30):21987–97. doi: 10.1074/jbc.M704321200
191. Tapon N, Nagata K, Lamarche N, Hall A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J (1998) 17(5):1395–404. doi: 10.1093/emboj/17.5.1395
192. Tilborghs S, Corthouts J, Verhoeven Y, Arias D, Rolfo C, Trinh XB, et al. The role of nuclear factor-kappa b signaling in human cervical cancer. Crit Rev Oncol Hematol (2017) 120:141–50. doi: 10.1016/j.critrevonc.2017.11.001
193. Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene (1999) 18(49):6938–47. doi: 10.1038/sj.onc.1203221
194. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch (2005) 446(5):475–82. doi: 10.1007/s00428-005-1264-9
195. Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS Jr. A requirement for NF-kappaB activation in bcr-abl-mediated transformation. Genes Dev (1998) 12(7):968–81. doi: 10.1101/gad.12.7.968
196. Nair A, Venkatraman M, Maliekal TT, Nair B, Karunagaran D. NF-kappaB is constitutively activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix. Oncogene (2003) 22(1):50–8. doi: 10.1038/sj.onc.1206043
197. Mukhopadhyay T, Roth JA, Maxwell SA. Altered expression of the p50 subunit of the NF-kappa b transcription factor complex in non-small cell lung carcinoma. Oncogene (1995) 11(5):999–1003.
198. Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. (1997) 100(12):2952–60. doi: 10.1172/JCI119848
Keywords: NF-kB, B cell, B cell development and differentiation, B cell deficiency, memory, immune memory, B cell memory subsets, B cell memory
Citation: Guldenpfennig C, Teixeiro E and Daniels M (2023) NF-kB’s contribution to B cell fate decisions. Front. Immunol. 14:1214095. doi: 10.3389/fimmu.2023.1214095
Received: 28 April 2023; Accepted: 03 July 2023;
Published: 18 July 2023.
Edited by:
Till Adhikary, University of Marburg, GermanyReviewed by:
Chaohong Liu, Huazhong University of Science and Technology, ChinaCopyright © 2023 Guldenpfennig, Teixeiro and Daniels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Daniels, ZGFuaWVsc21hQG1pc3NvdXJpLmVkdQ==; ZGFuaWVsc21hQG1pc3NvdXJpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.