 Ahmad Reza Panahi Meymandi1
Ahmad Reza Panahi Meymandi1 Behnia Akbari1
Behnia Akbari1 Tahereh Soltantoyeh1
Tahereh Soltantoyeh1 Jamshid Hadjati1
Jamshid Hadjati1 Daniel J. Klionsky2Behnam Badie3*
Daniel J. Klionsky2Behnam Badie3* Hamid Reza Mirzaei1,4,5*
Hamid Reza Mirzaei1,4,5*- 1Department of Medical Immunology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
- 2Life Sciences Institute and Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI, United States
- 3Division of Neurosurgery, City of Hope Beckman Research Institute, Duarte, California, United States
- 4Department of Genetics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, United States
- 5Institute for Immunology and Immune Health, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, United States
Despite chimeric antigen receptor (CAR) T cell therapy’s extraordinary success in subsets of B-cell lymphoma and leukemia, various barriers restrict its application in solid tumors. This has prompted investigating new approaches for producing CAR T cells with superior therapeutic potential. Emerging insights into the barriers to CAR T cell clinical success indicate that autophagy shapes the immune response via reprogramming cellular metabolism and vice versa. Autophagy, a self-cannibalization process that includes destroying and recycling intracellular components in the lysosome, influences T cell biology, including development, survival, memory formation, and cellular metabolism. In this review, we will emphasize the critical role of autophagy in regulating and rewiring metabolic circuits in CAR T cells, as well as how the metabolic status of CAR T cells and the tumor microenvironment (TME) alter autophagy regulation in CAR T cells to restore functional competence in CAR Ts traversing solid TMEs.
1 Introduction
In recent years, chimeric antigen receptor (CAR) T cell therapy has emerged as a groundbreaking approach for treating advanced hematological malignancies, revolutionizing cancer immunotherapy (1). CAR T cells are designed to recognize tumor-associated antigens on the surface of cancer cells and initiate intracellular signaling pathways that activate T cells, promote their proliferation, and enable the killing of tumor cells. The approval of the first anti-CD19 CAR T cell therapy by the US Food and Drug Administration (FDA) in 2017 marked a significant milestone in the field (2, 3). However, despite the success of CAR T cell therapy, several challenges remain, including toxicity, limited efficacy in solid tumors, reduced persistence, and inadequate tumor infiltration (4). To overcome these limitations, researchers are exploring novel strategies to engineer CAR T cells with enhanced anti-tumor activity. Among these approaches, metabolic regulation of CAR T cells has emerged as a promising avenue to reprogram these cells, making them more resistant to cytotoxicity and exhaustion while promoting their persistence.
Autophagy, an evolutionarily conserved self-destructive cellular process, is an active mechanism that occurs during metabolic deprivation (5). The term “autophagy” originates from the Greek words meaning “eating of self.” During autophagy, cellular cargo is delivered to lysosomes, where it is degraded and recycled, either selectively or non-selectively. This process is crucial for maintaining energy balance within cells (6). Autophagy is triggered under conditions of physiological stress, such as amino acid starvation, and serves as a survival mechanism by recycling cytoplasmic macromolecules. Among the three main types of autophagy (microautophagy, macroautophagy, and chaperone-mediated autophagy), macroautophagy is particularly relevant to T cell metabolism. Therefore, this review will primarily focus on macroautophagy. Macroautophagy, hereafter referred to as autophagy, is the most common form of autophagy observed in cells. It involves the formation of transient double-membrane structures, known as phagophores, around cytoplasmic components, including organelles, aggregated proteins, or microbes. Phagophores develop into autophagosomes, which subsequently fuse with lysosomes for the degradation and recycling of their contents (7). Autophagy plays a critical role in regulating various aspects of T cell biology, including T cell development, survival, and the formation of memory T (Tmem) cells (8). Recently, the role of autophagy in modulating T cell fate and function, mainly by influencing T cell metabolism, has gained increasing attention. T cells undergo metabolic reprogramming upon activation to meet their energy requirements. Autophagy serves as a link between T cell signaling and metabolic processes in response to T cell receptor (TCR) and interleukin 2 (IL-2) stimulation. In this review, we provide an overview of the intricate interplay between autophagy and metabolism in CAR T cells. We also discuss potential strategies for targeting autophagy and metabolism to engineer metabolically fit CAR T cells with enhanced anti-tumor potential.
2 Overview of molecular mechanisms of autophagy in T Cells
Autophagy has emerged as a crucial regulatory mechanism in the immune system, significantly influencing various aspects of T cell function. This cellular pathway governs the fate of different T cell subsets, and its activity is modulated by several factors and cellular complexes. Autophagy is activated in response to diverse scenarios, such as the engulfment and elimination of intracellular proteins, organelles, and pathogens through the formation of autophagic membrane structures (9). The molecular machinery involved in autophagy comprises a series of proteins, many of which are designated as ATG (autophagy-related) proteins. The initiation, expansion, and maturation of phagophores, the initial structures of autophagy, are tightly regulated. The ULK1/Atg1 complex plays a pivotal role in forming the phagophore structure (10). This complex, consisting of ULK1/ULK2, RB1CC1/FIP200, ATG13, and ATG101, activates the BECN1 (beclin 1, autophagy-related)-PIK3C3/VPS34 phosphatidylinositol 3-kinase (PtdIns3K) class III complex, which generates phosphatidylinositol-3-phosphate (PtdIns3P). The recruitment of PtdIns3P-binding molecules is integral to the process of phagophore formation (11). Two ubiquitin-like conjugation systems are essential for autophagy: the ATG12–ATG5-ATG16L1 system and the ATG8-family proteins, which include MAP1LC3/LC3 or GABARAP (in this review, we refer to LC3 for simplicity) (12). These conjugation systems facilitate the lipidation of LC3 and the subsequent formation of autophagosomes. During LC3 lipidation, LC3 is proteolytically processed from its precursor form, LC3-I, to the lipidated form, LC3-II, which functions as a docking site for cargo receptors on phagophore membranes (13). The cargo receptors connect the autophagy machinery to specific cargoes, and subsequently, the phagophore expands and seals, giving rise to the autophagosome (14). The autophagosome then fuses with the lysosome, forming an autolysosome (15). Within the autolysosome, the cargoes are degraded by lysosomal enzymes, and the breakdown products are released through permeases for recycling (16). This process is shown in Figure 1.
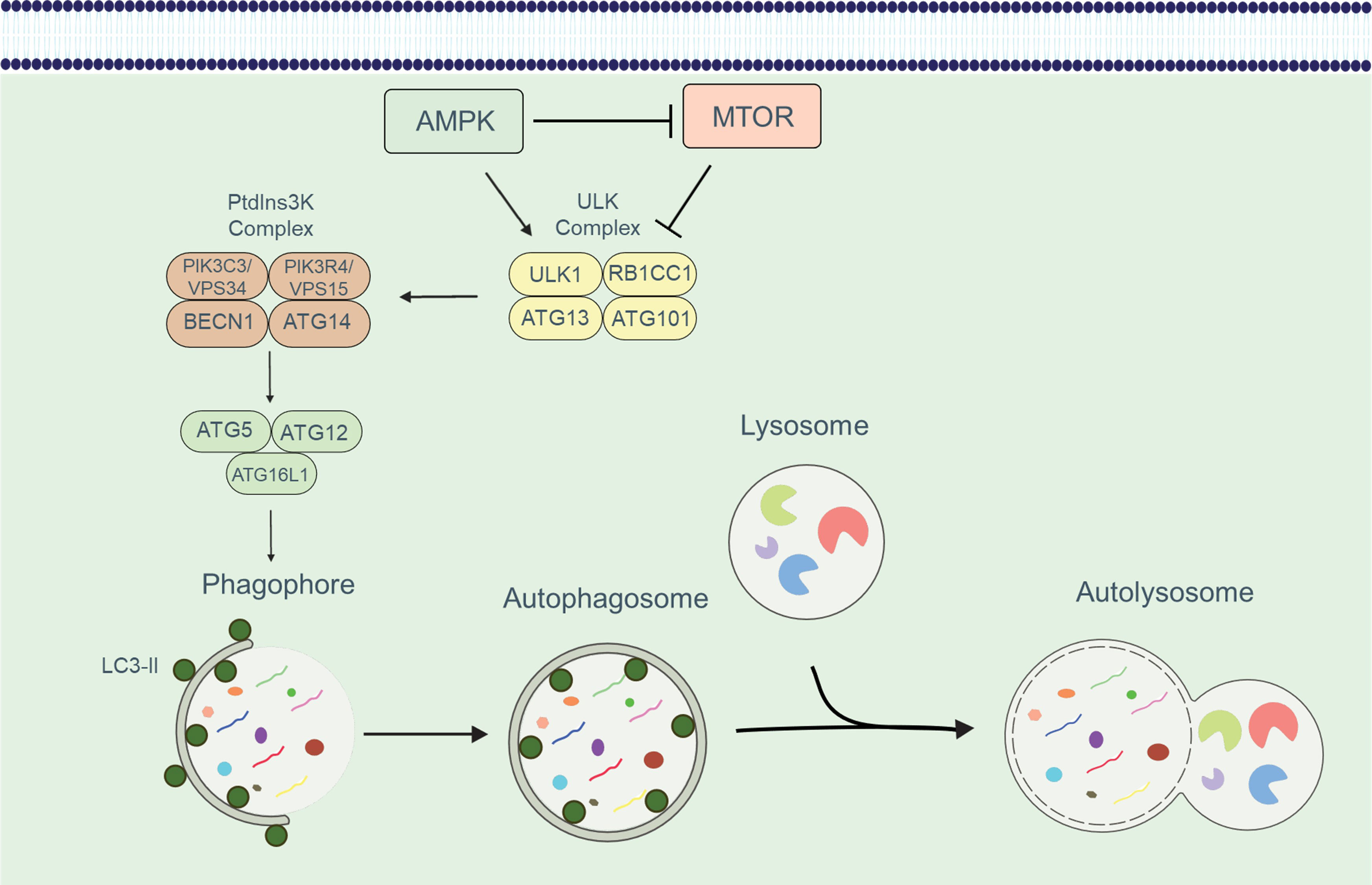
Figure 1 An overview of the autophagic process. The development of the phagophore and the recruitment of the ATG16L1 complex is induced by the sequential activation of the ULK and PtdIns3K class III complexes. The ATG16L1 complex along with LC3-II enables the phagophore to expand and form an autophagosome. Autophagosome formation and cargo isolation are aided by the LC3/Atg8 conjugation system. Following isolation, the cargo is degraded in an autolysosome, which forms when autophagosomes and lysosomes fuse.
3 Role of autophagy in T cell immunobiology
Autophagy is critical in various aspects of T cell immunobiology, including T cell survival, proliferation, TCR signaling, and T cell memory formation (Figure 2). In this section, we elaborate on the vital role of autophagy in T cell immunobiology.
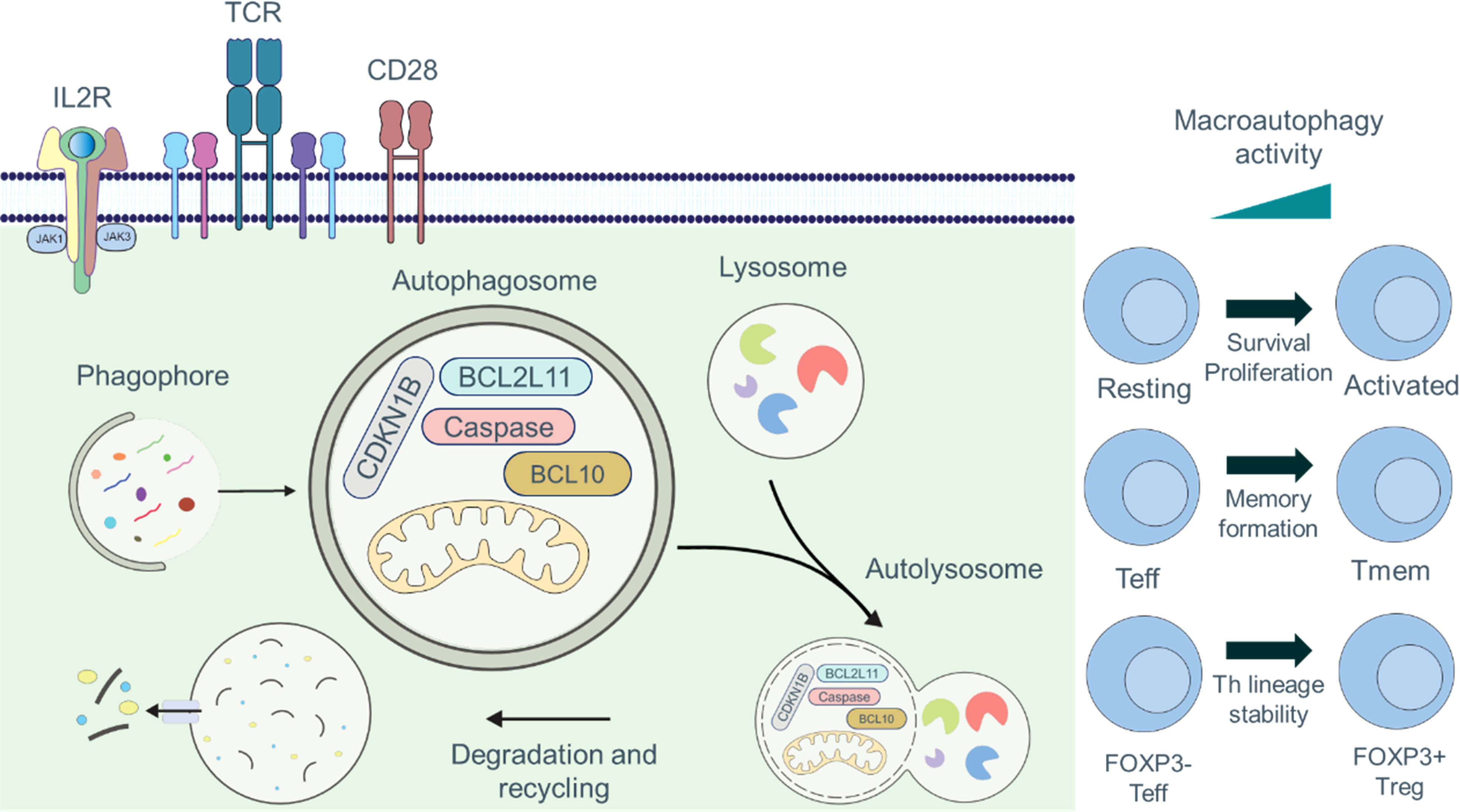
Figure 2 Roles of autophagy in T cell fate and function. Engagement of the TCR and CD28 signaling pathways is required for full activation of T cells, which can be further augmented by signaling through IL2R. Autophagy is maintained at basal levels in naïve and resting T cells, and the cargo of autophagosomes includes organelles, such as mitochondria, thus acting as an important quality control mechanism. Autophagy is induced by TCR- and IL2R-signaling. During activation, the cargo composition changes to primarily cytosolic components, and activation-induced degradation of certain proteins (CDKN1B, caspases, BCL2L11/Bim, or BCL10) has been described to regulate proliferation, survival, and activation. Autophagy plays crucial functions in controlling T cell metabolism, recycling cellular basic components, TCR signal transduction, and breaking down macromolecules to produce signaling metabolites. Additionally, macroautophagy is essential for T cell activation, cell survival, proliferation, and the production of CD8+ Tmem cells.
3.1 T cell survival
Previous studies have shown that prolonged persistence of T cells is associated with durable remission in patients with cancers, marking T cell survival as one of the key players in adoptive cellular therapy. Autophagy is essential for T cell survival, and its activity increases significantly upon T cell activation (17, 18). Autophagy-deficient T cell models have provided insights into the homeostatic role of autophagy in T cell proliferation and survival (19–21). Studies using T cell-specific Atg3, Atg5, and Atg7-deficient mice have demonstrated increased apoptosis rates and higher expression of CASP9 (caspase 9) (19, 20, 22). Similarly, BECN1/beclin-1-deficient T cell models exhibit elevated apoptosis rates with increased levels of proCASP3, CASP8, BCL2, and BCL2L11/Bim (23). Autophagy, particularly mitophagy, also regulates mitochondrial turnover as a quality control mechanism to maintain mitochondrial homeostasis (20, 22). Autophagy-deficient T cells show increased mitochondrial and endoplasmic reticulum content, leading to T cell death due to accumulation of reactive oxygen species (ROS) resulting from impaired removal of damaged organelles (19, 20, 22, 24). As a result of autophagy-related changes in mitochondrial turnover and pro-apoptotic factors, autophagy-deficient models exhibit reduced numbers of CD4+ and CD8+ T cells.
3.2 T cell proliferation
Besides defects in survival, defects in proliferation may also explain reduced CD4+ and CD8+ T cell numbers in autophagy-deficient cells, as autophagy regulates T cell proliferation (19, 25). T lymphocytes deficient in Atg5 and Atg7 fail to efficiently proliferate upon TCR stimulation (18, 19). Autophagy-deficient T cells are unable to enter the S phase after TCR stimulation. This defect is attributed to the accumulation of CDKN1B/p27, a negative T cell cycle regulator that is usually degraded by autophagy. Genetic deletion of a single Cdkn1b allele restores proliferation, indicating that autophagy regulates T cell proliferation mainly through selective degradation of CDKN1B (25). In summary, autophagy is essential for degrading organelles, apoptotic proteins, and cell cycle regulators, thereby ensuring proper T cell proliferation and survival.
3.3 TCR signaling
Following TCR stimulation, many downstream signaling events are required to initiate and maintain activation, and at least one major TCR signaling event is modulated by autophagy. BCL10, a key mediator of TCR-to-NFKB/NFκB signaling, is recognized by the autophagic cargo receptor SQSTM1/p62 and degraded upon effector T (Teff) cell activation. Inhibition of autophagy leads to the accumulation of BCL10, suggesting that autophagy prevents excessive TCR signaling, which could result in inappropriate T cell responses (26). In addition, autophagy-mediated degradation of PTPN1 removes its inhibitory effects on downstream signaling pathways of the TCR. Consequently, impaired autophagy results in PTPN1 accumulation, leading to reduced T cell priming response and diminished subsequent stimulation response (27).
3.4 T cell memory formation
Aside from regulating T cell proliferation, survival, and TCR signaling, autophagy regulates CD8+ T cell memory formation (28, 29). Atg7-deficient CD8+ T cells in mice show impaired memory function (28, 29). Studies have also found that defects in CD8+ T cell memory formation are related to failure to induce metabolic switches, such as upregulation of fatty acid oxidation, which is essential for CD8+ T cell memory formation (28–30). Consequently, these studies shed light on the role of autophagy in modulating T cell homeostasis.
4 Metabolic signature in T cell immunobiology
Metabolism of naïve T cells depends on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to provide energy. The switch from resting naïve T cells to highly proliferative Teff cells needs extensive metabolic reprogramming. As mitochondrial oxidative phosphorylation and ROS rise, aerobic glycolysis is also induced rapidly (31–33). Upon antigen recognition, extensive metabolic reprogramming occurs. Activation of the PI3K-AKT-mTOR pathway leads to the activation of transcription factors such as HIF1A/HIF-1α and MYC/c-Myc, which in turn upregulate the expression of SLC2A1/GLUT1 (solute carrier family 2 member 1) to promote glycolysis (34). This metabolic switch is known as the “Warburg effect,” where glycolysis is enhanced even in the presence of sufficient oxygen (35). In aerobic glycolysis, glucose is metabolized into pyruvate and converted into lactate independently of mitochondria. This allows the naïve T (Tn) cell to meet its metabolic needs for rapid proliferation and cytokine production to differentiate into Teff cells. The metabolic pattern of Tmem cells is similar to that of Tn cells, but oxidative phosphorylation is slightly higher, which allows them to be quickly activated after encountering an antigen (36). Unlike Teff cells, Tmem cells do not rely mainly on aerobic glycolysis but prefer OXPHOS. This process is fueled partially by mitochondrial catabolism of intracellular fatty acids (36, 37). Additionally, Tmem cells have increased mitochondrial mass and spare respiratory capacity, providing metabolic advantages for surviving and recalling after exposure to antigen (31, 36). Figure 3 illustrates the metabolic signature in T cell immunobiology.
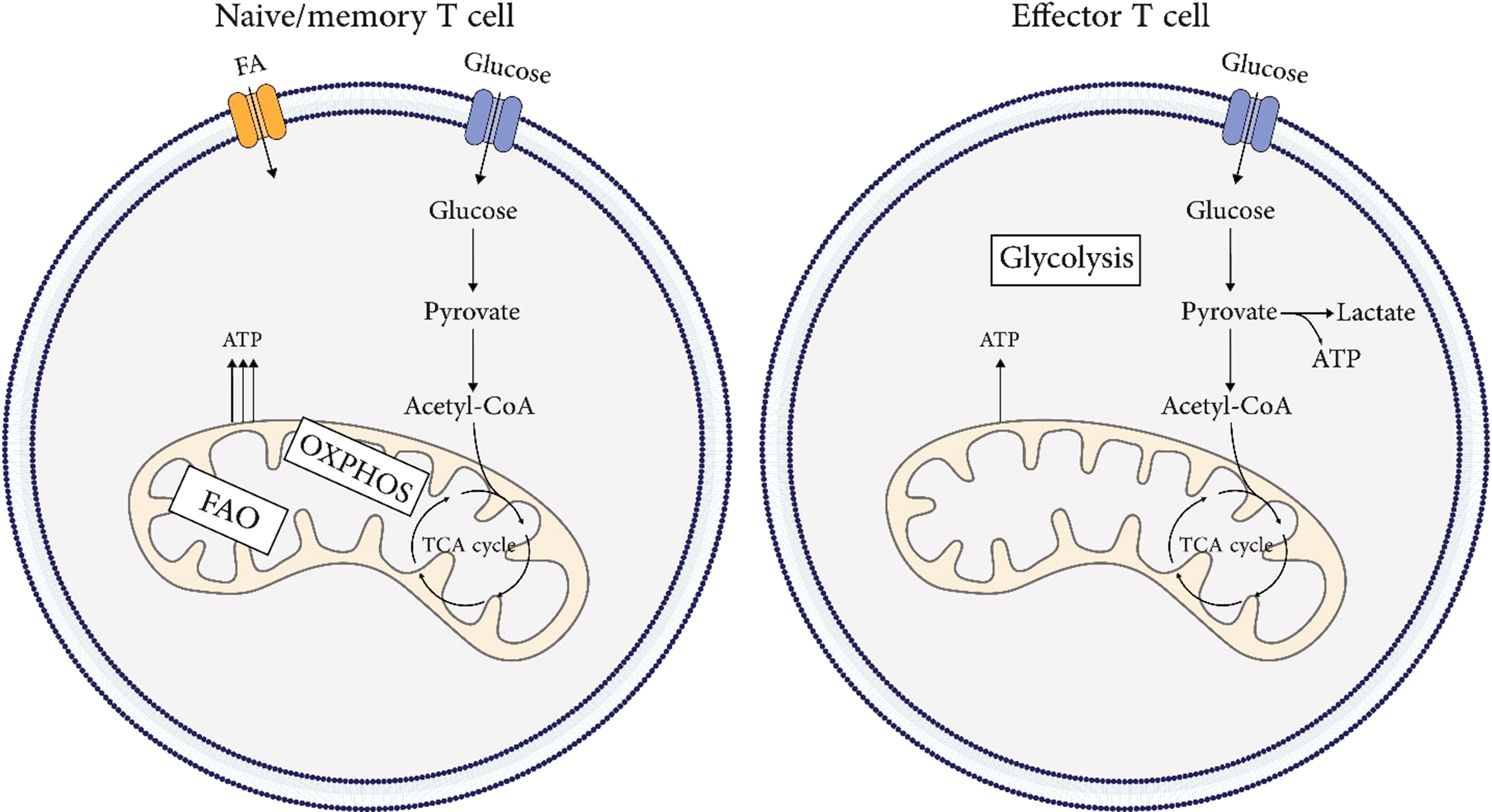
Figure 3 Metabolic signature in T cells immunobiology. Metabolism of naïve and, memory T cells depends on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to provide energy. The metabolic pattern of memory T cells is similar to that of naïve T cells, but oxidative phosphorylation is slightly higher, which allows them to be quickly activated after encountering an antigen. The switch from resting naïve or memory T cells to highly proliferative effector T cells needs extensive metabolic reprogramming. As mitochondrial oxidative phosphorylation and ROS rise, aerobic glycolysis is also induced rapidly.
5 Crosstalk between autophagy and metabolism in (CAR) T Cells
Common nutrient-sensing pathways integrate autophagy and metabolism in CAR T cells. The MTOR (mechanistic target of rapamycin kinase) and AMP-activated protein kinase (AMPK) signaling pathways work together to coordinate metabolic pathways and autophagic activity based on environmental conditions, nutrient availability, and energy status (Figure 4) (38–40). During cellular differentiation, both metabolism and autophagy need to accurately sense nutrient levels and respond accordingly to optimize nutrient utilization, storage, and recycling (41, 42). MTOR complex 1 (MTORC1) plays a critical role in sensing various stimuli, including growth factors and nutrients. The PI3K-AKT signaling axis promotes anabolism by activating MTOR networks, which modulate metabolic landscapes through transcriptional reprogramming (43). Transcription factors such as HIF1A, SREBF, and MYC/c-Myc are involved in this reprogramming process (34, 44).
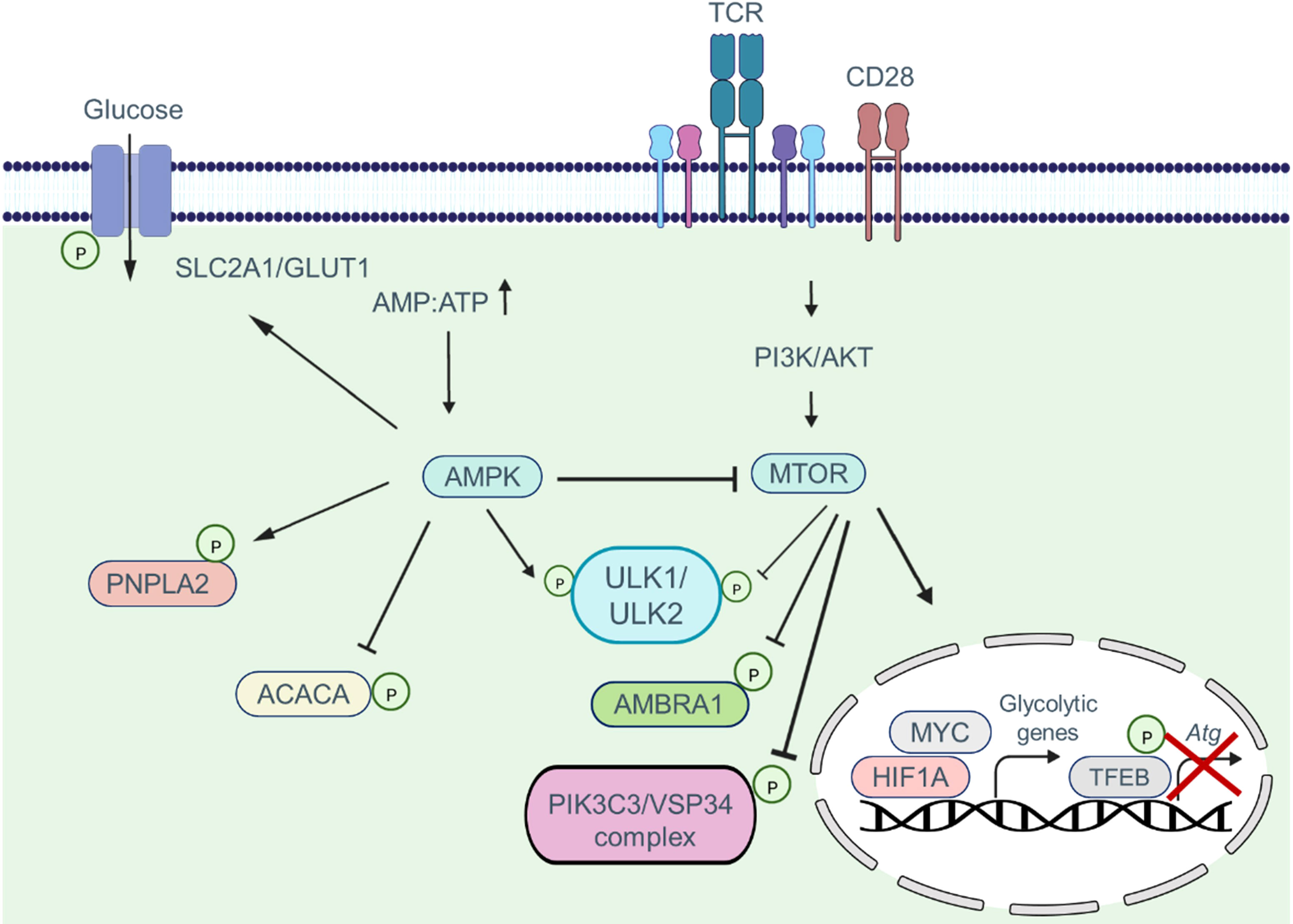
Figure 4 The combination of MTOR and AMPK signaling pathways coordinate and regulate metabolic pathways via the autophagic activity of T cells. MTOR activation suppresses autophagy by phosphorylating autophagy pathway components such as the ULK1/ULK2 complex, AMBRA1, and the PIK3C3/VPS34 complex. Moreover, MTOR signaling promotes phosphorylation and deactivation of the transcription factor TFEB, which is involved in the transcription of Atg genes. In addition, when the MTOR pathway is activated, the transcription factors MYC and HIF1A enter the nucleus and induce the transcription of glycolytic genes. AMPK activation, in contrast, inhibits the MTOR pathway and induces phosphorylation of autophagy pathway components, which in turn enhances the autophagic activity within cells. Furthermore, AMPK shifts metabolism toward OXPHOS phosphorylating and activating the SLC2A1/GLUT1 channel, as well as the PNPLA2/ATGL and ACACA/ACC1 enzymes.
Energy depletion triggers the activation of AMPK. When the ATP : AMP ratio decreases, AMP allosterically stabilizes and activates AMPK by binding to its phosphorylated form (45). AMPK directly phosphorylates metabolic enzymes and transporters, including PNPLA2/ATGL, SLC2A1/GLUT1, and ACACA/ACC1, to regulate energy balance (46–48). MTORC1 and AMPK control autophagy by regulating ULK1 kinase. In nutrient-rich conditions, MTORC1 phosphorylates ULK1 and ULK2, inhibiting autophagy (38). MTORC1 also phosphorylates AMBRA1, inhibiting the activity of the PIK3C3/VSP34 complex and influencing autophagy transcriptional regulation through TFEB phosphorylation and repression (49, 50). However, when nutrients are limited, the inhibitory effect of MTORC1 on ULK1/ULK2 is alleviated. Decreasing the ATP : AMP ratio activates AMPK, leading to the phosphorylation and activation of ULK1/ULK2 (51). ULK1/ULK2, in turn, promotes autophagy and may play a crucial role in providing alternative nutrient sources during metabolic stress (38). The interplay between T cell function, metabolism, and autophagy suggests their close association and highlights the importance of autophagy in regulating T cell metabolism, which subsequently impacts T cell function.
5.1 Autophagy signature governing (CAR) T cell metabolism
Autophagy can influence metabolic pathways (52), and the autophagic pathway plays a crucial role in successful metabolic reprogramming during immune cell differentiation. In recent years, studies have demonstrated the active role of cellular metabolism in determining T cell fate and function (53, 54). Additionally, autophagy is an essential catabolic process in T cells, and it can modulate the differentiation and function of CAR T cells by regulating metabolism. Teff cells deficient in autophagy produce less ATP during activation, exhibit decreased glycolysis and mitochondrial respiration, and show impaired cytokine secretion and cell proliferation (18). Autophagy degrades proteins, lipids, and glycogen, providing energy substrates and inhibiting this catabolic process can hinder efficient T cell activation (55). Recent research suggests that autophagy can regulate T cell metabolism by activating MTORC1. TAX1BP1 regulates autophagy, where autophagy-mediated degradation of cytosolic proteins provides L-cysteine to activate MTORC1 and induce metabolic changes (56). CD4+ T cells lacking TAX1BP1 fail to initiate the glycolytic anabolic changes necessary for meeting the energy demands of activation, resulting in reduced expression of HK2 (Hexokinase 2) and SLC2A1/GLUT1 (56).
Autophagy is crucial for meeting the metabolic demands of CAR T cell activation and regulating T cell metabolism during CD8+ Teff to Tmem differentiation. The presence of central memory T cells (Tcm) and stem cell-like memory T cells is vital for the success of CAR T cell therapy (57). Several studies have demonstrated the connection between autophagy and memory T cell differentiation, survival, and function (29, 58, 59). T cells lacking Atg5 shift significantly toward an effector memory phenotype and produce higher levels of IFN-γ and TNF-α (59). Mechanistically, Atg5-/-CD8+ T cells exhibit increased glucose metabolism, resulting in changes in histone methylation, increased H3K4me3 density, and transcriptional activation of metabolic and effector target genes (59). Autophagy-deficient CD8+ T cells show defects in memory formation due to disrupted metabolism, particularly in mitochondrial FAO (29). Unbiased metabolite data reveals dysregulated mitochondrial FAO, critical for memory T cell formation (30), in Atg5 or Atg7-deficient T cells at the peak of clonal expansion, precisely when antigen-specific T cells transition to the memory phase (29). Autophagy’s ability to degrade lipid droplets also contributes to the switch from glycolysis to FAO required for efficient CD8+ memory formation (60). Autophagy-deficient T cells may also exhibit impaired mitochondrial turnover, leading to other metabolic defects (22). A study by Yang et al. discovered that pik3c3/vps34 deletion reduces mitochondrial activity during T cell activation (61). The precise utilization and generation of substrates for mitochondrial FAO by memory T cells, and the direct regulation of mitochondrial FAO by autophagy in antigen-specific effector CD8+ T cells, present exciting subjects for future research. Although these studies indicate the ability of autophagy to reprogram CD8+ T cell metabolism, modify epigenetic marks, and limit effector functions, they report different functional consequences regarding tumor control resulting from autophagy activation in T cells. The discrepancies may arise from different functional outcomes observed in long-term (knockout models) versus acute loss of autophagy. Still, these studies collectively support the central role of autophagy in modulating anti-tumor CD8+ T cell responses. However, the precise mechanisms through which autophagy influences T cell memory formation are not yet fully understood. Autophagy likely acts as a double-edged sword in regulating T cell memory formation, with outcomes dependent on physiological and environmental conditions.
During CAR T cell production, metabolic reprogramming occurs when these cells are activated with anti-CD3 and anti-CD28 magnetic beads, transitioning from fatty acid phosphorylation to glycolysis. During this activation process, T cells that require less glucose tend to differentiate into Tmem cells, while those requiring more glucose tend to differentiate into Teff cells (62). Furthermore, in vitro stimulation of T cells with anti-CD3 and anti-CD28 antibodies induces autophagy (63). Hence, autophagy may modulate the metabolic program of CAR T cells even during CAR T cell manufacturing. The effects of autophagy modulation on CAR T cell metabolism, fate, and function are summarized in Table 1.
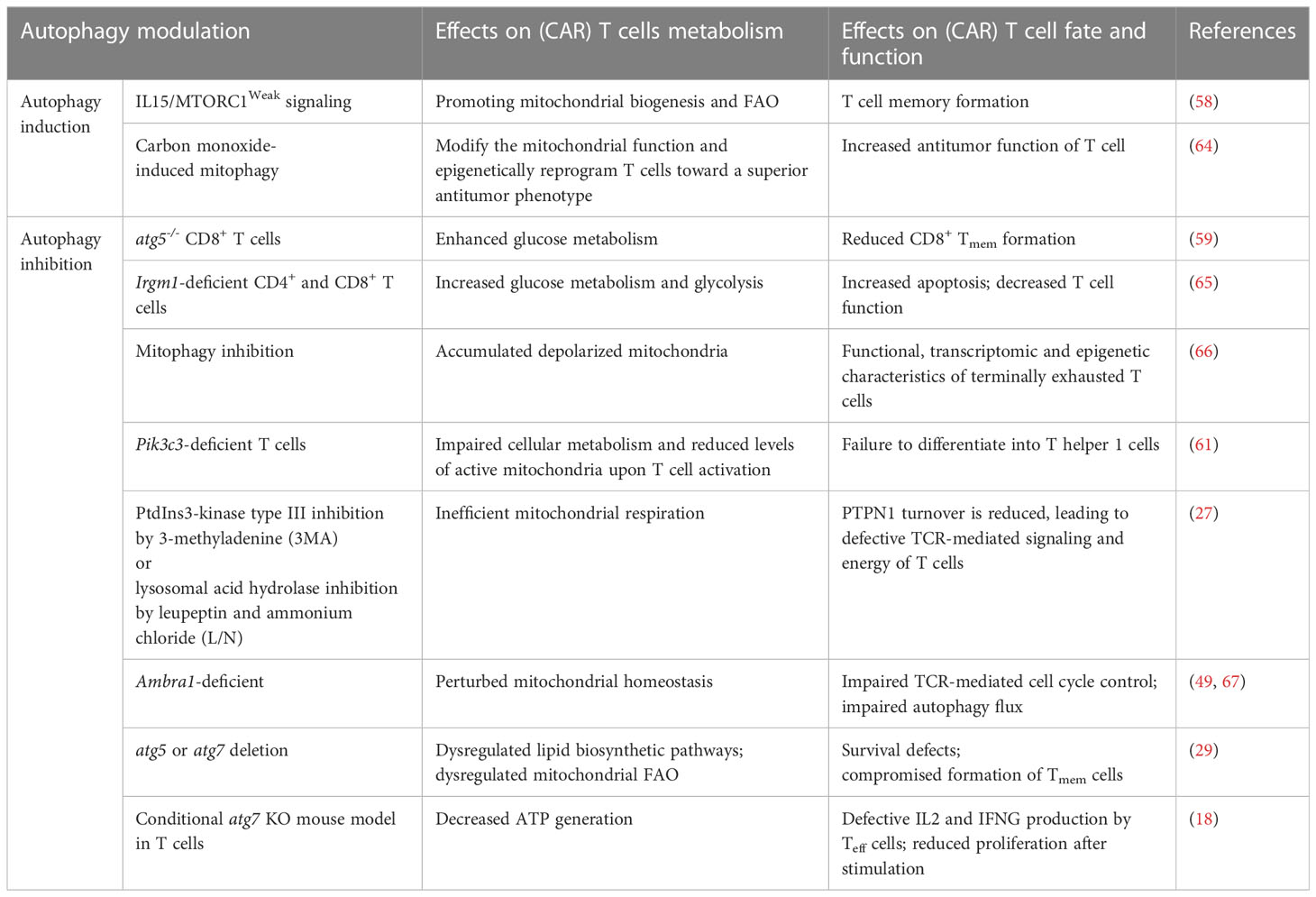
Table 1 Effects of autophagy modulation on (CAR) T cell metabolism, fate, and function.
5.2 Metabolic signature governing (CAR) T cell autophagy
Appropriate regulation of autophagy activity is necessary to control CAR T cell function. Metabolic changes induced by the TME can affect autophagy in CAR T cells. Under nutrient-depleted microenvironments, autophagy promotes metabolic adaptation by facilitating the availability of biomolecules required by CAR T cells (Figure 5). Xia et al. demonstrated in a study that tumor-infiltrating T cells have dysfunctional autophagy and reduced levels of RB1CC1/FIP200, a protein essential for autophagosome formation. The decrease in RB1CC1 expression is caused by tumor-derived lactate, which disrupts the balance between pro- and anti-apoptotic factors, enhancing tumor immune evasion (68).
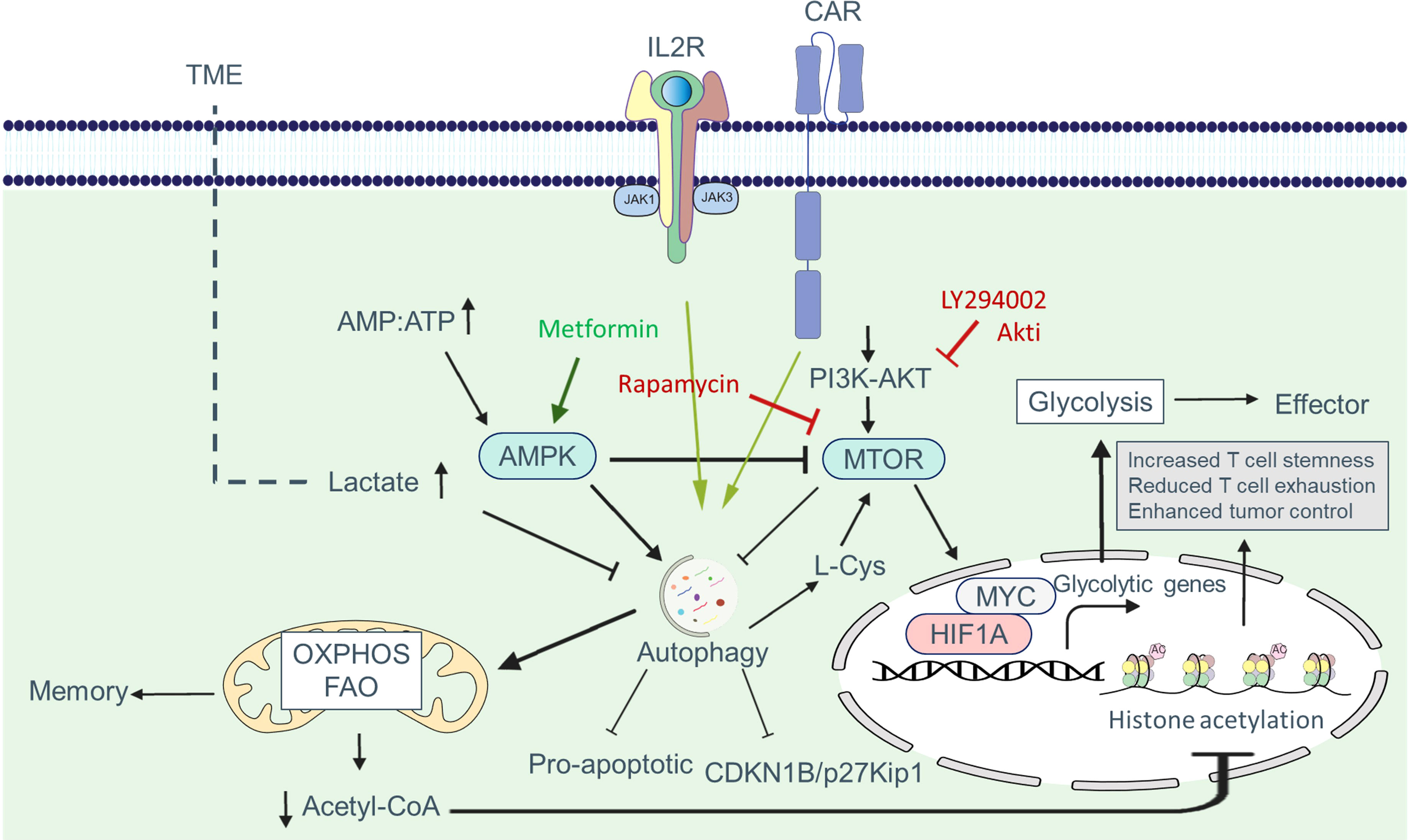
Figure 5 Autophagy-metabolism axis in CAR T cells. Autophagy is triggered in CAR T cells in response to tumor-associated antigens, allowing these cells to better adjust to metabolic demands for proliferation and activation. One of the signaling pathways increased in CAR T cells is the PI3K-AKT-MTOR pathway, which enhances the expression of glycolytic genes such as SLC2A1/GLUT1 via the transcription factors MYC and HIF1A. Increased glycolysis supplies the metabolic mediators required for cell growth and allows cells to develop an effector phenotype. Autophagy can be inhibited in part by MTOR. In the presence of nutrient deficiency in the tumor microenvironment, the AMP : ATP ratio rises, resulting in AMPK activation. AMPK shifts metabolism to OXPHOS and FAO by blocking MTOR and activating autophagy, which produces more ATP and leads to memory formation. Tumor metabolites such as lactate can hamper the adaptation of CAR T cells in the TME by inhibiting autophagy. The reduction of acetyl-CoA following OXPHOS reduces histone acetylation at effector and exhaustion loci, which helps reduce exhaustion, increase stemness and increase the antitumor activity of CAR T cells.
T cells possess nutrient-sensing systems comprising sensors, transporters, and signaling proteins that enable them to detect and respond to fluctuations in nutrient availability. Recent research has focused on understanding how T cells sense nutrients and how alterations in nutrient availability, such as glucose, amino acids, and fatty acids, can affect autophagy in these cells. Potassium overload or nutrient deficiency in the TME limits nutrient uptake by T cells, leading to autophagy via MTORC1 inhibition. Vodnala et al. found that increased potassium levels in the TME reduce nutrient uptake by CD8+ T cells, activating autophagy and promoting metabolic reprogramming towards OXPHOS, which preferentially utilizes acetyl coenzyme A. Reduced acetyl coenzyme A content results in decreased histone acetylation at effector and exhaustion loci, leading to increased T cell stemness, reduced T cell exhaustion, and enhanced tumor control (69). Moreover, autophagy activation in CD8+ T cells within the TME reduces T cell exhaustion and improves tumor control (69). Further studies can provide insights into the role of other tumor microenvironmental factors in altering nutrient sensing in T cells.
Autophagy is crucial for maintaining mitochondrial integrity and is essential for metabolic reprogramming to produce Teff cells. Regulatory T cells (Treg cells) infiltrating tumors express high levels of ARG2 (arginase type II), leading to the degradation of intracellular arginine and decreased MTOR activation by arginine, contributing to high levels of autophagy (70). Loss of autophagy in Treg cells switches metabolic pathways from oxidative phosphorylation to glycolysis with active MTORC1 and MYC, resulting in FOXP3 instability (70, 71). Autophagy-deficient Treg cells are more prone to apoptosis and functional problems (71). Recent studies have shown that PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) activity increases the rate of glycolysis and is related to the Warburg effect. Knockin and knockout experiments have positioned the glycolytic enzyme PFKFB3 upstream of the autophagy machinery. The metabolic consequence of PFKFB3 deficiency in T cells is the weakening of autophagic activity (72, 73). Therefore, metabolic changes in CAR T cells can also affect autophagic pathways (Figure 5). The effects of metabolic modulation on CAR T cell autophagy, fate, and function are summarized in Table 2.
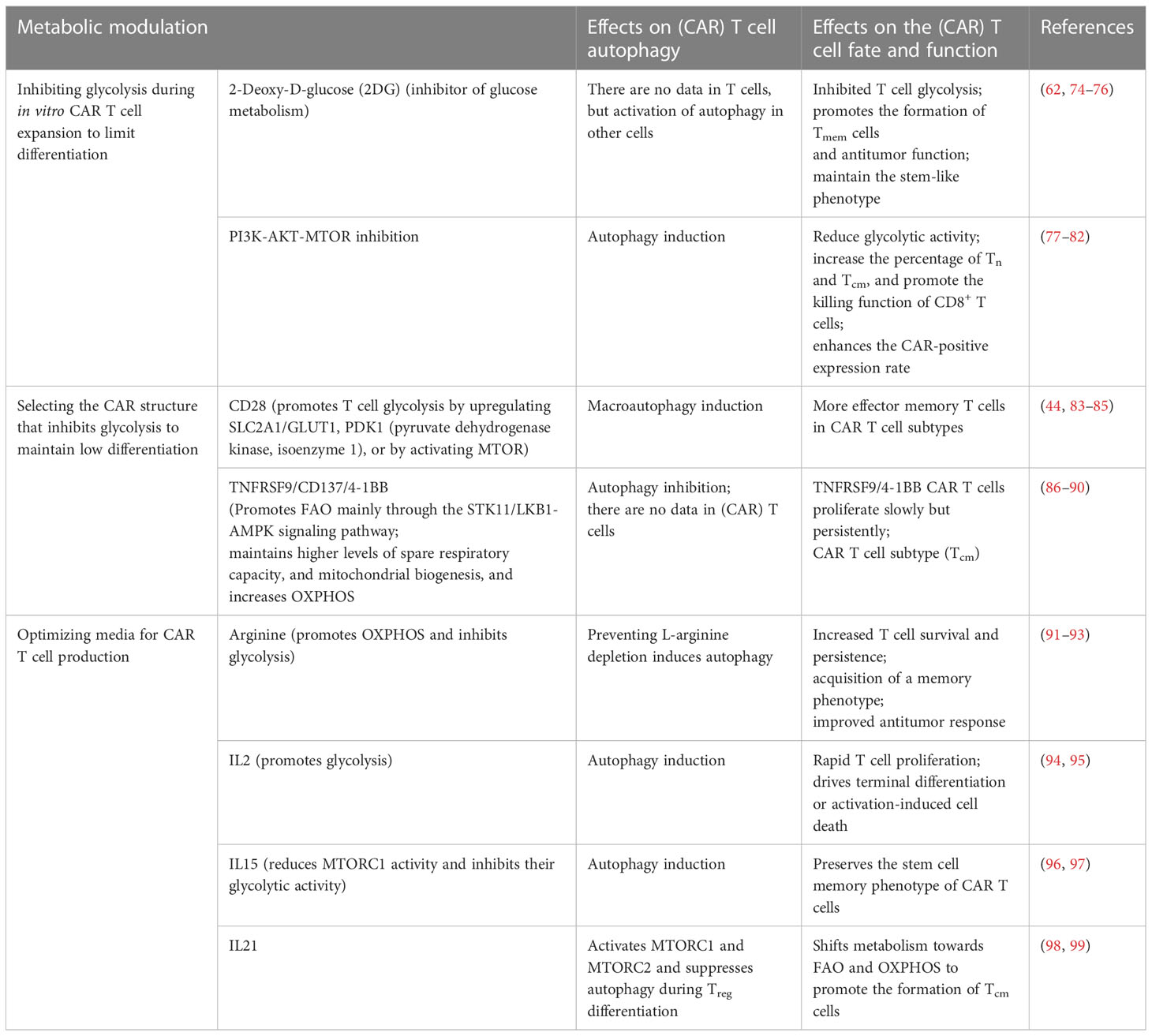
Table 2 Effects of metabolic modulation on (CAR) T cells autophagy, fate, and function.
6 Therapeutic implications of the autophagy-metabolism axis in CAR T cell therapy
6.1 Targeting the autophagy-metabolism axis in CAR T cells
6.1.1 CAR construct design
CAR constructs are designed with an extracellular domain comprising an antigen-specific single-chain variable fragment/scFv of an antibody linked to an intracellular signaling domain of a T cell receptor. Synthetic CARs can incorporate co-stimulatory domains that activate different signaling pathways upon antigen recognition (100). Designing a CAR with a co-stimulatory domain that regulates autophagy and metabolism in CAR T cells can influence their function. For example, the CD28 co-stimulatory domain enhances aerobic glycolysis and promotes effector memory differentiation in CAR T cells (86). CD28 stimulation activates the AKT-MTOR signaling pathway and HIF1A transcription factor, leading to increased glycolysis through enhanced glucose uptake and expression of glycolytic enzymes. Additionally, T cells redirected with CD3 and CD28 co-stimulation show upregulation of positive autophagy regulators and downregulation of negative autophagy regulators. Autophagy has been shown to enhance T cell survival by preventing cell death (19, 23). Therefore, CD28 co-stimulation supports highly proliferating and metabolically active CAR T cells.
In contrast to CD28, the TNFRSF9/CD137/4-1BB co-stimulatory domain promotes mitochondrial biogenesis and oxidative metabolism (86). CAR T cells with TNFRSF9 exhibit longer persistence and a higher Tcm phenotypes than those with CD28 (86). Thus, TNFRSF9 enhances central memory differentiation and improves the proliferation and persistence of CAR T cells in vitro. However, it is important to note that human TNFRSF9 can be degraded through the autophagic pathway (101), which may limit the potential of autophagy induction in CAR T cells containing TNFRSF9. Further studies are needed to elucidate the role of TNFRSF9 in autophagy regulation in CAR T cells.
Fourth-generation CAR T cells are designed to incorporate transgenic cytokines to enhance proliferation and persistence (102). Commonly used cytokines include IL12, IL15, and IL18, which affect T cell metabolism. For instance, IL15-treated CAR T cells exhibit reduced MTORC1 activity, leading to a decrease in glycolysis. Furthermore, IL15-treated CAR T cells express more genes related to FAO and display elevated levels of OXPHOS. These metabolic changes are accompanied by autophagy activation, enhancing CAR T cell anticancer activity in vivo by preserving stem cell memory and improving proliferative efficiency (103–105). These observations indicate that CAR constructs with different co-stimulatory domains activate distinct metabolic and autophagy pathways in T cells, thereby influencing their fitness within the TME (106).
6.1.2 Modulation of signaling pathways
Targeting components of the autophagy-metabolism axis using specific compounds is a promising approach to modulate CAR T cell biology and improve their antitumor potential. Studies have shown that the PI3K-AKT-MTOR pathway promotes the differentiation of terminal effector T cells rather than Tcm cells, reducing CAR T cell persistence (107). To address this, researchers utilize inhibitors targeting the glycolysis pathway, PI3K, AKT, and MTOR to decrease T cell glycolysis levels ex vivo, resulting in a more functional T cell product in vivo (108). Recent reports have highlighted the essential role of T cell-intrinsic mitochondrial regulation by autophagy in sustaining immunity against tumors (66). Inhibiting the PI3K-AKT-MTOR pathway can enhance autophagy, a well-described homeostatic process that promotes T cell memory and mitochondrial fitness (29, 109). The effects of modulating autophagy-metabolism signaling pathways on CAR T cells are summarized in Table 3.
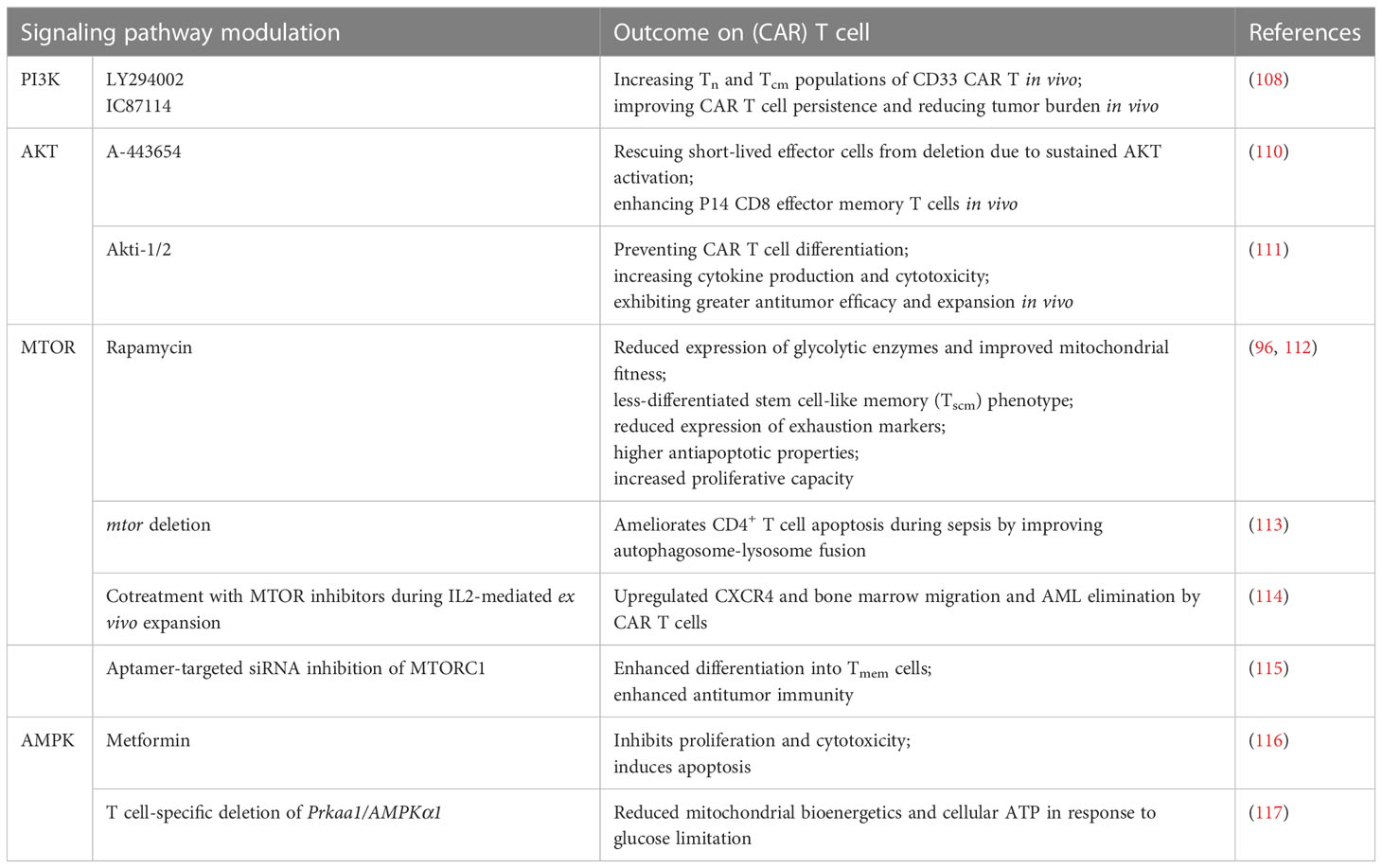
Table 3 Effects of autophagy-metabolism signaling pathway modulation on (CAR) T cells.
6.1.3 Genetic manipulation
Genetic modifications targeting the autophagy-metabolism axis can enhance the antitumoral function of CAR T cells. Studies have shown that in bone marrow chimeras with ATG5-deficient donor cells, there is a significant increase in IFN-γ and TNFα-producing CD8+ T cells in tumor-infiltrating lymphocytes, leading to improved tumor control (59). Similarly, CD8+ T cells lacking ATG5 display an effector memory phenotype and produce more IFN-γ and TNFα (118). Autophagy-competent mice receiving subtherapeutic doses of atg5-/- T cells also demonstrate controlled tumor growth (118). Therefore, genetic ablation of Atg5 can increase the metabolic activity and tumor-killing ability of CAR T cells. Consequently, Atg5 ablation provides a novel therapeutic opportunity to improve the efficacy of CAR T cell immunotherapy in solid tumors. Researchers have also designed CRISPR-Cas9 gene editing to knock out the autophagy function in CAR T cells as a novel immunotherapy approach to treat ovarian cancer (119).
7 Targeting the autophagy-metabolism axis as a CAR T cell combination therapy
7.1 Improving CAR T cell trafficking
The TME is known to impede the infiltration and function of CAR T cells in solid tumors. Autophagy has been shown to regulate the expression of chemokines in tumor cells that mediate immune cell migration to the tumor (120, 121). Inhibiting autophagy in tumors may modify the TME and promote the production of TH1-type chemokines, facilitating the trafficking of CAR T cells (120, 122). A study in lung tumor models demonstrated that simultaneous treatment with chemotherapy and MEK inhibitors promotes mitophagy and recognition of mitochondrial DNA by TLR9 (120, 121). TLR9 signaling leads to the expression of CXCL10 in tumor cells and the recruitment of CD8+ T cells to the tumors (121). Therefore, targeting autophagy in tumor cells can be utilized to increase the migration of T cells to the tumor and improve the antitumor response in combination with immunotherapies such as immune checkpoint blockade (ICB) or CAR T cell therapy.
7.2 Lactate
Increasing lactate levels in the TME can inhibit autophagy in CAR T cells (68). Therefore, the use of lactate dehydrogenase (LDH) and monocarboxylate transporter (MCT) inhibitors (123) in combination with CAR T cell therapy may enhance autophagy, thereby improving the survival and proliferation of CAR T cells.
7.3 Immune checkpoint blockade
CD274/PD-L1 molecules are overexpressed in several types of cancer (124). The interaction between CD274/PD-L1 and PDCD1/PD1 on T cells prevents cytotoxic T cell activation, allowing cancer cells to evade the immune system (124). The CD274/PD-L1/PD1 axis is critical in regulating T cell metabolism. PDCD1/PD1 blocks glycolysis by inhibiting the PI3K-AKT-MTOR pathways and downregulating SLC2A1/GLUT1 expression, which is crucial for T cell activation (125). Additionally, PDCD1/PD1 activates AMPK, a kinase that regulates fatty acid oxidation and induces autophagy by activating ULK1 (126). In contrast, CD274/PD-L1 induces glycolysis in tumor cells, depleting glucose from the TME (127), which is essential for CAR T cell activity. Therefore, inhibitors targeting the PDCD1/PD1-CD274/PD-L1 axis can combined with CAR T cells to affect the autophagy-metabolism pathway, thereby improving the antitumor function of CAR T cells.
7.4 Metabolic reprogramming
Metabolic reprogramming of the tumor microenvironment can significantly impact CAR T cell function. One approach to modulating the TME is to target specific metabolic pathways in tumor cells. For example, glycolysis or glutamine metabolism inhibitors can alter the metabolic landscape of the TME, potentially enhancing CAR T cell efficacy (33, 128). Furthermore, the manipulation of specific metabolic checkpoints, such as the inhibition of key enzymes involved in lipid metabolism or amino acid utilization, may provide opportunities to enhance CAR T cell function within the TME (129, 130). Strategies aimed at metabolic reprogramming of both the TME and CAR T cells hold promise for improving the therapeutic outcomes of CAR T cell combination therapy.
7.5 Hypoxia and autophagy
Hypoxia, a condition of reduced oxygen availability, is a common feature of the TME and has been associated with immunosuppression and resistance to therapy. Autophagy plays a crucial role in cellular adaptation to hypoxic stress, including the survival of tumor cells. Targeting the interplay between hypoxia and autophagy within the TME presents a potential strategy to enhance CAR T cell therapy. Modulating hypoxia-inducible factors (HIFs), which regulate both metabolism and autophagy, can influence the function of CAR T cells in hypoxic tumor regions (131, 132). Combination therapies that target hypoxia, such as hypoxia-activated prodrugs or inhibitors of HIF signaling, in conjunction with autophagy modulation, may promote CAR T cell persistence and antitumor activity.
8 Conclusions
This review investigates the autophagy-metabolism axis in (CAR) T cells. Autophagy is present in naïve T cells at a basal level; however, upon TCR stimulation, autophagy increases to support the metabolic demands for activation and proliferation. Various aspects of (CAR) T cell performance against the tumor, including survival, proliferation, phenotype, and exhaustion are affected by the interaction of autophagy and metabolism. In general, inhibition of autophagy in (CAR) T cells leads to increased glycolysis, inefficient mitochondrial respiration, reduced ATP production, and decreased FAO. These metabolic changes contribute to an effector phenotype. However, inhibition of autophagy may increase apoptotic proteins and cell cycle inhibitors that compromise the survival and proliferation of (CAR) T cells.
Conversely, the induction of autophagy is associated with increased OXPHOS, and FAO, which helps (CAR) T cells to differentiate toward the memory phenotype. Also, activation of autophagy can lead to increased survival and proliferation of (CAR) T cells by destroying pro-apoptotic proteins and cell cycle inhibitors. Nutrient-deficient conditions in the tumor microenvironment can cause metabolic changes in CAR T cells. The metabolic stress caused by the TME impairs effector T cell function and induces apoptosis. An elevated level of autophagy in CAR T cells may boost T cell fitness and survival in the TME; therefore, enhancing autophagy in CAR T cells before transfusion to patients might improve the effectiveness of the therapy (133). Furthermore, blocking autophagy in tumors may improve CAR T therapy efficacy by increasing tumor-associated antigen expression. Additionally, the metabolites produced by tumor cells can modulate autophagy in CAR T cells by influencing the components of the autophagy pathways. However, despite recent progress in this area, many aspects concerning autophagy, metabolism, and CAR T cell function remain unknown. Further studies are warranted to gain a comprehensive understanding of this axis in CAR T cells, ultimately identifying targets that can be manipulated to enhance the antitumor function of these cells. Continued research in this field has the potential to advance the development of more effective CAR T cell therapies.
Author contributions
HM conceived the idea. AP, BA and TS wrote the first draft of the manuscript and drew the figures. JH, DK, BB, and HM gave critical comments and revised the manuscript. All authors read and approved the final version of this work.
Funding
This publication was partly supported by Tehran University of Medical Sciences (grants nos. 50756 and 50760, awarded to HM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CAR, chimeric antigen receptor; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; Tcm, central memory T cells Teff, effector T cell; Tmem, memory T cell; TME, tumor microenvironment; Tn, naïve T cell; Treg, regulatory T cell.
References
1. Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M. The application of CAR-T cell therapy in hematological Malignancies: advantages and challenges. Acta Pharm Sin B (2018) 8(4):539–51. doi: 10.1016/j.apsb.2018.03.001
2. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
3. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med (2017) 377(26):2545–54. doi: 10.1056/NEJMoa1708566
4. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):1–11. doi: 10.1038/s41408-021-00459-7
5. Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell (2016) 3(12):588. doi: 10.15698/mic2016.12.546
6. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
7. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
8. Wang L, Das JK, Kumar A, Peng HY, Ren Y, Xiong X, et al. Autophagy in T-cell differentiation, survival and memory. Immunol Cell Biol (2021) 99(4):351–60. doi: 10.1111/imcb.12422
9. Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol (2019) 35(1):453–75. doi: 10.1146/annurev-cellbio-100818-125300
10. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol (2010) 22(2):132–9. doi: 10.1016/j.ceb.2009.12.004
11. Stjepanovic G, Baskaran S, Lin MG, Hurley JH. Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol Cell (2017) 67(3):528–34.e3. doi: 10.1016/j.molcel.2017.07.003
12. Walczak M, Martens S. Dissecting the role of the Atg12–Atg5-Atg16 complex during autophagosome formation. Autophagy (2013) 9(3):424–5. doi: 10.4161/auto.22931
13. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol (2014) 16(6):495–501. doi: 10.1038/ncb2979
14. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol (2010) 12(9):836–41. doi: 10.1038/ncb0910-836
15. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy (2021) 17(10):2680–8. doi: 10.1080/15548627.2020.1823124
16. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther (2022) 7(1):196. doi: 10.1038/s41392-022-01046-3
17. Botbol Y, Patel B, Macian F. Common γ-chain cytokine signaling is required for macroautophagy induction during CD4+ T-cell activation. Autophagy (2015) 11(10):1864–77. doi: 10.1080/15548627.2015.1089374
18. Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol (2010) 185(12):7349–57. doi: 10.4049/jimmunol.1000576
19. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204(1):25–31. doi: 10.1084/jem.20061303
20. Jia W, He Y-W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol (2011) 186(9):5313–22. doi: 10.4049/jimmunol.1002404
21. Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci (2012) 109(22):8670–5. doi: 10.1073/pnas.1205305109
22. Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol (2009) 182(7):4046–55. doi: 10.4049/jimmunol.0801143
23. Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ (2012) 19(1):144–52. doi: 10.1038/cdd.2011.78
24. Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy (2009) 5(5):625–35. doi: 10.4161/auto.5.5.8133
25. Jia W, He MX, McLeod IX, Guo J, Ji D, He YW. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy (2015) 11(12):2335–45. doi: 10.1080/15548627.2015.1110666
26. Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity (2012) 36(6):947–58. doi: 10.1016/j.immuni.2012.04.008
27. Mocholi E, Dowling SD, Botbol Y, Gruber RC, Ray AK, Vastert S, et al. Autophagy is a tolerance-avoidance mechanism that modulates TCR-mediated signaling and cell metabolism to prevent induction of T cell anergy. Cell Rep (2018) 24(5):1136–50. doi: 10.1016/j.celrep.2018.06.065
28. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife (2014) 3:e03706. doi: 10.7554/eLife.03706
29. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi: 10.1038/ni.3025
30. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
31. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA (2013) 110(35):14336–41. doi: 10.1073/pnas.1221740110
32. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
33. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
34. Gnanaprakasam JN, Wang R. Myc in regulating immunity: metabolism and beyond. Genes (Basel) (2017) 8(3). doi: 10.3390/genes8030088
35. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
36. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi: 10.1016/j.immuni.2011.12.007
37. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
38. Metur SP, Klionsky DJ. Adaptive immunity at the crossroads of autophagy and metabolism. Cell Mol Immunol (2021) 18(5):1096–105. doi: 10.1038/s41423-021-00662-3
39. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi: 10.1111/j.1600-065X.2012.01152.x
40. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493(7432):346–55. doi: 10.1038/nature11862
41. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol (2007) 7(10):767–77. doi: 10.1038/nri2161
42. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi: 10.1038/nri3532
43. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci (2013) 126(8):1713–9. doi: 10.1242/jcs.125773
44. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
45. Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. Structural basis for AMP binding to mamMalian AMP-activated protein kinase. Nature (2007) 449(7161):496–500. doi: 10.1038/nature06161
46. Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J (2016) 283(16):2987–3001. doi: 10.1111/febs.13698
47. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
48. Shi Y, Shen HM, Gopalakrishnan V, Gordon N. Epigenetic regulation of autophagy beyond the cytoplasm: a review. Front Cell Dev Biol (2021) 9:675599. doi: 10.3389/fcell.2021.675599
49. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol (2013) 15(4):406–16. doi: 10.1038/ncb2708
50. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy (2012) 8(6):903–14. doi: 10.4161/auto.19653
51. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi: 10.1038/ncb2152
52. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330(6009):1344–8. doi: 10.1126/science.1193497
53. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259. doi: 10.1146/annurev-immunol-032712-095956
54. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33(4):168–73. doi: 10.1016/j.it.2012.01.010
55. Yang J, Zhou R, Ma Z. Autophagy and energy metabolism. Adv Exp Med Biol (2019) 1206:329–57. doi: 10.1007/978-981-15-0602-4_16
56. Whang MI, Tavares RM, Benjamin DI, Kattah MG, Advincula R, Nomura DK, et al. The ubiquitin binding protein TAX1BP1 mediates autophagosome induction and the metabolic transition of activated T cells. Immunity (2017) 46(3):405–20. doi: 10.1016/j.immuni.2017.02.018
57. Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol Immunother (2018) 67(7):1053–66. doi: 10.1007/s00262-018-2155-7
58. Ara A, Wu Z, Xu A, Ahmed KA, Leary SC, Islam MF, et al. The critical role of ampkα1 in regulating autophagy and mitochondrial respiration in il-15-stimulated mtorc1(weak) signal-induced t cell memory: an interplay between yin (ampkα1) and yang (mtorc1) energy sensors in t cell differentiation. Int J Mol Sci (2022) 23(17). doi: 10.3390/ijms23179534
59. DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8+ T cell anti-tumor immunity. Cell Rep (2019) 27(2):502–13.e5. doi: 10.1016/j.celrep.2019.03.037
60. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458(7242):1131–5. doi: 10.1038/nature07976
61. Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J, et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function: PIK3C3/VPS34 in T cell metabolism and function. Autophagy (2021) 17(5):1193–204. doi: 10.1080/15548627.2020.1752979
62. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
63. Alsaleh G, Panse I, Swadling L, Zhang H, Richter FC, Meyer A, et al. Autophagy in T cells from aged donors is maintained by spermidine and correlates with function and vaccine responses. Elife (2020) 9:e57950. doi: 10.7554/eLife.57950.sa2
64. Chakraborty P, Parikh RY, Choi S, Tran D, Gooz M, Hedley ZT, et al. Carbon monoxide activates PERK-regulated autophagy to induce immunometabolic reprogramming and boost antitumor T-cell function. Cancer Res (2022) 82(10):1969–90. doi: 10.1158/0008-5472.CAN-21-3155
65. Alwarawrah Y, Danzaki K, Nichols AG, Fee BE, Bock C, Kucera G, et al. Irgm1 regulates metabolism and function in T cell subsets. Sci Rep (2022) 12(1):850. doi: 10.1038/s41598-021-04442-x
66. Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol (2020) 21(12):1540–51. doi: 10.1038/s41590-020-0793-3
67. Akatsuka H, Kuga S, Masuhara K, Davaadorj O, Okada C, Iida Y, et al. AMBRA1 is involved in T cell receptor-mediated metabolic reprogramming through an ATG7-independent pathway. Biochem Biophys Res Commun (2017) 491(4):1098–104. doi: 10.1016/j.bbrc.2017.08.019
68. Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci Immunol (2017) 2(17):eaan4631. doi: 10.1126/sciimmunol.aan4631
69. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science (2019) 363(6434):eaau0135. doi: 10.1126/science.aau0135
70. Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN, et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight (2019) 4(24). doi: 10.1172/jci.insight.129756
71. Becher J, Simula L, Volpe E, Procaccini C, La Rocca C, D'Acunzo P, et al. AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3-FOXP3 axis. Dev Cell (2018) 47(5):592–607.e6. doi: 10.1016/j.devcel.2018.11.010
72. Yang Z, Goronzy JJ, Weyand CM. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy (2014) 10(2):382–3. doi: 10.4161/auto.27345
73. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med (2013) 210(10):2119–34. doi: 10.1084/jem.20130252
74. Xi H, Kurtoglu M, Liu H, Wangpaichitr M, You M, Liu X, et al. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother Pharmacol (2011) 67(4):899–910. doi: 10.1007/s00280-010-1391-0
75. Matsuda F, Fujii J, Yoshida S-i. Autophagy induced by 2-deoxy-D-glucose suppresses intracellular multiplication of Legionella pneumophila in A/J mouse macrophages. Autophagy (2009) 5(4):484–93. doi: 10.4161/auto.5.4.7760
76. Foskolou IP, Barbieri L, Vernet A, Bargiela D, Cunha PP, Velica P, et al. The S enantiomer of 2-hydroxyglutarate increases central memory CD8 populations and improves CAR-T therapy outcome. Blood Adv (2020) 4(18):4483–93. doi: 10.1182/bloodadvances.2020002309
77. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristicsAkt inhibition improves T-cell antitumor immunity. Cancer Res (2015) 75(2):296–305. doi: 10.1158/0008-5472.CAN-14-2277
78. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi: 10.1038/nature08155
79. Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced therapeutic efficacy and memory of tumor-specific CD8 T cells by ex vivo PI3K-δ InhibitionPI3K-δ Inhibition enhances efficacy and memory CD8 T cells. Cancer Res (2017) 77(15):4135–45. doi: 10.1158/0008-5472.CAN-16-1925
80. Feng L, Liao H, Liu J, Xu C, Zhong K, Zhu H, et al. Inhibition of PI3K/Akt/mTOR pathway by ammonium chloride induced apoptosis and autophagy in MAC-T cell. Res Vet Sci (2021) 136:622–30. doi: 10.1016/j.rvsc.2021.01.020
81. Wang H, Dong R, Fan WW, Zheng XC, Li AM, Wang WD. Timosaponin A−III induces autophagy of T−cell acute lymphoblastic leukemia Jurkat cells via inhibition of the PI3K/Akt/mTOR pathway. Oncol Rep (2019) 41(5):2937–44. doi: 10.3892/or.2019.7072
82. Zhang Q, Ding J, Sun S, Liu H, Lu M, Wei X, et al. Akt inhibition at the initial stage of CAR-T preparation enhances the CAR-positive expression rate, memory phenotype and in vivo efficacy. Am J Cancer Res (2019) 9(11):2379.
83. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/S1074-7613(02)00323-0
84. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
85. Arnold CR, Pritz T, Brunner S, Knabb C, Salvenmoser W, Holzwarth B, et al. T cell receptor-mediated activation is a potent inducer of macroautophagy in human CD8+ CD28+ T cells but not in CD8+ CD28– T cells. Exp Gerontol (2014) 54:75–83. doi: 10.1016/j.exger.2014.01.018
86. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44(2):380–90. doi: 10.1016/j.immuni.2016.01.021
87. Choi BK, Lee DY, Lee DG, Kim YH, Kim SH, Oh HS, et al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cell Mol Immunol (2017) 14(9):748–57. doi: 10.1038/cmi.2016.02
88. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med (2018) 215(4):1091–100. doi: 10.1084/jem.20171068
89. Chen R, Xu Y, Zhong W, Li B, Yang P, Wang ZQ, et al. Activation of CD137 signaling enhances vascular calcification through c-Jun N-terminal kinase-dependent disruption of autophagic flux. Mediators Inflamm (2018) 2018:8407137. doi: 10.1155/2018/8407137
90. Li XY, Chen R, Zhong W, Li B, Shao C, Wang ZQ, et al. CD137 signaling promotes the formation of plaque calcification via inhibiting the fusion of autophagy and lysosomal in apo E(-/-) mice. Zhonghua Xin Xue Guan Bing Za Zhi (2017) 45(12):1078–85. doi: 10.3760/cma.j.issn.0253-3758.2017.12.013
91. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–842.e13. doi: 10.1016/j.cell.2016.09.031
92. García-Navas R, Munder M, Mollinedo F. Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy (2012) 8(11):1557–76. doi: 10.4161/auto.21315
93. Kishton RJ, Sukumar M, Restifo NP. Arginine arms T cells to thrive and survive. Cell Metab (2016) 24(5):647–8. doi: 10.1016/j.cmet.2016.10.019
94. Carrio R, Bathe OF, Malek TR. Initial antigen encounter programs CD8+ T cells competent to develop into memory cells that are activated in an antigen-free, IL-7-and IL-15-rich environment. J Immunol (2004) 172(12):7315–23. doi: 10.4049/jimmunol.172.12.7315
96. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory PhenotypeSuperior antitumor activity of CAR-T cells cultured in IL15. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.CIR-18-0466
97. Zhu L, Xie X, Zhang L, Wang H, Jie Z, Zhou X, et al. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat Commun (2018) 9(1):1–14. doi: 10.1038/s41467-018-05097-5
98. Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget (2018) 9(17):13125. doi: 10.18632/oncotarget.24442
99. Kato H, Perl A. The IL-21-mTOR axis blocks Treg differentiation and function by suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol (2018) 70(3):427-38. doi: 10.1002/art.40380
100. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor DesignMaking better chimeric antigen receptors. Cancer Discov (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
101. Sun R. POST-TRANSLATIONAL REGULATION OF 4-1BB, AN EMERGING TARGET FOR CANCER IMMUNOTHERAPY. Purdue University Graduate School Thesis (2022). doi: 10.25394/PGS.19653570.v1.
102. Petersen CT, Krenciute G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front Oncol (2019) 9:69. doi: 10.3389/fonc.2019.00069
103. Jenkins Y, Zabkiewicz J, Ottmann O, Jones N. Tinkering under the hood: Metabolic optimisation of car-t cell therapy. Antibodies (2021) 10(2):17. doi: 10.3390/antib10020017
104. Xu A, Bhanumathy KK, Wu J, Ye Z, Freywald A, Leary SC, et al. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci (2016) 6(1):30. doi: 10.1186/s13578-016-0098-2
105. Zhang S, Zhao J, Bai X, Handley M, Shan F. Biological effects of IL-15 on immune cells and its potential for the treatment of cancer. Int Immunopharmacol (2021) 91:107318. doi: 10.1016/j.intimp.2020.107318
106. Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the metabolism to improve the efficacy of CAR T-cell immunotherapy. Cells (2020) 10(1):14. doi: 10.3390/cells10010014
107. Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T cell in adoptive cancer immunotherapy: challenges and ways forward. Front Immunol (2020) 11:702. doi: 10.3389/fimmu.2020.00702
108. Zheng W, O'Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia (2018) 32(5):1157–67. doi: 10.1038/s41375-017-0008-6
109. Semenza GL. Mitochondrial autophagy: life and breath of the cell. Autophagy (2008) 4(4):534–6. doi: 10.4161/auto.5956
110. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol (2012) 188(9):4305–14. doi: 10.4049/jimmunol.1103568
111. Urak R, Walter M, Lim L, Wong CW, Budde LE, Thomas S, et al. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J Immunother Cancer (2017) 5(1):1–13. doi: 10.1186/s40425-017-0227-4
112. Shen L, Xiao Y, Tian J, Lu Z. Remodeling metabolic fitness: Strategies for improving the efficacy of chimeric antigen receptor T cell therapy. Cancer Lett (2022) 529:139–52. doi: 10.1016/j.canlet.2022.01.006
113. Wang H, Bai G, Chen J, Han W, Guo R, Cui N. mTOR deletion ameliorates CD4 + T cell apoptosis during sepsis by improving autophagosome-lysosome fusion. Apoptosis (2022) 27(5-6):401–8. doi: 10.1007/s10495-022-01719-y
114. Maiti A, Daver NG. Lowering mTORC1 drives CAR T-cells home in acute myeloid leukemia. Clin Cancer Res (2021) 27(21):5739–41. doi: 10.1158/1078-0432.CCR-21-2574
115. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest (2014) 124(1):188–97. doi: 10.1172/JCI69856
116. Mu Q, Jiang M, Zhang Y, Wu F, Li H, Zhang W, et al. Metformin inhibits proliferation and cytotoxicity and induces apoptosis via AMPK pathway in CD19-chimeric antigen receptor-modified T cells. OncoTargets Ther (2018) 11:1767. doi: 10.2147/OTT.S154853
117. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi: 10.1016/j.immuni.2014.12.030
118. Carleton G, Lum JJ. Autophagy metabolically suppresses CD8+ T cell antitumor immunity. Autophagy (2019) 15(9):1648–9. doi: 10.1080/15548627.2019.1628545
119. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci (2019) 15(12):2548. doi: 10.7150/ijbs.34213
120. Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev (2011) 25(14):1510–27. doi: 10.1101/gad.2051011
121. Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell (2022) 40(2):136–152.e12. doi: 10.1016/j.ccell.2021.12.009
122. Levy J, Cacheux W, Bara MA, L'Hermitte A, Lepage P, Fraudeau M, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat Cell Biol (2015) 17(8):1062–73. doi: 10.1038/ncb3206
123. Wang Z-H, Peng WB, Zhang P, Yang XP, Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine (2021) 73:103627. doi: 10.1016/j.ebiom.2021.103627
124. Wang Y, Wu L, Tian C, Zhang Y. PD-1-PD-L1 immune-checkpoint blockade in Malignant lymphomas. Ann Hematol (2018) 97(2):229–37. doi: 10.1007/s00277-017-3176-6
125. Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J (Sudbury Mass) (2014) 20(4):265. doi: 10.1097/PPO.0000000000000059
126. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
127. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer (2019) 18(1):1–17. doi: 10.1186/s12943-018-0928-4
128. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
129. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev (2010) 236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x
130. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature (2016) 529(7584):37–42. doi: 10.1038/nature16187
131. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
132. Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest (2010) 120(1):127–41. doi: 10.1172/JCI40027
Keywords: CAR T cell, metabolism, autophagy, tumor microenvironment, adoptive cellular therapy (ACT)
Citation: Panahi Meymandi AR, Akbari B, Soltantoyeh T, Hadjati J, Klionsky DJ, Badie B and Mirzaei HR (2023) Crosstalk between autophagy and metabolic regulation of (CAR) T cells: therapeutic implications. Front. Immunol. 14:1212695. doi: 10.3389/fimmu.2023.1212695
Received: 26 April 2023; Accepted: 04 August 2023;
Published: 22 August 2023.
Edited by:
Mingzhu Zheng, Southeast University, ChinaCopyright © 2023 Panahi Meymandi, Akbari, Soltantoyeh, Hadjati, Klionsky, Badie and Mirzaei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Behnam Badie, YmJhZGllQGNvaC5vcmc=; Hamid Reza Mirzaei, aC1taXJ6YWVpQHR1bXMuYWMuaXI=